HigB of Pseudomonas aeruginosa Enhances Killing of Phagocytes by Up-Regulating the Type III Secretion System in Ciprofloxacin Induced Persister Cells
 Mei Li1
Mei Li1  Yuqing Long1
Yuqing Long1  Ying Liu1
Ying Liu1  Yang Liu2
Yang Liu2  Ronghao Chen1
Ronghao Chen1  Jing Shi1
Jing Shi1  Lu Zhang1
Lu Zhang1  Yongxin Jin1
Yongxin Jin1  Liang Yang2,3
Liang Yang2,3  Fang Bai4
Fang Bai4  Shouguang Jin1,5*
Shouguang Jin1,5*  Zhihui Cheng1*
Zhihui Cheng1*  Weihui Wu1*
Weihui Wu1*- 1State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Molecular Microbiology and Technology of the Ministry of Education, Department of Microbiology, College of Life Sciences, Nankai University, Tianjin, China
- 2Singapore Centre for Environmental Life Sciences Engineering, Nanyang Technological University, Singapore, Singapore
- 3School of Biological Sciences, Division of Structural Biology and Biochemistry, Nanyang Technological University, Singapore, Singapore
- 4State Key Laboratory of Medicinal Chemical Biology and College of Pharmacy and Life Sciences, Nankai University, Tianjin, China
- 5Department of Molecular Genetics and Microbiology, College of Medicine, University of Florida, Gainesville, FL, USA
Bacterial persister cells are dormant and highly tolerant to lethal antibiotics, which are believed to be the major cause of recurring and chronic infections. Activation of toxins of bacterial toxin-antitoxin systems inhibits bacterial growth and plays an important role in persister formation. However, little is known about the overall gene expression profile upon toxin activation. More importantly, how the dormant bacterial persisters evade host immune clearance remains poorly understood. Here we demonstrate that a Pseudomonas aeruginosa toxin-antitoxin system HigB-HigA is required for the ciprofloxacin induced persister formation. Transcriptome analysis of a higA::Tn mutant revealed up regulation of type III secretion systems (T3SS) genes. Overexpression of HigB increased the expression of T3SS genes as well as bacterial cytotoxicity. We further demonstrate that wild type bacteria that survived ciprofloxacin treatment contain higher levels of T3SS proteins and display increased cytotoxicity to macrophage compared to vegetative bacterial cells. These results suggest that P. aeruginosa accumulates T3SS proteins during persister formation, which can protect the persister cells from host clearance by efficiently killing host immune cells.
Introduction
Bacterial persisters are rare cells in a bacterial population that are tolerant to lethal antibiotics. Formation of persisters has been observed in almost all bacterial species investigated (Lewis, 2010). Persistence as a phenotypic switch is pre-existing in bacterial populations, with the characteristic of dormancy or slow growth. Reinoculation of the persister cells results in a similar heterogeneous population in which a subpopulation is tolerant to antibiotics. Formation of persister cells is influenced by environmental stresses and growth phases (Balaban et al., 2004; Keren et al., 2004; Dörr et al., 2009, 2010; Bernier et al., 2013; Helaine et al., 2014).
Toxin–antitoxin (TA) systems, composed of a toxin and a cognate antitoxin, play important roles in persister formation (Kim et al., 2011; Germain et al., 2013; Maisonneuve et al., 2013; Helaine et al., 2014; Verstraeten et al., 2015). Toxins can inhibit bacterial protein synthesis, DNA replication, cell wall synthesis or depolarize membrane, resulting in slow growth or dormant persister cells (Page and Peti, 2016). Toxins can be activated by various stimulations. For example, environmental stresses, such as starvation, induce the synthesis of bacterial alarmones guanosine tetraphosphate (ppGpp) and guanosine pentaphosphate (pppGpp), which trigger the degradation of antitoxins by proteases, resulting in activation of toxins (Nguyen et al., 2011; Maisonneuve et al., 2013). In addition, fluoroquinolones and oxidative stresses can activate toxins and induce persister formation through SOS response (Dörr et al., 2009, 2010; Wu et al., 2012).
Bacterial persisters are believed to be responsible for recurrent and chronic infections, due to the failure of antibiotics to eradicate the bacterial pathogens (Lewis, 2007). However, the mechanism by which the rare dormant persister cells evade the killing by host phagocytes remains poorly understood. Although it is believed that persister cells embedded in biofilm are shielded from host phagocytes (Leid, 2009), whether free persister cells are capable of surviving the attack of immune cells is not known. Numerous studies have revealed roles of TA systems in the regulation of bacterial gene expression, including virulence factors (Bertram and Schuster, 2014; Wen et al., 2014). Therefore, studies on the functions of TA systems will facilitate the understanding of the physiology of persister cells as well as their survival strategies within the host environments.
Pseudomonas aeruginosa is an opportunistic pathogen that causes acute and chronic infections in human (Balasubramanian et al., 2013). In P. aeruginosa strain PA14, two potential toxin-antitixoin systems have been identified, namely RelE-RelB and HigB-HigA (Williams et al., 2011; Wood and Wood, 2016). It has been demonstrated that HigB is a RNase, which cleaves mRNAs (Hurley and Woychik, 2009; Schureck et al., 2015, 2016a,b; Wood and Wood, 2016). In this study, we demonstrate that the toxin HigB contributes to persister formation. RNA-seq results revealed up regulation of type III secretion system (T3SS) genes in a higA::Tn mutant. The T3SS is a needle-like apparatus conserved in Gram negative pathogenic bacteria, through which effector proteins are directly translocated into the host cells, altering cell signaling or causing cell death. In P. aeruginosa, the T3SS plays an essential role in bacterial pathogenesis by killing phagocytes (Hauser, 2009; Diaz and Hauser, 2010). Consistent with the T3SS gene expression pattern, the higA::Tn mutant displayed higher cytotoxicity than the wild type strain. As expected, overexpression of the HigB resulted in a similar phenotype. These results imply a high level of cytotoxicity of the persister cells. Indeed, compared to vegetative cells, cells that survived short term ciprofloxacin treatment displayed increased cytotoxicity, which depends on HigB mediated up regulation of the T3SS. To our knowledge, this is the first demonstration of a connection between the HigB-HigA system and the T3SS. Our results suggest that T3SS proteins get accumulated during the process of persister formation, enabling the bacterial persisters to survive host clearance by actively killing the host immune cells.
Results
HigA Negatively Regulates the higB-higA Operon
A recent study identified the open reading frame of HigB in PA14 and demonstrated its growth inhibitory function (Wood and Wood, 2016). In most type II TA systems, toxin and antitoxin genes form one operon and the antitoxin binds to and represses its own promoter (Wood and Wood, 2016). To test whether higB and higA are in the same operon, we designed a pair of primers annealing to the 5′ end of higB and 3′ end of higA coding region, respectively (Figure 1A), and performed RT-PCR. Total RNA was isolated from PA14 and a higA mutant from the PA14 transponson insertion mutant library (Liberati et al., 2006). A 384-bp PCR product was amplified using cDNA from the higA::Tn mutant (Figure 1B, lane 4), and the size was the same as that when genomic DNA was used as the template (Figure 1B, lane 2). Substantially less PCR product was obtained when cDNA from wild type PA14 was used as the template (Figure 1B, lane 3), suggesting a lower HigB mRNA level. To confirm the transcriptional level of higB and higA, we performed quantitative RT PCR with previously reported PA1769 and proC as internal controls for normalization (Savli et al., 2003; Son et al., 2007). Since HigB might cleave mRNAs and affect the expression of multiple genes, we included the 16S rRNA (PA0668.1) (Ruzin et al., 2007), which might not be a target of HigB, as another internal control. Similar mRNA fold of changes (within 1.5-fold difference) were observed when proC and the 16S rRNA were used as internal controls. Therefore, we used the 16S rRNA as the internal control in this study. At both exponential and stationary growth phases, the mRNA levels of higB and higA in the higA::Tn mutant were higher than those in wild type PA14 (Figures 1C,D). In addition, a previous microarray analysis has demonstrated an up regulation of higB in a higA mutant (Wood and Wood, 2016). In combination, these results suggest that higB and higA are in the same operon, which is negatively regulated by the HigA.
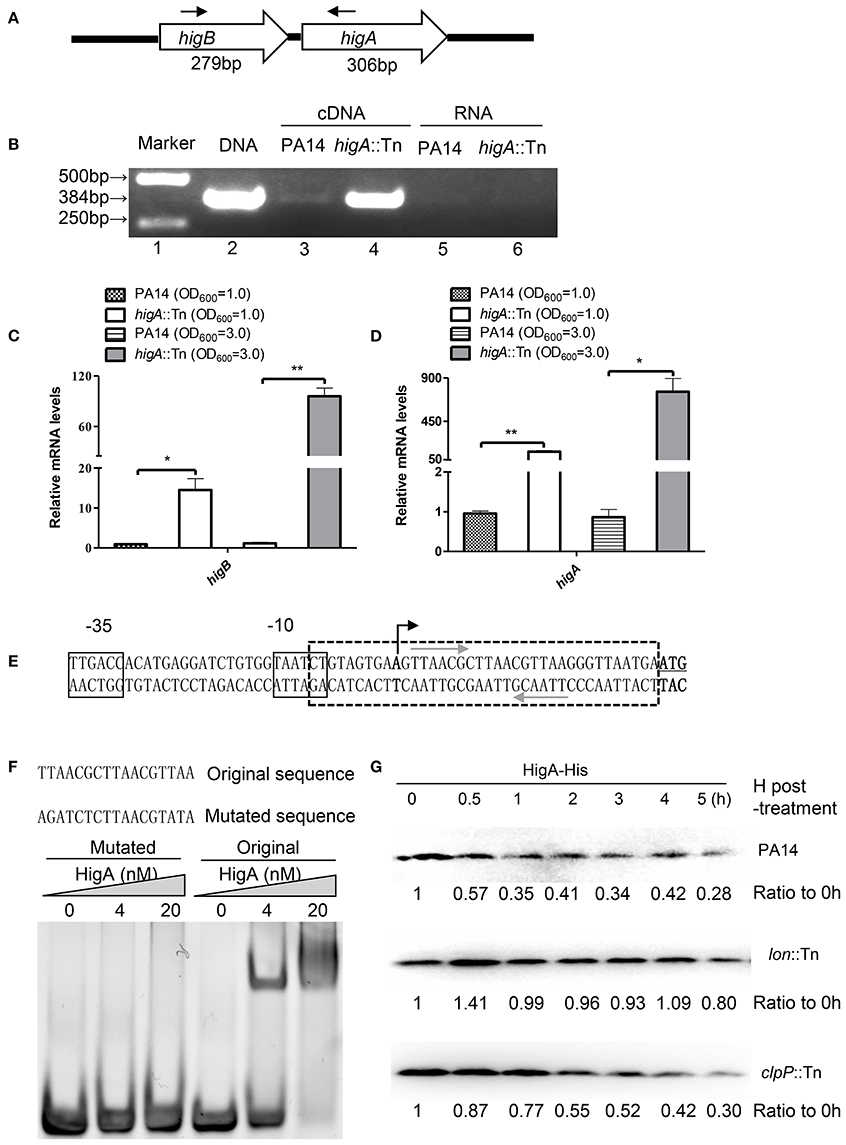
Figure 1. HigA negatively regulates the higB-higA operon. (A) Sketch map of the higB-higA operon. Arrows indicate the directions and locations of the primers for RT-PCR. (B) Total RNA was isolated from PA14 and the higA::Tn mutant. cDNA was synthesized and used as templates in PCR. RNAs were used in RT-PCR as negative controls. (C,D) The relative mRNA levels of higB and higA genes in PA14 and the higA::Tn mutant. Total RNA was isolated and the mRNA levels were determined by quantitative RT-PCR with the 16S rRNA as the internal control. Data represents the mean ± standard deviation from three independent experiments performed in triplicate. *p < 0.05; **p < 0.01 compared to wild type PA14 by Student's t-test. (E) Promoter region of the higB-higA operon. The predicted −10 and −35 elements of the promoter are boxed. The transcriptional start site is indicated by a black arrow, and the start codon of higB is underlined. The palindromic sequences of hypothetical HigA binding sites are indicated by gray arrows. (F) EMSA displaying binding of HigA to the higB-higA promoter. Purified HigA-His protein was incubated with the 38-bp DNA fragment indicated by the box with dashed lines in (E) or altered sequence. The mixtures were electrophoresed and observed by ethidium bromide staining. (G) Cleavage of HigA by the Lon protease. Wild type PA14, the clpP::Tn and lon::Tn mutants carrying pMMB67EH-higA-His were cultured in the presence of 1 mM IPTG for 1 h. Then 50 μg/ml spectinomycin was added to the medium. At indicated time points, the HigA-His levels were determined by Western blot analysis with an anti-His antibody. The relative density of each band was determined with Image J.
To examine whether HigA binds to the promoter of its own operon, we first determined the transcriptional start site by a 5′ RACE analysis. The start site was located at 29 bp upstream of the start codon for higB (Figure 1E). Of note, we found a palindromic sequence downstream of the −10 region (Figure 1E), which might be the binding site of HigA. Electrophoretic mobility shift assay (EMSA) revealed an interaction between the fragment and HigA, and mutation of the palindromic sequence abolished the interaction (Figure 1F). These results suggest that HigA directly binds to and represses the promoter of the higB-higA operon.
The Lon Protease Contributes to the Degradation of HigA
To identify which protease is involved in the degradation of HigA, a C-terminus His-tagged HigA (HigA-His) driven by a tac promoter was introduced into wild type PA14. After 60 min of culture in the presence of IPTG, spectinomycin was added to block protein synthesis, then the stability of HigA-His was monitored. In wild type PA14 and a clpP::Tn mutant, the HigA-His was gradually degraded. However, the protein was stable in a lon::Tn mutant (Figure 1G, Figure S1A), suggesting a role of the Lon protease in the degradation of HigA.
HigB-HigA Regulates Persister Formation in PA14
To test the role of HigB-HigA in persister formation, the higA::Tn mutant was examined for a time-dependent killing by ciprofloxacin. Compared to the wild type PA14, the higA::Tn mutant displayed 100-fold higher survival rate, which was restored to the wild type level by complementation with an intact higA gene (Figure 2A). However, a ΔhigB mutant displayed a similar survival rate as the wild type PA14 (Figure 2B), which we suspect might be due to redundant TA systems in P. aeruginosa. It has been demonstrated that sublethal level of ciprofloxacin induces persister formation (Dörr et al., 2009, 2010). Thus, we examined the role of HigB in ciprofloxacin induced persister formation as previously described (Dörr et al., 2009, 2010). Pre-exposure to 0.025 μg/ml (1/10 MIC) ciprofloxacin increased the survival rate of wild type PA14 by approximately 5-fold, suggesting an induction of persister formation (Figure 2B). However, deletion of higB or the higB-higA operon abolished such induction (Figure 2B). The expression of higB and higA was induced by the ciprofloxacin treatment (Figures 2C,D) and overexpression of HigB increased bacterial survival rate by approximately 1000-fold (Figure 2E). In addition, the minimal inhibitory concentration (MIC) of ciprofloxacin was not altered by the mutation of higA or overexpression of higB (data not shown). In combination, these results demonstrate that HigB contributes to persister formation.
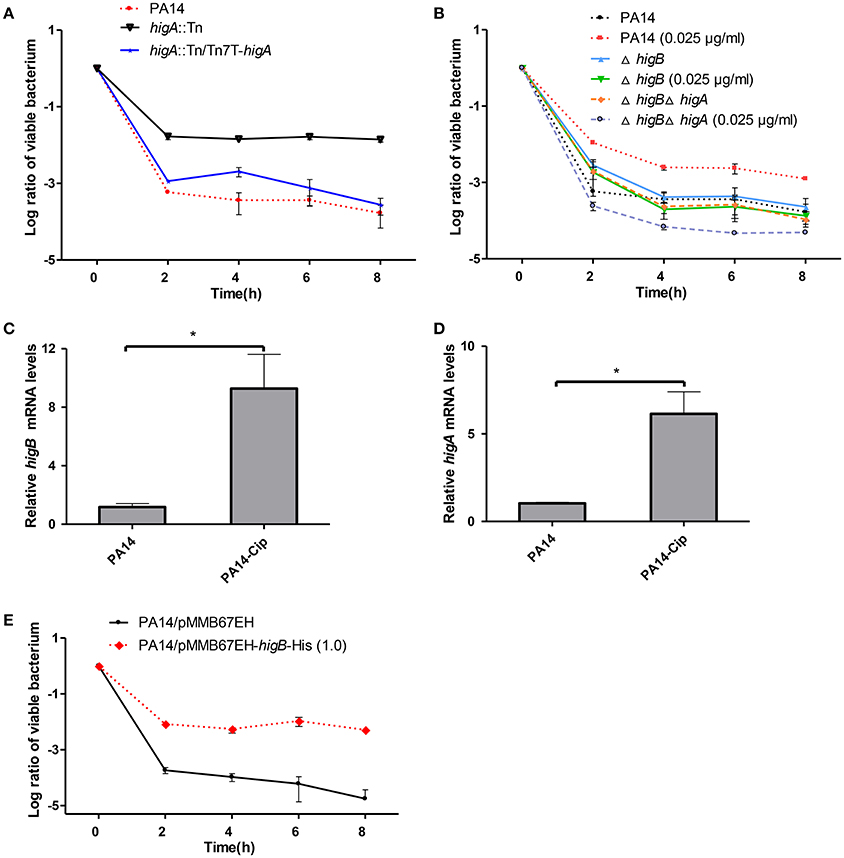
Figure 2. Role of HigA-HigB in persister formation. (A) Wild type PA14, a higA::Tn mutant and a complemented strain were treated with 0.25 μg/ml ciprofloxacin. At indicated time points the survival rates were determined by plating assay. (B) Wild type PA14, the ΔhigA and ΔhigAΔhigB mutants were cultured in the presence or absence of 0.025 μg/ml ciprofloxacin for 2 h and then treated with 0.25 μg/ml ciprofloxacin. The survival rates were determined by plating assay. (C,D) Wild type PA14 was treated with 0.025 μg/ml ciprofloxacin for 2 h and the mRNA levels of higB or higA were determined by quantitative RT-PCR. Error bars represent the standard error. *p < 0.05, by Student's t-test. (E) Wild type PA14 carrying a Ptac driven higB gene or the empty vector were cultured with 1 mM IPTG for 2 h, followed by treatment with 0.25 μg/ml ciprofloxacin. The survival rates were determined by plating. Error bars represent the standard errors. The graphs are representatives of three independent experiments.
Transcriptome Analysis of the higA::Tn Mutant
RNA-seq analyses were employed to explore the effect of higA inactivation on bacterial global gene expression at both exponential and stationary growth phases. Compared to wild type PA14, expression of 193 genes was altered in the higA mutant at both growth phases (Table S1). Of note, all of the T3SS genes were up regulated in the higA mutant (Table 1), suggesting a regulatory role of the TA system on the T3SS.
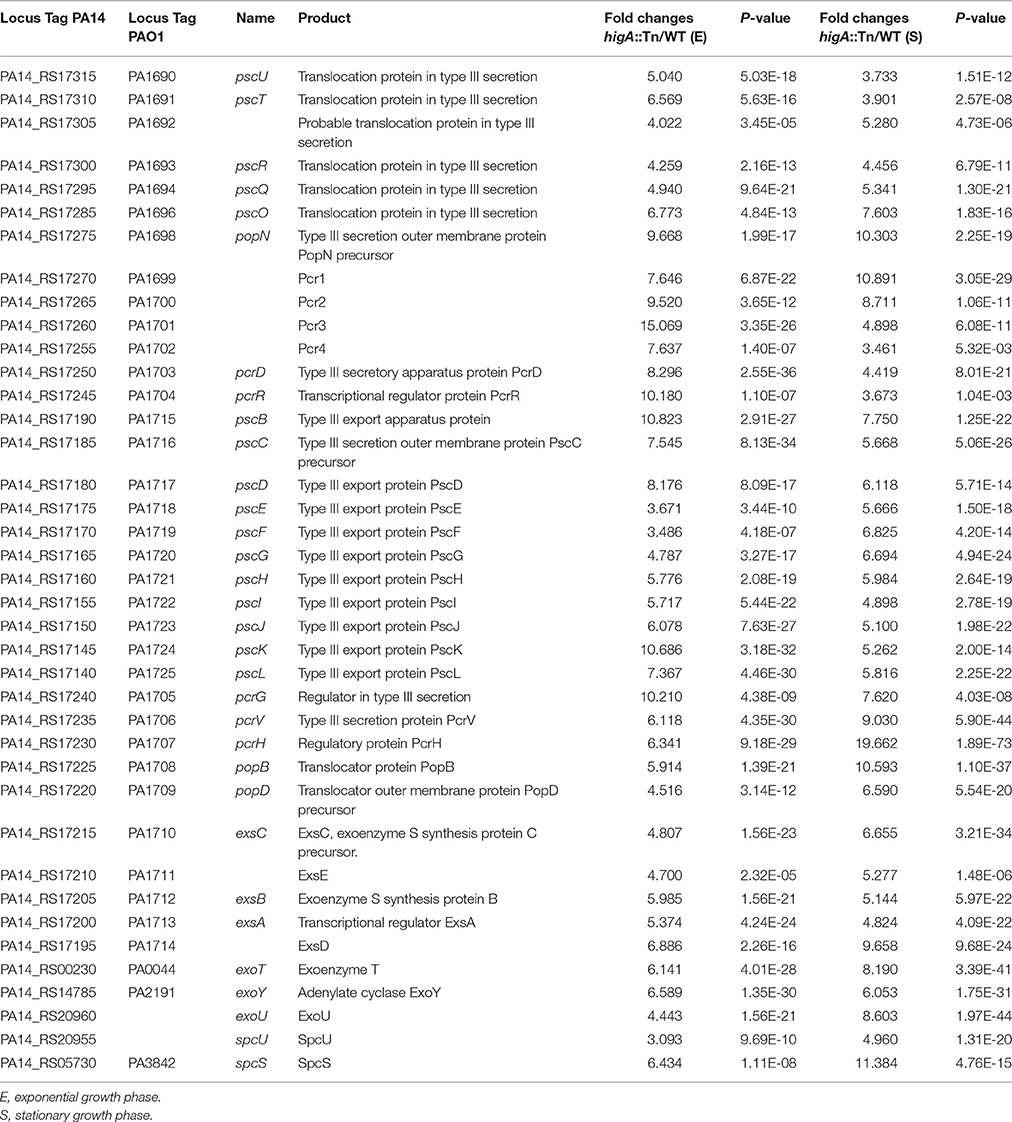
Table 1. mRNA levels of T3SS genes in the higA::Tn mutant compared to those in wild type PA14.
Increased Expression of T3SS Genes and Cytotoxicity of the higA::Tn Mutant
To confirm the elevated expression of T3SS genes in the higA::Tn mutant, the mRNA levels of exsA, exsC (two positive regulatory genes), pcrV (required for translocation of effector proteins) and exoU (encodes for an effector protein) were examined. Mutation of higA resulted in higher mRNA levels of all of these genes, which were restored to the wild type levels by complementation with a higA gene (Figure 3A, Figure S1B). As the higA::Tn mutant grew slower than the wild type strain, translation of the T3SS genes might be impeded. To test this possibility, we examined PcrV protein levels by immunostaining in strains harboring a mcherry gene driven by the promoter of higB-higA (PhigB-mcherry). Compared to the wild type strain, the higA::Tn mutant expressed higher levels of PcrV and mCherry proteins (Figure S1C). Next, we constructed a C-terminal His-tagged ExoU driven by its native promoter (PexoU-ExoU-His). Consistent with the above results, the levels of ExoU-His protein in the higA::Tn mutant were higher at both exponential (Figure 3B) and stationary growth phases (Figure S1D).
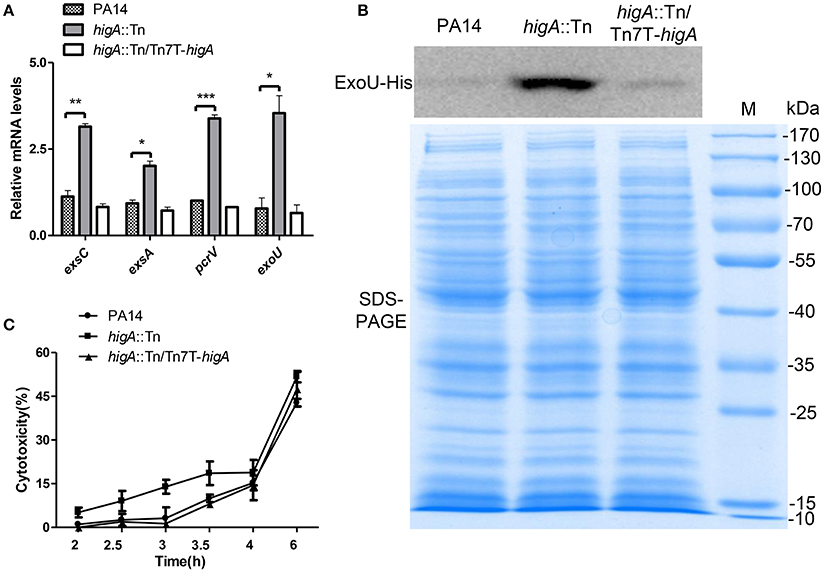
Figure 3. Expression levels of T3SS genes in wild type PA14, the higA::Tn mutant and complemented strain. (A) Relative mRNA levels of exsC, exsA, pcrV, and exoU in indicated strains at exponential growth phase (OD600 = 0.8~1.0). Error bars represent the standard errors. *p < 0.05, **p < 0.01, ***p < 0.005 compared to wild type PA14 by Student's t-test. (B) Bacteria carrying an exoU-His driven by its native promoter (PexoU-exoU-His) were grown in LB at 37°C. At the OD600 of 1.0, bacteria were collected. Samples from equivalent bacterial cells were loaded into SDS-PAGE gels and stained with Coomassie blue or probed with an anti-His antibody. (C) Raw264.7 cell were infected by indicated strains at an MOI of 10. At indicated time points, the relative cytotoxicity was determined by the LDH release assay.
To test whether the increased expression of T3SS genes leads to higher cytotoxicity, we performed LDH release assay. Compared to wild type PA14, the higA::Tn mutant caused quicker cell death to either macrophages (Raw264.7) (Figure 3C) or epithelial cells (HeLa) (Figure S1E). In addition, when HeLa cells were infected with strains containing the ExoU-His, more ExoU was translocated into the cells by the higA::Tn mutant (Figures S1F,G). Altogether, these results demonstrate that mutation of the higA results in up regulation of T3SS genes and consequently higher cytotoxicity.
Activation of HigB Increases the Expression of T3SS Genes and Cytotoxicity
HigB functions as a RNase, which is directly inhibited by HigA (Wood and Wood, 2016). Thus, we examined the role of HigB in the expression of T3SS genes and cytotoxicity. First, a ΔhigBΔhigA double mutant was constructed, which displayed similar levels of T3SS gene expression and cytotoxicity as the wild type PA14 (Figures S2A,B). Second, a C-terminal His-tagged HigB driven by a regulatable tac promoter (Ptac-higB-His) or the empty vector was introduced into wild type PA14 (Figure S2C). Addition of IPTG increased the mRNA levels of exsC, exsA, pcrV, and exoU, with the highest levels in the presence of 0.5 mM IPTG. In the presence of 1.0 mM IPTG, the mRNA levels of those genes were reduced, which might be due to strong growth inhibition as a consequence of high level expression of the HigB (Figures 4A–D). To further confirm the expression level of ExoU, we transferred the plasmid carrying PexoU-ExoU-His into the above strains. Consistently, the protein level of ExoU was increased by the overexpression of HigB (Figure 4E).
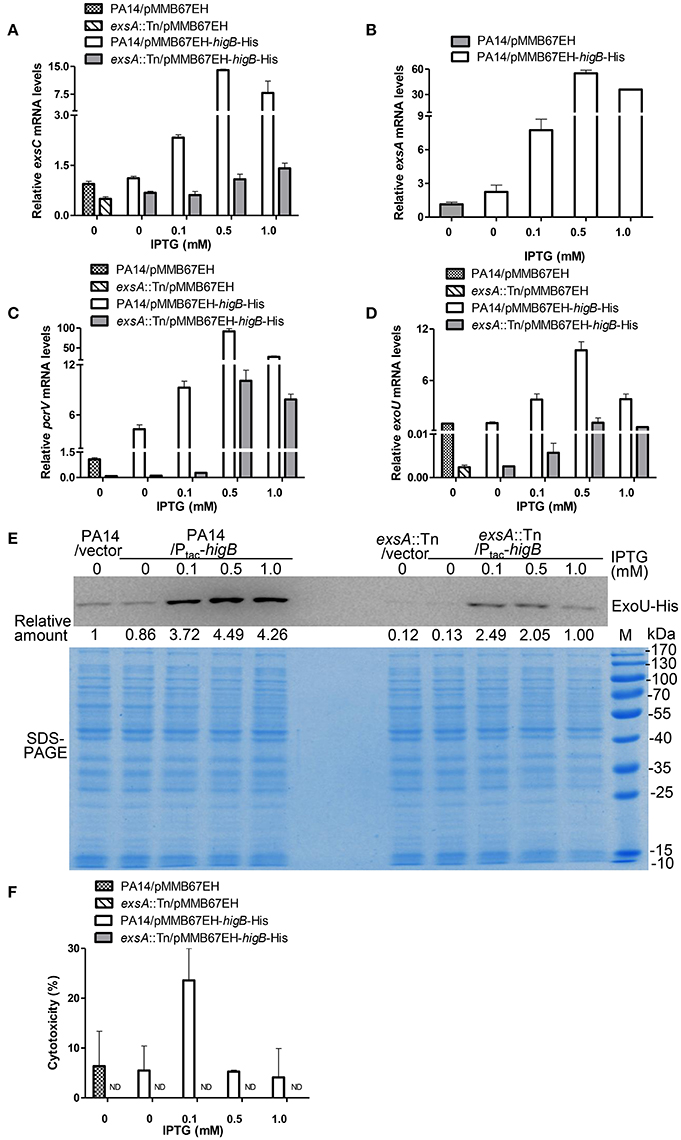
Figure 4. HigB promotes T3SS mediated cytotoxicity. Wild type PA14 and an exsA::Tn mutant containing pMMB67EH-higB-His or pMMB67EH was grown in the presence of indicated concentrations of IPTG for 3 h. Quantitative RT-PCR was used to determine the relative mRNA levels of exsC (A), exsA (B), pcrV (C), and exoU (D). (E) PA14 or exsA::Tn containing exoU-His driven by its native promoter (PexoU-exoU-His) and pMMB67EH-higB or pMMB67EH was grown in the presence of indicated concentrations of IPTG for 3 h. Samples from equivalent bacterial cells were loaded into SDS-PAGE gels and stained with Coomassie blue or or probed with an anti-His antibody. The relative density of each band was determined by Image J. (F) Raw264.7 cells were infected with the bacteria at an MOI of 10 for 3.5 h, followed by LDH release assay. ND, not detectable.
Next, we determined the correlation between bacterial cytotoxicity and expression levels of HigB. Bacteria grown in the presence 0.1 mM IPTG displayed the highest cytotoxicity to both Raw264.7 (Figure 4F) and HeLa cells (Figure S2D). However, further increase of the IPTG concentration reduced the cytotoxicity (Figure 4F, Figure S2D), although the mRNA levels of the T3SS genes were higher than or similar to those in the presence 0.1 mM IPTG (Figures 4A–D). Mutation of exsA severely reduced the HigB mediated increase of the T3SS gene expression and cytotoxicity (Figures 4A–F, Figure S2D). These results suggest that HigB promotes bacterial cytotoxicity through the T3SS. However, too high level of HigB might repress the overall bacterial fitness, which impedes the translocation of T3SS effector proteins.
Cytotoxicity of Persister Cells
Our results from the higA::Tn mutant and the HigB overexpressing strain demonstrate that activation of HigB increases persister formation and T3SS mediated cytotoxicity. A more important question is whether persister cells harbor higher levels of T3SS proteins and are more cytotoxic than their isogenic vegetative cells.
In the bacterial survival assay, we noticed lysis of bacterial cells during ciprofloxacin treatment, presumably due to production and release of pyocins (Penterman et al., 2014; Sun et al., 2014). Based on this phenotype, we developed a method to collect persister cell by washing the ciprofloxacin treated bacteria with 0.3M sucrose, which could efficiently remove lysed cell debris. To assess the effectiveness of this method, we treated a wild type PA14 strain containing a gfp gene driven by the higB promoter (PhigB-gfp) with 0.025 μg/ml ciprofloxin for 2 h to induce persister formation. Then the cells were incubated with 0.25 μg/ml ciprofloxacin for 6 h, resulting in a survival rate of 0.01% as determined by plating assay. Such treated bacterial cells were harvested by centrifugation and washed twice with 0.3M sucrose, followed by propidium iodide (PI) staining. As presented in Figure S3A, 93 ± 0.5% collected cells were PI negative, suggesting an effective isolation of persister cells. In addition, bacteria with strong green fluorescence were negative for PI staining, or vice versa (Figure S3A), indicating an up regulation of HigB in the persister cells. To examine the levels of PcrV in the persister cells, the collected bacterial cells were subjected to immunostaining with an anti-PcrV antibody. 79.7 ± 3.7% GFP positive cells were positive for PcrV (Figure S3B). In combination, these results demonstrate elevated levels of HigB and PcrV in persister cells.
Next, we examined the cytotoxicity of persister cells. Persister cells of wild type PA14 were collected as aforementioned and used to infect Raw264.7 cells. However, the persister cells displayed minimal cytotoxicity compared to vegetative cells (Figures S3C,D). We suspected that the 6-h exposure to ciprofloxacin might result in highly dormant cells that are unable to inject T3SS effectors. Therefore, we reduced the treatment time to 30 min, which resulted in 25% bacterial survival rate. As represented in Figure 5A, 83 ± 2.8% cells collected after ciprofloxacin treatment were PI negative. 89 ± 6.6% cells were GFP positive but GFP and PI double positive cell was barely observed (Figure 5A, lower panels), suggesting high levels of HigB in survived cells. Among the cells, 77 ± 10.1% were double positive for GFP and PcrV (Figure 5B, lower panels), whereas bacteria grown in LB were negative for GFP or PcrV (Figure 5B, upper panels). These results suggest that the expression levels of HigB and PcrV were higher in the survived bacterial cells than those in vegetative cells. These surviving bacteria displayed higher cytotoxicity to Raw264.7 cells (Figure 5C). Addition of anti-PcrV antibody, which has been demonstrated to protect cells from T3SS mediated cytotoxicity (Warrener et al., 2014), protected the Raw264.7 cells from killing by the bacteria survived of the ciprofloxacin treatment (Figure 5C).
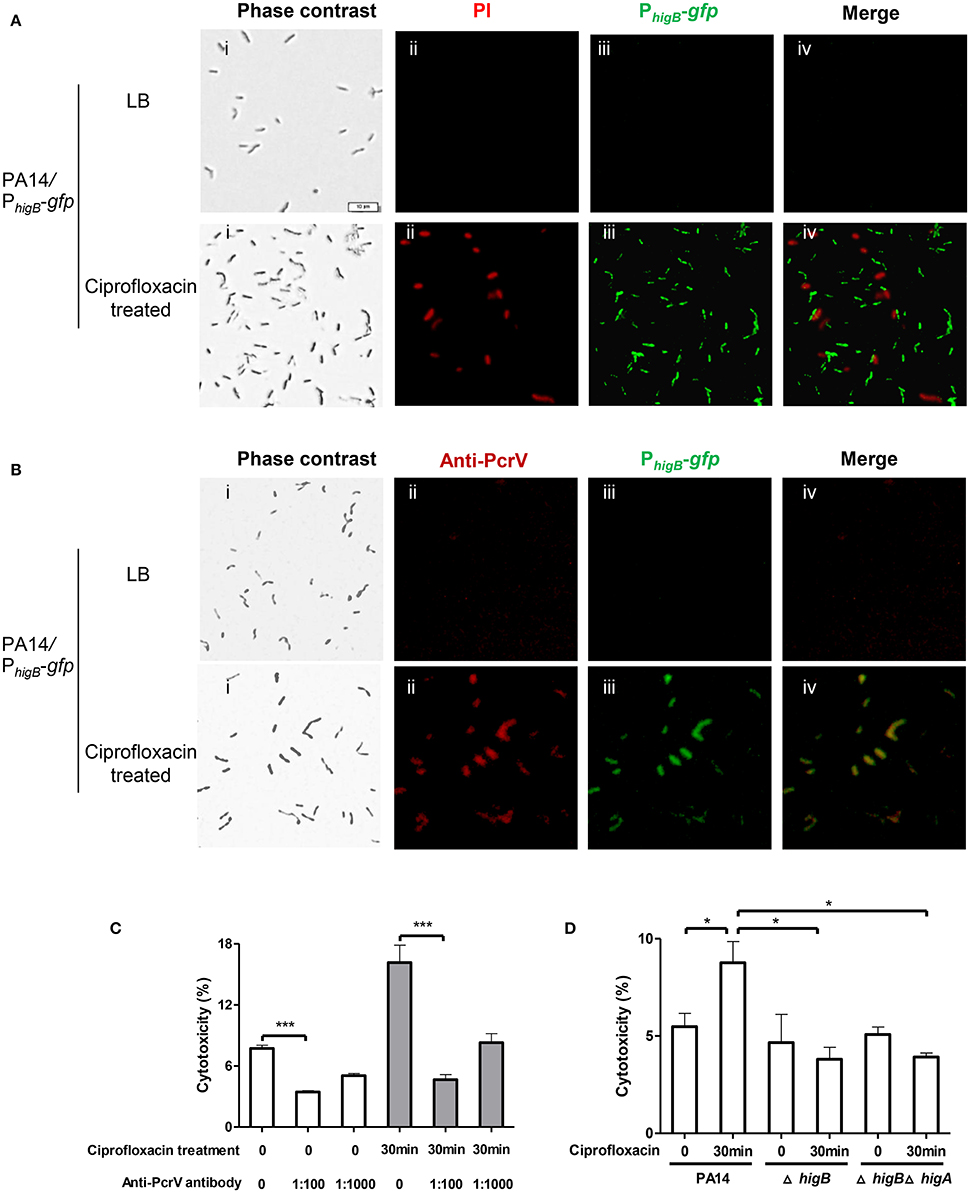
Figure 5. Levels of HigB, PcrV and cytotoxicity of bacterial cells that survived ciprofloxacin treatment. PA14/PhigB-gfp was cultured in the presence of 0.025 μg/ml ciprofloxacin for 2 h and then treated with 0.125 μg/ml ciprofloxacin for 30 min. The bacteria were washed with PBS twice, stained with PI (A) or fixed and permeabilized and then stained with rabbit anti-PcrV, followed by Alex Fluor 594–labeled goat anti–rabbit immunoglobulin. Bar = 10 μm (B). Quantification of fluorescence positive cells was based on analysis of about 100 cells from three different samples. (C) Wild type PA14 was cultured in the presence of 0.025 μg/ml ciprofloxacin for 2 h and then treated with 0.125 μg/ml ciprofloxacin for 30 min. Raw264.7 cells were infected with the surviving bacteria or bacteria grown in LB at an MOI of 10 for 3.5 h. The anti-PcrV antibody was added to the medium at indicated dilutions. The relative cytotoxicity was determined by the LDH release assay. Error bars represent the standard errors. (D) Wild type PA14, the ΔhigA or ΔhigAΔhigB mutant was cultured in the presence of 0.025 μg/ml ciprofloxacin for 2 h and then treated with 0.125 μg/ml ciprofloxacin for 30 min. Raw264.7 cells were infected with the survived bacteria or bacteria grown in LB at an MOI of 10 for 3.5 h. The relative cytotoxicity was determined by the LDH release assay. Error bars represent the standard errors. Each graph represents the results of three independent experiments. *p < 0.05, ***p < 0.005 by Student's t-test.
To exclude the possibility that the macrophages were killed from stimulation by large amount of LPS or other bacterial ligands in the collected bacterial samples, we tested an exsA::Tn mutant strain. After the same ciprofloxacin treatment, the survival rate of the mutant was similar to that of wild type PA14, however, the bacteria displayed minimum cytotoxicity (Figures S3C,D). Next, we incubated wild type bacteria at 50°C for 30 min, which also resulted in 25% survival rate. The surviving bacteria barely caused cell death (Figures S3C,D). In combination, the above results demonstrate that compared to vegetative cells, bacteria that survived the 30 min ciprofloxacin treatment contain higher level of T3SS proteins, which leads to increased cytotoxicity.
HigB Contributes to the Increased T3SS Gene Expression and Cytotoxicity of Bacteria Survived Ciprofloxacin Treatment
To examine the role of HigB in the expression of T3SS genes in bacteria survived ciprofloxacin treatment, we treated either the ΔhigB mutant or the ΔhigBΔhigA mutant with 0.025 μg/ml ciprofloxin for 2 h followed by incubation with 0.125 μg/ml ciprofloxacin for 30 min. The expression levels of HigB and PcrV were examined by fluorescence microscopy as described above. Similar to wild type PA14, the promoter activity of higB was increased in the surviving bacteria (Figures S4, S5). The stronger fluoresce in the ΔhigBΔhigA mutant further confirmed the negative regulatory role of the HigA on the higB-higA operon (Figures S4, S5). The levels of PcrV were significantly lower in the ΔhigB or ΔhigBΔhigA mutant than that in the wild type PA14 (Figures S4, S5). Consistently, the ΔhigB or ΔhigBΔhigA mutant cells survived ciprofloxacin treatment displayed lower cytotoxicity compared to the counterpart of wild type cells (Figure 5D). Therefore, HigB plays an important role in the up regulation of T3SS genes and increased cytotoxicity in survived bacteria.
Discussion
In this study, we demonstrated that HigB is involved in ciprofloxacin induced persister formation and up regulation of the T3SS genes in P. aeruginosa. Mutation of higA or overexpression of higB did not alter the MIC of ciprofloxacin to the bacteria. Our RNA-seq results demonstrated no significant change in the expression levels of the multidrug efflux pumps in the higA mutant. Quantitative RT PCR results confirmed that the expression level of the major multidrug efflux pump MexAB-OprM was not altered in the higA mutant or the higB overexpression strain (data not shown) (Dreier and Ruggerone, 2015). However, the bacterial survival rate was significantly increased by the mutation of higA or overexpression of higB after ciprofloxacin treatment (Figures 2A,E), suggesting a role of HigA-HigB in persister formation.
Through a microarray analysis, Wood et al. found that mutation of higA reduced the expression of pyochelin biosynthesis genes (Wood and Wood, 2016). Our RNA-seq analysis of the higA::Tn mutant revealed similar expression pattern of those genes (Table S1). In addition, the whole T3SS gene clusters were up regulated, which we demonstrate to be dependent on HigB. The P. aeruginosa In M. tuberculosis, overexpression of HigB reduced the levels of a subset of mRNAs and increased HigB is the cleavage of tmRNA, which is involved in the rescue of ribosomes stalled on mRNAs (Christensen and Gerdes, 2003; Schuessler et al., 2013). It has been demonstrated in E. coli and M. tuberculosis that HigB associates with ribosome and cleaves mRNA at A-rich sequences (Hurley and Woychik, 2009; Schureck et al., 2015, 2016a,b). In addition, mutation of the higA gene did not lead to bacterial death (Wood and Wood, 2016 and our study). These results indicate that HigB might have a specific range of target mRNAs. As many genes contain A-rich codons, it is difficult to judge the target mRNAs solely based on the sequence. One of the possibilities is that the recognition of target mRNA or subsequent cutting by the HigB is affected by the movement of ribosome, i.e., the longer the ribosome stall at the A-rich codons, the more likely the mRNA is cleaved by HigB. As it has been demonstrated that ribosome stalling is affected by the amino acid sequence as well as environmental stimulations (Jin et al., 2016; Wilson et al., 2016), it will be interesting to examine the HigB mediated cleavage of the A-rich codons (such as AAA) with different neighboring sequences or under different conditions.
In this study, we used various concentrations of IPTG to induce ectopic expression of HigB in wild type PA14. With increasing expression of the HigB, the levels of T3SS gene expression and bacterial cytotoxicity rose and then dropped. Consistent with these observations, wild type PA14 that survived 0.5-h ciprofloxacin treatment displayed higher T3SS mediated cytotoxicity. However, bacteria that survived 6-h ciprofloxacin treatment displayed minimal cytotoxicity, although the PcrV level was higher than that in untreated bacteria. We hypothesize that the HigB recognizing sites might be overrepresented in the mRNA of a T3SS negative regulator, rendering it more vulnerable to HigB mediated cleavage. Of note, overexpression of HigB increased the expression levels of T3SS genes in an exsA::Tn mutant (Figures 4A–F). These results suggest that the HigB targeted T3SS regulator might repress the expression of T3SS genes independent of ExsA.
On the other hand, with higher levels of HigB, mRNAs with less HigB recognizing sites are also cleaved, thus reducing the overall biological fitness and the bacterial ability to respond to host cell contact. In addition, the assembly of T3SS apparatus or translocation of T3SS effectors might be impeded, thus leading to reduced cytotoxicity. Given the complicated environment in the host, each bacterium might encounter different levels of antibiotics. It might be possible that during persister formation, the levels of activated HigB are heterologous among the bacterial population. Moderate activation of HigB increases the expression of T3SS and bacterial cytotoxicity, while further up regulation and activation of HigB render the bacteria dormant and highly tolerant to antibiotics. It has been demonstrated in an animal model that T3SS-negative bacteria are protected from host clearance by the isogenic wild type strain, which actively kills phagocytes through the T3SS (Hauser, 2009; Diaz and Hauser, 2010; Czechowska et al., 2014). Therefore, we suspect that in the bacterial population that survived antibiotic treatment, bacteria with higher cytotoxicity might protect highly dormant cells from host immune cells, thus enable the survival of the persister cells. In biofilm, HigB in a small portion of bacteria might be activated, leading to persister formation as well as up regulation of T3SS genes. We previously found that the biomass of P. aeruginosa biofilm was reduced by ciprofloxacin treatment, suggesting dispersal of the biofilm (Sun et al., 2014). Therefore, it might be possible that bacteria inside biofilm are getting exposed to phagocytes during antibiotic treatment. In this scenario, the highly dormant persister cells might be protected by the T3SS proficient cells. It will be interesting to observe the expression levels of HigB and T3SS genes in individual cells inside biofilm with or without antibiotic treatment.
Recently, Pu et al. demonstrated that up regulation of drug efflux genes and increased efflux activity in persister cells of E. coli (Pu et al., 2016). Together with our results, these findings suggest that persister cells might be armed with various defense and offense factors that enable them to actively defend against environmental stresses before entering into deeper dormancy state. Thus, exploration of the gene expression profiles of persister cells will shed light on their surviving strategies in various host environments.
Materials and Methods
Bacterial Strains and Plasmids
The bacterial strains used in this study are listed in Table S2. Bacteria were cultured in Luria–Bertani (LB) broth (10 g/l tryptone, 5 g/l Nacl, 5 g/l yeast extract, pH 7.0–7.5) or LB agar (LB broth containing 15 g/l agar) under aerobic conditions at 37°C. When needed, the medium was supplemented with tetracycline (100 μg/ml) (BBI life sciences, Shanghai, China), gentamicin (100 μg/ml) (BBI life sciences), streptomycin (50 μg/ml) (BBI life sciences), or carbenicillin (150 μg/ml) (BBI life sciences) for P. aeruginosa, and ampicillin (100 μg/ml) (BBI life sciences) for E. coli.
Plasmids used in this study are listed in Table S2. For DNA manipulation, standard protocols or manufacture instructions of commercial products were followed. Chromosomal gene mutations were generated as described previously (Hoang et al., 1998).
Reverse Transcription and Quantitative RT PCR
Total RNA was isolated from bacteria at indicated time points with an RNeasy Minikit (Tiangen Biotech, Beijing, China). The cDNA was synthesized from total RNA using random primers and PrimeScript Reverse Transcriptase (TaKaRa, Dalian, China). Specific Primers (Table S2) were used for quantitative RT PCR. For quantitative RT PCR, cDNA was mixed with 4 pmol of forward and reverse primers and SYBR Premix Ex Taq™ II (TaKaRa) in a total reaction volume of 20 μl. The results were determined using a CFX Connect Real-Time system (Bio-Rad, USA).
5′ Race Analysis
The transcriptional start site of the higB-higA operon was determined by 5′ RACE (rapid amplification of cDNA ends). The cDNA was synthesized from total RNA using primer higA-R and higB-R. cDNA was purified with a DNA Clean kit (Sangon Biotech, Shanghai, China) and tailed with poly (dC) using terminal deoxynucleotidyl transferase (TaKaRa), then amplified by PCR. The obtained PCR product was ligated into a T-vector (TaKaRa), then sequenced.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) was performed as described with minor modification (Sun et al., 2014). Briefly, a 38-bp DNA fragment corresponding to sequence up-stream of higB start codon or the 38-bp DNA fragment with palindrome sequence scrambled as a negative control was synthesized. DNA fragments (300 ng) were incubated with 0, 4 or 20 nM purified recombinant HigA protein at 30°C for 30 min in a 20-μl reaction (10 mM Tris-HCl, pH 7.6, 4% glycerol, 1 mM EDTA, 5 mM CaCl2, 100 mM NaCl, 10 mM-β-Mercaptoethanol). Samples were loaded onto a 8% native polyacrylamide gel in 0.5 × Tris-borate-EDTA (TBE) buffer (0.044 M Tris base, 0.044 M boric acid, 0.001 M EDTA, pH 8.0) that had been prerun for 1 h, electrophoresed on ice at 90 V for 2 h followed by DNA staining in 0.5 × TBE containing 0.5 μg/ml ethidium bromide. Bands were visualized with a molecular imager ChemiDoc™ XRS + (Bio-Rad).
Antitoxin Stabilization Assays
Overnight culture of wild type PA14, a clpP::Tn or lon::Tn mutant harboring pMMB67EH-higA-His plasmid was sub-cultured in fresh LB broth to an OD600 of 0.5, then induced with 1 mM IPTG for 1 h, followed by treatment with 50 μg/ml streptomycin. Bacteria were collected at 0, 0.5, 1, 2, 3, 4, and 5 h, boiled in 1 × SDS loading buffer, then subjected to SDS-PAGE. Proteins was transferred onto a PVDF membrane and incubated with mouse anti-His antibody (1:2000) (Millipore, USA) at room temperature for 1 h. After washing with 1 × phosphate buffered saline (1 × PBS: 274 mM NaCl, 5.4 mM KCl, 20 mM Na2HPO4, 4 mM KH2PO4, pH 7.4) for four times, the membranes were incubated with a horseradish peroxidase-conjugated goat anti-mouse IgG (1:2000) (Promega, USA) at room temperature for 1 h. Signals were detected with the ECL-plus kit (Millipore) and visualized with a Bio-Rad molecular imager ChemiDoc™ XRS+.
Persistence Assay
Persistence of P. aeruginosa was measured by time-dependent killing experiments (Dörr et al., 2010). To test the persistence level induced by sublethal level of ciprofloxacin, overnight bacterial culture was sub-cultured in fresh LB broth and grown to an OD600 of 0.4 with or without 0.025 μg/ml ciprofloxacin. Then the bacterial cultures were exposed to 0.25 μg/ml ciprofloxacin. To test the effect of higA mutation or higB overexpression on persister formation, indicated strains were grown to an OD600 of 0.4, followed by treatment with 0.25 μg/ml ciprofloxacin. At indicated time points, the live bacterial number was determined by serial dilution and plating. The plate was incubated at 37°C for 24 h before colony counting.
RNA Sequencing and Data Analysis
PA14 and the higA::Tn mutant were cultured in LB broth at 37°C and harvested at log phage (OD600 of 0.8–1.0) and stationary phase (OD600 of 2.5–3.0). Total RNA was extracted with an RNeasy Protect Bacteria Mini Kit with on-column DNase I digestion (Qiagen, Shanghai, China). A Turbo DNA-free vigorous protocol was used for a second round of DNase treatment (Ambion). 16S, 23S, and 5S rRNA were removed using the Ribo-Zero Magnetic Kit (Bacteria) (Epicentre).
Gene expression analysis was conducted via Illumina RNA sequencing (RNA-Seq technology). RNA-Seq was conducted for 3 biological replicates of each sample. The rRNA-depleted RNA was fragmented to 150–200 bp in sizes, then first and second strand cDNA were synthesized, followed by end repair and adapter ligation. After 12 cycles of PCR enrichment, the quality of the libraries was assessed using a Bioanalyzer (Agilent Technologies). The libraries were sequenced using an Illumina HiSeq 2500 platform with a paired-end protocol and read lengths of 100-nt.
The sequencing data was analyzed using the method described previously (Chua et al., 2014). Sequence reads were mapped onto PA14 reference genome (NC_008463) using a CLC genomics Workbench 8.0 (CLC Bio-Qiagen, Aarhus, Denmark). The count data of expression values were then analyzed using a DESeq package of R/Bioconductor. The differentially expressed genes were identified by performing a negative binomial test using the DESeq, with the cut-off of fold-change larger than 2. The raw sequence reads were normalized by dividing with size factors, then Log2 (N + 1) transformed.
Immunofluorescence Assay
Bacteria with or without ciprofloxacin treatment were cytocentrifuged onto glass slides and fixed with 4% paraformaldehyde at room temperature for 30 min. Then bacteria were washed with 1 × PBS three times and permeabilized with 0.2% Triton X-100 in 1 × PBS at room temperature for 5 min. After washed twice with PBS, the bacteria were incubated with rabbit anti-PcrV serum (1:50) in PBSG (1 × PBS containing 0.1% gelatin) at 37°C for 1 h. The cells were washed twice with PBSG and incubated with the secondary antibody, green or red-conjugated goat anti- rabbit IgG (1:100) (Thermo Fisher Scientific, USA) in PBSG at 37°C for 1 h. To determine the viability, bacteria were stained with 1 μg/ml PI in 1 × PBS at room temperature for 15 min after 0.5 or 6 h ciprofloxacin treatment. Then cells were analyzed by a BX53 fluorescence microscope (Olympus, Japan).
Cell Culture and Cytotoxicity Assays
Raw264.7 cells and HeLa cells were cultured in DMEM medium with 10% fetal bovine serum (FBS) at 37°C in 5% CO2, and 95% air, supplemented with 1% penicillin/streptomycin and ciprofloxacin (10 μg/ml). Overnight bacterial culture was sub-cultured in fresh LB broth to OD600 of 0.8 before infection. Bacteria were washed once and resuspended in 1 × PBS. Raw264.7 and HeLa cells were infected with bacteria at a multiplicity of infection (MOI) of 10 or 40, respectively, in DMEM medium without FBS and antibiotics. At the end of incubation, lactate dehydrogenase (LDH) present in the supernatant was measured using the LDH cytotoxicity assay kit (Beyotime, Haimen, China). Cells treated with LDH release agent C0017-1 were used as a control of total release (100% LDH release). The background level (0% LDH release) was determined with DMEM medium. The percentage of cytotoxicity was calculated following the manufacturer's instruction.
Effector Delivery Assay
HeLa cells were infected with strains containing PexoU-exoU-His at an MOI of 40. 1.5 h post infection, the cells were washed 3 times with 1 × PBS and lysed with 0.25% Trion-X 100. The Cell lysates were subjected to 10% SDS-PAGE. Proteins were transferred onto a PVDF membrane. The protein amounts of actin and ExoU were determined by Western blot analysis using mouse anti-His antibody or rabbit anti-β actin antibody (1:2000) (Cell Signaling Technology, USA).
Protective Effect of Anti-PcrV Antibody on Raw264.7 Cells
Overnight bacterial cultures were sub-cultured in fresh LB broth to OD600 of 0.4 with 0.025 μg/ml ciprofloxacin, then the bacterial cultures were exposed to 0.125 μg/ml ciprofloxacin. Bacteria with or without ciprofloxacin treatment were washed with 1 × PBS, then added to 104 Raw264.7 cells in 200 μl culture medium with various concentrations (0, 1:100, 1:1000) of either normal rabbit IgG or rabbit anti-PcrV IgG. Each mixture was incubated at 37°C for 3.5 h. Cytotoxicity was measured by LDH release assay as described above.
Author Contributions
Conceived and designed the experiments: WW, ML, SJ, ZC. Performed the experiments: ML, YuL, YiL, JS, RC, LZ, YJ, LY, YaL. Analyzed the data: ML, WW, ZC, SJ, FB, LY, YaL. Wrote the paper: ML, WW, ZC, SJ.
Funding
This work was supported by National Science Foundation of China (31670130, 31370168 and 31370167); Program of international S&T cooperation (2015DFG32500) and Science and Technology Committee of Tianjin (15JCYBJC53900 and 15JCZDJC33000). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fcimb.2016.00125
Figure S1. Expression levels of T3SS genes in wild type PA14, a higA::Tn mutant and a complemented strain. (A) Cleavage of HigA by the Lon protease. Wild type PA14, the clpP::Tn and lon::Tn mutants carrying pMMB67EH-higA-His were cultured in the presence of 1 mM IPTG for 1 h. Then 50 μg/ml spectinomycin was added to the medium. At indicated time points, the HigA-His levels were determined by Western blot analysis with an anti-His antibody. Density of each band was quantified with Image J. This graph represents the results of three independent experiments.(B) Relative mRNA levels of exsC, exsA, pcrV and exoU in indicated strains at stationary growth phase (OD600 = 2.5~3.0). Data represents the mean ± standard deviation from three independent experiments performed in triplicates. *p < 0.05, **p < 0.01, ***p < 0.005 compared to wild type PA14 by Student's t-test. (C) Fluorescence microscopy of PA14 and the higA::Tn mutant containing PhigB-mcherry. Bacteria were grown in LB to an OD600 of 3.0, collected and washed with PBS twice. The bacteria were fixed, permeabilized and then stained with rabbit anti-PcrV followed by Alex Fluor 594–labeled goat anti–rabbit immunoglobulin. Bar = 20 μm. (D) Bacteria carrying an exoU-His driven by its native promoter (PexoU-exoU-His) were grown in LB at 37°C. At stationary growth phase, bacteria were collected. Samples from equivalent bacterial cells were loaded into SDS-PAGE gels and stained with Coomassie blue or probed with an anti-His antibody. (E) HeLa cells were infected with indicated strains at an MOI of 40 for 3 h, followed by the LDH release assay. The values and bars represent the means and standard deviations of triplicate measurements. *p < 0.05 by Student's t-test. HeLa cells were infected with strains containing PexoU-exoU-His (F) or Plac-gfp-His (G) at an MOI of 40. 1.5 h after infection, the cells were washed 3 times with PBS and lysed with 0.25% Trion-X 100. The intracellular levels of ExoU-His and GFP-His were determined by Western blot analysis.
Figure S2. HigB promotes expression levels of T3SS genes and cytotoxicity. (A) Relative mRNA levels of T3SS genes in wild type PA14, the higA::Tn and ΔhigBΔhigA mutants. Bacteria were grown to an OD600 of 1.0, followed by total RNA isolation. The mRNA levels of exsC, exsA, pcrV, and exoU were determined by quantitative RT-PCR. (B) Raw264.7 cell cells were infected with indicated strains at an MOI of 10 for 3.5 h. The relative cytotoxicity was determined by the LDH release assay. (C) PA14 or the exsA::Tn mutant containing pMMB67EH-higB-His or pMMB67EH was grown in the presence of indicated concentrations of IPTG for 3 h. The levels of HigB in bacterial cell lysates were determined by Western blot analysis. The loading control was displayed in Figure 4E. The relative density of each band was determined by Image J. (D) HeLa cells were infected with the indicated bacteria at an MOI of 40 for 3 h, followed by LDH release assay. ND, not detectable. The values and bars represent the means and standard deviations of triplicate measurements. *p < 0.05, **p < 0.01, ***p < 0.005 by Student's t-test.
Figure S3. Fluorescence microscopy of PA14 containing PhigB-gfp. At an OD600 of 0.3, bacteria were incubated with 0.025 μg/ml ciprofloxacin for 2 h and then treated with 0.25 μg/ml ciprofloxacin for 6 h in LB. The ciprofloxacin treated and untreated bacteria were collected and washed with PBS twice. The bacterial cells were stained with PI (A) or immunostained with rabbit anti-PcrV followed by Alex Fluor 594–labeled goat anti–rabbit immunoglobulin. Bar = 10 μm (B). Quantification of fluorescence positive cells was based on analysis of about 100 cells from three different samples. (C) PA14 or the exsA::Tn mutant were cultured in the presence or absence of 0.025 μg/ml ciprofloxacin for 2 h and then treated with 0.125 μg/ml ciprofloxacin for 30 min or 6 h. Or, the PA14 cells were incubated at 50°C for 30 min. Live bacteria were collected. Raw264.7 cells were infected with the indicated bacteria at an MOI of 10 for 3.5 h. (D) HeLa cells were infected with the indicated bacteria at MOI of 40 for 3.0 h. The relative cytotoxicity levels were determined by LDH release assay. ND, not detectable. *p < 0.05, compared to each of the other samples by Student's t-test.
Figure S4. Fluorescence microscopy of ΔhigB mutant containing PhigB-gfp. At an OD600 of 0.3, a ΔhigB mutant containing PhigB-gfp were incubated with 0.025 μg/ml ciprofloxacin for 2 h and then treated with 0.125 μg/ml ciprofloxacin for 30 min in LB. The ciprofloxacin treated and untreated bacteria were collected and washed with PBS twice. The bacterial cells were stained with PI (A) or immunostained with rabbit anti-PcrV followed by Alex Fluor 594–labeled goat anti–rabbit immunoglobulin (B). Bar = 10 μm.
Figure S5. Fluorescence microscopy of ΔhigBΔhigA mutant containing PhigB-gfp. At the OD600 of 0.3, a ΔhigBΔhigA mutant containing PhigB-gfp were incubated with 0.025 μg/ml ciprofloxacin for 2 h and then treated with 0.125 μg/ml ciprofloxacin for 30 min in LB. The ciprofloxacin treated and untreated bacteria were collected and washed with PBS twice. The bacterial cells were stained with PI (A) or immunostained with rabbit anti-PcrV followed by Alex Fluor 594–labeled goat anti–rabbit immunoglobulin (B). Bar = 10 μm.
Table S1. PA14 Transcriptome analysis: differentially regulated genes.
Table S2. Bacterial strains, plasmids and primers used in this study.
References
Balaban, N. Q., Merrin, J., Chait, R., Kowalik, L., and Leibler, S. (2004). Bacterial persistence as a phenotypic switch. Science 305, 1622–1625. doi: 10.1126/science.1099390
Balasubramanian, D., Schneper, L., Kumari, H., and Mathee, K. (2013). A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res. 41, 1–20. doi: 10.1093/nar/gks1039
Bernier, S. P., Lebeaux, D., Defrancesco, A. S., Valomon, A., Soubigou, G., Coppée, J.-Y., et al. (2013). Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 9:e1003144. doi: 10.1371/journal.pgen.1003144
Bertram, R., and Schuster, C. F. (2014). Post-transcriptional regulation of gene expression in bacterial pathogens by toxin-antitoxin systems. Front Cell Infect. Microbiol. 4:6. doi: 10.3389/fcimb.2014.00006
Christensen, S. K., and Gerdes, K. (2003). RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 48, 1389–1400. doi: 10.1046/j.1365-2958.2003.03512.x
Chua, S. L., Liu, Y., Yam, J. K. H., Chen, Y., Vejborg, R. M., Tan, B. G. C., et al. (2014). Dispersed cells represent a distinct stage in the transition from bacterial biofilm to planktonic lifestyles. Nat. Commun. 5:4462. doi: 10.1038/ncomms5462
Czechowska, K., McKeithen-mead, S., Al Moussawi, K., and Kazmierczak, B. I. (2014). Cheating by type 3 secretion system-negative Pseudomonas aeruginosa during pulmonary infection. Proc. Natl. Acad. Sci. U.S.A. 111, 7801–7806. doi: 10.1073/pnas.1400782111
Diaz, M. H., and Hauser, A. R. (2010). Pseudomonas aeruginosa cytotoxin ExoU is injected into phagocytic cells during acute pneumonia. Infect. Immun. 78, 1447–1456. doi: 10.1128/IAI.01134-09
Dörr, T., Lewis, K., and Vulić, M. (2009). SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5:e1000760. doi: 10.1371/journal.pgen.1000760
Dörr, T., Vulić, M., and Lewis, K. (2010). Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8:e1000317. doi: 10.1371/journal.pbio.1000317
Dreier, J., and Ruggerone, P. (2015). Interaction of antibacterial compounds with RND efflux pumps in Pseudomonas aeruginosa. Front. Microbiol. 8:660. doi: 10.3389/fmicb.2015.00660
Germain, E., Castro-Roa, D., Zenkin, N., and Gerdes, K. (2013). Molecular mechanism of bacterial persistence by HipA. Mol. Cell 52, 248–254. doi: 10.1016/j.molcel.2013.08.045
Hauser, A. R. (2009). The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7, 654–665. doi: 10.1038/nrmicro2199
Helaine, S., Cheverton, A. M., Watson, K. G., Faure, L. M., Matthews, S. A., and Holden, D. W. (2014). Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343, 204–208. doi: 10.1126/science.1244705
Hoang, T. T., Karkhoff-Schweizer, R. R., Kutchma, A. J., and Schweizer, H. P. (1998). A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86. doi: 10.1016/S0378-1119(98)00130-9
Hurley, J. M., and Woychik, N. A. (2009). Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J. Biol. Chem. 284, 18605–18613. doi: 10.1074/jbc.M109.008763
Jin, Y., Jin, S., and Wu, W. (2016). Regulation of bacterial gene expression by ribosome stalling and rescuing. Curr. Genet. 62, 309–12. doi: 10.1007/s00294-015-0545-3
Keren, I., Kaldalu, N., Spoering, A., Wang, Y., and Lewis, K. (2004). Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230, 13–18. doi: 10.1016/S0378-1097(03)00856-5
Kim, J.-S., Heo, P., Yang, T.-J., Lee, K.-S., Cho, D.-H., Kim, B. T., et al. (2011). Selective killing of bacterial persisters by a single chemical compound without affecting normal antibiotic-sensitive cells. Antimicrob. Agents Chemother. 55, 5380–5383. doi: 10.1128/AAC.00708-11
Lewis, K. (2007). Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56. doi: 10.1038/nrmicro1557
Lewis, K. (2010). Persister cells. Annu. Rev. Microbiol. 64, 357–372. doi: 10.1146/annurev.micro.112408.134306
Liberati, N. T., Urbach, J. M., Miyata, S., Lee, D. G., Drenkard, E., Wu, G., et al. (2006). An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U.S.A. 103, 2833–2838. doi: 10.1073/pnas.0511100103
Maisonneuve, E., Castro-Camargo, M., and Gerdes, K. (2013). (p) ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154, 1140–1150. doi: 10.1016/j.cell.2013.07.048
Nguyen, D., Joshi-Datar, A., Lepine, F., Bauerle, E., Olakanmi, O., Beer, K., et al. (2011). Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986. doi: 10.1126/science.1211037
Page, R., and Peti, W. (2016). Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 12, 208–214. doi: 10.1038/nchembio.2044
Penterman, J., Singh, P. K., and Walker, G. C. (2014). Biological cost of pyocin production during the SOS response in Pseudomonas aeruginosa. J. Bacteriol. 196, 3351–3359. doi: 10.1128/JB.01889-14
Pu, Y., Zhao, Z., Li, Y., Zou, J., Ma, Q., Zhao, Y., et al. (2016). Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell 62, 284–294. doi: 10.1016/j.molcel.2016.03.035
Ruzin, A., Keeney, D., and Bradford, P. A. (2007). AdeABC multidrug efflux pump is associated with decreased susceptibility to tigecycline in Acinetobacter calcoaceticus–Acinetobacter baumannii complex. J Antimicrob. Chemother. 59, 1001–1004. doi: 10.1093/jac/dkm058
Savli, H. Kolayli, F., Karadenizli, A., Gundes, S., Ozbek, U., and Vahaboglu, H. (2003). Expression stability of six housekeeping genes: a proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR. J. Med. Microbiol. 52, 403–408. doi: 10.1099/jmm.0.05132-0
Schuessler, D. L., Cortes, T., Fivian-Hughes, A. S., Lougheed, K. E., Harvey, E., Buxton, R. S., et al. (2013). Induced ectopic expression of HigB toxin in Mycobacterium tuberculosis results in growth inhibition, reduced abundance of a subset of mRNAs and cleavage of tmRNA. Mol. Microbiol. 90, 195–207. doi: 10.1111/mmi.12358
Schureck, M. A., Dunkle, J. A., Maehigashi, T., Miles, S. J., and Dunham, C. M. (2015). Defining the mRNA recognition signature of a bacterial toxin protein. Proc. Natl. Acad. Sci. U.S.A. 112, 13862–13867. doi: 10.1073/pnas.1512959112
Schureck, M. A., Maehigashi, T., Miles, S. J., Marquez, J., and Dunham, C. M. (2016a). mRNA bound to the 30S subunit is a HigB toxin substrate. RNA 22, 1261–1270. doi: 10.1261/rna.056218.116
Schureck, M. A., Repack, A., Miles, S. J., Marquez, J., and Dunham, C. M. (2016b). Mechanism of endonuclease cleavage by the HigB toxin. Nucleic Acids Res. 44, 7944–7953. doi: 10.1093/nar/gkw598
Son, M. S., Matthews, W. J. Jr., Kang, Y., Nguyen, D. T., and Hoang, T. T. (2007). In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect. Immun. 75, 5313–5324. doi: 10.1128/IAI.01807-06
Sun, Z., Shi, J., Liu, C., Jin, Y., Li, K., Chen, R., et al. (2014). PrtR homeostasis contributes to Pseudomonas aeruginosa pathogenesis and resistance against ciprofloxacin. Infect. Immun. 82, 1638–1647. doi: 10.1128/IAI.01388-13
Verstraeten, N., Knapen, W. J., Kint, C. I., Liebens, V., Van den bergh, B., Dewachter, L., et al. (2015). Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell 59, 9–21. doi: 10.1016/j.molcel.2015.05.011
Warrener, P., Varkey, R., Bonnell, J. C., Digiandomenico, A., Camara, M., Cook, K., et al. (2014). A novel anti-PcrV antibody providing enhanced protection against Pseudomonas aeruginosa in multiple animal infection models. Antimicrob. Agents Chemother. 58, 4384–4391. doi: 10.1128/AAC.02643-14
Wen, Y., Behiels, E., and Devreese, B. (2014). Toxin–Antitoxin systems: their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 70, 240–249. doi: 10.1111/2049-632X.12145
Williams, J. J., Halvorsen, E. M., Dwyer, E. M., Difazio, R. M., and Hergenrother, P. J. (2011). Toxin–antitoxin (TA) systems are prevalent and transcribed in clinical isolates of Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus. FEMS Microbiol. Lett. 322, 41–50. doi: 10.1111/j.1574-6968.2011.02330.x
Wilson, D. N., Arenz, S., and Beckmann, R. (2016). Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr. Opin. Struc. Biol. 37, 123–133. doi: 10.1016/j.sbi.2016.01.008
Wood, T. L., and Wood, T. K. (2016). The HigB/HigA toxin/antitoxin system of Pseudomonas aeruginosa influences the virulence factors pyochelin, pyocyanin, and biofilm formation. Microbiology Open 5, 499–511. doi: 10.1002/mbo3.346
Keywords: toxin/antitoxin, type III secretion system, persistence, Pseudomonas aeruginosa, gene regulation
Citation: Li M, Long Y, Liu Y, Liu Y, Chen R, Shi J, Zhang L, Jin Y, Yang L, Bai F, Jin S, Cheng Z and Wu W (2016) HigB of Pseudomonas aeruginosa Enhances Killing of Phagocytes by Up-Regulating the Type III Secretion System in Ciprofloxacin Induced Persister Cells. Front. Cell. Infect. Microbiol. 6:125. doi: 10.3389/fcimb.2016.00125
Received: 27 July 2016; Accepted: 27 September 2016;
Published: 14 October 2016.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
Arne Rietsch, Case Western Reserve University, USAWilliam D. Picking, University of Kansas, USA
Copyright © 2016 Li, Long, Liu, Liu, Chen, Shi, Zhang, Jin, Yang, Bai, Jin, Cheng and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weihui Wu, wuweihui@nankai.edu.cn
Zhihui Cheng, zhihuicheng@nankai.edu.cn
Shouguang Jin, sjin@ufl.edu