The Tumor Suppressor Kinase LKB1: Metabolic Nexus
 Mohammed Bourouh
Mohammed Bourouh Paola A. Marignani
Paola A. Marignani- Department of Biochemistry and Molecular Biology, Faculty of Medicine, Dalhousie University Halifax, Halifax, NS, Canada
Liver kinase B1 (LKB1) is a multitasking tumor suppressor kinase that is implicated in multiple malignancies such as lung, gastrointestinal, pancreatic, and breast. LKB1 was first identified as the gene responsible for Peutz-Jeghers syndrome (PJS) characterized by hamartomatous polyps and oral mucotaneous pigmentation. LKB1 functions to activate AMP-activated protein kinase (AMPK) during energy stress to shift metabolic processes from active anabolic pathways to active catabolic pathways to generate ATP. Genetic loss or inactivation of LKB1 promotes metabolic reprogramming and metabolic adaptations of cancer cells that fuel increased growth and division rates. As a result, LKB1 loss is associated with increased aggressiveness and treatment options for patients with LKB1 mutant tumors are limited. Recently, there has been new insights into the role LKB1 has on metabolic regulation and the identification of potential vulnerabilities in LKB1 mutant tumors. In this review, we discuss the tumor suppressive role of LKB1 and the impact LKB1 loss has on metabolic reprograming in cancer cells, with a focus on lung cancer. We also discuss potential therapeutic avenues to treat malignancies associated with LKB1 loss by targeting aberrant metabolic pathways associated with LKB1 loss.
Introduction
Metabolism is the outcome of key processes and reactions that generate energy to maintain cellular life. Metabolic processes serve to produce adenosine triphosphate (ATP) to meet the energetic demands of a cell, and the intermediates from these processes are used to generate biomolecules (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). Metabolic reactions can either be anabolic (to buildup) or catabolic (to breakdown) and both these processes must be balanced to maintain the energy supply of cells while preserving biomolecules to sustain cellular function.
A common characteristic of cancer cells is an insatiable demand for energy in order to meet the needs for growth and proliferation. Cancer cells will take control over multiple signaling networks to reprogram metabolic pathways that enable cancer cells to synthesize biomolecules and adapt to survive under elevated reactive oxygen species (ROS) (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). The mechanism behind metabolic reprogramming involves genetic adaptations through mutation of tumor suppressor genes and oncogenes that allow metabolic processes to be deregulated, leading to increased proliferation rate and survival of cancer cells. One such gene is the tumor suppressor serine/threonine kinase liver kinase B1 (LKB1), also known as serine-threonine kinase 11 (STK11).
LKB1 is implicated in multiple malignancies where it is often lost or inactivated. LKB1 was first identified as the gene responsible for Peutz-Jeghers syndrome (PJS), a dominant disorder characterized by benign hamartomatous polyps in the gastrointestinal tract and mucocutaneous melanin pigmentation (Peutz, 1921; Jeghers et al., 1949; Hemminki et al., 1997). Germline mutations in LKB1 that lead to the development of PJS result in loss of function of LKB1 through truncations, deletions, or direct mutations to the kinase domain abolishing LKB1 kinase activity (Mehenni et al., 1998; Tiainen et al., 1999; Ylikorkala et al., 1999; Boudeau et al., 2003a). PJS patients have an increased risk of developing different malignancies, primarily in the gastrointestinal tract; colon, gastric, and intestinal cancers (Giardiello et al., 2000; Karuman et al., 2001), and are also susceptible to malignancies of the breast, lung, uterus, ovaries, cervix, and testes (Avizienyte et al., 1998; Nishioka et al., 1999; Boardman et al., 2000; Sanchez-Cespedes et al., 2002; Shen et al., 2002).
While LKB1 mutations in PJS are associated with an increased risk of developing cancer, LKB1 somatic mutations leading to malignancies are rare. It is surprising then, that the exception is in non-small cell lung cancer (NSCLC), where LKB1 loss is implicated in 30% of cases (Sanchez-Cespedes, 2007; Ding et al., 2008; Gill et al., 2011). Furthermore, LKB1 haploinsufficiency has been observed in the pancreas (Morton et al., 2010), breast (Shen et al., 2002), endometrial (Contreras et al., 2008) and liver adenocarcinoma (Kim and Chen, 2004) although very infrequent.
The tumor suppressor function of LKB1 has largely been attributed to the phosphorylation and activation of the energy sensor AMP-activated protein kinase (AMPK) in response to nutrient availability and energy stress. Here, the LKB1-AMPK axis shifts cellular metabolism from active anabolic pathways to active catabolic pathways to correct the energy imbalance (Hardie, 2005).
When LKB1 activity is abolished, the mechanism regulating metabolic pathways is eliminated. Loss of LKB1 leads to increased glucose uptake and increased activity of aerobic glycolysis, commonly known as the Warburg effect (Warburg et al., 1927). Furthermore, loss of LKB1 also leads to increased ROS that needs to be quenched to prevent damage to macromolecules. The survival of cancer cells relies on meeting the energy demand and adapting to the increased ROS produced. In this review, we discuss the tumor suppressive role of LKB1 as a metabolic nexus, and how it is implicated in metabolic regulation, focusing on lung cancer. We also discuss the impact loss of LKB1 has on metabolic reprograming and tumor progression and potential therapeutic avenues to treat LKB1 deficient cancers by targeting aberrant metabolic pathways.
LKB1 Mutations in Lung Cancer
LKB1 is spontaneously mutated most frequently in lung cancer patients is associated with increased aggressiveness (Calles et al., 2015; Lin et al., 2021). LKB1 loss or inactivation is observed in 30% of lung adenocarcinoma. Lung adenocarcinoma (LUAD) is the most common type of lung cancer, accounting for 45% of cases. LUAD cases are stratified based on oncogenic mutation with ∼60% of LUAD cases associated with KRAS and EGFR mutations (Sun et al., 2007; Collisson et al., 2014). EGFR mutations are more common in never smokers with the most frequently observed mutations being exon 19 deletions, and the point mutation codon 858 (L858R). KRAS oncogenic mutations are present in smokers and are often the result of base substitutions at codons 12 (91%), 13 (6%), and 61 (2%). KRAS codon 12 mutations result in amino acid substitutions of glutamine to either cysteine (G12C, 44%), valine (G12V, 23%), or aspartic acid (G12D, 17%) being the most common (Ostrem et al., 2013; Cox et al., 2014). KRAS is a small GTPase that promotes activation of the MAPK pathway to promote cell growth and survival (Burotto et al., 2014). Lung adenocarcinoma patients that present with KRAS mutations have a higher mutational burden and co-occurring mutations with tumor suppressors genes. Different co-occurring mutations with KRAS exhibit unique tumor behaviors and have different gene expression profiles (Skoulidis et al., 2015).
The most studied Kras lung cancer mouse model is the LSL-KrasG12D mouse model. Using a transcriptional STOP element flanked by loxP sites, expression of KrasG12D can be induced in multiple ways: tissue and cell-specific promoters driving Cre expression, inhalation of adeno-Cre (Ad5-CMV-Cre), or intratracheal administration of Ad5-CMV-Cre. With Ad5-CMV-Cre administration, lung cancer development can be followed in a time-dependent manner. Furthermore, the clonality of cancer development can also be studied. Expression of Kras in the lung caused characteristics of early adenocarcinoma, where lungs presented with atypical adenomatous hyperplasia (AAH), epithelial hyperplasia (EH), and adenomas. Although this mouse model recapitulated early disease histopathologies of lung cancer, it did not recapitulate late stages. When loss of function mutations of Tp53 were combined with Kras oncogenic mutations, late-stage disease progression of lung adenocarcinoma was observed; nuclear atypia, elicit stromal desmoplasia, invasion, and metastasis (Jackson et al., 2005). It was not until Lkb1 was co-mutated with Kras that lung adenocarcinoma disease progression exhibited a similar pattern and severity to human disease.
Mutations in other tumor suppressors (RB1, CDKN2A, SMARCA4/BRG1) are also frequently implicated in lung cancer. TP53 is the most frequently mutated tumor suppressor in many cancers including lung cancer where it is mutated in ∼70% of cases (Sanchez-Cespedes, 2007). KRAS mutant lung adenocarcinoma is often associated with a mutation in CDKN2A or LKB1. Mouse models of lung cancer typically use three different genotypes to recapitulate LUAD; Kras;Tp53 (KP), Kras;Cdkn2a (KC), and Kras;Lkb1 (KL). Each model exhibits different characteristics, severity, and aggressiveness of lung adenocarcinomas with different microenvironments, gene expression signatures, and responses to therapies (Skoulidis et al., 2015).
Early work on the KL mouse model characterized LUAD development compared to the KP genotype. The KL genotype is the only genetic combination to fully recapitulate human lung adenocarcinoma in mice, showing all histological subgroups: squamous cell carcinoma, large cell carcinoma, adenocarcinoma, and adenosquamous carcinoma (Ji et al., 2007; Chong and Jänne, 2013; Shaw and Engelman, 2013). Adenosquamous and squamous subtypes are not seen in KP or KC models. Lkb1 ectopic expression in KL, KC, or KP tumors significantly reduced growth and induced apoptosis, further demonstrating the functional classification of Lkb1 as a tumor suppressor gene (Ji et al., 2007).
The LKB1 Kinase
LKB1 is a conserved, ubiquitously expressed multitasking serine/threonine kinase with tumor suppressor function (Marignani et al., 2010). LKB1 is a member of the Ca2+-calmodulin dependent protein kinase family (Marignani, 2005) with orthologues in frogs, mice, worms, and flies (Smith et al., 1999; Watts et al., 2000; Martin and St Johnston, 2003). The human LKB1 gene maps to chromosome 19p13.3 and is 23 kb long, consisting of 10 exons of which exons 1–9 are coding and exon 10 is non-coding. LKB1 is transcribed in the telomere to centromere direction and generates a 50 kDa protein. LKB1 is ubiquitously expressed in mice and humans with tissue-specific differences in overall abundance (Towler et al., 2008). In mice, Lkb1 protein is most abundant in embryonic and extra-embryonic tissues. Later in development, Lkb1 protein is concentrated in heart, esophagus, pancreas, kidney, colon, lung, small intestines, and stomach tissues (Luukko et al., 1999; Rowan et al., 1999). In adult mice, Lkb1 protein levels are most abundant in epithelial tissues, follicles, and corpus luteum of the ovary, seminiferous tubules of the testis, skeletal muscle monocytes, and glial cells (Rowan et al., 1999; Conde et al., 2007).
LKB1 is primarily found in a complex with the pseudokinase STE20-related adaptor (STRAD) (Baas et al., 2003) and the scaffolding protein Mouse protein 25 (MO25) (Boudeau et al., 2003b; Marignani et al., 2007). Unlike other kinases, LKB1 does not become catalytically active through T-loop threonine phosphorylation of the kinase domain, but instead when bound to adaptor proteins STRAD and MO25 (Baas et al., 2003; Boudeau et al., 2003b; Boudeau et al., 2006; Marignani et al., 2007; Dorfman and Macara, 2008). STRAD is a homolog of the STE20 family of kinases but lacks multiple critical residues required for a functional kinase domain (Dorfman and Macara, 2008). Although STRAD lacks a functional kinase domain, STRAD adopts an active conformation when bound to ATP (Zeqiraj et al., 2009). The binding of STRAD to MO25 enhances the affinity of STRAD to ATP. Furthermore, STRAD binding to ATP and MO25 are required for LKB1 catalytic activation (Baas et al., 2003; Boudeau et al., 2003b; Boudeau et al., 2006; Dorfman and Macara, 2008). LKB1 when in complex with STRAD and MO25 increases LKB1 catalytic activity by approximately 10 fold compared to LKB1 alone (Boudeau et al., 2003b) (Figure 1).
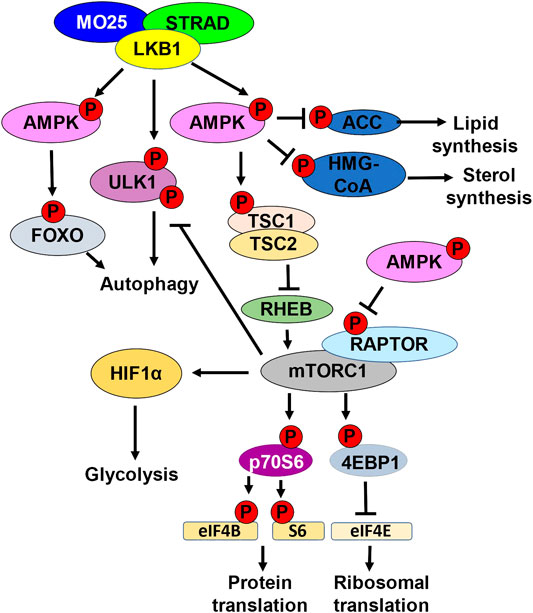
FIGURE 1. LKB1 metabolic nexus. Schematic representation of downstream LKB1 signaling. LKB1 in complex with STRAD and MO25 phosphorylates and activates AMPK. AMPK phosphorylates and inhibits ACC, inhibiting lipid synthesis. AMPK phosphorylates and inhibits HMG-CoA, inhibiting sterol synthesis. Active AMPK also regulates autophagy by phosphorylating and activating FOXO. LKB1 can directly promote autophagy by phosphorylating ULK1. LKB1 activation of AMPK also inhibits mTORC1 kinase activation. mTORC1 kinase phosphorylates and activates p70S6. p70S6 phosphorylates and activates eIF4B and S6 kinase, promoting protein translation. mTORC1 also phosphorylates and inhibits 4EBP1, the inhibitor of eIF4E. eIF4e activation leads to increased ribosomal translation. mTORC1 can also inhibits autophagy by phosphorylating ULK1. mTORC1 promotes glycolysis by upregulating HIF1α. AMPK directly inhibits mTORC1 by phosphorylating RAPTOR. AMPK can also indirectly inhibit mTORC1 by phosphorylating and activating TSC1/TSC2. Active TSC1/2 complex inhibits RHEB.
In addition to regulating the activity of LKB1, STRAD also regulates LKB1 subcellular localization, particularly nuclear-cytoplasmic localization. Individually, STRAD and MO25 can freely diffuse through nuclear pores and thus are localized both in the nucleus and cytoplasm. When STRAD and MO25 are co-expressed, they exhibit exclusively cytoplasmic localization. LKB1 contains an N-terminal nuclear localization signal (NLS) directing LKB1 to the nucleus via an importin α/β mechanism. STRAD competes with importin α/β for binding to LKB1, and therefore binding of STRAD to LKB1 sequesters LKB1 in the cytoplasm. Co-expression of LKB1 with STRAD and MO25 causes LKB1 to localize in the cytoplasm (Baas et al., 2003; Boudeau et al., 2003b). STRAD promotes LKB1 nuclear export in a CRM1 and exportin7 dependent manner (Dorfman and Macara, 2008).
Oncogenic mutations generally occur in the kinase domain of LKB1 and abolish kinase activity. LKB1 introduction to cancer cell lines that do not express LKB1 results in a G1 arrest (Tiainen et al., 1999; Tiainen et al., 2002). Expression of catalytic deficient LKB1 mutants, where the mutations are found within the kinase domain, does not result in a G1 arrest, suggesting that catalytic activity is required for tumor suppressor function (Scott et al., 2007). Furthermore, LKB1 mutations that abolish the binding to STRAD-MO25 also do not exhibit cell cycle arrest functions suggesting that the binding of STRAD-MO25 to LKB1 is required for the cell cycle arrest function of LKB1 (Tiainen et al., 1999; Tiainen et al., 2002; Scott et al., 2007). Interestingly, the expression of LKB1 with a catalytically deficient point mutation, LKB1D194A, resulted in the expression of genes important in cell cycle progression (CYCD1, RB1, CYCE, and CYCA). This suggests that catalytic deficient mutants of LKB1 can promote cell growth through kinase-independent functions (Scott et al., 2007).
Characterization of LKB1 Using Mouse Models
The early developmental link between LKB1 and PJS motivated the generation of Lkb1 loss of function alleles in mice to study the function of Lkb1 in disease. Homozygous loss of Lkb1 in mice resulted in embryonic lethality at midgestation. This lethality was attributed to abnormal Vegf regulation, where Vegf was expressed at higher levels compared to wild-type mice. Vegf expression is regulated in part through hypoxia-induced factor (Hif1α), suggesting that loss of Lkb1 results in metabolic stress (Ylikorkala et al., 2001).
To better characterize Lkb1 function in vivo, conditional knockout alleles were generated to study the consequence of Lkb1 loss in tissue specific manner. Conditional Lkb1 knockout (KO) alleles were generated to induce Lkb1 KO using Cre-recombinase (Bardeesy et al., 2002). Characterization of these alleles confirmed earlier observations that Lkb1 loss is embryonic lethal and that Lkb1 heterozygotes developed hamartomatous polyps like PJS patients (Bardeesy et al., 2002). Furthermore, DePinho’s group determined that Lkb1 heterozygous mice were more susceptible to carcinogenesis when exposed to 7,12-dimethylbenz(a)anthracene (DMBA) (Bardeesy et al., 2002).
This mouse model was used to identify the role of Lkb1 in energy metabolism. The Alessi group studied the effects of Lkb1 KO in muscle tissues by inducing Lkb1 KO using Cre driven under the muscle creatine kinase (MCK) promoter. Expression of MCK-Cre excised Lkb1 in heart and skeletal muscle. They found that the AMP:ATP ratios were significantly elevated compared to control mice. This suggested that Lkb1 was involved in correcting the metabolic imbalance and that when Lkb1 activity is lost, muscle cells were not able to generate ATP to correct the imbalance (Sakamoto et al., 2005). This work led to the connection between LKB1 and energy metabolism. Later, it was discovered that LKB1 promotes the phosphorylation and activation of the AMPK family of kinases (Hawley et al., 2003; Lizcano et al., 2004).
LKB1 Activates the AMPK Family of Kinases
LKB1 functions upstream of the AMPK of kinases family, which consists of AMPK, and 12 other related kinases termed the AMPK-related kinases (ARKs); novel (Nu) AMP related kinase 1 and 2 (NUAK1,2), salt inducible kinase 1–3 (SIK1-3), microtubule affinity regulating kinases 1–4 (MARK1-4), brain selective kinases 1 and 2 (BRSK1,2), and maternal embryonic leucine zipper kinase (MELK) (Scott et al., 2007).
Before the association of LKB1 with energy metabolism, the C.elegans ortholog of LKB1 (Par-4) was implicated in cell polarity. Par-4 is asymmetrically localized during the first embryonic division, providing key signals for fate decisions in subsequent cell divisions (Watts et al., 2000). A role for LKB1 in regulating cell polarization was later observed in mammalian cells, suggesting evolutionary conserved function. Furthermore, the discovery that LKB1 phosphorylates and activates the ARK kinases in addition to AMPK provided insights into the potential mechanism behind the cell polarity related function of LKB1 (Lizcano et al., 2004). Both the MARK and BRSK kinases regulate microtubule dynamics and contribute to regulate cell polarity through LKB1 phosphorylation and activation (Kojima et al., 2007; Nakano and Takashima, 2012). MARK proteins phosphorylate and inhibit microtubule associated proteins (MAPs) causing microtubule depolymerization and reorganization (Kojima et al., 2007). A genetic screen in HEK293T cells identified the Hippo pathway protein YAP as a mediator of LKB1-MARK axis, implicating LKB1 in regulate organ size (Mohseni et al., 2014).
The tumor suppressor function of LKB1 is also partially mediated to the ARK kinases. LKB1 phosphorylation and activation of NUAK1 promotes cell cycle arrest in response to UV induced DNA damage. LKB1 and NUAK1 can phosphorylate cyclin-dependent kinase inhibitor 1A (CDKN1A), which inhibits cyclin-CDK complexes preventing G1/S transition and leads to a cell cycle arrest in a TP53 dependent mechanism (Zeng and Berger, 2006; Esteve-Puig et al., 2014). NUAK 1 and 2 are also implicated in glucose tolerance and attenuation of insulin signaling in muscle cells and regulate cell motility and muscle contraction through activation of myosin (Koh et al., 2010; Zagórska et al., 2010; Vallenius et al., 2011).
Finally, the LKB1-SIK axis plays a role in regulating metabolism. SIK phosphorylation of cAMP response element binding protein (CREB) and CREB regulated transcription co-activator (CRTC) regulates multiple biological processes including metabolism, cell differentiation, and proliferation (Gao et al., 2018). LKB1 activation of SIK kinases inhibits gluconeogenesis in liver cells, and the LKB1-SIK axis promotes growth and differentiation of adipose tissue (Patel et al., 2014; Darling and Cohen, 2021). Furthermore, LKB1-SIK axis can promote GLUT4 mediated glucose import in muscle and adipose tissues by phosphorylating and inhibiting CRTC2/3 (Stringer et al., 2015). SIK1 and SIK3 inhibit the expression of lipogenic genes, thereby reducing lipogenesis (Du et al., 2008; Sun et al., 2020).
The different and numerous ARK kinases highlight the diverse and cell specific functions of LKB1. LKB1 can directly phosphorylate and activate all members of the AMPK family except MELK. In this way, LKB1 acts as a master regulatory kinase acting through the AMPK family of kinases in a cell specific manner to regulate multiple pathways related to metabolism, cell polarity, migration, division, and transcription (Ylikorkala et al., 2001).
LKB1-AMPK in Metabolic Regulation
The best characterized, and primary target of LKB1 to regulate energy metabolism is AMPK, which is the focus of this review. LKB1 functions upstream of AMPK, the central regulator in maintaining intracellular ATP levels. Upon activation under energy stress, when the AMP:ATP ratio is high, AMPK acts as a metabolic switch to inhibit anabolic (fatty acid and protein synthesis) pathways and promotes the activation of catabolic pathways (glycolysis, fatty acid oxidation, and autophagy) to correct the energy imbalance (Ciccarese et al., 2019). AMPK is a heterotrimeric protein kinase composed of a catalytic a subunit, and two regulatory subunits ß and γ. AMP binds the γ subunit promoting a conformational change to remove allosteric inhibition of AMPK and promote its activation with additional Thr172 phosphorylation by upstream kinases. The primary kinase responsible for AMPK activation was identified to be LKB1, providing the first insights that the tumor suppressor function of LKB1 was mediated through AMPK to regulate energy metabolism (Hawley et al., 2003; Woods et al., 2003; Hardie and Alessi, 2013).
One of the direct readouts to assess AMPK activity is phosphorylation of acetyl-CoA carboxylase (ACC) which is the first enzyme in de novo lipid synthesis. AMPK-mediated phosphorylation of ACC is an inhibitory post-translational modification, thereby inhibiting fatty acid synthesis (Carling et al., 1987). AMPK also phosphorylates HMG-CoA reductase which plays a role in sterol synthesis (Sato et al., 1993). Fatty acid synthesis is important for cancer cell progression and LKB1 mutant tumors exhibit elevated gene expression signature of genes involved in lipid synthesis (Carling et al., 1987; Bhatt et al., 2019) (Figure 1).
The LKB1-AMPK axis also inhibits the central integrator of energy and mitogenic signals to cell growth and division, the mammalian target of rapamycin complex 1 (mTORC1) (Corradetti et al., 2004). mTORC1 with its adaptor regulatory-associated protein of mTOR (RAPTOR), is a kinase responsible for promoting the phosphorylation of eukaryotic initiation factor 4E (EIF4E) binding protein (4EBP1), an inhibitor of EIF4E elongation factor, and p70 ribosomal S6 kinase 1 (p70S6). mTORC1 promotes the translation of mRNAs required for cell growth and division, including ribosomal proteins and proteins involved in translation (Thoreen et al., 2012) (Figure 1).
AMPK can indirectly inhibit mTORC1 by phosphorylating and activating tuberous sclerosis complex 2 (TSC2) (Corradetti et al., 2004). TSC2 functions as a heterodimer with TSC1, and together functions to indirectly inhibit mTOR through inhibition of the GTPase RAS homolog enriched in the brain (RHEB). When RHEB is active, RHEB binds and activates mTORC1 using its GTPase activity to induce a conformational change to the mTORC1 complex (Tee et al., 2003). TSC1/TSC2 bind and inhibit RHEB, thereby inhibiting mTORC1. AMPK can also directly inhibit mTORC1 by directly phosphorylating RAPTOR (Jewell et al., 2019). AMPK phosphorylation of RAPTOR promotes 14–3–3 protein binding, inhibiting mTORC1 from interacting with downstream targets 4EBP1 and p70S6 (Nojima et al., 2003) (Figure 1).
A consequence of LKB1 loss is enhanced mTORC1 activation. mTORC1 promotes cell growth and metabolic changes such as increasing glycolysis and inhibiting autophagy. LKB1-AMPK directly, and indirectly through mTORC1, regulate autophagy. LKB1 directly phosphorylates ULK1 to promote autophagy and plays a role in mitochondrial biogenesis. mTORC1 phosphorylates and inhibits ULK1 inhibiting autophagy, and this is dysregulated in LKB1 mutant cells. Furthermore, AMPK can promote the transcription of genes involved in autophagy through activation of the transcription factor FOXO (Kim et al., 2011) (Figure 1). Through the regulation of mTORC1, LKB1 and AMPK provide an important regulatory link between cell metabolism and cell growth and division.
LKB1 Loss Promotes Metabolic Changes
The LKB1-AMPK-mTORC1 axis is often deregulated in cancer cells, resulting in metabolic changes to support cancer cell growth and division. Cancer cells are often in a state of energy imbalance and have elevated energy requirements to manage high growth and division rates. Although producing less ATP, cancer cells show increased glycolysis rates even in the presence of oxygen. This shift to aerobic glycolysis demonstrates the Warburg effect (Pavlova and Thompson, 2016). LKB1 plays an important role in regulating glycolysis. Cells mutant for LKB1 exhibit increased glucose uptake and increased extracellular acidification rate (ECAR). ECAR is determined by measuring lactate levels, which is produced by lactate dehydrogenase (LDH) to regenerate NAD+ for glycolysis. Lactate is then exported out of the cell causing acidification of the extracellular space. In the lung cancer cell line A549, which is deficient for LKB1, transient expression of LKB1 resulted in a reduction of ECAR by 20% (Faubert et al., 2014). This suggests that LKB1 is an important regulator of glycolysis and that LKB1 loss impacts glycolysis rate in cancer cells.
The metabolic reprogramming that occurs under LKB1 loss is also observed in a Her2 positive Lkb1 mutant breast cancer mouse model, Stk11−/−/NIC. Loss of Lkb1 reduces the latency of Her2 positive breast cancer from 197 to 147 days. Loss of Lkb1 is associated with elevated mTORC1 activation and reduced AMPK activity (Andrade-Vieira et al., 2013). LKB1 acts as a metabolic nexus, connecting AMPK and mTORC1 signaling with downstream metabolic pathways. One such link is how LKB1 loss impacts glycolysis. AMPK and mTORC1 are important regulators of glycolysis and since Lkb1 acts upstream of AMPK and mTOR, the Marignani group examined the metabolic changes that occur when LKB1 is lost. Stk11−/−/NIC tumors exhibit increased branch chain amino acids (BCAA) valine and isoleucine and elevated glutamine and methionine levels and showed lower levels of glutathione, a ROS scavenger suggesting increased ROS stress which promotes tumor development. Furthermore, LDH and pyruvate dehydrogenase (PDH) levels are elevated, indicating elevated glycolysis (Andrade-Vieira et al., 2013).
In this model of breast cancer, loss of Lkb1 synergizes with activated Her2, which promotes downstream activation of PI3K and AKT, leading to elevated mTORC1 activity. Elevated mTORC1 promotes metabolic reprogramming, increasing glycolysis by promoting the translation of glucose transporters, and glycolytic enzymes. The resulting increase in glycolysis is observed through elevated lactate (Andrade-Vieira et al., 2013) (Figure 2).
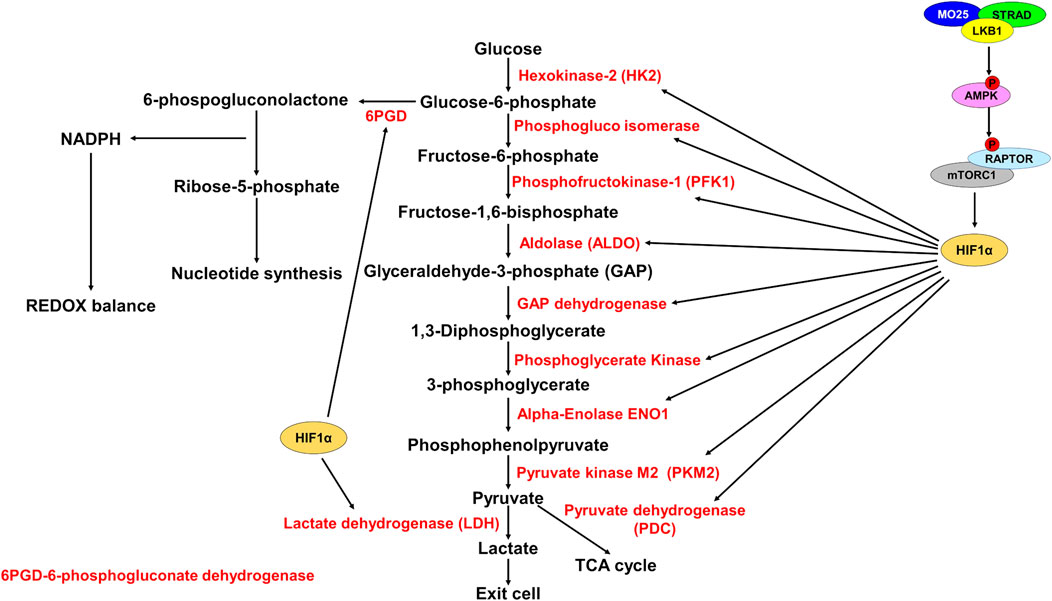
FIGURE 2. HIF1α promotes expression of glycolytic enzymes. Schematic representation of glycolysis and the pentose phosphate pathway (PPP). LKB1 phosphorylates and activates AMPK which then phosphorylates and inhibits RAPTOR, thereby inhibiting mTORC1. mTORC1 promotes HIF1α upregulation. Active HIF1α promotes expression of glycolytic enzymes (red) increasing glycolysis rate. HIF1α also promotes expression of 6PGD to shunt glucose-6-phosphate to the PPP to generate NADPH for REDOX balance and for synthesis of ribose-5-phosphate for nucleotide synthesis.
Glycolysis
Glycolysis is an important metabolic pathway that supports energy and biomolecule production, synthesis of ROS scavengers, and synthesis of nucleotides. Under normal physiological conditions, glucose is oxidized to pyruvate and shunted to the electron transport chain (ETC) for the generation of ATP by oxidative phosphorylation (OXPHOS). Under aerobic conditions, 1 molecule of glucose generates ∼36 ATP and under anaerobic conditions, generates 2 molecules of ATP (Spinelli and Haigis, 2018). Glycolysis generates significantly fewer ATP molecules than OXPHOS but occurs at a much faster rate, therefore increased glycolysis can supply ATP production quicker under anaerobic conditions (Rogatzki et al., 2015). To do this, regeneration of NAD+ is required to support glycolysis. LDH generates NAD+ and lactate from pyruvate and NADH, thus supporting the increased glycolysis and ATP generation requirement (Figure 2).
The importance of glycolysis is not only through energy production, but glycolytic intermediates are important for many anabolic processes such as nucleotide synthesis and generating ROS scavengers (Lin et al., 2015; Ju et al., 2017). Cancer cells characteristically show increased utilization of glucose by glycolysis and a shift from OXPHOS to glycolysis to support their growth and division (Warburg et al., 1927). This idea has recently been questioned as OXPHOS was shown to still be utilized for energy production in some cancers (Davidson et al., 2016). OXPHOS is supported by lactate import and utilization through the TCA cycle, while glucose is used to support glycolysis and the pentose phosphate pathway (PPP) shunt to produce ROS scavenger NADPH. NADPH manages ROS stress and is important in generating ribose-5-phosphate for nucleotide biosynthesis (Lin et al., 2015; Ju et al., 2017) (Figures 2,3).
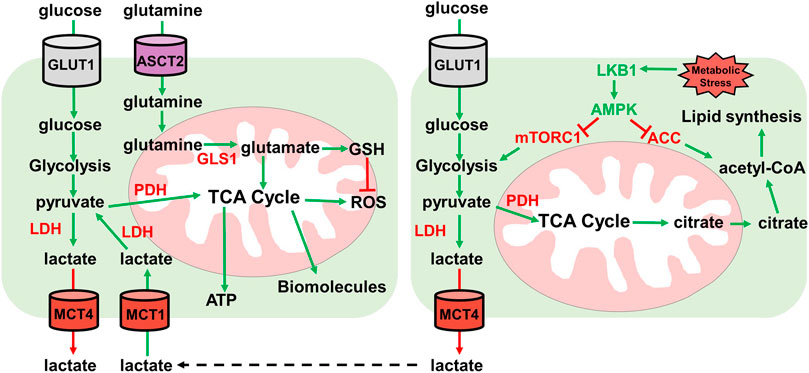
FIGURE 3. Lactate and glutamine metabolism. Overview of lactate and glutamine metabolism. Glucose is imported by GLUT1 transporter. Glucose is then metabolized by glycolysis into pyruvate. Pyruvate then either enters the TCA cycle through pyruvate dehydrogenase (PDH) for ATP and biomolecule synthesis, or pyruvate is converted to lactate by lactate dehydrogenase (LDH). Lactate is then exported by MCT4 lactate exporter. Extracellular lactate can be imported by MCT1 and converted to pyruvate by lactate dehydrogenase for utilization in the TCA cycle for ATP and biomolecule synthesis. Glutamine is imported by ASCT2 where it then enters the mitochondria and is converted to glutamate. Glutamate can either enter the TCA cycle or is converted to GSH for ROS neutralization. Under metabolic stress conditions, the LKB1-AMPK pathway is activated. LKB1 activates AMPK which inhibits ACC and mTORC1. AMPK inhibition of ACC prevents lipid synthesis and mTORC1 inhibition by AMPK reduces glycolysis. Low glycolysis rate reduces TCA cycle generation of citrate which is used to generate acetyl-CoA for lipid synthesis.
LKB1 Regulates Glycolysis
Elevated glycolysis rate is a hallmark of cancer, and tumors deficient in LKB1 show elevated glycolysis rates, elevated glucose import, and increased expression of glycolytic enzymes. The effect LKB1 loss has on glycolysis and glucose uptake is mediated by mTORC1 and concomitant oncogenic mutations. In LKB1 mutant tumors, AMPK activity is diminished which leads to elevated mTORC1 activation (van Veelen et al., 2011). mTORC1 promotes the upregulation of the transcription factor HIF1α that becomes active under hypoxic conditions to promote aerobic respiration and ATP synthesis (Keith et al., 2012) (Figure 2).
HIF1α is regulated both at the protein level and translational level. Under normoxic physiological conditions, HIF1α is targeted for degradation by the E3 ligase von Hipple-Lindau tumor suppressor (Tanimoto et al., 2000). Under hypoxic conditions, HIF1α is stabilized where it translocates to the nucleus and induces gene transcription. Furthermore, mTORC1 promotes the upregulation of HIF1α. This was demonstrated in mouse embryonic fibroblasts (MEFs) by deleting Tsc2 to promote mTORC1 activity. Tsc2−/− MEFs show elevated Hif1α levels, and this was dependent on mTORC1 targets 4Ebp1 and S6k1 (Düvel et al., 2010). In breast cancer cells, mTORC1 promotes HIF1α translation (Sakamoto et al., 2014). The increased mTORC1 activity also correlated with increased glucose uptake and utilization, which was suppressed by depleting HIF1α.
LKB1 mutant tumors show elevated HIF1α expression leading to increased glycolysis and lactate production (Andrade-Vieira et al., 2013; Faubert et al., 2014; Nam et al., 2016). Knockdown of HIF1α in LKB1 mutant cells causes reduced growth and cell death under nutrient stress (Faubert et al., 2014; Nam et al., 2016). Gastrointestinal hamartomatous polyps from Lkb1 heterozygous mice also show increased expression of the glucose transporter Glut1, and the first enzyme in glycolysis hexokinase-2 (HK2) (Shackelford et al., 2009). Both GLUT1 and HK2 are transcriptional targets of HIF1α (Figure 2).
Oncogenic mutations also synergize with LKB1 loss to promote glycolysis and glucose uptake. LKB1 is frequently concomitant with KRAS in lung cancers and KRAS oncogenic mutants upregulate glycolysis. KRAS was shown to promote HIF1α expression in colon cancer cells. Furthermore, Kerr et al. showed that KRAS copy number impacts glucose utilization (Kerr and Martins, 2018). Increasing the copy number of KRAS leads to increased glucose utilization, increased expression of GLUT transporters and glycolytic enzymes feeding the TCA cycle, and in later stages, glycolysis mediates management of ROS. Davidson et al. showed that in lung cancer models of KrasG12D and Tp53 (KP mice), KrasG12D promotes increased glucose metabolism to generate pyruvate and lactate, consistent with observations that Kras oncogenic mutations yield elevated lactate (Davidson et al., 2016). Pyruvate is then shunted to the TCA cycle through pyruvate dehydrogenase (PDH), indicated by increased citrate levels. This indicates that in Kras models of lung cancer, glucose is utilized to generate both pyruvate and lactate, and pyruvate is shunted to the TCA cycle. LKB1 loss can synergize with KRAS oncogenic mutations by increasing glucose uptake and glycolysis, leading to increased lactate production.
Lactate as an Energy Source in Lung Tumors
Lactate has become more important in energy metabolism than once thought (Hui et al., 2017). Under normal metabolism where OXPHOS is functional, pyruvate is oxidized to acetyl-CoA by the pyruvate dehydrogenase complex (PDC). In hypoxic conditions, pyruvate is converted to lactate by LDH. Cancer cells display elevated LDH expression, leading to elevated lactate formation and export. This creates an acidic tumor microenvironment which creates a favorable environment for invasion and metastasis and modulates immune cell functions (Rizwan et al., 2013; Xie et al., 2014; Brand et al., 2016). Furthermore, circulating lactate can be imported and utilized as an energy source and substrate for lipogenesis (Brooks, 2009; Chen et al., 2016). Lactate is imported using the lactate transporter monocarboxylate transporter 1 (MCT1). Inhibiting MCT1 reduces oxidative respiration and promotes an increase in glycolysis in both cancer cell lines in culture and xenograft models (Sonveaux et al., 2008; Pavlides et al., 2009) (Figure 3).
Lactate incorporation in tumor metabolism was seen in human NSCLC patients. NSCLC patients were infused with labeled glucose and incorporation of metabolic intermediates was measured from tumor samples. Results showed elevated lactate labeling compared to glycolytic metabolites (Faubert et al., 2017). Furthermore, LKB1 mutant xenografts (H460 and HCC15 cells) in mice showed a similar phenotype with increased labeling of lactate (Faubert et al., 2017). In these xenograft models, infusion with labeled lactate showed labeled TCA intermediates, suggesting carbon from lactate can be incorporated into the TCA cycle (Faubert et al., 2017).
This study showed that in H460 and HCC15 cells, lactate shuttled in and out of the cells through lactate transporters. MCT1 knockout HCC15 cells did not affect viability or division but displayed an increase in oxygen consumption rate (OCR) and decreased ECAR, indicating decreased glycolysis and decreased lactate production and export. This was also seen in HCC15 MCT1 knockout xenograft models that when exposed to labeled lactate, showed reduced metabolite labeling of pyruvate and TCA intermediates. Like observations in cultured cells, MCT1 knockout HC115 cell xenografts also displayed reduced glycolysis (Faubert et al., 2017).
This study illustrated the importance of lactate in LKB1 mutant NSCLC tumors. Lactate can be utilized as an energy source through incorporation into the TCA cycle for ATP generation and TCA intermediates can act as precursors for synthesis of various amino acids. Extracellular lactate from neighboring cells can be imported and incorporated into the TCA through pyruvate (Figure 3). LKB1 mutant lung cancer cells show elevated LDHA/B and the lactate transporter MCT1/4. The lactate export and import mechanisms highlight the uncoupling of glycolysis from the TCA cycle. Lactate cannot simply be incorporated into the TCA cycle, but rather extracellular lactate is imported for entry into the TCA cycle (Wu et al., 2016; Faubert et al., 2017).
LKB1 Regulates ROS Balance: PPP and Glutamine Metabolism
The metabolic reprogramming caused by loss of LKB1 activity results in elevated ROS and metabolic stress. Cellular metabolism generates reactive oxygen species that need to be quenched to prevent damage to DNA, proteins, and RNAs (Finkel, 2011). The increased aerobic glycolysis in cancer cells shunts metabolites towards the PPP to generate nucleotides via Ru-5-P and NADPH for ROS scavenging and lipid synthesis (Patra and Hay, 2014). NADPH is also an important mediator of glutathione ROS scavenging. Glutathione (GSH) and thioredoxins (TRX), two proteins that neutralize ROS, are synthesized using NADPH-dependent mechanisms (Nathan and Ding, 2010) (Figure 3). Depleting PPP genes causes increased ROS through defective ROS scavenging and leads to cancer cell death in colorectal cells (Ju et al., 2017). In lung cancer cell lines, depleting 6-phosphogluconate dehydrogenase (6PGD), decreases lipogenesis, RNA biosynthesis and increases ROS (Lin et al., 2015) (Figure 2). Furthermore, in A549 and H460 cells, both LKB1 mutant cell lines, PPP-related genes are elevated indicating a reliance on the PPP pathway (Martín-Bernabé et al., 2014).
Glutamine is a common metabolite used in ROS scavenging and biosynthesis of biomolecules. Glutamine is essential for cancer cell growth in culture and is the most utilized amino acid. Glutamine can be shunted to the TCA cycle to provide acetyl-CoA or for the synthesis of other biomolecules. This is important in conditions where glucose metabolism generates lactate and not acetyl-CoA like in LKB1 deficient tumors (Wise et al., 2011; Mullen et al., 2012). KRAS oncogene mutations synergize with LKB1 loss to promote glutamine metabolism to combat ROS (Trachootham et al., 2006; Galan-Cobo et al., 2019). Glutamine can be used for amino acid and nucleic acid synthesis, further pointing to the essentiality for growth in cell culture (Caiola et al., 2016; Gwinn et al., 2018). In lung tumors in vivo however, glutamine does not enter the TCA cycle. GLS1, which is required for metabolizing glutamine in mitochondria, is not required for oncogenic KRAS-dependent growth (Davidson et al., 2016). As mentioned above, lactate is the primary carbon source for the TCA cycle (Faubert et al., 2017) (Figure 3).
Glutamine in KRAS oncogenic mutant tumors is necessary for mediating ROS neutralization. KL tumors are frequently concomitant for KEAP2, the inhibitor of oxidative stress response transcription factor NRF2. When KEAP2 mutations are concomitant with KL mutations, accelerated tumor growth is observed (Romero et al., 2017; Galan-Cobo et al., 2019). Furthermore, when LKB1 is expressed in LKB1 deficient A549 cells, cell death is observed due to increased glutamine transformation and are less sensitive than control A549 cells to glutamine inhibitors (Galan-Cobo et al., 2019). LKB1 mutant cells promote NRF2 dependent GCL expression, the primary enzyme to generate γ-Gly-Gly from glutamine and cysteine to increase GSH pools. Knockdown of NRF2 in A549 cells causes decreased GSH formation from glutamine (Mitsuishi et al., 2012). The dependence of LKB1 deficient cells to glutamine is also seen in polycystic kidney disease where LKB1 mutant kidneys show increased dependence on glutamine to provide a synthesis of non-essential amino acids and GSH for ROS neutralization (Flowers et al., 2018).
Hexosamine Biosynthesis
Another pathway that is elevated in LKB1 mutant tumors is the hexosamine biosynthesis pathway (HBP). The HBP produces UDP-N-acetylglucosamine (UDP-GlcNAc) which is important for protein glycosylation. The HBP integrates intermediates from glycolysis, lipid synthesis, glutamine metabolism, and nucleotide metabolism to generate UDP-GlcNAc (Vasconcelos-dos-Santos et al., 2015; Ferrer et al., 2016). UDP-GlcNAc is used by glycosyltransferases to generate glycoconjugates glycoproteins, glycolipids, and glycosaminoglycans. Glycosylation can modulate protein activity. For example, glycosylation inhibits phosphofructokinase 1 (PFK1) under hypoxic conditions, diverting glycolysis intermediates to PPP to decrease ROS (Sola-Penna et al., 2010).
Lung cancer cells have altered glycosylation and KL tumors display an elevated HBP gene expression profile. LKB1 acts to suppress HBP, acting as an inhibitor of glycosylation. Inhibiting glycosylation by inhibiting glutamine-fructose-6-phosphate transaminase 2 (GFPT2) causes cell death in KL tumor cells but not in KRASG12D tumor cells. This suggests a potential vulnerability in KL cells that can be exploited (Kim et al., 2020). KL mice treated with GFPT inhibitor Azaserine significantly suppressed tumor growth and this effect was specific to LKB1 loss. This suggests a potential therapeutic target in KL lung cancers.
Targeting LKB1 Mutant Cancers Through Metabolism
The metabolic changes that occur under LKB1 loss present an opportunity for therapeutic intervention by targeting aberrant metabolic pathways. LKB1 mutant tumors display elevated glycolysis, lactate metabolism, and fatty acid synthesis due to the loss of AMPK activity, and subsequent loss of mTORC1 inhibition (Carling et al., 1987; van Veelen et al., 2011; Keith et al., 2012; Bhatt et al., 2019). Investigating downstream pathways could identify potential targets for therapeutic intervention. This is the case for biguanides metformin and closely related phenformin. Metformin is used to treat type II diabetes and functions by inhibiting mitochondrial complex I of the ETC (El-Mir et al., 2000; Dykens et al., 2008). The inhibition of complex I uncouples mitochondrial membrane potential, and reduces ATP generation through OXPHOS, leading to activation of AMPK in an LKB1 dependent mechanism (El-Mir et al., 2000; Dykens et al., 2008). In a mouse model of NSCLC where Lkb1 is deficient, phenformin was able to reduce tumor growth (Shackelford et al., 2013). Although phenformin did not cause activation of AMPK, due to the lack of functional Lkb1, the disruption of OXPHOS and subsequent reduction in ATP caused metabolic stress and elevated ROS. Lkb1 deficient tumors are unable to tolerate high levels of ROS and therefore undergo apoptosis as a result (Shackelford et al., 2013).
Cancer cells rely on elevated glycolysis and overexpress genes related to glucose metabolism (Hanahan and Weinberg, 2011; Pavlides et al., 2009). LKB1 deficient tumors display elevated glycolysis signature mediated by elevated mTORC1 activity and HIF1α expression (Figure 2) (Andrade-Vieira et al., 2013; Faubert et al., 2014; Nam et al., 2016). Inhibitors against mTORC1 have limited therapeutic response in NSCLC. In NSCLC mouse models, the mTORC1 inhibitor rapamycin failed to induce a therapeutic response against KL tumors (Liang et al., 2010). mTORC1 inhibitors are often associated with resistance due to AKT-mTORC2 feedback loop, activating of mTORC1 (Wander et al., 2011). To circumvent potential resistance mechanisms associated with mTOR inhibitors, the Marignani’ group conducted a pre-clinical trial that investigated strategies to inhibit energy and growth simultaneously by targeting glycolysis and mTOR as a potential therapeutic strategy for HER2 positive breast cancer. In Stk11−/−/NIC mice, Lkb1 loss synergizes with enhanced Her2 activation to promote activation of both arms of mTOR, mTORC1 and mTORC2, resulting in enhanced cell growth fueled by enhanced metabolism. They showed that simultaneously targeting glycolysis and mTOR complexes directly with glucose analog 2-deoxyglucose (2-DG) and mTOR inhibitor AZD8055 reduced tumor growth significantly and provided a stronger effect in combination than either 2-DG or AZD8055 alone (Andrade-Vieira et al., 2014).
Targeting glycolytic enzymes could provide a therapeutic window without dramatically affecting normal cells, which do not rely on elevated glucose metabolism (Pavlides et al., 2009; Hanahan and Weinberg, 2011). WZB117 is a glucose transporter inhibitor that showed promising results in NSCLC xenograft models using LKB1 deficient A549 cells (Liu et al., 2012). WZB117 reduced ATP, GLUT1 and glycolytic enzyme protein levels (Liu et al., 2012). Furthermore, WZB117 significantly reduced tumor volume (Liu et al., 2012).
The hexokinase-2 inhibitor 2-DG has shown promising results in combination therapies (Maher et al., 2004; Andrade-Vieira et al., 2014). Hexokinase-2 catalyzes the phosphorylation of glucose, restricting its export and keeping glucose inside the cell (Bustamante and Pedersen, 1977; Bustamante et al., 1981). Paclitaxel and 2-DG significantly reduced tumor growth in NSCLC tumor xenograft model compared to single therapy alone (Maschek et al., 2004). By restricting the use of glucose by inhibiting hexokinase-2, other downstream metabolites are also reduced. Glycolysis intermediates cannot be shunted to the PPP to generate NADPH and Ru-5-BP for nucleotide synthesis restricting ROS scavenging and cell division (Figures 2,3). This will increase ROS stress, reduce tumor growth, and induce apoptosis.
LKB1 mutant tumors are susceptible to ROS stress and rely on multiple pathways to manage ROS stress. LKB1 mutant tumors display elevated PPP pathway activity to generate NADPH along with increased glutamine metabolism to generate glutathione to scavenge ROS. Treating LKB1 mutant tumors with compounds that increasing ROS or targeting KL tumors with glutamine inhibitors to reduce glutathione production inhibited tumor growth (Galan-Cobo et al., 2019). Glutamine metabolism is upregulated in KL tumors, where glutamine functions to generate glutathione to manage ROS stress (Figure 3) (Caiola et al., 2016; Davidson et al., 2016; Gwinn et al., 2018). Since LKB1 deficient tumors are sensitive to increased ROS stress, compounds that produce excess ROS stress could serve as potential therapeutic agents for LKB1 deficient tumors.
Another potential liability in LKB1 mutant tumors is lactate metabolism. Similar to the findings by the Marignani group, inhibition of glycolysis can also lead to reduced lactate production. Inhibitors of glycolytic enzymes or LDH can provide potential therapeutic avenues to explore. A549 cells deficient in LDHA showed reduced tumor formation after tail-vein injection (Xie et al., 2014). Inhibiting lactate production in KL tumors can restrain oxidative phosphorylation and amino acid synthesis by preventing TCA cycle regeneration. Furthermore, inhibiting lactate transporters could also be an effective approach. Knockout of MCT1 in LKB1 deficient cells reduced tumor growth in xenograft models, highlighting lactate metabolism as a potential therapeutic vulnerability (Faubert et al., 2017).
LKB1 mutant lung tumors display increased fatty acid deposits attributed to the elevated fatty acid synthesis pathways. Loss of AMPK activity causes ACC to become activated and promote fatty acid synthesis, while also impacting the expression of genes involved in fatty acid oxidation (Figures 1,3) (Carling et al., 1987; Bhatt et al., 2019). Targeting fatty acid synthesis by inhibiting ACC in mouse xenografts of LKB1 deficient cells showed reduced tumor growth (Svensson et al., 2016). Lipids are important components in signaling molecules and for generation of membranes, and fatty acid synthesis is critical for cancer cell growth and survival. Targeting other genes involved in fatty acid synthesis provides other potential targets that could attenuate LKB1 deficient tumor growth.
Exploiting metabolic vulnerabilities in LKB1 deficient lung cancer provides promising avenues for the development of novel, effective therapies. Many metabolic pathways are regulated by LKB1. Understanding the impact of LKB1 loss has on tumor metabolism provides a road map for identifying potential targets.
Conclusion
Metabolic adaptations by cancer cells are critical to meet the energetic needs and synthesis of macromolecules. Many cancer types show elevated glycolysis even in aerobic conditions. Glycolysis provides intermediates to support nucleotide synthesis and redox balance, both critical to supporting growth and division.
The tumor suppressor LKB1 plays an important role in regulating multiple metabolic pathways. By activating AMPK, LKB1 functions as a metabolic nexus, linking AMPK and mTORC1 signaling, to downstream metabolic pathways. LKB1 loss is associated with multiple malignancies and causes metabolic reprograming to increase glycolysis and lactate production, elevate hexosamine biosynthesis and glutamine metabolism. Understanding metabolic pathways regulated by LKB1 can highlight therapeutic avenues by targeting pathways dysregulated due to LKB1 loss and lead to better health outcomes in patients.
Author Contributions
Writing-original draft preparation, MB and PAM; writing-review and editing, MB and PAM; supervision- PAM; project administration PAM; funding acquisition, PAM.
Funding
Cancer Research Society, The Canadian Cancer Society Diane Campbell designated research fund (grant #706202). Natural Sciences and Engineering Research Council of Canada.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank members of the Marignani Lab for their support. The authors acknowledge that Dalhousie University campuses are located on original lands of the Mi’kma’ki, the ancestral and unceded territory of the Mi’kmaq People.
References
Andrade-Vieira, R., Goguen, D., Bentley, H. A., Bowen, C. V., and Marignani, P. A. (2014). Pre-clinical Study of Drug Combinations that Reduce Breast Cancer burden Due to Aberrant mTOR and Metabolism Promoted by LKB1 Loss. Oncotarget 5 (24), 12738–12752. doi:10.18632/oncotarget.2818
Andrade-Vieira, R., Xu, Z., Colp, P., and Marignani, P. A. (2013). Loss of Lkb1 Expression Reduces the Latency of ErbB2-Mediated Mammary Gland Tumorigenesis, Promoting Changes in Metabolic Pathways. Plos One 8 (2), e56567. doi:10.1371/journal.pone.0056567
Avizienyte, E., Roth, S., Loukola, A., Hemminki, A., Lothe, R. A., Stenwig, A. E., et al. (1998). Somatic Mutations in LKB1 Are Rare in Sporadic Colorectal and Testicular Tumors. Cancer Res. 58 (10), 2087–2090.
Baas, A. F., Boudeau, J., Sapkota, G. P., Smit, L., Medema, R., Morrice, N. A., et al. (2003). Activation of the Tumour Suppressor Kinase LKB1 by the STE20-like Pseudokinase STRAD. EMBO J. 22 (12), 3062–3072. doi:10.1093/emboj/cdg292
Bardeesy, N., Sinha, M., Hezel, A. F., Signoretti, S., Hathaway, N. A., Sharpless, N. E., et al. (2002). Loss of the Lkb1 Tumour Suppressor Provokes Intestinal Polyposis but Resistance to Transformation. Nature 419 (6903), 162–167. doi:10.1038/nature01045
Bhatt, V., Khayati, K., Hu, Z. S., Lee, A., Kamran, W., Su, X., et al. (2019). Autophagy Modulates Lipid Metabolism to Maintain Metabolic Flexibility for Lkb1-Deficient Kras-Driven Lung Tumorigenesis. Genes Dev. 33 (3-4), 150–165. doi:10.1101/gad.320481.118
Boardman, L. A., Couch, F. J., Burgart, L. J., Schwartz, D., Berry, R., McDonnell, S. K., et al. (2000). Genetic Heterogeneity in Peutz-Jeghers Syndrome. Hum. Mutat. 16 (1), 23–30. doi:10.1002/1098-1004(200007)16:1<23::aid-humu5>3.0.co;2-m
Boudeau, J., Baas, A. F., Deak, M., Morrice, N. A., Kieloch, A., Schutkowski, M., et al. (2003). MO25/Interact with STRAD/Enhancing Their Ability to Bind, Activate and Localize LKB1 in the Cytoplasm. EMBO J. 22 (19), 5102–5114. doi:10.1093/emboj/cdg490
Boudeau, J., Kieloch, A., Alessi, D. R., Stella, A., Guanti, G., and Resta, N. (2003). Functional Analysis of LKB1/STK11 Mutants and Two Aberrant Isoforms Found in Peutz-Jeghers Syndrome Patients. Hum. Mutat. 21 (2), 172. doi:10.1002/humu.9112
Boudeau, J., Miranda-Saavedra, D., Barton, G. J., and Alessi, D. R. (2006). Emerging Roles of Pseudokinases. Trends Cel Biol. 16 (9), 443–452. doi:10.1016/j.tcb.2006.07.003
Brand, A., Singer, K., Koehl, G. E., Kolitzus, M., Schoenhammer, G., Thiel, A., et al. (2016). LDHA-associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cel Metab. 24 (5), 657–671. doi:10.1016/j.cmet.2016.08.011
Brooks, G. A. (2009). Cell-cell and Intracellular Lactate Shuttles. J. Physiol. 587 (Pt 23), 5591–5600. doi:10.1113/jphysiol.2009.178350
Burotto, M., Chiou, V. L., Lee, J.-M., and Kohn, E. C. (2014). The MAPK Pathway across Different Malignancies: A New Perspective. Cancer 120 (22), 3446–3456. doi:10.1002/cncr.28864
Bustamante, E., Morris, H. P., and Pedersen, P. L. (1981). Energy Metabolism of Tumor Cells. Requirement for a Form of Hexokinase with a Propensity for Mitochondrial Binding. J. Biol. Chem. 256 (16), 8699–8704. doi:10.1016/s0021-9258(19)68900-3
Bustamante, E., and Pedersen, P. L. (1977). High Aerobic Glycolysis of Rat Hepatoma Cells in Culture: Role of Mitochondrial Hexokinase. Proc. Natl. Acad. Sci. U.S.A. 74 (9), 3735–3739. doi:10.1073/pnas.74.9.3735
Caiola, E., Brunelli, L., Marabese, M., Broggini, M., Lupi, M., and Pastorelli, R. (2016). Different Metabolic Responses to PI3K Inhibition in NSCLC Cells Harboring Wild-type and G12C Mutant KRAS. Oncotarget 7 (32), 51462–51472. doi:10.18632/oncotarget.9849
Calles, A., Sholl, L. M., Rodig, S. J., Pelton, A. K., Hornick, J. L., Butaney, M., et al. (2015). Immunohistochemical Loss of LKB1 Is a Biomarker for More Aggressive Biology in KRAS-Mutant Lung Adenocarcinoma. Clin. Cancer Res. 21 (12), 2851–2860. doi:10.1158/1078-0432.ccr-14-3112
Carling, D., Zammit, V. A., and Hardie, D. G. (1987). A Common Bicyclic Protein Kinase cascade Inactivates the Regulatory Enzymes of Fatty Acid and Cholesterol Biosynthesis. FEBS Lett. 223 (2), 217–222. doi:10.1016/0014-5793(87)80292-2
Chen, Y.-J., Mahieu, N. G., Huang, X., Singh, M., Crawford, P. A., Johnson, S. L., et al. (2016). Lactate Metabolism Is Associated with Mammalian Mitochondria. Nat. Chem. Biol. 12 (11), 937–943. doi:10.1038/nchembio.2172
Chong, C. R., and Jänne, P. A. (2013). The Quest to Overcome Resistance to EGFR-Targeted Therapies in Cancer. Nat. Med. 19 (11), 1389–1400. doi:10.1038/nm.3388
Ciccarese, F., Zulato, E., and Indraccolo, S. (2019). LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective. Oxid Med. Cel Longev 2019, 8730816. doi:10.1155/2019/8730816
Collisson, E. A., Campbell, J. D., Brooks, A. N., Berger, A. H., Lee, W., Chmielecki, J., et al. (2014). Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 511 (7511), 543–550. doi:10.1038/nature13385
Conde, E., Suarez-Gauthier, A., García-García, E., Lopez-Rios, F., Lopez-Encuentra, A., García-Lujan, R., et al. (2007). Specific Pattern of LKB1 and Phospho-Acetyl-CoA Carboxylase Protein Immunostaining in Human normal Tissues and Lung Carcinomas. Hum. Pathol. 38 (9), 1351–1360. doi:10.1016/j.humpath.2007.01.022
Contreras, C. M., Gurumurthy, S., Haynie, J. M., Shirley, L. J., Akbay, E. A., Wingo, S. N., et al. (2008). Loss of Lkb1 Provokes Highly Invasive Endometrial Adenocarcinomas. Cancer Res. 68 (3), 759–766. doi:10.1158/0008-5472.can-07-5014
Corradetti, M. N., Inoki, K., Bardeesy, N., DePinho, R. A., and Guan, K.-L. (2004). Regulation of the TSC Pathway by LKB1: Evidence of a Molecular Link between Tuberous Sclerosis Complex and Peutz-Jeghers Syndrome. Genes Dev. 18 (13), 1533–1538. doi:10.1101/gad.1199104
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J., and Der, C. J. (2014). Drugging the Undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 13 (11), 828–851. doi:10.1038/nrd4389
Darling, N. J., and Cohen, P. (2021). Nuts and Bolts of the Salt-Inducible Kinases (SIKs). Biochem. J. 478 (7), 1377–1397. doi:10.1042/bcj20200502
Davidson, S. M., Papagiannakopoulos, T., Olenchock, B. A., Heyman, J. E., Keibler, M. A., Luengo, A., et al. (2016). Environment Impacts the Metabolic Dependencies of Ras-Driven Non-small Cell Lung Cancer. Cel Metab. 23 (3), 517–528. doi:10.1016/j.cmet.2016.01.007
Ding, L., Getz, G., Wheeler, D. A., Mardis, E. R., McLellan, M. D., Cibulskis, K., et al. (2008). Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature 455 (7216), 1069–1075. doi:10.1038/nature07423
Dorfman, J., and Macara, I. G. (2008). STRADα Regulates LKB1 Localization by Blocking Access to Importin-α, and by Association with Crm1 and Exportin-7. MBoC 19, 1614–1626. doi:10.1091/mbc.e07-05-0454
Du, J., Chen, Q., Takemori, H., and Xu, H. (2008). SIK2 Can Be Activated by Deprivation of Nutrition and it Inhibits Expression of Lipogenic Genes in Adipocytes. Obesity 16 (3), 531–538. doi:10.1038/oby.2007.98
Düvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol. Cel 39 (2), 171–183. doi:10.1016/j.molcel.2010.06.022
Dykens, J. A., Jamieson, J., Marroquin, L., Nadanaciva, S., Billis, P. A., and Will, Y. (2008). Biguanide-induced Mitochondrial Dysfunction Yields Increased Lactate Production and Cytotoxicity of Aerobically-Poised HepG2 Cells and Human Hepatocytes In Vitro. Toxicol. Appl. Pharmacol. 233 (2), 203–210. doi:10.1016/j.taap.2008.08.013
El-Mir, M.-Y., Nogueira, V., Fontaine, E., Avéret, N., Rigoulet, M., and Leverve, X. (2000). Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J. Biol. Chem. 275 (1), 223–228. doi:10.1074/jbc.275.1.223
Esteve-Puig, R., Gil, R., González-Sánchez, E., Bech-Serra, J. J., Grueso, J., Hernández-Losa, J., et al. (2014). A Mouse Model Uncovers LKB1 as an UVB-Induced DNA Damage Sensor Mediating CDKN1A (p21WAF1/CIP1) Degradation. Plos Genet. 10 (10), e1004721. doi:10.1371/journal.pgen.1004721
Faubert, B., Li, K. Y., Cai, L., Hensley, C. T., Kim, J., Zacharias, L. G., et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171 (2), 358–371. e9. doi:10.1016/j.cell.2017.09.019
Faubert, B., Vincent, E. E., Griss, T., Samborska, B., Izreig, S., Svensson, R. U., et al. (2014). Loss of the Tumor Suppressor LKB1 Promotes Metabolic Reprogramming of Cancer Cells via HIF-1α. Proc. Natl. Acad. Sci. U.S.A. 111 (7), 2554–2559. doi:10.1073/pnas.1312570111
Ferrer, C. M., Sodi, V. L., and Reginato, M. J. (2016). O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J. Mol. Biol. 428 (16), 3282–3294. doi:10.1016/j.jmb.2016.05.028
Finkel, T. (2011). Signal Transduction by Reactive Oxygen Species. J. Cel Biol 194 (1), 7–15. doi:10.1083/jcb.201102095
Flowers, E. M., Sudderth, J., Zacharias, L., Mernaugh, G., Zent, R., DeBerardinis, R. J., et al. (2018). Lkb1 Deficiency Confers Glutamine Dependency in Polycystic Kidney Disease. Nat. Commun. 9 (1), 814. doi:10.1038/s41467-018-03036-y
Galan-Cobo, A., Sitthideatphaiboon, P., Qu, X., Poteete, A., Pisegna, M. A., Tong, P., et al. (2019). LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 79 (13), 3251–3267. doi:10.1158/0008-5472.can-18-3527
Gao, W.-W., Tang, H.-M. V., Cheng, Y., Chan, C.-P., Chan, C.-P., and Jin, D.-Y. (2018). Suppression of Gluconeogenic Gene Transcription by SIK1-Induced Ubiquitination and Degradation of CRTC1. Biochim. Biophys. Acta (Bba) - Gene Regul. Mech. 1861 (3), 211–223. doi:10.1016/j.bbagrm.2018.01.021
Giardiello, F. M., Brensinger, J. D., Tersmette, A. C., Goodman, S. N., Petersen, G. M., Booker, S. V., et al. (2000). Very High Risk of Cancer in Familial Peutz-Jeghers Syndrome. Gastroenterology 119 (6), 1447–1453. doi:10.1053/gast.2000.20228
Gill, R. K., Yang, S.-H., Meerzaman, D., Mechanic, L. E., Bowman, E. D., Jeon, H.-S., et al. (2011). Frequent Homozygous Deletion of the LKB1/STK11 Gene in Non-small Cell Lung Cancer. Oncogene 30 (35), 3784–3791. doi:10.1038/onc.2011.98
Gwinn, D. M., Lee, A. G., Briones-Martin-del-Campo, M., Conn, C. S., Simpson, D. R., Scott, A. I., et al. (2018). Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 33 (1), 91–107. e6. doi:10.1016/j.ccell.2017.12.003
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of Cancer: the Next Generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hardie, D. G., and Alessi, D. R. (2013). LKB1 and AMPK and the Cancer-Metabolism Link - Ten Years after. BMC Biol. 11 (1), 36. doi:10.1186/1741-7007-11-36
Hardie, D. G. (2005). New Roles for the LKB1→AMPK Pathway. Curr. Opin. Cel Biol. 17 (2), 167–173. doi:10.1016/j.ceb.2005.01.006
Hawley, S. A., Boudeau, J., Reid, J. L., Mustard, K. J., Udd, L., Mäkelä, T. P., et al. (2003). Complexes between the LKB1 Tumor Suppressor, STRAD Alpha/beta and MO25 Alpha/beta Are Upstream Kinases in the AMP-Activated Protein Kinase cascade. J. Biol. 2, 28. doi:10.1186/1475-4924-2-28
Hemminki, A., Tomlinson, I., Markie, D., Järvinen, H., Sistonen, P., Björkqvist, A.-M., et al. (1997). Localization of a Susceptibility Locus for Peutz-Jeghers Syndrome to 19p Using Comparative Genomic Hybridization and Targeted Linkage Analysis. Nat. Genet. 15 (1), 87–90. doi:10.1038/ng0197-87
Hui, S., Ghergurovich, J. M., Morscher, R. J., Jang, C., Teng, X., Lu, W., et al. (2017). Glucose Feeds the TCA Cycle via Circulating Lactate. Nature 551 (7678), 115–118. doi:10.1038/nature24057
Jackson, E. L., Olive, K. P., Tuveson, D. A., Bronson, R., Crowley, D., Brown, M., et al. (2005). The Differential Effects of Mutant P53 Alleles on Advanced Murine Lung Cancer. Cancer Res. 65 (22), 10280–10288. doi:10.1158/0008-5472.can-05-2193
Jeghers, H., McKusick, V. A., and Katz, K. H. (1949). Generalized Intestinal Polyposis and Melanin Spots of the Oral Mucosa, Lips and Digits; a Syndrome of Diagnostic Significance. N. Engl. J. Med. 241, 1031–1036. doi:10.1056/NEJM194912292412601
Jewell, J. L., Fu, V., Hong, A. W., Yu, F. X., Meng, D., Melick, C. H., et al. (2019). GPCR Signaling Inhibits mTORC1 via PKA Phosphorylation of Raptor. Elife 8, e43038. doi:10.7554/eLife.43038
Ji, H., Ramsey, M. R., Hayes, D. N., Fan, C., McNamara, K., Kozlowski, P., et al. (2007). LKB1 Modulates Lung Cancer Differentiation and Metastasis. Nature 448 (7155), 807–810. doi:10.1038/nature06030
Ju, H.-Q., Lu, Y.-X., Wu, Q.-N., Liu, J., Zeng, Z.-L., Mo, H.-Y., et al. (2017). Disrupting G6PD-Mediated Redox Homeostasis Enhances Chemosensitivity in Colorectal Cancer. Oncogene 36 (45), 6282–6292. doi:10.1038/onc.2017.227
Karuman, P., Gozani, O., Odze, R. D., Zhou, X. C., Zhu, H., Shaw, R., et al. (2001). The Peutz-Jegher Gene Product LKB1 Is a Mediator of P53-dependent Cell Death. Mol. Cel 7 (6), 1307–1319. doi:10.1016/s1097-2765(01)00258-1
Keith, B., Johnson, R. S., and Simon, M. C. (2012). HIF1α and HIF2α: Sibling Rivalry in Hypoxic Tumour Growth and Progression. Nat. Rev. Cancer 12 (1), 9–22. doi:10.1038/nrc3183
Kerr, E. M., and Martins, C. P. (2018). Metabolic Rewiring in Mutant Kras Lung Cancer. FEBS J. 285 (1), 28–41. doi:10.1111/febs.14125
Kim, J. E., and Chen, J. (2004). Regulation of Peroxisome Proliferator-Activated Receptor-γ Activity by Mammalian Target of Rapamycin and Amino Acids in Adipogenesis. Diabetes 53 (11), 2748–2756. doi:10.2337/diabetes.53.11.2748
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2011). AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cel Biol 13, 132–141. doi:10.1038/ncb2152
Kim, J., Lee, H. M., Cai, F., Ko, B., Yang, C., Lieu, E. L., et al. (2020). The Hexosamine Biosynthesis Pathway Is a Targetable Liability in KRAS/LKB1 Mutant Lung Cancer. Nat. Metab. 2 (12), 1401–1412. doi:10.1038/s42255-020-00316-0
Koh, H.-J., Toyoda, T., Fujii, N., Jung, M. M., Rathod, A., Middelbeek, R. J.-W., et al. (2010). Sucrose Nonfermenting AMPK-Related Kinase (SNARK) Mediates Contraction-Stimulated Glucose Transport in Mouse Skeletal Muscle. Proc. Natl. Acad. Sci. U.S.A. 107 (35), 15541–15546. doi:10.1073/pnas.1008131107
Kojima, Y., Miyoshi, H., Clevers, H. C., Oshima, M., Aoki, M., and Taketo, M. M. (2007). Suppression of Tubulin Polymerization by the LKB1-Microtubule-Associated Protein/Microtubule Affinity-Regulating Kinase Signaling. J. Biol. Chem. 282 (32), 23532–23540. doi:10.1074/jbc.m700590200
Liang, M.-C., Ma, J., Chen, L., Kozlowski, P., Qin, W., Li, D., et al. (2010). TSC1 Loss Synergizes with KRAS Activation in Lung Cancer Development in the Mouse and Confers Rapamycin Sensitivity. Oncogene 29 (11), 1588–1597. doi:10.1038/onc.2009.452
Lin, C., Lin, X., Lin, K., Tan, J., Wei, C., and Liu, T. (2021). LKB1 Expression and the Prognosis of Lung Cancer: A Meta-Analysis. Medicine 100 (46), e27841. doi:10.1097/md.0000000000027841
Lin, R., Elf, S., Shan, C., Kang, H.-B., Ji, Q., Zhou, L., et al. (2015). 6-Phosphogluconate Dehydrogenase Links Oxidative PPP, Lipogenesis and Tumour Growth by Inhibiting LKB1-AMPK Signalling. Nat. Cel Biol 17 (11), 1484–1496. doi:10.1038/ncb3255
Liu, Y., Cao, Y., Zhang, W., Bergmeier, S., Qian, Y., Akbar, H., et al. (2012). A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell GrowthIn VitroandIn Vivo. Mol. Cancer Ther. 11 (8), 1672–1682. doi:10.1158/1535-7163.mct-12-0131
Lizcano, J. M., Göransson, O., Toth, R., Deak, M., Morrice, N. A., Boudeau, J., et al. (2004). LKB1 Is a Master Kinase that Activates 13 Kinases of the AMPK Subfamily, Including MARK/PAR-1. EMBO J. 23 (4), 833–843. Epub 2004 Feb 19. doi:10.1038/sj.emboj.7600110
Luukko, K., Ylikorkala, A., Tiainen, M., and Mäkelä, T. P. (1999). Expression of LKB1 and PTEN Tumor Suppressor Genes during Mouse Embryonic Development. Mech. Dev. 83 (1-2), 187–190. doi:10.1016/s0925-4773(99)00050-7
Maher, J. C., Krishan, A., and Lampidis, T. J. (2004). Greater Cell Cycle Inhibition and Cytotoxicity Induced by 2-Deoxy-D-Glucose in Tumor Cells Treated under Hypoxic vs Aerobic Conditions. Cancer Chemother. Pharmacol. 53 (2), 116–122. doi:10.1007/s00280-003-0724-7
Marignani, P. A., and Sanchez-Cespedes, M. (2010). “The Tumour Suppressor Role of LKB1 in Human Cancer,” in Emerging Signalling Pathways in Tumor Biology: Transworld Research Network. Editor P. A. Lazo, 71–94.
Marignani, P. A. (2005). LKB1, the Multitasking Tumour Suppressor Kinase. J. Clin. Pathol. 58 (1), 15–19. doi:10.1136/jcp.2003.015255
Marignani, P. A., Scott, K., Bagnulo, R., Cannone, D., Ferrari, E., Stella, A., et al. (2007). Novel Splice Isoforms of STRADα Differentially Affect LKB1 Activity, Complex Assembly and Subcellular Localization. Cancer Biol. Ther. 6 (10), 1627–1631. doi:10.4161/cbt.6.10.4787
Martin, S. G., and St Johnston, D. (2003). A Role for Drosophila LKB1 in Anterior-Posterior axis Formation and Epithelial Polarity. Nature 421 (6921), 379–384. doi:10.1038/nature01296
Martín-Bernabé, A., Cortés, R., Lehmann, S. G., Seve, M., Cascante, M., and Bourgoin-Voillard, S. (2014). Quantitative Proteomic Approach to Understand Metabolic Adaptation in Non-small Cell Lung Cancer. J. Proteome Res. 13 (11), 4695–4704.
Maschek, G., Savaraj, N., Priebe, W., Braunschweiger, P., Hamilton, K., Tidmarsh, G. F., et al. (2004). 2-Deoxy-d-glucose Increases the Efficacy of Adriamycin and Paclitaxel in Human Osteosarcoma and Non-small Cell Lung Cancers In Vivo. Cancer Res. 64 (1), 31–34. doi:10.1158/0008-5472.can-03-3294
Mehenni, H., Gehrig, C., Nezu, J.-i., Oku, A., Shimane, M., Rossier, C., et al. (1998). Loss of LKB1 Kinase Activity in Peutz-Jeghers Syndrome, and Evidence for Allelic and Locus Heterogeneity. Am. J. Hum. Genet. 63 (6), 1641–1650. doi:10.1086/302159
Mitsuishi, Y., Taguchi, K., Kawatani, Y., Shibata, T., Nukiwa, T., Aburatani, H., et al. (2012). Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 22 (1), 66–79. doi:10.1016/j.ccr.2012.05.016
Mohseni, M., Sun, J., Lau, A., Curtis, S., Goldsmith, J., Fox, V. L., et al. (2014). A Genetic Screen Identifies an LKB1-MARK Signalling axis Controlling the Hippo-YAP Pathway. Nat. Cel Biol 16 (1), 108–117. doi:10.1038/ncb2884
Morton, J. P., Jamieson, N. B., Karim, S. A., Athineos, D., Ridgway, R. A., Nixon, C., et al. (2010). LKB1 Haploinsufficiency Cooperates with Kras to Promote Pancreatic Cancer through Suppression of P21-dependent Growth Arrest. Gastroenterology 139 (2), 586–597. e6. doi:10.1053/j.gastro.2010.04.055
Mullen, A. R., Wheaton, W. W., Jin, E. S., Chen, P.-H., Sullivan, L. B., Cheng, T., et al. (2012). Reductive Carboxylation Supports Growth in Tumour Cells with Defective Mitochondria. Nature 481 (7381), 385–388. doi:10.1038/nature10642
Nakano, A., and Takashima, S. (2012). LKB1 andAMP‐activated Protein Kinase: Regulators of Cell Polarity. Genes Cells 17 (9), 737–747. doi:10.1111/j.1365-2443.2012.01629.x
Nam, K., Oh, S., and Shin, I. (2016). Ablation of CD44 Induces Glycolysis-To-Oxidative Phosphorylation Transition via Modulation of the C-Src-Akt-LKB1-Ampkα Pathway. Biochem. J. 473 (19), 3013–3030. doi:10.1042/bcj20160613
Nathan, C., and Ding, A. (2010). SnapShot: Reactive Oxygen Intermediates (ROI). Cell 140 (6), 951. doi:10.1016/j.cell.2010.03.008
Nishioka, Y., Kobayashi, K., Sagae, S., Sugimura, M., Ishioka, S., Nagata, M., et al. (1999). Mutational Analysis of STK11 H Gene in Ovarian Carcinomas. Jpn. J. Cancer Res. 90 (6), 629–632. doi:10.1111/j.1349-7006.1999.tb00793.x
Nojima, H., Tokunaga, C., Eguchi, S., Oshiro, N., Hidayat, S., Yoshino, K.-i., et al. (2003). The Mammalian Target of Rapamycin (mTOR) Partner, Raptor, Binds the mTOR Substrates P70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 278 (18), 15461–15464. doi:10.1074/jbc.c200665200
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A., and Shokat, K. M. (2013). K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 503 (7477), 548–551. doi:10.1038/nature12796
Patel, K., Foretz, M., Marion, A., Campbell, D. G., Gourlay, R., Boudaba, N., et al. (2014). The LKB1-Salt-Inducible Kinase Pathway Functions as a Key Gluconeogenic Suppressor in the Liver. Nat. Commun. 5 (1), 4535. doi:10.1038/ncomms5535
Patra, K. C., and Hay, N. (2014). The Pentose Phosphate Pathway and Cancer. Trends Biochem. Sci. 39 (8), 347–354. doi:10.1016/j.tibs.2014.06.005
Pavlides, S., Whitaker-Menezes, D., Castello-Cros, R., Flomenberg, N., Witkiewicz, A. K., Frank, P. G., et al. (2009). The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle 8 (23), 3984–4001. doi:10.4161/cc.8.23.10238
Pavlova, N. N., and Thompson, C. B. (2016). The Emerging Hallmarks of Cancer Metabolism. Cel Metab. 23 (1), 27–47. doi:10.1016/j.cmet.2015.12.006
Peutz, J. (1921). A Very Remarkable Case of Familial Polyposis of Mucous Membrane of Intestinal Tract and Accompanied by peculiar Pigmentations of Skin and Mucous Membrane [Dutch]. Nederlands Tijdschrift voor Geneeskunde 10, 134–146.
Rizwan, A., Serganova, I., Khanin, R., Karabeber, H., Ni, X., Thakur, S., et al. (2013). Relationships between LDH-A, Lactate, and Metastases in 4T1 Breast Tumors. Clin. Cancer Res. 19 (18), 5158–5169. doi:10.1158/1078-0432.ccr-12-3300
Rogatzki, M. J., Ferguson, B. S., Goodwin, M. L., and Gladden, L. B. (2015). Lactate Is Always the End Product of Glycolysis. Front. Neurosci. 9, 22. doi:10.3389/fnins.2015.00022
Romero, R., Sayin, V. I., Davidson, S. M., Bauer, M. R., Singh, S. X., LeBoeuf, S. E., et al. (2017). Keap1 Loss Promotes Kras-Driven Lung Cancer and Results in Dependence on Glutaminolysis. Nat. Med. 23 (11), 1362–1368. doi:10.1038/nm.4407
Rowan, A., Tomlinson, I., Bataille, V., MacKie, R., Healy, E., Bicknell, D., et al. (1999). Somatic Mutations in the Peutz-Jegners (LKB1/STKII) Gene in Sporadic Malignant Melanomas. J. Invest. Dermatol. 112 (4), 509–511. doi:10.1046/j.1523-1747.1999.00551.x
Sakamoto, K., McCarthy, A., Smith, D., Green, K. A., Grahame Hardie, D., Ashworth, A., et al. (2005). Deficiency of LKB1 in Skeletal Muscle Prevents AMPK Activation and Glucose Uptake during Contraction. EMBO J. 24 (10), 1810–1820. doi:10.1038/sj.emboj.7600667
Sakamoto, T., Weng, J. S., Hara, T., Yoshino, S., Kozuka-Hata, H., Oyama, M., et al. (2014). Hypoxia-Inducible Factor 1 Regulation through Cross Talk between mTOR and MT1-MMP. Mol. Cel Biol 34 (1), 30–42. doi:10.1128/mcb.01169-13
Sanchez-Cespedes, M., Parrella, P., Esteller, M., Nomoto, S., Trink, B., Engles, J. M., et al. (2002). Inactivation of LKB1/STK11 Is a Common Event in Adenocarcinomas of the Lung. Cancer Res. 62 (13), 3659–3662.
Sanchez-Cespedes, M. (2007). A Role for LKB1 Gene in Human Cancer beyond the Peutz-Jeghers Syndrome. Oncogene 26 (57), 7825–7832. doi:10.1038/sj.onc.1210594
Sato, R., Goldstein, J. L., and Brown, M. S. (1993). Replacement of Serine-871 of Hamster 3-Hydroxy-3-Methylglutaryl-CoA Reductase Prevents Phosphorylation by AMP-Activated Kinase and Blocks Inhibition of Sterol Synthesis Induced by ATP Depletion. Proc. Natl. Acad. Sci. U.S.A. 90 (20), 9261–9265. doi:10.1073/pnas.90.20.9261
Scott, K. D., Nath-Sain, S., Agnew, M. D., and Marignani, P. A. (2007). LKB1 Catalytically Deficient Mutants Enhance Cyclin D1 Expression. Cancer Res. 67 (12), 5622–5627. doi:10.1158/0008-5472.can-07-0762
Shackelford, D. B., Abt, E., Gerken, L., Vasquez, D. S., Seki, A., Leblanc, M., et al. (2013). LKB1 Inactivation Dictates Therapeutic Response of Non-small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell 23 (2), 143–158. doi:10.1016/j.ccr.2012.12.008
Shackelford, D. B., Vasquez, D. S., Corbeil, J., Wu, S., Leblanc, M., Wu, C.-L., et al. (2009). mTOR and HIF-1α-Mediated Tumor Metabolism in an LKB1 Mouse Model of Peutz-Jeghers Syndrome. Proc. Natl. Acad. Sci. U.S.A. 106 (27), 11137–11142. doi:10.1073/pnas.0900465106
Shaw, A. T., and Engelman, J. A. (2013). ALKin Lung Cancer: Past, Present, and Future. Jco 31 (8), 1105–1111. doi:10.1200/jco.2012.44.5353
Shen, Z., Wen, X. F., Lan, F., Shen, Z. Z., and Shao, Z. M. (2002). The Tumor Suppressor Gene LKB1 Is Associated with Prognosis in Human Breast Carcinoma. Clin. Cancer Res. 8 (7), 2085–2090.
Skoulidis, F., Byers, L. A., Diao, L., Papadimitrakopoulou, V. A., Tong, P., Izzo, J., et al. (2015). Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 5 (8), 860–877. doi:10.1158/2159-8290.cd-14-1236
Smith, D. P., Spicer, J., Smith, A., Swift, S., and Ashworth, A. (1999). The Mouse Peutz-Jeghers Syndrome Gene Lkbl Encodes a Nuclear Protein Kinase. Hum. Mol. Genet. 8 (8), 1479–1485. doi:10.1093/hmg/8.8.1479
Sola-Penna, M., Da Silva, D., Coelho, W. S., Marinho-Carvalho, M. M., and Zancan, P. (2010). Regulation of Mammalian Muscle Type 6-Phosphofructo-1-Kinase and its Implication for the Control of the Metabolism. IUBMB Life 62 (11), 791–796. doi:10.1002/iub.393
Sonveaux, P., Végran, F., Schroeder, T., Wergin, M. C., Verrax, J., Rabbani, Z. N., et al. (2008). Targeting Lactate-Fueled Respiration Selectively Kills Hypoxic Tumor Cells in Mice. J. Clin. Invest. 118 (12), 3930–3942. doi:10.1172/JCI36843
Spinelli, J. B., and Haigis, M. C. (2018). The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cel Biol 20 (7), 745–754. doi:10.1038/s41556-018-0124-1
Stringer, D. M., Zahradka, P., and Taylor, C. G. (2015). Glucose Transporters: Cellular Links to Hyperglycemia in Insulin Resistance and Diabetes. Nutr. Rev. 73 (3), 140–154. doi:10.1093/nutrit/nuu012
Sun, Y., Connors, K., and Yang, D-Q. (2007). AICAR Induces Phosphorylation of AMPK in an ATM-dependent, LKB1-independent Manner. Mol. Cell Biochem. 306 (1-2), 239–245. doi:10.1007/s11010-007-9575-6
Sun, Z., Jiang, Q., Li, J., and Guo, J. (2020). The Potent Roles of Salt-Inducible Kinases (SIKs) in Metabolic Homeostasis and Tumorigenesis. Sig Transduct Target. Ther. 5 (1), 150. doi:10.1038/s41392-020-00265-w
Svensson, R. U., Parker, S. J., Eichner, L. J., Kolar, M. J., Wallace, M., Brun, S. N., et al. (2016). Inhibition of Acetyl-CoA Carboxylase Suppresses Fatty Acid Synthesis and Tumor Growth of Non-small-cell Lung Cancer in Preclinical Models. Nat. Med. 22 (10), 1108–1119. doi:10.1038/nm.4181
Tanimoto, K., Makino, Y., Pereira, T., and Poellinger, L. (2000). Mechanism of regulation of the hypoxia-inducible factor-1alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 19 (16), 4298–4309. doi:10.1093/emboj/19.16.4298
Tee, A. R., Manning, B. D., Roux, P. P., Cantley, L. C., and Blenis, J. (2003). Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. 13 (15), 1259–1268. doi:10.1016/s0960-9822(03)00506-2
Thoreen, C. C., Chantranupong, L., Keys, H. R., Wang, T., Gray, N. S., and Sabatini, D. M. (2012). A Unifying Model for mTORC1-Mediated Regulation of mRNA Translation. Nature 485 (7396), 109–113. doi:10.1038/nature11083
Tiainen, M., Vaahtomeri, K., Ylikorkala, A., and Makela, T. P. (2002). Growth Arrest by the LKB1 Tumor Suppressor: Induction of p21WAF1/CIP1. Hum. Mol. Genet. 11 (13), 1497–1504. doi:10.1093/hmg/11.13.1497
Tiainen, M., Ylikorkala, A., and Mäkelä, T. P. (1999). Growth Suppression by Lkb1 Is Mediated by a G 1 Cell Cycle Arrest. Proc. Natl. Acad. Sci. U.S.A. 96 (16), 9248–9251. doi:10.1073/pnas.96.16.9248
Towler, M. C., Fogarty, S., Hawley, S. A., Pan, D. A., Martin, D. M. A., Morrice, N. A., et al. (2008). A Novel Short Splice Variant of the Tumour Suppressor LKB1 Is Required for Spermiogenesis. Biochem. J. 416 (1), 1–14. doi:10.1042/bj20081447
Trachootham, D., Zhou, Y., Zhang, H., Demizu, Y., Chen, Z., Pelicano, H., et al. (2006). Selective Killing of Oncogenically Transformed Cells through a ROS-Mediated Mechanism by β-phenylethyl Isothiocyanate. Cancer Cell 10 (3), 241–252. doi:10.1016/j.ccr.2006.08.009
Vallenius, T., Vaahtomeri, K., Kovac, B., Osiceanu, A.-M., Viljanen, M., and Mäkelä, T. P. (2011). An Association between NUAK2 and MRIP Reveals a Novel Mechanism for Regulation of Actin Stress Fibers. J. Cel Sci. 124 (3), 384–393. doi:10.1242/jcs.072660
van Veelen, W., Korsse, S. E., van de Laar, L., and Peppelenbosch, M. P. (2011). The Long and Winding Road to Rational Treatment of Cancer Associated with LKB1/AMPK/TSC/mTORC1 Signaling. Oncogene 30 (20), 2289–2303. doi:10.1038/onc.2010.630
Vasconcelos-dos-Santos, A., Oliveira, I. A., Lucena, M. C., Mantuano, N. R., Whelan, S. A., Dias, W. B., et al. (2015). Biosynthetic Machinery Involved in Aberrant Glycosylation: Promising Targets for Developing of Drugs against Cancer. Front. Oncol. 5, 138. doi:10.3389/fonc.2015.00138
Wander, S. A., Hennessy, B. T., and Slingerland, J. M. (2011). Next-generation mTOR Inhibitors in Clinical Oncology: How Pathway Complexity Informs Therapeutic Strategy. J. Clin. Invest. 121 (4), 1231–1241. doi:10.1172/jci44145
Warburg, O., Wind, F., and Negelein, E. (1927). The Metabolism of Tumors in the Body. J. Gen. Physiol. 8 (6), 519–530. doi:10.1085/jgp.8.6.519
Watts, J. L., Morton, D. G., Bestman, J., and Kemphues, K. J. (2000). The C. elegans Par-4 Gene Encodes a Putative Serine-Threonine Kinase Required for Establishing Embryonic Asymmetry. Development 127 (7), 1467–1475. doi:10.1242/dev.127.7.1467
Wise, D. R., Ward, P. S., Shay, J. E. S., Cross, J. R., Gruber, J. J., Sachdeva, U. M., et al. (2011). Hypoxia Promotes Isocitrate Dehydrogenase-dependent Carboxylation of α-ketoglutarate to Citrate to Support Cell Growth and Viability. Proc. Natl. Acad. Sci. U.S.A. 108 (49), 19611–19616. doi:10.1073/pnas.1117773108
Woods, A., Johnstone, S. R., Dickerson, K., Leiper, F. C., Fryer, L. G. D., Neumann, D., et al. (2003). LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase cascade. Curr. Biol. 13 (22), 2004–2008. doi:10.1016/j.cub.2003.10.031
Wu, H., Ying, M., and Hu, X. (2016). Lactic Acidosis Switches Cancer Cells from Aerobic Glycolysis Back to Dominant Oxidative Phosphorylation. Oncotarget 7 (26), 40621–40629. doi:10.18632/oncotarget.9746
Xie, H., Hanai, J.-i., Ren, J.-G., Kats, L., Burgess, K., Bhargava, P., et al. (2014). Targeting Lactate Dehydrogenase-A Inhibits Tumorigenesis and Tumor Progression in Mouse Models of Lung Cancer and Impacts Tumor-Initiating Cells. Cel Metab. 19 (5), 795–809. doi:10.1016/j.cmet.2014.03.003
Ylikorkala, A., Avizienyte, E., Tomlinson, I. P., Tiainen, M., Roth, S., Loukola, A., et al. (1999). Mutations and Impaired Function of LKB1 in Familial and Non-familial Peutz-Jeghers Syndrome and a Sporadic Testicular Cancer. Hum. Mol. Genet. 8 (1), 45–51. doi:10.1093/hmg/8.1.45
Ylikorkala, A., Rossi, D. J., Korsisaari, N., Luukko, K., Alitalo, K., Henkemeyer, M., et al. (2001). Vascular Abnormalities and Deregulation of VEGF in Lkb1 -Deficient Mice. Science 293 (5533), 1323–1326. doi:10.1126/science.1062074
Zagórska, A., Deak, M., Campbell, D. G., Banerjee, S., Hirano, M., Aizawa, S., et al. (2010). New Roles for the LKB1-NUAK Pathway in Controlling Myosin Phosphatase Complexes and Cell Adhesion. Sci. Signal. 3 (115), ra25–ra. doi:10.1126/scisignal.2000616
Zeng, P.-Y., and Berger, S. L. (2006). LKB1 Is Recruited to the p21/WAF1 Promoter by P53 to Mediate Transcriptional Activation. Cancer Res. 66 (22), 10701–10708. doi:10.1158/0008-5472.can-06-0999
Keywords: LKB1, AMPK, mTOR, tumor suppressor, cancer metabolism, glycolysis, lung cancer
Citation: Bourouh M and Marignani PA (2022) The Tumor Suppressor Kinase LKB1: Metabolic Nexus. Front. Cell Dev. Biol. 10:881297. doi: 10.3389/fcell.2022.881297
Received: 22 February 2022; Accepted: 07 April 2022;
Published: 28 April 2022.
Edited by:
Philipp Kaldis, Lund University, SwedenReviewed by:
Montse Sanchez-Cespedes, Josep Carreras Leukaemia Research Institute (IJC), SpainElvire Pons-Tostivint, Université de Nantes, France
Copyright © 2022 Bourouh and Marignani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola A. Marignani, paola.marignani@dal.ca