A Five-Component Biginelli-Diels-Alder Cascade Reaction
 Taber S. Maskrey
Taber S. Maskrey Madeline C. Frischling
Madeline C. Frischling Mikhaila L. Rice
Mikhaila L. Rice Peter Wipf
Peter Wipf- Department of Chemistry, University of Pittsburgh, Pittsburgh, PA, United States
A new multi-component condensation was discovered during the reaction of a urea, β-keto ester, and formaldehyde. In the presence of catalytic indium bromide, a Biginelli dihydropyrimidinone intermediate was further converted to a five-component condensation product through a formal hetero Diels-Alder reaction. The product structure was confirmed by NMR and NOE analysis, and the proposed stepwise mechanism was supported by the reaction of the Biginelli intermediate with ethyl 2-methylene-3-oxobutanoate.
Introduction
Dihydropyrimidinones (DHPMs) represent an attractive class of biologically active small molecules. DHPMs have been shown to have neuroprotective, antiparasitic, antiviral, antitumor, anti-inflammatory, and anticancer activities (de Fátima et al., 2015). Monastrol, for example, is one of the most studied DHPMs based on its antiproliferative activity (Figure 1). In 1999, monastrol was found to disrupt mitosis (Mayer et al., 1999; Kapoor et al., 2000). Subsequently, several potent analogs with increased anticancer potency have also been synthesized (Ragab et al., 2017).
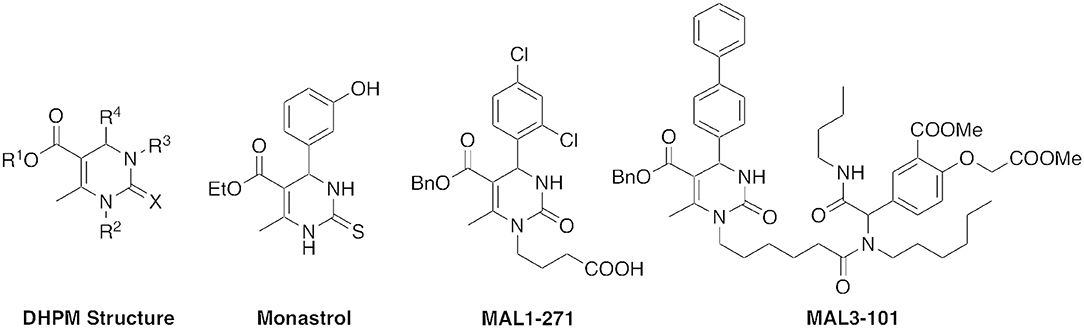
Figure 1. DHPM structure and biologically active analogs.
The DHPM structure is the product of a reaction between a urea, β-keto ester, and an aldehyde, commonly termed the Biginelli reaction. The creation of a structurally diverse library of pharmacologically active compounds is an attractive possibility with this three-component condensation (Wan and Pan, 2012). As part of our studies on the preparation of bioactive DHPMs, in particular heat shock protein 70 (Hsp70) antagonists and agonists such as MAL3-101 and MAL1-271 (Figure 1), we explored the scope of Biginelli reactions in solution, fluorous media, and on solid support, and developed analogs with potent anticancer, antiviral, and neuroprotective properties (Wipf and Cunningham, 1995; Studer et al., 1997; Huryn et al., 2011; Manos-Turvey et al., 2016).
Standard Biginelli reactions frequently use catalytic amounts of Brønsted acids, such as hydrochloric acid (HCl) (Nagarajaiah et al., 2016). Various Lewis acid catalysts, including InCl3 (Ranu et al., 2000), FeCl3 (Lu and Bai, 2002), BF3•OEt2 (Hu et al., 1998), ZnCl2 (Sun et al., 2004), GaCl3 (Yuan et al., 2017), and InBr3 have also been used successfully (Phucho et al., 2009). While working to advance the scope of our own DHPM library, we noted the use of 10% InBr3 as a catalyst at reflux in ethanol for 7 h to produce DHPMs (Fu et al., 2002; Martins et al., 2004). We decided to explore these reaction conditions further as they gave good yields even with formaldehyde as a reaction component. Initially, we hoped to be able to introduce substituents at the 4-position of the DHPM ring through an oxidative photochemical reaction related to the previously reported Friedel-Crafts amidoalkylation with the visible light catalyst, Ru(bpy)3Cl2 (Dai et al., 2012) (Figure 2). However, we found that InBr3 conditions promoted a new five-component condensation with formaldehyde, and decided to first investigate the scope of this process.
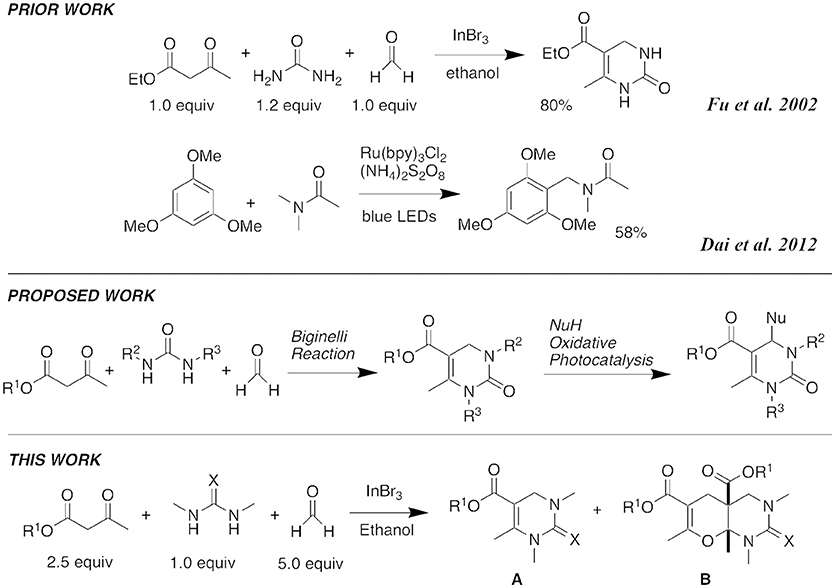
Figure 2. Literature precedence and synthetic plans.
Results and Discussion
The originally reported InBr3 reaction conditions utilized an excess of urea and equal equivalents of β-keto ester and aldehyde. For our initial test reactions, we chose N,N′-dimethylurea as the limiting reagent. The reaction of N,N′-dimethylurea, 1.8 equivalents of ethyl acetoacetate as the β-keto ester, and 3 equivalents of formaldehyde in 95% ethanol at reflux for 7 h yielded not only the expected DHPM product 1A but also a higher molecular weight product that we did not anticipate (Figure 3). Through NMR and NOE analyses, we were able to assign its fused bicyclic structure as 1B. We hypothesized that this secondary product occurred through a five-component condensation reaction, and an in situ formal hetero Diels-Alder reaction of the expected DHPM Biginelli intermediate was in agreement with the product structure.
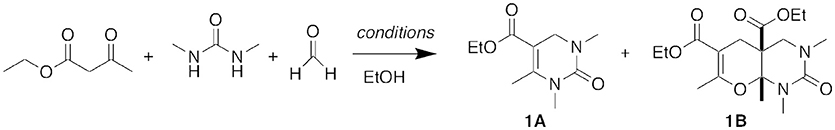
Figure 3. Five-component condensation reaction. For conditions, see Table 1.
Prior reports of the product of a Biginelli reaction being utilized as a dienophile in a hetero Diels-Alder reaction are rare. Sharma and coworkers demonstrated the reaction of an isolated Biginelli heterocycle with N-arylidine-N′-methylformamidines and N-arylidine guanidine in THF (Sharma et al., 2005). Our present synthesis represents the first one pot reaction to form these highly functionalized bicyclic products from readily available precursors.
We subsequently explored a range of reaction conditions in an attempt to improve overall yield and selectivity (Figure 3). For these optimizations, we used our original reaction components N,N′-dimethylurea, ethyl acetoacetate, and formaldehyde (Table 1). The traditional Brønsted acid catalyst, HCl, gave no conversion with formaldehyde (entry 2). We experimented with other Lewis acids (entries 3–6) to catalyze the reaction but saw no conversion with FeCl3 or ZnCl2. Conversion was modest with both InCl3 and AlCl3. Increasing the proportions of the β-keto ester and the aldehyde to 2.5 and 5 equivalents, respectively, improved the yield of both products 1A and 1B (entry 7). However, increasing or decreasing the reactant concentration (entries 8 and 9) did not improve the overall yield. Doubling the reaction time to 14.5 h also did not increase the conversion to product 1B (entry 10 vs. entry 7). After a shortened reaction time of 2.0 h, only product 1A was isolated in 28% yield (entry 11). Accordingly, entry 7 represented our optimized conditions: 1 equivalent of urea, 2.5 equivalents of β-keto ester, 5.0 equivalents of aldehyde, and 0.1 equivalents of InBr3 at reflux conditions in ethanol (0.2 M) for 7 h provided 45% of DHPM 1A and 48% of pyranopyrimidinone 1B.
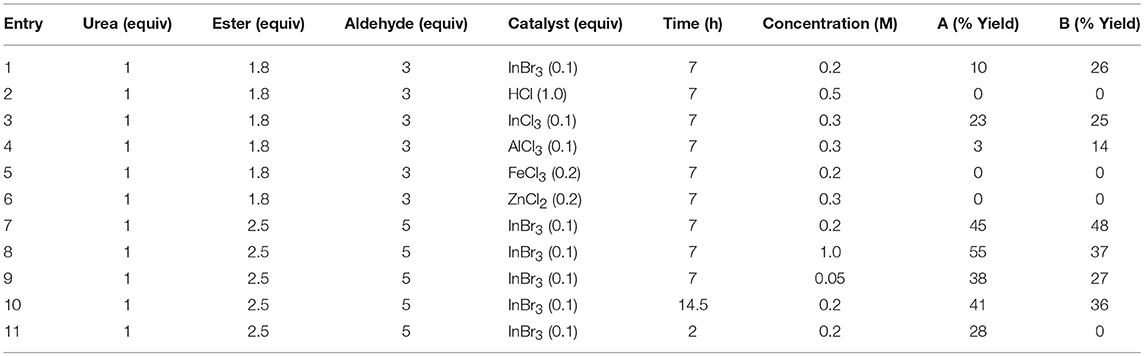
Table 1. Investigation of reaction time, concentration, equivalents, and catalyst with formaldehyde, N,N′-dimethylurea, and ethyl acetoacetate (Figure 3).
We then used these optimized conditions to further explore the scope of the reaction (Table 2). Formaldehyde was used as the aldehyde component in all reactions since preliminary trials with other aldehydes and ketones were not successful in producing any fused bicycles B. We also used symmetrical ureas exclusively to avoid possible regioisomers (exploratory reactions confirmed a lack of regioselectivity with unsymmetrical ureas; for example, while 1-methylurea and 1-(4-methoxyphenyl)-3-methylurea provided both the Biginelli DHPM and the Diels-Alder products, we were unable to separate the regioisomers formed in an approximately 1:1 ratio). Curiously, the reaction with the N,N′-dimethylthiourea stopped at the Biginelli product 2A, which was isolated in 61% yield (entry 2). Methyl acetoacetate with thiourea 2 (entry 3) also gave similar results, but product 3A was formed in lower yield, likely due to trans-esterification with ethanol. Utilizing benzyl acetoacetate, we were able to isolate both Biginelli (4A, 5A) and five-component condensation products (4B, 5B) in good overall yields with the urea and thiourea derivatives (entries 4 and 5). However, the yield of the five-component condensation Diels-Alder product 4B was considerably higher with the urea derivative than the pyranopyrimidinethione 5B resulting from the thiourea. With allyl acetoacetate and N,N′-dimethylurea, we were able to isolate the five-component condensation product 6B, but not the corresponding Biginelli intermediate, as it proved unstable. We also attempted to replace the traditional β-keto ester with 5,5-dimethyl-1,3-cyclohexanedione (dimedone) to probe the feasibility of adding an additional ring in the hetero Diels-Alder reaction (Figure 4). Unfortunately, with both N,N′-dimethylurea and its thiourea equivalent, we were only able to isolate Biginelli products 7A and 8A, in significantly lower yields than for the β-keto ester conversions.
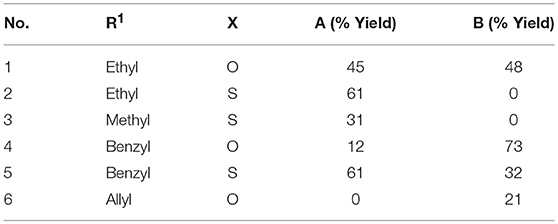
Table 2. Reaction scope with formaldehyde, N,N′-dimethyl(thio)urea (X=O,S), and alkyl (R1) acetoacetates. For products A and B, see Figure 2.
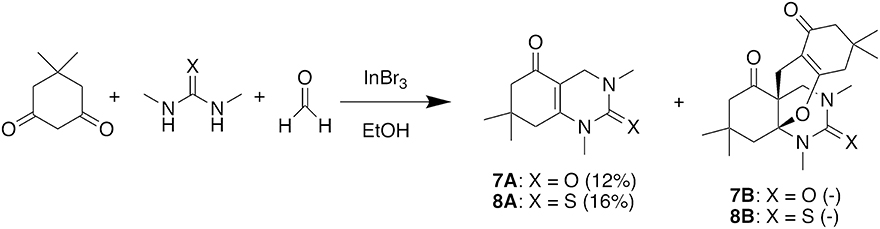
Figure 4. Attempted five-component condensation reaction with a cyclic 1,3-diketone.
To confirm our hypothesis that the five-component condensation product formed through a hetero Diels-Alder reaction with the DHPM (Biginelli) intermediate, we reacted Biginelli product 1A with ethyl 2-methylene-3-oxobutanoate (Figure 5). After stirring in sulfolane at room temperature for 18 h, we isolated the corresponding hetero Diels-Alder Product, 1B, in 11% yield. We were also able to repeat this conversion with the Biginelli intermediate 5A to form the hetero Diels-Alder Product 9B.
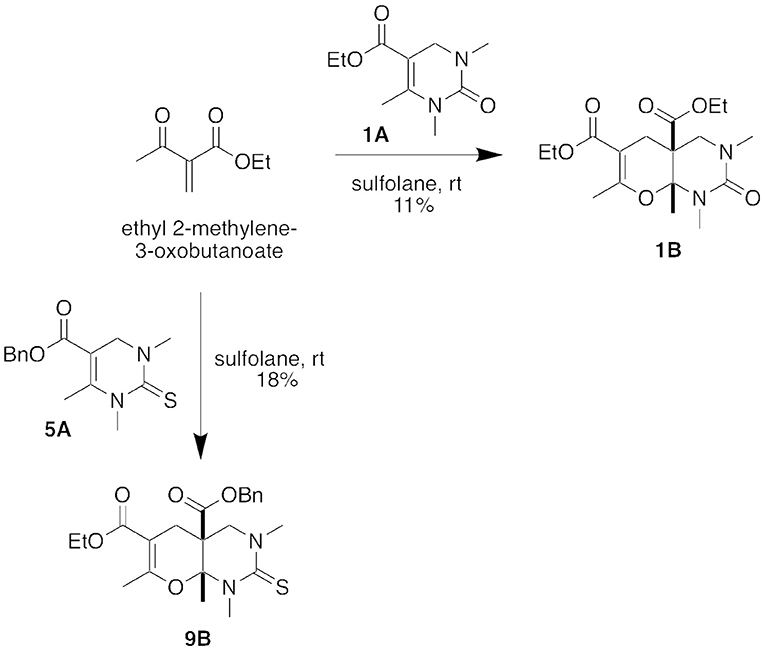
Figure 5. Stepwise conversions of intermediate DHPM (Biginelli) products in a hetero Diels-Alder reaction.
Interestingly, when the five-component condensation product 1B was subjected to Krapcho dealkylation conditions with LiCl in DMSO, we isolated the DHPM product, 1A, in 87% yield, presumable through a retro Diels-Alder reaction (Figure 6) (Krapcho et al., 1967).

Figure 6. Retro Diels-Alder reaction under Krapcho dealkylation conditions.
Figure 7 illustrates our proposed mechanism for the five-component condensation reaction. In the presence of a Lewis acid, condensation of the urea with the aldehyde and subsequent loss of water generates a sufficiently reactive electrophile for attack by the β-keto ester, which then undergoes cyclization to give 1A. The excess β-keto ester and formaldehyde react to form a methylene group at the α-position, and this electrophile then undergoes a hetero Diels-Alder reaction with the intermediate DHPM 1A to provide the fused heterocyclic 1B as a single stereoisomer. We determined the configuration of the methyl group to be cis to the ester at the two quaternary ring fusion atoms by converting the ester to the primary alcohol 10 with LiBH4, and then using a NOE analysis to confirm that the methyl group protons showed a >10% percent double resonance enhancement with the newly formed CH2 protons.
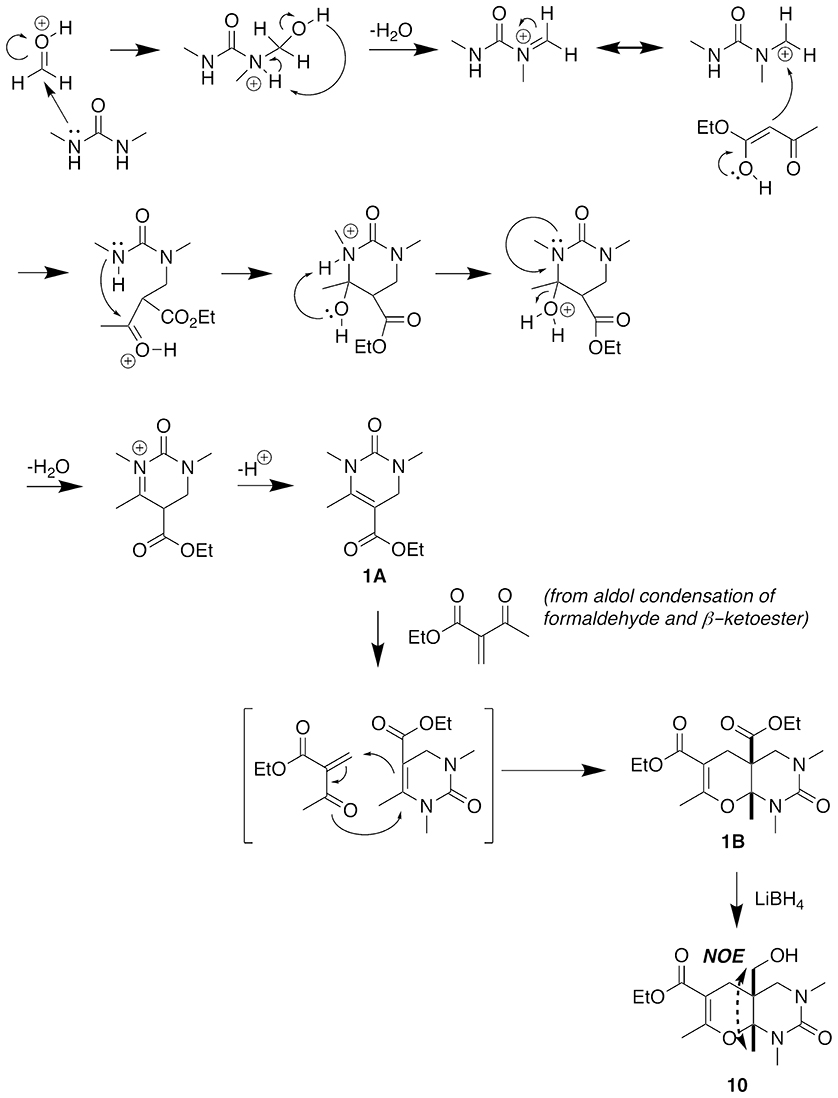
Figure 7. Proposed mechanism of five-component condensation.
Conclusions
We have discovered and optimized experimental conditions for a novel one pot, five-component condensation reaction with a β-keto ester, urea, and formaldehyde. The reaction appears to proceed through an intermediate DHPM (Biginelli) product. The DHPM then reacts with the condensation product of the β-keto ester and formaldehyde through a formal hetero Diels-Alder reaction. While the scope of this new process is still limited to formaldehyde and symmetrical N,N′-dialkylated ureas, it provides easy access to bicyclic ring systems that were previously inaccessible through a single transformation.
Materials and Methods
General
All reagents were obtained commercially unless otherwise noted. All glassware was dried in an oven at 140°C for 2 h prior to use. Reactions were monitored by TLC analysis (EMD Millipore pre-coated silica gel 60 F254 plates, 250 μM layer thickness) and visualization was accomplished with a 254 nM UV light or staining with a PMA solution (5 g of phosphomolybdic acid in 100 mL of 95% EtOH), p-anisaldehyde solution (2.5 mL of p-anisaldehyde, 2 mL of AcOH, and 3.5 mL of conc. H2SO4 in 100 mL of 95% EtOH), or a KMnO4 solution (1.5 g of KMnO4 and 1.5 g of K2CO3 in 100 mL of a 0.1% NaOH solution). Some purification by chromatography was performed using a SiO2 Büchi flash chromatography system. 1H/13C NMR spectra were recorded on either a Bruker Avance 300/75 MHz, Bruker Avance 400/100 MHz, Bruker Avance 500/135 MHz, or Bruker Avance 600/150 MHz instruments. Chemical shifts were reported in parts per million with the residual solvent peak used as the internal standard (1H/13C: CDCl3, 7.26, 77.0 ppm; CD3OD, 3.31, 49.3 ppm; DMSO, 2.50, 39.5 ppm). Chemical shifts were tabulated as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quarter, dd = doublet of doublet, dt = doublet of triplet, dq = doublet of quartet, m = multiplet, b = broad, app = apparent), coupling constants, and integration. All 1D NMR spectra were processed using Bruker Topspin NMR. IR spectra were obtained on an Identity IR-ATR spectrometer. Melting points (uncorrected) were determined using a Mel-Temp instrument. HRMS data were obtained on a Thermo Scientific Exactive HRMS coupled to a Thermo Scientific Accela HPLC system using a 2.1 x 50 mm 3.5 μm Waters XTerra C18 column eluting with MeCN/H2O containing 0.1% formic acid.
General 5-Component Condensation Reaction Procedure
Representative procedure for ethyl 1,3,6-trimethyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1A) and diethyl(4aSR,8aRS)-1,3,7,8a-tetramethyl-2-oxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (1B). A solution of ethyl acetoacetate (1.21 mL, 9.43 mmol, 2.5 equiv), paraformaldehyde (0.478 g, 15.3 mmol, 5 equiv), and N,N′-dimethylurea (0.456 g, 5.07 mmol, 1 equiv) in 95% ethanol (25 mL) was stirred at room temperature for 10 min. Indium(III) bromide (0.180 g, 0.507 mmol, 0.1 equiv) was added and the reaction mixture was heated under reflux for 7 h, cooled to room temperature, filtered through basic Al2O3 (EtOAc) and concentrated under reduced pressure. The residue was purified by chromatography on SiO2 (hexanes/EtOAc, 3:1 to 1:3) to afford 1A (0.481 g, 2.26 mmol, 45%) as a light yellow wax and 1B (0.855 g, 2.41 mmol, 48%) as a yellow oil. This procedure was followed for all products in Table 2 and Figure 4, and spectral properties are presented below.
Ethyl 1,3,6-trimethyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1A). IR (ATR) 2981.1, 2908.3, 1672.9, 1622.7, 1414.1, 1262.5, 1204.0, 1077.4, 1026.8, 749.3 cm−1; 1H NMR (400 MHz, CD3OD) δ 4.16 (q, J = 7.1 Hz, 2 H), 4.01 (d, J = 1.1 Hz, 2 H), 3.17 (s, 3 H), 2.92 (s, 3 H), 2.46 (t, J = 1.3 Hz, 3 H), 1.28 (t, J = 7.1 Hz, 3 H); 13C NMR (100 MHz, CD3OD) δ 165.9, 154.8, 150.8, 98.3, 59.8, 48.5, 34.2, 29.8, 14.8, 13.2; HRMS (HESI) m/z calcd for C10H17N2O3 [M+H]+ 213.1234, found 213.1234.
Diethyl(4aSR,8aRS)-1,3,7,8a-tetramethyl-2-oxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (1B). IR (ATR) 2987.8, 2931.8, 1705.4, 1629.0, 1498.5, 1442.6, 1343.8, 1233.9, 1086.6, 1041.9, 984.1, 870.4, 844.3, 753.0 cm−1; 1H NMR (400 MHz, CD3OD) δ 4.22–4.14 (m, 4 H), 3.53 (d, J = 12.7 Hz, 1 H), 3.30 (d, J = 12.7 Hz, 1 H), 2.95 (s, 3 H), 2.92 (s, 3 H), 2.69 (dq, J = 17.8, 1.7 Hz, 1 H), 2.45 (dq, J = 17.8, 1.6 Hz, 1 H), 2.24 (t, J = 1.6 Hz, 3 H), 1.61 (s, 3 H), 1.28 (t, J = 7.2 Hz, 3 H), 1.25 (t, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CD3OD) δ 170.6, 167.2, 160.2, 154.9, 99.8, 87.8, 61.4, 59.9, 49.3, 44.0, 34.9, 27.6, 27.5, 19.5, 18.5, 13.5, 13.1; HRMS (HESI) m/z calcd for C17H27N6O2 [M+H]+ 355.1864, found 355.1862.
Ethyl 1,3,6-trimethyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (2A). IR (ATR) 2978.4, 1670.0, 1636.5, 1524.6, 1442.6, 1356.9, 1267.4, 1207.8, 1168.6, 1140.7, 1088.5, 1015.8 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.20 (q, J = 7.1 Hz, 2 H), 4.06 (d, J = 1.0 Hz, 2 H), 3.55 (s, 3 H), 3.42 (s, 3 H), 2.45 (t, J = 1.0 Hz, 3 H), 1.30 (t, J = 7.1 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 180.8, 165.3, 149.2, 101.3, 60.5, 47.9, 42.9, 37.9, 16.2, 14.3; HRMS (HESI) m/z calcd for C10H17N2O2S [M+H]+ 229.1005, found 229.1004.
Methyl 1,3,6-trimethyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (3A). IR(ATR) 2929.7, 2828.8, 1698.0, 1619.2, 1516.3, 1459.9, 1425.5, 1366.5, 1317.3, 1261.1, 1169.4, 1091.3, 990.9, 803.2, 765.3 cm−1; 1H-NMR (400 MHz, CDCl3) δ 4.09 (s, 2 H), 3.77 (s, 3 H), 3.58 (s, 3 H), 3.45 (s, 3 H), 2.48 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 180.8, 165.7, 149.6, 101.0, 51.6, 47.9, 42.9, 37.9, 16.3; HRMS (HESI) m/z calcd for C9H15N2O2S [M+H]+ 215.0849, found 215.0848.
Benzyl 1,3,6-trimethyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4A). IR (ATR) 2933.0, 1662.6, 1554.5, 1485.5, 1450.1, 1192.9, 1127.6, 1077.3, 1027.0, 691.5 cm−1; 1H-NMR (300 MHz, CD3OD) δ 7.37–7.34 (m, 5 H), 5.17 (s, 2 H), 4.04 (bd, J = 1.2 Hz, 2 H), 3.17 (s, 3 H), 2.91 (s, 3 H), 2.47 (t, J = 1.2 Hz, 3 H); 13C NMR (75 MHz, CDCl3) δ 180.8, 165.0, 149.9, 136.0, 128.6, 128.3, 128.1, 101.8, 66.3, 47.9, 42.9, 37.9, 16.4; HRMS (HESI) m/z calcd for C15H19N2O3 [M+H]+ 275.1390, found 275.1388.
Dibenzyl(4aSR,8aRS)-1,3,7,8a-tetramethyl-2-oxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (4B). IR (ATR) 2937.4, 2881.5, 1712.9, 1629.0, 1506.0, 1448.2, 1379.3, 1347.6, 1232.0, 1077.3, 982.3, 883.5, 756.7 cm−1; 1H NMR (300 MHz, CD3OD) δ 7.35–7.32 (m, 10 H), 5.19, 5.15 (AB, J = 16.4 Hz, 2 H), 5.15 (s, 2 H), 3.54 (d, J = 12.9 Hz, 1 H), 3.27 (d, J = 12.9 Hz, 1 H), 2.90 (s, 3 H), 2.84 (s, 3 H), 2.73 (dq, J = 17.8, 1.6 Hz, 1 H), 2.48 (dq, J = 17.8, 1.6 Hz, 1 H), 2.20 (t, J = 1.6 Hz, 3 H), 1.54 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 170.6, 167.0, 161.2, 154.1, 136.2, 135.0, 128.6, 128.5 (2C), 128.1 (2C), 128.0, 99.6, 87.6, 67.3, 66.0, 49.7, 44.1, 35.7, 28.1, 27.8, 20.7, 19.7; HRMS (HESI) m/z calcd for C27H31N2O6 [M+H]+ 479.2177, found 479.2176.
Benzyl 1,3,6-trimethyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5A). IR (ATR) 2933.7, 1712.9, 1629.0 1450.1, 1358.8, 1254.4, 1215.2, 1163.0, 1094.1, 1066.1, 1028.8, 734.4 cm−1; 1H-NMR (600 MHz, CDCl3) δ 7.39–7.32 (m, 5 H), 5.19 (s, 2 H), 4.08 (d, J = 1.0 Hz, 2 H), 3.55 (s, 3 H), 3.41 (s, 3 H), 2.47 (d, J = 1.0 Hz, 3 H); 13C NMR (150 MHz, CDCl3) δ 180.8, 165.0, 149.9, 136.0, 128.6, 128.30, 128.17, 100.9, 66.3, 47.9, 43.0, 38.0, 16.4; HRMS (HESI) m/z calcd for C15H19N2O2S [M+H]+ 291.1162, found 291.1160.
Dibenzyl(4aSR,8aRS)-1,3,7,8a-tetramethyl-2-thioxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (5B). IR (ATR) 2933.7, 1712.9, 1629.0, 1450.1, 1340.1, 1232.0, 1215.2, 1077.3, 1002.8, 995.3, 734.4, 695.2 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.35–7.32 (m, 10 H), 5.19–5.08 (m, 4 H), 3.79 (d, J = 13.6 Hz, 1 H), 3.39 (s, 3 H), 3.34 (s, 3 H), 3.31 (d, J = 13.5 Hz, 1H), 2.63 (dd, J = 17.8, 1.5 Hz, 1 H), 2.36 (dd, J = 17.8, 1.5 Hz, 1 H), 2.23 (t, J = 1.5 Hz, 3 H), 1.51 (s, 3 H); 13C NMR (100 MHz, CD3OD) δ 178.3, 169.0, 166.8, 160.1, 136.4, 135.4, 128.3, 128.2 (2C), 128.1 (2C), 128.0, 127.9 (2C), 127.8, 101.1, 86.5, 68.0, 67.1, 65.7, 51.5, 43.1, 42.7, 34.1, 26.0, 20.2, 18.1, 13.9; HRMS (HESI) m/z calcd for C27H31N2O5S [M+H]+ 495.1948, found 495.1945.
Diallyl(4aSR,8aRS)-1,3,7,8a-tetramethyl-2-oxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (6B). IR (ATR) 2934.3, 1728.4, 1709.3, 1646.5, 1500.4, 1444.8, 1410.4, 1345.0, 1234.1, 1213.6, 1102.4, 1081.7, 1039.7, 986.7, 881.9, 753.5 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 6.00–5.84 (m, 2 H), 5.32–5.21 (m, 4 H), 4.61 (dd, J = 16.0 Hz, 4.0 Hz, 4 H), 3.51 (d, J = 12.4 Hz, 1 H), 3.22 (d, J = 12.4 Hz, 1 H), 2.80 (s, 3 H), 2.68 (s, 3 H), 2.66 (d, J = 17.6 Hz, 1 H), 2.40 (d, J = 17.6 Hz, 1 H), 2.20 (s, 3 H), 1.52 (s, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 170.5, 166.6, 160.6, 153.8, 133.5, 132.5, 118.1, 118.0, 99.9, 88.1, 65.8, 64.7, 49.3, 44.2, 35.7, 28.3, 27.7, 20.7, 19.7; HRMS (HESI) m/z calcd for C19H27N2O6 [M+H]+ 379.1864, found 379.1862.
1,3,7,7-Tetramethyl-4,6,7,8-tetrahydroquinazoline-2,5(1H,3H)-dione (7A). IR (ATR) 2960.4, 2890.4, 1722.3, 1620.5, 1574.3, 1438.2, 1380.7, 1268.3, 1224.5, 1127.4, 1079.6, 1006.6 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.04 (s, 2 H), 3.20 (s, 3 H), 2.98 (s, 3 H), 2.39 (s, 2 H), 2.24 (s, 2 H), 1.11 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 194.3, 153.5, 153.2, 105.6, 49.2, 45.6, 40.2, 35.8, 32.8, 30.4, 28.6; HRMS (HESI) m/z calcd for C12H19N2O2 [M+H]+ 223.1441, found 223.1440.
1,3,7,7-Tetramethyl-2-thioxo-2,3,4,6,7,8-hexahydroquinazolin-5(1H)-one (8A). IR (ATR) 2950.5, 2922.5, 2868.5, 1724.1, 1629.0, 1450.1, 1261.8, 1215.2, 1127.6, 1064.3, 982.3 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.12 (s, 2 H), 3.60 (s, 3 H), 3.44 (s, 3 H), 2.43 (s, 2 H), 2.26 (s, 2 H), 1.11 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 194.6, 180.1, 151.0, 107.4, 49.3, 45.8, 43.8, 40.6, 37.4, 33.1, 28.6; HRMS (HESI) m/z calcd for C12H19N2OS [M+H]+ 239.1213, found 239.1211.
Diels-Alder Reaction of DHPM Intermediate to Form 5-Component Condensation Product:
Ethyl 2-methyl-3-oxo-2-(phenylthio)butanoate. A stirred solution of N-chlorosuccinimide (6.21 g, 45.1 mmol) in CH2Cl2 (66 mL) was treated with thiophenol (0.40 mL, 3.8 mmol) and heated to reflux. After the reaction mixture changed color from yellow to orange, indicating the initiation of the reaction, additional thiophenol (4.24 mL, 41.2 mmol) was added dropwise to maintain a gentle reflux. The mixture was then allowed to cool to RT. After 1 h, the mixture was further cooled to 4°C on an ice bath, and ethyl 2-methylacetoacetate (6.81 mL, 46.7 mmol) was added over a 30 min period. After warming to RT and stirring for an additional 30 min, HCl was removed by bubbling nitrogen through the product solution and the solvent was removed under reduced pressure. The residue was suspended in petroleum ether (44 mL) and filtered. The resulting solid filtrate was washed with 5 portions (44 mL each) of petroleum ether. The combined organic fractions were combined and concentrated to give ethyl 2-methyl-3-oxo-2-(phenylthio)butanoate (11.6 g, 46.1 mmol) as a dark yellow liquid in quantitative yield with a small impurity. This product was used for the next step without further purification: 1H NMR (400 MHz, CDCl3) δ 7.51–7.30 (m, 5 H), 4.28–4.19 (m, 2 H), 2.37 (s, 3 H), 1.49 (s, 3 H), 1.32–1.26 (m, 3 H).
Ethyl 2-methyl-3-oxo-2-(phenylsulfinyl)butanoate. A solution of MCPBA (4.89 g, 19.8 mmol) in CH2Cl2 (51 mL) was added dropwise to a cold (0°C) solution of ethyl 2-methyl-3-oxo-2-(phenylthio)butanoate (5.01 g, 19.9 mmol) in CH2Cl2 (51 mL). The reaction mixture was stirred for 50 min, filtered through a plug of neutral Al2O3 (CH2Cl2), and concentrated under reduced pressure to yield ethyl 2-methyl-3-oxo-2-(phenylsulfinyl)butanoate (5.31 g, 19.8 mmol, 98%) as a yellow liquid that was used for the next step without further purification: IR (ATR) 1,030 cm−1.
Ethyl 2-methylene-3-oxobutanoate. A solution of ethyl 2-methyl-3-oxo-2-(phenylsulfinyl)butanoate (5.21 g, 19.4 mmol) in sulfolane (8 mL) was heated at 60°C and 0.1 mm Hg in a Kugelrohr distillation setup for 2 h. The collection vial was cooled to −78°C in a dry ice bath. The tube was vented with nitrogen gas, and the first distillate of ethyl 2-methylene-3-oxobutanoate was collected as a colorless liquid (0.66 g). The distillation was repeated with the remaining solution at 90°C for 1 h and 110°C for 1 h to give a second batch of ethyl 2-methylene-3-oxobutanoate together with residual sulfolane (1.22 g), for an overall yield of 68% (1.87 g, 13.2 mmol). Characteristic signals for ethyl 2-methylene-3-oxobutanoate: 1H NMR (300 MHz, CDCl3) δ 6.44 (d, J = 0.9 Hz, 1 H), 6.41 (d, J = 0.9 Hz, 1 H), 4.30 (q, J = 7.2 Hz, 2 H), 2.43 (s, 3 H), 1.33 (t, J = 7.2 Hz, 3 H).
(Diethyl(4aSR,8aRS)-1,3,7,8a-tetramethyl-2-oxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (1B). Ethyl 1,3,6-trimethyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1A, 0.096 g, 0.45 mmol) was added to ethyl 2-methylene-3-oxobutanoate containing residual sulfolane (0.244 g, 1.72 mmol). The reaction mixture was stirred at room temperature for 18 h and then concentrated under reduced pressure. The residue was purified by gradient chromatography on SiO2 (hexanes/EtOAc, 4:1 to 1:1) to afford 1B (0.0682 g, 0.192 mmol, 11%) as a yellow oil that was spectroscopically identical to 1B previously obtained in the one-pot Biginelli-Diels-Alder reaction.
4a-Benzyl 6-ethyl (4aSR,8aRS)-1,3,7,8a-tetramethyl-2-thioxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (9B). Benzyl 1,3,6-trimethyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5A, 0.043 g, 0.15 mmol) was added to ethyl 2-methylene-3-oxobutanoate containing residual sulfolane (0.021 g, 0.15 mmol). The mixture was stirred at room temperature for 18 h. Another 5 equivalents of ethyl 2-methylene-3-oxobutanoate containing residual sulfolane (0.105 g, 0.738 mmol) was added and the reaction mixture was stirred at room temperature for an additional 48 h. The mixture was concentrated under reduced pressure and the resulting residue was purified by chromatography on SiO2 (hexanes/EtOAc, 4:1 to 2:1) to afford 9B (0.0113 g, 0.0261 mmol, 18%) as a yellow oil: IR (ATR) 2974.7, 2933.7, 1705.4, 1643.9, 1524.6, 1450.1, 1340.1, 1232.0, 1077.3, 734.4, 700.8 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.34–7.25 (m, 5 H), 5.15, 5.12 (AB, J = 12.0 Hz, 2 H), 4.19–4.08 (m, 2 H), 3.78 (d, J = 13.6 Hz, 1 H), 3.38 (s, 3 H), 3.32–3.28 (m, 4 H), 2.60–2.55 (m, 1 H), 2.35–2.29 (m, 1 H), 2.20 (t, J = 1.6 Hz, 3 H), 1.48 (s, 3 H), 1.27–1.23 (m, 3 H); 13C NMR (100 MHz, CDCl3): δ 178.5, 170.0, 166.9, 159.6, 134.9, 128.7, 128.6, 128.1, 101.3, 86.1, 67.6, 61.7, 60.3, 52.2, 43.9, 43.1, 34.9, 21.5, 19.2, 14.3, 14.0; HRMS (HESI) m/z calcd for C22H29N2O5S [M+H]+ 433.1792, found 433.1788.
Conversion of 5-Component Condensation Product to DHPM Under Krapcho Conditions
Ethyl 1,3,6-trimethyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1A). A mixture of diethyl (4aSR,8aRS)-1,3,7,8a-tetramethyl-2-oxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (1B, 0.050 g, 0.14 mmol), LiCl (0.016 g, 0.37 mmol), DMSO (2 mL), and water (4 drops) was heated at 160°C and the reaction was monitored by TLC analysis (2:1, hexanes:EtOAc). After 4 h, the reaction mixture was cooled to room temperature and extracted with EtOAc. The organic layer was dried (MgSO4), filtered, and concentrated to yield ethyl 1,3,6-trimethyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1A, 0.026 g, 0.12 mmol, 87%) as a light yellow oil that was spectroscopically identical to 1A previously obtained in the one-pot Biginelli-Diels-Alder reaction.
Structural Confirmation by Conversion to Alcohol
Ethyl (4aRS,8aRS)-4a-(hydroxymethyl)-1,3,7,8a-tetramethyl-2-oxo-1,3,4,4a,5,8a-hexahydro-2H-pyrano[2,3-d]pyrimidine-6-carboxylate (10). A mixture of 4 M LiBH4 in THF (0.16 mL, 0.63 mmol) and diethyl (4aSR,8aRS)-1,3,7,8a-tetramethyl-2-oxo-1,3,4,8a-tetrahydro-2H-pyrano[2,3-d]pyrimidine-4a,6(5H)-dicarboxylate (1B, 150 mg, 0.42 mmol) in Et2O (2.1 mL) was heated at 35°C for 3 h. The reaction was monitored by TLC analysis (2:1, EtOAc:hexanes). After 3 h, the reaction mixture was quenched with 1 M HCl with ice-cooling. The solution was diluted with water and extracted with CH2Cl2 (3 × 5 mL). The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure to yield 10 (0.0794 g, 0.254 mmol, 60%) as white solid: Mp 114.8–116.9°C; IR (ATR) 3367.1, 2923.7, 1702.4, 1621.6, 1506.7, 1445.3, 1406.9, 1377.5, 1346.8, 1291.2, 1250.2, 1159.3, 1110.5, 985.0, 884.9, 838.9, 753.7 cm−1; 1H NMR (400 MHz, DMSO) δ 5.01 (t, J = 5.4 Hz, 1 H), 4.08 (q, J = 7.2 Hz, 2 H), 3.42 (dd, J = 10.8, 5.2 Hz, 1 H), 3.27 (dd, J = 10.8, 5.2 Hz, 1 H), 3.12 (d, J = 12.0 Hz, 1 H), 2.94 (d, J = 12.0 Hz, 1 H), 2.81 (s, 3 H), 2.80 (s, 3 H), 2.32 (bd, J = 16.4 Hz, 1 H), 2.15 (s, 3 H), 2.13 (bd, J = 16.4 Hz, 1 H), 1.37 (s, 3 H), 1.21 (t, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, DMSO) δ 167.6, 159.9, 154.3, 100.0, 89.9, 62.1, 59.9, 48.7, 37.6, 35.9, 28.1, 27.1, 19.8, 19.7, 14.8; HRMS (HESI) m/z calcd for C15H25N2O5 [M+H]+ 313.1758, found 313.1759.
Author Contributions
TM, MF, and MR worked on the presented work under the guidance of PW. The manuscript was written by TM and PW with input from all authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Jessica M. Williams for her contributions during the preliminary reaction screening efforts. The authors also thank Boehringer-Ingelheim Pharmaceuticals Inc., Ridgefield CT, for discretionary funding of this project.
References
Dai, C., Meschini, F., Narayanam, J. M., and Stephenson, C. R. (2012). Friedel–crafts amidoalkylation via thermolysis and oxidative photocatalysis. J. Organ. Chem. 77, 4425–4431. doi: 10.1021/jo300162c
de Fátima, Â., Braga, T. C., da S. Neto, L., Terra, B. S., Oliveira, B. G., da Silva, D. L., et al. (2015). A mini-review on biginelli adducts with notable pharmacological properties. J. Adv. Res. 6, 363–673. doi: 10.1016/j.jare.2014.10.006
Fu, N. Y., Yuan, Y. F., Cao, Z., Wang, S. W., Wang, J. T., and Peppe, C. (2002). Indium(III) bromide-catalyzed preparation of dihydropyrimidinones: improved protocol conditions for the Biginelli reaction. Tetrahedron 58, 4801–4807. doi: 10.1016/S0040-4020(02)00455-6
Hu, E. H., Sidler, D. R., and Dolling, U.-H. (1998). Unprecedented catalytic three component one-pot condensation reaction:an efficient synthesis of 5-alkoxycarbonyl-4-aryl-3,4-dihydropyrimidin-2(1H)-ones. J. Organ. Chem. 63, 3454–3457. doi: 10.1021/jo970846u
Huryn, D. M., Brodsky, J. L., Brummond, K. M., Chambers, P. G., Eyer, B., Ireland, A. W., et al. (2011). Chemical methodology as a source of small-molecule checkpoint inhibitors and heat shock protein 70 (Hsp70) modulators. Proc. Natl. Acad. Sci. U.S.A. 108, 6757–6762. doi: 10.1073/pnas.1015251108
Kapoor, T. M., Mayer, T. U., Coughlin, M. L., and Mitchison, T. J. (2000). Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell Biol. 150, 975–988. doi: 10.1083/jcb.150.5.975
Krapcho, A. P., Glynn, A. G., and Grenon, B. J. (1967). The decarbethoxylation of geminal dicarbethoxy compounds. Tetrahedron Lett. 8, 215–217. doi: 10.1016/S0040-4039(00)90519-7
Lu, J., and Bai, Y. (2002). Catalysis of the Biginelli reaction by ferric and nickel chloride hexahydrates. One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Synthesis 4, 466–470. doi: 10.1055/s-2002-20956
Manos-Turvey, A., Al-Ashtal, H. A., Needham, P. G., Hartline, C. B., Prichard, M. N., Wipf, P., et al. (2016). Dihydropyrimidinones and -thiones with improved activity against human polyomavirus family members. Bioorg. Med. Chem. Lett. 26, 5087–5091. doi: 10.1016/j.bmcl.2016.08.080
Martins, M. A. P., Teixeira, M. V. M., Cunico, W., Scapin, E., Mayer, R., Pereira, C. M. P., et al. (2004). Indium(III) bromide catalyzed one-pot synthesis of trichloromethylated tetrahydropyrimidinones. Tetrahedron Lett. 45, 8991–8994. doi: 10.1016/j.tetlet.2004.10.048
Mayer, T. U., Kapoor, T. M., Haggarty, S. J., King, R. W., Schreiber, S. L., and Mitchison, T. J. (1999). Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 286, 971–974. doi: 10.1126/science.286.5441.971
Nagarajaiah, H., Mukhopadhyay, A., and Moorthy, J. N. (2016). Biginelli reaction: an overview. Tetrahedron Lett. 57, 5135–5149. doi: 10.1016/j.tetlet.2016.09.047
Phucho, I. T., Nongpiur, A., Tumtin, S., Nongrum, R., and Nongkhlaw, R. L. (2009). Recent progress in the chemistry of dihydropyrimidinones. Rasayan J. Chem. 2, 662–676.
Ragab, F. A. F., Abou-Seri, S. M., Abdel-Aziz, S. A., Alfayomy, A. M., and Aboelmagd, M. (2017). Design, synthesis and anticancer activity of new monastrol analogues bearing 1,3,4-oxadiazole moiety. Eur. J. Med. Chem. 138, 140–151. doi: 10.1016/j.ejmech.2017.06.026
Ranu, B. C., Hajra, A., and Jana, U. (2000). Indium(III) chloride-catalyzed one-pot synthesis of dihydropyrimidinones by a three-component coupling of 1,3-dicarbonyl compounds, aldehydes, and urea: an improved procedure for the Biginelli reaction. J. Organ. Chem. 65, 6270–6272. doi: 10.1021/jo000711f
Sharma, P., Kumar, A., Rane, N., and Gurram, V. (2005). Hetero Diels–Alder reaction: a novel strategy to regioselective synthesis of pyrimido[4,5-d]pyrimidine analogues from Biginelli derivative. Tetrahedron 61, 4237–4248. doi: 10.1016/j.tet.2005.02.066
Studer, A., Jeger, P., Wipf, P., and Curran, D. P. (1997). Fluorous Synthesis: fluorous protocols for the Ugi and Biginelli multicomponent condensations. J. Organ. Chem. 62, 2917–2924.
Sun, Q., Wang, Y. Q., Ge, Z. M., Cheng, T. M., and Li, R. T. (2004). A highly efficient solvent-free synthesis of dihydropyrimidinones catalyzed by zinc chloride. Synthesis 7, 1047–1051. doi: 10.1002/chin.200437148
Wan, J. P., and Pan, Y. (2012). Recent advance in the pharmacology of dihydropyrimidinone. Mini Rev. Med. Chem. 12, 337–349. doi: 10.2174/138955712799829267
Wipf, P., and Cunningham, A. (1995). A solid phase protocol of the Biginelli dihydropyrimidine synthesis suitable for combinatorial chemistry. Tetrahedron Lett. 36, 7819–7822. doi: 10.1016/0040-4039(95)01660-A
Keywords: Biginelli reaction, dihydropyrimidinone, multi-component condensation, 5-CC, hetero Diels-Alder reaction, InBr3
Citation: Maskrey TS, Frischling MC, Rice ML and Wipf P (2018) A Five-Component Biginelli-Diels-Alder Cascade Reaction. Front. Chem. 6:376. doi: 10.3389/fchem.2018.00376
Received: 26 June 2018; Accepted: 03 August 2018;
Published: 24 August 2018.
Edited by:
Andrea Basso, Università di Genova, ItalyReviewed by:
Alessandro Massi, University of Ferrara, ItalyGian Cesare Tron, Università degli Studi del Piemonte Orientale, Italy
Copyright © 2018 Maskrey, Frischling, Rice and Wipf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Wipf, pwipf@pitt.edu