Promoting Effect of Cerium Oxide on the Catalytic Performance of Yttrium Oxide for Oxidative Coupling of Methane
 Masaaki Haneda
Masaaki Haneda Yuya Katsuragawa
Yuya Katsuragawa Yuichiro Nakamura
Yuichiro Nakamura Atsuya Towata3
Atsuya Towata3- 1Advanced Ceramics Research Center, Nagoya Institute of Technology, Tajimi, Japan
- 2Frontier Research Institute for Materials Science, Nagoya Institute of Technology, Nagoya, Japan
- 3Magnetic Powder Metallurgy Research Center, National Institute of Advanced Industrial Science and Technology, Nagoya, Japan
The promoting effect of CeO2 on the catalytic performance of Y2O3, which is moderately active catalyst, for the oxidative coupling of methane (OCM) reaction was investigated. The addition of CeO2 into Y2O3 by coprecipitation method caused a significant increase in not only CH4 conversion but also C2 (C2H6/C2H4) selectivity in the OCM reaction. C2 yield at 750 °C was increased from 5.6% on Y2O3 to 10.2% on 3 mol% CeO2/Y2O3. Further increase in the CeO2 loading caused an increase in non-selective oxidation of CH4 to CO2. A good correlation between the catalytic activity for the OCM reaction and the amount of H2 consumption for the reduction of surface/subsurface oxygen species in the H2-TPR profile was observed, suggesting the possibility that highly dispersed CeO2 particles act as catalytically active sites in the OCM reaction. The 16O/18O isotopic exchange reaction suggested that the beneficial role of CeO2 in the OCM reaction is to promote the formation of active oxygen species via the simple hetero-exchange mechanism, resulting in the promotion of CH4 activation.
Introduction
Methane, which is the main constituent of natural gas, is an abundant hydrocarbon resource for energy and chemicals. Since methane is the most stable hydrocarbon, the selective oxidation to the chemicals seems to be difficult compared with other hydrocarbons. Therefore, in many cases, methane is first partially oxidized to synthesis gas (CO + H2), and then converted to the chemicals such as methanol, acetic acid, light hydrocarbons and so on. This is well-known process as C1 chemistry.
On the other hand, as for the direct conversion of methane to the chemicals, oxidative coupling of methane (OCM) is regarded as energy efficient and simple chemical process for the production of higher hydrocarbons, especially, ethane and ethylene (Lunsford, 1995). The development of highly active OCM catalyst has been continuously performed over the past few decades (Hutchings et al., 1989; Arndt et al., 2011; Takanabe, 2012; Farrell et al., 2016) since the pioneering works of Keller and Bhasin (1982). The OCM reaction proceeds via the formation of methyl radicals formed by the reaction of methane with active oxygen species and subsequent coupling the methyl radicals (Dubois and Cameron, 1990). Therefore, designing the catalyst surface to create the active oxygen species such as O−, O, and O2− for the OCM reaction must be the important strategy to develop highly active catalysts (Kumar et al., 2016).
Cerium oxide (CeO2) is well-known material showing reversible oxygen release/absorb properties, so-called OSC (oxygen storage capacity) (Trovarelli, 1996), leading us to the expectation that CeO2 can promote the activation of methane and then improve the catalytic performance for the OCM reaction. The promoting effect of CeO2 on the OCM reaction has already been reported by many researchers. For example, Tang et al. (2011) reported that the addition of CeO2 into Li/MgO caused an increase in the CH4 conversion at lower reaction temperature with little change in C2 selectivity. They explained the promoting effect of CeO2 by oxygen activation caused via the electron transfer between Ce4+/Ce3+ and Li/MgO. A similar promoting effect of CeO2 was reported for Na/CaO (Pacheco Filho et al., 2000) and Li/MgO (Elkins et al., 2016). In case of alkali-doped catalysts, Li+O− and Na+O− sites are proposed to participate in oxygen activation (Ito et al., 1985; Lin et al., 1986). Therefore, the role of CeO2 is suspected to improve the catalytic ability of Li+O− and Na+O− sites through the electronic interaction. On the other hand, Rane et al. (2006) investigated the additive effect of rare earth elements (La, Sm, Ce, Nd, and Yb) on the OCM activity of CaO, and found that CeO2 is not good additive for the formation of C2 products. This is probably due to the high ability of CeO2 for the complete oxidation of CH4 to CO2 (Ferreira et al., 2012; Xu et al., 2018).
Recently, we have investigated that similarity of catalytically active sites for oxidative coupling of methane (OCM) and NO decomposition, and found that the catalysts showing the activity for the latter reaction are also active for the former reaction (Haneda et al., 2018). We also reported that the addition of small amount of CeO2 into Ba/Y2O3 causes a significant increase in the NO decomposition activity (Doi et al., 2015), suggesting the possibility that CeO2 behaves as effective promoter for the OCM reaction. In this study, we prepared CeO2-promoted Y2O3 as the OCM catalyst by various methods, and found that CeO2/Y2O3 prepared by coprecipitation method showed the highest OCM activity. The active state and the role of CeO2 in the OCM reaction are discussed on the basis of its catalyst characterizations.
Experimental
Catalyst Preparation
A plural CeO2-promoted Y2O3 catalyst was prepared by coprecipitation, homogeneous precipitation, impregnation and hydrothermal methods. The loading of CeO2 was fixed at 3 mol%, except for the case of coprecipitation method. The details of CeO2/Y2O3 catalysts as well as Y2O3 itself prepared in the present study are summarized in Table 1.

Table 1. Physico-chemical properties of CeO2/Y2O3.
Yttrium Oxide (Y2O3)
Y2O3 was prepared by precipitation method using yttrium(III) nitrate and ammonia aqueous solution, followed by drying and calcination at 800°C for 5 h in air.
Coprecipitation Method [CeO2/Y2O3(CP)]
An aqueous solution of ammonia (10% NH4OH, FUJIFILM Wako Pure Chemical Corporation) as a precipitation agent was added to an aqueous solution of yttrium(III) nitrate [Y(NO3)3·6H2O, FUJIFILM Wako Pure Chemical Corporation], and ammonium cerium(IV) nitrate [(NH4)2Ce(NO3)6, FUJIFILM Wako Pure Chemical Corporation] at room temperature. The precipitate thus obtained was washed with distilled water, followed by drying and calcination at 800°C for 5 h in air. The loading of CeO2 was changed from 1 to 10 mol%. The samples are abbreviated as CeO2(x)/Y2O3(CP), where x is the loading of CeO2.
Homogeneous Precipitation Method [CeO2/Y2O3(HP)]
The precipitation of yttrium and cerium hydroxides was obtained by adding urea [CO(NH2)2, FUJIFILM Wako Pure Chemical Corporation] to an aqueous solution of yttrium(III) nitrate and cerium(III) nitrate [Ce(NO3)3·6H2O, FUJIFILM Wako Pure Chemical Corporation] and stirring at 90°C for 24 h, and then washed with distilled water, followed by drying and calcination at 800°C for 5 h in air.
Impregnation Method [CeO2/Y2O3(I)]
The deposition of Ce ions onto Y2O3 was carried out by impregnating Y2O3 powder, which was prepared by precipitation method as mentioned above, with an aqueous solution of cerium(III) nitrate, followed by drying and calcination at 800°C for 5 h in air.
Hydrothermal Method [CeO2/Y2O3(HT)]
The oleate solution prepared by dissolving potassium oleate (C17H33COOK, 19% solution, FUJIFILM Wako Pure Chemical Corporation) with distilled water was added to an aqueous solution of yttrium(III) nitrate and ammonium cerium(IV) nitrate at room temperature under vigorously-stirred condition, followed by addition of ammonia aqueous solution. The mixture solution thus obtained was transferred to a Teflon vessel and then treated at 200°C for 6 h in an autoclave. The product thus obtained was washed with distilled water, followed by drying and calcination at 800°C for 5 h in air.
Catalyst Characterizations
X-ray diffraction (XRD) patterns were recorded using a Rigaku MiniFlex diffractometer with Cu Kα radiation at 30 kV and 15 mA. The scanning was done from 2θ = 15–65° at a speed of 1 deg min−1. The BET surface area of the catalysts was determined by N2 physisorption at liquid nitrogen temperature using a BELSORP mini-II, after evacuating the samples at 300°C for 1 h. Raman spectra were measured with a Micro-RAM300/NK (Lambda Vision) equipped with a TE-cooled charge coupled device (CCD) detector and a green laser (λ = 532 nm) under the ambient atmosphere. Direct observation of CeO2/Y2O3(CP) samples by TEM was performed with a JEM-2100 (JEOL) operating at an acceleration voltage of 200 kV. H2-TPR measurement was conducted to estimate the reducibility of the catalysts (Haneda et al., 2015). The TPR profiles were obtained from room temperature to 800°C in a 30 cm3min−1 flow of 5% H2/Ar at a heating rate of 10 °C min−1. The consumption of H2 was monitored using a thermal conductivity detector (TCD).
The 16O/18O isotopic exchange reaction was carried out in a flow reactor system. An adequate amount of the sample (30 mg) was installed in a quartz reactor, and then pretreated in a flow of 5% O2/He at 600°C for 1 h. After cooling to 100°C in flowing 5% O2/He, the sample was purged with He at 100°C for 30 min. The reaction gas composed of 1% 18O2/He was fed to the catalyst at a rate of 20 cm3 min−1, and then the temperature was increased up to 600 °C at a heating rate of 10°C min−1. The masses of 32 (16O2), 34 (18O16O), and 36 (18O2) were continuously monitored by a quadrupole mass spectrometer (PFEFFER OminiStar).
Activity Measurement
The catalytic activity for oxidative coupling of methane was evaluated using a fixed-bed continuous flow reactor. The reaction gas composed of 44.4% CH4 and 11.1% O2 diluted with He as the balance gas was fed to a 0.1 g catalyst that had been pretreated in situ in a flow of He at 650°C for 2 h at a rate of 45 cm3 min−1. The effect of variation of CH4/O2 ratio was also investigated, where O2 concentration was varied over the range of 5.5–22.2% and CH4 concentration was fixed at 44.4%. The reaction temperature was increased from 650 to 800°C in steps of 50°C, and the steady-state catalytic activity was measured at each temperature. The effluent gas was analyzed with the use of on-line gas chromatograph equipped with TCD and FID as a detector (Shimadzu GC-2014ATTF) using a Molecular Sieve 13X column (for analysis of O2, CH4, and CO), a Porapak QS column (for analysis of CO2), and a DC-200 (20%)/Shimalite column (for analysis of C2H4 and C2H6).
Results and Discussion
Activity of CeO2/Y2O3 Prepared by Different Method
Figure 1A shows the XRD patterns of 3 mol% CeO2/Y2O3 prepared by different method. Distinct XRD peaks indexed to the cubic phase of Y2O3 were observed for all the samples. No peaks due to CeO2 were detected because of the low loading (3 mol%), suggesting the presence of CeO2 nanoparticles. It is of interest that a shift of XRD peaks due to Y2O3 toward lower angle was observed for CeO2/Y2O3(HP) (Figure 1B–b), suggesting the formation of CeO2-Y2O3 solid solution. This can be explained by the fact that the radius of Ce3+ ion (0.114 nm)/Ce4+ ion (0.097 nm) is larger than that of Y3+ ion (0.09 nm) (Shannon, 1976). Homogeneous precipitation including Ce3+ and Y3+ ions may be produced under the conditions, where the pH of solution is gradually increased via the hydrolysis of urea.
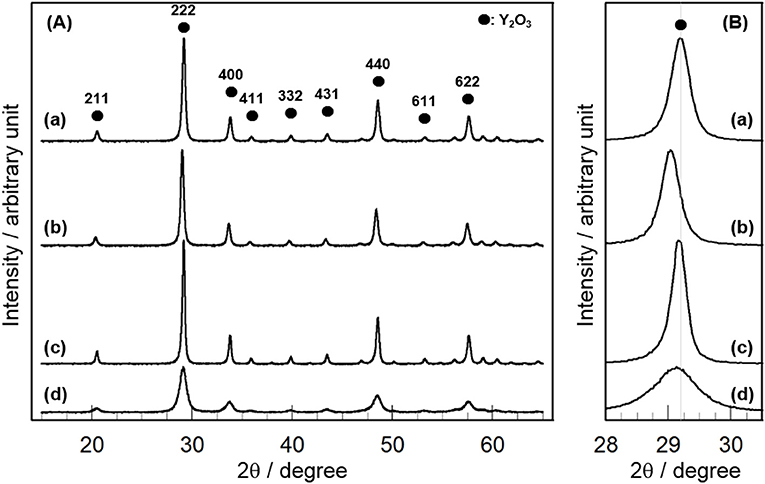
Figure 1. XRD patterns of (a) CeO2/Y2O3(CP), (b) CeO2/Y2O3(HP), (c) CeO2/Y2O3(I), (d) CeO2/Y2O3(HT) given in the wide range of 2θ = 16 − 64° (A) and in the expanded range of 2θ = 28.0 − 30.5° (B).
Table 1 summarizes the crystallite size of Y2O3, which was calculated from the XRD peak due to the (222) plane (2θ = 29.1°) using Scherrer's equation without the subtraction of the instrument broadening, where shape factor of “K” was used as 0.9, and the BET surface area of CeO2/Y2O3. No significant difference in the crystallite size of Y2O3 was observed for CeO2/Y2O3(CP), CeO2/Y2O3(HP) and CeO2/Y2O3(I). In accordance with this, these samples gave similar BET surface area in the range of 23–28 m2g−1. On the other hand, the BET surface area of CeO2/Y2O3(HT) was lower than those of other samples, although the crystallite size of Y2O3 was evaluated to be small. This conflict might be ascribed to the formation of aggregated Y2O3 crystallites with less-porous structure via hydrothermal process (Haneda et al., 2018).
Figure 2 shows the catalytic activity of 3 mol% CeO2/Y2O3 for the OCM reaction. It appears that the addition of CeO2 into Y2O3 by coprecipitation (CP), impregnation (I), and hydrothermal (HT) processes caused an increase in not only CH4 conversion (Figure 2A) but also C2 selectivity (Figure 2B) in the entire temperature range, suggesting that CeO2 is effective additive for the OCM reaction. Since the participation of oxygen species such as O−, O, O2−, and O formed on the catalyst surface in the activation of CH4 molecule for the OCM reaction has been established by many researchers (Keller and Bhasin, 1982; Borchert and Baerns, 1997; Sekine et al., 2009; Takanabe, 2012; Liang et al., 2014), active oxygen species would be efficiently supplied from the lattice of CeO2. On the other hand, as seen in Figure 2, CeO2/Y2O3(HP) showed lower CH4 conversion and C2 selectivity than Y2O3. This is probably ascribed to the formation of CeO2-Y2O3 solid solution revealed from XRD measurements (Figure 1Bb), resulting in ineffective supply of active oxygen species. Taking into account the fact that CeO2/Y2O3(HP) possesses similar physico-chemical properties, such as BET surface area and crystallite size (Table 1), with CeO2/Y2O3(CP) and CeO2/Y2O3(I), it is suspected the presence of the important factors affecting the catalytic activity of CeO2/Y2O3 for the OCM reaction.
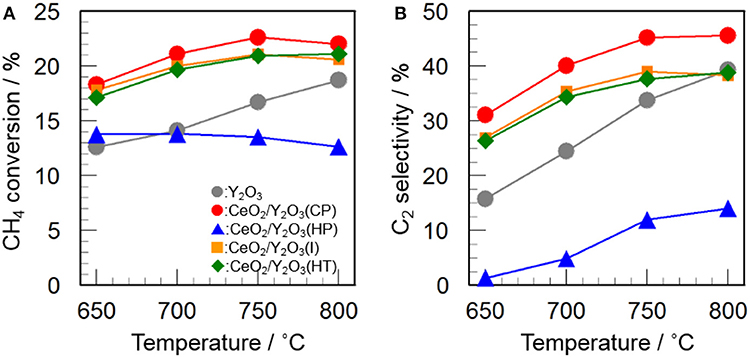
Figure 2. Catalytic activity of 3 mol% CeO2/Y2O3 for the OCM reaction. (A) CH4 conversion and (B) C2 selectivity.
Effect of CeO2 Loading on the Activity of CeO2/Y2O3(CP)
Structural Characterization of CeO2/Y2O3(CP)
In order to gain information on the activity controlling factor, the catalytic performance of CeO2(x)/Y2O3(CP) with different CeO2 loading was examined. Structural characterizations were carried out. In Table 2 are summarized the BET surface area of CeO2(x)/Y2O3(CP). It appears that no significant difference in the BET surface area was obtained in the range of 19–27 m2g−1. Figure 3A shows the XRD patterns of CeO2(x)/Y2O3(CP). No other peaks, except for those assignable to Y2O3, were detected irrespective of CeO2 loading. It is also noteworthy that a shift of XRD peaks by the addition of CeO2 was not recognized. Figure 3B shows the Raman spectra of CeO2(x)/Y2O3(CP). Raman bands ascribed to Fg + Ag mode of cubic Y2O3 with C-type structure (Repelin et al., 1995; Yashima et al., 1997; Ubaldini and Carnasciali, 2008) were observed for all the samples, and their intensities were gradually decreased with an increase in CeO2 loading. From XRD and Raman measurements, CeO2 particles were revealed to present as highly dispersion state on the surface and/or in the bulk of Y2O3.
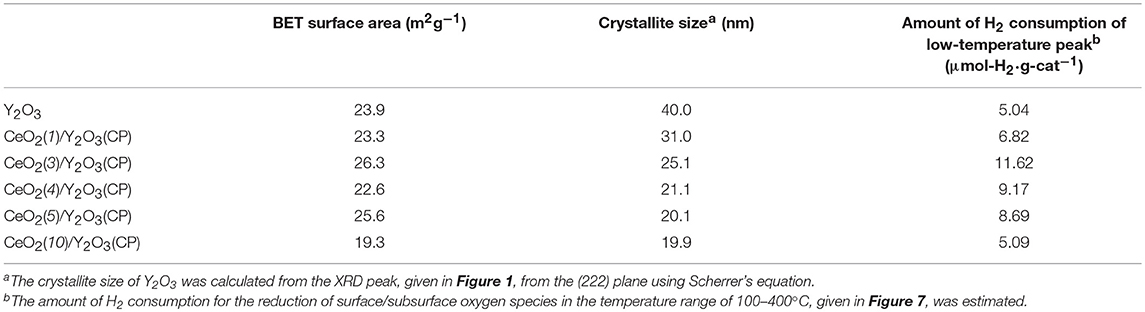
Table 2. BET surface area, crystallite size of Y2O3 and the amount of H2 consumption in H2-TPR of CeO2(x)/Y2O3(CP).
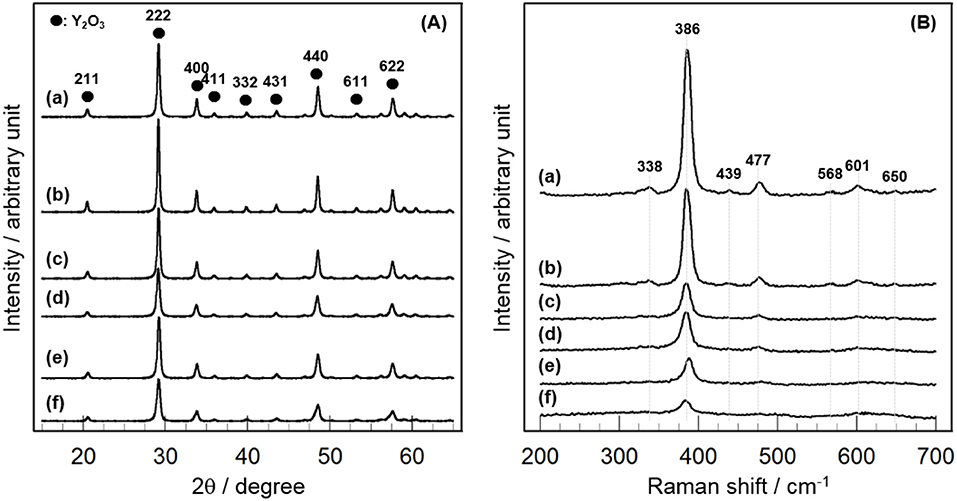
Figure 3. Structural characterization of CeO2(x)/Y2O3(CP) by (A) XRD and (B) Raman. (a) Y2O3, (b) CeO2(1)/Y2O3(CP), (c) CeO2(3)/Y2O3(CP), (d) CeO2(4)/Y2O3(CP), (e) CeO2(5)/Y2O3(CP), (f) CeO2(10)/Y2O3(CP).
In order to gain information on the dispersion state of CeO2 particles, the morphology of CeO2(x)/Y2O3(CP) with the CeO2 loadings (x) of 0, 3, and 10 mol% was observed by TEM analysis. As can be seen in Figures 4a–c, the addition of CeO2 into Y2O3 did not cause a significant change in the particle morphology, whereas the particle size was gradually decreased. This is in accordance with the crystallite size of Y2O3 (Table 2). These results suggest that the aggregated particles observed in TEM images mainly consist of Y2O3. Figures 4d–f show the TEM images of CeO2(x)/Y2O3(CP) observed at high magnification. Unfortunately, CeO2 and Y2O3 particles were not clearly distinguished each other because of low CeO2 loading (< 10 mol%) and the same crystal system (cubic). However, it appears from the comparison of Figures 4d,e that the surface morphology of Y2O3 was changed by addition of 3 mol% CeO2. Namely, Y2O3 consists of the particles with smooth surface (Figure 4d), whereas the surface of Y2O3 particles in CeO2(3)/Y2O3(CP) seems to be rough (Figure 4e), suggesting the surface interaction between CeO2 and Y2O3. In other words, CeO2 nanoparticles of which the size is smaller than the detection limit by TEM and XRD might be dispersed on the surface of Y2O3 particles. On the other hand, as seen in Figure 4f, CeO2(10)/Y2O3(CP) seems to be composed of a plurality of particles with different surface morphology. CeO2 nanoparticles might be present not only independently but also weakly interacting with Y2O3.

Figure 4. TEM images of (a,d) Y2O3, (b,e) CeO2(3)/Y2O3(CP), (c,f) CeO2(10)/Y2O3(CP).
Catalytic Activity of CeO2/Y2O3(CP) for the OCM Reaction
Figure 5 shows the effect of CeO2 loading on the catalytic activity of CeO2(x)/Y2O3(CP) for the OCM reaction. As mentioned before, the addition of CeO2 into Y2O3 caused an increase in not only CH4 conversion but also C2 selectivity in the entire temperature range. As can be seen in Figure 5A, CH4 conversion was drastically increased by addition of 3 mol% CeO2. Further increase in CeO2 loading up to 10 mol% did not cause any change in the CH4 conversion. In accordance with previous reports (Primet and Garbowski, 2002), CeO2 seems to be good additive for the activation of CH4 molecules. On the other hand, as seen in Figure 5B, the effect of CeO2 loading on the C2 selectivity was slightly different from that on the CH4 conversion. The C2 selectivity was gradually increased with an increase in the CeO2 loading up. The maximum C2 selectivity was achieved at 3 mol% CeO2 loading. However, further increase in CeO2 loading caused a gradual decrease in the C2 selectivity. The optimum CeO2 loading for the formation of C2H6/C2H4 as a product was found to be 3 mol%. As seen in Table 2, the highest BET surface area was obtained for CeO2(3)/Y2O3(CP). This is in agreement with the catalytic activity. However, although CeO2(5)/Y2O3(CP) was found to possess similar BET surface area with CeO2(3)/Y2O3(CP), the former catalytic activity was clearly lower than that of the latter catalyst. This suggests that the BET surface area is not only the important factor to determine the catalytic activity.
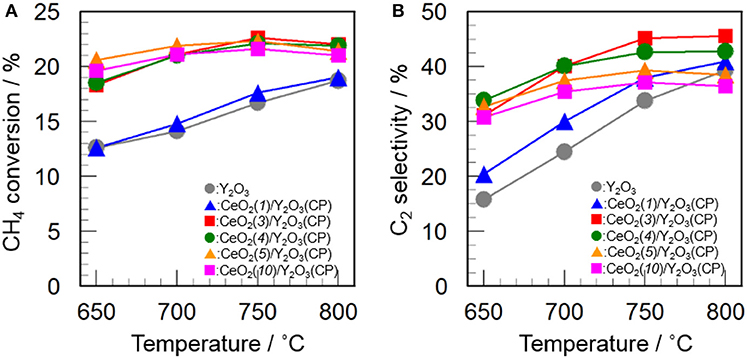
Figure 5. Effect of CeO2 loading on the activity of CeO2(x)/Y2O3(CP) for the OCM reaction. (A) CH4 conversion and (B) C2 selectivity.
Since the CH4/O2 ratio is an important parameter to optimize the reaction conditions, the effect of CH4/O2 ratio on the catalytic activity of CeO2(3)/Y2O3(CP) was examined. As can be seen in Figure 6, an increase in the CH4/O2 ratio caused a decrease in the CH4 conversion as well as an increase in the C2 selectivity. This indicates that the reaction gas containing higher concentration of O2 is favored for the activation of CH4, resulting in high CH4 conversion, while is also favored for the formation of undesirable CO2, leading to low C2 selectivity. The trade-off between CH4 conversion and C2 selectivity is well-known phenomena in the OCM reaction (Ghose et al., 2014; Godini et al., 2014; Ivanov et al., 2014). From the kinetic point of view, the activated CH4 molecule (CH3· radical) may preferentially react with O2 species in the presence of excess O2. It would be important to explore the optimum reaction conditions to achieve high C2 productivity.
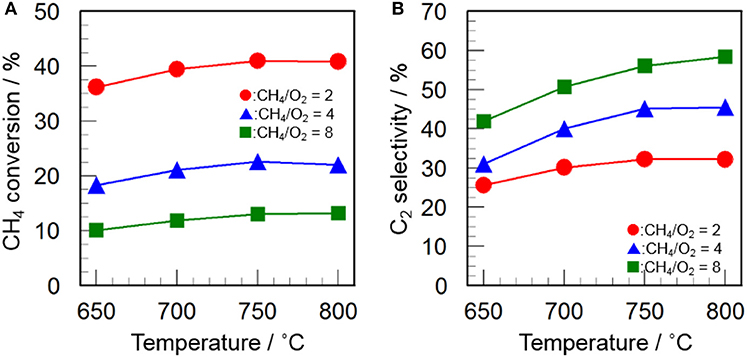
Figure 6. Effect of CH4/O2 ratio on (A) CH4 conversion and (B) C2 selectivity in the OCM reaction over CeO2(3)/Y2O3(CP). (Red circles) CH4/O2 = 2, (blue triangles) CH4/O2 = 4, (green squares) CH4/O2 = 8.
Active Sites for the OCM Reaction Over CeO2/Y2O3(CP)
XRD and Raman measurements revealed that CeO2 particles are present as highly dispersion state on the surface and/or in the bulk of Y2O3 (Figure 3). In addition, TEM observation suggested that the dispersion state of CeO2 particles is different depending on CeO2 loading (Figure 4). In case of CeO2(3)/Y2O3(CP), CeO2 nanoparticles might be dispersed on the surface of Y2O3 particles, while CeO2 nanoparticles might be present not only independently but also weakly interacting with Y2O3 in CeO2(10)/Y2O3(CP). Therefore, the dispersion state of CeO2 would be related to the catalytic activity. It is well known that the reduction of CeO2 particles gradually proceeds from surface to bulk in the wide range from low to high temperature region, respectively (Yao and Yao, 1984; Trovarelli, 1996; Imagawa et al., 2011). In order to obtain an information on the dispersion state of CeO2, H2-TPR measurements were carried out. As given in Figure 7a, very broad H2 consumption peaks in the temperature range of 100–700°C were observed in the H2-TPR profile of Y2O3. This is probably due to the reduction of oxygen species interacting with the defects sites in the lattice of Y2O3 with C-type structure. No significant change in the H2-TPR profile was observed for CeO2(1)/Y2O3(CP) (Figure 7b). This is in agreement with the tendency of the catalytic activity (Figure 5). It is noteworthy that the addition of 3 mol% CeO2 caused an appearance of distinct H2 consumption peaks at around 200 and 600°C (Figure 7c). The area of H2 consumption peak at around 600°C, which can be ascribed to the reduction of oxygen species in the bulk of CeO2 (Yao and Yao, 1984; Trovarelli, 1996; Imagawa et al., 2011), was gradually increased with an increase in CeO2 loading up to 10 mol%. In contrast, the low-temperature peak at around 200 °C due to the reduction of surface/subsurface oxygen species on CeO2 particles (Yao and Yao, 1984; Trovarelli, 1996; Imagawa et al., 2011) was clearly decreased for CeO2(10)/Y2O3(CP). These results suggest that the size of CeO2 particles dispersed on the surface and/or in the bulk of Y2O3 was significantly increased with an increase in CeO2 loading.
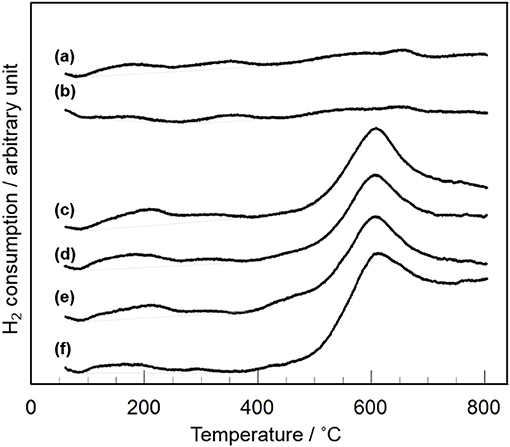
Figure 7. H2-TPR profiles of CeO2(x)/Y2O3(CP) with different CeO2 loading. (a) Y2O3, (b) CeO2(1)/Y2O3(CP), (c) CeO2(3)/Y2O3(CP), (d) CeO2(4)/Y2O3(CP), (e) CeO2(5)/Y2O3(CP), (f) CeO2(10)/Y2O3(CP).
Table 2 summarizes the amount of H2 consumption for the reduction of surface/subsurface oxygen species in the temperature range of 100–400°C. It is of interest that the maximum amount of H2 consumption was obtained for CeO2(3)/Y2O3(CP), and then decreased with CeO2 loading. Figure 8 shows the relationship between the amount of H2 consumption for the reduction of surface/subsurface oxygen species and the catalytic activity for the OCM reaction at 750 °C. It appears that the CH4 conversion and C2 selectivity were linearly increased with an increase in the amount of H2 consumption. This clearly indicates that oxygen species on the surface/subsurface of highly dispersed CeO2 particles can effectively activate CH4 molecule, resulting in the selective formation of C2H6/C2H4. On the basis of XRD, Raman, TEM, and H2-TPR measurements, we can conclude that the creation of highly dispersed CeO2 particles is one of the strategies for the catalyst design leading to the development of highly active OCM catalysts.
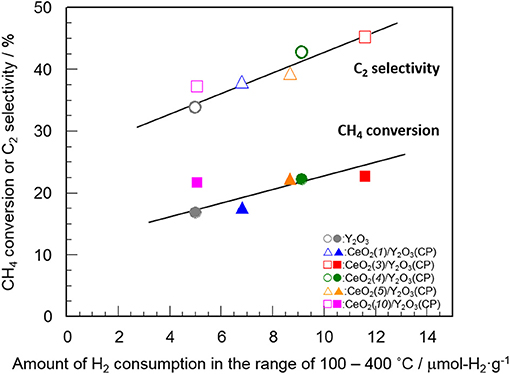
Figure 8. Relationship between the amount of H2 consumption for the reduction of surface/subsurface oxygen species and the catalytic activity for the OCM reaction at 750°C.
Catalytic Role of CeO2 in the OCM Reaction
Since CeO2 is well-known material showing reversible oxygen release/absorb properties (Trovarelli, 1996), the 16O/18O isotopic exchange reaction was carried out to clarify the possibility of O2 activation as catalytically role of CeO2 in the OCM reaction. Martin and Duprez (1996) and Duprez (2006) investigated the oxygen mobility of various kinds of oxide by using the 16O/18O isotopic exchange reaction, and proposed two mechanisms depending on the type of oxide. One group is non-reducible oxide such as Al2O3, SiO2, ZrO2 and MgO, and the other is reducible oxide such as CeO2 and CeO2-ZrO2. In the former case, the simple hetero-exchange that occurs with the participation of only one oxygen of the oxide at each step of the following equations (1) and (2):
Here, the consecutive evolution of 18O16O and 16O2 can be observed. On the other hand, the isotopic exchange reaction over reducible oxides takes place via the multiple-hetero exchange mechanism, where the reaction between a molecule dioxygen (18O2) in gas phase and two atomic oxygens (16O) of the solid occurs the following equation (3) or (4):
In this case, 16O2 and 16O18O seem to be simultaneously evolved.
Figure 9 shows the profiles of 16O2 and 16O18O evolution and 18O2 consumption in the 16O/18O isotopic exchange reaction over Y2O3 and CeO2(3)/Y2O3(CP). It appears that both catalysts gave similar profiles for each O2 species. The evolution of 16O18O and the consumption of 18O2 were simultaneously observed, followed by the 16O2 evolution at higher temperature. This suggests that the simple hetero-exchange mechanism is favored on Y2O3 and CeO2/Y2O3 with lower CeO2 loading, although the O2 activation over CeO2-containing sample was suspected to be governed by the multiple-hetero exchange mechanism. This is probably due to the high dispersion state of CeO2 nanoparticles on the surface of Y2O3, as revealed by H2-TPR measurement (Figure 7).
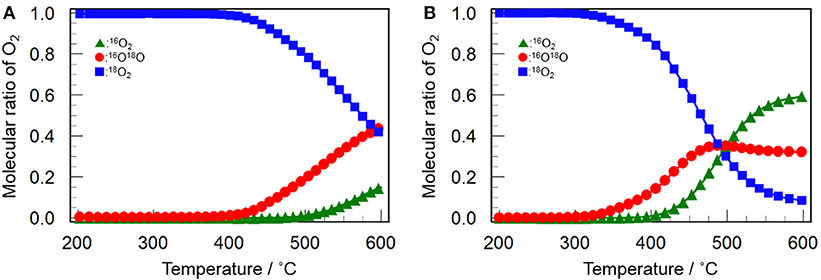
Figure 9. Change in the molecular ratio of 16O2 (green triangles), 16O18O (red circles) and 18O2 (blue squares) during the isotopic exchange reaction of O2 over (A) Y2O3 and (B) CeO2(3)/Y2O3(CP) as a function of temperature.
Dong et al. (2004a,b) investigated the oxygen mobility of CeO2-ZrO2 based catalyst with different structural homogeneity, and reported that oxygen species activated at lower temperature via the multiple-hetero exchange mechanism is highly active for the complete oxidation reaction. It can be expected that oxygen species with moderate activity, which was evolved via the simple hetero-exchange mechanism, plays a beneficial role in CH4 activation (Kumar et al., 2016), leading to the selective oxidation to C2H6/C2H4. Taking into account the fact that the temperature for the evolution of 16O18O and 16O2 was significantly lowered by addition of CeO2 into Y2O3 (Figure 9), we can conclude that the catalytically role of CeO2 in the OCM reaction is to promote the formation of active oxygen species via the simple hetero-exchange mechanism.
Conclusion
The catalytic activity of Y2O3 for the OCM reaction was effectively improved by addition of CeO2. Among the catalysts tested here, CeO2/Y2O3 prepared by coprecipitation method showed the highest activity. The optimum CeO2 loading was 3 mol%. From the structural characterizations by XRD, Raman and TEM, CeO2 particles were found to be dispersed on the surface of Y2O3 without the formation of CeO2-Y2O3 solid solution. H2-TPR measurements revealed that the amount of H2 consumption for the reduction of CeO2 surface was increased with an increase in CeO2 loading and then reached the maximum at 3 mol%. Highly dispersed CeO2 particles was suspected to act as catalytically active sites in the OCM reaction. From the 16O/18O isotopic exchange reaction, the beneficial role of CeO2 in the OCM reaction was concluded to promote the formation of active oxygen species via the simple hetero-exchange mechanism.
Author Contributions
MH: worked on the experimental setup; YK and YN: conducted the experiments; AT: carried out the TEM analysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by a Grant–in–Aid for Scientific Research (No. 16K14046, 18K05294) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
Arndt, S., Laugel, G., Levchenko, S., Horn, R., Baerns, M., Scheffler, M., et al. (2011). A critical assessment of Li/MgO-based catalysts for the oxidative coupling of methane. Catal. Rev. Sci. Eng. 53, 424–514. doi: 10.1080/01614940.2011.613330
Borchert, H., and Baerns, M. (1997). The effect of oxygen-anion conductivity of metal–oxide doped lanthanum oxide catalysts on hydrocarbon selectivity in the oxidative coupling of methane. J. Catal. 168, 315–320. doi: 10.1006/jcat.1997.1662
Doi, Y., Haneda, M., and Ozawa, M. (2015). Promoting effect of CeO2 on the catalytic activity of Ba-Y2O3 for direct decomposition of NO. Bull. Chem. Soc. Jpn. 88, 117–123. doi: 10.1246/bcsj.20140230
Dong, F., Suda, A., Tanabe, T., Nagai, Y., Sobukawa, H., Shinjoh, H., et al. (2004a). Characterization of the dynamic oxygen migration over Pt/CeO2-ZrO2 catalysts by 18O/16O isotopic exchange reaction. Catal. Today 90, 223–229. doi: 10.1016/j.cattod.2004.04.030
Dong, F., Suda, A., Tanabe, T., Nagai, Y., Sobukawa, H., Shinjoh, H., et al. (2004b). Dynamic oxygen mobility and a new insight into the role of Zr atoms in three-way catalysts of Pt/CeO2-ZrO2. Catal. Today 93–95, 827–832. doi: 10.1016/j.cattod.2004.06.076
Dubois, J. L., and Cameron, C. J. (1990). Common features of oxidative coupling of methane cofeed catalysts. Appl. Catal. 67, 49–71. doi: 10.1016/S0166-9834(00)84431-0
Duprez, D. (2006). “Oxygen and hydrogen surface mobility in supported metal catalysts study by 18O/16O and 2H/1H exchange”, in Isotopes in Heterogeneous Catalysis, eds J. S. J. Hargreaves, S. D. Jackson, and G. Webb (London: Imperial College Press), 133–182.
Elkins, T. W., Roberts, S. J., and Hagelin-Weaver, H. E. (2016). Effects of alkali and alkaline-earth metal dopants on magnesium oxide supported rare-earth oxide catalysts in the oxidative coupling of methane. Appl. Catal. A 528, 175–190. doi: 10.1016/j.apcata.2016.09.011
Farrell, B. L., Igenegbai, V. O., and Linic, S. (2016). A viewpoint on direct methane conversion to ethane and ethylene using oxidative coupling on solid catalysts. ACS Catal. 6, 4340–4346. doi: 10.1021/acscatal.6b01087
Ferreira, V. J., Tavares, P., Figueiredo, J. L., and Faria, J. L. (2012). Effect of Mg, Ca, and Sr on CeO2 based catalysts for the oxidative coupling of methane: investigation on the oxygen species responsible for catalytic performance. Ind. Eng. Chem. Res. 51, 10535–10541. doi: 10.1021/ie3001953
Ghose, R., Hwang, H. T., and Varma, A. (2014). Oxidative coupling of methane using catalysts synthesized by solution combustion method: Catalyst optimization and kinetic studies. Appl. Catal. A, 472, 39–46. doi: 10.1016/j.apcata.2013.12.004
Godini, H. R., Gili, A., Görke, O., Arndt, S., Simon, U., Thomas, A., et al. (2014). Sol–gel method for synthesis of Mn–Na2WO4/SiO2 catalyst for methane oxidative coupling. Catal. Today 236, 12–22. doi: 10.1016/j.cattod.2014.01.005
Haneda, M., Kaneko, T., Kamiuchi, N., and Ozawa, M. (2015). Improved three-way catalytic activity of bimetallic Ir-Rh catalyst supported on CeO2-ZrO2. Catal. Sci. Technol. 5, 1792–1800. doi: 10.1039/c4cy01502a.
Haneda, M., Tanaka, M., Doi, Y., and Bion, N. (2018). Oxidative coupling of methane over Ba-doped Y2O3 catalyst – Similarity with active site for direct decomposition of NO. Mol. Catal. 457, 74–81.doi: 10.1016/j.mcat.2018.07.010
Hutchings, G. J., Scurrell, M. S., and Woodhouse, J. R. (1989). Oxidative coupling of methane using oxide catalysts. Chem. Soc. Rev. 18, 251–283. doi: 10.1039/cs9891800251
Imagawa, H., Suda, A., Yamamura, K., and Sun, S. (2011). Monodisperse CeO2 nanoparticles and their oxygen storage and release properties. J. Phys. Chem. C 115, 1740–1745. doi: 10.1021/jp109878
Ito, T., Wang, J., Lin, C. H., and Lunsford, J. H. (1985). Oxidative dimerization of methane over a lithium-promoted magnesium oxide catalyst. J. Am. Chem. Soc. 107, 5062–5068. doi: 10.1021/ja00304a008
Ivanov, D. V., Isupova, L. A., Gerasimov, E., Yu., Dovlitova, L. S., Glazneva, T. S., et al. (2014). Oxidative methane coupling over Mg, Al, Ca, Ba, Pb-promoted SrTiO3 and Sr2TiO4: influence of surface composition and microstructure. Appl. Catal. A 485, 10–19. doi: 10.1016/j.apcata.2014.07.024
Keller, G. E., and Bhasin, M. M. (1982). Synthesis of ethylene via oxidative coupling of methane. I. Determination of active catalysts. J. Catal. 73, 9–19. doi: 10.1016/0021-9517(82)90075-6
Kumar, G., Lau, S. L. J., Krcha, M. D., and Janik, M. J. (2016). Correlation of methane activation and oxide catalyst reducibility and its implications for oxidative coupling, ACS Catal. 6, 1812–1821. doi: 10.1021/acscatal.5b02657
Liang, Y., Li, Z., Nourdine, M., Shahid, S., and Takanabe, K. (2014). Methane coupling reaction in an oxy-steam stream through an OH radical pathway by using supported alkali metal catalysts. ChemCatChem 6, 1245–1251. doi: 10.1002/cctc.201400018
Lin, C. H., Campbell, K. D., Wang, J. X., and Lunsford, J. H. (1986). Oxidative dimerization of methane over lanthanum oxide. J. Phys. Chem. 90, 534–537. doi: 10.1021/j100276a004
Lunsford, J. H. (1995). The catalytic oxidative coupling of methane. Angew. Chem. Int. 34, 970–980. doi: 10.1002/anie.199509701
Martin, D., and Duprez, D. (1996). Mobility of surface species on oxides. 1. Isotopic exchange of 18O2 with 16O of SiO2, Al2O3, ZrO2, MgO, CeO2, and CeO2-Al2O3. Activation by noble metals. Correlation with oxide basicity. J. Phys. Chem. 100, 9429–9438. doi: 10.1021/jp9531568
Pacheco Filho, J. G. A., Eon, J. G., and Schmal, M. (2000). Oxidative coupling of methane on Ce/Na/CaO catalysts. Catal. Lett. 68, 197–202. doi: 10.1023/A:1019064310606
Primet, M., and Garbowski, E. (2002). “Fundamentals and application of ceria in combustion reactions”, in Catalysis by Ceria and Related Materials, ed A. Trovarelli (London: Imperial College Press), 407–429.
Rane, V. H., Chaudhari, S. T., and Choudhary, V. R. (2006). Comparison of the surface and catalytic properties of rare earth-promoted CaO catalysts in the oxidative coupling of methane. J. Chem. Technol. Biotechnol. 81, 208–215. doi: 10.1002/jctb.1387
Repelin, Y., Proust, C., Husson, E., and Beny, J. M. (1995). Vibrational spectroscopy of the C-form of yttrium sesquioxide. J. Solid State Chem. 118, 163–169. doi: 10.1006/jssc.1995.1326
Sekine, Y., Tanaka, K., Matsukata, M., and Kikuchi, E. (2009). Oxidative coupling of methane on Fe-doped La2O3 catalyst. Energy Fuels 23, 613–616. doi: 10.1021/ef800665r.
Shannon, R. D. (1976). Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta. Cryst. A 32, 751–767. doi: 10.1107/S0567739476001551
Takanabe, K. (2012). Catalytic conversion of methane: Carbon dioxide reforming and oxidative coupling. J. Jpn. Petrol. Inst. 55, 1–12. doi: 10.1627/jpi.55.1
Tang, L., Yamaguchi, D., Wong, L., Burke, N., and Chiang, K. (2011). The promoting effect of ceria on Li/MgO catalysts for the oxidative coupling of methane. Catal. Today 178, 172–180. doi: 10.1016/j.cattod.2011.07.014
Trovarelli, A. (1996). Catalyti properties of ceria and CeO2-containing materials. Catal. Rev. Sci. Eng. 38, 439–520. doi: 10.1080/01614949608006464
Ubaldini, A., and Carnasciali, M. M. (2008). Raman characterization of powder of cubic RE2O3 (RE = Nd, Gd, Dy, Tm, and Lu), Sc2O3 and Y2O3. J. Alloy. Compd. 454, 374–378. doi: 10.1016/j.jallcom.2006.12.067
Xu, J., Peng, L., Fang, X., Fu, Z., Liu, W., Xu, X., et al. (2018). Developing reactive catalysts for low temperature oxidative coupling of methane: on the factors deciding the reaction performance of Ln2Ce2O7 with different rare earth A sites. Appl. Catal. A 552, 117–128. doi: 10.1016/j.apcata.2018.01.004
Yao, H. C., and Yao, Y. F. Y. (1984). Ceria in automotive exhaust catalysts. I. Oxygen storage. J. Catal. 86, 254–265. doi: 10.1016/0021-9517(84)90371-3
Keywords: oxidative coupling of methane, cerium oxide, yttrium oxide, 16O/18O isotopic exchange, H2-TPR
Citation: Haneda M, Katsuragawa Y, Nakamura Y and Towata A (2018) Promoting Effect of Cerium Oxide on the Catalytic Performance of Yttrium Oxide for Oxidative Coupling of Methane. Front. Chem. 6:581. doi: 10.3389/fchem.2018.00581
Received: 17 July 2018; Accepted: 06 November 2018;
Published: 22 November 2018.
Edited by:
Cristina Artini, Università di Genova, ItalyCopyright © 2018 Haneda, Katsuragawa, Nakamura and Towata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masaaki Haneda, haneda.masaaki@nitech.ac.jp