Mitochondria-Associated Endoplasmic Reticulum Membranes in Breast Cancer
 Hongjiao Yu
Hongjiao Yu Chaonan Sun
Chaonan Sun Qing Gong
Qing Gong Du Feng2*
Du Feng2*- 1Department of Biochemistry and Molecular Biology, Guangzhou Medical University-Guangzhou Institutes of Biomedicine and Health (GMU-GIBH) Joint School of Life Sciences, Guangzhou Medical University, Guangzhou, China
- 2Guangzhou Municipal and Guangdong Provincial Key Laboratory of Protein Modification and Degradation, State Key Laboratory of Respiratory Disease, School of Basic Medical Sciences, Guangzhou Medical University, Guangzhou, China
Mitochondria-associated ER membranes (MAMs) represent a crucial intracellular signaling hub, that regulates various cellular events including Ca2+ homeostasis, lipid metabolism, mitochondrial function, and cellular survival and death. All of these MAM-mediated cellular events contribute to carcinogenesis. Indeed, altered functions of MAMs in several types of cancers have been documented, in particular for breast cancer. Over the past years, altered expression of many MAM-resident proteins have been reported in breast cancer. These MAM-resident proteins play an important role in regulation of breast cancer initiation and progression. In the current review, we discuss our current knowledge about the functions of MAMs, and address the underlying mechanisms through which MAM-resident proteins regulate breast cancer. A fuller understanding of the pathways through which MAMs regulate breast cancer, and identification of breast cancer-specific MAM-resident proteins may help to develop novel therapeutic strategies for breast cancer.
Introduction
The interconnection between mitochondria and endoplasmic reticulum was the first inter-organelle contact site discovered in the late 1950's (Bernhard and Rouiller, 1956). Several subsequent fractionation studies have successfully isolated mitochondria-associated endoplasmic reticulum membranes (MAMs) as a distinct and purified structure from liver cells (Wanson et al., 1975; Pickett et al., 1981; Katz et al., 1983). These elegant studies provide the experimental evidence for the existence of MAMs. Both of endoplasmic reticulum (ER) and mitochondria play crucial roles in regulation of cellular homeostasis and cell death. The discovery of MAMs therefore leads to a growing research interest in characterization of the biological function of MAMs in cells. Over the past few decades, extensive researches have reported that MAMs act as signaling hubs that control ER and mitochondrial biology, and disturbances of MAMs result in dysfunctions in a wide range of cellular processes including apoptosis, inflammation, and autophagy (Morciano et al., 2018). These cellular activities have crucial roles in several pathologies including but not limited to cardiovascular disease and cancer. Recently, we have extensively reviewed the functional and mechanistic roles of MAMs in inflammation and cardiovascular disease (Liu et al., 2020). In the current review, we will highlight the multiple physiological functions of MAMs in mitochondrial Ca2+ signaling, metabolism and autophagy, and discuss how alterations in MAMs contribute to carcinogenesis.
Structure and Biochemical Composition of MAMs
It is now widely accepted that MAM are 10–25 nm wide regions of juxtaposed membranes of ER and mitochondria tethered by proteins (Csordas et al., 2006), without complete fusion or loss of organelle identity. The biological functions of the MAMs are tightly regulated by the number, length and thickness of contacts between ER and mitochondria. The technique of biochemical purification of MAMs from different mammalian tissues and cultured cells has been greatly improved in the past years, and allows subsequent analysis of the composition of MAMs. The enrichment of several proteins including acyl-coenzyme A: cholesterol acyltransferase 1 (ACAT1/SOAT1), and the existence of Ca2+ nanodomains in MAMs have been identified (Rusinol et al., 1994; Vance, 2014). These data suggest that MAMs may have critical roles in regulation of lipid synthesis and Ca2+ trafficking. Since then, several studies have used high-throughput proteomics to identify novel proteins present in MAM purified from different cellular models (Poston et al., 2013; Cho et al., 2017; Hung et al., 2017). However, only two proteins, thioredoxin-related transmembrane protein (TMX1) and calnexin are commonly seen in the MAMs fraction from these studies (Simmen and Herrera-Cruz, 2018). It should be noted that not all proteins localized in the MAMs can be identified in the dynamic cellular environment. Nevertheless, a better characterization of the cell-specific MAM proteome with more novel techniques will definitely increase our current knowledge of the biochemical composition and biological functions of MAMs. Besides, Yang and colleagues presented a novel technique to detect MAM distribution and dynamics using split GFP protein as a reporter for labeling the ER membrane and mitochondrial outer membrane (Yang et al., 2018).
Biological Functions of MAMs
In the past years, accumulating evidences have increased our knowledge of inter-organellar communication in the regulation of key cellular processes, such as metabolism and cell death. It is now widely recognized that the interaction between mitochondria and ER through the MAMs is critical in the various aspects of cellular health. In the next sections, we will discuss some of the vital cellular functions and biological processes regulated by MAMs.
MAMs in Regulation of Ca2+ Signaling
Ca2+ is an important intracellular second messenger, which is required for functional mitochondrial metabolism, bioenergetics and cell survival. MAMs have been shown to regulate a range of cell metabolism and cell fate determination through modulating cellular Ca2+ signaling (Figure 1, Rizzuto et al., 2009). ER is the main intracellular Ca2+ storage organelle in the cells. The tight tether between the ER membranes and mitochondria allows Ca2+ to be rapidly transferred between the two intracellular organelles (Rizzuto et al., 1998; Filippin et al., 2003). Indeed, the accumulation of Ca2+ in mitochondria is largely dependent on ER. Intriguingly, many ER associated Ca2+ handling proteins are enriched in MAMs, including inositol 1,4,5-trisphosphate receptors (IP3R) and the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA). The IP3R together with the ryanodine receptors have been reported to be the principal ER Ca2+ channel (Parys and De Smedt, 2012). It appears that isoform 3 of the IP3Rs co-localizes most strongly with MAMs and acts as a primary regulator for transmitting Ca2+ signals to mitochondria (Mendes et al., 2005). It has been reported that the IP3Rs directly interact with the mitochondrial voltage-dependent anion channel 1 (VDAC1) located at the outer mitochondrial membrane (OMM) and associated to the MAMs (Szabadkai et al., 2006). The molecular chaperone glucose-regulated protein 75 (GRP75) regulates the interaction between VDAC1 and the IP3Rs, and it serves as a bridge which allows the transfer of Ca2+ from the ER to the Ca2+ permeable OMM. Unlike OMM, the inner mitochondrial membrane (IMM) is not permeable for Ca2+. Ca2+ located in the intermembrane space (IMS) is transferred to the mitochondrial matrix through the mitochondrial Ca2+ uniporter (MCU) (Baughman et al., 2011; De Stefani et al., 2011). The MCU complex is a multi-protein complex consisting of MCU, MCUb, EMRE, MICU1 and 2, MCUR1 and SLC25A3 (Mammucari et al., 2018). Each partner of the MCU multi-protein complex tightly controls Ca2+ concentration in the mitochondrial matrix, thereby regulating cell survival, death and metabolism.
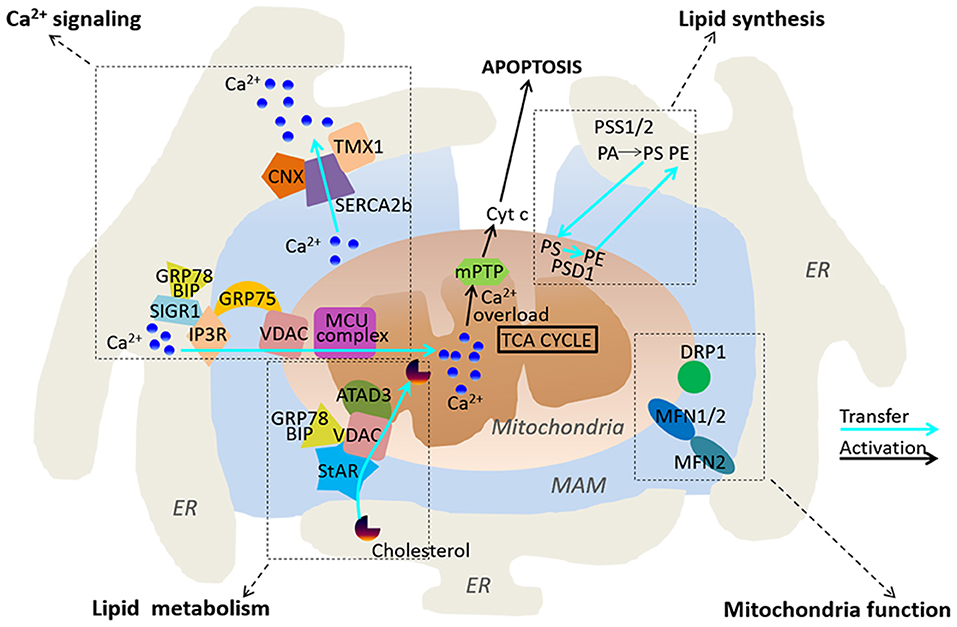
Figure 1. Functional roles of MAMs. MAMs play regulatory roles in Ca2+ signaling, lipid synthesis and metabolism, and mitochondria functions.
The SERCA pump localized in the ER membrane regulates ER Ca2+ levels through actively pumping Ca2+ into the ER from cytosol, thereby leading to a Ca2+ gradient between cytosol and the ER. Interestingly, SERCA2b, an isoform of SERCA, exhibits the highest Calcium ions affinity, and is enriched in MAMs. It has been reported that calnexin and TMX1 regulate SERCA2b activity through a direct interaction with SERCA2b in a palmitoylation-dependent manner. Palmitoylation of calnexin switches its function from quality control of protein folding to ER-Ca2+ signaling by increasing the activity of SERCA2b, whereas TMX1 counteracts the interaction between Calnexin and SERCA2b, which inhibits SERCA2b and promotes Ca2+ influx to mitochondria (Gutierrez and Simmen, 2018). Another important protein present in MAMs is Mitofusin-2 (MFN2). It belongs to the GTPase protein family and plays a crucial role in regulation of mitochondrial fusion. MFN regulates the stability of ER-mitochondria interaction, Ca2+ and lipid transfer. It has been reported that loss of MFN2 reduces ER-mitochondria contact sites, and leads to impaired mitochondrial Ca2+ uptake (De Brito and Scorrano, 2008). However, there are discrepant results reporting that reduced or ablated MFN2 expression increases numbers of ER–mitochondria close contacts, and promote Ca2+ transfer between the two organelles (Filadi et al., 2015). The precise role of MFN2 in ER-mitochondria interaction therefore remains to be further investigated.
MAMs Act as an Important Regulator of Lipid Synthesis and Metabolism
ER is the primary site for lipid synthesis. MAMs tightly regulate the lipid trafficking between ER and mitochondria (Figure 1). A number of proteins associated with lipid metabolism are enriched in MAMs, including phosphatidylserine synthases (PSS1 and 2), which is required for the synthesis of essential lipids such as phosphatidylserine (PS) and phosphatidylcholine (PC) (Futerman, 2006; Van Meer et al., 2008). The transfer of PS from ER to mitochondria via the MAMs is a key rate-limiting step in phosphatidylethanolamine (PE) synthesis. Previous studies have reported that reduced phosphatidylserine decarboxylase (PSD) activity causes accumulation of PS in MAMs (Voelker, 1984). These data indicate that MAMs play an important role in the transfer of PS between the ER and mitochondria. The long chain acyl-coA synthetase (ACSL4/FACL4) is a common marker used for characterization of MAMs. It regulates the ligation of fatty acids to coenzyme A and other cholesterol metabolites (Lewin et al., 2001). It has been previously reported that ACSL4 is localized in MAMs, and plays a key role in regulation of mitochondrial fusion during steroidogenesis in Leydig cells (Poderoso et al., 2013). In addition, cholesterol relative to the bulk of ER are enriched in MAMs and dysregulation of MAM cholesterol composition alters ER-mitochondria contacts (Fujimoto et al., 2012). Intriguingly, several enzymes that are associated with cholesterol metabolism and transport are found in MAMs, including ACAT1/SOAT1, and steroidogenic acute regulatory protein (StAR) (Rusinol et al., 1994; Prasad et al., 2015). Caveolin-1 (CAV1) is an important regulator of cholesterol intracellular transport and membrane organization, and it has been recently identified as a key component of MAMs. Genetic deletion of Cav1 in mice reduces the stability of ER-mitochondria contact sites and an accumulation of free cholesterol in MAMs (Sala-Vila et al., 2016). Altogether, MAM appears to form a key molecular platform that plays an important role in regulation of lipid synthesis and metabolism.
MAMs Regulates Mitochondria Function
Mitochondria morphology and dynamics are crucial in regulation of its diverse activity including mitochondrial respiration and metabolism, Ca2+ homeostasis and apoptosis. In response to various stimuli or stress, the morphology, mobility, and fusion-fission equilibrium of mitochondria are dramatically changed (Eisner et al., 2018). The underlying molecular mechanisms of mitochondrial fusion and fission have been extensively reviewed elsewhere (Wai and Langer, 2016), and will not be discussed in detail in this review.
As a microdomain between ER and mitochondria, MAMs not only regulate lipid synthesis and metabolism in ER, but also play a critical role in regulation of mitochondrial function. Indeed, several previous studies have revealed MAMs as a key regulator of mitochondrial dynamic and morphology (Figure 1). Dynamin-related protein 1 (DRP1) and MFN2 are well-established regulators of mitochondrial shape and fusion/fission balance. These two proteins have been found to be enriched in MAMs (De Brito and Scorrano, 2008; Saotome et al., 2008). As a most well-studied MAM-resident protein, MFN2 has been reported to regulate MAM structure and function. MFN2 locates at both OMM and the ER membranes, and it co-works with the OMM-associated MFN1 to establish homotypic and heterotypic ER–mitochondria interactions. Mitochondrial fission is induced at ER-mitochondria contact sites where ER tubules physically wrap around a part of the mitochondrial network and promotes mitochondria fission through the recruitment of DRP1 on the OMM (Friedman et al., 2011). The Miro family of proteins Miro1 and 2 are the master regulators of mitochondrial motility. They are located at the OMM and harbor two EF-hand Ca2+ binding domains, thereby sensing high levels of Ca2+ to regulate mitochondrial motility (Saotome et al., 2008). Miro1 and 2 have been shown to localize at the ER–mitochondria contact sites, where they sense local Ca2+ to connect the mitochondria to kinesin 1 (Fransson et al., 2006; Saotome et al., 2008).
The Ca2+ concentration in mitochondria plays a fundamental role in regulation of ATP production, which is required for a wide range of cellular processes. The Krebs Cycle, also known as the citric acid cycle, occurs within the mitochondria, which produces ATP through oxidative phosphorylation. The activities of key dehydrogenases of the Krebs Cycle are dependent on Ca2+, including the pyruvate dehydrogenase, the α-ketoglutarate, the isocitrate dehydrogenases and FAD-glycerol phosphate dehydrogenase (Hansford and Chappell, 1967; Rizzuto et al., 2004; Szabadkai and Duchen, 2008). The rapid exchange of Ca2+ in MAMs therefore has a critical role in regulation of mitochondrial bioenergetics, and subsequently cell fate determination (Cardenas et al., 2010). This view has been supported by several lines of evidences. Loss or gain-of-function studies have reported that MFN2, a key regulator of the ER–mitochondria contacts, significantly alters mitochondrial bioenergetics, which is independent of fusogenic function of MFN2 (Schrepfer and Scorrano, 2016). Deletion of MFN2 reduces several mitochondrial activities such as glucose oxidation, cellular respiration, mitochondrial membrane potential and proton leak, and mitochondrial coenzyme Q levels (Mourier et al., 2015), while overexpression of MFN2 increases mitochondrial metabolism (Lewis et al., 2016). Interestingly, MFN2 also transiently protects against starvation-induced autophagy and apoptosis through inducing mitochondrial hyperfusion and elongation (Lewis et al., 2016).
MAMs and Cell Death
Although transportation of Ca2+ in MAMs benefits to mitochondrial bioenergetics, massive and prolonged mitochondrial Ca2+ overload can open the mitochondrial permeability transition pore (Szabadkai and Duchen, 2008). As a result, proapoptotic and caspase-activating factors in mitochondria are released in the cytoplasm such as including cytochrome c (Cyt c). Cytochrome c in the cytoplasm exacerbates Ca2+ release from IP3R by binding to it, thus avoiding the Ca2+-dependent inhibition of the receptor and amplifying caspase activation to promote apoptosis (Figure 1, Boehning et al., 2003). Further studies have demonstrated that tightening of the ER–mitochondria interaction increases mitochondrial Ca2+ uptake, leading to apoptotic cell death, while loosening of the ER–mitochondria interaction promotes mitochondrial respiration and cellular survival (Csordas et al., 2006; Marchi et al., 2014). MAMs may also induce apoptosis or ferroptosis through increasing reactive oxygen species (ROS), and subsequent the accumulation of lipid peroxidation products in cells (Verfaillie et al., 2012; Yang et al., 2014).
The proapoptotic BCL2 protein family members such as BAK and BAX mediate Ca2+ traffic from ER to mitochondria and exert a key role in regulating cell death (Scorrano et al., 2003). Over the past years an increasing amount of cancer-related proteins have been reported to be abundant in MAMs and to exert their antiapoptotic or proapoptotic functions through the manipulation of Ca2+ transfer and signaling between ER and mitochondria. IP3R, which plays a central role in the regulation of ER-mitochondria Ca2 transfer, is also a target under control of several oncogenes and tumor suppressors (Bittremieux et al., 2016). One of the main regulators of IP3R, the Akt serine threonine kinase, preferentially phosphorylates IP3Rs for its isoform 3 (IP3R3) in MAMs and blocks apoptosis by inhibition of IP3R-mediated ER-mitochondria Ca2+ release (Szado et al., 2008; Marchi et al., 2012). The antiapoptotic function of Akt is regulated by its inhibitors (PTEN and PML) and its mTORC2 activator, which are also enriched in MAM. The PTEN (phosphatase and tensin homolog) tumor suppressor is the dominant inhibitor of PI3K/AKT signaling pathways while the promyelocytic leukemia protein tumor suppressor (PML) functions in several apoptotic pathways. mTORC2/Akt at MAMs control apoptosis through interaction with the IP3R-Grp75–VDAC complex and regulation of IP3R3 phosphorylation and Ca2+ efflux from the ER to mitochondria (Betz et al., 2013).
The oncogenic transcription factor STAT3 has recently been determined enriched in ER and MAM. Constitutively active STAT3 induces cell resistance to apoptosis via reduced ER-mitochondria Ca2+ transfer and IP3R3 degradation mediated by IP3R3/STAT3 interaction (Avalle et al., 2019). The transcription factor p53 is one of the major tumor suppressors. It has been recently revealed that localization in ER and MAM enables p53 to mediate Ca2+ dependent apoptosis independent of its DNA-binding domain through interaction with SERCA. This interaction induces a change in SERCA oxidative state and facilitates Ca2+ uptake in the ER and ultimately impacts on sensitivity to apoptosis under stress conditions (Giorgi et al., 2015).
The long isoform of the cellular FLICE-inhibitory protein (c-FLIPL) has also been reported to localize at ER and MAM. As a well-known inhibitor of the extrinsic cell death initiator caspase-8, ablation of c-FLIPL disrupts ER-mitochondria juxtaposition, reducing ER-mitochondria Ca2+ transfer and affecting sensitivity to Ca2+ dependent apoptosis (Marini et al., 2015).
MAM appears as a crucial platform in autophagosome formation and plays an important role in autophagy. During starvation-induced autophagy, proteins involved in the autophagic machinery process such as ATG5 and ATG14, together with critical proteins involved in autophagosome formation such as Vps34 and Beclin 1, are recruited to MAM fraction (Hamasaki et al., 2013). Beclin 1 belongs to the class III phosphatidylinositol 3 kinase complex (PI3K complex III) and is essential for autophagy induction as well as autophagosome formation. Function of Beclin 1 is tightly regulated through direct binding of autophagy inducing or inhibiting regulators, such as autophagy/Beclin 1 regulator 1 (AMBRA1) and BCL2 (Kang et al., 2011). Interestingly, Ca2+ signaling appears to act as an activator and meanwhile as an inhibitor of autophagy (Bootman et al., 2018). IP3R is a key negative regulator of autophagy which is directly targeted by Beclin 1 and promotes the formation of a complex with Beclin 1/BCL2. The pool of soluble Beclin 1 protein for autophagy induction is therefore decreased by the BCL2-mediated sequestration of Beclin 1 (Vicencio et al., 2009). However, further studies demonstrated that IP3R enhances autophagic pathway through formation of IP3R/Beclin 1 complex. After starvation, increased Beclin 1 interacts with IP3R and promotes IP3R-mediated Ca2+ signaling by enhanced ER-Ca2+ stores content through upregulation of GRP78/BiP and calreticulin (Decuypere et al., 2011).
MAM also plays a critical role in inflammation pathways through recruitment of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) to the MAM faction from cytosol. Stimulus including extracellular ATP and impaired Ca2+ flux from ER to mitochondria trigger the production of ROS and ultimately activate the NLRP3 inflammasome. Activated NLRP3 inflammasome recruits apoptosis-associated speck-like protein containing a CARD (ASC) and relocates to the MAM fraction, which finally results in caspase 1-dependent release of proinflammatory cytokines IL-1β and IL-18 and leads to Gasdermin D-driven pyroptotic cell death (Tschopp, 2011). Details on NLPR3 activation and regulation have been excellently reviewed elsewhere and will not be discussed here (Missiroli et al., 2018; Swanson et al., 2019).
Functional Alterations of MAMs and its Regulatory Role in Breast Cancer
As discussed above, given the fact that MAMs play critical roles in regulation of cell homeostasis, survival and apoptosis, it is not surprising that MAMs are functionally altered during carcinogenesis. Indeed, several tumor suppressors or oncogenes have been found to target MAMs to regulate carcinogenesis. Here, we discuss the emerging evidence showing the critical roles of MAMs in breast cancer development.
Breast cancer is one of the most common cancers in the world. MAMs have critical roles in the onset of breast cancer, which reprogram normal cell signaling toward malignancy. Abnormal expression and localization of MAM-resident proteins have been widely reported in breast cancer (Figure 2). For example, the stress-activated chaperone sigma-1 receptor (Sig1R) at the ER-mitochondria interface has been reported to be expressed higher in metastatic potential cancer cells compared to that in normal tissues (Aydar et al., 2006; Gueguinou et al., 2017). It has been previously reported that Sig1R binds the MAM chaperone BiP/GRP78 under physiological conditions. However, Sig1R dissociates from BiP and binds IP3R3 upon activation of IP3Rs, which stabilize IP3R3 at the MAMs and enhance IP3R3-mediated Ca2+ fluxes to the mitochondria to regulate cell survival (Hayashi and Su, 2007). Importantly, in response to chronic ER stress involving prolonged ER Ca2+ depletion, Sig1R can translocate from MAMs to the peripheral ER, causing reduced cell damage and death (Hayashi and Su, 2003, 2007). In addition, Sig1R also involves in cell invasiveness in breast cancer. Sig1R can form a functional molecular platform with the calcium-activated K+ channel SK3 and Orai1, which drives Ca2+ influx to promote the migration of cancer cells (Gueguinou et al., 2017).
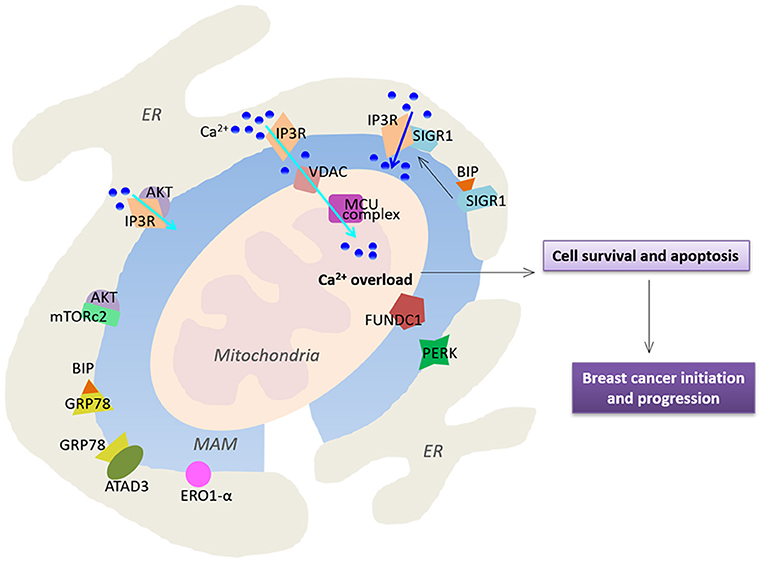
Figure 2. Functional alterations of MAMs in breast cancer. MAMs-resident proteins play a crucial role in regulation of Ca2+ homeostasis, thereby affecting cell survival and apoptosis. Cyan arrows highlight Ca2+ transport while the blue arrow indicates a larger Ca2+ fluxes. See text for further details.
As mentioned before, MAMs have critical roles in regulation of Ca2+ signaling in cells. Indeed, Ca2+ fluxes toward the mitochondria via MCU are crucial for tumor growth and metastatic behavior. It has been previously reported that silencing of MCU inhibits cell migration and invasion. In triple-negative breast cancer, MCU prevents tumor progression in MDA-MB-231 xenografts and regulates metastasis via a key regulator hypoxia-inducible factor 1 (HIF1) which controls gene reprogramming (Tosatto et al., 2016)
Furthermore, IP3R3 is highly enriched in MAMs, and is recognized as a MAM marker (Wieckowski et al., 2009). IP3R3 transmits Ca2+-mediated proapoptotic signals to the mitochondria, and also plays an important role in regulation of cellular bioenergetics and metabolism in breast cancer (Mendes et al., 2005). Inhibition of IP3R3 expression leads to autophagic breast cancer cell death, and mitotic catastrophe in tumorigenic cells but not in normal cells (Bultynck, 2016; Cardenas et al., 2016; Singh et al., 2017a). Corroborating with these observations, higher expression of IP3R3 in human malignant tissues and higher concentrations of metabolites in serum samples have been reported in breast cancer patients (Singh et al., 2017b). On the other hand, IP3R3 function is also regulated by a wider range of oncogenes and tumor suppressors such as the oncogene Akt kinase (Khan et al., 2006; Szado et al., 2008). Altered PI3K/Akt/mTOR pathway is frequently seen in human breast cancers (Stemke-Hale et al., 2008; Gonzalez-Angulo et al., 2011). Selective phosphorylation of IP3R3 by AKT reduces ER-mitochondria Ca2+ transfer and attenuates apoptotic responses (Marchi et al., 2012). The mechanistic TOR complex 2 (mTORc2) is located in MAMs, where it phosphorylates Akt at position S473 (Sarbassov et al., 2005; Betz et al., 2013). The essential role of mTORc2-Akt signaling in maintaining proper MAM functionality has been demonstrated. Loss of mTORc2 induces disruption of MAM architecture and mitochondrial defects (Betz et al., 2013). Consistently, it has been reported that expression of the mTORc2 core component Rictor is significantly up-regulated in invasive breast cancer specimens, which promotes Akt-dependent tumor progression in HER2-amplified breast cancers (Morrison Joly et al., 2016).
The role of MAM chaperone BiP/GRP78 in the development of breast cancer has also been reported. ATAD3a is a mitochondria and MAM resident protein with unknown function. GRP78 has been reported to cooperate with ATAD3a to stabilizing WASF3, a protein that facilitates actin polymerization, thereby enhancing invasion and metastasis (Teng et al., 2016). ATAD3a regulates ER-mitochondria contact site formation and cholesterol substrate delivery to the mitochondria (Issop et al., 2015). Importantly, dysregulation in lipid composition of MAMs results in severe consequences, affecting Ca2+ homeostasis, ER-mitochondria contact sites, and mitochondrial functions (Vance, 2014), which are known to regulate cell apoptosis and tumors. Importantly, a number of previous studies have reported that enhanced lipogenesis in cancer cells promotes mass growth (Zaidi et al., 2013).
Endoplasmic reticulum oxidoreductin 1-α (ERO1-α) is an oxidizing enzyme that is enriched in MAMs (Anelli et al., 2012). ERO1-α controls oxidative folding and ER redox homeostasis, and regulates ER Ca2+ flux and subsequent mitochondrial Ca2+ accumulation, which has been reported to be highly expressed in several types of cancer (Kakihana et al., 2012). Notably, ERO1-α has been reported to be a novel predictor for poor prognosis of breast cancer (Kutomi et al., 2013). ERO1-α mediated ER-mitochondria Ca2+ flux activates the procaspase activating compound-1 (PAC-1) to induce ER stress and mitochondrial permeabilization, thereby promoting apoptosis in a variety of cancer cell types (Seervi et al., 2013). Interestingly, it has been reported that ERO1-α expression is correlated with programmed cell death ligand 1 (PD-L1) in triple-negative breast cancer cells. Ablation of ERO1-α causes significantly reduced PD-L1–mediated T-cell apoptosis, indicating ERO1-α may be involved in tumor-mediated immunosuppression (Tanaka et al., 2017).
RNA-dependent protein kinase (PKR)–like ER kinase (PERK), a critical ER stress sensor at MAMs, has been shown to play a key role in maintaining the ER-mitochondria juxtaposition and ROS-mediated mitochondrial apoptosis (Verfaillie et al., 2012; Van Vliet et al., 2017). PERK-dependent signaling involves in tumor initiation and expansion to preserve redox homeostasis and promote tumor growth in MDA-MB-468 and T47D cell lines (Bobrovnikova-Marjon et al., 2010). Silencing of PERK expression reduces tumor growth and restores sensitivity to chemotherapy in resistant tumor xenografts (Salaroglio et al., 2017).
FUN14 domain-containing protein 1 (FUNDC1) is a novel outer mitochondrial membrane protein localized in MAMs (Wu et al., 2016, 2017). It regulates the formation of MAMs and elicits Ca2+ release from ER into mitochondria and cytosol in mouse cardiomyocytes (Wu et al., 2017). Several studies have shown that overexpression of FUNDC1 induces mitophagy in various cancer cell lines (Hirota et al., 2015; Hui et al., 2019; Roperto et al., 2019). Interestingly, a recent study has reported that FUNDC1 enhances breast cancer proliferation and migration via Ca2+ influx-mediated NFATC1 activation and translocation into the nucleus to activate BMI1 transcription (Wu et al., 2019). Notably, FUNDC1 expression is positively associated with worse disease progression in breast cancer (Wu et al., 2019).
Conclusion and Future Perspectives
In conclusion, MAMs are critical regulators of intracellular signaling pathways and regulate various biological functions such as Ca2+ homeostasis, lipid metabolism, mitochondrial function, and cell death. Over the past years, targeting of cell death pathways is a commonly used therapeutic strategy in the treatment of breast cancer. Cell-death-pathway-involved proteins in MAMs such as BCL2, p53 tumor suppressor, NLRP3 and their related proteins have been widely investigated for prognosis and targeted anti-cancer therapy. With increasing evidences support a critical role of MAMs in the tumorigenesis of breast cancer, understanding the mechanisms and functions of the mitochondria-ER interface are particularly important and urgent. It is likely that there might be different ER-mitochondria contact sites in different types of cells, which leaves many unanswered questions. One crucial question is that whether there are properties of MAM-resident proteins that are uniquely associated with one specific type of cancer.
Functional MAM alterations appear to represent a new therapeutic strategy to target cancer cells by suppressing their survival and invasion, however, it will be a major challenge to limit these effects to cancer cells without affecting normal cells, identification of potential breast cancer-specific MAM proteins is therefore of decisive importance for the accurate therapeutic target in cancer treatment. Specific targeting of cancer-altered MAM function, dynamics or structures also open new perspectives toward other types of human metabolic disorders that involve MAM pathways.
Author Contributions
As the first authors, HY and CS make the same important contributions to design concept, collected and analyzed materials, wrote articles and drew. DF and QG make the final review and finalization of the articles to be published as the corresponding authors. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Guangzhou Municipal Science and Technology Project (No. 201904010067); Natural Science Foundation of Guangdong Province (No. 2018A030313560, 2020A1515010009).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anelli, T., Bergamelli, L., Margittai, E., Rimessi, A., Fagioli, C., Malgaroli, A., et al. (2012). Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 16, 1077–1087. doi: 10.1089/ars.2011.4004
Avalle, L., Camporeale, A., Morciano, G., Caroccia, N., Ghetti, E., Orecchia, V., et al. (2019). STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca(2+) fluxes and apoptotic responses. Cell Death Differ 26, 932–942. doi: 10.1038/s41418-018-0171-y
Aydar, E., Onganer, P., Perrett, R., Djamgoz, M. B., and Palmer, C. P. (2006). The expression and functional characterization of sigma (sigma) 1 receptors in breast cancer cell lines. Cancer Lett. 242, 245–257. doi: 10.1016/j.canlet.2005.11.011
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Bernhard, W., and Rouiller, C. (1956). Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J. Biophys. Biochem. Cytol. 2, 73–78. doi: 10.1083/jcb.2.4.73
Betz, C., Stracka, D., Prescianotto-Baschong, C., Frieden, M., Demaurex, N., and Hall, M. N. (2013). Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. U.S.A. 110, 12526–12534. doi: 10.1073/pnas.1302455110
Bittremieux, M., Parys, J. B., Pinton, P., and Bultynck, G. (2016). ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca(2+) signaling. Biochim. Biophys. Acta 1863, 1364–1378. doi: 10.1016/j.bbamcr.2016.01.002
Bobrovnikova-Marjon, E., Grigoriadou, C., Pytel, D., Zhang, F., Ye, J., Koumenis, C., et al. (2010). PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29, 3881–3895. doi: 10.1038/onc.2010.153
Boehning, D., Patterson, R. L., Sedaghat, L., Glebova, N. O., Kurosaki, T., and Snyder, S. H. (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 5, 1051–1061. doi: 10.1038/ncb1063
Bootman, M. D., Chehab, T., Bultynck, G., Parys, J. B., and Rietdorf, K. (2018). The regulation of autophagy by calcium signals: do we have a consensus? Cell Calcium 70, 32–46. doi: 10.1016/j.ceca.2017.08.005
Bultynck, G. (2016). Onco-IP3Rs feed cancerous cravings for mitochondrial Ca(2.). Trends Biochem. Sci. 41, 390–393. doi: 10.1016/j.tibs.2016.03.006
Cardenas, C., Miller, R. A., Smith, I., Bui, T., Molgo, J., Muller, M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. doi: 10.1016/j.cell.2010.06.007
Cardenas, C., Muller, M., Mcneal, A., Lovy, A., Jana, F., Bustos, G., et al. (2016). Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep. 14, 2313–2324. doi: 10.1016/j.celrep.2016.02.030
Cho, I. T., Adelmant, G., Lim, Y., Marto, J. A., Cho, G., and Golden, J. A. (2017). Ascorbate peroxidase proximity labeling coupled with biochemical fractionation identifies promoters of endoplasmic reticulum-mitochondrial contacts. J. Biol. Chem. 292, 16382–16392. doi: 10.1074/jbc.M117.795286
Csordas, G., Renken, C., Varnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
De Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De Stefani, D., Raffaello, A., Teardo, E., Szabo, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Decuypere, J. P., Welkenhuyzen, K., Luyten, T., Ponsaerts, R., Dewaele, M., Molgo, J., et al. (2011). Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 7, 1472–1489. doi: 10.4161/auto.7.12.17909
Eisner, V., Picard, M., and Hajnoczky, G. (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 20, 755–765. doi: 10.1038/s41556-018-0133-0
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U.S.A. 112, E2174–E2181. doi: 10.1073/pnas.1504880112
Filippin, L., Magalhaes, P. J., Di Benedetto, G., Colella, M., and Pozzan, T. (2003). Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J. Biol. Chem. 278, 39224–39234. doi: 10.1074/jbc.M302301200
Fransson, S., Ruusala, A., and Aspenstrom, P. (2006). The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 344, 500–510. doi: 10.1016/j.bbrc.2006.03.163
Friedman, J. R., Lackner, L. L., West, M., Dibenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Fujimoto, M., Hayashi, T., and Su, T. P. (2012). The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 417, 635–639. doi: 10.1016/j.bbrc.2011.12.022
Futerman, A. H. (2006). Intracellular trafficking of sphingolipids: relationship to biosynthesis. Biochim. Biophys. Acta 1758, 1885–1892. doi: 10.1016/j.bbamem.2006.08.004
Giorgi, C., Bonora, M., Sorrentino, G., Missiroli, S., Poletti, F., Suski, J. M., et al. (2015). p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 112, 1779–1784. doi: 10.1073/pnas.1410723112
Gonzalez-Angulo, A. M., Ferrer-Lozano, J., Stemke-Hale, K., Sahin, A., Liu, S., Barrera, J. A., et al. (2011). PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol. Cancer Ther. 10, 1093–1101. doi: 10.1158/1535-7163.MCT-10-1089
Gueguinou, M., Crottes, D., Chantome, A., Rapetti-Mauss, R., Potier-Cartereau, M., Clarysse, L., et al. (2017). The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca(2+) homeostasis. Oncogene 36, 3640–3647. doi: 10.1038/onc.2016.501
Gutierrez, T., and Simmen, T. (2018). Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 70, 64–75. doi: 10.1016/j.ceca.2017.05.015
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Hansford, R. G., and Chappell, J. B. (1967). The effect of Ca2+ on the oxidation of glycerol phosphate by blowfly flight-muscle mitochondria. Biochem. Biophys. Res. Commun. 27, 686–692. doi: 10.1016/S0006-291X(67)80090-1
Hayashi, T., and Su, T. P. (2003). Intracellular dynamics of sigma-1 receptors (sigma(1) binding sites) in NG108-15 cells. J. Pharmacol. Exp. Ther. 306, 726–733. doi: 10.1124/jpet.103.051292
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Hirota, Y., Yamashita, S., Kurihara, Y., Jin, X., Aihara, M., Saigusa, T., et al. (2015). Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 11, 332–343. doi: 10.1080/15548627.2015.1023047
Hui, L., Wu, H., Wang, T. W., Yang, N., Guo, X., and Jang, X. J. (2019). Hydrogen peroxide-induced mitophagy contributes to laryngeal cancer cells survival via the upregulation of FUNDC1. Clin. Transl. Oncol. 21, 596–606. doi: 10.1007/s12094-018-1958-5
Hung, V., Lam, S. S., Udeshi, N. D., Svinkina, T., Guzman, G., Mootha, V. K., et al. (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife 6:e24463. doi: 10.7554/eLife.24463
Issop, L., Fan, J., Lee, S., Rone, M. B., Basu, K., Mui, J., et al. (2015). Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: role of ATAD3. Endocrinology 156, 334–345. doi: 10.1210/en.2014-1503
Kakihana, T., Nagata, K., and Sitia, R. (2012). Peroxides and peroxidases in the endoplasmic reticulum: integrating redox homeostasis and oxidative folding. Antioxid. Redox Signal. 16, 763–771. doi: 10.1089/ars.2011.4238
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580. doi: 10.1038/cdd.2010.191
Katz, J., Wals, P. A., Golden, S., and Raijman, L. (1983). Mitochondrial-reticular cytostructure in liver cells. Biochem. J. 214, 795–813. doi: 10.1042/bj2140795
Khan, M. T., Wagner, L. 2nd, Yule, D. I., Bhanumathy, C., and Joseph, S. K. (2006). Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 281, 3731–3737. doi: 10.1074/jbc.M509262200
Kutomi, G., Tamura, Y., Tanaka, T., Kajiwara, T., Kukita, K., Ohmura, T., et al. (2013). Human endoplasmic reticulum oxidoreductin 1-alpha is a novel predictor for poor prognosis of breast cancer. Cancer Sci. 104, 1091–1096. doi: 10.1111/cas.12177
Lewin, T. M., Kim, J. H., Granger, D. A., Vance, J. E., and Coleman, R. A. (2001). Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 276, 24674–24679. doi: 10.1074/jbc.M102036200
Lewis, S. C., Uchiyama, L. F., and Nunnari, J. (2016). ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353:aaf5549. doi: 10.1126/science.aaf5549
Liu, H., Liu, X., Zhuang, H., Fan, H., Zhu, D., Xu, Y., et al. (2020). Mitochondrial contact sites in inflammation-induced cardiovascular disease. Front. Cell Dev. Biol. 8:692. doi: 10.3389/fcell.2020.00692
Mammucari, C., Raffaello, A., Vecellio Reane, D., Gherardi, G., De Mario, A., and Rizzuto, R. (2018). Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch. 470, 1165–1179. doi: 10.1007/s00424-018-2123-2
Marchi, S., Giorgi, C., Oparka, M., Duszynski, J., Wieckowski, M. R., and Pinton, P. (2014). Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol. Cell Oncol. 1:e956469. doi: 10.4161/23723548.2014.956469
Marchi, S., Marinello, M., Bononi, A., Bonora, M., Giorgi, C., Rimessi, A., et al. (2012). Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. 3:e304. doi: 10.1038/cddis.2012.45
Marini, E. S., Giampietri, C., Petrungaro, S., Conti, S., Filippini, A., Scorrano, L., et al. (2015). The endogenous caspase-8 inhibitor c-FLIPL regulates ER morphology and crosstalk with mitochondria. Cell Death Differ 22, 1131–1143. doi: 10.1038/cdd.2014.197
Mendes, C. C., Gomes, D. A., Thompson, M., Souto, N. C., Goes, T. S., Goes, A. M., et al. (2005). The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 280, 40892–40900. doi: 10.1074/jbc.M506623200
Missiroli, S., Patergnani, S., Caroccia, N., Pedriali, G., Perrone, M., Previati, M., et al. (2018). Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 9:329. doi: 10.1038/s41419-017-0027-2
Morciano, G., Marchi, S., Morganti, C., Sbano, L., Bittremieux, M., Kerkhofs, M., et al. (2018). Role of mitochondria-associated ER membranes in calcium regulation in cancer-specific settings. Neoplasia 20, 510–523. doi: 10.1016/j.neo.2018.03.005
Morrison Joly, M., Hicks, D. J., Jones, B., Sanchez, V., Estrada, M. V., Young, C., et al. (2016). Rictor/mTORC2 drives progression and therapeutic resistance of HER2-amplified breast cancers. Cancer Res. 76, 4752–4764. doi: 10.1158/0008-5472.CAN-15-3393
Mourier, A., Motori, E., Brandt, T., Lagouge, M., Atanassov, I., Galinier, A., et al. (2015). Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell Biol. 208, 429–442. doi: 10.1083/jcb.201411100
Parys, J. B., and De Smedt, H. (2012). Inositol 1,4,5-trisphosphate and its receptors. Adv. Exp. Med. Biol. 740, 255–279. doi: 10.1007/978-94-007-2888-2_11
Pickett, C. B., Rosenstein, N. R., and Jeter, R. L. (1981). The physical association between rat liver mitochondria and rough endoplasmic reticulum. II. Possible role RER-MT complexes play in the biosynthesis of cytochrome P-450. Exp. Cell Res. 132, 225–234. doi: 10.1016/0014-4827(81)90098-7
Poderoso, C., Duarte, A., Cooke, M., Orlando, U., Gottifredi, V., Solano, A. R., et al. (2013). The spatial and temporal regulation of the hormonal signal. Role of mitochondria in the formation of a protein complex required for the activation of cholesterol transport and steroids synthesis. Mol. Cell Endocrinol. 371, 26–33. doi: 10.1016/j.mce.2012.12.024
Poston, C. N., Krishnan, S. C., and Bazemore-Walker, C. R. (2013). In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteomics 79, 219–230. doi: 10.1016/j.jprot.2012.12.018
Prasad, M., Kaur, J., Pawlak, K. J., Bose, M., Whittal, R. M., and Bose, H. S. (2015). Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. J. Biol. Chem. 290, 2604–2616. doi: 10.1074/jbc.M114.605808
Rizzuto, R., Duchen, M. R., and Pozzan, T. (2004). Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE 2004:re1. doi: 10.1126/stke.2152004re1
Rizzuto, R., Marchi, S., Bonora, M., Aguiari, P., Bononi, A., De Stefani, D., et al. (2009). Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim. Biophys. Acta 1787, 1342–1351. doi: 10.1016/j.bbabio.2009.03.015
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. doi: 10.1126/science.280.5370.1763
Roperto, S., Russo, V., De Falco, F., Rosati, A., Catoi, C., and Roperto, F. (2019). FUNDC1-mediated mitophagy in bovine papillomavirus-infected urothelial cells. Vet. Microbiol. 234, 51–60. doi: 10.1016/j.vetmic.2019.05.017
Rusinol, A. E., Cui, Z., Chen, M. H., and Vance, J. E. (1994). A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 269, 27494–27502. doi: 10.1016/S0021-9258(18)47012-3
Salaroglio, I. C., Panada, E., Moiso, E., Buondonno, I., Provero, P., Rubinstein, M., et al. (2017). PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 16:91. doi: 10.1186/s12943-017-0657-0
Sala-Vila, A., Navarro-Lerida, I., Sanchez-Alvarez, M., Bosch, M., Calvo, C., Lopez, J. A., et al. (2016). Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci. Rep. 6:27351. doi: 10.1038/srep27351
Saotome, M., Safiulina, D., Szabadkai, G., Das, S., Fransson, A., Aspenstrom, P., et al. (2008). Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 20728–20733. doi: 10.1073/pnas.0808953105
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. doi: 10.1126/science.1106148
Schrepfer, E., and Scorrano, L. (2016). Mitofusins, from Mitochondria to Metabolism. Mol. Cell 61, 683–694. doi: 10.1016/j.molcel.2016.02.022
Scorrano, L., Oakes, S. A., Opferman, J. T., Cheng, E. H., Sorcinelli, M. D., Pozzan, T., et al. (2003). BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300, 135–139. doi: 10.1126/science.1081208
Seervi, M., Sobhan, P. K., Joseph, J., Ann Mathew, K., and Santhoshkumar, T. R. (2013). ERO1alpha-dependent endoplasmic reticulum-mitochondrial calcium flux contributes to ER stress and mitochondrial permeabilization by procaspase-activating compound-1 (PAC-1). Cell Death Dis. 4:e968. doi: 10.1038/cddis.2013.502
Simmen, T., and Herrera-Cruz, M. S. (2018). Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cell Biol. 53, 61–69. doi: 10.1016/j.ceb.2018.04.014
Singh, A., Chagtoo, M., Tiwari, S., George, N., Chakravarti, B., Khan, S., et al. (2017a). Inhibition of inositol 1, 4, 5-trisphosphate receptor induce breast cancer cell death through deregulated autophagy and cellular bioenergetics. J. Cell. Biochem. 118, 2333–2346. doi: 10.1002/jcb.25891
Singh, A., Sharma, R. K., Chagtoo, M., Agarwal, G., George, N., Sinha, N., et al. (2017b). 1H NMR metabolomics reveals association of high expression of inositol 1, 4, 5 trisphosphate receptor and metabolites in breast cancer patients. PLoS ONE 12:e0169330. doi: 10.1371/journal.pone.0169330
Stemke-Hale, K., Gonzalez-Angulo, A. M., Lluch, A., Neve, R. M., Kuo, W. L., Davies, M., et al. (2008). An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 68, 6084–6091. doi: 10.1158/0008-5472.CAN-07-6854
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. doi: 10.1038/s41577-019-0165-0
Szabadkai, G., Bianchi, K., Varnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi: 10.1083/jcb.200608073
Szabadkai, G., and Duchen, M. R. (2008). Mitochondria: the hub of cellular Ca2+ signaling. Physiology 23, 84–94. doi: 10.1152/physiol.00046.2007
Szado, T., Vanderheyden, V., Parys, J. B., De Smedt, H., Rietdorf, K., Kotelevets, L., et al. (2008). Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 105, 2427–2432. doi: 10.1073/pnas.0711324105
Tanaka, T., Kutomi, G., Kajiwara, T., Kukita, K., Kochin, V., Kanaseki, T., et al. (2017). Cancer-associated oxidoreductase ERO1-alpha promotes immune escape through up-regulation of PD-L1 in human breast cancer. Oncotarget 8, 24706–24718. doi: 10.18632/oncotarget.14960
Teng, Y., Ren, X., Li, H., Shull, A., Kim, J., and Cowell, J. K. (2016). Mitochondrial ATAD3A combines with GRP78 to regulate the WASF3 metastasis-promoting protein. Oncogene 35, 333–343. doi: 10.1038/onc.2015.86
Tosatto, A., Sommaggio, R., Kummerow, C., Bentham, R. B., Blacker, T. S., Berecz, T., et al. (2016). The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol. Med. 8, 569–585. doi: 10.15252/emmm.201606255
Tschopp, J. (2011). Mitochondria: sovereign of inflammation? Eur. J. Immunol. 41, 1196–1202. doi: 10.1002/eji.201141436
Van Meer, G., Voelker, D. R., and Feigenson, G. W. (2008). Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. doi: 10.1038/nrm2330
Van Vliet, A. R., Giordano, F., Gerlo, S., Segura, I., Van Eygen, S., Molenberghs, G., et al. (2017). The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol. Cell 65, 885–899.e886. doi: 10.1016/j.molcel.2017.01.020
Vance, J. E. (2014). MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609. doi: 10.1016/j.bbalip.2013.11.014
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19, 1880–1891. doi: 10.1038/cdd.2012.74
Vicencio, J. M., Ortiz, C., Criollo, A., Jones, A. W., Kepp, O., Galluzzi, L., et al. (2009). The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 16, 1006–1017. doi: 10.1038/cdd.2009.34
Voelker, D. R. (1984). Phosphatidylserine functions as the major precursor of phosphatidylethanolamine in cultured BHK-21 cells. Proc. Natl. Acad. Sci. U.S.A. 81, 2669–2673. doi: 10.1073/pnas.81.9.2669
Wai, T., and Langer, T. (2016). Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 27, 105–117. doi: 10.1016/j.tem.2015.12.001
Wanson, J. C., Drochmans, P., May, C., Penasse, W., and Popowski, A. (1975). Isolation of centrolobular and perilobular hepatocytes after phenobarbital treatment. J. Cell Biol. 66, 23–41. doi: 10.1083/jcb.66.1.23
Wieckowski, M. R., Giorgi, C., Lebiedzinska, M., Duszynski, J., and Pinton, P. (2009). Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590. doi: 10.1038/nprot.2009.151
Wu, L., Zhang, D., Zhou, L., Pei, Y., Zhuang, Y., Cui, W., et al. (2019). FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMed. 41, 384–394. doi: 10.1016/j.ebiom.2019.02.032
Wu, S., Lu, Q., Wang, Q., Ding, Y., Ma, Z., Mao, X., et al. (2017). Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136, 2248–2266. doi: 10.1161/CIRCULATIONAHA.117.030235
Wu, W., Lin, C., Wu, K., Jiang, L., Wang, X., Li, W., et al. (2016). FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 35, 1368–1384. doi: 10.15252/embj.201593102
Yang, W. S., Sriramaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, Z., Zhao, X., Xu, J., Shang, W., and Tong, C. (2018). A novel fluorescent reporter detects plastic remodeling of mitochondria-ER contact sites. J. Cell Sci. 131:jcs208686. doi: 10.1242/jcs.208686
Keywords: mitochodnria, ER, MAM, breast cancer, carcinogenesis
Citation: Yu H, Sun C, Gong Q and Feng D (2021) Mitochondria-Associated Endoplasmic Reticulum Membranes in Breast Cancer. Front. Cell Dev. Biol. 9:629669. doi: 10.3389/fcell.2021.629669
Received: 15 November 2020; Accepted: 08 January 2021;
Published: 28 January 2021.
Edited by:
Simone Patergnani, University of Ferrara, ItalyReviewed by:
Claudia Giampietri, Sapienza University of Rome, ItalySoumasree De, University of Bern, Switzerland
Copyright © 2021 Yu, Sun, Gong and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Gong, 443292273@qq.com; Du Feng, Fenglab@gzhmu.edu.cn
†These authors share first authorship