Neutrophil extracellular traps-mediated Beclin-1 suppression aggravates atherosclerosis by inhibiting macrophage autophagy
 Masataka Sano1,2†
Masataka Sano1,2†  Yasuhiro Maejima1*† Shun Nakagama1 Yuka Shiheido-Watanabe1
Yasuhiro Maejima1*† Shun Nakagama1 Yuka Shiheido-Watanabe1  Natsuko Tamura1
Natsuko Tamura1  Kenzo Hirao3 Mitsuaki Isobe4
Kenzo Hirao3 Mitsuaki Isobe4  Tetsuo Sasano1
Tetsuo Sasano1- 1Department of Cardiovascular Medicine, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 2Department of Professional Development, Tokyo Medical and Dental University Hospital, Tokyo, Japan
- 3Department of Cardiovascular Medicine, AOI Universal Hospital, Kawasaki, Japan
- 4Sakakibara Heart Institute, Tokyo, Japan
A growing body of evidence suggests that neutrophil extracellular traps (NETs) critically contribute to the development of atherosclerosis. However, the detailed mechanism of how NETs promote atherogenesis remains unknown. In this study, we explored the role of NETs for promoting atherosclerosis by modulating the activity of autophagy in macrophages. NETs were effectively induced by a nicotine administration to the HL-60 cell-derived neutrophil-like cells. Treatment with NETs markedly suppressed both autophagosome formation and autophagosome–lysosome fusion in 7-ketocholesterol-treated macrophages, which are accompanied by the enhancement of inflammasome activity. NETs upregulate epidermal growth factor receptor (EGFR) activity, which enhances Beclin-1 phosphorylation of the tyrosine residues of Beclin-1 by EGFR, inhibits the PI3 kinase activity of the Beclin1–Vps34 complex, and suppresses autophagosome formation in macrophages. Furthermore, NET-induced activation of EGFR allows Rubicon to increase its expression, thereby suppressing autophagosome-lysosome fusion. In vivo experiments revealed that the suppression of NET formation by ablating peptidyl arginine deiminase-4 in neutrophil leukocytes resulted in the attenuation of atherosclerotic plaques in a nicotine-administered HFD-fed ApoE−/−mice. Taken together, these results suggest that NET-mediated EGFR–Beclin-1 signaling in the macrophages promotes atherogenesis by autophagy inhibition-mediated inflammasome activation.
Introduction
Atherosclerosis causes lethal consequences including the stroke and coronary artery diseases (Roth et al., 2020). Growing body of evidence suggest that the atherosclerotic lesions are developed under the predisposing conditions including hyperlipidemia, hypertension, diabetes, and smoking habits, which result in the inflammatory response of the arterial walls. However, it remains unknown that through the precise molecular mechanisms how to induce the atherosclerosis through a detrimental stimuli-induced inflammatory response.
Neutrophil Extracellular Traps (NETs) are immunoregulatory mechanisms that are formed when the peptidyl arginine deiminase-4 (Pad4) of neutrophil leukocytes is activated in response to an external stimuli, which causes induction of a nuclear histone citrullination, thereby promoting the chromatin release into the extracellular space (Li et al., 2010). NETs include enzymes such as neutrophil elastase (NE), cathepsin-G, myeloperoxidase (MPO), and nucleosomes, a complex of DNA and citrullinated histones (Yang et al., 2016). Actually, NETs are discovered as one of the host defense systems for disarming and killing bacteria in the extracellular spaces (Brinkmann et al., 2004). On the other hand, increasing lines of evidence suggests that the NETs play an important role in the induction and progression of the various immune-mediated diseases such as rheumatoid arthritis, bronchial asthma, malignant neoplasms (Papayannopoulos, 2018), and atherosclerosis (Warnatsch, Ioannou, Wang and Papayannopoulos, 2015). Indeed, previous preclinical studies demonstrated that the suppression of the NETs formation significantly reduced the asprogression of atherosclerosis in ApoE-deficient (ApoE−/−) mice (Knight et al., 2014; Jiménez-Alcázar et al., 2017; Franck et al., 2018). However, the details regarding which NETs are activated by any of the stimuli to develop and progress the atherosclerotic lesions and plaque instability remains unknown.
Autophagy is an intracellular mechanism by which the aggregated proteins and damaged organelles are enveloped in a lipid bilayer called autophagosome and transported to lysosomes for a removal (Mizushima, Levine, Cuervo and Klionsky, 2008). Autophagy plays a role in maintaining the cellular homeostasis and regulating the defense responses to stress, where it plays a protective role by controlling the quality of the intracellular proteins and removing danger signals (Sciarretta, Maejima, Zablocki and Sadoshima, 2018). One of the critical regulators of autophagy is Beclin-1, a mammalian ortholog of the Atg6 in yeast (Liang et al., 1999). Beclin-1 forms three distinct molecular complexes: Beclin-1-Vps34-Vps15-Atg14L complex, Beclin-1-Vps34-Vps15-UVRAG complex, and Beclin-1-Vps34-Vps15-UVRAG-Rubicon complex (Matsunaga et al., 2009; Rong et al., 2009). Among them, different from the other two complexes, a Beclin-1 complex containing Rubicon plays a suppressive role in the regulation of the autophagosome–lysosome fusion, the final step in autophagy, which is mediated by SNARE proteins, including Vamp8 and Syntaxin-17 (Itakura et al., 2012; Nah et al., 2021). Specifically, Rubicon negatively regulates the autophagosome–lysosome fusion by the sequestration of UVRAG from the Beclin-1-associated complex, which results in the attenuation of Vps34 kinase activity (Matsunaga et al., 2009; Zhong et al., 2009). The activity of Beclin-1 is tightly regulated by various post-translational modifications such as phosphorylation (Maejima, Isobe and Sadoshima, 2016). We and other investigators have shown that the serine/threonine kinases phosphorylate Beclin-1, thereby modulating the autophagy machinery by regulating the Beclin-1-associated class III PI3 kinase activity (Wei, Pattingre, Sinha, Bassik and Levine, 2008; Zalckvar, Berissi, Eisenstein and Kimchi, 2009; Fogel et al., 2013; Maejima et al., 2013). Beth Levine and her groups discovered that the activated epidermal growth factor receptor (EGFR) phosphorylates multisite tyrosine residues of Beclin-1 in Tyr229, Tyr233, and Tyr352, which decreases its function and suppresses autophagy (Wei et al., 2013). Such a discovery revealed the molecular link between an oncogenic receptor tyrosine kinase and the core autophagy machinery that critically contributes to tumor progression in cancer cells.
Increasing evidence reveals that autophagy in the macrophages plays a critical role in alleviating the atherosclerosis by degrading inflammasome, a molecular complex that provokes the inflammatory response due to an innate immunity (Liao et al., 2012; Razani et al., 2012; Ito et al., 2021). In addition, it has been shown that the NETs license macrophages for inflammasome activation, thereby developing the atherosclerotic lesions (Kolaczkowska et al., 2013). Furthermore, it has been shown that NETs inhibit autophagy in the macrophages and other cells (Warnatsch et al., 2015).
Based on such previous findings, we hypothesized that NETs would aggravate the atherosclerosis by modulating autophagy machinery and innate inflammatory response in the macrophages. In the present study, we investigated the mechanism of how NETs regulate macrophage autophagy for the development of atherosclerosis in a hyperlipidemic ApoE−/−mice.
Materials and methods
Experimental animals
We used 12-week-old male mice of C57BL/6J wild-type (WT) (CLEA Japan, Tokyo, Japan), ApoE−/−(#002052), Pad4-floxed (Pad4fl/fl; #026708) and Mrp8-Cre transgenic (Tg-Mrp8-Cre; #021614) (Jackson Laboratory, Bar Harbor, ME, United States). To generate a neutrophil-specific Pad4-deficient ApoE−/−(ApoE−/−; Neu-Pad4−/−) mice, Pad4fl/fl mice were crossed with Tg-Mrp8-Cre mice, and then crossed them with an ApoE−/−mice. These animals were housed in a pathogen-free animal care facility under the standard laboratory conditions (27°C, 40%–60% humidity, 12-h light/12-h dark cycle) and allowed full access to fresh water and a standard rodent chow (CLEA Japan, Tokyo, Japan). ApoE−/−mice and ApoE−/−; Neu-Pad4−/−mice were fed a high-fat diet (HFD) of Western type containing 21% pork lard and 0.15% cholesterol (CLEA Japan, Tokyo, Japan) with or without a nicotine tartrate dihydrate (24303-84, Nakalai Tesque, Kyoto, Japan) in drinking water (100 μg/ml) for 20 weeks. All the animal care and experimental procedures were approved by the Tokyo Medical and Dental University Guide for the Care and Use of the Laboratory Animals (Permit Number: A2021-023A), and the Guide for the Care and Use of the Laboratory Animals published by the US National Institutes of Health.
Generation of neutrophil extracellular traps and their purification
HL-60 cells (ATCC: CCL-240) were cultured in the RPMI-1641 (Sigma-Aldrich, St. Louis, MO, United States) supplemented with the 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) and maintained them at 37°C in a 5% CO2 incubator. To differentiate HL-60 cells into a neutrophil-like cells, a total of 200 µM of all-trans retinoic acid (ATRA, #186-01114, Fujifilm-Wako, Tokyo, Japan) was treated into HL-60 cells and incubated them for 72 h, as described previously (Kawakami et al., 2014). NETs were isolated as previously described with a minor modification (Najmeh, Cools-Lartigue, Giannias, Spicer, and Ferri, 2015). Briefly, 50 µM of phorbol 12-myristate 13-acetate (PMA, #P8139, Sigma-Aldrich, St. Louis, MO, United States) or 10 µM of the nicotine was added to the HL-60-derived neutrophil-like cells and incubated for 2 h. After aspirating the supernatant off, the surface of the culture dishes was gently washed with 5 ml of phosphate-buffered saline (PBS). The collected samples with the PBS were spun down at 15,000 rpm × 5 min, then the pellet was resuspended in 100 µl of PBS after removing the supernatant. The induced NETs were determined by the immunofluorescence, scanning electron microscopy, and immunoblotting. After determining the NET DNA concentration as previously described (Warnatsch et al., 2015), the concentration of NETs was adjusted to 500 µg/ml by adding PBS for the co-culture experiments.
Co-culture experiments of macrophages and neutrophil extracellular traps
THP-1 cells (ATCC: TIB-202) were cultured in RPMI-1641 (Sigma-Aldrich, St. Louis, MO, United States) supplemented with the 10% FBS and 1% P/S and maintained them at 37°C in a 5% CO2 incubator. To differentiate the THP-1 cells into macrophages, a total of 50 nM of PMA was treated to THP-1 cells and incubated them for 48 h. Autophagy was induced by adding the 70 µM of 7-ketocholesterol (7KC). To evaluate the effect of the NETs on macrophages, a total of 5 or 50 µg of NETs were added with or without a treatment with the EGFR tyrosine kinase inhibitor AG1478 (#S2728, Selleck Chemicals, Houston TX, United States), and then the samples were collected after 30 min or 4 h.
Immunoblot analyses
Cultured cells were lysed in the lysis buffer [1% Triton X-100 (Sigma-Aldrich), 50-mM Tris, 150-mM NaCl, and 2-mM EDTA diluted with distilled water], phosphatase inhibitor cocktail tablets (PhosSTOP EASYpack; Roche Diagnostics, Indianapolis, IN, United States), and protease inhibitor cocktail (Sigma-Aldrich) on ice for 10 min. The mixture was centrifuged at 15,000 rpm for 10 min, and the supernatant was analyzed via immunoblotting. Equal amounts (10 µg per lane) of the total proteins from each lysate were separated by a sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred into nitrocellulose membranes, which were incubated with the primary antibodies, washed, and then reacted with the secondary antibodies [HRP-linked anti-rabbit IgG (#7074) or HRP-linked anti-mouse IgG (#7076); Cell Signaling] at a room temperature. Specific signals were visualized via chemiluminescence using the SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, Waltham, MA, United States), and immunoreactive bands were visualized by using the quantified iBright1500 imaging system (Thermo Scientific, Waltham, MA, United States). Primary antibodies against the following proteins were used: LC3 (#M186-3; MBL, Nagoya, Japan), p62/SQSTM1 (#PM045; MBL), NLRP3 (#A017374 Atlas Antibodies, Bromma, Sweden), Cleaved IL-1β (Asp116) (#83186; Cell Signaling), Beclin-1 (#3738; Cell Signaling), NE (#ab131260; Abcam), Citrullinated Histone H3 (#ab5103; Abcam), EGFR (#4267; Cell Signaling), phospho-EGFR (Tyr1068) (#3777; Cell Signaling), DYKDDDDK (=Flag, #14793; Cell Signaling), Vamp8 (#ab76021; Abcam), Syntaxin-17 (#ab229646; Abcam), Rubicon (#M170-3; MBL), and β-actin (#4970; Cell Signaling).
Enzyme-linked immunosorbent assay
The culture media of the THP-1-derived macrophages were analyzed via ELISA for the levels of the IL-1β (Human IL-1β ELISA MAX Deluxe Set; BioLegend, San Diego, CA, United States), according to the manufacturer’s protocols. Briefly, the culture media of the THP-1-derived macrophages or standards were added to 96-well plates coated with the capture antibody (anti-IL-1β) and incubated at room temperature for 2 h. The solutions of a detection antibody and Avidin-HRP were sequentially added to the plates, washing and incubating between each adding step. After adding substrate solution to each well of the plates, read absorbance at 450 nm.
Immunoprecipitation assay
Differentiated THP-1 cells transduced with an adenovirus harboring Flag-Beclin-1 for 48 h with or without treatment with 7KC, NETs (50 µg), and AG1478 centrifuged at 1,000 × g for 10 min, and the pellets were resuspended in 500 µl of lysis buffer (50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 1 mM PMSF, 10 µg/ml aprotinin, protease inhibitor cocktail, and phosphatase inhibitor) for 10 min on ice. After centrifugation at 15,000 × g for 10 min, the supernatants were incubated with anti-Flag M2 magnetic beads (Sigma, M8823-1ML, Lot# SLBJ 3044V) for 1 h at 4°C, which were then washed three times with 500 µl of Tris-buffered saline with 0.1% Tween 20 at 4°C. The beads were resuspended in a 2 × sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer, resolved by SDS-PAGE, and analyzed by immunoblotting with an anti-tyrosine phosphorylation antibody (#ab10321; Abcam).
Evaluation of the whole aortic plaque lesions
Mice were euthanized through an overdose of three types of the mixed anesthetic agents (medetomidine, midazolam, and butorphanol at the concentrations of 0.15, 2.0, and 2.5 mg/kg, respectively). The whole aortas were excised from the carcasses. The whole aortas were opened longitudinally from an ascending aorta to the iliac bifurcation, pinned en face, and stained for lipids with the Oil Red O. The stained area was identified as the atherosclerotic lesion area and evaluated as a percentage of the total aortic area by using the ImageJ software (National Institutes of Health, Bethesda, MD, United States).
Immunocytochemistry
HL-60-derived neutrophil-like cells with or without the PMA or nicotine treatment seeded on the chamber slides were fixed with the 4% paraformaldehyde (PFA) for 10 min. After washing with the PBS and permeabilization with the 1% Triton X-100, the cells were incubated with a blocking buffer [5% bovine serum albumin (BSA), 0.1% Triton X-100] and then with the primary antibodies [Citrullinated Histone H3, #ab5103, Abcam; Myeloperoxidase (MPO), #ab208670, Abcam] overnight at 4°C. After washing with the PBS, Alexa Fluor-conjugated (488) secondary antibody was applied with the blocking buffer. Finally, coverslips were mounted on the slides using a Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs, Burlingame, CA, United States), and photographed.
Immunohistochemistry
Thin-sliced paraffin-embedded histological sections were processed with antigen retrieval procedures after deparaffinization. To reduce nonspecific binding, sections were incubated with 10% normal bovine serum at room temperature. To stain for macrophages or neutrophil leukocytes, sections were incubated with antibodies against CD11b (#ab128797, Abcam) or MPO (#ab208670, Abcam) at 4°C overnight. After washing with PBS, the tissues were incubated with HRP-labeled polymer anti-mouse antibodies (Dako, K4007) for 30 min at room temperature, and the conjugates were detected using Histofine Simple Stain AEC solution (#H1506, Nichirei Corporation, Tokyo, Japan). Sections were counterstained with hematoxylin. Immunofluorescence double staining for the histological sections was performed to examine the localization of NETs in the atherosclerotic plaques using the same protocol of immunocytochemistry described above. Specimens were observed under confocal microscopy.
Scanning electron microscopy
The HL-60-derived neutrophil-like cells with or without the PMA or nicotine treatment were fixed with 2.5% glutaraldehyde for 2–5 h, washed overnight in 0.1 M PBS, post-fixed with the 2% OsO4 for 2 h, and then dehydrated in the ascending concentrations of ethanol (50%, 70%, 80%, 90%, 95%, and 100%). Then, the specimens were washed with a 3-methylbutyl acetate for 20 min, dried to the critical point, attached to an aluminum mount using a sliver paste, coated with the 7 nm of the titanium, and examined under a scanning electron microscope (S-4500/EMAX-7000, Hitachi) at the different magnifications.
Transmission electron microscopy
The THP-1-derived macrophages treated with or without a 7KC and/or NETs were fixed in the 2.5% glutaraldehyde in 0.1 M phosphate buffer for 2 h, washed with a 0.1 M PB, post-fixed in a 1% osmium tetroxide in the 0.1 M PB for 2 h, dehydrated in a graded series of ethanol, and embedded in the Epon 812 (TAAB Laboratory Equipment, Aldermaston, United Kingdom). Semi-thin sections were cut at a thickness of 1 μm and stained with a toluidine blue. Ultrathin 90-nm sections were collected on the copper grids, double-stained with the uranyl acetate and lead citrate, and then observed under a transmission electron microscope (H7011; Hitachi High-tech, Tokyo, Japan).
Phosphatidylinositol 3-kinase assay in-situ
To measure the abundance of a membrane-associated phosphatidyl inositol-3-phosphate to evaluate the activity of the Beclin-1-Vps34 complex in-situ, we previously generated an Ad-GFP-2×FYVE, which harbors the FYVE (Fab1, YOTB, Vac1p, and EEA1) domain (Maejima et al., 2013). The amino acid sequence we used for this adenovirus construction was as follows: NH2-WVPDSQAPNCMKCEARFTFTKRRHHCRACGKVFCASCCSLKCKLLYMDRKEARVCVICHSVL-COOH. The THP-1-derived macrophages grown on coverslips were transduced with an Ad-GFP-2×FYVE. After 48 h, media were changed into a complete or glucose-free, and cells were treated with or without a 7KC and/or NETs for 2 h. Cells were fixed with a 4% PFA in PBS for 15 min. The number of GFP-2×FYVE dots in the cytoplasm was counted in five independent visual fields.
Evaluation of fluorescent LC3 puncta
The THP-1-derived macrophages grown on the coverslips were transduced with an adenovirus harboring mRFP-GFP-LC3 (Ad-tf-LC3) at 15 MOI (Hariharan et al., 2010). Twenty-four hours after the adenovirus transduction, the cells were washed with a PBS, fixed with 4% PFA, mounted on the slides using a Vectashield with DAPI (Vector Labs, Burlingame, CA, United States), and viewed under a fluorescence microscope. The number of the GFP and mRFP dots was determined by a manual counting of fluorescent puncta in the five fields from three different cell preparations. The number of dots per cell was obtained by dividing the total number of dots by the number of the nuclei in each microscopic field.
Statistics
All the statistical analyses were conducted using the GraphPad Prism (GraphPad Software, Inc., San Diego, CA, United States). The normality of distribution was determined using a D’Agostino-Pearson normality test and confirmed that all data were normally distributed. All statistical data are expressed as mean ± standard error of the mean (SEM). All statistical analyses were performed using an unpaired Student’s t-test, one-way analysis of variance (ANOVA) or two-way ANOVA, followed by the post-hoc Bonferroni–Dunn method for multiple pairwise comparisons. In all cases, the results were considered statistically significant at a p-value < 0.05.
Results
Nicotine induces neutrophil extracellular traps that contains abundant neutrophil elastase
Previous reports have shown that the nicotine induces NETs from an isolated human and mouse neutrophil leukocytes (Hosseinzadeh, Thompson, Segal and Urban, 2016; Lee et al., 2017). To establish an experimental system to obtain great amounts of NETs, we determined whether nicotine could induce NET formation from a differentiated HL-60 cells. To this end, we differentiated the human HL-60 cells into neutrophil-like cells by treating HL-60 cells with 200 µM of ATRA for 72 h (Figure 1A). Treatment with 50 µM of PMA, a well-known inducer of NETs, to neutrophil-like cells could effectively induce NETs (Figure 1B and Supplementary Figure S1). Similarly, treatment with 10 µM of nicotine to neutrophil-like cells resulted in the induction of NET formation (Figure 1B and Supplementary Figure S1). Consistently, immunoblot analyses revealed that the treatment with 10 µM of nicotine to neutrophil-like cells caused Histone H3 citrullination (Figure 1C). Furthermore, the abundance of NE, one of the major components of the NETs, was induced by the addition of nicotine (Figure 1C). These results suggest that the treatment with nicotine to neutrophil leukocytes promotes Histone H3 citrullination, thereby provoking NET formation, which results in the enhancement of NE release to the extracellular space.
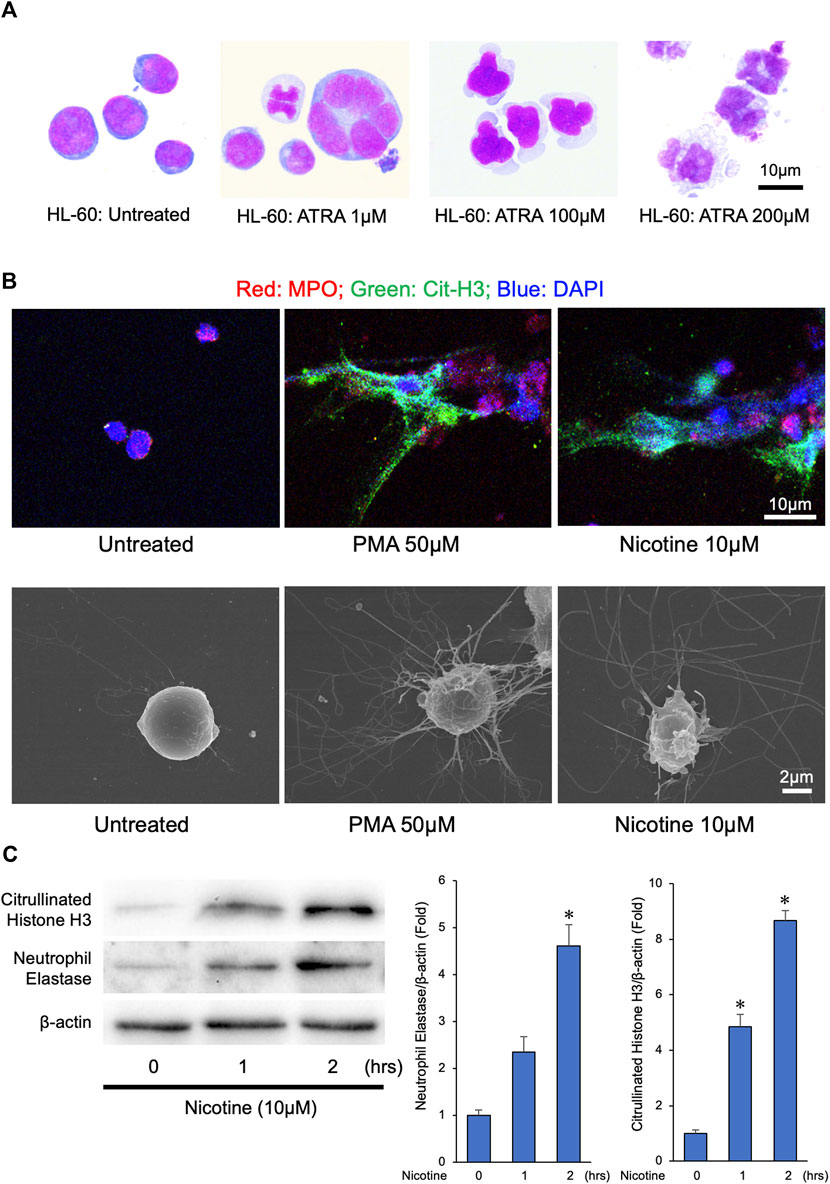
FIGURE 1. NETs were released from differentiated HL-60 in response to phorbol 12-myristate 13-acetate (PMA) or nicotine stimulation. (A) Human HL-60 cells were differentiated into neutrophil-like cells by treating HL-60 cells with ATRA (200 µM) for 72 h (B) NETs were induced by PMA (50 µM) or nicotine (10 µM) administration to differentiated HL-60 cells. Upper panels: Representative confocal immunofluorescence images of differentiated HL-60 cells with or without PMA or nicotine administration stained for myeloperoxidase (MPO, red), citrullinated histone 3 (Cit-H3, green), and DNA (DAPI, blue). Lower panels: Representative scanned electron microscopic images of the differentiated HL-60 cells with or without the PMA or nicotine administration. (C) Left panel: Representative immunoblot images of neutrophil elastase, citrullinated Histone H3, and β-actin. Right panels: Densitometric analysis of immunoblots. *p < 0.05 vs. 0-h, n = 3 in each group. All data are expressed as mean ± SEM.
Neutrophil extracellular traps attenuate autophagosome formation and enhance inflammasome activity in macrophages
To analyze the effect of NETs on autophagosome formation in the macrophages, we conducted a couple of experiments in which the NETs released from neutrophil-like cells were added to the macrophages derived from human THP-1 cells. To mimic the role of macrophage autophagy in the pathological status of atherosclerosis, we used 7KC, an atherosclerotic lesioned oxysterol that promotes oxidative stress, inflammation, and autophagy, as an inducer of autophagosome formation (Liao et al., 2012; Ito et al., 2021). When macrophages were treated with a 7KC, the number of autophagosomes in the macrophages were increased, which was confirmed by a transmission electron microscopic examination (Figure 2A). Immunoblot analyses showed that the 7-KC treatment significantly increased the accumulation of LC3-II protein in the macrophages (Figure 2B), suggesting that the 7KC significantly promotes autophagosome formation in the macrophages. The addition of NETs significantly reduced the 7KC-induced autophagosome formation in a concentration-dependent manner as evidenced by the number of autophagosomes observed via transmission electron microscopy (Figure 2A). Immunoblot analyses consistently demonstrated that the treatment with a NETs significantly decreased the amount of LC3-II that was enhanced by a 7KC (Figure 2B). Treatment of macrophages with a 7-KC increased the expression of inflammasome components such as NLRP3 and cleaved IL-1β, and the addition of NETs further increased the amount of these inflammasome components in a concentration-dependent manner (Figures 2B,C). These results indicate that the NETs suppress autophagosome formation and enhance the inflammasome activity induced by a 7KC in the macrophages.
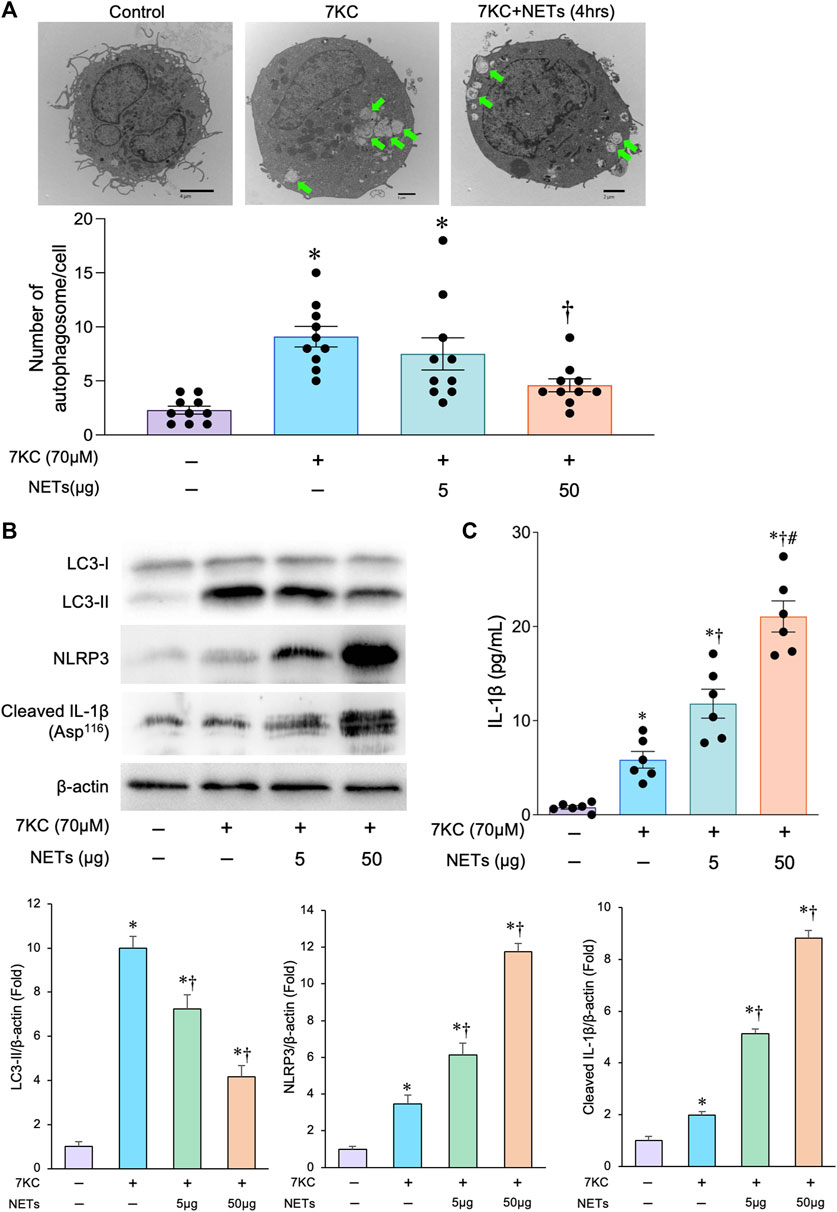
FIGURE 2. Effects of 7KC and NETs on macrophage autophagy in-vitro. (A) Upper panels: Representative transmission electron microscopic images of autophagosomes (arrows) in the macrophages differentiated from THP-1 cells with and without the administration of 7KC and or NETs. Lower panel: Quantitative analysis of the number of autophagosomes with and without the administration of 7KC and or NETs. *p < 0.05 vs. control group (Purple bar), †p < 0.05 vs. 7KC-treated group (Blue bar), n = 10 in each group. Data are expressed as dot plots and bars, with lines indicating the mean ± SEM. (B) Upper panel: Representative immunoblot images of LC3, NLRP3, cleaved IL-1β, and β-actin. Lower panels: Densitometric analysis of immunoblots. *p < 0.05 vs. control group (Purple bar), †p < 0.05 vs. 7KC-treated group (Blue bar), n = 3 in each group. All data are expressed as mean ± SEM. (C) Macrophages differentiated from the THP-1 cells with and without the administration of the 7KC and or NETs were analyzed for IL-1β secretion in the supernatants by the ELISA. *p < 0.05 vs. control group (Purple bar), †p < 0.05 vs. 7KC-treated group (Blue bar), #p < 0.05 vs. 7KC + NETs (5 μg)-treated group (Green bar), n = 6 in each group. Data are expressed as dot plots and bars, with lines indicating the mean ± SEM.
Suppression of neutrophil extracellular traps formation resulted in the attenuation of atherosclerotic plaques in high-fat diet-fed ApoE−/−mice
Histopathological analyses of atherosclerotic lesions of the HFD-fed ApoE−/−mice revealed the localization of both the macrophages containing numerous autophagosomes and neutrophil leukocytes in the atherosclerotic plaques, suggesting that both cells would be contributing to the development of atherosclerotic lesions (Figure 3A and Supplementary Figure S2A). To investigate the effect of NETs on atherosclerosis in vivo, we generated neutrophil-specific Pad4 knockout mice (ApoE−/−; Neu-Pad4−/−) by crossing Pad4fl/fl mice with Tg-Mrp8-Cre mice and then crossing them with an ApoE−/−mice. Both the ApoE−/−; Neu-Pad4−/−mice and ApoE−/−mice were fed with a HFD and given nicotine to induce NET formation in vivo (Supplementary Figure S2B). NETs could be detected in atherosclerotic lesions from HFD-fed ApoE−/−mice and ApoE−/−; Neu-Pad4−/−mice treated with nicotine (Figure 3B). Feeding of HFD increased the serum low-density lipoprotein cholesterol (LDL-C) level in ApoE−/−mice (Table 1). On the contrary, neither neutrophil-specific Pad4 ablation nor nicotine administration significantly changed the LDL-C level in the serum of experimental mice (Table 1). When nicotine was administered to the HFD-fed ApoE−/−mice, the area of the atherosclerotic lesions was markedly enlarged (Wang et al., 2017). On the other hand, the area of the atherosclerosis was significantly smaller in ApoE−/−; Neu-Pad4−/−mice fed with a HFD than in ApoE−/−mice, regardless of whether the nicotine was administered or not (Figure 3C). These results indicate that the nicotine treatment increases the size of atherosclerotic lesions in a HFD-loaded ApoE−/−mice, possibly through promoting the NETs formation.
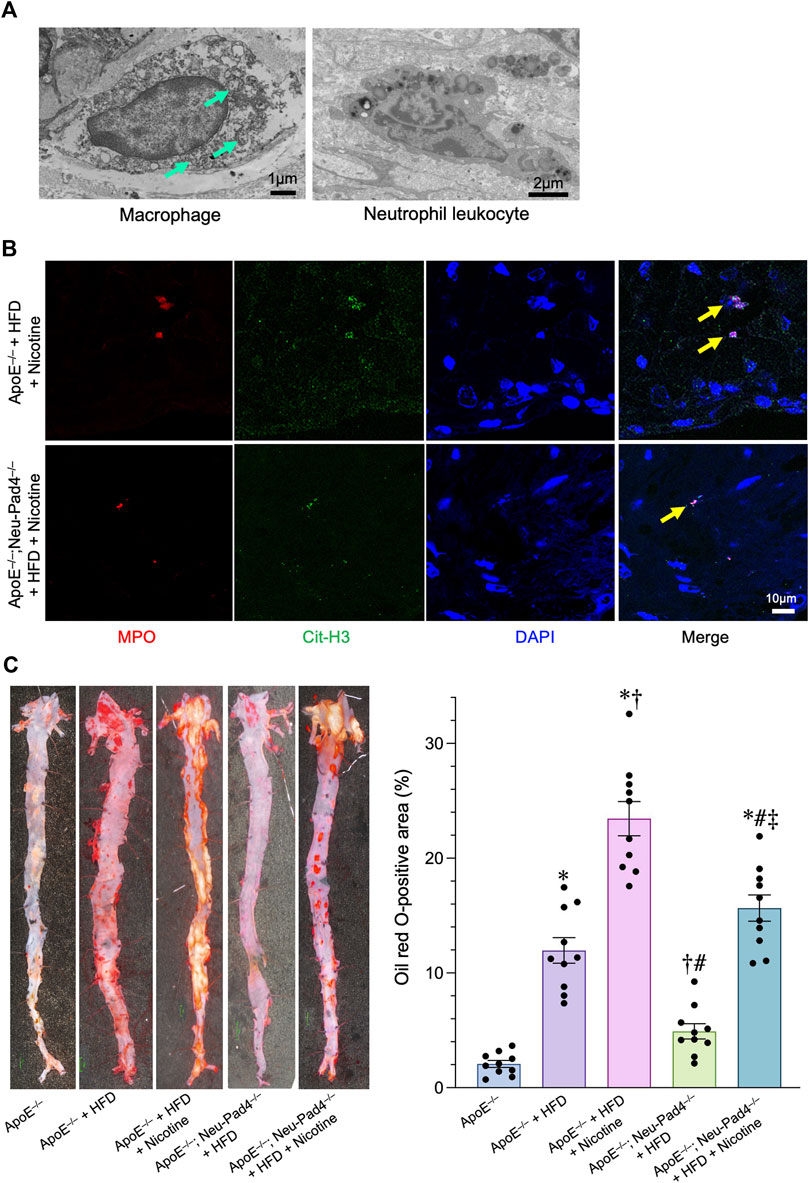
FIGURE 3. Role of NETs in the progression of atherosclerosis in high-fat diet (HFD)-fed apolipoprotein E knockout (ApoE−/−) mice. (A) Upper left panel: A representative transmission electron microscopic image of autophagosomes (arrows) in the macrophage derived from the atheroma of HFD-fed ApoE−/−mice. Upper right panel: A representative transmission electron microscopic image of the neutrophil leukocyte derived from the atheroma of HFD-fed ApoE−/−mice. (B) Representative confocal immunofluorescence images of aortic sections from the indicated mice stained for myeloperoxidase (MPO, red), citrullinated histone 3 (Cit-H3, green), and DNA (DAPI, blue). (C) Right panel: representative images of the aorta en face derived from HFD-fed ApoE−/−mice, ApoE−/−; Neu-Pad4−/−mice, or untreated ApoE−/−mice, stained with Oil Red O. Right panel: quantification of aortic en face plaque area in HFD-fed ApoE−/−mice, ApoE−/−; Neu-Pad4−/−mice, or untreated ApoE−/−mice, as shown in the representative images. *p < 0.05 vs. Wild-type (Blue bar), †p < 0.05 vs. ApoE−/−+ HFD (Purple bar), #p < 0.05 vs. ApoE−/−+ HFD + Nicotine (Pink bar), ‡p < 0.05 vs. ApoE−/−; Neu-Pad4−/−+ HFD (Green bar), n = 10 in each group. Data are expressed as dot plots and bars, with lines indicating the mean ± SEM.
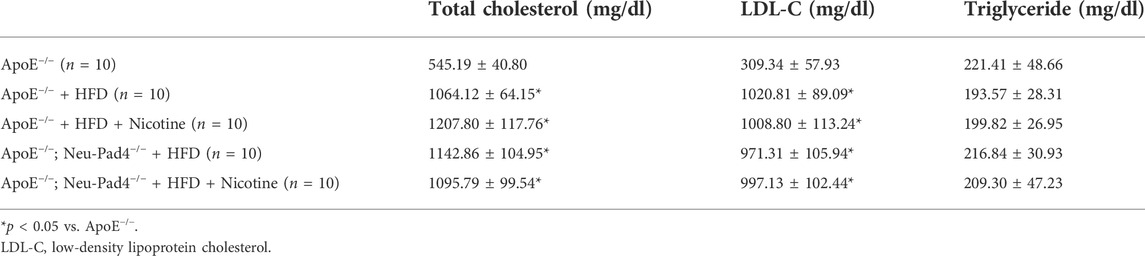
TABLE 1. The serum lipid profile.
Neutrophil extracellular traps suppress Beclin-1-associated phosphatidylinositol 3-kinase activation by promoting Beclin-1 phosphorylation
It has been shown that the NE, a major component of NETs, mediates ligand-dependent EGFR activation (DiCamillo et al., 2002; Kuwahara et al., 2006). To investigate the effect of NETs on EGFR of the macrophages, we added NETs to the cultured macrophages, and found that the addition of NETs promoted the phosphorylation of EGFR in the macrophages (Figure 4A). This reaction was significantly suppressed by the treatment with 10 μM of AG1478, a potent EGFR tyrosine kinase inhibitor. We next evaluated whether the NETs could phosphorylate Beclin-1 based on the previous finding that activated the EGFR phosphorylates tyrosine residues of Beclin-1, which decreases its function and suppresses the autophagy. To this end, we conducted immunoprecipitation assays using a macrophage-derived lysates overexpressing Flag-Beclin-1 with an anti-Flag tag-conjugated magnetic beads and immunoblotted with anti-tyrosine phosphorylation antibody and found that the tyrosine residues of Beclin-1 were markedly phosphorylated when the NETs were added. Co-administration of the AG1478, a potent tyrosine kinase inhibitor with NETs resulted in the attenuation of Beclin-1 phosphorylation (Figure 4B). To evaluate the effect of NETs on Beclin-1-dependent PI3 kinase activity, macrophages were transduced with the Ad-GFP-2×FYVE, a Phosphatidylinositol 3-phospate probe. Treatment with a 7KC significantly increased the number of GFP-2×FYVE dots in the macrophages. Conversely, co-treatment with a NETs significantly decreased the number of GFP-2×FYVE dots. On the other hand, the inhibition of EGFR activity with AG1478 administration significantly increased the number of the GFP-2×FYVE dots (Figure 4C). Altogether, these data suggests that the NETs inhibit the kinase activity of Beclin-1-dependent PI3 kinase activity possibly through phosphorylating the tyrosine residues of Beclin-1 in the macrophages.
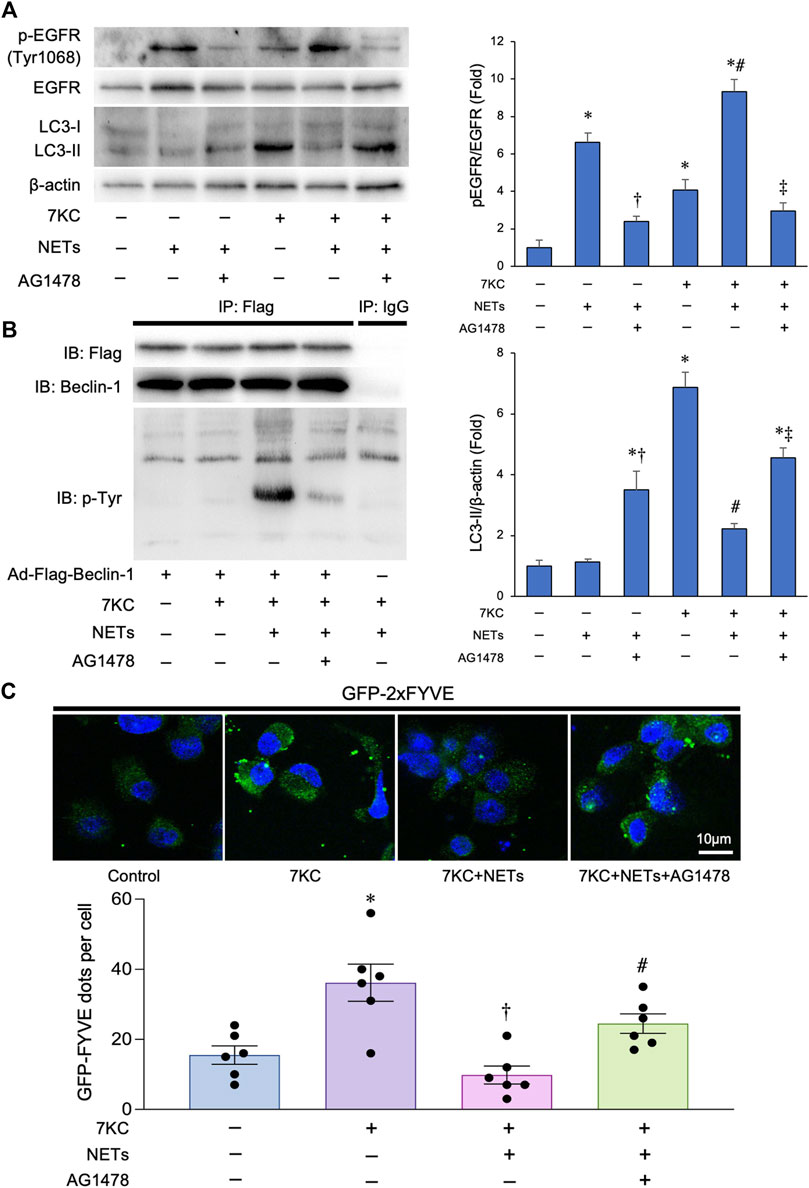
FIGURE 4. Effects of NETs for Beclin 1 phosphorylation and Beclin 1-associated PI3-K activation in macrophages. (A) Left panel: Representative immunoblot images of LC3, EGFR, phospho-EGFR (Tyr1068), and β-actin. Right panels: Densitometric analysis of immunoblots. *p < 0.05 vs. control group, †p < 0.05 vs. NET-treated group, #p < 0.05 vs. 7KC-treated group, ‡p < 0.05 vs. 7KC + NET-treated group, n = 3 in each group. All data are expressed as mean ± SEM. (B) Macrophages derived from the THP-1 cells were treated with a 7KC after Ad-Flag-Beclin-1 transduction and then treated with or without the NETs or AG1478, a potent EGFR tyrosine kinase inhibitor. 48 h after the transduction, lysates were extracted for co-immunoprecipitation with phospho-Tyrosine-specific antibody or control IgG, followed by probing with Beclin-1 antibody. Representative images are shown. (C) Membrane-associated phosphoinositide 3-kinase (PI3-K) assay in-situ. Macrophages derived from the THP-1 cells were treated with a 7KC after Ad-GFP-2×FYVE transduction, and then treated with or without NETs or AG1478. Upper: Representative images of GFP-2×FYVE dots are shown. Lower: Quantitative analysis of the number of GFP-2×FYVE dots is shown. *p < 0.05 vs. control group (Blue bar), †p < 0.05 vs. 7KC-treated group (Purple bar), #p < 0.05 vs. 7KC + NETs-treated group (Pink bar), n = 6 in each group. Data are expressed as the dot plots and bars, with the lines indicating the mean ± SEM.
Neutrophil extracellular traps negatively regulate autophagosome–lysosome fusion through upregulating Rubicon expression
To investigate the effect of NETs on autophagosome and lysosome fusion reaction in the macrophages (i.e., autophagic flux), we conducted an autophagic flux assay using the macrophages transduced with Ad-tf-LC3. Merged images demonstrated that the increase of the red puncta by treating with a 7KC, was significantly greater than that in the yellow puncta in the macrophages as compared to the untreated one (Figure 5A). On the other hand, persistent treatment (4 h) of NETs with a 7KC significantly suppressed the number of red/yellow puncta ratio in the macrophages (Figure 5A). In addition, there was a significant accumulation of the p62/SQSTM1, a selective substrate for autophagy, by a persistent treatment of NETs with a 7KC in the macrophages (Figure 5B), suggesting that the NETs inhibit autophagic flux in the macrophages. To further investigate how to suppress the autophagic flux by a treatment with NETs in the macrophages, the expression levels of representative proteins involved in the autophagic flux were evaluated by immunoblotting. Among the SNARE proteins required for the fusion of autophagosomes and lysosomes, the level of Vamp8 was not changed by the addition of the NETs, while the level of Syntaxin-17 was slightly increased by a treatment with the NETs (Figure 5B). On the other hand, the expression of Rubicon, a protein that forms a complex with Beclin-1 for inhibiting the autophagosome/lysosome fusion, was increased markedly (Figure 5B).
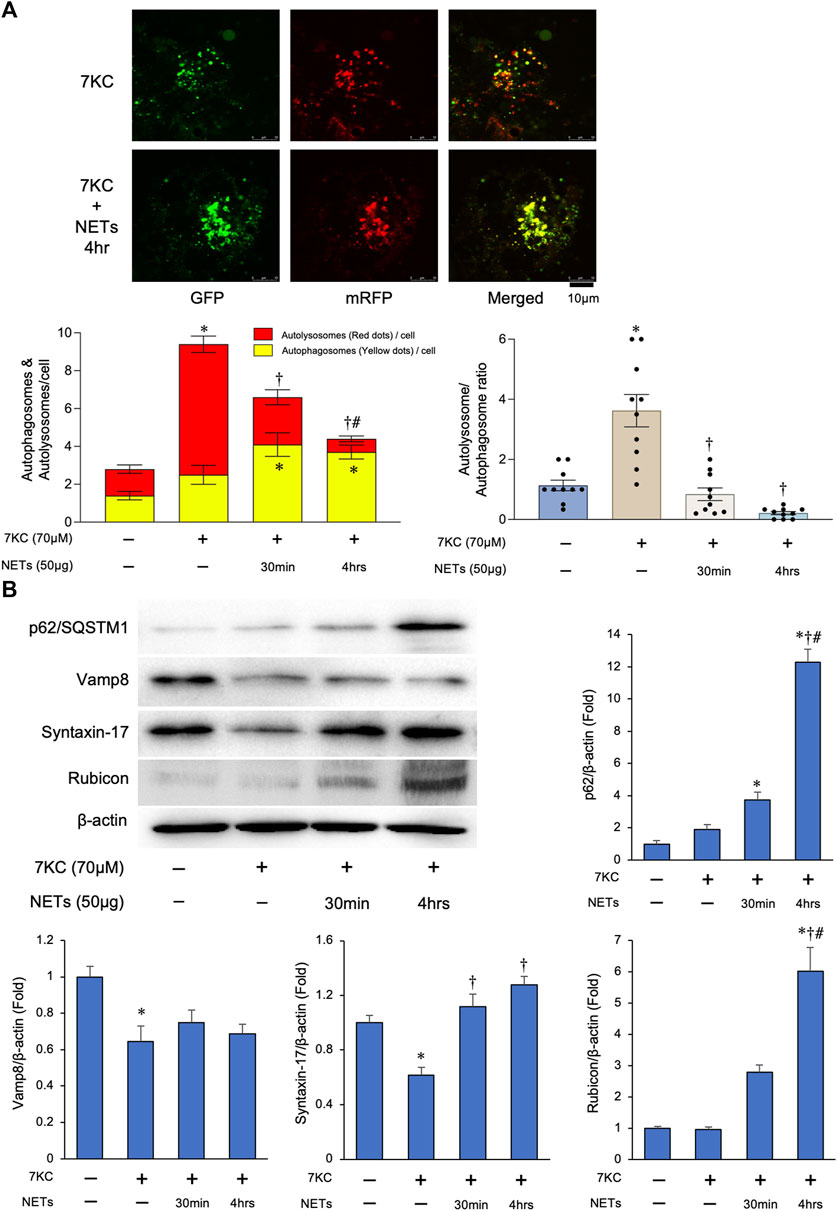
FIGURE 5. NETs negatively regulate autophagosome–lysosome fusion through upregulating Rubicon expression. (A) Autophagy flux assay. Macrophages derived from the THP-1 cells were treated with a 7KC after Ad-tf-LC3 transduction and then treated with or without the NETs. Upper panels: Representative images of the fluorescent LC3 puncta. Puncta with a yellow color in the merged images indicate the autophagosomes, and puncta with red color in the merged images indicate autolysosomes. Left lower panel: Quantitative analysis of the mean number of autophagosomes represented by the yellow puncta in the merged images and autolysosomes represented by the red puncta in the merged images per cell is shown. *p < 0.05 vs. control group, †p < 0.05 vs. 7KC-treated group, #p < 0.05 vs. 7KC + NETs (30 min)-treated group, n = 10 in each group. Data are expressed as the bars, with the lines indicating the mean ± SEM. Right lower panel: Quantitative analysis of the mean autophagosome/autolysosome ratio per cell is shown. *p < 0.05 vs. control group (Blue bar), †p < 0.05 vs. 7KC-treated group (Brown bar), n = 10 in each group. Data are expressed as dot plots and bars, with lines indicating the mean ± SEM. (B) Left upper panel: Representative immunoblot images of the p62/SQSTM1, Vamp8, Syntaxin-17, Rubicon, and β-actin. Right and Lower panels: Densitometric analysis of immunoblots. *p < 0.05 vs. control group, †p < 0.05 vs. 7KC-treated group, #p < 0.05 vs. NET (30 min)-treated group, n = 3 in each group. All data are expressed as mean ± SEM.
Collectively, all these results indicate that the suppression of autophagic flux in the macrophages by a NETs is possibly mediated through the upregulation of a Rubicon by the treatment with NETs.
Discussion
In the present study, we found that the NETs induced by nicotine, a common pro-atherosclerotic agent, negatively regulate autophagy in the macrophages, which causes an activation of the inflammasome activity. We also found that the NETs formation is critically associated with the atherogenesis in the HFD-loaded ApoE−/−mice. Finally, we revealed that the suppression of autophagy by NETs is mediated through a tyrosine phosphorylation of Beclin-1, and increased expression of Rubicon possibly through an EGFR activation in the macrophages (Figure 6).
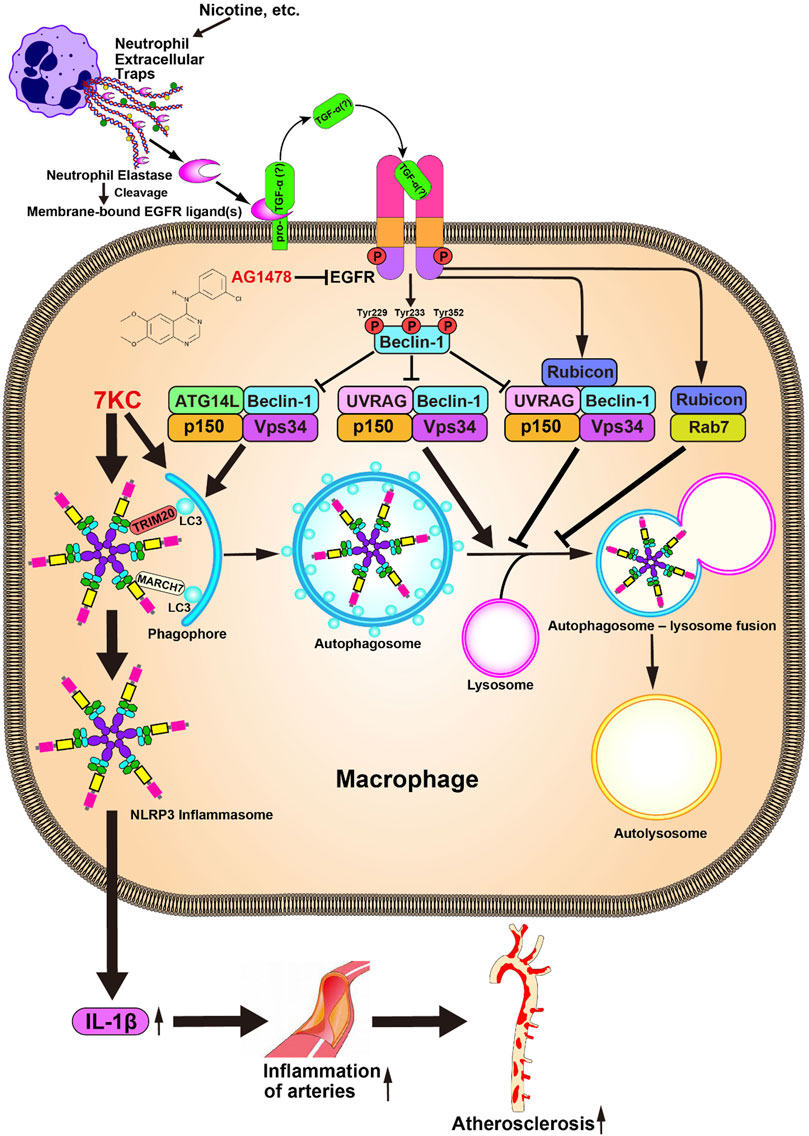
FIGURE 6. Proposed model for the deterioration of atherosclerosis by NETs. NETs induced by the various stimuli, including the nicotine, promote EGFR activity possibly through facilitating conversion of a membrane-bound EGFR ligand such as TGF-α, to its active soluble form by its proteolytic activity. Activated EGFR phosphorylates Beclin-1, thereby suppressing the Beclin-1-dependent PI3 kinase activity, which results in the suppression of an autophagy machinery. Attenuation of the autophagy activity causes enhancement of the inflammasome activity, which results in the aggravation of the atherosclerosis.
Although it has been shown that the NETs play an important role in promoting the atherosclerosis (Warnatsch et al., 2015), the cause of NETs induction other than the microbe infection has not been well investigated. In the present study, we found that the treatment with a nicotine to neutrophil leukocyte could induce NETs formation, which may support the epidemiological association of smoking habits with the development of atherosclerosis.
As mentioned above, we hypothesized that the NETs may worsen atherosclerosis by inhibiting autophagy machinery, thereby enhancing inflammasome activity in the macrophages. Here we show that the administration of NETs activates the EGFR, thereby promoting the phosphorylation of Beclin-1 in its tyrosine residues. Increasing lines of evidence suggest that the NE releases the soluble EGFR ligands, thereby activating the EGFR-mediated intracellular signaling (Jin et al., 2021). In cultured keratinocytes, the application of NE facilitates cell proliferation by promoting the conversion of a membrane-bound pro-TGF-α to its active soluble form by its proteolytic activity, thereby enhancing the EGFR activity (Meyer-Hoffert et al., 2004). Thus, it should be reasonable that the NETs promote EGFR phosphorylation. Indeed, we found that NET administration facilitates the phosphorylation of both EGFR and Beclin-1 at tyrosine residues, which is abolished by co-administration with EGFR tyrosine kinase inhibitor in the macrophages. Concurrently, we found that the level of Rubicon was markedly increased in the macrophages by treatment with NETs. However, its detailed mechanism remains unknown. Li et al. (2021) recently reported an intriguing finding that the EGFR suppresses autophagy by upregulating the Rubicon in podocytes (Li et al., 2021). Based on these findings, NET-derived NE would promote EGFR activation, thereby facilitating both Beclin-1 phosphorylation and Rubicon expression, which in turn inhibit autophagy machinery in the macrophages. Thus, in the present study, we could add a novel finding regarding the role of EGFR as an autophagy modulator, which contributes to the progression of atherosclerosis.
One of the critical tasks of autophagy is removing dangerous objects in the cells such as damaged mitochondria and excessively activated inflammasome, which plays an important role in the progression of the atherosclerosis by enhancing a sterile inflammation triggered by the damage-associated molecular patterns (Liao et al., 2012; Razani et al., 2012; Ito et al., 2021). Specifically, the inflammasomes are degraded by a selective type of autophagy, termed as “precision autophagy,” by recognizing with the specialized receptors such as TRIM20 and MARCH7 (Kimura et al., 2015; Yan et al., 2015). Indeed, we recently revealed that the NLRP3-inflammasome is decomposed by a TRIM20-mediated precision autophagy in the 7KC-treated macrophages (Ito et al., 2021). In this study, we found that inhibition of autophagy by NETs contributes to the maintenance of the inflammasome activity, suggesting that the NETs may have a crucial role in inhibiting the precision autophagy.
Our experiments have several potential limitations. Our study demonstrated that the area of atherosclerotic plaque significantly decreased in a nicotine-treated HFD-fed ApoE−/−; Neu-Pad4−/−mice compared to those in HFD-fed ApoE−/−mice. This result indicates that the nicotine-mediated atherogenesis is partially mediated through the NETs formation. However, there should be other mediators that promote NET formation, which results in atherosclerosis. To verify the role of NETs in animal models, proteinase 3-deficient (PR3−/−) and/or NE−/−mice would be crossed with ApoE−/−mice because deleting these enzymes might more effectively abrogate NET formation (Warnatsch et al., 2015). In addition, to validate whether NET-mediated suppression of macrophage autophagy plays an important role for developing atherosclerosis in vivo, loss-of-function animal models such as macrophage-specific Atg5 knockout mice should be crossed with ApoE−/−; Neu-Pad4−/−mice. Similarly, cell experiments should be conducted using primary macrophages derived from macrophage-specific Atg5-knockout mice. Furthermore, as 7KC promotes not only autophagy but also oxidative stress and apoptosis, a line of experiments using pure pro-autophagic inducers should be conducted to evaluate the effect of NETs on macrophage autophagy in the presence of pathological conditions other than atherosclerosis. It is also insufficient to detect Beclin-1 phosphorylation by blotting with total phospho-tyrosine antibody. Ideally, the effect of NETs on Beclin-1 should be examined by blotting with a specific antibody that can detect Beclin-1 phosphorylation in Tyr229, Tyr233, and Tyr352 (Wei et al., 2013). From this perspective, further studies are needed in the future to increase the reliability of the current findings.
In conclusion, our current study revealed that the NET-mediated EGFR–Beclin-1 signaling in the macrophages promotes atherogenesis by accelerating the inflammasome activities, which is mediated through suppressing the autophagy. This suggests that the NETs-mediated EGFR–Beclin-1 signaling could be a novel candidate for developing the anti-atherosclerotic agent.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by the Tokyo Medical and Dental University Guide for the Care and Use of the Laboratory Animals (Permit Number: A2021-023A).
Author contributions
YM established the concept and designed and coordinated the study. MS, SN, YS-W, NT, KH, MI, and TS contributed to the conception or design of the study. MS, YM, SN, YS-W, and NT conducted the experiments. MS and YM analyzed data and conducted statistical analyses. YM wrote the manuscript, and MS, KH, MI, and TS participated in editing it. All authors contributed to the interpretations and conclusions presented. The authors read and approved the final manuscript.
Funding
This work was supported in part by a grant from the Smoking Research Foundation and JSPS KAKENHI Grant-in-Aid for Scientific Research (C) (17K09570 to YM).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Masanori Konishi (in-vitro experiments; Former members of the Department of Cardiovascular Medicine, Tokyo Medical and Dental University), Noriko Tamura (Pathological experiments, Department of Cardiovascular Medicine, Tokyo Medical and Dental University), and Yuriko Sakamaki (Electron microscopic experiments; Tissue Analyzing Unit in Research Core Center of Tokyo Medical and Dental University) for their excellent technical assistance during the research work. Also, we would like to thank Enago (www.enago.com) for English language editing.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.876147/full#supplementary-material
References
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. doi:10.1126/science.1092385
DiCamillo, S. J., Carreras, I., Panchenko, M. V., Stone, P. J., Nugent, M. A., Foster, J. A., et al. (2002). Elastase-released epidermal growth factor recruits epidermal growth factor receptor and extracellular signal-regulated kinases to down-regulate tropoelastin mRNA in lung fibroblasts. J. Biol. Chem. 277, 18938–18946. doi:10.1074/jbc.M200243200
Fogel, A. I., Dlouhy, B. J., Wang, C., Ryu, S. W., Neutzner, A., Hasson, S. A., et al. (2013). Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol. Cell. Biol. 33, 3675–3688. doi:10.1128/MCB.00079-13
Franck, G., Mawson, T. L., Folco, E. J., Molinaro, R., Ruvkun, V., Engelbertsen, D., et al. (2018). Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: Implications for superficial erosion. Circ. Res. 123, 33–42. doi:10.1161/CIRCRESAHA.117.312494
Hariharan, N., Maejima, Y., Nakae, J., Paik, J., Depinho, R. A., Sadoshima, J., et al. (2010). Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 107, 1470–1482. doi:10.1161/circresaha.110.227371
Hosseinzadeh, A., Thompson, P. R., Segal, B. H., and Urban, C. F. (2016). Nicotine induces neutrophil extracellular traps. J. Leukoc. Biol. 100, 1105–1112. doi:10.1189/jlb.3AB0815-379RR
Itakura, E., Kishi-Itakura, C., and Mizushima, N. (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 151, 1256–1269. doi:10.1016/j.cell.2012.11.001
Ito, Y., Maejima, Y., Nakagama, S., Shiheido-Watanabe, Y., Tamura, N., Sasano, T., et al. (2021). Rivaroxaban, a direct oral factor xa inhibitor, attenuates atherosclerosis by alleviating factor xa-PAR2-mediated autophagy suppression. JACC. Basic Transl. Sci. 6, 964–980. doi:10.1016/j.jacbts.2021.09.010
Jiménez-Alcázar, M., Rangaswamy, C., Panda, R., Bitterling, J., Simsek, Y. J., Long, A. T., et al. (2017). Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 358, 1202–1206. doi:10.1126/science.aam8897
Jin, W., Yin, H., Li, H., Yu, X. J., Xu, H. X., Liu, L., et al. (2021). Neutrophil extracellular DNA traps promote pancreatic cancer cells migration and invasion by activating EGFR/ERK pathway. J. Cell. Mol. Med. 25, 5443–5456. doi:10.1111/jcmm.16555
Kawakami, T., He, J., Morita, H., Yokoyama, K., Kaji, H., Tanaka, C., et al. (2014). Rab27a is essential for the formation of neutrophil extracellular traps (NETs) in neutrophil-like differentiated HL60 cells. PLoS One 9, e84704. doi:10.1371/journal.pone.0084704
Kimura, T., Jain, A., Choi, S. W., Mandell, M. A., Schroder, K., Johansen, T., et al. (2015). TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell. Biol. 210, 973–989. doi:10.1083/jcb.201503023
Knight, J. S., Luo, W., O'Dell, A. A., Yalavarthi, S., Zhao, W., Subramanian, V., et al. (2014). Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 114, 947–956. doi:10.1161/CIRCRESAHA.114.303312
Kolaczkowska, E., and Kubes, P. (2013). Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175. doi:10.1038/nri3399
Kuwahara, I., Lillehoj, E. P., Lu, W., Singh, I. S., Isohama, Y., Miyata, T., et al. (2006). Neutrophil elastase induces IL-8 gene transcription and protein release through p38/NF-{kappa}B activation via EGFR transactivation in a lung epithelial cell line. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L407–L416. doi:10.1152/ajplung.00471.2005
Lee, J., Luria, A., Rhodes, C., Raghu, H., Lingampalli, N., Sharpe, O., et al. (2017). Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatol. Oxf. 56, 644–653. doi:10.1093/rheumatology/kew449
Li, P., Li, M., Lindberg, M. R., Kennett, M. J., Xiong, N., Wang, Y., et al. (2010). PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862. doi:10.1084/jem.20100239
Li, Y., Pan, Y., Cao, S., Sasaki, K., Wang, Y., Niu, A., et al. (2021). Podocyte EGFR inhibits autophagy through upregulation of Rubicon in type 2 diabetic nephropathy. Diabetes 70, 562–576. doi:10.2337/db20-0660
Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., et al. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676. doi:10.1038/45257
Liao, X., Sluimer, J. C., Wang, Y., Subramanian, M., Brown, K., Pattison, J. S., et al. (2012). Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell. Metab. 15, 545–553. doi:10.1016/j.cmet.2012.01.022
Maejima, Y., Isobe, M., and Sadoshima, J. (2016). Regulation of autophagy by Beclin 1 in the heart. J. Mol. Cell. Cardiol. 95, 19–25. doi:10.1016/j.yjmcc.2015.10.032
Maejima, Y., Kyoi, S., Zhai, P., Liu, T., Li, H., Ivessa, A., et al. (2013). Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 19, 1478–1488. doi:10.1038/nm.3322
Matsunaga, K., Saitoh, T., Tabata, K., Omori, H., Satoh, T., Kurotori, N., et al. (2009). Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell. Biol. 11, 385–396. doi:10.1038/ncb1846
Meyer-Hoffert, U., Wingertszahn, J., and Wiedow, O. (2004). Human leukocyte elastase induces keratinocyte proliferation by epidermal growth factor receptor activation. J. Invest. Dermatol. 123, 338–345. doi:10.1111/j.0022-202X.2004.23202.x
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi:10.1038/nature06639
Nah, J., Zablocki, D., and Sadoshima, J. (2021). The roles of the inhibitory autophagy regulator Rubicon in the heart: A new therapeutic target to prevent cardiac cell death. Exp. Mol. Med. 53, 528–536. doi:10.1038/s12276-021-00600-3
Najmeh, S., Cools-Lartigue, J., Giannias, B., Spicer, J., and Ferri, L. E. (2015). Simplified human neutrophil extracellular traps (NETs) isolation and handling. J. Vis. Exp. (98), e52687. doi:10.3791/52687
Papayannopoulos, V. (2018). Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147. doi:10.1038/nri.2017.105
Razani, B., Feng, C., Coleman, T., Emanuel, R., Wen, H., Hwang, S., et al. (2012). Autophagy links inflammasomes to atherosclerotic progression. Cell. Metab. 15, 534–544. doi:10.1016/j.cmet.2012.02.011
Rong, Y. P., Bultynck, G., Aromolaran, A. S., Zhong, F., Parys, J. B., De Smedt, H., et al. (2009). The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. U. S. A. 106, 14397–14402. doi:10.1073/pnas.0907555106
Roth, G. A., Mensah, G. A., Johnson, C. O., Addolorato, G., Ammirati, E., Baddour, L. M., et al. (2020). Global burden of cardiovascular diseases and risk factors, 1990-2019: Update from the GBD 2019 study. J. Am. Coll. Cardiol. 76, 2982–3021. doi:10.1016/j.jacc.2020.11.010
Sciarretta, S., Maejima, Y., Zablocki, D., and Sadoshima, J. (2018). The role of autophagy in the heart. Annu. Rev. Physiol. 80, 1–26. doi:10.1146/annurev-physiol-021317-121427
Wang, C., Chen, H., Zhu, W., Xu, Y., Liu, M., Zhu, L., et al. (2017). Nicotine accelerates atherosclerosis in apolipoprotein E-deficient mice by activating α7 nicotinic acetylcholine receptor on mast cells. Arterioscler. Thromb. Vasc. Biol. 37, 53–65. doi:10.1161/ATVBAHA.116.307264
Warnatsch, A., Ioannou, M., Wang, Q., and Papayannopoulos, V. (2015). Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349, 316–320. doi:10.1126/science.aaa8064
Wei, Y., Pattingre, S., Sinha, S., Bassik, M., and Levine, B. (2008). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell. 30, 678–688. doi:10.1016/j.molcel.2008.06.001
Wei, Y., Zou, Z., Becker, N., Anderson, M., Sumpter, R., Xiao, G., et al. (2013). EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 154, 1269–1284. doi:10.1016/j.cell.2013.08.015
Yan, Y., Jiang, W., Liu, L., Wang, X., Ding, C., Tian, Z., et al. (2015). Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 160, 62–73. doi:10.1016/j.cell.2014.11.047
Yang, H., Biermann, M. H., Brauner, J. M., Liu, Y., Zhao, Y., Herrmann, M., et al. (2016). New insights into neutrophil extracellular traps: Mechanisms of formation and role in inflammation. Front. Immunol. 7, 302. doi:10.3389/fimmu.2016.00302
Zalckvar, E., Berissi, H., Eisenstein, M., and Kimchi, A. (2009). Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 5, 720–722. doi:10.4161/auto.5.5.8625
Keywords: Beclin 1, autophagy, neutrophil extracelluar traps, macrophage, atheroscelrosis
Citation: Sano M, Maejima Y, Nakagama S, Shiheido-Watanabe Y, Tamura N, Hirao K, Isobe M and Sasano T (2022) Neutrophil extracellular traps-mediated Beclin-1 suppression aggravates atherosclerosis by inhibiting macrophage autophagy. Front. Cell Dev. Biol. 10:876147. doi: 10.3389/fcell.2022.876147
Received: 15 February 2022; Accepted: 30 June 2022;
Published: 18 July 2022.
Edited by:
Haidong Xu, Soochow University Medical College, ChinaReviewed by:
Hai Huang, Comprehensive Cancer Center, The Ohio State University, United StatesYaozu Xiang, Tongji University, China
Runze Qiu, Nanjing Medical University, China
Copyright © 2022 Sano, Maejima, Nakagama, Shiheido-Watanabe, Tamura, Hirao, Isobe and Sasano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yasuhiro Maejima, ymaeji.cvm@tmd.ac.jp
†These authors have contributed equally to this work