Staphylococcus aureus temperate bacteriophage: carriage and horizontal gene transfer is lineage associated
- 1 Division of Clinical Sciences, Centre for Infection, St George’s University of London, London, UK
- 2 Bacterial Microarray Group, Division of Clinical Sciences, St George’s University of London, London, UK
Staphylococcus aureus is a major cause of human and animal infections. Bacteriophage are a class of mobile genetic element (MGE) that carry virulence genes and disseminate them horizontally, including Panton–Valentine leukocidin (PVL), the immune evasion cluster (IEC) associated with human specificity, and enterotoxin A the major toxin associated with food poisoning. S. aureus isolates group into major clonal complex (CC) lineages that largely evolve independently due to possession of different restriction–modification (RM) systems. We aimed to better understand the horizontal and vertical transmission dynamics of virulence and resistance genes by bacteriophage by using (i) bioinformatic approaches to analyze bacteriophage genomes from the first 79 sequenced S. aureus isolates and (ii) S. aureus microarrays to analyze the distribution of bacteriophage and virulence genes in S. aureus isolates from a broader range of lineages. The distribution of eight bacteriophage families was highly variable but lineage associated. Nevertheless, there was evidence of frequent acquisition and loss and not just vertical transmission. Most bacteriophage genes were dispensable, and extensive mosaicism was seen. Surprisingly, virulence genes were tightly associated with specific phage families. This data suggests S. aureus bacteriophage evolve rapidly, and the horizontal gene transfer (HGT) of virulence genes encoded by bacteriophage is restricted by bacteriophage family and the lineage of the host bacterium, delaying the evolution of fully resistant and virulent strains.
Introduction
Staphylococcus aureus is a frequent colonizer of humans and mammals and a prevalent cause of skin and hospital associated infections. Methicillin resistant S. aureus (MRSA) is an opportunistic and highly adaptive pathogen that is a major cause of hospital acquired (HA) infections and is emerging to be a common cause of community acquired (CA) and livestock acquired (LA) infections. The emergence of MRSA is a major global concern because resistance to every antibiotic currently available is reported, and therefore the number of antibiotics that are effective in control and treatment of infections is limited (Gould, 2005; Chambers and DeLeo, 2009; Jensen and Lyon, 2009). Of further concern is the emergence of fully vancomycin resistant S. aureus (VRSA), reducing our options to prevent and treat severe MRSA infections (Weigel et al., 2003).
The adaptation and evolution of these S. aureus is largely due to the acquisition of mobile genetic elements (MGEs) that carry virulence and resistance genes. Such MGEs include bacteriophage, staphylococcal cassette chromosomes (SCCs), plasmids, S. aureus pathogenicity islands (SaPIs), and transposons that can move between bacteria by horizontal gene transfer (HGT) mechanisms (Lindsay, 2010; Malachowa and DeLeo, 2010). MRSA clones have emerged through independent acquisitions of SCCmec elements harboring mecA genes by different S. aureus lineages in different countries, whilst fully VRSA have emerged though acquisition of transposons harboring the vanA gene (Deurenberg and Stobberingh, 2008). The immune evasion cluster (IEC) locus encodes human-specific immune evasion proteins (Chp, Sak, and Scn) that contribute to adaptation to the human host and are carried on bacteriophage integrated into the chromosome (known as a prophage; van Wamel et al., 2006). The key toxins Panton–Valentine leukotoxin (PVL, encoded by lukFS-PV genes) and Staphylococcal enterotoxin A (sea) are also found on an integrated prophage (Betley and Mekalanos, 1985; Kaneko et al., 1998). The dissemination of MGEs through S. aureus populations is concerning as they can alter the pathogenicity, resistance, and host range of bacteria generating strains that cause novel clinical challenges.
Horizontal transfer of DNA occurs in bacteria by conjugation, transduction, and/or transformation. In S. aureus transformation does not occur efficiently, and conjugative plasmids and transposons do not have a wide distribution. Bacteriophage transduction is therefore likely to be the most important HGT mechanism for S. aureus. Some bacteriophage can be hitchhiked by SaPIs for transfer (Lindsay et al., 1998), whilst other bacteriophage called generalized transducing bacteriophage, are able to package host bacterial DNA and deliver it to other bacteria. Bacteriophage therefore increase the genome plasticity of S. aureus, and are thought to facilitate the adaptation of the pathogen to different conditions during infection (Goerke et al., 2006a,b, 2009). S. aureus bacteriophage belonging to the Siphoviridae family, can be classified into families (φ1–φ8) based on the integrase gene (int) sequence they possess and the integration site (Lindsay and Holden, 2004).
The major barriers of HGT in bacteria are restriction–modification (RM) systems. S. aureus possess two RM systems; a type III RM system that prevents uptake of DNA originating from different bacterial species (Corvaglia et al., 2010) and a type I RM system that prevents uptake of DNA originating from other species but also between S. aureus lineages (Waldron and Lindsay, 2006). Therefore, each S. aureus lineage has a unique type I RM system that causes lineages to evolve independently, as well as possessing a unique but highly conserved combination of surface genes, secreted genes, and their regulators (Witney et al., 2005; McCarthy and Lindsay, 2010). Other barriers of HGT mechanisms have been described, but are less well characterized in S. aureus (Dempsey et al., 2005; Thomas and Nielsen, 2005).
Our overall aim was to investigate the carriage of and genome variation of bacteriophage in order to better understand the horizontal and vertical transmission dynamics in S. aureus. In this study we (i) characterize carriage of prophage amongst the first 79 sequenced S. aureus genomes and investigate the amount of variation in prophage genomes (ii) investigate the evolutionary relationship of bacteriophage originating from different S. aureus lineage backgrounds, and (iii) extend previous microarray analysis to investigate lineage distributions of bacteriophage and virulence genes in 254 S. aureus isolates.
Materials and Methods
Bacteriophage Classification
Bacteriophage are classified into families based on their integration (int) gene which also defines the site of prophage integration in the S. aureus chromosome (Lindsay and Holden, 2006). Eight different families are prevalent in S. aureus and their key features are described in Table 1, including alternative nomenclatures. Standard nomenclature includes the phage family followed by the bacterial host name; for example, φ3 (MRSA252) is the φ3 bacteriophage originating from the sequenced MRSA252 genome (Lindsay and Holden, 2006).
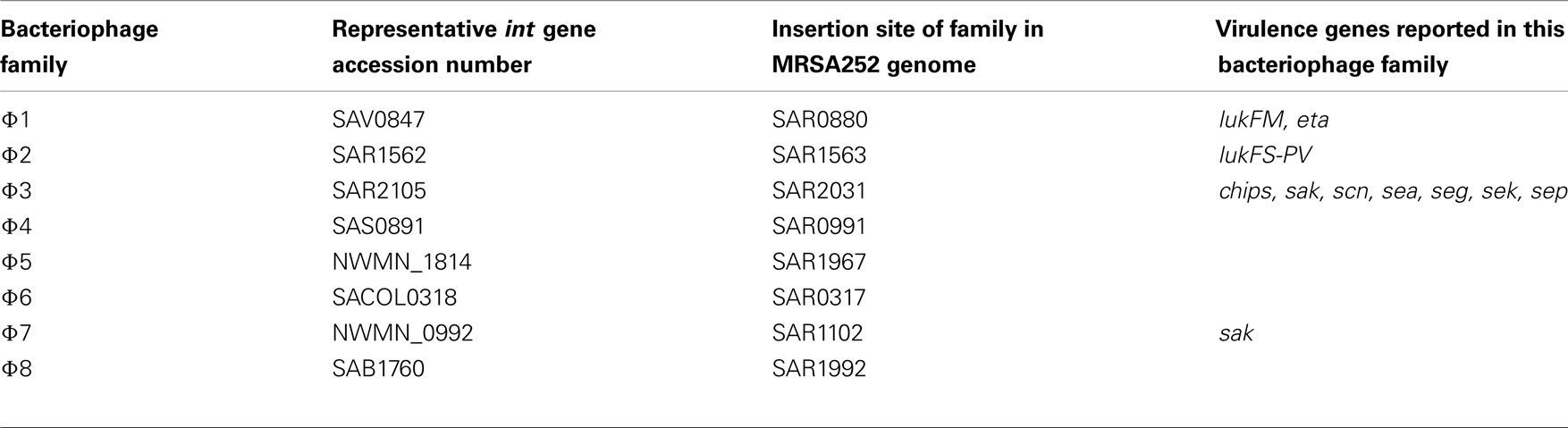
Table 1. Classification of S. aureu s bacteriophage.
Staphylococcus aureus Genomes
Sequence data is currently available for the genomes of 74 Staphylococcus aureus isolates on the GenBank database1. In addition, genome sequences from an another four S. isolates (EMRSA15, LGA251, BB155, MSHR 1132) were made available prior to publication from Matt Holden at The Wellcome Trust Sanger centre, UK2. The name and lineage distribution of bacteriophage in all genomes is shown in Table 2. The genomes represent 20 different lineages/clonal complexes (CCs), and 26 different sequence types (STs). Only 28 genomes have been fully sequenced, closed and annotated, whilst the remainder are found in contigs.

Table 2. Distribution of prophage and virulence genes in sequenced S. aureu s genomes.
Isolation of Bacteriophage Genomes from S. aureus Genomes
Carriage of bacteriophage in sequenced genomes was identified using a BLAST search of each bacteriophage integrase (int) gene and the flanking regions of insertion sites (Table 1). The Artemis Comparison Tool (ACT) was used to compare all full-length S. aureus genome sequences against the MRSA252 sequence in order to identify φ2 and φ3 prophage genomes and their coding domain sequences (CDS; Carver et al., 2005). For S. aureus genomes that were not completed, contigs that matched φ2 or φ3 bacteriophage were detected by BLAST analysis of the full φ2 (MRSA252) prophage sequence and φ3 (MRSA252) prophage sequence against each genome respectively. Matching contigs were then aligned against the MRSA252 genome in ACT and all CDS in φ2 or φ3 genome were identified. It should be noted that some bacteriophage sequences mapped across more than one contig and therefore such bacteriophage genomes may not be complete. Genomes where full-length φ2 or φ3 sequence mapped over more than three contigs were not been included in analysis. In total, 31 φ2 prophage (10 of which mapped across two contigs) and 48 φ3 (17 of which mapped across two contigs) prophage sequences were isolated from S. aureus genome sequences for comparative analysis. All ORFs from each prophage sequence were isolated.
Phylogenetic Analysis
Alignments of all φ2 and all φ3 prophage genomes were generated separately. Firstly, we generated separate pools of φ2 genes and φ3 genes using a strategy we have previously reported for microarray design (Witney et al., 2005). Briefly, one prophage genome was chosen as a reference and all of its genes were deposited in a gene pool. A second prophage genome was then compared against the gene pool and all genes that have a relatively poor BLAST hit score against previous genes were added to the gene pool. The process was repeated for all prophage genomes of a family. We next used BLAST analysis to generate a binary code matrix that reported presence and absence of each unique gene in each prophage genome for φ2 and φ3 bacteriophage families separately. Finally, phylogenetic analysis was performed using MrBayes v3.1.1 (Huelsenbeck and Ronquist, 2001). Analyses were run for 30,000,000 iterations (sampled every 1000 iterations) and convergence of Monte-Carlo Markov chain (MCMC) was assessed by the SD between chains falling below 0.05. The first 25% of sampled trees were discarded so that parameter estimates were only drawn from data generated after chain convergence. Phylogenetic trees were drawn and edited in FigTree v1.3.13.
Microarray Analysis
A total of 254 human and animal S. aureus isolates of UK origin were analyzed using the seven-strain S. aureus microarray (SAM-7; Witney et al., 2005). One hundred and sixty-one human carriage and invasive isolates have been previously described and represent the major dominant lineages of S. aureus from the Oxford UK community in 1999 (Feil et al., 2003; Lindsay et al., 2006), and an additional 37 isolates represent MRSA from hospitals in London, UK (Edgeworth et al., 2007). The 56 animal isolates have previously been described and originate from cows (n = 37), horses (n = 13), sheep (n = 2), goats (n = 2), and a camel (n = 1) in the UK (Sung et al., 2008).
The microarray assays were performed as previously described (Lindsay et al., 2006). Briefly, DNA was extracted using genomic-tip 100/G columns (Qiagen) or Bacterial Genomic Prep (Edge Biosystems), and 4 μg of test strain was labeled with Cy3 dye using DNA polymerase I large fragment (Klenow; Invitrogen), and 4 μg of reference (MRSA252) DNA was labeled using Cy5 dye. The two samples were pooled and hybridized to a S. aureus microarray overnight, prior to washing and scanning (Lindsay et al., 2006). Affymetrix 428 scanner was used to scan the microarrays and images were converted to raw data by BlueFuse for Microarrays 2.0 (BlueGnome, Cambridge, UK). All data analysis was performed in GeneSpring 7.0 (Agilent). Raw data from the previous experiments (Lindsay et al., 2006; Edgeworth et al., 2007; Sung et al., 2008) were normalized as a single experiment in GeneSpring using locally weighted scatterplot smoothing (LOWESS). The data we use here is deposited in BμG@Sbase (accession number: E-BUGS-62 and E-BUGS-34) and also ArrayExpress (accession number: E-BUGS-62 and E-BUGS-34). Condition tree clustering using the Spearman correlation was performed as a function in GeneSpring, according to Lindsay et al. (2006).
Results
Distribution of Prophage Families in Sequenced S. aureus Genomes
All eight bacteriophage int genes were identified in at least one sequenced S. aureus genome (Table 2). The frequency of carriage of bacteriophage families in 74 sequenced genomes varied considerably; φ1 (24% carriage), φ2 (53%), φ3 (82%), φ4 (6%), φ5 (22%), φ6 (18%), φ7 (13%), and φ8 (3%).
There was variation in the number of bacteriophage carried in a single isolate; most isolates carried between one and three prophage, but one isolate did not contain a prophage (TCH130) whilst others carried up to four (A6224, A8796, CGS03, CF-Marseille, JH1, JH9, and Newman). There was no association between number of bacteriophage carried and S. aureus lineage. No more than one copy of each prophage family was found in each genome sequence.
Certain bacteriophage families were prevalent in particular S. aureus lineages, for example φ1 bacteriophage were prevalent in CC5 (15/22 isolates) genomes, whilst φ2 bacteriophage were more common amongst CC1 (3/4 isolates), CC8 (8/12 isolates), and CC30 (14/19 isolates) lineages.
Clustering of Bacteriophage Populations
We assessed the relationship of 31 φ2 prophage and 48 φ3 prophage using Bayesian phylogenetic analyses of 221 and unique 324 genes respectively. Clusters of related prophage appear in both the φ2 and φ3 phylogenies, and Bayesian posterior probabilities (BPP) were consistently high (BPP ≥ 0.75; Figures 1A and 2A), meaning that the phylogeny of the tree and relationships of prophage are well supported. We note that we use the phylogenies to make conclusions on the relationship between prophage, and not on the temporal evolution of bacteriophage.
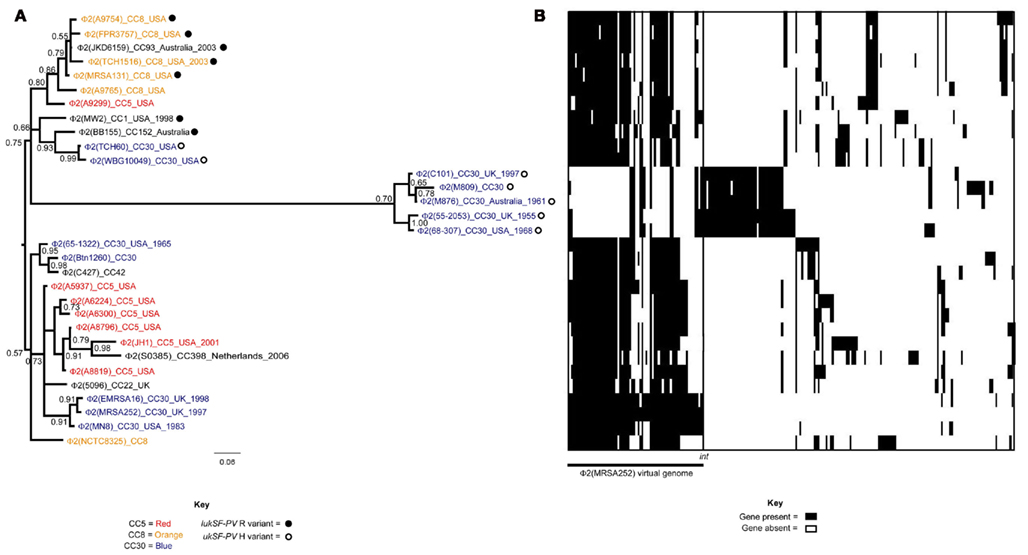
Figure 1. Evolutionary relationships of φ2 S. aureus prophage. (A) Phylogenetic analysis of 31 φ2 prophage is shown. Each prophage is named by the family it belongs to φ2, and then the name of the sequenced S. aureus genome it originates from (in parentheses). Additional information is giving on the clonal complex (CC) of the S. aureus isolate, as well as the country of origin and year of isolation if data was available. Branches are colored by CC lineage if more than one prophage originates from the same CC. The distribution of the lukSF-PV R and H variants is additionally mapped onto the phylogeny. (B) Presence/absence of 221 φ2 genes is shown for each of the φ2 prophage. Each horizontal line represents the genome of the adjacent φ2 prophage in the phylogeny. Each vertical line represents a φ2 gene. The color in the middle depicts if the gene is present (black) or absent (white). The genes are organized into virtual genomes, and the first genome shown in φ2 (MRSA252).
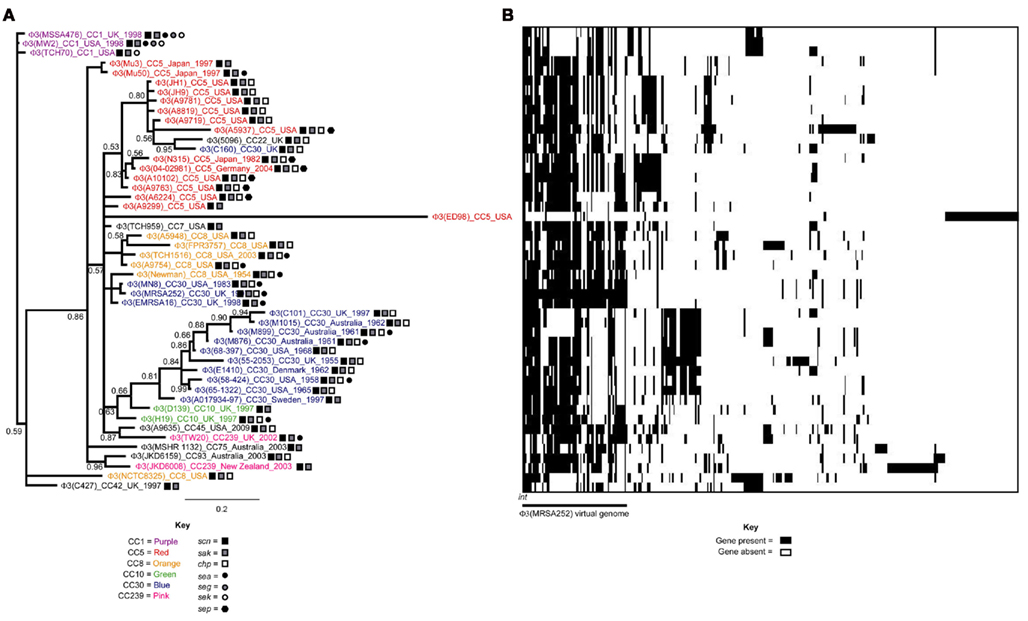
Figure 2. Evolutionary relationships of φ3 S. aureus prophage. (A) Phylogenetic analysis of 48 φ2 prophage is shown. Each prophage is named by the family it belongs to φ3, and then the name of the sequenced S. aureus genome it originates from (in parentheses). Additional information is giving on the clonal complex (CC) of the S. aureus isolate, as well as the country of origin and year of isolation if data was available. Branches are colored by CC lineage if more than one prophage originates from the same CC. The distribution of the virulence genes chp, sak, scn, sea, seg, sek, and sep is additionally mapped onto the phylogeny. (B) Presence/absence of 324 φ3 genes is shown for each of the φ3 prophage. Each horizontal line represents the genome of the adjacent φ3 prophage in the phylogeny. Each vertical line represents a φ3 gene. The color in the middle depicts if the gene is present (black) or absent (white). The genes are organized into virtual genomes, and the first genome shown in φ3 (MRSA252).
For both the φ2 and φ3 phylogenies prophage generally cluster according to lineage. For example, φ2 prophage from CC5 isolates cluster together (BPP = 0.94), as do φ2 prophage from CC8 isolates (BPP = 1.00; Figure 1A). This could be explained by vertical transmission, where the prophage are stable in the chromosome, pass to daughter cells on replication, and slowly evolve with the host genome.
However, there is also evidence of acquisition and loss of prophage. Firstly, the absence of prophage in some isolates of the same lineage suggests loss (Table 2). Secondly, some lineages have isolates carrying prophage from separate clusters of bacteriophage; such as the three separate clusters of φ2 found in CC5 (BPPs = 0.80, 0.83, and 0.86; Figure 2A). These clusters are typically composed of bacteriophage originating from S. aureus isolates in the same geographic region and/or time period. For example, the three distinct clusters of φ3 prophage found in CC5 isolates are from (i) Japan (BPP = 0.86) (ii) USA (BPP = 0.80) and (iii) USA/Germany/Japan (BPP = 0.83; Figure 2A). This could be explained by vertical transmission or horizontal transmission with exchange occurring between isolates in temporal and geographical contact. However, an interesting case are CC5 isolates N315 and Mu50, isolated from the same hospital at the same time, and carrying unique φ3 prophage, again arguing for HGT. Thirdly, we found cases where prophage from different S. aureus lineages clustered together; arguing that HGT of bacteriophage between lineages does occur but is infrequent. One such example is the close relationship of φ2 (JKD6159) from CC93 with φ2 bacteriophage originating from the CC8 lineage (Figure 1A). Likewise, φ3 (5096) of MRSA CC22 is closely related to φ3 (C160) of CC30; both S. aureus isolates originate from the UK (Figure 2A).
Diversification of Bacteriophage Genomes
The distribution of known virulence genes in prophage originating from sequenced genomes is shown (Table 2). In all cases, virulence factors were found strongly associated with only one family of prophage. Notably, PVL was found only on φ2 prophage, while IEC (chp, sak, and scn) and enterotoxin genes were only found on φ3 prophage. There are few or no virulence genes on φ1, φ4, φ5, φ6, φ7, or φ8 prophage, possibly because the identity and function of putative secreted proteins are yet to be characterized.
There was a good distribution between the clustering of prophage and the presence of virulence genes (Figures 1A and 2A). The PVL genes lukSF-PV were present in 14 of the 31 φ2 prophage included in our phylogenetic analysis, and these prophage clustered into two well-supported clades (BPP = 0.70 and 0.75; Figure 1A). This suggests that PVL-carrying φ2 bacteriophage from CA-MRSA are more closely related to each other than to other φ2 bacteriophage. Two sequence variants of PVL have been previously described, the H variant and the R variant (O’Hara et al., 2008); however the PVL-carrying φ2 prophage did not cluster into these separate clusters (Figure 1A). This suggests exchange of lukSF-PV variants between prophage clusters. There was also a good association between the clustering of the φ3 bacteriophage and the distribution of virulence factor genes (Figure 1B). For example, 4/5 bacteriophage with the virulence gene profile scn, sak, chp and sep clustered together (BPP = 0.83). However, there was also evidence that closely related bacteriophage had differing combinations of virulence genes, such as the absence of lukSF-PV in φ2 (A9299) and φ2 (A9765), and the acquisition of the staphylococcal enterotoxin A (sea) gene by φ3 (Mu50) in comparison to φ3 (Mu3) (Figures 1A and 2A). This suggests that virulence genes can be frequently transferred horizontally between bacteriophage, as well as lost.
The distribution of φ2 genes and φ3 genes amongst φ2 and φ3 bacteriophage are shown in Figures 1B and 2B, respectively. Closely related prophage possessed similar combinations of genes. For example, the φ3 prophage from CC1 isolates formed a single clade and their genomes were highly homologous (Figure 2B). In contrast, different clades were more extensively diversified and often differed by the presence/absence of single genes and/or runs of adjacent genes. For example, the two clades of PVL-carrying φ2 prophage possessed different combinations of genes (Figure 1B). This data supports the finding that (i) bacteriophage have mosaic genomes, and (ii) that bacteriophage diversification is driven by recombination that causes frequent shuffling of genes.
If virulence genes are strongly associated with specific families this suggests that recombination or exchange of genes does not occur frequently between bacteriophage from different families. We assessed the distribution of φ2 genes amongst φ3 genomes, and vice-versa, and found that 139 genes of a total of 545 genes were common to both the φ2 gene pool and φ3 gene pool (Data not shown). This suggests that genes are exchanged between different bacteriophage families, but that this may be less frequent than exchange within families.
Bacteriophage Dynamics in Localized Populations
The sequenced strains represent selected isolates from around the world, typically virulent and/or multi-drug resistant, and are not representative of locally evolving S. aureus populations. We therefore looked for variation in bacteriophage carriage and genomic diversity in a population of 161 carriage and invasive S. aureus isolates from the same region of Oxford and the same time period. We used the seven-strain whole genome sequence microarray which included the genes for five phage families. Overall, the carriage of bacteriophage genes in this population showed similar dynamics to the sequenced strain collection. As we extended this study to include hospital MRSA and animal S. aureus, the same dynamics were also evident.
The distribution of bacteriophage int genes was highly variable amongst S. aureus isolates suggesting that bacteriophage loss and movement is frequent (Figure 3). Carriage of some bacteriophage families were more frequent in some CC lineages than others. φ1 were associated with lineages CC51 and CC151. φ3 were associated with multiple lineages (CC5, CC6, CC8, CC12, CC22, CC25, CC30, CC45, CC97, and CC239). φ4/6 were associated with lineages CC151, CC239, and CC771. φ5 were associated with the CC771 lineage. Interestingly, φ2 were not associated to any lineage, but were frequent amongst CC30 isolates. φ7 and φ8 were not represented on SAM-7 as they do not appear in the first seven sequenced S. aureus genomes. In a few instances bacteriophage carriage was completely associated to lineage; for example φ3 bacteriophage is carried in all CC239 and CC25 isolates (Figure 1).
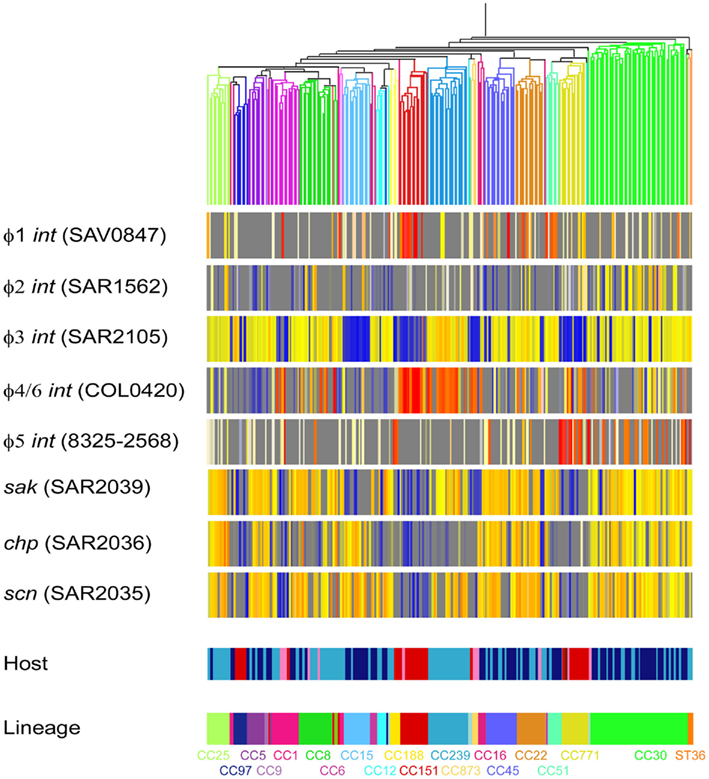
Figure 3. Microarray data from 254 S. aureus (198 human isolates and 56 animal isolates). Isolates are clustered by Spearman correlation using 728 CV gene, as reported in Lindsay et al. (2006). Isolates of S. aureus are represented by vertical lines, and further information about the origin of each isolate is shown at the bottom of the figure (Host and lineage). Human isolates are colored light blue (invasive) and dark blue (carriage). Animal isolates are colored red (cow), pink (horse), brown (sheep), and light pink (camel). DNA probes that represent bacteriophage int genes and immune evasion cluster (IEC) genes are represented by horizontal lines. The color in the middle depicts presence of absence of the gene in an isolate; gene presence is shown as red/yellow and gene absence is shown as blue/gray.
The virulence genes of the IEC (chp, sak, and scn) appeared to be carried by φ3 bacteriophage. φ3 can carry different IEC types, or different combinations of chp, sak, and scn. For example, CC30 isolates carry chp, sak, and scn, whilst CC239 isolates carry sak and scn but not chp (Figure 1). This further suggests that HGT of bacteriophage is infrequent between S. aureus lineages, as we would expect to see multiple IEC profiles within a lineage if HGT between lineages was frequent.
Overall, the data supported the findings that (i) prophage are highly variable, (ii) particular prophage families are associated with particular lineages, (iii) there is evidence of vertical as well as frequent horizontal transfer of phage between S. aureus (iv) HGT occurs within lineages at higher frequency than between lineages, (v) frequent recombination or mosaicism occurs within prophage families, (vi) virulence genes are strongly associated with particular phage families and therefore to particular S. aureus lineages.
Discussion
Staphylococcus aureus prophage carry important genes that impact on the ability to colonize the human nose, and to cause disease. It is interesting to consider why the bacterium has evolved to carry these genes on a MGE and not to systematically incorporate them into the stable regions of the chromosome. Presumably there is some cost to the bacterium to keep these elements, yet they are widely distributed and appear to move frequently. Prophage induction and HGT is triggered by stress including antibiotic exposure and oxygen radicals, both expected to be seen by S. aureus during infection (Maiques et al., 2006). It is therefore feasible that prophage contribute to the flexibility of the S. aureus genome, allowing isolates to rapidly adapt to new niches and environmental threats. Indeed, there are multiple reports of bacteriophage moving into and out of S. aureus isolates during colonization or infection (Moore and Lindsay, 2001; Goerke et al., 2004, 2006a,b; McCarthy et al., 2012), and prophage can be induced from clinical S. aureus isolates indicating that integrated bacteriophage remain functional (Goerke et al., 2004; Wirtz et al., 2009).
A prophage that is highly prevalent in a S. aureus population must be transmitted vertically, or must move frequently and be selected for. φ3 is the major prophage in human S. aureus isolates (Sung et al., 2008), and is selected for in human hosts as it encodes human-specific immune evasion proteins Chp, Sak, and Scn (van Wamel et al., 2006). This study shows that φ3 can be lost from the S. aureus genome, but has also been acquired on multiple occasions in the sequenced S. aureus isolates. Interestingly, φ3 has rearranged its genome frequently, can dispose of nearly all of its genes, but continues to favor the carriage of at least two of the IEC genes. Our data therefore argues for both vertical and frequent horizontal transmission of this bacteriophage. Horizontal transmission of MGEs is limited by the SauI RM system and it is anticipated that different S. aureus lineages carry bacteriophage at different frequencies (Waldron and Lindsay, 2006). The data presented here supports this conclusion.
Other prophage (φ4, φ5, φ6, φ7, and φ8) were less prevalent amongst both the sequenced S. aureus isolates and the isolates included in our microarray analyses. This perhaps suggests their genes are not as important for survival in the major environmental reservoirs, such as host species. Yet, they transfer at high frequency and are maintained in diverse populations, suggesting efficient transfer mechanisms and selection under certain niche environments. Identification of such niche–environment virulence genes could provide novel therapeutic targets to limit and control S. aureus populations. It is interesting to consider that PVL, carried only on φ2 prophage, is found at very high frequency in CA-MRSA populations (Vandenesch et al., 2003). This could argue that PVL φ2 prophage are transferred vertically. However, our data suggests substantial variation in φ2 prophage encoding this toxin, suggesting it is transferred and is selected for in CA-MRSA disease causing isolates. The missing link is the data regarding PVL prevalence in CA-MRSA colonizing isolates; if prevalent in colonizing isolates it may play a role in colonization, and if not, it may play an important role in invasive disease. More data from Asian CA-MRSA prophage will also be valuable in determining the role of this toxin.
The strong association of particular virulence factors with particular bacteriophage families is difficult to explain. Our data supports previous studies suggesting that Staphylococcal bacteriophage genomes are highly flexible, mosaic, and exchange DNA (Kwan et al., 2005; Goerke et al., 2009; Belcaid et al., 2010). Our data suggests exchange of genes occurs more often within than between bacteriophage families. Since most strains carry multiple prophage but only one copy from each family, it might be assumed that DNA was exchanged across bacteriophage family boundaries at higher frequency but that does not appear to be the case. It is interesting to question how this genome mosaicism has been generated. It has previously been proposed to be the result of frequent random recombination events followed by selection of the fittest bacteriophage (Hendrix, 2002). Virulence genes may additionally be associated to bacteriophage families if (i) these proteins play a role in the bacteriophage cycle, or (ii) the carriage of a virulence gene compensates the negative affect that a bacteriophage may have on expression of chromosomally encoded virulence genes, for example φ3 bacteriophage integrated into the β-hemolysin (hlb) gene.
Because virulence genes are confined to bacteriophage families, and the distribution of bacteriophage appear to be somewhat restrained by lineage, particular virulence genes are associated with particular lineages. This may delay the spread of virulence genes on bacteriophage from one CC lineage to all members of the S. aureus population. However, the evolution of new types of MRSA in recent years reminds us that particular lineages are gradually accumulating a wider range of MGEs, and individual isolates can exchange these MGEs within the lineage population. Understanding how this evolution is constrained and recognizing evolutionary leaps can enable the rapid response to emerging clones that will be required to control multi-resistant and virulent isolates in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Jason Hinds, Kate Gould, and Denise Waldron from the Bacterial Microarray Group at St George’s (BμG@S; http://www.bugs.sgul.ac.uk), funded by The Wellcome Trust, for assistance with all microarray studies. We would like to thank Gwenan Knight for assistance with figure processing. We would like to thank Matt Holden for early access to a number of sequenced genomes. This work was supported by the PILGRIM FP7 Grant from the EU.
Footnotes
References
Belcaid, M., Bergeron, A., and Poisson, G. (2010). Mosaic graphs and comparative genomics in phage communities. J. Comput. Biol. 17, 1315–1326.
Betley, M. J., and Mekalanos, J. J. (1985). Staphylococcal enterotoxin A is encoded by phage. Science 229, 185–187.
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M. A., Barrell, B. G., and Parkhill, J. (2005). ACT: the Artemis Comparison Tool. Bioinformatics 21, 3422–3423.
Chambers, H. F., and DeLeo, F. R. (2009). Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7, 629–641.
Corvaglia, A. R., François, P., Hernandez, D., Perron, K., Linder, P., and Schrenzel, J. (2010). A type III-like restriction endonuclease functions as a major barrier to horizontal gene transfer in clinical Staphylococcus aureus strains. Proc. Natl. Acad. Sci. U.S.A. 107, 11954–11958.
Dempsey, R. M., Carroll, D., Kong, H., Higgins, L., Keane, C. T., and Coleman, D. C. (2005). Sau42I, a BcgI-like restriction-modification system encoded by the Staphylococcus aureus quadruple-converting phage Phi42. Microbiology 151, 1301–1311.
Deurenberg, R. H., and Stobberingh, E. E. (2008). The evolution of Staphylococcus aureus. Infect. Genet. Evol. 8, 747–763.
Edgeworth, J. D., Yadegarfar, G., Pathak, S., Batra, R., Cockfield, J. D., Wyncoll, D., Beale, R., and Lindsay, J. A. (2007). An outbreak in an intensive care unit of a strain of methicillin-resistant Staphylococcus aureus sequence type 239 associated with an increased rate of vascular access device-related bacteremia. Clin. Infect. Dis. 44, 493–501.
Feil, E. J., Cooper, J. E., Grundmann, H., Robinson, D. A., Enright, M. C., Berendt, T., Peacock, S. J., Smith, J. M., Murphy, M., Spratt, B. G., Moore, C. E., and Day, N. P. (2003). How clonal is Staphylococcus aureus? J. Bacteriol. 185, 3307–3316.
Goerke, C., Koller, J., and Wolz, C. (2006a). Ciprofloxacin and trimethoprim cause phage induction and virulence modulation in Staphylococcus aureus. Antimicrob. Agents Chemother. 50, 171–177.
Goerke, C., Wirtz, C., Flückiger, U., and Wolz, C. (2006b). Extensive phage dynamics in Staphylococcus aureus contributes to adaptation to the human host during infection. Mol. Microbiol. 61, 1673–1685.
Goerke, C., Matias y Papenberg, S., Dasbach, S., Dietz, K., Ziebach, R., Kahl, B. C., and Wolz, C. (2004). Increased frequency of genomic alterations in Staphylococcus aureus during chronic infection is in part due to phage mobilization. J. Infect. Dis. 189, 724–734.
Goerke, C., Pantucek, R., Holtfreter, S., Schulte, B., Zink, M., Grumann, D., Bröker, B. M., Doskar, J., and Wolz, C. (2009). Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 191, 3462–3468.
Gould, I. M. (2005). The clinical significance of methicillin-resistant Staphylococcus aureus. J. Hosp. Infect. 61, 277–282.
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755.
Jensen, S. O., and Lyon, B. R. (2009). Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 4, 565–582.
Kaneko, J., Kimura, T., Narita, S., Tomita, T., and Kamio, Y. (1998). Complete nucleotide sequence and molecular characterization of the temperate staphylococcal bacteriophage phiPVL carrying Panton-Valentine leukocidin genes. Gene 215, 57–67.
Kwan, T., Liu, J., DuBow, M., Gros, P., and Pelletier, J. (2005). The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. U.S.A. 102, 5174–5179.
Lindsay, J. A. (2010). Genomic variation and evolution of Staphylococcus aureus. Int. J. Med. Microbiol. 300, 98–103.
Lindsay, J. A., and Holden, M. T. (2004). Staphylococcus aureus: superbug, super genome? Trends Microbiol. 12, 378–385.
Lindsay, J. A., and Holden, M. T. (2006). Understanding the rise of the superbug: investigation of the evolution and genomic variation of Staphylococcus aureus. Funct. Integr. Genomics 6, 186–201.
Lindsay, J. A., Moore, C. E., Day, N. P., Peacock, S. J., Witney, A. A., Stabler, R. A., Husain, S. E., Butcher, P. D., and Hinds, J. (2006). Microarrays reveal that each of the ten dominant lineages of Staphylococcus aureus has a unique combination of surface-associated and regulatory genes. J. Bacteriol. 188, 669–676.
Lindsay, J. A., Ruzin, A., Ross, H. F., Kurepina, N., and Novick, R. P. (1998). The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 29, 527–543.
Maiques, E., Úbeda, C., Campoy, S., Salvador, N., Lasa, I., Novick, R. P., Barbé, J., and Penadés, J. R. (2006). β-Lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J. Bacteriol. 188, 2726–2729.
Malachowa, N., and DeLeo, F. R. (2010). Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 67, 3057–3071.
McCarthy, A. J., Breathnach, A. S., and Lindsay, J. A. (2012). Detection of mobile genetic element (MGE) variation between colonising and infecting hospital-associated (HA)-MRSA isolates. J. Clin. Microbiol. doi: 10.1128/JCM.05938-11
McCarthy, A. J., and Lindsay, J. A. (2010). Genetic variation in Staphylococcus aureus surface and immune evasion genes is lineage associated: implications for vaccine design and host-pathogen interactions. BMC Microbiol. 10, 173. doi:10.1186/1471-2180-10-173
Moore, P. C., and Lindsay, J. A. (2001). Genetic variation among hospital isolates of methicillin-sensitive Staphylococcus aureus: evidence for horizontal transfer of virulence genes. J. Clin. Microbiol. 39, 2760–2767.
O’Hara, F. P., Guex, N., Word, J. M., Miller, L. A., Becker, J. A., Walsh, S. L., Scangarella, N. E., West, J. M., Shawar, R. M., and Amrine-Madsen, H. (2008). A geographic variant of the Staphylococcus aureus Panton-Valentine leukocidin toxin and the origin of community-associated methicillin-resistant S. aureus USA300. J. Infect. Dis. 197, 187–194.
Sung, J. M., Lloyd, D. H., and Lindsay, J. A. (2008). Staphylococcus aureus host specificity: comparative genomics of human versus animal isolates by multi-strain microarray. Microbiology 154, 1949–1959.
Thomas, C. M., and Nielsen, K. M. (2005). Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721.
van Wamel, W. J., Rooijakkers, S. H., Ruyken, M., van Kessel, K. P., and van Strijp, J. A. (2006). The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315.
Vandenesch, F., Naimi, T., Enright, M. C., Lina, G., Nimmo, G. R., Heffernan, H., Liassine, N., Bes, M., Greenland, T., Reverdy, M. E., and Etienne, J. (2003). Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerging Infect. Dis. 9, 978–984.
Waldron, D. E., and Lindsay, J. A. (2006). Sau1: a novel lineage-specific type I restriction-modification system that blocks horizontal gene transfer into Staphylococcus aureus and between S. aureus isolates of different lineages. J. Bacteriol. 188, 5578–5585.
Weigel, L. M., Clewell, D. B., Gill, S. R., Clark, N. C., McDougal, L. K., Flannagan, S. E., Kolonay, J. F., Shetty, J., Killgore, G. E., and Tenover, F. C. (2003). Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302, 1569–1571.
Wirtz, C., Witte, W., Wolz, C., and Goerke, C. (2009). Transcription of the phage-encoded Panton-Valentine leukocidin of Staphylococcus aureus is dependent on the phage life-cycle and on the host background. Microbiology 155, 3491–3499.
Witney, A. A., Marsden, G. L., Holden, M. T., Stabler, R. A., Husain, S. E., Vass, J. K., Butcher, P. D., Hinds, J., and Lindsay, J. A. (2005). Design, validation, and application of a seven-strain Staphylococcus aureus PCR product microarray for comparative genomics. Appl. Environ. Microbiol. 71, 7504–7514.
Keywords: Staphylococcus aureus, MRSA, bacteriophage, mobile genetic element, evolution, horizontal gene transfer, transmission, PVL
Citation: McCarthy AJ, Witney AA and Lindsay JA (2012) Staphylococcus aureus temperate bacteriophage: carriage and horizontal gene transfer is lineage associated. Front. Cell. Inf. Microbio. 2:6. doi: 10.3389/fcimb.2012.00006
Received: 16 December 2011;
Paper pending published: 05 January 2012;
Accepted: 23 January 2012;
Published online: 08 February 2012.
Edited by:
Martin John McGavin, University of Western Ontario, CanadaReviewed by:
Indranil Biswas, University of Kansas, USAJose R. Penades, ConsejoSuperior de Investigaciones Científicas, Spain
Copyright: © 2012 McCarthy, Witney and Lindsay. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Alex J. McCarthy, Division of Clinical Sciences, Centre for Infection, St George’s University of London, Cranmer Terrace, London SW17 0RE, UK. e-mail: amccarth@sgul.ac.uk