The Multifunctional Contribution of FGF Signaling to Cardiac Development, Homeostasis, Disease and Repair
 Farhad Khosravi
Farhad Khosravi Negah Ahmadvand
Negah Ahmadvand Saverio Bellusci
Saverio Bellusci Heinrich Sauer
Heinrich Sauer- 1Department of Physiology, Justus Liebig University Giessen, Giessen, Germany
- 2Cardio-Pulmonary Institute, Justus Liebig University Giessen, Giessen, Germany
The current focus on cardiovascular research reflects society’s concerns regarding the alarming incidence of cardiac-related diseases and mortality in the industrialized world and, notably, an urgent need to combat them by more efficient therapies. To pursue these therapeutic approaches, a comprehensive understanding of the mechanism of action for multifunctional fibroblast growth factor (FGF) signaling in the biology of the heart is a matter of high importance. The roles of FGFs in heart development range from outflow tract formation to the proliferation of cardiomyocytes and the formation of heart chambers. In the context of cardiac regeneration, FGFs 1, 2, 9, 16, 19, and 21 mediate adaptive responses including restoration of cardiac contracting rate after myocardial infarction and reduction of myocardial infarct size. However, cardiac complications in human diseases are correlated with pathogenic effects of FGF ligands and/or FGF signaling impairment. FGFs 2 and 23 are involved in maladaptive responses such as cardiac hypertrophic, fibrotic responses and heart failure. Among FGFs with known causative (FGFs 2, 21, and 23) or protective (FGFs 2, 15/19, 16, and 21) roles in cardiac diseases, FGFs 15/19, 21, and 23 display diagnostic potential. The effective role of FGFs on the induction of progenitor stem cells to cardiac cells during development has been employed to boost the limited capacity of postnatal cardiac repair. To renew or replenish damaged cardiomyocytes, FGFs 1, 2, 10, and 16 were tested in (induced-) pluripotent stem cell-based approaches and for stimulation of cell cycle re-entry in adult cardiomyocytes. This review will shed light on the wide range of beneficiary and detrimental actions mediated by FGF ligands and their receptors in the heart, which may open new therapeutic avenues for ameliorating cardiac complications.
Introduction
The ever-increasing threat of cardiac diseases accompanied by the modern, unhealthy lifestyle has prompted investigations on heart pathophysiology in order to develop novel therapeutic theories and options alleviating cardiac pathogenic symptoms and restoring physiological function. This progress, at least in part, profits from the understanding of cell signaling pathways governing embryonic cardiac development and cardiac physiology and pathophysiology. In the last two decades, cellular signaling networks in the heart, including fibroblast growth factors (FGFs) and their interplay with other growth factors and signaling contributors such as IGF1/2, VEGF, BMPs [as members of transforming growth factor-β (TGF-β) superfamily], Wnts, Notch and erythropoietin have been investigated in detail (Bruneau, 2013; Mascheck et al., 2015; Meganathan et al., 2015; Wilsbacher and McNally, 2016). Intriguingly, regardless of a 30–60% amino acid sequence homology between individual FGF family members, they contribute to distinct, and even contradictory actions, being either protective or pathogenic. Our current knowledge of FGF-dependent cardiac physiology is mainly based on loss- and gain-of-function studies provided through genetically modified animal models (transgenic or knockout), where the expression of FGFs and/or FGF receptors (FGFR) are impaired (Itoh et al., 2016). In addition, embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) serve as in vitro models for investigations on the implication of ligand–receptor interactions in cardiac cell differentiation (Kawai et al., 2004; Chan et al., 2010; Yamasaki et al., 2013; Mascheck et al., 2015).
FGF Superfamily
Fibroblast growth factors, consisting of a family of 22 identified members of pleiotropic proteins in human and mouse, mediate pivotal functions in the heart, ranging from development to homeostasis and disease. Since FGFs are among cardiac secreted proteins required for heart physiological and pathological responses, they are considered cardiomyokines (Doroudgar and Glembotski, 2011; Itoh et al., 2016). Based on a phylogenetic analysis, FGFs are categorized into 7 subfamilies, which are either intracellular or secreted factors with autocrine, paracrine, and/or endocrine functions (Ornitz and Itoh, 2015). The intracellular FGFs act in an intracrine manner and include members of the FGF11 subfamily (FGFs 11, 12, 13, and 14; Wang et al., 2011).
Mechanism of Action for Secreted FGFS
Secreted FGF ligands form complex structures with cofactors (also termed coreceptors) and FGFRs enabling them to regulate cell signaling and mediate a wide variety of functions. These FGF members serve as ligands through specific interaction with one of the seven alternatively spliced tyrosine kinase receptors (FGFRs 1b, 1c, 2b, 2c, 3b, 3c, and 4) which are expressed on the cell surface and are encoded from four genes (FGFR1-4; Zhang et al., 2006). Binding to FGFRs is modulated by extracellular cofactors such as Klothos (in the endocrine subfamily of FGF 15/19) and heparin/heparan sulfates (in autocrine/paracrine members of the FGF 1, 4, 7, 8, and 9 subfamilies; Rapraeger et al., 1991; Goetz et al., 2012; Itoh et al., 2015). Cofactors (such as heparan sulfate) mediate the formation of a complex between FGF and FGFR, and this complex activates FGFR dimerization and underlying cell signaling cascades, including phosphoinositide 3-kinase (PI3K)/serine/threonine protein kinase B (AKT), Ras/mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription (STAT), and phospholipase C (PLC)γ (Itoh et al., 2016).
In the characterization of FGF superfamily member effects, a putative redundancy in the function of distinct FGF ligands (e.g., FGFs 3 and 8 with FGF10) and receptors (e.g., between FGFR1c and 2c in interaction with FGF9 subfamily) in the process of heart development and the adult phase needs to be considered (Lavine et al., 2005; Marguerie et al., 2006; Watanabe et al., 2010; Urness et al., 2011; Krejci et al., 2016).
FGF Signaling in Heart Development
In the early stages of cardiac development, FGF signaling plays a central role for normal morphogenesis through profound effects on the second heart field (SHF) and the cardiac neuronal crest (CNC) cells which determine the outflow tract (OFT) formation (Hubert et al., 2018). Specifically, FGFs 2, 3, 4, 8, 10, 16, and 20 contribute to the communication between and within SHF cardiac progenitor stem cells, and this modulates the proliferation of SHF progenitor cells together with myocardial specification and finally the development of OFT (Table 1; Park et al., 2008; Felker et al., 2018). In the late phases of cardiac development, the myocardium grows and remodels coinciding with a proliferation shift from SHF progenitor cells to cardiomyocytes. These lead to the formation of ventricular and arterial heart chambers. FGF ligands were reported to be autocrinally or paracrinally involved in the modulation of fetal cardiomyocyte proliferation. FGFs 2, 4, 7, 8, 9, 10, 15/19 also appear to be involved in these cardiac morphologic events (Table 1).

Table 1. Roles of secreted FGFs in heart development.
Differential Expression of FGFs in Cardiac Development
In the early phase of heart development, FGF members are differentially expressed in distinct anatomical regions. For instance, Fgf10 is expressed in the pharyngeal mesoderm [between embryonic day (E.) 8.5 and E.10.5], and therefore is known as an exclusive marker of SHF (Kelly et al., 2001). The expression of Fgf8 is detectable at E.9.5 in the adjacent pharyngeal endoderm and ectoderm in addition to the SHF (Mesbah et al., 2012). FGF3 (between E.8.0 and E.10.0) is present in the pharyngeal endoderm. Fgf3 is also expressed at E8.0 in the ectoderm (Urness et al., 2011). FGF9 subfamily members (i.e., FGFs 9, 16, and 20 between E.10.5 and E.12.5) originate from the epicardium (i.e., FGF9 at E.10.5) and the endocardium (i.e., FGFs 9, 16, and 20 at E.10.5 and E.12.5; Lavine et al., 2005; Hotta et al., 2008; Lu et al., 2008b).
Mechanism of Action for FGFs in Cardiac Development
To fulfill these various and divergent functions, the contribution of other downstream pathways, i.e., PI3K/AKT (Luo et al., 2015) and Ras/extracellular signal-regulated kinase (ERK; Hutson et al., 2010) is a matter of significance for the understanding of FGF signaling (Figure 1). Importantly, the FGF-MAPK axis contributes to the continuance of cardiopharyngeal multipotency in cardiomyocyte-generating progenitor stem cells (Wang et al., 2019). Cardiac fibroblasts originate predominantly from the epicardium (up to 80% in the adult heart), the endocardium, SHF and neural crest. The differentiation of SHF progenitors from human pluripotent stem cells, which can later generate cardiac fibroblasts, is attributed to the effect of FGF2 as well as Wnt signaling cascades through the suppression of glycogen synthase kinase 3β (GSK3β; Zhang et al., 2019). This potential of FGF2 and Wnt is widely used in the maintenance of pluripotency in human ESCs (Taelman et al., 2019). Other proofs for the implication of FGF members in cardiomyogenic differentiation were provided in mouse ESC and iPSC models. For instance, FGF2 in combination with BMP2 promotes cardiomyocyte differentiation from ESCs and iPSCs (Kawai et al., 2004; Yamasaki et al., 2013). In this regard, FGF10 through interaction with its receptor FGFR2, expressed on stem cells, mediates the differentiation of cardiomyocytes (Chan et al., 2010).
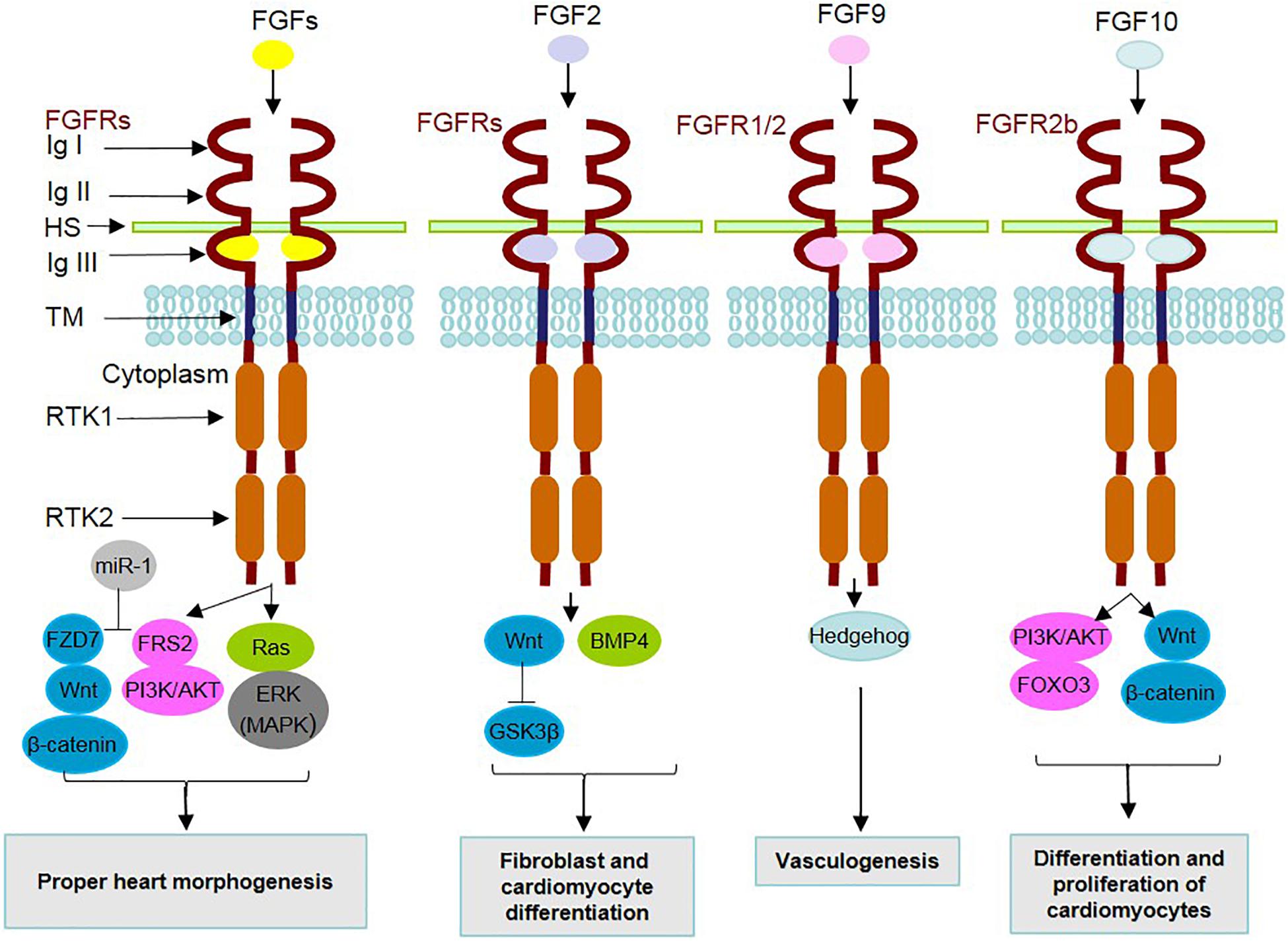
Figure 1. Mechanism of action of secreted FGFs and identified underlying signaling contributors in heart development. During heart development, FGF ligands, through binding to FGF receptors (FGFRs) and cofactors [such as heparan sulfate (HS)] and dimerization, contribute to the activation/inhibition of related underlying intracellular signaling pathways. The FGFR family is composed of 7 FGFRs encoded by 4 genes expressing the alternative splicing forms 1b, 1c, 2b, 2c, 3b, 3c, and 4. FGFRs contain extracellular immunoglobulin-like domains (Ig I–III), a transmembrane domain (TM) and receptor tyrosine kinase domains (RTKs 1 and 2). MicroRNA-1 (miR-1) is positively involved in FGF actions through inhibition of FRS2 and FZD7. A summary of the effects related to the interactions of FGFs with underlying signaling mediators in cardiac development is listed in the boxes.
Regarding the interplay of FGF signaling with other underlying intracellular pathways (Figure 1), a mediating role is attributed to adaptor proteins such as FRS2 (at E.9.5), which transduce FGF signal to MAPK/PI3K (Zhang et al., 2008). Additionally, microRNA-1 (miR-1) is a mediator functioning in the differentiation of cardiomyocytes, by exerting inhibitory effects on FRS2 and FZD7, which acts as a linker of FGF to the Wnt signaling cascade (Lu et al., 2013).
During SHF and OFT development, the Wnt/β-catenin pathway is involved in myocardial progenitor cell differentiation through the modulation of Fgf 3, 10, 16, and 20 expression levels (at E.9.5; Cohen et al., 2007). The expression of Fgf10 in the SHF is positively regulated by transcription factors (e.g., ISL1 and TBX1) in progenitor cells and negatively (e.g., NKX2-5) in cardiomyocytes (at E.9.5; Cohen et al., 2007; Watanabe et al., 2012). TBX1 plays the same activating role for FGF8 expression in the OFT (at E.9.5; Hu et al., 2004). Both FGFs 8 and 10 are involved in the proliferation of SHF progenitor cells, and thereby in the development of the arterial pole (at E.10.5 and between E.15.5 and E.18.5; Watanabe et al., 2010). Homozygous deletion of Fgf10 in embryos, which is lethal after birth, mainly because of lung aplasia, results in cardiac malformation and ventricular transposition in the thoracic cavity (at E.17.5 and E.18.5; Marguerie et al., 2006; Rochais et al., 2014; Chao et al., 2017). However, normal arterial pole elongation and septation in the absence of Fgf10 (at E.17.5) reflect its negligible effect on normal arterial development. Notably, the deletion of Fgfr2b, the major receptor of FGF10, impairs SHF deployment (at E.10.5), thus being different from the milder cardiac defects accompanied by Fgf10 deletion (Marguerie et al., 2006). This and other severe heart malformations in the OFT and right ventricle occurring in the absence of both Fgf10 and Fgf8 indicate a functional redundancy between FGF10 and other mesodermal FGFs, such as FGF8, which engage the same FGFR2b receptor in the early stages of cardiac development (at E.9.5; Marguerie et al., 2006; Watanabe et al., 2010). Similarly, functional overlap of the FGF family members (FGF10 and FGF3) emerges during heart tube extension which is necessary for the coordination of cardiac progenitor cells. However, deletion of these two Fgf genes showed no impact on progenitor cell specification (at E.8.0–E.10.5; Urness et al., 2011).
FGF8 plays a critical role in cardiac loop formation and the survival of migratory CNCs (at E.10.5). The ablation of Fgf8 perturbs OFT septation (at E.16.5; Abu-Issa et al., 2002). Another essential function of FGF8 is in the development of the anterior heart field (between E.7.75 and E.10.5; Ilagan et al., 2006). Heparan sulfate, as a glycosaminoglycan and a major component present in the cardiac extracellular matrix, is necessary for the proper function of FGF8 in alignment and formation of OFT, together with the sufficient recruitment of SHF and neural crest cells for OFT remodeling (Zhang R. et al., 2015).
FGFR4 and its ligands (FGFs 4 and 8) are involved in the proper left-right patterning of cardiac laterality, which is impaired in congenital heart disease associated with heterotaxy syndrome (Sempou et al., 2018; Guzzetta et al., 2020). The functions of FGFs 4 and 8 in mesoderm migration and embryonic axis patterning are dependent on Hedgehog signaling activation (at E.9.5; Guzzetta et al., 2020).
FGF9 subfamily members (i.e., FGFs 9, 16, and 20) control cardiomyocyte differentiation and proliferation in the myocardium (Lavine et al., 2005; Hotta et al., 2008; Lu et al., 2008b). This effect on cardiac progenitor cells is mediated through interaction with two receptors (FGFR1c and 2c) with functional redundancy. Knockout of Fgf9 reduces cardiac size and proliferation rate of cardiomyocytes (at E.12.5; Lavine et al., 2005). FGF16 is mainly encoded in embryonic cardiomyocytes (at E.14.5) and functions through interaction with heparin sulfate-FGFR1c (Hotta et al., 2008). Findings on the FGF16-associated implications seem to vary in different genetic backgrounds of selected mouse models (Lu et al., 2010). On the one hand, Fgf16 (−/−) is lethal in Swiss black mice (at E.11.5) with severe cardiac malformations (Lu et al., 2008a). On the other hand, these mutants in C57 black 6 mice survive, yet with modest attenuation of cardiac weight and cardiomyocyte cell numbers (Hotta et al., 2008). Intriguingly, similarities with the cardiac phenotype in Fgf9 (−/−) mice shed light on a possible synergistic effect of FGF9 and FGF16 on embryonic cardiomyocytes (Hotta et al., 2008). However, according to a recent study, FGFs 9 and 16 exert proliferative action on distinct targets (human cardiac fibroblasts versus cardiac progenitor cells, respectively) and receptors, and only FGF16 is specifically able to promote proliferation of mouse naïve cardiac progenitor cells and cardiomyocytes originated from human induced pluripotent stem cells (hiPSCs; Jennbacken et al., 2019).
In the last decade, a regulatory role in cardiomyocyte proliferation has been conferred to FGF10 after findings that ventricles are morphologically altered in this mutant model (E18.5). This study indicated, that FGF10 modulates cardiomyocyte proliferation in the fetal right ventricle in an autocrine fashion. FGF10 ligand and its receptor (FGFR2b) mediate this function through phosphorylation of the forkhead box O3 (FOXO3) transcription factor leading to diminished expression of cyclin-dependent kinase inhibitor p27kip1 (Rochais et al., 2014). Myocardium-generated FGF10 affects epicardial cells paracrinally, activating their receptors (FGFR1 and FGFR2), and thereby contributes to the migration of cells toward the compact myocardium. Thus, FGF10 loss of function indirectly perturbs cardiomyocyte proliferating capacity and morphologically leads to the formation of a smaller thin-walled heart (at E17.5). The effect of FGF10 on cardiac wall formation requires functional redundancy between FGFs 7 and 10 over FGFR2b (Vega-Hernández et al., 2011).
FGF15 is present in the pharyngeal endoderm. For normal OFT formation, ortholog members of the endocrine subfamily, i.e., FGFs 15 (in mice)/19 (in humans) play a morphogenesis-related role. In fact, FGF15/19 is critical in developing pharyngeal arches, and the absence of FGF15 results in profound defects in OFT morphology and aorta alignment (at E.18.5; Vincentz et al., 2005). In a possible paracrine function, FGF15/19 may act independently from the typical endocrine cascade engaging FGFR4-βKlotho in cardiac development (Itoh et al., 2016). In this regard, however, the requirement of FGFR4-βKlotho for activation of this signaling in the heart needs to be further investigated (Yu et al., 2000; Ito et al., 2005; Dongiovanni et al., 2020).
Regardless of the known FGF4 contribution to the formation of the cardiac valve leaflet, little is known about other FGF4 functions in the heart (Sugi et al., 2003). Importantly, FGF4 may be involved in the maturation of cardiomyocytes and their aggregation with cardiac fibroblasts, therefore mediating functions in heartbeat synchronization (Jang et al., 2020).
The regulatory role of FGF during heart development was identified to be associated with the function of KLF2, which is a member of the Krüppel-like factor (KLF) family, in the communication between endocardium and myocardium. KLF2 acts as an important transcription factor expressed in the endocardium, and functions on the integrity of the myocardial wall which is correlated to FGF signaling (Rasouli et al., 2018).
FGF Role in the Vascular Network Formation
Concurrent with the loss of Fgf9 vasculogenesis and development of coronary vessels is impaired. Mechanistically, this function of FGF9 in coronary development (at E.12.5–E.13.5) is related to the induction of the Hedgehog pathway (Lavine et al., 2006). Another process of epicardial/endocardial to myocardial FGF signal transduction in cardiovascular development occurs during the development of coronary vessels, induced by FGF ligands secreted from the endocardium and the epicardium. In this matter, myocardial originated FGFs 1, 2, and 7 stimulate epicardial epithelial-mesenchymal transition (EMT; Morabito et al., 2001). To fulfill this function in the coronary vasculature, Hedgehog signaling downstream of FGFR1 and FGFR2 is induced together with the expression of angiopoietin-2 and vascular endothelial growth factors (Vegf-A, -B, and -C), as proangiogenic growth factors (Lavine et al., 2006). FGFs are involved in vascular network formation, and this signaling is effective for neovasculogenesis and angiogenesis. In this regard, FGF1 and particularly FGF2 are known as inducers of angiogenesis, and this function is mediated by interaction with integrins (Mori et al., 2013, 2017; Sauer et al., 2013).
FGF Signaling in Cardiac Homeostasis
During cardiac homeostasis, FGFs play roles in the morphology and physiology of the heart. These actions range from cardiomyocyte proliferation to myocardial excitability together with angiogenesis and metabolic regulation of energy balance.
FGF Functions in Modulation of Cardiomyocyte Proliferation
In the postnatal developmental phase and adult heart, new cardiomyocytes still can proliferate, yet in a substantially decreased degree compared to embryonic heart development (Bergmann et al., 2015). This reduced cardiomyocyte renewal potential, which is albeit crucial for maintaining adult heart homeostasis, comprises annually 0.5–2% of total cardiomyocytes (Eschenhagen et al., 2017).
Fibroblast growth factor ligands (FGFs 1, 2, and 10) contribute to the modulation of adult cardiomyocyte proliferation (Table 2). The role of FGF2 in this matter is correlated to interaction with the specific receptor FGFR1 (Sheikh et al., 1999).
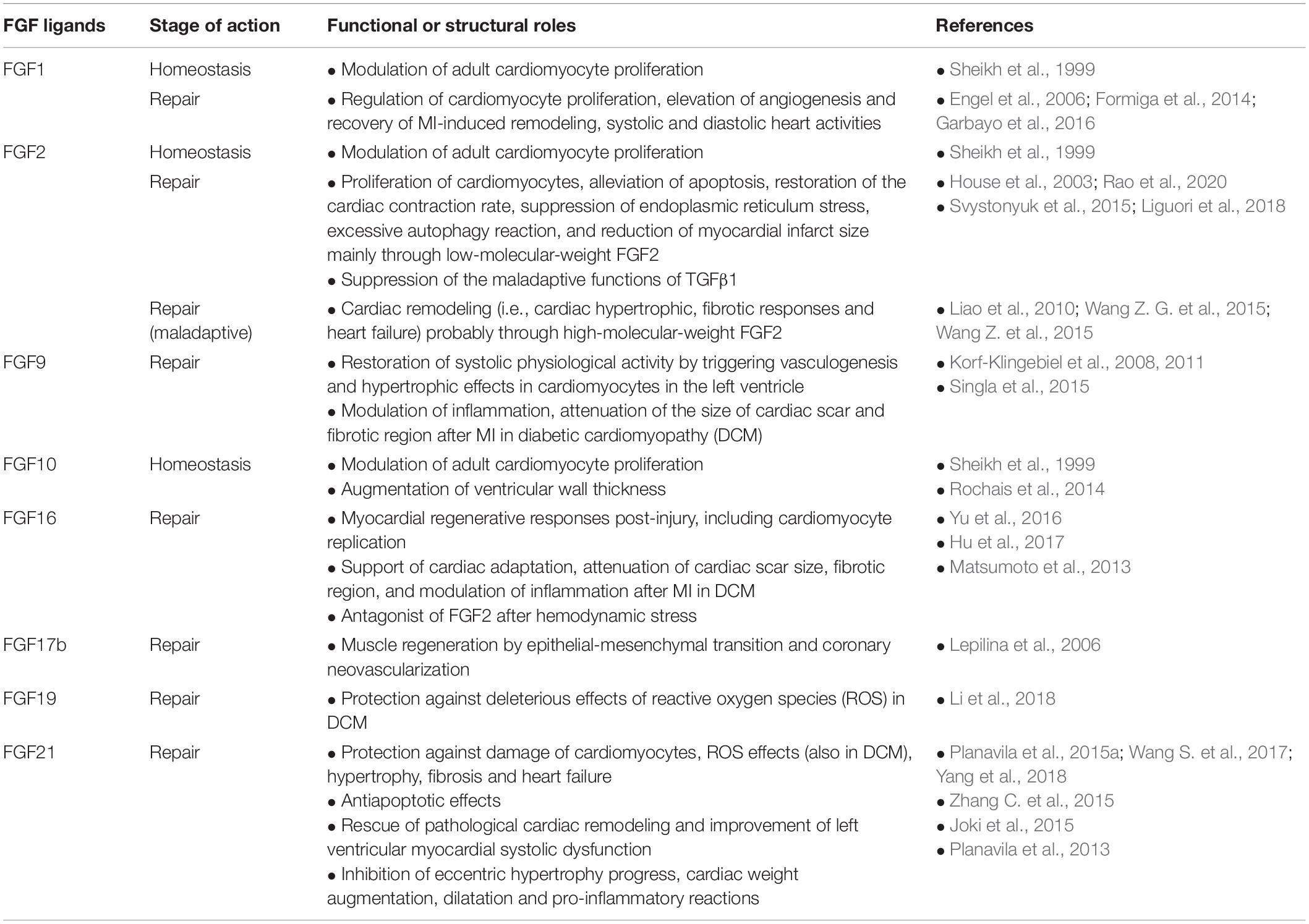
Table 2. Cardioprotective and maladaptive effects of secreted FGFs on homeostasis and repair of heart detected in experimental injury models.
Fibroblast growth factor signaling during cardiac homeostasis in the adult zebrafish leads to the addition of cells from the epicardium to the myocardial wall of ventricles (Wills et al., 2008). Seemingly, FGF10 is involved in adult mice in the enlargement of ventricular wall thickness through modulation of cardiomyocyte cell cycle re-entry. This controlling function of FGF10 in cardiac cell proliferation is initiated by interaction with FGFR2b in the embryonic stage and with FGFR1b in the adult stage (Rochais et al., 2014).
Intracellular FGFs and Myocardial Excitability
The intracellular FGF11 subfamily members (including FGFs 11, 12, 13, and 14) regulate the performance of voltage-dependent sodium channel-related family members, as well as calcium channels. Thereby, they contribute to the generation of action potential and myocardial excitability (Hartung et al., 1997; Liu et al., 2003; Hennessey et al., 2013; Yang et al., 2016). Consequently, their absence can lead to cardiomyopathies and arrhythmias (Liu et al., 2003; Wang et al., 2011; Wang X. et al., 2017). In addition to the above-mentioned positive role in cardiac development and homeostasis, however, detrimental remodeling effects are attributed to FGF13 during cardiac hypertrophy. Mechanistically, to exert these adverse roles, FGF13 negatively regulates caveolae-associated cardioprotection (Wei et al., 2017). Added to this, FGF13 activates the inflammatory and pro-apoptotic NF-κB-p53 axis in cardiomyocytes (Sun J. et al., 2020). Despite a few mouse model studies, intracellular FGFs have still received less research attention.
FGF Effects on Angiogenesis
A wide range of FGFs including FGFs 1, 2, 5, 7, 8, 16, and 18 are present in vascular smooth muscle cells and endothelial cells. Although Fgfr1 and Fgfr2 are highly expressed in endothelial cells and were identified to be involved in the repair through injury-related angiogenesis, a double knockout condition for these two receptors did not lead to a significant alteration in the homeostatic function of the vascular system (Oladipupo et al., 2014; House et al., 2016). This finding, however, is contrary to the reported severe FGF inhibitor-associated defects, even in homeostasis, emphasizing the significance of FGF signaling for physiological vascular integrity and activity (Murakami et al., 2008; De Smet et al., 2014). These FGF studies on endothelial cells shed light on another functional redundancy between FGFRs.
Metabolic Functions of FGF21
In homeostasis, FGF21, which is another member of the FGF15/19 subfamily, exerts multifunctional roles in the metabolism of lipid and glucose (Yan et al., 2015). FGF21 is highly expressed in the liver, and endocrinally targets distant cells through blood cells (Kharitonenkov et al., 2005; Itoh et al., 2016). In addition to the liver, FGF21 expression is detected in adipose tissues (white and brown), pancreas, skeletal muscles, and heart, however, at a lower level (Yan et al., 2015). In fact, FGF21 functionally modulates energy balance and adaptation to oxidative stress (Gómez-Sámano et al., 2017). Moreover, FGF21 mediates functions in the heart via paracrine and autocrine fashions. A paracrine effect is attributed to FGF21 forming a complex with βKlotho-FGFR1c (Figure 2). Added to this, FGF21 possesses a low affinity to the heparan sulfate cofactor (Itoh et al., 2016).
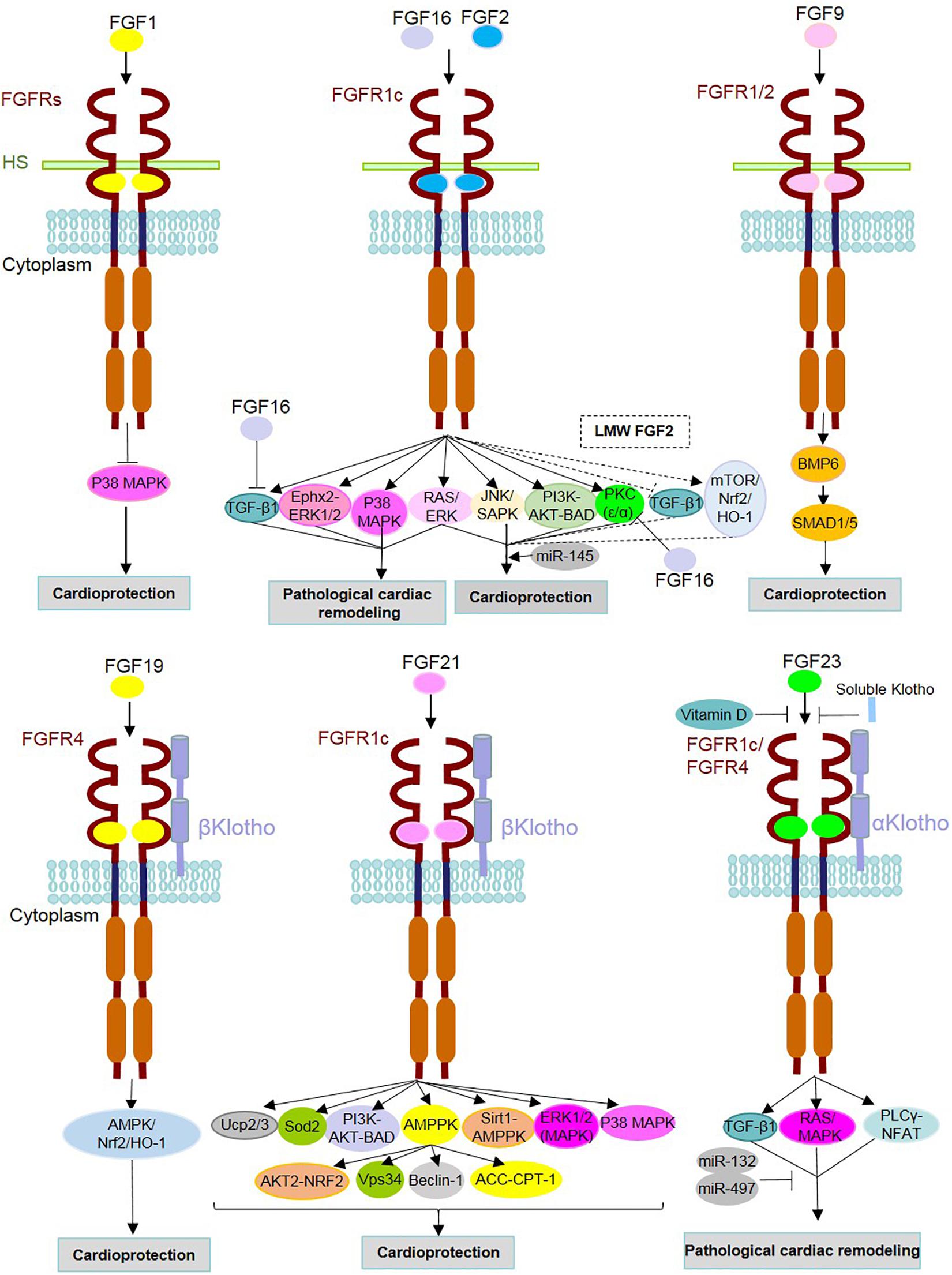
Figure 2. Mechanism of action of FGFs and identified underlying signaling contributors in heart homeostasis, repair and diseases. FGF ligands (FGFs) are involved either in the protection or pathogenesis of the heart through binding with FGF receptors (FGFRs) and cofactors [i.e., heparan sulfate (HS) and α/β Klothos]. After dimerization, they contribute to the activation/inhibition of underlying intracellular signaling pathways. FGF16 and FGF2 can target the same receptor (FGFR1c), but with opposite functions. As an antagonist of FGF2, FGF16 inhibits TGFβ1 and PKC isoforms (PKC-α and PKC-ε). FGF2 is known to play both adaptive and maladaptive roles in cardiac events, and this could be due to the distinct effects of its isoforms [low- and high-molecular-weight isoforms (LMW and HMW)]. Dashed lines indicate signaling pathways and effects related to LMW FGF2. Vitamin D and soluble Klotho suppress the detrimental effects of FGF23-FGFR interactions. MicroRNAs (miR) mediate cardioprotective functions such as miR-145 in association with the Ras-MAPK and PI3K-AKT-related effects of FGF2 as well as miRs-132 and miR-497 through inhibition of FGF23-involved adverse cardiac events.
Roles of FGF in Cardiac Pathophysiology and Repair
Fibroblast growth factors contribute to cardiac pathophysiology (Figure 2). This association was further revealed in genetically engineered rodent and cell culture models which elicit pathological conditions by triggering cardiac remodeling responses (e.g., cardiac hypertrophy, fibrosis and, as a later severe consequence, heart failure). FGFs are involved in the adaptive responses after myocardial infarction (MI) and induced hypertrophy. Moreover, FGFs are known to protect the heart against oxidative stress and diabetic cardiomyopathy. Additionally, FGFs contribute to maladaptive functions mediated by TGFβ1. In addition to these findings in experimental models, a variety of human diseases with cardiac complications affected by FGFs are discussed as follows.
FGF Contributions to Adaptive Responses in Myocardial Infarction and Induced Hypertrophy
FGFs (mainly FGF2 as well as FGFs 1, 9, 17b, and 21) play important roles in cardiac repair after MI and their mechanism of action in injury models have been well characterized. In addition to cardiomyocytes, FGF2 is expressed in non-cardiomyocytes (i.e., mainly cardiac fibroblasts; Zhang et al., 2014) and exerts a paracrine role in the heart through a complex with heparan sulfate and FGFRs (predominantly FGFR1c; Tacer et al., 2010; Ornitz and Itoh, 2015). While knockout mice in the absence of Fgf2 genes survive normally, their repair-related responses after MI are impaired. In contrast to these adverse effects and in a condition where Fgf2 is overexpressed, left ventricular function is preserved after MI through induction of proliferation (in fibroblasts and endothelial cells) and hypertrophy (in cardiomyocytes; Virag et al., 2007). The contribution of FGF2 in these cardiac hypertrophic and fibrotic responses was also suggested in hypertrophy-inducing challenges caused by isoproterenol (House et al., 2010), as well as angiotensin II (Ang II) in a two-kidney one-clip model(Pellieux et al., 2001). However, the underlying signaling target of FGF2 for mediating these functions is controversially discussed ranging from p38 MAPK (Pellieux et al., 2001) to ERK in a mechanism that is independent of any correlation with the p38 MAPK signaling pathway (House et al., 2010). Added to these protection-associated pathways, FGF2 may induce ε isoform of protein kinase C (PKC) and, consequently, phosphorylation of connexin-43 as a major component of the cardiac gap junction (Srisakuldee et al., 2009). Later studies further strengthened the correlation of FGF2 with Ras-MAPK and PI3K-AKT (Wang Z. et al., 2015). In this regard, FGF2 and FGF21 elicit similar protective features in the heart as a result of an injury induced by ischemia−reperfusion or hypoxia-reoxygenation, which mimics MI (House et al., 2015; Ren et al., 2019). In an ischemia−reperfusion injury model, overexpression of Fgf2 restores cardiac contracting rate and minimizes myocardial infarct size (House et al., 2003).
FGF2 in association with downstream cascades (Ras-MAPK and PI3K-AKT) and miR-145 suppresses endoplasmic reticulum stress, excessive autophagy reaction and expression of mitochondrial malfunction-associated proteins, but elevates the removal of ubiquitinated protein (Wang Z. et al., 2015; Wang Z. G. et al., 2015). In contrast, FGF21 in a hypoxia-reoxygenation model recruited autophagic flux, together with elevation in the expression of autophagy modulator genes (Beclin-1 and Vps34), to limit cardiomyocyte damage (Hu et al., 2018; Ren et al., 2019).
FGF2 may be involved in the cardioprotective function of soluble epoxide hydrolase (Ephx2), a metabolizing enzyme required for the epoxidation of arachidonic acid. Since the hypertrophic potential of FGF2 is effectively counteracted upon gene inactivation of Ephx2, the cardiac remodeling role of FGF2 is attributed to the function of Ephx2, and subsequent activation of ERK1/2, but not AKT or p38 (Zhang et al., 2014). A similar interaction between FGF2 and ERK1/2 in cardiac fibroblasts plays a role in the development of lipopolysaccharide (LPS)-triggered cardiac adverse events, leading to fibrosis and heart failure (Chen et al., 2019). Along with an elevated number of cardiac mast cells in both ventricles under hypertrophic conditions, overexpression of Fgf2 may denote its pro-fibrotic role in the progress of myocardial fibrosis (Kotov et al., 2020).
Inconsistent findings regarding the positive and inverse correlation of FGF2 and cardiac remodeling may be related to distinct functions of FGF2 isoforms including low- and high-molecular-weight isoforms (LMW and HMW; Okada-Ban et al., 2000). For instance, unlike the cardioprotective effect of LMW FGF2, HMW FGF2 mediates adverse cardiac functions under conditions of myocardial ischemia−reperfusion (Liao et al., 2010).
A mimicry myocardial ischemia-caused injury revealed that an endocrine interplay of upregulated hepatocyte- and adipocyte-derived FGF21 with activation of a βKlotho-FGFR1 complex, and consequently downstream activation of PI3K-AKT1-BAD in cardiomyocytes, ameliorates myocardial actions such as left ventricular function improvement, and exerts a long-term cardioprotective effect (Liu et al., 2013). Seemingly, FGF21 infusion rescues the pathological cardiac remodeling post-MI in a wild-type mouse model, and FGF21 exerts this protective action through an adipokine mediator, termed adiponectin. In other words, the adenoviral vector-based overexpression of FGF21 supports adaptation after MI with manifestations such as left ventricular systolic and dilation improvement (Joki et al., 2015). Another effective hormonal contribution of FGF21 and adiponectin, which are highly secreted due to methionine restriction (MR) in diet, prevents cardiac damages of hyperhomocysteinemia derived from dietary MR in mice (Ables et al., 2015).
Although a low level of FGF9 is encoded in adult cardiomyocytes, exogenous exposure of FGF9 from bone marrow or a transgenic-based stimulation of FGF9 expression in adult myocardium compensates MI-mediated cardiac defects. As a result, systolic physiological activity is restored, thus attenuating the mortality risk of MI (Korf-Klingebiel et al., 2008, 2011). To fulfill this compensatory role after MI, FGF9 triggers vasculogenesis through proliferative effects in endothelial cells and hypertrophic effects in cardiomyocytes, and in general, in the left ventricle. FGF9 mediates these functions through secretion of BMP6, which subsequently activates SMAD1/5 (Korf-Klingebiel et al., 2011).
Positive modulatory effects of FGF1 and FGF2 in the repair of induced ischemia-reperfusion injury and MI were revealed to be based on cardiomyocyte proliferation, stimulation of angiogenesis and the recovery of MI-induced remodeling, systolic and diastolic activities of the heart (Engel et al., 2006; Formiga et al., 2014; Garbayo et al., 2016; Rao et al., 2020). This repairing function of FGF1 is further improved by co-treatment of FGF1 with an inhibitor of p38 MAPK. This inhibitor hampers the activation of p38 MAPK correlated with the suppression of cardiomyocyte proliferation (Engel et al., 2005, 2006). Comparable effects of FGF1 were observed in combination with neuregulin1 (NRG1), which is a receptor tyrosine kinase agonist in the epidermal growth factor receptor family (Fuller et al., 2008; Formiga et al., 2014; Garbayo et al., 2016).
In injury models, FGF-FGFR signal transduction acts in coronary neovascularization required for muscle regeneration. This role in regeneration was evident in zebrafish following cardiac injury. In this process, an epithelial-mesenchymal transition occurs, where epicardial fgfr2 and fgfr4 expression is promoted when Fgf17b from the myocardium in its proximity is released (Lepilina et al., 2006). This function of FGFRs in neovascularization and vascular remodeling was further supported in mouse injury models upon transgenic deletion of Fgfr1/2 from endothelial cells. However, the absence of FGFR1/2 did not impair normal cardiovascular function during homeostasis (Oladipupo et al., 2014; House et al., 2016). Another FGF-dependent induction of angiogenesis was reported in Fgfr2 overexpressing endothelial cells after MI. The action of Fgfr2 overexpression together with the reduction in cardiomyocyte apoptosis after MI is correlated to upregulation of Fgf2 and its autocrine effect (Matsunaga et al., 2009). Anti-apoptotic and pro-angiogenic protective responses following FGF2 treatment after MI in mice were induced through activation of the AKT-hypoxia-inducible factor-1 alpha (HIF-1α)-VEGF axis (Rao et al., 2020). In cardiac regeneration of the zebrafish after injury, the active presence of the FGF-AKT signaling cascade is required for the survival of cardiomyocytes (Tahara et al., 2021). Nevertheless, in these injury model-based studies, differences in postnatal heart regeneration between zebrafish with a complete regeneration capacity and mammals in which the injury in the myocardium scar is mainly covered by fibrotic tissue, have to be considered (Kikuchi and Poss, 2012).
Cardioprotective Response of FGFs Against Oxidative Stress
Oxidative stress is another cardiac insult induced by reactive oxygen species (ROS), and causes deleterious structural and functional effects resulting in the development of heart failure, particularly after MI (Giordano, 2005). A protective role of FGF21 was evidenced against isoproterenol and LPS stimulated ROS generation in cardiac cells and subsequent hypertrophic and pro-inflammatory reactions (Planavila et al., 2015b). Upon gene inactivation of Fgf21 (−/−), isoproterenol, which is a β adrenoreceptor agonist, induces progress of eccentric hypertrophy accompanied by pro-inflammatory reactions, suppression of fatty acid oxidation and augmentation of cardiac weight and dilatation. These phenotypes are restored after administration of FGF21 (Planavila et al., 2013). Indeed, FGF21 secretion from cardiomyocytes is substantially increased in response to cardiac stress (e.g., in MI; Planavila et al., 2015b). Mechanistically, LPS-induced expression of Fgf21 is regulated by the sirtuin-1 (Sirt1) pathway, and the FGF21 mediated reduction of ROS generation is attributable to the activation of antioxidative cascades, e.g., by upregulation of mitochondrial uncoupling proteins (Ucp) 2 and 3, together with superoxide dismutase-2 (Sod2; Planavila et al., 2015a). Thus, FGF21 is involved in protection against injuries from both systemic (in an endocrine fashion of enhanced production by hepatocytes post-MI) and local (paracrine signals by cardiomyocytes) resources (Planavila et al., 2013). Paracrine action of FGF21 in cardiomyocytes, particularly against hypertrophy, is associated with βKlotho-FGFR1c and leads to activation of the MAPK cascade (Planavila et al., 2013, 2015b).
Collectively, to combat cardiopathogenic stress, FGF21 negatively and positively modulates the expression of genes contributing to oxidative and antioxidative signaling cascades, respectively, thereby addressing cardiac remodeling and heart failure (Planavila et al., 2015a; Gómez-Sámano et al., 2017).
Cardioprotective Response of FGFs in Experimental Diabetic Cardiomyopathy
Fibroblast growth factors 9, 16, 19, and 21 protect the diabetic heart against maladaptive cardiac responses and modulate inflammatory reactions.
In a MI model and under a diabetes-inducing genetic modification (db/db), FGF9 and FGF16 suppressed pro-inflammatory responses (i.e., monocyte infiltration, M1 macrophage generation differentiated from monocytes and pro-inflammatory cytokine production). Conversely, anti-inflammatory reactions (i.e., differentiation of M2 macrophages from monocytes and anti-inflammatory cytokines) were augmented. Thus, FGFs 9 and 16 play a cardioprotective role through modulation of inflammation and support cardiac adaptation (e.g., attenuation in the size of the cardiac scar and fibrotic region) post-MI in diabetic patients (Singla et al., 2015; Hu et al., 2017).
Fgf21 ablation in type 1 diabetic mice leads to cardiac remodeling, lipid accumulation and oxidative stress. As a result of this, diabetic cardiomyopathy (DCM)- a severe abnormality in the structure and function of the myocardium- emerges earlier and is more severely deteriorating (Yan et al., 2015). In line with these findings, suppression of FGF21 induced fibrosis and hypertrophy in the heart, while the presence of FGF21 protected against these responses (Chen et al., 2018). Moreover, FGF21 was shown to inhibit the induction of apoptosis of cardiomyocytes occurring during type-1 diabetes. The antiapoptotic effect of FGF 21 was attributed to the activation of p38 MAPK, ERK1/2, and AMP-activated protein kinase (AMPK) pathways (Zhang C. et al., 2015).
Cardiac expression of FGF21 in type-1 diabetic mice and subsequent cardiac defensive actions against maladaptive cardiac events and inflammation are boosted by the administration of fenofibrate. Fenofibrate is a prominent medication for the control of systemic lipid levels and is considered an agonist of peroxisome-proliferator-activated receptor α (PPARα). The fenofibrate-induced cardioprotective function of FGF21 in type-1 diabetes is correlated to Sirt1, another upregulated target of fenofibrate, which rescues disrupted autophagy (Zhang et al., 2016). Similarly, treatment of type-2 diabetes patients with fenofibrate elevated FGF21 serum levels (Ong et al., 2012). FGF21 exerts this protective role against DCM in type 2 diabetes employing AMPK-associated anti-oxidative (AMPK-AKT2-NRF2) and lipid-diminishing (AMPK-ACC-CPT1) pathways (Yang et al., 2018). Similar protection against the deleterious effects of ROS in DCM is mediated by FGF19. To fulfill this anti-oxidative function, a target pathway consisting of AMPK/nuclear erythroid factor 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) is activated (Li et al., 2018).
Interplay of FGFs With TGFβ1-Induced Structural Heart Disorders
Profound extracellular matrix structural alteration, hypertrophy and fibrosis in the heart are induced after continuous exposure toward TGFβ1 and are mediated by miR-132. FGFs mediate contradictory actions on the adverse cardiac responses caused by TGFβ1 which include suppressing (LMW FGF2 and FGF16) versus boosting (FGFs 2 and 23) effects on TGFβ1 functions.
The inhibition of TGFβ1 maladaptive functions using FGF2 in human cardiac myofibroblasts proposes another cardioprotective role for FGF2 against progressive cardiac disorders, such as chamber remodeling and tissue fibrosis (Svystonyuk et al., 2015). Regarding the above-mentioned different actions of FGF2 isoforms, an anti-fibrotic function is induced by LMW FGF2 through antagonizing TGF-β1, thereby compensating the differentiation of fibroblasts to myofibroblasts and attenuating the generation of extracellular matrix (Liguori et al., 2018).
Conversely, FGF2 promotes the expression of Tgf-β1 and thereby hypertrophic reaction and fibrosis. Intriguingly, this is abrogated in the presence of FGF16. In fact, cardiac remodeling upon exposure toward Ang II occurs in the lack of Fgf16, which is correlated to the activation of TGF-β1 as its underlying target (Matsumoto et al., 2013). Thus, Fgf16, which is predominantly encoded in cardiomyocytes, functions as an antagonist for FGF2, which is released from cardiac fibroblasts after hemodynamic stress in the context of cardiac adaptive responses (Kaye et al., 1996; Pellieux et al., 2001; Matsumoto et al., 2013). The antagonizing effect of FGF16 on FGF2 is mediated through competition over FGFR1c in cardiomyocytes (Matsumoto et al., 2013). By this virtue, FGF16 suppressed the proliferative role of FGF2 on cardiomyocytes. Mechanistically, FGF16 exerts inhibitory effects on FGF2-induced PKC isoforms (PKC-α and PKC-ε), but no limiting impact on other targets of FGF2 (e.g., p38 MAPK, ERK1/2, or JNK/SAPK; Lu et al., 2008b). To elicit myocardial regenerative responses post-injury, including cardiomyocyte replication, FGF16 is modulated by GATA4 – as a major cardiac transcription factor controlling the expression of specific genes in the heart (Yu et al., 2016).
In contrast, FGF23 in collaboration with TGFβ1, but not in its absence, induces myocardial fibrosis. In an animal model of experimental hypertrophy that triggers myocardial fibrosis, Fgf23 and Fgfr1 are highly expressed. Indeed, FGF23 originating from cardiomyocytes elicits a boosting effect on fibrosis in a paracrine manner through FGFR1 on cardiac fibroblasts (Kuga et al., 2020). In another hypertrophic and fibrotic model triggered by Ang II, the observed adverse structural and functional cardiac events are attributed to FGF23 activating the TGFβ1-miR-132 axis (Ding et al., 2019).
Cardiopathophysiological Roles of FGFs in Human Diseases
Impairment of FGF signaling is correlated with congenital and metabolic diseases, as well as a range of cancer types (Carter et al., 2015). In the context of cardiac disorders, either FGFs (e.g., 2, 21, and 23) are major contributors in the development of these diseases, or their serum levels are altered as a consequence of cardiac diseases (e.g., 15/19, 21, and 23). This introduces them as serum biomarkers for the prediction and diagnosis of cardiac diseases (Itoh et al., 2016; Emrich et al., 2019). In the following a range of FGF-related cardiac complications is explained (Table 3):
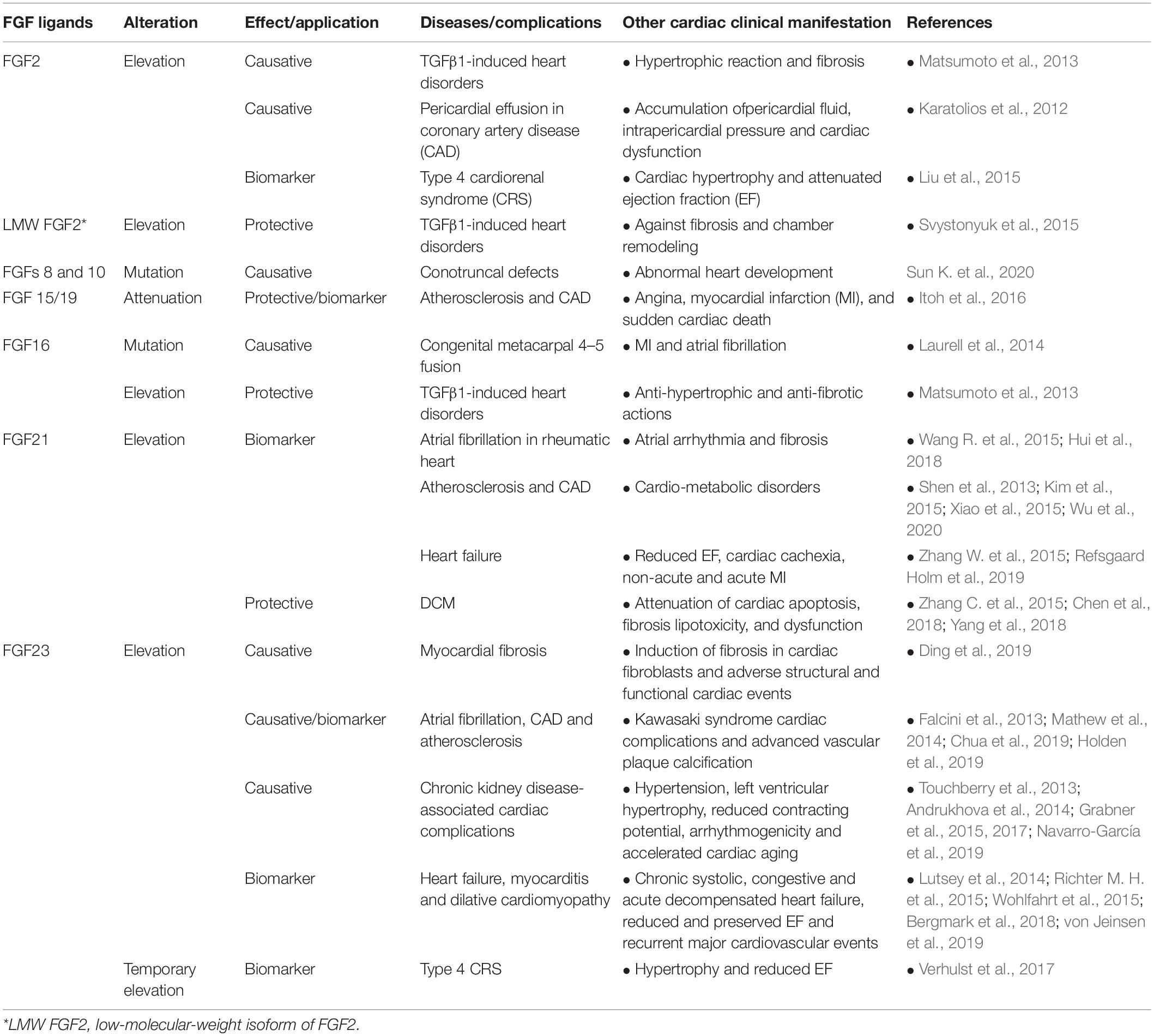
Table 3. Secreted FGFs involved in/against cardiac pathogenicity and their potential as serum biomarker in diseases.
Pericardial Effusion
Pericardial effusion is mainly caused by inflammatory responses, and manifests in the malformation and accumulation of pericardial fluid, resulting in augmented intrapericardial pressure and consequently cardiac dysfunction (Karatolios et al., 2012). As a possible immune reaction under pathogenic conditions, FGF2 can be generated by immune cells (e.g., T-lymphocytes and macrophages; Kuwabara et al., 1995; Peoples et al., 1995). Thus, a higher level of FGF2 together with other inflammatory cytokines in the serum and pericardium of patients proposes a causative role for FGF2 in the pathogenesis of pericardial effusion (Karatolios et al., 2012).
Atrial Fibrillation
This atrial complication, which is associated with arrhythmia in atrial chambers, can develop to atrial fibrosis as a severe cardiac remodeling. Atrial fibrillation also occurs in rheumatic heart patients that is correlated to the elevation of FGFs 21 and 23 in their serum and atrial tissues. The negative modulatory effect of FGFs in the maintenance and progress of atrial fibrillation and fibrosis suggested the potential use of FGFs 21 and 23 as diagnostic and therapeutic biomarkers (Wang R. et al., 2015; Chua et al., 2019). Notably, atherosclerosis patients showed a positive correlation between the higher concentration of FGFs 21 and 23 in the serum and the incidence of atrial fibrillation (Mathew et al., 2014; Hui et al., 2018).
Congenital Heart Diseases (CHD)
Metacarpal 4–5 fusion (MF4) disease is identified as a hereditary disorder characterized by the fusion of the fourth and fifth metacarpal bone. Among its clinical manifestations, MI and atrial fibrillation occur which are attributed to FGF16 mutations in the X-chromosome. Indeed, this finding reveals the link of nonsense mutations in FGF16 with CHD (Laurell et al., 2014). Moreover, mutations in FGF8 and 10 are correlated to conotruncal defects, which impair OFT development and are associated with a high mortality rate (Sun K. et al., 2020).
Cardiac Disorders Concomitant of Kidney Diseases
Life-threatening cardiovascular complications such as left ventricular hypertrophy are prevalently associated with chronic kidney disease (CKD) (Grabner et al., 2015). As a causative contributor to this cardiac pathogenicity a substantial increase in FGF23 levels was reported (Jimbo and Shimosawa, 2014; Wyatt and Drüeke, 2016). In kidney homeostasis, circulating FGF23 is predominantly derived from bone and modulates the metabolism of vitamin D and phosphate as well as sodium and chloride through interaction with αKlotho-FGFR1c (Andrukhova et al., 2014; Erben, 2016). Importantly, this hormonal FGF23 elevates sodium uptake and thereby can cause cardiovascular dysfunctions, e.g., hypertension and hypertrophy (Andrukhova et al., 2014). Added to its hypertrophic effect, FGF23 substantially increases intracellular calcium, induces arrhythmogenicity, and attenuates the contracting potential of cardiomyocytes. Notably, these adverse effects on cardiac cell function are prevented by the administration of soluble Klotho (sKlotho; Touchberry et al., 2013; Navarro-García et al., 2019). The sKlotho in the circulation, mainly provided by the kidney, is proteolytically cleaved from transmembrane Klotho and may interact with FGF23-FGFR1. However, the association of sKlotho with FGF23 is yet controversial (Seiler et al., 2013; Marçais et al., 2017; Memmos et al., 2019).
Additionally, the attenuated concentration of sKlotho in type 2 diabetic and hemodialysis patients (in the early and late stages of CKD, respectively) along with the elevated risk of cardiac events may imply this cardioprotective effect of sKlotho (Marçais et al., 2017; Memmos et al., 2019; Silva et al., 2019).
In addition, the expression of Fgf23 and Tgf-β1 and, as a result, the detrimental cardiac outcomes are suppressed by administration of Klotho which inhibits the TGFβ1-miR-132 axis, thus suggesting therapeutic potential against cardiac remodeling (Ding et al., 2019). In this regard, the therapeutic administration of soluble αKlotho could be a treatment strategy to counteract Fgf23 and Tgf-β1-mediated adverse cardiac injury consequences, such as fibrosis and remodeling. Indeed, this coreceptor of FGF23 suppressed the apoptotic and fibrotic action of isoproterenol-caused injury on cardiomyocytes and endothelial cells, however, mechanistically independent of FGF23 contribution (Chen, 2020).
Cohort studies on end-stage renal disease patients with severe cardiac complications indicated enhanced serum levels of FGF23 and calcium. FGF23 elevation is accompanied by an inverse association between miR-497 and FGF23, thus suggesting another protective axis against detrimental cardiac effects of FGF23 (Xu et al., 2019; Liu et al., 2020).
In addition to this indirect endocrine effect, FGF23 can also be released from cardiomyocytes and acts in a paracrine manner (Leifheit-Nestler et al., 2016). In CKD patients, FGF23 is highly upregulated leading to left ventricular hypertrophy by interaction with FGFR4, and subsequently stimulating hypertrophy-inducing cascades of PLCγ-calcineurin-nuclear factor of activated T-cells (NFAT) and Ras/MAPK, however, without the contribution of Klotho cofactor (Faul et al., 2011; Leifheit-Nestler et al., 2016). This FGFR4-dependent hypertrophic effect of FGF23 was further confirmed in rats and mice since hypertrophy was suppressed either in presence of FGF23 blocking antibody or deletion of Fgfr4 (−/−; Grabner et al., 2015).
Intriguingly, in a 5/6 nephrectomized rat renal injury model, FGF23-induced adverse left ventricular remodeling and consequently accelerated cardiac aging are reversible (Grabner et al., 2017). Seemingly, the administration of the vitamin D metabolite calcitriol (1,25-dihydroxyvitamin D), to the renal injury model diminishes this excessive FGF23-FGFR4 signal and thereby inhibits subsequent cardiovascular abnormalities. This indicates a synergic effect of vitamin D deficiency and FGF23 elevation in the development of cardiac hypertrophy (Leifheit-Nestler et al., 2017). These findings open a new avenue for a putative therapeutic strategy by blocking FGF23-FGFR4 in combination with vitamin D or its receptor to prevent or alleviate the massive cardiovascular injuries in CKD patients (Grabner et al., 2017; Leifheit-Nestler et al., 2017).
Despite strong evidence on the effect of elevated FGF23 in cardiac complications followed by kidney disorders, two cohort studies on hemodialysis patients together with non-cardiovascular patients did not manifest any causal correlation between FGF23 levels and cardiovascular disease symptoms (Marthi et al., 2018; Takashi et al., 2020).
Type 4 Cardiorenal Syndrome (CRS)
This chronic class of CRS syndrome is identified by heart dysfunction and adverse remodeling as a consequence of a primary CKD (Clementi et al., 2013). Cardiac hypertrophy and attenuated ejection fraction (EF) in type 4 CRS patients are attributed to augmented oxidative stress. Mechanistically, this oxidative stress causes cardiac injury in a mimicry type 4 CRS rat model by activation nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and subsequently ERK1/2 phosphorylation, leading to an increase in the expression of FGF2. The type 4 CRS injury and phenotypes are reversed by apocynin, a widely used antioxidant that inhibits NADPH oxidase (Liu et al., 2015). Additionally, type 4 CRS coincides with the temporary elevation of FGF23 serum levels (Verhulst et al., 2017).
Coronary Artery Anomalies
FGF15/19 contributes to the regulation of lipid and glucose metabolism and thereby, together with adiponectin, prevent atherosclerosis and other metabolic diseases (Wu and Li, 2011). Coronary artery disease (CAD) as the most prevalent cardiovascular disease with high mortality is an ischemia-related disease that can be accompanied by symptoms such as angina, MI, and sudden cardiac death (Itoh et al., 2016). The attenuated serum level of FGF19 and adiponectin in CAD patients imply a negative correlation of these factors and the severity of CAD, suggesting this factor as a serum biomarker of CAD (Hao et al., 2013).
FGF23 is elevated in CAD patients with a history of type 2 diabetes or a high risk of atherosclerosis (Lutsey et al., 2014; Tuñón et al., 2016; Holden et al., 2019). FGF23 is involved in atherosclerosis-related damage through advanced vascular plaque calcification (Holden et al., 2019). FGF23 levels are increased in the serum of patients with Kawasaki syndrome, as another type of coronary artery abnormality which is associated with severe cardiovascular complications in children (Falcini et al., 2013).
Similarly, FGF21 is elevated in the serum of CAD patients, correlated with pathogenic impaired metabolism of lipids (Lin et al., 2010; Shen et al., 2013; Kim et al., 2015). Excessive serum levels of FGF21 were reported in atherosclerosis patients with ischemic heart disease as well as in non-alcoholic fatty liver disease, type 2 diabetes, and metabolic disorders (Shen et al., 2013; Kim et al., 2015; Xiao et al., 2015; Wu et al., 2020).
FGF2 concentration is increased in the pericardial fluid (compared to the serum) of CAD patients (Karatolios et al., 2012). As mentioned above, FGF2 exerts a beneficial role in heart restoration after MI in mice, prompting its translation into clinical trials (House et al., 2003). However, intramyocardial injection of recombinant FGF2 in CAD patients did not significantly improve symptoms (Simons et al., 2002; Kukuła et al., 2011).
Heart Failure Syndrome Signs and Symptoms
According to the dysfunction level of the left ventricular EF heart failure syndrome can be further divided into two subtypes with preserved EF (≤50%) and reduced EF (≤40%; Tschöpe et al., 2018). An independent association was reported between elevated levels of FGF23 in the serum of heart failure patients with reduced EF or preserved EF (Koller et al., 2015; Almahmoud et al., 2018; Bergmark et al., 2019; Roy et al., 2020). This coincides with a substantial attenuation of plasmatic Klotho (Bergmark et al., 2019). A high concentration of FGF23 was reported in the serum of patients with chronic systolic and congestive heart failure (Wohlfahrt et al., 2015; von Jeinsen et al., 2019).
In up to 20% of heart failure patients with reduced EF, which is accompanied by an inflammatory losing weight syndrome, namely cardiac cachexia, the serum level of FGF21 is also increased (Refsgaard Holm et al., 2019). Augmented circulating FGF21 levels are also evident in other patients with non-acute and acute MI (Zhang W. et al., 2015).
Notably, a similar trend of elevated FGF23 concentration was detected in the serum of patients with recurrent major cardiovascular events, myocarditis, dilative cardiomyopathy and all-cause mortality (Lutsey et al., 2014; Richter M. H. et al., 2015; Bergmark et al., 2018). Mechanistically, the adverse FGF23 effect is associated with macrophage-oncostatin M (OSM) signaling, which is a prominent mediator of heart failure (Richter M. et al., 2015). This correlation is suggested since OSM stimulates the production of FGF23 in cardiomyocytes, but not in non-cardiomyocytes. OSM is a cytokine of the interleukin-6 family, secreted from infiltrated macrophages, and, unlike FGF23, is not considered as a proper biomarker due to its low serum concentration (Richter M. et al., 2015). Newly developed antibody therapies against the OSM receptor (Oβ) may open strategies to block the macrophage-OSM-Oβ cascade and suppress cardiac damages caused by increased FGF23 (Pöling et al., 2014; Richter M. et al., 2015).
Acute Decompensated Heart Failure (ADHF)
A substantially increased degree of FGF23 was detected in the serum of ADHF patients, but not in their myocardium, indicating an endocrine mechanism of FGF23 action in heart failure (Andersen et al., 2016). Thus, FGF23 can be considered a predictive marker of ADHF (Andersen et al., 2016; Emrich et al., 2019).
Diabetic Cardiomyopathy (DCM)
In line with the aforementioned association of FGFs and DCM in experimental models, this correlation was corroborated in clinical studies. In this regard, elevated levels of FGF21 in the serum of DCM patients elicited a cardioprotective function (Ong et al., 2012; Cheng et al., 2016).
The Regenerative Potential of FGFS Through Induction of Cardiomyocyte Proliferation
Cardiovascular diseases are a major cause of death in the industrialized world. Indeed, unraveling the contribution of fetal FGF signaling to cardiac repair and especially cardiomyocyte proliferation may open new avenues to support cardiac regenerative processes.
A gradual attenuation in the proliferative capacity of cardiomyocytes during embryogenesis which exaggerates postnatally (after 1–3 days) with a significant and rapid reduction in the proliferative capacity, massively limits the adult cardiac regeneration rate (Li et al., 1996; Porrello et al., 2011; Ye et al., 2018). The idea of cardiac cell expansion has been tempting in the last decades, aiming for regenerative therapies applicable in cardiac treatment post-MI. Common strategies for cardiac regenerative medicine range from progenitor stem cell therapy (through injection or engineered tissue implantation) to stimulation of cell cycle re-entry in adult cardiomyocytes and direct reprogramming of them (El Agha et al., 2016; Tzahor and Poss, 2017). To this end, the proliferative potential of FGFs (e.g., FGFs 1, 2, 8, 10, and 16) on cardiomyocytes or their progenitor cells during development and homeostasis has been a center of research attention (Sheikh et al., 1999; Engel et al., 2006; Watanabe et al., 2010; Jennbacken et al., 2019).
FGF1 in combination with p38 MAPK inhibitor or NRG1 can promote the proliferation of cardiomyocytes (Engel et al., 2005, 2006; Formiga et al., 2014; Garbayo et al., 2016). This may occur by the interplay between FGF and MAPK signaling cascades in the alleviation of the limited mammalian cardiac repair potential.
A common problem in growth factor employment for clinical therapies is their low bioavailability, rapid elimination and degradation after treatment. To tackle the short stability and half-time issues, a drug delivery system was developed using biomaterials combined with FGFs. Preclinical studies in rats and pigs used biocompatible polymeric poly(lactic-co-glycolic acid) and polyethylene glycol microparticles loaded with FGF1. This beneficial delivery method prevented perturbation in the regenerative function of FGF1 post-MI after administration (Garbayo et al., 2016; Pascual-Gil et al., 2017).
FGF Potential in Cardiomyocyte Cell Cycle Reentry
Notably, FGF10 has been shown to modulate prenatal cardiomyocyte proliferation, while in the postnatal heart the proliferation together with the expression of FGF10 is suppressed. This suggests FGF10 as a putative candidate for the induction of cell cycle re-entry of cardiomyocytes, applicable for cardiac regenerative purposes. During cardiac injury in adult mice, FGF10 promoted the renewal of cardiomyocytes supporting the cardiac repairing function of FGF10 (Rochais et al., 2014). In contrast, a transgenic neonatal mouse model in which Fgf10 in the myocardium was overexpressed was unable in epithelial to mesenchymal transition and improvement of the neonatal cardiac repair capacity after a cryoinjury, regardless of inducing effects on the epicardial cell number (Rubin et al., 2013).
FGF Application in Cell Replacement-Based Therapies
The stimulatory effects of FGFs can be employed to upscale cardiomyocyte cell numbers for stem cell-based clinical intervention and pharmaceutical tests. In the future, these ligands can be further used to enhance cardiomyocyte specification from cardiac progenitor cells, i.e., ESCs and iPSCs. For instance, FGF10 itself and FGF2 in the presence of BMP2 have been shown to be effective in cardiac cell differentiation, and FGF16 in the proliferation of cardiomyocytes from mouse and human ESCs and iPSCs (Kawai et al., 2004; Chan et al., 2010; Yamasaki et al., 2013; Jennbacken et al., 2019).
Another therapeutic model using the cell replacement approach is based on transplantation of engineered cardiac patches transferring ESCs- or hiPSCs-derived cardiomyocytes to the injured heart (Kolossov et al., 2006; Fan et al., 2020). Treatment of these patches with FGF1 and an inhibitor of GSK3 (CHIR99021), which activates Wnt signaling, leads to elevated hiPSCs derived cardiomyocyte cell divisions and increased grafting rate together with improved cardiac function following MI (Fan et al., 2020). Thus, FGF signaling could be exploited in the worldwide challenge of finding proper stem cell-based therapy for heart failure.
To improve cardiac repair after MI, secretome of progenitor cells containing FGFs in combination with other growth factors can be employed (Timmers et al., 2008; Sharma et al., 2017). A recent secretomic analysis study indicated a range of FGFs (1, 4, 9, 16, and 18) with presumed proliferative capacity. FGF16 may exert this capacity by specific targeting of cardiac progenitor cells, and thereby may be beneficial in the recovery from cardiac injuries (Jennbacken et al., 2019).
FGF Application in Direct Reprogramming Based Therapies
Direct reprogramming as another advanced therapeutic strategy aims to counteract MI detrimental effects and to replace lost cardiomyocytes (Yamakawa et al., 2015). FGFs combined with cardiac transcription factors (Gata4, Mef2c, Tbx5, and Hand2) serve in the transdifferentiation of somatic cells (e.g., fibroblasts) to cardiac progenitor cells (Li et al., 2015) or cardiomyocytes, thus bypassing the intermediate conversion step to pluripotent stem cells (Sadahiro et al., 2015). Co-administration of FGFs 2 and 10 together with VEGF induced the reprogramming of fibroblasts to cardiomyocyte-like cells through interplay with PI3K-AKT and p38 MAPK pathways (Yamakawa et al., 2015). Transplantation of FGF2-induced reprogrammed cardiac progenitor cells from dermal fibroblasts in a rat model post-MI effectively recovered cardiac damages (Li et al., 2015).
Anti Cardiotoxicity Potential of FGFS in Therapies
Strong evidence has been provided by animal studies on the stimulation of cardioprotective function or minimizing the detrimental cardiac effects through modulation of FGF-FGR-coreceptors complex members which may be translated into human therapies.
The aforementioned cardioprotective capacity of FGFs 2, 16, and 21 can be used to counteract or limit the adverse cardiac side effects of drugs. In this regard, in a cardiomyocyte cell line and mouse model, severe cardiopathogenic damages of doxorubicin, a chemotherapy medication for cancer treatment, are alleviated (Wang et al., 2013, 2018; Koleini et al., 2017; Wang S. et al., 2017). This co-administration of doxorubicin with FGF21 induced anti-oxidative, anti-apoptotic, and anti-inflammatory (including enhanced Sirt1 function) responses (Wang S. et al., 2017). FGF2 mediates this anti-cardiotoxic action by its LMV isoforms through activation of antioxidant and detoxification cascades (mTOR/Nrf-2/HO-1 pathway; Koleini et al., 2017, 2019). This protective effect induced by the administration of exogenous LMW-FGF2 is replicated by endogenous LMW-FGF2 only if HMW FGF2 is neutralized or eliminated (Koleini et al., 2019).
Conclusion
Continued progress in the characterization of FGF family members with multipotential roles in cardiac events strengthened our knowledge regarding the necessity of these contributions for heart pathophysiology (Figure 3). These effects on the heart range from involvement in embryonic development to homeostasis after birth and in adulthood, from protective responses under pathophysiological attacks to correlation with maladaptations leading to detrimental cardiac effects. Importantly, an essential protective potential against cardiac insults could be conferred to FGFs 1, 2, 9, 16, 19, and 21. In attempts to employ this potential for regenerative purposes, FGFs 1, 2, 10, and 16 have been promisingly tested for the induction of cardiomyocyte differentiation and proliferation from pluripotent stem cells as well as transdifferentiation of cardiac fibroblasts. Interestingly, FGFs involved in the pathological events of the heart are either defensive contributors (e.g., FGFs 2, 15/19, 16, and 21) or considered as causative agents of cardiac diseases and complications (e.g., FGFs 2, 21, and 23) and diagnostic biomarkers (e.g., FGFs 15/19, 21, and 23). FGF ligands, receptors and their antagonists can serve to preclude or alleviate cardiac complications and promote repair. However, this promising body of evidence is predominantly inferred from experimental models. In the future, the beneficial effects of FGFs on the heart should be further investigated in the clinical context. This will result in the creation of new stem cell-based therapies through up-scaling of cardiomyocytes, and the discovery of therapies counteracting detrimental cardiac effects of pathological FGFs by specific blockers.
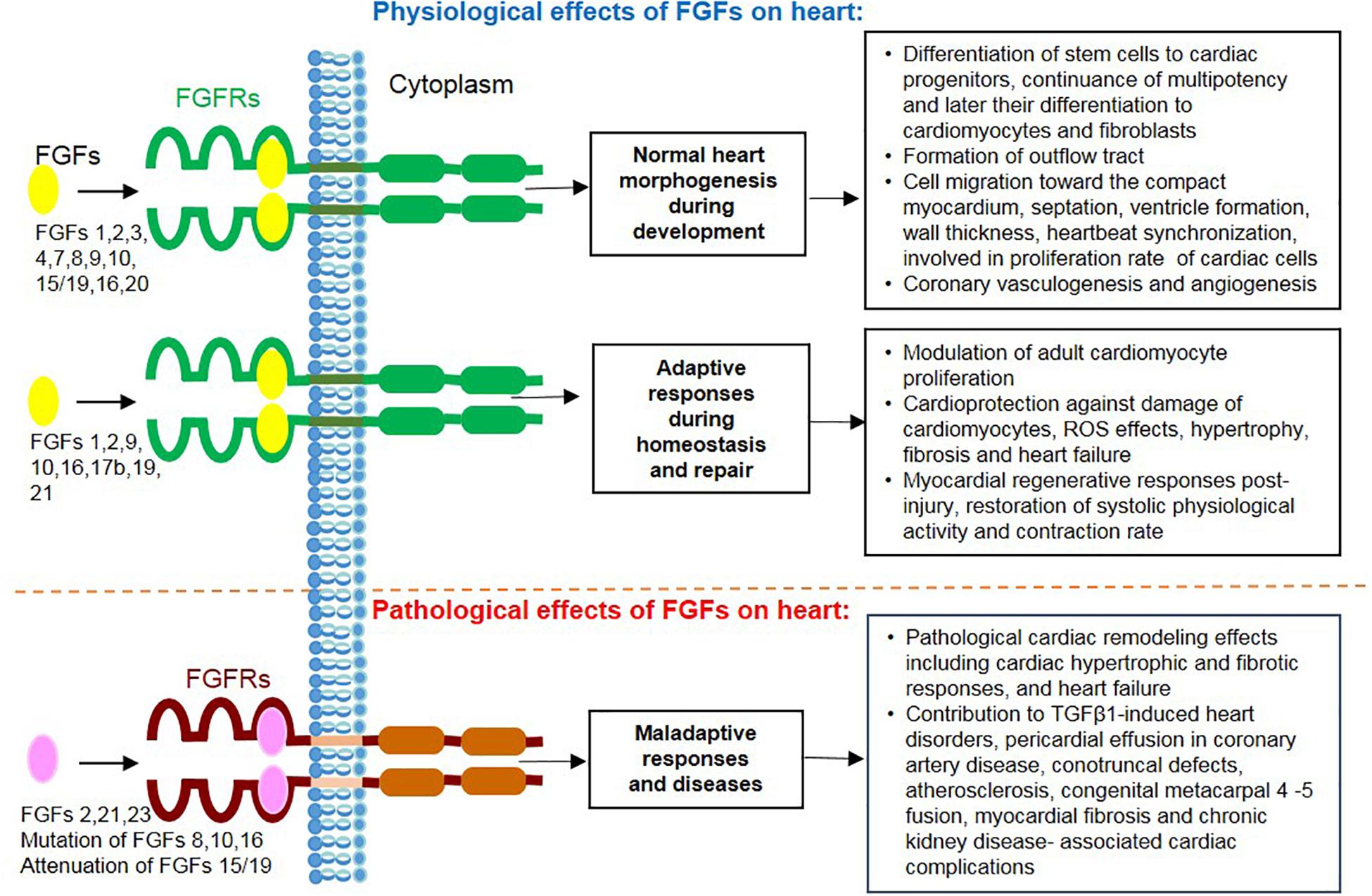
Figure 3. Overview of actions mediated by secreted FGF ligands on the heart including physiological effects during cardiac development, homeostasis and repair together with their pathological and maladaptive effects during heart disease.
Author Contributions
FK made an original plan, wrote the manuscript, and discussed it. NA, SB, and HS wrote the manuscript and discussed it. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a research grant of the University Medical Center Giessen and Marburg (UKGM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ables, G. P., Ouattara, A., Hampton, T. G., Cooke, D., Perodin, F., Augie, I., et al. (2015). Dietary methionine restriction in mice elicits an adaptive cardiovascular response to hyperhomocysteinemia. Sci. Rep. 5:8886. doi: 10.1038/srep08886
Abu-Issa, R., Smyth, G., Smoak, I., Yamamura, K. I., and Meyers, E. N. (2002). Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 129, 4613–4625.
Almahmoud, M. F., Soliman, E. Z., Bertoni, A. G., Kestenbaum, B., Katz, R., Lima, J. A. C., et al. (2018). Fibroblast growth factor-23 and heart failure with reduced versus preserved ejection fraction: MESA. J. Am. Heart Assoc. 7:e008334. doi: 10.1161/JAHA.117.008334
Andersen, I. A., Huntley, B. K., Sandberg, S. S., Heublein, D. M., and Burnett, J. C. (2016). Elevation of circulating but not myocardial FGF23 in human acute decompensated heart failure. Nephrol. Dial. Transplant. 31, 767–772. doi: 10.1093/ndt/gfv398
Andrukhova, O., Slavic, S., Smorodchenko, A., Zeitz, U., Shalhoub, V., Lanske, B., et al. (2014). FGF23 regulates renal sodium handling and blood pressure. EMBO Mol. Med. 6, 744–759. doi: 10.1002/emmm.201303716
Bergmann, O., Zdunek, S., Felker, A., Salehpour, M., Alkass, K., Bernard, S., et al. (2015). Dynamics of cell generation and turnover in the human heart. Cell 161, 1566–1575. doi: 10.1016/j.cell.2015.05.026
Bergmark, B. A., Udell, J. A., Morrow, D. A., Cannon, C. P., Steen, D. L., Jarolim, P., et al. (2018). Association of fibroblast growth factor 23 with recurrent cardiovascular events in patients after an acute coronary syndrome A secondary analysis of a randomized clinical trial. JAMA Cardiol. 3, 473–480. doi: 10.1001/jamacardio.2018.0653
Bergmark, B. A., Udell, J. A., Morrow, D. A., Jarolim, P., Kuder, J. F., Solomon, S. D., et al. (2019). Klotho, fibroblast growth factor-23, and the renin–angiotensin system — an analysis from the PEACE trial. Eur. J. Heart Fail. 21, 462–470. doi: 10.1002/ejhf.1424
Bruneau, B. G. (2013). Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb. Perspect. Biol. 5:a008292. doi: 10.1101/cshperspect.a008292
Carter, E. P., Fearon, A. E., and Grose, R. P. (2015). Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 25, 221–233. doi: 10.1016/j.tcb.2014.11.003
Chan, S. S. K., Li, H. J., Hsueh, Y. C., Lee, D. S., Chen, J. H., Hwang, S. M., et al. (2010). Fibroblast growth factor-10 promotes cardiomyocyte differentiation from embryonic and induced pluripotent stem cells. PLoS One 5:e14414. doi: 10.1371/journal.pone.0014414
Chao, C. M., Yahya, F., Moiseenko, A., Tiozzo, C., Shrestha, A., Ahmadvand, N., et al. (2017). Fgf10 deficiency is causative for lethality in a mouse model of bronchopulmonary dysplasia. J. Pathol. 241, 91–103. doi: 10.1002/path.4834
Chen, C., Meng, Z., Zheng, Y., Hu, B., and Shen, E. (2018). Fibroblast growth factor 21 inhibition aggravates cardiac dysfunction in diabetic cardiomyopathy by improving lipid accumulation. Exp. Ther. Med. 15, 75–84. doi: 10.3892/etm.2017.5375
Chen, L. C., Shibu, M. A., Liu, C. J., Han, C. K., Ju, D. T., Chen, P. Y., et al. (2019). ERK1/2 mediates the lipopolysaccharide-induced upregulation of FGF-2, uPA, MMP-2, MMP-9 and cellular migration in cardiac fibroblasts. Chem. Biol. Interact. 306, 62–69. doi: 10.1016/j.cbi.2019.04.010
Chen, W. Y. (2020). Soluble alpha-klotho alleviates cardiac fibrosis without altering cardiomyocytes renewal. Int. J. Mol. Sci. 21:2186. doi: 10.3390/ijms21062186
Cheng, P., Zhang, F., Yu, L., Lin, X., He, L., Li, X., et al. (2016). Physiological and pharmacological roles of FGF21 in cardiovascular diseases. J. Diabetes Res. 2016:1540267. doi: 10.1155/2016/1540267
Chua, W., Purmah, Y., Cardoso, V. R., Gkoutos, G. V., Tull, S. P., Neculau, G., et al. (2019). Data-driven discovery and validation of circulating blood-based biomarkers associated with prevalent atrial fibrillation. Eur. Heart J. 40, 1268–1276. doi: 10.1093/eurheartj/ehy815
Clementi, A., Virzì, G. M., Goh, C. Y., Cruz, D. N., Granata, A., Vescovo, G., et al. (2013). Cardiorenal syndrome type 4: a review. Cardiorenal Med. 3, 63–70. doi: 10.1159/000350397
Cohen, E. D., Wang, Z., Lepore, J. J., Min, M. L., Taketo, M. M., Epstein, D. J., et al. (2007). Wnt/β-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J. Clin. Invest. 117, 1794–1804. doi: 10.1172/JCI31731
De Smet, F., Tembuyser, B., Lenard, A., Claes, F., Zhang, J., Michielsen, C., et al. (2014). Fibroblast growth factor signaling affects vascular outgrowth and is required for the maintenance of blood vessel integrity. Chem. Biol. 21, 1310–1317. doi: 10.1016/j.chembiol.2014.07.018
Ding, J., Tang, Q., Luo, B., Zhang, L., Lin, L., Han, L., et al. (2019). Klotho inhibits angiotensin II-induced cardiac hypertrophy, fibrosis, and dysfunction in mice through suppression of transforming growth factor-β1 signaling pathway. Eur. J. Pharmacol. 859:172549. doi: 10.1016/j.ejphar.2019.172549
Dongiovanni, P., Crudele, A., Panera, N., Romito, I., Meroni, M., De Stefanis, C., et al. (2020). β-Klotho gene variation is associated with liver damage in children with NAFLD. J. Hepatol. 72, 411–419. doi: 10.1016/j.jhep.2019.10.011
Doroudgar, S., and Glembotski, C. C. (2011). The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol. Med. 17, 207–214. doi: 10.1016/j.molmed.2010.12.003
El Agha, E., Kosanovic, D., Schermuly, R. T., and Bellusci, S. (2016). Role of fibroblast growth factors in organ regeneration and repair. Semin. Cell Dev. Biol. 53, 76–84. doi: 10.1016/j.semcdb.2015.10.009
Emrich, I. E., Brandenburg, V., Sellier, A. B., Schauerte, J., Wiedenroth, J., Untersteller, K., et al. (2019). Strength of fibroblast growth factor 23 as a cardiovascular risk predictor in chronic kidney disease weaken by ProBNP adjustment. Am. J. Nephrol. 49, 203–211. doi: 10.1159/000497125
Engel, F. B., Hsieh, P. C. H., Lee, R. T., and Keating, M. T. (2006). FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc. Natl. Acad. Sci. U.S.A. 103, 15546–15551. doi: 10.1073/pnas.0607382103
Engel, F. B., Schebesta, M., Duong, M. T., Lu, G., Ren, S., Madwed, J. B., et al. (2005). p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 19, 1175–1187. doi: 10.1101/gad.1306705
Erben, R. G. (2016). Update on FGF23 and Klotho signaling. Mol. Cell. Endocrinol. 432, 56–65. doi: 10.1016/j.mce.2016.05.008
Eschenhagen, T., Bolli, R., Braun, T., Field, L. J., Fleischmann, B. K., Frisén, J., et al. (2017). Cardiomyocyte regeneration: a consensus statement. Circulation 136, 680–686. doi: 10.1161/CIRCULATIONAHA.117.029343
Falcini, F., Rigante, D., Masi, L., Covino, M., Franceschelli, F., Leoncini, G., et al. (2013). Fibroblast growth factor 23 (FGF23) gene polymorphism in children with Kawasaki syndrome (KS) and susceptibility to cardiac abnormalities. Ital. J. Pediatr. 39:69. doi: 10.1186/1824-7288-39-69
Fan, C., Tang, Y., Zhao, M., Lou, X., Pretorius, D., Menasche, P., et al. (2020). CHIR99021 and fibroblast growth factor 1 enhance the regenerative potency of human cardiac muscle patch after myocardial infarction in mice. J. Mol. Cell. Cardiol. 141, 1–10. doi: 10.1016/j.yjmcc.2020.03.003
Faul, C., Amaral, A. P., Oskouei, B., Hu, M. C., Sloan, A., Isakova, T., et al. (2011). FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 121, 4393–4408. doi: 10.1172/JCI46122
Felker, A., Prummel, K. D., Merks, A. M., Mickoleit, M., Brombacher, E. C., Huisken, J., et al. (2018). Continuous addition of progenitors forms the cardiac ventricle in zebrafish. Nat. Commun. 9:2001. doi: 10.1038/s41467-018-04402-6
Formiga, F. R., Pelacho, B., Garbayo, E., Imbuluzqueta, I., Díaz-Herráez, P., Abizanda, G., et al. (2014). Controlled delivery of fibroblast growth factor-1 and neuregulin-1 from biodegradable microparticles promotes cardiac repair in a rat myocardial infarction model through activation of endogenous regeneration. J. Control. Release 173, 132–139. doi: 10.1016/j.jconrel.2013.10.034
Fuller, S. J., Sivarajah, K., and Sugden, P. H. (2008). ErbB receptors, their ligands, and the consequences of their activation and inhibition in the myocardium. J. Mol. Cell. Cardiol. 44, 831–854. doi: 10.1016/j.yjmcc.2008.02.278
Garbayo, E., Gavira, J. J., De Yebenes, M. G., Pelacho, B., Abizanda, G., Lana, H., et al. (2016). Catheter-based intramyocardial injection of FGF1 or NRG1-loaded MPs improves cardiac function in a preclinical model of ischemia-reperfusion. Sci. Rep. 6:25932. doi: 10.1038/srep25932
Giordano, F. J. (2005). Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 115, 500–508. doi: 10.1172/JCI200524408
Goetz, R., Ohnishi, M., Ding, X., Kurosu, H., Wang, L., Akiyoshi, J., et al. (2012). Klotho coreceptors inhibit signaling by paracrine fibroblast growth factor 8 subfamily ligands. Mol. Cell. Biol. 32, 1944–1954. doi: 10.1128/mcb.06603-11
Gómez-Sámano, M. Á., Grajales-Gómez, M., Zuarth-Vázquez, J. M., Navarro-Flores, M. F., Martínez-Saavedra, M., Juárez-León, Ó. A., et al. (2017). Fibroblast growth factor 21 and its novel association with oxidative stress. Redox Biol. 11, 335–341. doi: 10.1016/j.redox.2016.12.024
Grabner, A., Amaral, A. P., Schramm, K., Singh, S., Sloan, A., Yanucil, C., et al. (2015). Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 22, 1020–1032. doi: 10.1016/j.cmet.2015.09.002
Grabner, A., Schramm, K., Silswal, N., Hendrix, M., Yanucil, C., Czaya, B., et al. (2017). FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Sci. Rep. 7:1993. doi: 10.1038/s41598-017-02068-6
Guzzetta, A., Koska, M., Rowton, M., Sullivan, K. R., Jacobs-Li, J., Kweon, J., et al. (2020). Hedgehog–FGF signaling axis patterns anterior mesoderm during gastrulation. Proc. Natl. Acad. Sci. U.S.A. 117, 15712–15723. doi: 10.1073/pnas.1914167117
Hao, Y., Zhou, J., Zhou, M., Ma, X., Lu, Z., Gao, M., et al. (2013). Serum levels of fibroblast growth factor 19 are inversely associated with coronary artery disease in Chinese individuals. PLoS One 8:e72345. doi: 10.1371/journal.pone.0072345
Hartung, H., Feldman, B., Lovec, H., Coulier, F., Birnbaum, D., and Goldfarb, M. (1997). Murine FGF-12 and FGF-13: expression in embryonic nervous system, connective tissue and heart. Mech. Dev. 64, 31–39. doi: 10.1016/S0925-4773(97)00042-7
Hennessey, J. A., Wei, E. Q., and Pitt, G. S. (2013). Fibroblast growth factor homologous factors modulate cardiac calcium channels. Circ. Res. 113, 381–388. doi: 10.1161/CIRCRESAHA.113.301215
Holden, R. M., Hétu, M. F., Li, T. Y., Ward, E., Couture, L. E., Herr, J. E., et al. (2019). The heart and kidney: abnormal phosphate homeostasis is associated with atherosclerosis. J. Endocr. Soc. 3, 159–170. doi: 10.1210/js.2018-00311
Hotta, Y., Sasaki, S., Konishi, M., Kinoshita, H., Kuwahara, K., Nakao, K., et al. (2008). Fgf16 is required for cardiomyocyte proliferation in the mouse embryonic heart. Dev. Dyn. 237, 2947–2954. doi: 10.1002/dvdy.21726
House, S. L., Bolte, C., Zhou, M., Doetschman, T., Klevitsky, R., Newman, G., et al. (2003). Cardiac-specific overexpression of fibroblast growth factor-2 protects against myocardial dysfunction and infarction in a murine model of low-flow ischemia. Circulation 108, 3140–3148. doi: 10.1161/01.CIR.0000105723.91637.1C
House, S. L., Castro, A. M., Lupu, T. S., Weinheimer, C., Smith, C., Kovacs, A., et al. (2016). Endothelial fibroblast growth factor receptor signaling is required for vascular remodeling following cardiac ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 310, H559–H571. doi: 10.1152/ajpheart.00758.2015
House, S. L., House, B. E., Glascock, B., Kimball, T., Nusayr, E., Schultz, J. E. J., et al. (2010). Fibroblast growth factor 2 mediates isoproterenol-induced cardiac hypertrophy through activation of the extracellular regulated kinase. Mol. Cell. Pharmacol. 2, 143–154. doi: 10.4255/mcpharmacol.10.20
House, S. L., Wang, J., Castro, A. M., Weinheimer, C., Kovacs, A., and Ornitz, D. M. (2015). Fibroblast growth factor 2 is an essential cardioprotective factor in a closed-chest model of cardiac ischemia-reperfusion injury. Physiol. Rep. 3:e12278. doi: 10.14814/phy2.12278
Hu, S., Cao, S., Tong, Z., and Liu, J. (2018). FGF21 protects myocardial ischemia-reperfusion injury through reduction of miR-145-mediated autophagy. Am. J. Transl. Res. 10, 3677–3688.
Hu, T., Yamagishi, H., Maeda, J., McAnally, J., Yamagishi, C., and Srivastava, D. (2004). Tbx1 regulates fibroblast growth factors in the anterior heart field through reinforcing autoregulatory loop involving forkhead transcription factors. Development 131, 5491–5502. doi: 10.1242/dev.01399
Hu, Y., Li, L., Shen, L., Gao, H., Yu, F., Yin, W., et al. (2017). FGF-16 protects against adverse cardiac remodeling in the infarct diabetic heart. Am. J. Transl. Res. 9, 1630–1640.
Hubert, F., Payan, S. M., and Rochais, F. (2018). FGF10 signaling in heart development, homeostasis, disease and repair. Front. Genet. 9:599. doi: 10.3389/fgene.2018.00599
Hui, T. H., McClelland, R. L., Allison, M. A., Rodriguez, C. J., Kronmal, R. A., Heckbert, S. R., et al. (2018). The relationship of circulating fibroblast growth factor 21 levels with incident atrial fibrillation: the Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 269, 86–91. doi: 10.1016/j.atherosclerosis.2017.12.026
Hutson, M. R., Zeng, X. L., Kim, A. J., Antoon, E., Harward, S., and Kirby, M. L. (2010). Arterial pole progenitors interpret opposing FGF/BMP signals to proliferate or differentiate. J. Cell Sci. 123:e1. doi: 10.1242/jcs.079756
Ilagan, R., Abu-Issa, R., Brown, D., Yang, Y. P., Jiao, K., Schwartz, R. J., et al. (2006). Fgf8 is required for anterior heart field development. Development 133, 2435–2445. doi: 10.1242/dev.02408
Ito, S., Fujimori, T., Furuya, A., Satoh, J., Nabeshima, Y., and Nabeshima, Y. I. (2005). Impaired negative feedback suppression of bile acid synthesis in mice lacking βKlotho. J. Clin. Invest. 115, 2202–2208. doi: 10.1172/JCI23076
Itoh, N., Ohta, H., and Konishi, M. (2015). Endocrine FGFs: evolution, physiology, pathophysiology, and pharmacotherapy. Front. Endocrinol. 6:154. doi: 10.3389/fendo.2015.00154
Itoh, N., Ohta, H., Nakayama, Y., and Konishi, M. (2016). Roles of FGF signals in heart development, health, and disease. Front. Cell Dev. Biol. 4:110. doi: 10.3389/fcell.2016.00110
Jang, Y., Choi, S. C., Lim, D. S., Kim, J. H., Kim, J., and Park, Y. (2020). Modulating cardiomyocyte and fibroblast interaction using layer-by-layer deposition facilitates synchronisation of cardiac macro tissues. Soft Matter 16, 428–434. doi: 10.1039/c9sm01531k
Jennbacken, K., Wågberg, F., Karlsson, U., Eriksson, J., Magnusson, L., Chimienti, M., et al. (2019). Phenotypic screen with the human secretome identifies FGF16 as inducing proliferation of iPSC-derived cardiac progenitor cells. Int. J. Mol. Sci. 20:6037. doi: 10.3390/ijms20236037
Jimbo, R., and Shimosawa, T. (2014). Cardiovascular risk factors and chronic kidney disease - FGF23: a key molecule in the cardiovascular disease. Int. J. Hypertens. 2014:381082. doi: 10.1155/2014/381082
Joki, Y., Ohashi, K., Yuasa, D., Shibata, R., Ito, M., Matsuo, K., et al. (2015). FGF21 attenuates pathological myocardial remodeling following myocardial infarction through the adiponectin-dependent mechanism. Biochem. Biophys. Res. Commun. 459, 124–130. doi: 10.1016/j.bbrc.2015.02.081
Karatolios, K., Moosdorf, R., Maisch, B., and Pankuweit, S. (2012). Cytokines in pericardial effusion of patients with inflammatory pericardial disease. Mediators Inflamm. 2012:382082. doi: 10.1155/2012/382082
Kawai, T., Takahashi, T., Esaki, M., Ushikoshi, H., Nagano, S., Fujiwara, H., et al. (2004). Efficient cardiomyogenic differentiation of embryonic stem cell by fibroblast growth factor 2 and bone morphogenetic protein 2. Circ. J. 68, 691–702. doi: 10.1253/circj.68.691
Kaye, D., Pimental, D., Prasad, S., Mäki, T., Berger, H. J., McNeil, P. L., et al. (1996). Role of transiently altered sarcolemmal membrane permeability and basic fibroblast growth factor release in the hypertrophic response of adult rat ventricular myocytes to increased mechanical activity in vitro. J. Clin. Invest. 97, 281–291. doi: 10.1172/JCI118414
Kelly, R. G., Brown, N. A., and Buckingham, M. E. (2001). The arterial pole of the mouse heart forms from fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell 1, 435–440. doi: 10.1016/S1534-5807(01)00040-5
Kharitonenkov, A., Shiyanova, T. L., Koester, A., Ford, A. M., Micanovic, R., Galbreath, E. J., et al. (2005). FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635. doi: 10.1172/JCI23606
Kikuchi, K., and Poss, K. D. (2012). Cardiac regenerative capacity and mechanisms. Annu. Rev. Cell Dev. Biol. 28, 719–741. doi: 10.1146/annurev-cellbio-101011-155739
Kim, W. J., Kim, S. S., Lee, H. C., Song, S. H., Bae, M. J., Yi, Y. S., et al. (2015). Association between serum fibroblast growth factor 21 and coronary artery disease in patients with type 2 diabetes. J. Korean Med. Sci. 30, 586–590. doi: 10.3346/jkms.2015.30.5.586
Koleini, N., Nickel, B. E., Wang, J., Roveimiab, Z., Fandrich, R. R., Kirshenbaum, L. A., et al. (2017). Fibroblast growth factor-2-mediated protection of cardiomyocytes from the toxic effects of doxorubicin requires the mTOR/Nrf-2/HO-1 pathway. Oncotarget 8, 87415–87430. doi: 10.18632/oncotarget.20558
Koleini, N., Santiago, J. J., Nickel, B. E., Sequiera, G. L., Wang, J., Fandrich, R. R., et al. (2019). Elimination or neutralization of endogenous high-molecular-weight FGF2 mitigates doxorubicin-induced cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 316, H279–H288. doi: 10.1152/ajpheart.00587.2018
Koller, L., Kleber, M. E., Brandenburg, V. M., Goliasch, G., Richter, B., Sulzgruber, P., et al. (2015). Fibroblast growth factor 23 is an independent and specific predictor of mortality in patients with heart failure and reduced ejection fraction. Circ. Heart Fail. 8, 1059–1067. doi: 10.1161/CIRCHEARTFAILURE.115.002341
Kolossov, E., Bostani, T., Roell, W., Breitbach, M., Pillekamp, F., Nygren, J. M., et al. (2006). Engraftment of engineered ES cell-derived cardiomyocytes but not BM cells restores contractile function to the infarcted myocardium. J. Exp. Med. 203, 2315–2327. doi: 10.1084/jem.20061469
Korf-Klingebiel, M., Kempf, T., Sauer, T., Brinkmann, E., Fischer, P., Meyer, G. P., et al. (2008). Bone marrow cells are a rich source of growth factors and cytokines: implications for cell therapy trials after myocardial infarction. Eur. Heart J. 29, 2851–2858. doi: 10.1093/eurheartj/ehn456
Korf-Klingebiel, M., Kempf, T., Schlüter, K. D., Willenbockel, C., Brod, T., Heineke, J., et al. (2011). Conditional transgenic expression of fibroblast growth factor 9 in the adult mouse heart reduces heart failure mortality after myocardial infarction. Circulation 123, 504–514. doi: 10.1161/CIRCULATIONAHA.110.989665
Kotov, G., Landzhov, B., Stamenov, N., Stanchev, S., and Iliev, A. (2020). Changes in the number of mast cells, expression of fibroblast growth factor-2 and extent of interstitial fibrosis in established and advanced hypertensive heart disease. Ann. Anat. 232:151564. doi: 10.1016/j.aanat.2020.151564
Krejci, E., Pesevski, Z., Nanka, O., and Sedmera, D. (2016). Physiological role of fgf signaling in growth and remodeling of developing cardiovascular system. Physiol. Res. 65, 425–435. doi: 10.33549/physiolres.933216
Kuga, K., Kusakari, Y., Uesugi, K., Semba, K., Urashima, T., Akaike, T., et al. (2020). Fibrosis growth factor 23 is a promoting factor for cardiac fibrosis in the presence of transforming growth factor-β1. PLoS One 15:e0231905. doi: 10.1371/journal.pone.0231905
Kukuła, K., Chojnowska, L., Da̧browski, M., Witkowski, A., Chmielak, Z., Skwarek, M., et al. (2011). Intramyocardial plasmid-encoding human vascular endothelial growth factor A165/basic fibroblast growth factor therapy using percutaneous transcatheter approach in patients with refractory coronary artery disease (VIF-CAD). Am. Heart J. 161, 581–589. doi: 10.1016/j.ahj.2010.11.023
Kuwabara, K., Ogawa, S., Matsumoto, M., Koga, S., Clauss, M., Pinsky, D. J., et al. (1995). Hypoxia-mediated induction of acidic/basic fibroblast growth factor and platelet-derived growth factor in mononuclear phagocytes stimulates growth of hypoxic endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 92, 4606–4610. doi: 10.1073/pnas.92.10.4606
Laurell, T., Nilsson, D., Hofmeister, W., Lindstrand, A., Ahituv, N., Vandermeer, J., et al. (2014). Identification of three novel fgf16 mutations in x-linked recessive fusion of the fourth and fifth metacarpals and possible correlation with heart disease. Mol. Genet. Genomic Med. 2, 402–411. doi: 10.1002/mgg3.81
Lavine, K. J., White, A. C., Park, C., Smith, C. S., Choi, K., Long, F., et al. (2006). Fibroblast growth factor signals regulate a wave of Hedgehog activation that is essential for coronary vascular development. Genes Dev. 20, 1651–1666. doi: 10.1101/gad.1411406
Lavine, K. J., Yu, K., White, A. C., Zhang, X., Smith, C., Partanen, J., et al. (2005). Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. Cell 8, 85–95. doi: 10.1016/j.devcel.2004.12.002
Leifheit-Nestler, M., Grabner, A., Hermann, L., Richter, B., Schmitz, K., Fischer, D. C., et al. (2017). Vitamin D treatment attenuates cardiac FGF23/FGFR4 signaling and hypertrophy in uremic rats. Nephrol. Dial. Transplant. 32, 1493–1503. doi: 10.1093/ndt/gfw454
Leifheit-Nestler, M., Siemer, R. G., Flasbart, K., Richter, B., Kirchhoff, F., Ziegler, W. H., et al. (2016). Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol. Dial. Transplant. 31, 1088–1099. doi: 10.1093/ndt/gfv421
Lepilina, A., Coon, A. N., Kikuchi, K., Holdway, J. E., Roberts, R. W., Burns, C. G., et al. (2006). A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127, 607–619. doi: 10.1016/j.cell.2006.08.052
Li, F., Wang, X., Capasso, J. M., and Gerdes, A. M. (1996). Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 28, 1737–1746. doi: 10.1006/jmcc.1996.0163
Li, X., Wu, D., and Tian, Y. (2018). Fibroblast growth factor 19 protects the heart from oxidative stress-induced diabetic cardiomyopathy via activation of AMPK/Nrf2/HO-1 pathway. Biochem. Biophys. Res. Commun. 502, 62–68. doi: 10.1016/j.bbrc.2018.05.121
Li, X.-H., Li, Q., Jiang, L., Deng, C., Liu, Z., Fu, Y., et al. (2015). Generation of functional human cardiac progenitor cells by high-efficiency protein transduction. Stem Cells Transl. Med. 4, 1415–1424. doi: 10.5966/sctm.2015-0136
Liao, S., Bodmer, J. R., Azhar, M., Newman, G., Coffin, J. D., Doetschman, T., et al. (2010). The influence of FGF2 high molecular weight (HMW) isoforms in the development of cardiac ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 48, 1245–1254. doi: 10.1016/j.yjmcc.2010.01.014
Liguori, T. T. A., Liguori, G. R., Moreira, L. F. P., and Harmsen, M. C. (2018). Fibroblast growth factor-2, but not the adipose tissue-derived stromal cells secretome, inhibits TGF-β1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Sci. Rep. 8:16633. doi: 10.1038/s41598-018-34747-3
Lin, Z., Wu, Z., Yin, X., Liu, Y., Yan, X., Lin, S., et al. (2010). Serum levels of FGF-21 are increased in coronary heart disease patients and are independently associated with adverse lipid profile. PLoS One 5:e15534. doi: 10.1371/journal.pone.0015534
Liu, C. J., Dib-Hajj, S. D., Renganathan, M., Cummins, T. R., and Waxman, S. G. (2003). Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem. 278, 1029–1036. doi: 10.1074/jbc.M207074200
Liu, D., Zhou, S., and Mao, H. (2020). MicroRNA-497/fibroblast growth factor-23 axis, a predictive indictor for decreased major adverse cardiac and cerebral event risk in end-stage renal disease patients who underwent continuous ambulatory peritoneal dialysis. J. Clin. Lab. Anal. 34:e23220. doi: 10.1002/jcla.23220
Liu, S. Q., Roberts, D., Kharitonenkov, A., Zhang, B., Hanson, S. M., Li, Y. C., et al. (2013). Endocrine protection of ischemic myocardium by FGF21 from the liver and adipose tissue. Sci. Rep. 3:2767. doi: 10.1038/srep02767
Liu, Y., Liu, Y., Liu, X., Chen, J., Zhang, K., Huang, F., et al. (2015). Apocynin Attenuates cardiac injury in type 4 cardiorenal syndrome via suppressing cardiac fibroblast growth factor-2 with oxidative stress inhibition. J. Am. Heart Assoc. 4:e001598. doi: 10.1161/JAHA.114.001598
Lu, S. Y., Jin, Y., Li, X., Sheppard, P., Bock, M. E., Sheikh, F., et al. (2010). Embryonic survival and severity of cardiac and craniofacial defects are affected by genetic background in fibroblast growth factor-16 null mice. DNA Cell Biol. 29, 407–415. doi: 10.1089/dna.2010.1024
Lu, S. Y., Sheikh, F., Sheppard, P. C., Fresnoza, A., Duckworth, M. L., Detillieux, K. A., et al. (2008a). FGF-16 is required for embryonic heart development. Biochem. Biophys. Res. Commun. 373, 270–274. doi: 10.1016/j.bbrc.2008.06.029
Lu, S. Y., Sontag, D. P., Detillieux, K. A., and Cattini, P. A. (2008b). FGF-16 is released from neonatal cardiac myocytes and alters growth-related signaling: a possible role in postnatal development. Am. J. Physiol. Cell Physiol. 294, C1242–C1249. doi: 10.1152/ajpcell.00529.2007
Lu, T. Y., Lin, B., Li, Y., Arora, A., Han, L., Cui, C., et al. (2013). Overexpression of microRNA-1 promotes cardiomyocyte commitment from human cardiovascular progenitors via suppressing WNT and FGF signaling pathways. J. Mol. Cell. Cardiol. 63, 146–154. doi: 10.1016/j.yjmcc.2013.07.019
Luo, W., Zhao, X., Jin, H., Tao, L., Zhu, J., Wang, H., et al. (2015). Akt1 signaling coordinates bmp signaling and β-catenin activity to regulate second heart field progenitor development. Development 142, 732–742. doi: 10.1242/dev.119016
Lutsey, P. L., Alonso, A., Selvin, E., Pankow, J. S., Michos, E. D., Agarwal, S. K., et al. (2014). Fibroblast growth factor-23 and incident coronary heart disease, heart failure, and cardiovascular mortality: the atherosclerosis risk in communities study. J. Am. Heart Assoc. 3:e000936. doi: 10.1161/JAHA.114.000936
Marçais, C., Maucort-Boulch, D., Drai, J., Dantony, E., Carlier, M. C., Blond, E., et al. (2017). Circulating klotho associates with cardiovascular morbidity and mortality during hemodialysis. J. Clin. Endocrinol. Metab. 102, 3154–3161. doi: 10.1210/jc.2017-00104
Marguerie, A., Bajolle, F., Zaffran, S., Brown, N. A., Dickson, C., Buckingham, M. E., et al. (2006). Congenital heart defects in Fgfr2-IIIb and Fgf10 mutant mice. Cardiovasc. Res. 71, 50–60. doi: 10.1016/j.cardiores.2006.03.021
Marthi, A., Donovan, K., Haynes, R., Wheeler, D. C., Baigent, C., Rooney, C. M., et al. (2018). Fibroblast growth factor-23 and risks of cardiovascular and noncardiovascular diseases: a meta-analysis. J. Am. Soc. Nephrol. 29, 2000–2013. doi: 10.1681/ASN.2017121334
Mascheck, L., Sharifpanah, F., Tsang, S. Y., Wartenberg, M., and Sauer, H. (2015). Stimulation of cardiomyogenesis from mouse embryonic stem cells by nuclear translocation of cardiotrophin-1. Int. J. Cardiol. 193, 23–33. doi: 10.1016/j.ijcard.2015.05.019
Mathew, J. S., Sachs, M. C., Katz, R., Patton, K. K., Heckbert, S. R., Hoofnagle, A. N., et al. (2014). Fibroblast growth factor-23 and incident atrial fibrillation: the multi-ethnic study of atherosclerosis (MESA) and the cardiovascular health study (CHS). Circulation 130, 298–307. doi: 10.1161/CIRCULATIONAHA.113.005499
Matsumoto, E., Sasaki, S., Kinoshita, H., Kito, T., Ohta, H., Konishi, M., et al. (2013). Angiotensin II-induced cardiac hypertrophy and fibrosis are promoted in mice lacking Fgf16. Genes Cells 18, 544–553. doi: 10.1111/gtc.12055
Matsunaga, S., Okigaki, M., Takeda, M., Matsui, A., Honsho, S., Katsume, A., et al. (2009). Endothelium-targeted overexpression of constitutively active FGF receptor induces cardioprotection in mice myocardial infarction. J. Mol. Cell. Cardiol. 46, 663–673. doi: 10.1016/j.yjmcc.2009.01.015
Meganathan, K., Sotiriadou, I., Natarajan, K., Hescheler, J., and Sachinidis, A. (2015). Signaling molecules, transcription growth factors and other regulators revealed from in-vivo and in-vitro models for the regulation of cardiac development. Int. J. Cardiol. 183, 117–128. doi: 10.1016/j.ijcard.2015.01.049
Memmos, E., Sarafidis, P., Pateinakis, P., Tsiantoulas, A., Faitatzidou, D., Giamalis, P., et al. (2019). Soluble Klotho is associated with mortality and cardiovascular events in hemodialysis. BMC Nephrol. 20:217. doi: 10.1186/s12882-019-1391-1
Mesbah, K., Rana, M. S., Francou, A., van duijvenboden, K., Papaioannou, V. E., Moorman, A. F., et al. (2012). Identification of a Tbx1/Tbx2/Tbx3 genetic pathway governing pharyngeal and arterial pole morphogenesis. Hum. Mol. Genet. 21, 1217–1229. doi: 10.1093/hmg/ddr553
Morabito, C. J., Dettman, R. W., Kattan, J., Collier, J. M., and Bristow, J. (2001). Positive and negative regulation of epicardial-mesenchymal transformation during avian heart development. Dev. Biol. 234, 204–215. doi: 10.1006/dbio.2001.0254
Mori, S., Hatori, N., Kawaguchi, N., Hamada, Y., Shih, T. C., Wu, C. Y., et al. (2017). The integrin-binding defective FGF2 mutants potently suppress FGF2 signalling and angiogenesis. Biosci. Rep. 37:BSR20170173. doi: 10.1042/BSR20170173
Mori, S., Tran, V., Nishikawa, K., Kaneda, T., Hamada, Y., Kawaguchi, N., et al. (2013). A dominant-negative FGF1 mutant (the R50E Mutant) suppresses tumorigenesis and angiogenesis. PLoS One 8:e57927. doi: 10.1371/journal.pone.0057927
Murakami, M., Nguyen, L. T., Zhang, Z. W., Moodie, K. L., Carmeliet, P., Stan, R. V., et al. (2008). The FGF system has a key role in regulating vascular integrity. J. Clin. Invest. 118, 3355–3366. doi: 10.1172/JCI35298
Navarro-García, J. A., Delgado, C., Fernández-Velasco, M., Val-Blasco, A., Rodríguez-Sánchez, E., Aceves-Ripoll, J., et al. (2019). Fibroblast growth factor-23 promotes rhythm alterations and contractile dysfunction in adult ventricular cardiomyocytes. Nephrol. Dial. Transplant. 34, 1864–1875. doi: 10.1093/ndt/gfy392
Okada-Ban, M. B., Thiery, J. P., and Jouanneau, J. (2000). Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 32, 263–267. doi: 10.1016/S1357-2725(99)00133-8
Oladipupo, S. S., Smith, C., Santeford, A., Park, C., Sene, A., Wiley, L. A., et al. (2014). Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc. Natl. Acad. Sci. U.S.A. 111, 13379–13384. doi: 10.1073/pnas.1324235111
Ong, K. L., Rye, K. A., O’Connell, R., Jenkins, A. J., Brown, C., Xu, A., et al. (2012). Long-term fenofibrate therapy increases fibroblast growth factor 21 and retinol-binding protein 4 in subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 97, 4701–4708. doi: 10.1210/jc.2012-2267
Ornitz, D. M., and Itoh, N. (2015). The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266. doi: 10.1002/wdev.176
Park, E. J., Watanabe, Y., Smyth, G., Miyagawa-Tomita, S., Meyers, E., Klingensmith, J., et al. (2008). An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development 135, 3599–3610. doi: 10.1242/dev.025437
Pascual-Gil, S., Simón-Yarza, T., Garbayo, E., Prósper, F., and Blanco-Prieto, M. J. (2017). Cytokine-loaded PLGA and PEG-PLGA microparticles showed similar heart regeneration in a rat myocardial infarction model. Int. J. Pharm. 523, 531–533. doi: 10.1016/j.ijpharm.2016.11.022
Pellieux, C., Foletti, A., Peduto, G., Aubert, J. F., Nussberger, J., Beermann, F., et al. (2001). Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J. Clin. Invest. 108, 1843–1851. doi: 10.1172/JCI13627
Peoples, G. E., Blotnick, S., Takahashii, K., Freeman, M. R., Klagsbrun, M., and Eberlein, T. J. (1995). T lymphocytes that infiltrate tumors and atherosclerotic plaques produce heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor: a potential pathologic role. Proc. Natl. Acad. Sci. U.S.A. 92, 6547–6551. doi: 10.1073/pnas.92.14.6547
Planavila, A., Redondo, I., Hondares, E., Vinciguerra, M., Munts, C., Iglesias, R., et al. (2013). Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat. Commun. 4:2019. doi: 10.1038/ncomms3019
Planavila, A., Redondo-Angulo, I., Ribas, F., Garrabou, G., Casademont, J., Giralt, M., et al. (2015a). Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc. Res. 106, 19–31. doi: 10.1093/cvr/cvu263
Planavila, A., Redondo-Angulo, I., and Villarroya, F. (2015b). FGF21 and cardiac physiopathology. Front. Endocrinol. 6:133. doi: 10.3389/fendo.2015.00133
Pöling, J., Gajawada, P., Richter, M., Lörchner, H., Polyakova, V., Kostin, S., et al. (2014). Therapeutic targeting of the oncostatin M receptor-β prevents inflammatory heart failure. Basic Res. Cardiol. 109:396. doi: 10.1007/s00395-013-0396-3
Porrello, E. R., Mahmoud, A. I., Simpson, E., Hill, J. A., Richardson, J. A., Olson, E. N., et al. (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. doi: 10.1126/science.1200708
Rao, Z., Shen, D., Chen, J., Jin, L., Wu, X., Chen, M., et al. (2020). Basic fibroblast growth factor attenuates injury in myocardial infarction by enhancing hypoxia-inducible factor-1 Alpha accumulation. Front. Pharmacol. 11:1193. doi: 10.3389/fphar.2020.01193
Rapraeger, A. C., Krufka, A., and Olwin, B. B. (1991). Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science 252, 1705–1708. doi: 10.1126/science.1646484
Rasouli, S. J., El-Brolosy, M., Tsedeke, A. T., Bensimon-Brito, A., Ghanbari, P., Maischein, H. M., et al. (2018). The flow responsive transcription factor Klf2 is required for myocardial wall integrity by modulating Fgf signaling. eLife 7:e38889. doi: 10.7554/eLife.38889
Refsgaard Holm, M., Christensen, H., Rasmussen, J., Johansen, M. L., Schou, M., Faber, J., et al. (2019). Fibroblast growth factor 21 in patients with cardiac cachexia: a possible role of chronic inflammation. ESC Heart Fail. 6, 983–991. doi: 10.1002/ehf2.12502
Ren, Z., Xiao, W., Zeng, Y., Liu, M. H., Li, G. H., Tang, Z. H., et al. (2019). Fibroblast growth factor-21 alleviates hypoxia/reoxygenation injury in H9c2 cardiomyocytes by promoting autophagic flux. Int. J. Mol. Med. 43, 1321–1330. doi: 10.3892/ijmm.2019.4071
Richter, M., Lautze, H. J., Walther, T., Braun, T., Kostin, S., and Kubin, T. (2015). The failing heart is a major source of circulating FGF23 via oncostatin M receptor activation. J. Heart Lung Transplant. 34, 1211–1214. doi: 10.1016/j.healun.2015.06.007
Richter, M. H., Lautze, H., Skwara, W., Schönburg, M., Beiras-Fernandez, A., Werner, I., et al. (2015). Activation of oncostatin M receptor-β in cardiomyocytes increases serum levels of FGF23 during heart failure. J. Heart Lung Transplant. 34:S174. doi: 10.1016/j.healun.2015.01.475
Rochais, F., Sturny, R., Chao, C. M., Mesbah, K., Bennett, M., Mohun, T. J., et al. (2014). FGF10 promotes regional foetal cardiomyocyte proliferation and adult cardiomyocyte cell-cycle re-entry. Cardiovasc. Res. 104, 432–442. doi: 10.1093/cvr/cvu232
Roy, C., Lejeune, S., Slimani, A., de Meester, C., Ahn As S. A., Rousseau, M. F., et al. (2020). Fibroblast growth factor 23: a biomarker of fibrosis and prognosis in heart failure with preserved ejection fraction. ESC Heart Fail. 7, 2494–2507. doi: 10.1002/ehf2.12816
Rubin, N., Darehzereshki, A., Bellusci, S., Kaartinen, V., and Lien, C. L. (2013). FGF10 signaling enhances epicardial cell expansion during neonatal mouse heart repair. J. Cardiovasc. Dis. Diagnosis 1:101. doi: 10.4172/2329-9517.1000101
Sadahiro, T., Yamanaka, S., and Ieda, M. (2015). Direct cardiac reprogramming: progress and challenges in basic biology and clinical applications. Circ. Res. 116, 1378–1391. doi: 10.1161/CIRCRESAHA.116.305374
Sauer, H., Ravindran, F., Beldoch, M., Sharifpanah, F., Jedelská, J., Strehlow, B., et al. (2013). A 2-macroglobulin enhances vasculogenesis/angiogenesis of mouse embryonic stem cells by stimulation of nitric oxide generation and induction of fibroblast growth factor-2 expression. Stem Cells Dev. 22, 1443–1454. doi: 10.1089/scd.2012.0640
Seiler, S., Wen, M., Roth, H. J., Fehrenz, M., Flügge, F., Herath, E., et al. (2013). Plasma Klotho is not related to kidney function and does not predict adverse outcome in patients with chronic kidney disease. Kidney Int. 83, 121–128. doi: 10.1038/ki.2012.288
Sempou, E., Lakhani, O. A., Amalraj, S., and Khokha, M. K. (2018). Candidate Heterotaxy Gene FGFR4 is essential for patterning of the left-right organizer in Xenopus. Front. Physiol. 9:1705. doi: 10.3389/fphys.2018.01705
Sharma, S., Mishra, R., Bigham, G. E., Wehman, B., Khan, M. M., Xu, H., et al. (2017). A deep proteome analysis identifies the complete secretome as the functional unit of human cardiac progenitor cells. Circ. Res. 120, 816–834. doi: 10.1161/CIRCRESAHA.116.309782
Sheikh, F., Fandrich, R. R., Kardami, E., and Cattini, P. A. (1999). Overexpression of long or short FGFR-1 results in FGF-2-mediated proliferation in neonatal cardiac myocyte cultures. Cardiovasc. Res. 42, 696–705. doi: 10.1016/S0008-6363(99)00008-5
Shen, Y., Ma, X., Zhou, J., Pan, X., Hao, Y., Zhou, M., et al. (2013). Additive relationship between serum fibroblast growth factor 21 level and coronary artery disease. Cardiovasc. Diabetol. 12:124. doi: 10.1186/1475-2840-12-124
Silva, A. P., Mendes, F., Carias, E., Gonçalves, R. B., Fragoso, A., Dias, C., et al. (2019). Plasmatic klotho and fgf23 levels as biomarkers of ckd-associated cardiac disease in type 2 diabetic patients. Int. J. Mol. Sci. 20:1536. doi: 10.3390/ijms20071536
Simons, M., Annex, B. H., Laham, R. J., Kleiman, N., Henry, T., Dauerman, H., et al. (2002). Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation 105, 788–793. doi: 10.1161/hc0802.104407
Singla, D. K., Singla, R. D., Abdelli, L. S., and Glass, C. (2015). Fibroblast growth factor-9 enhances M2 macrophage differentiation and attenuates adverse cardiac remodeling in the infarcted diabetic heart. PLoS One 10:e0120739. doi: 10.1371/journal.pone.0120739
Srisakuldee, W., Jeyaraman, M. M., Nickel, B. E., Tanguy, S., Jiang, Z. S., and Kardami, E. (2009). Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc. Res. 83, 672–681. doi: 10.1093/cvr/cvp142
Sugi, Y., Ito, N., Szebenyi, G., Myers, K., Fallon, J. F., Mikawa, T., et al. (2003). Fibroblast growth factor (FGF)-4 can induce proliferation of cardiac cushion mesenchymal cells during early valve leaflet formation. Dev. Biol. 258, 252–263. doi: 10.1016/S0012-1606(03)00099-X
Sun, J., Niu, C., Ye, W., An, N., Chen, G., Huang, X., et al. (2020). FGF13 is a novel regulator of NF-κB and potentiates pathological cardiac hypertrophy. iScience 23, 101627. doi: 10.1016/j.isci.2020.101627
Sun, K., Zhou, S., Wang, Q., Meng, Z., Peng, J., Zhou, Y., et al. (2020). Mutations in fibroblast growth factor (FGF8) and FGF10 identified in patients with conotruncal defects. J. Transl. Med. 18:283. doi: 10.1186/s12967-020-02445-2
Svystonyuk, D. A., Ngu, J. M. C., Mewhort, H. E. M., Lipon, B. D., Teng, G., Guzzardi, D. G., et al. (2015). Fibroblast growth factor-2 regulates human cardiac myofibroblast-mediated extracellular matrix remodeling. J. Transl. Med. 13:147. doi: 10.1186/s12967-015-0510-4
Tacer, K. F., Bookout, A. L., Ding, X., Kurosu, H., John, G. B., Wang, L., et al. (2010). Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 24, 2050–2064. doi: 10.1210/me.2010-0142
Taelman, J., Popovic, M., Bialecka, M., Tilleman, L., Warrier, S., Van Der Jeught, M., et al. (2019). WNT inhibition and increased FGF signaling promotes derivation of less heterogeneous primed human embryonic stem cells, compatible with differentiation. Stem Cells Dev. 28, 579–592. doi: 10.1089/scd.2018.0199
Tahara, N., Akiyama, R., Wang, J., Kawakami, H., Bessho, Y., and Kawakami, Y. (2021). The FGF-AKT pathway is necessary for cardiomyocyte survival for heart regeneration in zebrafish. Dev. Biol. 472, 30–37. doi: 10.1016/j.ydbio.2020.12.019
Takashi, Y., Wakino, S., Minakuchi, H., Ishizu, M., Kuroda, A., Shima, H., et al. (2020). Circulating FGF23 is not associated with cardiac dysfunction, atherosclerosis, infection or inflammation in hemodialysis patients. J. Bone Miner. Metab. 38, 70–77. doi: 10.1007/s00774-019-01027-7
Timmers, L., Lim, S. K., Arslan, F., Armstrong, J. S., Hoefer, I. E., Doevendans, P. A., et al. (2008). Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. 1, 129–137. doi: 10.1016/j.scr.2008.02.002
Touchberry, C. D., Green, T. M., Tchikrizov, V., Mannix, J. E., Mao, T. F., Carney, B. W., et al. (2013). FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am. J. Physiol. Endocrinol. Metab. 304, E863–E873. doi: 10.1152/ajpendo.00596.2012
Tschöpe, C., Birner, C., Böhm, M., Bruder, O., Frantz, S., Luchner, A., et al. (2018). Heart failure with preserved ejection fraction: current management and future strategies: expert opinion on the behalf of the Nucleus of the “Heart Failure Working Group” of the German Society of Cardiology (DKG). Clin. Res. Cardiol. 107, 1–19. doi: 10.1007/s00392-017-1170-6
Tuñón, J., Fernández-Fernández, B., Carda, R., Pello, A. M., Cristóbal, C., Tarín, N., et al. (2016). Circulating fibroblast growth factor-23 plasma levels predict adverse cardiovascular outcomes in patients with diabetes mellitus with coronary artery disease. Diabetes. Metab. Res. Rev. 32, 685–693. doi: 10.1002/dmrr.2787
Tzahor, E., and Poss, K. D. (2017). Cardiac regeneration strategies: staying young at heart. Science 356, 1035–1039. doi: 10.1126/science.aam5894
Urness, L. D., Bleyl, S. B., Wright, T. J., Moon, A. M., and Mansour, S. L. (2011). Redundant and dosage sensitive requirements for Fgf3 and Fgf10 in cardiovascular development. Dev. Biol. 356, 383–397. doi: 10.1016/j.ydbio.2011.05.671
Vega-Hernández, M., Kovacs, A., de Langhe, S., and Ornitz, D. M. (2011). FGF10/FGFR2b signaling is essential for cardiac fibroblast development and growth of the myocardium. Development 138, 3331–3340. doi: 10.1242/dev.064410
Verhulst, A., Neven, E., and D’Haese, P. C. (2017). Characterization of an animal model to study risk factors and new therapies for the cardiorenal syndrome, a major health issue in our aging population. Cardiorenal Med. 7, 234–244. doi: 10.1159/000462984
Vincentz, J. W., McWhirter, J. R., Murre, C., Baldini, A., and Furuta, Y. (2005). Fgf15 is required for proper morphogenesis of the mouse cardiac outflow tract. Genesis 41, 192–201. doi: 10.1002/gene.20114
Virag, J. A. I., Rolle, M. L., Reece, J., Hardouin, S., Feigl, E. O., and Murry, C. E. (2007). Fibroblast growth factor-2 regulates myocardial infarct repair: effects on cell proliferation, scar contraction, and ventricular function. Am. J. Pathol. 171, 1431–1440. doi: 10.2353/ajpath.2007.070003
von Jeinsen, B., Sopova, K., Palapies, L., Leistner, D. M., Fichtlscherer, S., Seeger, F. H., et al. (2019). Bone marrow and plasma FGF-23 in heart failure patients: novel insights into the heart–bone axis. ESC Heart Fail. 6, 536–544. doi: 10.1002/ehf2.12416
Wang, C., Hennessey, J. A., Kirkton, R. D., Wang, C., Graham, V., Puranam, R. S., et al. (2011). Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 109, 775–782. doi: 10.1161/CIRCRESAHA.111.247957
Wang, J., Nachtigal, M. W., Kardami, E., and Cattini, P. A. (2013). FGF-2 protects cardiomyocytes from doxorubicin damage via protein kinase C-dependent effects on efflux transporters. Cardiovasc. Res. 98, 56–63. doi: 10.1093/cvr/cvt011
Wang, J., Xiang, B., Dolinsky, V. W., Kardami, E., and Cattini, P. A. (2018). Cardiac Fgf-16 expression supports cardiomyocyte survival and increases resistance to doxorubicin cytotoxicity. DNA Cell Biol. 37, 866–877. doi: 10.1089/dna.2018.4362
Wang, R., Yi, X., Li, X., and Jiang, X. (2015). Fibroblast growth factor-21 is positively associated with atrial fibrosis in atrial fibrillation patients with rheumatic heart disease. Int. J. Clin. Exp. Pathol. 8, 14901–14908.
Wang, S., Wang, Y., Zhang, Z., Liu, Q., and Gu, J. (2017). Cardioprotective effects of fibroblast growth factor 21 against doxorubicin-induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell Death Dis. 8:e3018. doi: 10.1038/cddis.2017.410
Wang, W., Niu, X., Stuart, T., Jullian, E., Mauck, W. M., Kelly, R. G., et al. (2019). A single-cell transcriptional roadmap for cardiopharyngeal fate diversification. Nat. Cell Biol. 21, 674–686. doi: 10.1038/s41556-019-0336-z
Wang, X., Tang, H., Wei, E. Q., Wang, Z., Yang, J., Yang, R., et al. (2017). Conditional knockout of Fgf13 in murine hearts increases arrhythmia susceptibility and reveals novel ion channel modulatory roles. J. Mol. Cell. Cardiol. 104, 63–74. doi: 10.1016/j.yjmcc.2017.01.009
Wang, Z., Wang, Y., Ye, J., Lu, X., Cheng, Y., Xiang, L., et al. (2015). bFGF attenuates endoplasmic reticulum stress and mitochondrial injury on myocardial ischaemia/reperfusion via activation of PI3K/Akt/ERK1/2 pathway. J. Cell. Mol. Med. 19, 595–607. doi: 10.1111/jcmm.12346
Wang, Z. G., Wang, Y., Huang, Y., Lu, Q., Zheng, L., Hu, D., et al. (2015). bFGF regulates autophagy and ubiquitinated protein accumulation induced by myocardial ischemia/reperfusion via the activation of the PI3K/Akt/mTOR pathway. Sci. Rep. 5:9287. doi: 10.1038/srep09287
Watanabe, Y., Miyagawa-Tomita, S., Vincent, S. D., Kelly, R. G., Moon, A. M., and Buckingham, M. E. (2010). Role of mesodermal FGF8 and FGF10 overlaps in the development of the arterial pole of the heart and pharyngeal arch arteries. Circ. Res. 106, 495–503. doi: 10.1161/CIRCRESAHA.109.201665
Watanabe, Y., Zaffran, S., Kuroiwa, A., Higuchi, H., Ogura, T., Harvey, R. P., et al. (2012). Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nkx2-5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proc. Natl. Acad. Sci. U.S.A. 109, 18273–18280. doi: 10.1073/pnas.1215360109
Wei, E. Q., Sinden, D. S., Mao, L., Zhang, H., Wang, C., and Pitt, G. S. (2017). Inducible Fgf13 ablation enhances caveolae-mediated cardioprotection during cardiac pressure overload. Proc. Natl. Acad. Sci. U.S.A. 114, E4010–E4019. doi: 10.1073/pnas.1616393114
Wills, A. A., Holdway, J. E., Major, R. J., and Poss, K. D. (2008). Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development 135, 183–192. doi: 10.1242/dev.010363
Wilsbacher, L., and McNally, E. M. (2016). Genetics of cardiac developmental disorders: cardiomyocyte proliferation and growth and relevance to heart failure. Annu. Rev. Pathol. Mech. Dis. 11, 395–419. doi: 10.1146/annurev-pathol-012615-044336
Wohlfahrt, P., Melenovsky, V., Kotrc, M., Benes, J., Jabor, A., Franekova, J., et al. (2015). Association of fibroblast growth factor-23 levels and angiotensin-converting enzyme inhibition in chronic systolic heart failure. JACC Heart Fail. 3, 829–839. doi: 10.1016/j.jchf.2015.05.012
Wu, L., Qian, L., Zhang, L., Zhang, J., Zhou, J., Li, Y., et al. (2020). Fibroblast growth factor 21 is related to atherosclerosis independent of nonalcoholic fatty liver disease and predicts atherosclerotic cardiovascular events. J. Am. Heart Assoc. 9, e015226. doi: 10.1161/JAHA.119.015226
Wu, X., and Li, Y. (2011). Therapeutic utilities of fibroblast growth factor 19. Expert Opin. Ther. Targets 15, 1307–1316. doi: 10.1517/14728222.2011.624514
Wyatt, C. M., and Drüeke, T. B. (2016). Fibroblast growth factor receptor 4: The missing link between chronic kidney disease and FGF23-induced left ventricular hypertrophy? Kidney Int. 89, 7–9. doi: 10.1016/j.kint.2015.11.012
Xiao, Y., Liu, L., Xu, A., Zhou, P., Long, Z., Tu, Y., et al. (2015). Serum fibroblast growth factor 21 levels are related to subclinical atherosclerosis in patients with type 2 diabetes. Cardiovasc. Diabetol. 14:72. doi: 10.1186/s12933-015-0229-9
Xu, L., Hu, X., and Chen, W. (2019). Fibroblast growth factor-23 correlates with advanced disease conditions and predicts high risk of major adverse cardiac and cerebral events in end-stage renal disease patients undergoing continuous ambulatory peritoneal dialysis. J. Nephrol. 32, 307–314. doi: 10.1007/s40620-018-0557-4
Yamakawa, H., Muraoka, N., Miyamoto, K., Sadahiro, T., Isomi, M., Haginiwa, S., et al. (2015). Fibroblast growth factors and vascular endothelial growth factor promote cardiac reprogramming under defined conditions. Stem Cell Rep. 5, 1128–1142. doi: 10.1016/j.stemcr.2015.10.019
Yamasaki, S., Nabeshima, K., Sotomaru, Y., Taguchi, Y., Mukasa, H., Furue, M. K., et al. (2013). Long-term serial cultivation of mouse induced pluripotent stem cells in serum-free and feeder-free defined medium. Int. J. Dev. Biol. 57, 715–724. doi: 10.1387/ijdb.130173to
Yan, X., Chen, J., Zhang, C., Zhou, S., Zhang, Z., Chen, J., et al. (2015). FGF21 deletion exacerbates diabetic cardiomyopathy by aggravating cardiac lipid accumulation. J. Cell. Mol. Med. 19, 1557–1568. doi: 10.1111/jcmm.12530
Yang, H., Feng, A., Lin, S., Yu, L., Lin, X., Yan, X., et al. (2018). Fibroblast growth factor-21 prevents diabetic cardiomyopathy via AMPK-mediated antioxidation and lipid-lowering effects in the heart. Cell Death Dis. 9:227. doi: 10.1038/s41419-018-0307-5
Yang, J., Wang, Z., Sinden, D. S., Wang, X., Shan, B., Yu, X., et al. (2016). FGF13 modulates the gating properties of the cardiac sodium channel Nav1.5 in an isoform-specific manner. Channels 10, 410–420. doi: 10.1080/19336950.2016.1190055
Ye, L., D’Agostino, G., Loo, S. J., Wang, C. X., Su, L. P., Tan, S. H., et al. (2018). Early regenerative capacity in the porcine heart. Circulation 138, 2798–2808. doi: 10.1161/CIRCULATIONAHA.117.031542
Yu, C., Wang, F., Kan, M., Jin, C., Jones, R. B., Weinstein, M., et al. (2000). Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J. Biol. Chem. 275, 15482–15489. doi: 10.1074/jbc.275.20.15482
Yu, W., Huang, X., Tian, X., Zhang, H., He, L., Wang, Y., et al. (2016). GATA4 regulates Fgf16 to promote heart repair after injury. Development 143, 936–949. doi: 10.1242/dev.130971
Zhang, C., Huang, Z., Gu, J., Yan, X., Lu, X., Zhou, S., et al. (2015). Fibroblast growth factor 21 protects the heart from apoptosis in a diabetic mouse model via extracellular signal-regulated kinase 1/2-dependent signalling pathway. Diabetologia 58, 1937–1948. doi: 10.1007/s00125-015-3630-8
Zhang, H., Wang, T., Zhang, K., Liu, Y., Huang, F., Zhu, X., et al. (2014). Deletion of soluble epoxide hydrolase attenuates cardiac hypertrophy via down-regulation of cardiac fibroblasts-derived fibroblast growth factor-2. Crit. Care Med. 42, e345–e354. doi: 10.1097/CCM.0000000000000226
Zhang, J., Cheng, Y., Gu, J., Wang, S., Zhou, S., Wang, Y., et al. (2016). Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin. Sci. 130, 625–641. doi: 10.1042/CS20150623
Zhang, J., Lin, Y., Zhang, Y., Lan, Y., Lin, C., Moon, A. M., et al. (2008). Frs2α-deficiency in cardiac progenitors disrupts a subset of FGF signals required for outflow tract morphogenesis. Development 135, 3611–3622. doi: 10.1242/dev.025361
Zhang, J., Tao, R., Campbell, K. F., Carvalho, J. L., Ruiz, E. C., Kim, G. C., et al. (2019). Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 10:2238. doi: 10.1038/s41467-019-09831-5
Zhang, R., Cao, P., Yang, Z., Wang, Z., Wu, J. L., Chen, Y., et al. (2015). Heparan sulfate biosynthesis enzyme, Ext1, contributes to outflow tract development of mouse heart via modulation of FGF signaling. PLoS One 10:e0136518. doi: 10.1371/journal.pone.0136518
Zhang, W., Chu, S., Ding, W., and Wang, F. (2015). Serum level of fibroblast growth factor 21 is independently associated with acute myocardial infarction. PLoS One 10:e0129791. doi: 10.1371/journal.pone.0129791
Keywords: FGF (fibroblast growth factor), heart development, cardiac regeneration, cardiac diseases, cardiac adaptive and maladaptive responses, stem cells
Citation: Khosravi F, Ahmadvand N, Bellusci S and Sauer H (2021) The Multifunctional Contribution of FGF Signaling to Cardiac Development, Homeostasis, Disease and Repair. Front. Cell Dev. Biol. 9:672935. doi: 10.3389/fcell.2021.672935
Received: 26 February 2021; Accepted: 20 April 2021;
Published: 14 May 2021.
Edited by:
Jangho Kim, Chonnam National University, South KoreaReviewed by:
Felix B. Engel, University Hospital Erlangen, GermanyMyung Chul Lee, Harvard Medical School, United States
Copyright © 2021 Khosravi, Ahmadvand, Bellusci and Sauer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farhad Khosravi, farhad.khosravi@physiologie.med.uni-giessen.de