Rhinovirus and Innate Immune Function of Airway Epithelium
 Haleh Ganjian
Haleh Ganjian Charu Rajput
Charu Rajput Manal Elzoheiry
Manal Elzoheiry Umadevi Sajjan
Umadevi Sajjan- 1Department of Thoracic Medicine and Surgery, Lewis Katz Medical School, Temple University, Philadelphia, PA, United States
- 2Department of Physiology, Lewis Katz Medical School, Temple University, Philadelphia, PA, United States
Airway epithelial cells, which lines the respiratory mucosa is in direct contact with the environment. Airway epithelial cells are the primary target for rhinovirus and other inhaled pathogens. In response to rhinovirus infection, airway epithelial cells mount both pro-inflammatory responses and antiviral innate immune responses to clear the virus efficiently. Some of the antiviral responses include the expression of IFNs, endoplasmic reticulum stress induced unfolded protein response and autophagy. Airway epithelial cells also recruits other innate immune cells to establish antiviral state and resolve the inflammation in the lungs. In patients with chronic lung disease, these responses may be either defective or induced in excess leading to deficient clearing of virus and sustained inflammation. In this review, we will discuss the mechanisms underlying antiviral innate immunity and the dysregulation of some of these mechanisms in patients with chronic lung diseases.
Introduction
Healthy adults inhale about 7–8 L of air per minute or ~11 L of air per day. Therefore, the lung is by far the largest organ in humans that is in direct contact with the environment. Along with air, we also inhale microorganisms and environmental pollutants; therefore, the lung is equipped with several powerful defense mechanisms. Conductive airways, which act as a passageway for the air to move in and out of the lungs, moisten, equilibrate the temperature, filter the inhaled air, and protect the alveoli, which participates in gas exchange. The epithelium that lines the conductive airways, in addition to providing a physical barrier to separate the environment from the lung milieu, also plays a pivotal role in shaping the lung innate and adaptive immunity. Basal cells in the airway epithelium have wound healing properties, so that the airway epithelium repair and regenerate rapidly after the injury. Besides, airway epithelial cells also communicate with other innate immune cells, such as macrophages, neutrophils and dendritic cells, to promote pathogen clearance and limit lung inflammation. Therefore, the airway epithelium through its barrier and innate immune functions play an essential role in clearing the inhaled pathogens and harmful pollutants to maintain homeostasis in the lung.
Patients with chronic lung disorders such as chronic obstructive pulmonary disease (COPD), asthma and cystic fibrosis often show structurally and functionally altered airway epithelium. Such changes may affect the responses to respiratory infections, sometimes accelerating the progression of lung disease in these patients. This review will focus on the innate immune responses of airway epithelium to rhinovirus, which causes common colds in healthy subjects but exacerbates lung disease in patients with COPD, asthma and cystic fibrosis. We will also review the dysregulation of antiviral immunity of airway epithelial cells in patients with chronic lung disorders.
Conductive Airway Epithelium
The proximal conductive airways show pseudostratified morphology with different cell types (Figure 1). The luminal cells, which are in direct contact with the environment, include ciliated cells, mucus-secreting goblet cells, and club cells, another type of secretory cell. Basal cells are located under the luminal cells and closer to the basement membrane. Basal cells are specialized airway stem cells with a capacity to regenerate fully functional airway epithelium following injury (Rock et al., 2009). There are also submucosal glands in the proximal airways, which secrete mucus and water into the airway epithelial surface. In contrast, airway epithelium in the distal conductive airways is cuboidal and primarily consists of ciliated cells with few club cells and airway basal cells. Small airways are devoid of goblet cells and submucosal glands.
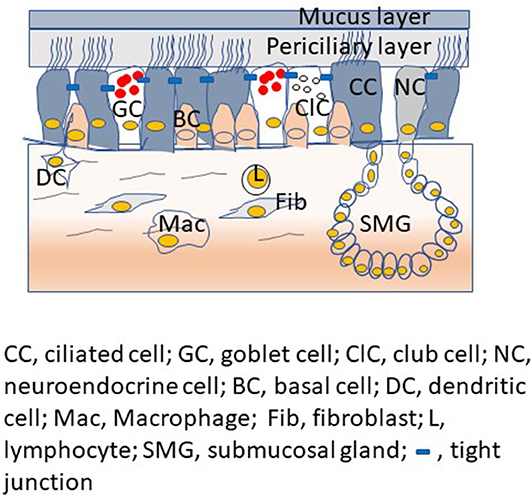
Figure 1. Proximal airway mucosa showing different type of airway epithelial cells with airway surface liquid, submucosal gland and professional innate immune cells in the submucosa.
Airway epithelium in the proximal airways is overlaid by airway surface fluid, which consists of a thin layer of periciliary less viscous liquid and a layer of semi-viscous mucous. The secretions of club cells and submucosal serous cells contribute to the periciliary fluid. On the other hand, the gel-forming mucins, MUC5AC and MUC5B expressed by goblet cells and the mucosal cells of submucosal glands contribute to the mucus layer (Ostedgaard et al., 2017). A recent study suggests that club cells may also be one of the sources for MUC5B in the proximal and distal airways (Okuda et al., 2019). Height and the viscosity of the periciliary fluid are critical factors as they determine the efficient unidirectional beating of cilia. The mucus layer, which is semi-viscous, traps the inhaled pathogens and other particulate materials and the cilia. Subsecuently, the mucus layer with its unidirectional movement sweep the trapped material toward pharynx for removal from the respiratory tract (Knowles and Boucher, 2002). There are also cell surface tethered mucin glycoproteins that interact with glycosaminoglycan keratin sulfate and this complex is assumed to act as a periciliary brush, creating another layer of protection and preventing the penetration of particulates and microorganisms (Button et al., 2012; Kesimer et al., 2013). The airway epithelial surface liquid also consists of a mixture of host defense molecules such as nitric oxide, lactoferrin, lysozyme, β-defensins, and cathelicidins. These molecules are secreted by airway epithelium, submucosal glands and other innate immune cells of the lungs (Singh et al., 2000; Ganz, 2002; Do et al., 2008). These host defense molecules are capable of killing bacteria, viruses and fungi directly or by limiting essential micronutrients required for successful colonization of pathogens; thus, these defense molecules function as a chemical shield against inhaled pathogens (Zasloff, 2002; Brogden, 2005). Furthermore, the tight and adherence junctional complexes located at the apicolateral border of airway epithelial cells also contribute to defense function by preventing paracellular invasion of inhaled pathogens. Paracellular permeability of airway epithelium is maintained through the cooperation of two mutually exclusive tight and adherence junctions. Tight junctions are located apically in the intercellular junctional complexes. These junctions inhibit water and solute flow, and regulate ion movement through the paracellular spaces (Schneeberger and Lynch, 1992; Pohl et al., 2009; Ganesan et al., 2013). Tight junctions also separate the apical from the basolateral cell surface domains to establish cell polarity (Shin et al., 2006). Adherence junctions mediate cell-to-cell adhesion and promote the formation of tight junctions. These intercellular junctions also prevent the inhaled microbes and allergens from crossing the airway epithelial barrier via paracellular route and activate professional innate immune cells present in the submucosa. Besides, intercellular junctions serve as platforms that regulate gene expression, cell proliferation, and differentiation (Balda and Matter, 2009; Koch and Nusrat, 2009).
In addition to acting as a physical barrier, airway epithelial cells actively participate in recognition of pathogens by various pattern recognition receptors (PRR), which are divided into transmembrane receptors and those localize to intracellular compartments. While the transmembrane receptors include Toll-like receptors (Mayer et al., 2007) and C-type lectins (Gavino et al., 2005; Heyl et al., 2014), the intracellular receptors include NOD like receptors (NLR) (Slevogt et al., 2007; Hirota et al., 2012; He et al., 2016; Han et al., 2019), RIG-I and MDA5 (Wang et al., 2009; Slater et al., 2010). Recognition of pathogen associated molecular patterns by PRR activates signal transduction pathways leading to the expression of antimicrobial factors such as defensins, cathelicidin, nitric oxide, interferons, and interferon-inducible factors (Vareille et al., 2011; Ganesan et al., 2013; Denney and Ho, 2018). These antimicrobial factors can kill the infecting pathogens directly without the involvement of other innate immune cells of the lungs. The airway epithelial cells upon recognition of pathogens also express pro-inflammatory chemokines and cytokines to recruit other innate immune cells to the airways to enhance elimination of the pathogens and resolve inflammation.
Airway epithelial cells being at the interface of the environment and the lung milieu are always in contact with the inhaled microbes and microbial products, and this may cause persistent inflammation of respiratory tissue even if the inhaled substance is not harmful. Since inflammation can cause tissue damage, frequent and persistent inflammatory responses may lead to airway remodeling and tissue destruction. To overcome this effect, airway epithelial cells also express intrinsic anti-inflammatory molecules such as inhibitory arachidonic acid metabolites, which include lipoxins, resolvins, and prostaglandin E2 (Bonnans et al., 2007; Levy and Serhan, 2014; Colby et al., 2016), suppressor of cytokine signaling (SOCS)1 and 3 (Niwa et al., 2018; Xander et al., 2019), anti-inflammatory cytokines (IL-10 and TGF-β), soluble cytokine receptors/receptor antagonists (sIL-1RN, sIL-13RA2, sTNFR2, IL-1RA), protease inhibitors (SLP1, SERPINA1, SERPINB1, TIMP-1), and immunosupressive cell surface molecules (B7-H1, B7-DC, IL-13RA2, and FASL) (Levine et al., 1997; Hamann et al., 1998; Kim et al., 2005; Matsumura et al., 2007). Many of these molecules are induced by pro-inflammatory cytokines and Th2 cytokines, suggesting that they may be regulated by negative feedback pathways to dampen the inflammation. Therefore, dysregulation or imbalance of inflammatory to anti-inflammatory signaling can result in persistent or overt lung inflammation, ultimately leading to irreversible tissue damage.
There is also ample evidence that airway epithelial cells communicate with other innate immune cells via secreted molecules to clear pathogens and maintain homeostasis. Under unstimulated conditions, bronchial epithelial cells secret transforming growth factor (TGF)-β, which in turn (a) inhibits LPS-induced TNF-α in monocytes and macrophages, (b) induces expression of IL-10 and arginase-1 in dendritic cells, an indication of alternative activation, and (c) inhibits the proliferation of T cells (Mayer et al., 2008). Bronchial epithelial cell expression of programmed death receptor-ligand1 (PD-L1) in response to respiratory syncytial virus activates and promotes proliferation and antiviral function of CD8+ve T cells in a co-culture model of bronchial epithelial cells and CD8+ve T cells (Telcian et al., 2011). In a co-culture model of bronchial epithelial cells and dendritic cells, epithelial cells secretes GM-CSF, GCSF, and VEGF in response to respiratory virus infection and this enhances type I IFN expression in plasmacytoid dendritic cells that is critical for viral clearance (Rahmatpanah et al., 2019). Under an allergic environment, airway epithelial cells express thymic stromal lymphopoietin (TSLP) and IL-33 in response to viral infection and promote generation of TH2 cells (Mehta et al., 2016). Airway epithelial cells express TSLP, IL-33, and IL-25 in response to LPS and house dust mite allergen and cause dendritic cell activation and allergic inflammation (Hammad et al., 2009). These findings indicate that the interactions of airway epithelial cells with other innate immune cells plays a critical role in shaping the overall local innate immunity in the lungs.
Rhinovirus
Rhinoviruses (RVs) belong to the Picornaviridae family and are responsible for the majority of the common colds (Makela et al., 1998). In patients with underlying lung disease such as COPD, asthma, and cystic fibrosis, RV exacerbates the respiratory illness (Lee et al., 2012; Etherington et al., 2014; George et al., 2014; Stolz et al., 2019). RVs are small (27 to 30 nm), non-enveloped viruses with an icosahedral capsid and a single-stranded positive-sense RNA (+ssRNA) genome made up of 7,200 bases (Palmenberg and Gern, 2015). So far, more than160 serotypes of RVs have been discovered. They have been divided based on phylogeny into three species, Rhinovirus-A (RV-A), Rhinovirus-B (RV-B), and Rhinovirus-C (RV-C). Whereas RV-A and RV-B have smoother, spherical capsid structure, RV-C has 60 dominant spike-like protrusions, or “fingers,” on the outer surface of the virion (Liu et al., 2016). RV-C has a large deletion in VP1, one of the structural proteins, and it lacks a protruding “plateau” around each of the 5-fold vertices, a characteristic feature of RV-A and RV-B (Basta et al., 2014; Liu et al., 2016).
RV-As include 80 serotypes, RV-Bs include 32 serotypes, and the recently found RV-C species contains at least 57 serotypes (http://www.picornaviridae.com). All three RV subgroups bind to plasma membrane glycoproteins to gain entry into the cells. Historically, RV-A and -B strains are classified into two groups depending on their cellular receptor utilization for internalization into the cells. Approximately 89 serotypes of RV-A and -B species belong to the major group and bind to human ICAM-1 (Greve et al., 1989); thus, showing the species specificity. The minor group RVs, which consist of at least 12 serotypes of RV-A, bind to the low-density lipoprotein receptor (LDLR) family with no species specificity. The receptor binding sites were mapped around the 5-fold axis of symmetry in viral capsid, but they have also shown to be different between the major and minor group RVs. The first domain of ICAM-1 binds the virus inside the canyon (a 2.5 nm depression) surrounding the dome at the vertex (Kolatkar et al., 1999). In contrast, the LDLR ligand-binding domain, composed of multiple ligand-binding repeats, attaches to the top of the star-like surface-exposed structure at the vertex (Hewat et al., 2000). These binding interactions are required for entry into the host cells by endocytosis. In addition to these receptors, RV-A and RV-B also interact with TLR2 on airway epithelial cells and macrophages, and this interaction modulates RV-induced innate immune responses (Unger et al., 2012; Ganesan et al., 2016; Bentley et al., 2019; Xander et al., 2019). Binding to TLR2 may not be necessary for viral entry into the host cells. More recently discovered RV-C binds to CDHR3, a member of the cadherin family of transmembrane proteins, which mediates RV-C entry into host cells. An asthma-related mutation (Cys529 → Tyr) in CDHR3 is associated with increased viral binding and progeny yields in vitro, and it is proposed to be due to increased cell surface expression of this receptor (Bonnelykke et al., 2014; Bochkov et al., 2015). Further, the extracellular domains 1-3 of CDHR3 was shown to mediate RV-C interaction with host cells (Watters and Palmenberg, 2018). However, the mechanisms of entry or replication of RV-C using CDHR3 are so far unknown.
The RV genome encodes four structural proteins, VP1, VP2, VP3, and VP4, which make up the viral capsid, and seven non-structural proteins, 2A, 2B, 2C, 3A, 3B, 3C, and 3D, which participate in RV genome replication and polypeptide processing. The viral life cycle involves viral capsid binding and entry into the airway epithelial cell, the release of viral genome into the cytoplasm, transcription and translation of the viral genome, polypeptide processing and subsequently virion assembly. The VP1 structural protein mediates viral binding to host cell surface (Palmenberg and Gern, 2015). RV-A and RV-B binding to their receptors initiates the process of receptor mediated endocytosis, which allow particle entry into the host cell and trigger genome uncoating within the endosome (Fuchs and Blaas, 2010). These receptors direct virus uptake via clathrin-dependent or independent endocytosis or via macropinocytosis (Fuchs and Blaas, 2010; Blaas, 2016). Acidic pH in the endosomes triggers uncoating of the virus and release of viral RNA into the cytoplasm. The viral genome is transcribed by viral 3D RNA polymerase. The genome is translated with host factors, and the polypeptide is subsequently processed by viral 2A and 3C proteases (Fuchs and Blaas, 2010). This is followed by virion assembly, genome packaging, and virus shedding, either by lytic or non-lytic processes. Not much is known about the replication cycle of RV-C.
Innate Immune Responses of Airway Epithelial Cells to Rhinovirus Infection
As discussed above, airway epithelial cells express cell surface and cytoplasmic PRRs. Amongst these PRRs, TLR2 is expressed on the cell surface, TLR3 and TLR7 in the endosomes and MDA5 and RIG-I in the cytoplasm. These PRRs are implicated in RV-induced innate immune responses in airway epithelial cells.
Replication-Dependent and Independent Pro-inflammatory Responses
RV primarily infects airway epithelial cells. RV binds to both ciliated and non-ciliated cells (Jakiela et al., 2008; Lopez-Souza et al., 2009; Faris et al., 2016; Griggs et al., 2017; Tan et al., 2018; Warner et al., 2019) in the airway epithelium. RV infection causes very little cellular damage suggesting that immediate innate defense mechanisms of airway epithelial cells can efficiently inhibit viral replication; thus, establishing an antiviral state. The airway epithelial cells express chemokines such as CXCL-1, CXCL-5, and IL-8 in response to RV interaction with its glycoprotein receptors and viral endocytosis (Newcomb et al., 2005; Sajjan et al., 2006; Bentley et al., 2007). Inhibition of phosphoinositide (PI)-3 kinase or TLR2 reduces RV-induced CXCL-8 expression (Newcomb et al., 2005; Bentley et al., 2007; Unger et al., 2012) (Figure 2). Interaction of viral genome with endosomal TLR7, which recognizes single-stranded RNA, may also induce pro-inflammatory cytokines (Triantafilou et al., 2011; Tan et al., 2018). At later stages of infection, expression of these chemokines is also stimulated via activation of TLR3, MDA5 and RIG-I signaling by dsRNA, an intermediate generated during viral replication (Hewson et al., 2005; Sajjan et al., 2006; Slater et al., 2010; Triantafilou et al., 2011). Airway epithelial cells also express and secrete growth factors, such as amphiregulin, epithelial cell growth factor (EGF) and epiregulin all of which activates EGF receptor signaling to increase IL-8 and ICAM-1 levels. Inhibition of EGF receptor, Erk1/2 or STATs attenuates RV-induced IL-8 in the airway epithelial cells (Liu et al., 2008). Upregulation of CXCL-8 and granulocyte colony stimulating factor was also observed in the nasal secretions of healthy human volunteers after the experimental infection with RV (Grunberg et al., 1997; Gern et al., 2000), and these responses correlated with neutrophil infiltration in the nasal cavity (Grunberg et al., 1997). Even though these early pro-inflammatory responses are necessary, they must be tightly regulated to prevent excessive inflammation and subsequent tissue damage. Dual specificity phosphatases, which inhibit various mitogen-activated protein kinase (MAPK), toll-interacting protein (Tollip), a negative regulator of TLR signaling pathway, and TLR2-activation mediated IRAK-1 degradation within a short period after RV infection may all contribute to limiting RV-induced inflammation (Unger et al., 2012; Manley et al., 2019; Dakhama et al., 2020).
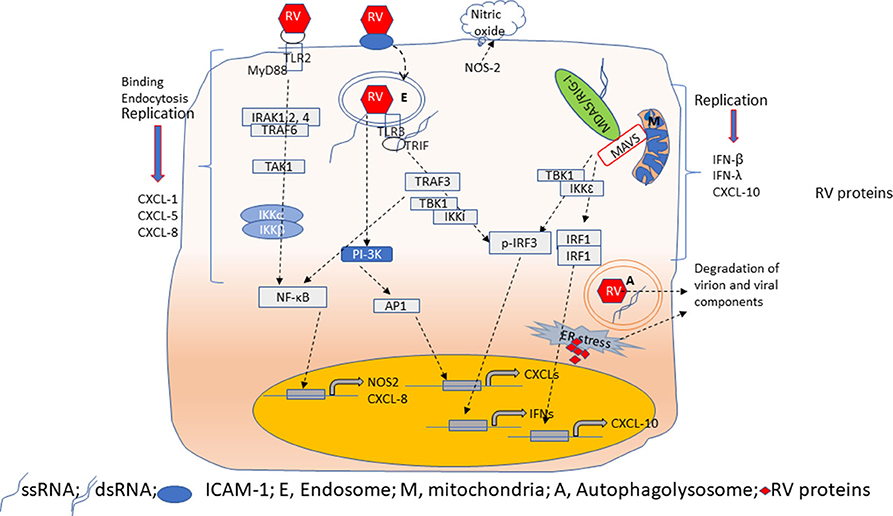
Figure 2. Overview of innate immune responses of airway epithelial cells to RV. Airway epithelial cells show replication-independent CXCL responses which occurs as a result of activation PI-3 kinase/AP1 following binding and endocytosis of RV. Interaction of RV with TLR2 may induce CXCL-8 and NOS-2 expression via MyD88 signaling pathway. NOS-2 generates nitric oxide by catalysis of arginine. CXCL responses are also elicited when TLR3 binds to RV dsRNA via NF-κB. In contrast, RV-stimulated IFN and CXCL-10 responses are primarily due to dsRNA generated during RV replication. Binding of dsRNA to TLR3 induces IFNs via TBK1/IKKi/IRF3 activation. On the other hand, cytoplasmic receptors MDA5 or RIG-I upon binding to dsRNA translocate and interact with MAVS expressed on mitochondria. This interaction triggers activation of TBK1/IKKε to induce expression of IFNs and CXCL-10 via IRF3 and IRF1/NF-κB, respectively. During viral replication, accumulation of RV proteins and RV genome activates ER stress and induces autophagy. These processes degrade RV proteins to limit virion assembly.
Replication-Dependent Responses
Airway epithelial cells express CXCL-10/IP-10, and type I and type III interferons (IFNs), such as IFN-β, IFN-λ1, λ2, and λ3 in response to replication- efficient RV infection (Figure 2). CXCL-10 recruits activated type 1 T lymphocytes and natural killer cells to the site of infection, which can recognize and kill the infected cells. IFNs, in addition to amplifying type I and III IFN responses, also stimulate expression of interferon-incuced genes such as viperin, MX-1, ISG-15, and double-stranded (ds)-RNA receptors (TLR3, MDA-5, and RIG-I) to establish a local antiviral state. Expression of CXCL-10 and antiviral IFNs in airway epithelial cells is stimulated primarily by dsRNA intermediate generated during RV replication; thus, replication-deficient RV does not stimulate these responses (Wang et al., 2009; Schneider et al., 2010). Airway epithelial cells constitutively express TLR3 in the endosomes; therefore, TLR3 may participate in the early detection of viral dsRNA leading to IRF3 and NF-κB activation followed by expression of CXCL-10 and IFNs (Wang et al., 2009; Slater et al., 2010). Cytoplasmic ds-RNA receptors, MDA5 and RIG-I are IFN-inducible genes. In airway epithelial cells, while RIG-I is constitutively expressed, MDA5 is expressed only after RV infection (Wang et al., 2009 #51; Gimenes-Junior et al., 2019a #21). Interaction of these receptors with viral dsRNA in the cytoplasm enhances expression of IFNs (Wang et al., 2009; Slater et al., 2010; Zaheer et al., 2014). Although RV-stimulated expression of both CXCL-10 and IFNs are induced by activation of dsRNA receptors, CXCL-10 expression is regulated by IRF-1, and NF-κB, but not IRF3 (Spurrell et al., 2005; Zaheer and Proud, 2010). In contrast, the dsRNA-induced expression of IFNs is regulated primarily by IRF3. Both MDA5 and RIG-I are helicases with the CARD domain and interact with the CARD domain of MAVS after recognition of dsRNA in the cytoplasm. This interaction activates IKKα and IKKε to stimulate the expression of CXCL-10 and IFNs espression, respectively, but the underlying reasons for this redundancy are not well-known. Since viral dsRNA is longer and MDA5 recognizes high molecular weight or long dsRNA, it is conceivable that MDA5 may play a significant role in stimulating IFN responses to RV infection. Consistent with this notion, we demonstrated that knockdown of MDA5, but not RIG-I significantly reduced RV-induced IFNs (Wang et al., 2009 #51). In support of our findings, a child with inherited MDA5 deficiency was found to have recurrent RV infection (Lamborn et al., 2017). Nasal epithelial cells from this patient showed an increase in viral transcripts compared to her parents' nasal epithelial cells when challenged with RV. Such an increase in viral transcripts was not observed when the patient's nasal cells were infected with RV. These observations indicate that MDA5 may play a pivotal role in controlling the RV replication in airway epithelial cells via the induction of IFNs.
In response to RV infection, although airway epithelial cells express both type I and type III interferons, the expression of type III IFNs such as IFN-λ1 (IL-29) and IFN-λ2 (IL-28A) is more robust. IFN-λs are expressed at both mRNA and protein levels (Schneider et al., 2010; Chattoraj et al., 2011). While receptors for type I IFNs are expressed in a variety of cell types, type III IFN receptors are expressed only on epithelial cells (Mordstein et al., 2008; Sommereyns et al., 2008). Mice lacking type III IFN receptor show greater susceptibility to influenza A virus, influenza B virus, respiratory syncytial virus, human metapneumovirus, and coronavirus (Mordstein et al., 2010). Therefore, it is conceivable that type III IFNs may also be essential for controlling viral replication in RV-infected airway epithelial cells. Airway epithelial cells express several IFN-inducible genes (ISG) following RV infection (Proud et al., 2008). So far, only viperin and ISG-15 have been investigated for their innate immune properties in the context of RV infection. Whereas knockdown of viperin increased viral load, knockdown of ISG-15 enhanced CXCL-10 production without affecting viral titer in RV-infected airway epithelial cells (Proud et al., 2008; Zaheer et al., 2014), indicating a role of ISGs in inhibiting viral replication and modulation of innate immunity.
Oxidative Stress and Innate Immunity
Another arm of innate immunity in airway epithelial cells is oxidative stress. Airway epithelial cells in response to RV infection express nitric oxide synthase−2 (NOS-2) in vitro and in subjects experiencing symptomatic colds (Sanders et al., 2001). NOS-2 generates nitric oxide, a potent antiviral agent. Human volunteers showed increase in nitric oxide generation in their nasal cavities after experimental infection with RV (Sanders et al., 2004). Exhaled nitric oxide correlated inversely with viral titer at 4 days post-RV infection in these volunteers. Later, nitric oxide was demonstrated to negatively regulate RV-induced CXCL-10 via NF-kB and IRF-1 downregulation (Spurrell et al., 2005; Koetzler et al., 2009; Zaheer et al., 2009); thus, implicating immunomodulatory role for nitric oxide in addition to antiviral property. Kaul et al. reported that replication-deficient RV induces reactive oxygen species via p47-phox while neutralization of reactive oxygen species reduced RV-stimulated IL-8 in these cells (Kaul et al., 2000). We demonstrated that RV transiently disrupts barrier function in polarized and mucociliary-differentiated airway epithelial cells in vitro and in a mouse model of RV infection in vivo (Sajjan et al., 2008). The disruption of barrier function was dependent on RV-induced oxidative stress via NADPH oxidase (NOX)1 and Nod-like receptor (NLR)X-1 (Comstock et al., 2011; Unger et al., 2014). Airway epithelial cells, which are already under oxidative stress due to preexisting conditions such as bacterial infection or persistent epithelial to mesenchymal transition show reduced IFN expression and higher viral loads in response to RV infection (Chattoraj et al., 2011; Yang et al., 2017). Further, induction of oxidative stress by pretreatment with hydrogen peroxide also attenuated antiviral responses in airway epithelial cells obtained from patients with asthma and COPD, but not healthy subjects. This is because the normal cells express antioxidant genes, such as superoxide dismutase 1 and 2 (Menzel et al., 2019). Our own studies indicate that pretreatment with antioxidant, diphenylene iodonium (DPI) suppresses RV-induced IFN responses in cells from healthy subjects (Sajjan et al., unpublished data). More recently, nasal epithelial cells were demonstrated to express higher IFNs than bronchial cells in response to RV infection, and this was due to increased expression of NRF2-mediated antioxidant response in bronchial epithelial cells (Mihaylova et al., 2018). Conversely, treatment of epithelial cells which are already under oxidative stress with DPI or MITOTEMPO increased IFN responses to normal levels and reduced the viral titers following RV infection (Chattoraj et al., 2011; Gimenes-Junior et al., 2019b). Reduced expression of IFNs in cells under oxidative stress is due to the unavailability of MAVS to the dsRNA receptor. These observations suggest that while induction of oxidative stress during viral infection improves antiviral responses and modulate innate immune responses, preexisting oxidative stress may reduce the antiviral IFN responses.
Endoplasmic Reticulum Stress in Innate Immunity
The endoplasmic reticulum (ER) supports various processes in the eukaryotic cells. ER membrane provides ample surface area for cellular reactions such as the production of proteins, steroids and lipids, as well as protein folding and secretion. During cell proliferation and differentiation, the ER may be overloaded by molecular chaperones, folding enzymes and massive protein products causing ER stress. Furthermore, pathophysiological stress signals such as viral infection, accumulation of misfolded proteins, hypoxia ER-Ca2 depletion can cause ER stress (Kaufman, 1999; Koumenis, 2006; Jheng et al., 2010; Song et al., 2019). To overcome the ER stress, the eukaryotic cells have developed an adaptive response known as unfolded protein response (UPR) to reduce the load of newly synthesized proteins and misfolded proteins by increased expression of molecular chaperones. Additionally, the proteins that fail to fold are degraded by ER degradation pathway. UPR related ER degradation involves proteasomal degradation and macroautophagy (Fujita et al., 2007; Korolchuk et al., 2010). The UPR consists of three parallel pathways involving 3 different misfolded protein sensors, ATF6, ER-resident integral membrane protein (IRE)1 and proteins kinase RNA (PKR)-like ER kinase (PERK). The precise mechanism of how these three sensors are activated is not well-understood. However, there is a general consensus that the ER luminol chaperone glucose-regulated protein (GRP)78/BIP is normally bound to these sensors to prevent the activation of UPR. When the GRP-78 senses or binds to misfolded proteins, it dissociates from the sensors to activate UPR signaling (Bertolotti et al., 2000; Shen et al., 2002; Kelsen, 2016). Activation of any arm of the UPR increases GRP78 expression via feedback mechanism and this indicates ER stress.
During the viral life cycle, ER stress may arise from the exploitation of ER membrane, accumulation of misfolded proteins, ER calcium imbalance, and depletion of ER membrane during virion release. Many positive strand RNA viruses, including RV have been shown to cause UPR-related ER stress (Su et al., 2002; Tardif et al., 2004; Jheng et al., 2010). Recently RV was shown to increase the expression of GRP78 as early as 6 h post-infection, suggesting RV induces ER stress and this was associated with the activation of PERK and ATF6 arms of UPR, and also an increase in the expression of viral structural protein, VP2 (Song et al., 2019). Moreover, RV 2B protein, which participates in the processing of RV polypeptide during viral replication co-localized with ER membrane. Conversely, RV-stimulated ER stress also induced apoptosis in the cells indicating that ER stress may be required for limiting viral replication. All the work was performed in H1HeLa cells, which are highly susceptible to RV infection; therefore, the consequences of RV-induced ER stress in relevant airway epithelial cells are not known. Enterovirus (EV)71, which is closely related to RV, was also demonstrated to induce ER stress via activation PERK. As observed with RV, EV71 also increased the expression of GRP78. However, overexpression of GRP78 significantly reduced the viral progeny and activation of PERK. These observations suggest that like in RV-infected H1HeLa cells, induction of ER stress and UPR activation by EV1 may be necessary for limiting viral replication; thus, comprising one of the arms of innate antiviral immunity.
Role of Autophagy in Antiviral Immunity
As described above, ER stress and autophagy are closely related and play an important role in maintaining homeostasis in all eukaryotic cells by mediating the removal of harmful cytoplasmic components, including misfolded proteins, and recycling of the nutrients (Yorimitsu and Klionsky, 2005; Senft and Ronai, 2015). During autophagy, aberrant cytoplasmic constituents and damaged organelles are enveloped in a double membrane vesicles. These vesicles will fuse with the lysosomes to form autophagolysosomes, the components in the autophagolysosomes will be degraded by hydrolases, and the nutrients such as amino acids are recycled (Mijaljica et al., 2011; Feng et al., 2014). In addition to ER stress response, autophagy is also initiated by oxidative stress, nutrient deprivation, and type I IFNs (Ambjorn et al., 2013; Schmeisser et al., 2013; He et al., 2018; O'Grady, 2019). The first step in autophagy is the formation of an isolation membrane at the preautophagosomal site and the membrane originates from ER (Axe et al., 2008). This step is followed by the elongation phase, which involves continuous processing by two ubiquitin-like protein-conjugation systems, the Atg12 and Atg8/LC3 system. Similar to ubiquitination, Atg12 is conjugated to Atg5 by Atg7 and Atg10. Atg7 and Atg3 mediates the conjugation of Atg8 to phosphatidylethanolamine. Both Atg12-Atg5 complex, and Atg8 localize to developing autophagosome and then Atg12-Atg5 complex facilitates lipidation of Atg8/LC3 and directs its subcellular localization. The latter may act as a scaffold protein that supports membrane expansion, and the amount of LC3 protein in the membrane those correlates with the size of autophagosome (Geng and Klionsky, 2008). The mammalian target of rapamycin complex 1 (mTORC1) is the master regulator of autophagy and regulated by nutrient starvation, cellular stressors and growth factors. During nutrient deprivation, AMP-activated kinase (AMPK) induces autophagy by suppressing mTORC1 (Zachari and Ganley, 2017). PI3-kinase/AKT signaling pathway activated by growth factors or cellular stress can also suppress mTOR to induce autophagy. Type I IFNs bind to their receptors leading to Janus kinase and non-receptor tyrosine protein kinase-mediated phosphorylation of insulin receptor substrate 1 and 2, which in turn activate PI3K-AKT. AKT not only inhibits mTOR but also stimulates the expression of a variety of autophagy genes (Mammucari et al., 2008).
Given the degradative function of autophagy, it is conceivable that viral components, viral particles and, host proteins necessary for viral replication can all be captured and removed by autophagy. Thus, autophagy may be an integral part of innate antiviral response. For example, picornaviruses can be detected by galectin 8, which induces autophagy to degrade the viral genome; thus, restricting the viral replication (Staring et al., 2017). P62, a protein that carries cargo to autophagosomes for degradation, binds to Sindbis virus capsid proteins and targets to autophagic degradation (Orvedahl et al., 2010). Induction of autophagy following RV infection has been demonstrated in H1HeLa cells and inhibition of autophagy with 3-methladenine increased viral titer (Jackson et al., 2005; Klein and Jackson, 2011), but the mechanisms underlying how RV subverts autophagy to its own benefit is yet to be determined. Other viruses in the Picornaviridae family such as poliovirus and coxsackievirus also subvert autophagy to promote viral replication either by degrading the P62 or by hijacking other host proteins that block galectin 8 (Shi et al., 2013). Similar mechanisms may be used by RV to evade autophagic degradation particularly in susceptible H1HeLa cells (Jackson et al., 2005; Klein and Jackson, 2011). Our on-going studies indicate that RV induces autophagy in airway epithelial and it is not known whether it is beneficial to host or to virus (Sajjan et al., unpublished observations).
Most of the studies to understand the innate immune responses are conducted with either non-epithelial H1HeLa cells or submerged cultures of bronchial epithelial cell lines. Although these cell culture systems are suitable for mechanistic studies, mucociliary-differentiated cultures which closely resemble airway epithelium in vivo may reflect the in vivo responses more accurately. Unlike submerged cultures, mucociliary-differentiated cell cultures are polarized, and thus separates basal (submucosal side) from the apical (luminal) side with regards to receptor expression and responses to infection. For example, nitric oxide is generated primarily by ciliated cells in response to infection; therefore, effects of nitric oxide in the context of RV infection may not be possible in submerged cultures. CDHR3, a receptor for RV-C is only expressed on ciliated cells; therefore, undifferentiated airway epithelial cells may not be useful in studying responses to RV-C unless the cells are genetically modified (Griggs et al., 2017; Everman et al., 2019). A recent study showed that in mucociliary-differentiated cells, RV induces different vectoriality of release for IFNs (mainly apical), CXCL-10, and IL-17C (mainly basolateral) (Jamieson et al., 2019; Warner et al., 2019). Receptors for IFNs are expressed on the apical surface; therefore, apically released IFNs will readily interact with their receptor to establish antiviral state in the neighboring uninfected cells. On the other hand, basolateral expressed CXCL-10 and IL-17C may play a role in recruitment of T cells, neutrophils, and immature monocytes to submucosa to protect submucosal tissue from the invading virus. CXCL-8, a potent neutrophil chemoattractant is expressed at both apical and basolateral surface after RV infection (Sajjan et al., unpublished observations) and this may create a cytokine gradient for the recruitment of neutrophils into the airway lumen. These studies indicate the advantages of using mucociliary-differentiated cultures in the studies related RV infection.
Interaction of Airway Epithelial Cells With Hematopoietic Cells During RV Infections
In the lung, airway epithelial cells function in concert with other innate immune cells. There are some studies demonstrating the interaction of airway epithelial cells with recruited hematopoietic cells in the context of RV infection. Although RV bind and enter the monocytes and macrophages, it does not replicate in these cells (Gern et al., 1996). However co-culturing of airway epithelial cells with monocytes increases ICAM-1 expression that promotes RV replication in monocytes, and this was mediated through secreted products by airway epithelial cells (Zhou et al., 2017). In response to RV, airway epithelial cells also secrete CXCL-1, CXCL-8, and CXCL-10 which are potent chemoattractants for neutrophils and immature monocytes, the latter cell type express high amounts of IL-6, CXCL-8, and IFNs in response to RV when co-cultured with epithelial cells (Zhou et al., 2017). Although the role of neutrophils in viral clearance is not known, neutrophils suppress IL-6 and CXCL-8 secretion from monocytes (Tang et al., 2016); therefore, neutrophils may limit RV-induced overt inflammation in vivo. Nasal epithelial cells express programmed death ligand (PD)-L1 and PD-L2 in response to RV (Heinecke et al., 2008). These ligands are co-stimulatory molecules and upon ligation to its receptor PD-1 inhibit activation of T and B cells. In a co-culture model of airway epithelial cells and T cells, the airway epithelial cells express PD-L1 and PD-L2 in response to RV, this in turn inhibits T cell activation and expression of IFN-γ, which is indeed necessary for killing virus-infected cells. On the other hand, PD-1 activity is required for the termination of the late phase of allergic inflammation activation (Deppong et al., 2006). Therefore, depending on the specific T cells being engaged, airway epithelial expression of PD-L1 and PD-L2 may be either detrimental or beneficial to host. More recently, bronchial epithelial cells from patients with asthma have been shown to express IL-25 in response to RV, and this can enhance Th2 inflammation (Beale et al., 2014). Secretion of TSLP from airway epithelial cells can drive expansion of type 2 innate lymphoid cell (ILC2) expansion and mucus metaplasia during experimental RV infection in baby mouse model of asthma (Hong et al., 2014; Han et al., 2017). Nasal mucosal epithelial cells express IL-15 following experimental RV infection in humans, which in turn activates CD8+ and natural killer cells to promote the expression of IFN-γ that may contribute to viral clearance (Jayaraman et al., 2014). We have demonstrated that in the absence of TLR2 signaling, RV stimulates exaggerated CXCL-10 expression in airway epithelial cells via activation of IL-33/ST2 signaling axis (Ganesan et al., 2016). COPD airway epithelial cells show heightened CXCL-10 expression in response to RV and this can be attributed to attenuated TLR2 signaling (Schneider et al., 2010; Xander et al., 2019). In a mouse model of COPD RV-induced CXCL-10 promotes excessive recruitment and activation of CD8+ve cells and intermediate macrophages to cause progression of lung disease (Gimenes-Junior et al., 2019a). These observations indicate that airway epithelial cells interact with variety of innate immune cells during RV infection and this can be either beneficial or detrimental to the host depending upon the microenvironment in the lungs.
Antiviral Immunity is Dysfunctional in Airway Epithelial Cells From Patients With Chronic Obstructive Pulmonary Disease
Epithelium in the airways of patients with COPD often show remodeling, including goblet cell and basal cell metaplasia and sometimes squamous metaplasia (Rock et al., 2010; Gohy et al., 2019). We and others have demonstrated that airway basal/stem cells isolated from COPD bronchi regenerate airway epithelium that is structurally different from the normal in showing basal cell and goblet cell hyperplasia (Schneider et al., 2010; Gohy et al., 2015; Ghosh et al., 2018; Jing et al., 2019). The regenerated COPD airway epithelial cell cultures produce more than normal IL-6, CXCL-1, and CXCL-8, indicating that they may maintain pro-inflammatory phenotype (Schneider et al., 2010). Such differences in regenerated airway epithelial phenotype may arise from the acquired defective repair mechanisms in airway basal cells. Additionally, it may also significantly affect innate immune responses. Consistent with this notion, we found that mucociliary-differentiated COPD airway epithelial cells show exaggerated pro-inflammatory and antiviral responses to RV infection (Schneider et al., 2010). Despite having higher type I and type III IFN responses as well as IFN-stimulated gene expression, COPD cells showed higher viral load. However, both normal and COPD cell cultures completely cleared the virus by 7 days post-infection. Since cigarette smoke is thought to be the primary risk factor for the development of COPD, some studies have used cigarette smoke-exposed airway epithelial cells to investigate the altered innate immune responses to RV. Normal airway epithelial cells acutely exposed to cigarette smoke extract showed downregulation in all the antiviral innate immune responses and an increase in viral load in response to RV infection (Eddleston et al., 2011; Proud et al., 2012; Hudy and Proud, 2013). Similar reduced antiviral responses and an increase in viral load was observed in mucociliary-differentiated cell cultures after acute exposure to RV (Berman et al., 2016). The observed discrepancy may be explained based on the acute vs. years of smoking-induced changes that occur in COPD patients.
One of the mechanisms underlying higher IFN responses is due to reduced expression of SIRT-1, which is required for limiting the amplification phase of IFN expression that occurs as a result of activation of IFN receptor signaling (Xander et al., 2019) (Figure 3). Furthermore, SIRT-1 expression following RV infection requires functional TLR2, and the TLR2 signaling pathway is dysregulated in COPD airway epithelial cells. Another important feature that we observed was at later stages of RV infection that is at 14 days post-infection, COPD, but not normal cell cultures showed further increase in the number of goblet cells and expression of mucin genes (Jing et al., 2019). This was due to persistent activation of NOTCH signaling in COPD cell cultures following RV infection. Interestingly, at this stage, there was no detectable RV indicating that RV infection may induce changes that are sustained in COPD cells even after the virus cleared. This may be due to defective anti-inflammatory or repair pathways, and needs further investigation.
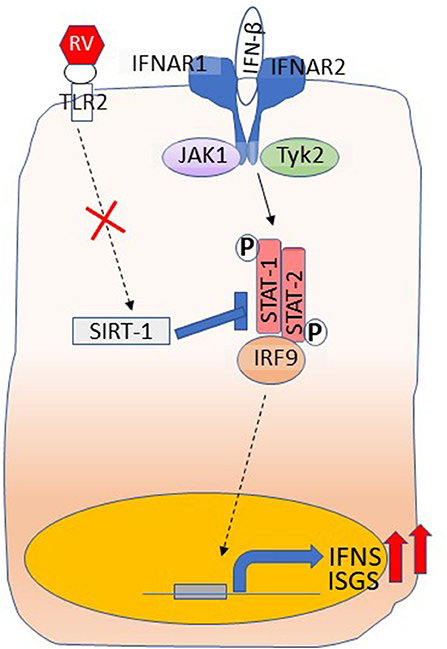
Figure 3. Dysregulated IFN responses in COPD airway epithelial cells. Type I and III IFNs bind to their respective receptors and stimulate the expression of ISGs as well amplify the expression of IFNs through activation of JAK-STAT pathway. RV interaction with TLR2 induces the expression of SIRT-1, which inhibits STAT activation to limit amplification of IFNs and the expression of ISGs and this arm is dysregulated in COPD. Shown in the figure is binding of IFN-β to its receptors, IFNAR1 and IFNAR2. Interaction of IFN-λs with its receptors IFNLR1 and IL10R2 activates JAK-STAT signaling similar to IFN-β.
Innate Immune Responses of Bronchial Epithelial Cells From Patients With Asthma and CF to RV Infection
Airway epithelium in patients with asthma and cystic fibrosis (CF) also show remodeling with goblet cell metaplasia and respond abnormally to RV infection. Based on the outcome of in vitro and in vivo studies, it is thought that RV may enhance Th2 cytokines via the expression of TSLP and IL-25 derived from airway epithelial cells; thus, exacerbating the lung disease in patients with asthma (Kato et al., 2007; Uller et al., 2010; Han et al., 2017). There are also conflicting data with regards to antiviral IFN responses to RV in patients with asthma. Some studies demonstrated that primary airway epithelial cells from asthma are defective in mounting antiviral IFNs, particularly type III IFNs expression in response to RV (Wark et al., 2005; Contoli et al., 2006). Later on, other studies showed no difference in IFN responses to RV infection between the normal and asthmatic airway epithelial cells (Bochkov et al., 2010; Bai et al., 2015; Ravi et al., 2019). As observed in COPD cells, airway epithelial cells from asthmatic subjects showed higher expression of certain genes, such as ICAM-1 Inhibin beta, LOXL2A, IL-6, and IL-1β at basal levels (Bochkov et al., 2010). Following RV infection, although there was no difference in the viral clearance between the normal and asthmatic airway epithelial cells, the latter showed some differentially regulated genes, which include CCL5, CXCL10, and CX3CL1, MUC5AC, CDHR3 (Bai et al., 2015). Moreover, bronchial cells from experimentally-infected asthmatic patients showed higher IFNs, which correlated with higher viral load in the nasopharyngeal cavity (Ravi et al., 2019). The observed differences between these studies may be due to polymorphisms that have been recently reported to affect innate immune responses to RV infection. Polymorphism in chromosome 17q21, which positively correlates with orosomucoid like 3 (ORMDL3) expression confers the major genetic susceptibility not only to onset of childhood asthma, but also to RV infection-associated wheezing (Moffatt et al., 2007; Caliskan et al., 2013). ORMDL3 inhibits serine palmitoyltransferase, and the expression of ORMDL3 in epithelial cells was associated with a reduction in RV-induced ER stress and IFN responses and concomitant increase in viral load (Liu et al., 2020). While the pharmocologic induction of ER stress reduced viral replication and increased IFN expression in ORMDL3 expressing cells, inhibition of serine palmitoyltransferase negated the effects of ORMDL3 knockdown. These results indicate that ORMDL3 may negatively regulate ER stress- and sphingolipid-associated antiviral immunity. However, how sphingolipid regulates antiviral immunity is not known.
IFN responses to RV infection in CF airway epithelial cells is also controversial. While one study demonstrated reduced IFN responses to RV infection (Vareille et al., 2012), we and others have demonstrated that RV induces higher than normal IFN responses in mucociliary-differentiated CF bronchial epithelial cells (Chattoraj et al., 2011; Dauletbaev et al., 2015). We further demonstrated that prior infection with Pseudomonas aeruginosa which causes chronic infection in CF airways, reduces RV-induced IFN responses in CF but not in normal airway epithelial cells, and this is due to enhanced oxidative stress.
Conclusions
Conductive airway epithelial cells, the primary target of RV, not only function as a physical barrier but also plays a pivotal role in mounting appropriate innate immune responses. Airway epithelial cells are equipped with layers of antiviral immune mechanisms. Nitric oxide that is generated immediately after an infection has potent antiviral activity and modulates inflammatory cytokine expression to avoid overt inflammation. Type I and type III IFNs-stimulated genes, in addition to interfering with viral replication also limit virus-induced pro-inflammatory cytokines to prevent excessive inflammation. Although controversial, some studies correlated reduced IFN expression with higher viral load in patients with asthma. Even though, prophylactic treatment with IFNs limits influenza viral replication in vitro (Watson et al., 2020 #215), it had no significant effect on asthma symptoms in a small clinical trial (Djukanovic et al., 2014 #214). In contrast, overexpression of these cytokines may trigger unwanted inflammation. Therefore, the airway epithelial cells have evolved to overcome this effect by expressing anti-inflammatory or inhibitors of signaling pathways. In the context of RV infection, TLR2 signaling appears to play an important role in limiting overexpression of RV-induced IFNs via SIRT-1 expression. SIRT-1 inhibits STAT-1/2, which occurs during the amplification of IFNs via interaction of IFNs with its receptors. This mechanism is dysregulated in COPD airway epithelial cells, which show heightened expression of IFNs in response to RV. Airway epithelial cells-derived chemokines recruit a variety of innate immune cells, which detect and clear virus-infected epithelial cells and apoptotic cells, establish antiviral state and resolve the acute inflammation caused by RV. If these protective innate immune responses of airway epithelial cells to RV are not tightly regulated, it can lead to excessive or persistent inflammation due to continued recruitment and activation of T cells, macrophages and neutrophils. The fact that airway epithelial cells express number of molecules, including DUSP, TOLLIP, IRAK-M, which negatively regulate MAPK and TLR pathways indicate that under normal situation, airway epithelial cells can balance pro-inflammatory to anti-inflammatory responses to RV. Although, ER stress and autophagy play important role in limiting replication in RV-infected H1 HeLa cells, whether these processes affect viral replication or expression of pro-inflammatory cytokines in airway epithelial cells is yet to be determined. It is also not known how ER stress and autophagy affect viral clearance or virus-induced inflammation in patients with chronic lung disease who already show increase in ER stress and autophagy.
Despite these new advances in the field of innate immune responses to RV, mechanisms underlying the RV-associated exacerbations in patients with chronic lung diseases are not completely understood. While COPD patients often show Th1 inflammation, majority of the patients with asthma show Th2 inflammation in response to viral infections. Therefore, the mechanisms underlying RV-associated inflammation in asthma and COPD may be very different and this should be considered while translating the findings to develop new therapies.
Author Contributions
HG wrote description on rhinovirus. CR and ME conducted literature search and provided description on autophagy and ER stress. US wrote the manuscript.
Funding
This work was supported by NIH grant AT007620 to US.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ambjorn, M., Ejlerskov, P., Liu, Y., Lees, M., Jaattela, M., and Issazadeh-Navikas, S. (2013). IFNB1/interferon-beta-induced autophagy in MCF-7 breast cancer cells counteracts its proapoptotic function. Autophagy 9, 287–302. doi: 10.4161/auto.22831
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701. doi: 10.1083/jcb.200803137
Bai, J., Smock, S. L., Jackson, G. R. Jr., MacIsaac, K. D., Huang, Y., Mankus, C., et al. (2015). Phenotypic responses of differentiated asthmatic human airway epithelial cultures to rhinovirus. PLoS ONE 10:e0118286. doi: 10.1371/journal.pone.0118286
Balda, M. S., and Matter, K. (2009). Tight junctions and the regulation of gene expression. Biochim. Biophys. Acta 1788, 761–767. doi: 10.1016/j.bbamem.2008.11.024
Basta, H. A., Sgro, J. Y., and Palmenberg, A. C. (2014). Modeling of the human rhinovirus C capsid suggests a novel topography with insights on receptor preference and immunogenicity. Virology 448, 176–184. doi: 10.1016/j.virol.2013.10.006
Beale, J., Jayaraman, A., Jackson, D. J., Macintyre, J. D. R., Edwards, M. R., Walton, R. P., et al. (2014). Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci. Transl. Med. 6:256ra134. doi: 10.1126/scitranslmed.3009124
Bentley, J. K., Han, M., Jaipalli, S., Hinde, J. L., Lei, J., Ishikawa, T., et al. (2019). Myristoylated rhinovirus VP4 protein activates TLR2-dependent proinflammatory gene expression. Am. J. Physiol. Lung Cell Mol. Physiol. 317:L57–L70. doi: 10.1152/ajplung.00365.2018
Bentley, J. K., Newcomb, D. C., Goldsmith, A. M., Jia, Y., Sajjan, U. S., and Hershenson, M. B. (2007). Rhinovirus activates interleukin-8 expression via a Src/p110beta phosphatidylinositol 3-kinase/Akt pathway in human airway epithelial cells. J. Virol. 81, 1186–1194. doi: 10.1128/JVI.02309-06
Berman, R., Jiang, D., Wu, Q., and Chu, H. W. (2016). Alpha1-Antitrypsin reduces rhinovirus infection in primary human airway epithelial cells exposed to cigarette smoke. Int. J. Chron. Obstruct. Pulmon. Dis. 11, 1279–1286. doi: 10.2147/COPD.S105717
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Blaas, D. (2016). Viral entry pathways: the example of common cold viruses. Wien. Med. Wochenschr. 166, 211–226. doi: 10.1007/s10354-016-0461-2
Bochkov, Y. A., Hanson, K. M., Keles, S., Brockman-Schneider, R. A., Jarjour, N. N., and Gern, J. E. (2010). Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal. Immunol. 3, 69–80. doi: 10.1038/mi.2009.109
Bochkov, Y. A., Watters, K., Ashraf, S., Griggs, T. F., Devries, M. K., Jackson, D. J., et al. (2015). Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc. Natl. Acad. Sci. U. S. A. 112, 5485–5490. doi: 10.1073/pnas.1421178112
Bonnans, C., Gras, D., Chavis, C., Mainprice, B., Vachier, I., Godard, P., et al. (2007). Synthesis and anti-inflammatory effect of lipoxins in human airway epithelial cells. Biomed. Pharmacother. 61, 261–267. doi: 10.1016/j.biopha.2007.02.016
Bonnelykke, K., Sleiman, P., Nielsen, K., Kreiner-Moller, E., Mercader, J. M., Belgrave, D., et al. (2014). A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 46, 51–55. doi: 10.1038/ng.2830
Brogden, K. A. (2005). Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250. doi: 10.1038/nrmicro1098
Button, B., Cai, L. H., Ehre, C., Kesimer, M., Hill, D. B., Sheehan, J. K., et al. (2012). A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337, 937–941. doi: 10.1126/science.1223012
Caliskan, M., Bochkov, Y. A., Kreiner-Moller, E., Bonnelykke, K., Stein, M. M., Du, G., et al. (2013). Rhinovirus wheezing illness and genetic risk of childhood-onset asthma. N. Engl. J. Med. 368, 1398–1407. doi: 10.1056/NEJMoa1211592
Chattoraj, S. S., Ganesan, S., Faris, A., Comstock, A., Lee, W. M., and Sajjan, U. S. (2011). Pseudomonas aeruginosa suppresses interferon response to rhinovirus infection in cystic fibrosis but not in normal bronchial epithelial cells. Infect. Immun. 79, 4131–4145. doi: 10.1128/IAI.05120-11
Colby, J. K., Abdulnour, R. E., Sham, H. P., Dalli, J., Colas, R. A., Winkler, J. W., et al. (2016). Resolvin D3 and aspirin-triggered resolvin D3 are protective for injured epithelia. Am. J. Pathol. 186, 1801–1813. doi: 10.1016/j.ajpath.2016.03.011
Comstock, A. T., Ganesan, S., Chattoraj, A., Faris, A. N., Margolis, B. L., Hershenson, M. B., et al. (2011). Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 85, 6795–6808. doi: 10.1128/JVI.02074-10
Contoli, M., Message, S. D., Laza-Stanca, V., Edwards, M. R., Wark, P. A., Bartlett, N. W., et al. (2006). Role of deficient type III interferon-lambda production in asthma exacerbations. Nat. Med. 12, 1023–1026. doi: 10.1038/nm1462
Dakhama, A., Al Mubarak, R., Pavelka, N., Voelker, D., Seibold, M., Ledford, J. G., et al. (2020). Tollip inhibits ST2 signaling in airway epithelial cells exposed to type 2 cytokines and rhinovirus. J. Innate. Immun. 12, 103–115. doi: 10.1159/000497072
Dauletbaev, N., Das, M., Cammisano, M., Chen, H., Singh, S., Kooi, C., et al. (2015). Rhinovirus load is high despite preserved interferon-beta response in cystic fibrosis bronchial epithelial cells. PLoS ONE 10:e0143129. doi: 10.1371/journal.pone.0143129
Denney, L., and Ho, L. P. (2018). The role of respiratory epithelium in host defence against influenza virus infection. Biomed J. 41, 218–233. doi: 10.1016/j.bj.2018.08.004
Deppong, C., Juehne, T. I., Hurchla, M., Friend, L. D., Shah, D. D., Rose, C. M., et al. (2006). Cutting edge: B and T lymphocyte attenuator and programmed death receptor-1 inhibitory receptors are required for termination of acute allergic airway inflammation. J. Immunol. 176, 3909–3913. doi: 10.4049/jimmunol.176.7.3909
Djukanovic, R., Harrison, T., Johnston, S. L., Gabbay, F., Wark, P., Thomson, N. C., et al. (2014). The effect of inhaled IFN-β on worsening of asthma symptoms caused by viral infections. A randomized trial. Am. J. Respir. Crit. Care Med. 190, 145–154. doi: 10.1164/rccm.201312-2235OC
Do, T. Q., Moshkani, S., Castillo, P., Anunta, S., Pogosyan, A., Cheung, A., et al. (2008). Lipids including cholesteryl linoleate and cholesteryl arachidonate contribute to the inherent antibacterial activity of human nasal fluid. J. Immunol. 181, 4177–4187. doi: 10.4049/jimmunol.181.6.4177
Eddleston, J., Lee, R. U., Doerner, A. M., Herschbach, J., and Zuraw, B. L. (2011). Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am. J. Respir. Cell Mol. Biol. 44, 118–126. doi: 10.1165/rcmb.2009-0266OC
Etherington, C., Naseer, R., Conway, S. P., Whitaker, P., Denton, M., and Peckham, D. G. (2014). The role of respiratory viruses in adult patients with cystic fibrosis receiving intravenous antibiotics for a pulmonary exacerbation. J. Cyst. Fibros. 13, 49–55. doi: 10.1016/j.jcf.2013.06.004
Everman, J. L., Sajuthi, S., Saef, B., Rios, C., Stoner, A. M., Numata, M., et al. (2019). Functional genomics of CDHR3 confirms its role in HRV-C infection and childhood asthma exacerbations. J. Allergy Clin. Immunol. 144, 962–971. doi: 10.1016/j.jaci.2019.01.052
Faris, A. N., Ganesan, S., Chattoraj, A., Chattoraj, S. S., Comstock, A. T., Unger, B. L., et al. (2016). Rhinovirus delays cell repolarization in a model of injured/regenerating human airway epithelium. Am. J. Respir. Cell Mol. Biol. 55, 487–499. doi: 10.1165/rcmb.2015-0243OC
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi: 10.1038/cr.2013.168
Fuchs, R., and Blaas, D. (2010). Uncoating of human rhinoviruses. Rev. Med. Virol. 20, 281–297. doi: 10.1002/rmv.654
Fujita, E., Kouroku, Y., Isoai, A., Kumagai, H., Misutani, A., Matsuda, C., et al. (2007). Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 16, 618–629. doi: 10.1093/hmg/ddm002
Ganesan, S., Comstock, A. T., and Sajjan, U. S. (2013). Barrier function of airway tract epithelium. Tissue Barriers 1:e24997. doi: 10.4161/tisb.24997
Ganesan, S., Pham, D., Jing, Y., Farazuddin, M., Hudy, M. H., Unger, B., et al. (2016). TLR2 activation limits rhinovirus-stimulated CXCL-10 by attenuating IRAK-1-dependent IL-33 receptor signaling in human bronchial epithelial cells. J. Immunol. 197, 2409–2420. doi: 10.4049/jimmunol.1502702
Ganz, T. (2002). Antimicrobial polypeptides in host defense of the respiratory tract. J. Clin. Invest. 109, 693–697. doi: 10.1172/JCI0215218
Gavino, A. C., Chung, J. S., Sato, K., Ariizumi, K., and Cruz, P. D. Jr. (2005). Identification and expression profiling of a human C-type lectin, structurally homologous to mouse dectin-2. Exp. Dermatol. 14, 281–288. doi: 10.1111/j.0906-6705.2005.00312.x
Geng, J., and Klionsky, D. J. (2008). The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. Protein modifications: beyond the usual suspects' review series. EMBO Rep. 9, 859–864. doi: 10.1038/embor.2008.163
George, S. N., Garcha, D. S., Mackay, A. J., Patel, A. R., Singh, R., Sapsford, R. J., et al. (2014). Human rhinovirus infection during naturally occurring COPD exacerbations. Eur. Respir. J. 44, 87–96. doi: 10.1183/09031936.00223113
Gern, J. E., Dick, E. C., Lee, W. M., Murray, S., Meyer, K., Hand zel, Z. T., et al. (1996). Rhinovirus enters but does not replicate inside monocytes and airway macrophages. J. Immunol. 156, 621–627.
Gern, J. E., Vrtis, R., Grindle, K. A., Swenson, C., and Busse, W. W. (2000). Relationship of upper and lower airway cytokines to outcome of experimental rhinovirus infection. Am. J. Respir. Crit. Care Med. 162, 2226–2231. doi: 10.1164/ajrccm.162.6.2003019
Ghosh, M., Miller, Y. E., Nakachi, I., Kwon, J. B., Baron, A. E., Brantley, A. E., et al. (2018). Exhaustion of airway basal progenitor cells in early and established chronic obstructive pulmonary disease. Am. J. Respir Crit. Care Med. 197, 885–896. doi: 10.1164/rccm.201704-0667OC
Gimenes-Junior, J., Owuar, N., Vari, H. R., Li, W., Xander, N., Kotnala, S., et al. (2019b). FOXO3a regulates rhinovirus-induced innate immune responses in airway epithelial cells. Sci. Rep. 9:18180. doi: 10.1038/s41598-019-54567-3
Gimenes-Junior, J., Srivastava, V., ReddyVari, H., Kotnala, S., Mishra, R., Farazuddin, M., et al. (2019a). Rhinovirus-induces progression of lung disease in a mouse model of COPD via IL-33/ST2 signaling axis. Clin. Sci. 133, 983–996. doi: 10.1042/CS20181088
Gohy, S., Carlier, F. M., Fregimilicka, C., Detry, B., Lecocq, M., Ladjemi, M. Z., et al. (2019). Altered generation of ciliated cells in chronic obstructive pulmonary disease. Sci. Rep. 9:17963. doi: 10.1038/s41598-019-54292-x
Gohy, S. T., Hupin, C., Fregimilicka, C., Detry, B. R., Bouzin, C., Gaide Chevronay, H., et al. (2015). Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur. Respir. J. 45, 1258–1272. doi: 10.1183/09031936.00135814
Greve, J. M., Davis, G., Meyer, A. M., Forte, C. P., Yost, S. C., Marlor, C. W., et al. (1989). The major human rhinovirus receptor is ICAM-1. Cell 56, 839–847. doi: 10.1016/0092-8674(89)90688-0
Griggs, T. F., Bochkov, Y. A., Basnet, S., Pasic, T. R., Brockman-Schneider, R. A., Palmenberg, A. C., et al. (2017). Rhinovirus C targets ciliated airway epithelial cells. Respir. Res. 18:84. doi: 10.1186/s12931-017-0567-0
Grunberg, K., Timmers, M. C., Smits, H. H., de Klerk, E. P., Dick, E. C., Spaan, W. J., et al. (1997). Effect of experimental rhinovirus 16 colds on airway hyperresponsiveness to histamine and interleukin-8 in nasal lavage in asthmatic subjects in vivo. Clin. Exp. Allergy 27, 36–45. doi: 10.1111/j.1365-2222.1997.tb00670.x
Hamann, K. J., Dorscheid, D. R., Ko, F. D., Conforti, A. E., Sperling, A. I., Rabe, K. F., et al. (1998). Expression of fas (CD95) and FasL (CD95L) in human airway epithelium. Am. J. Respir. Cell Mol. Biol. 19, 537–542. doi: 10.1165/ajrcmb.19.4.3100
Hammad, H., Chieppa, M., Perros, F., Willart, M. A., Germain, R. N., and Lambrecht, B. N. (2009). House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 15, 410–416. doi: 10.1038/nm.1946
Han, M., Bentley, J. K., Rajput, C., Lei, J., Ishikawa, T., Jarman, C. R., et al. (2019). Inflammasome activation is required for human rhinovirus-induced airway inflammation in naive and allergen-sensitized mice. Mucosal. Immunol. 12, 958–968. doi: 10.1038/s41385-019-0172-2
Han, M., Rajput, C., Hong, J. Y., Lei, J., Hinde, J. L., Wu, Q., et al. (2017). The innate cytokines IL-25, IL-33, and TSLP cooperate in the induction of type 2 innate lymphoid cell expansion and mucous metaplasia in rhinovirus-infected immature mice. J. Immunol. 199, 1308–1318. doi: 10.4049/jimmunol.1700216
He, L., Zhang, J., Zhao, J., Ma, N., Kim, S. W., Qiao, S., et al. (2018). Autophagy: the last defense against cellular nutritional stress. Adv. Nutr. 9, 493–504. doi: 10.1093/advances/nmy011
He, Y., Hara, H., and Nunez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 41, 1012–1021. doi: 10.1016/j.tibs.2016.09.002
Heinecke, L., Proud, D., Sanders, S., Schleimer, R. P., and Kim, J. (2008). Induction of B7-H1 and B7-DC expression on airway epithelial cells by the Toll-like receptor 3 agonist double-strand ed RNA and human rhinovirus infection: in vivo and in vitro studies. J. Allergy Clin. Immunol. 121, 1155–1160. doi: 10.1016/j.jaci.2008.02.009
Hewat, E. A., Neumann, E., Conway, J. F., Moser, R., Ronacher, B., Marlovits, T. C., et al. (2000). The cellular receptor to human rhinovirus 2 binds around the 5-fold axis and not in the canyon: a structural view. EMBO J. 19, 6317–6325. doi: 10.1093/emboj/19.23.6317
Hewson, C. A., Jardine, A., Edwards, M. R., Laza-Stanca, V., and Johnston, S. L. (2005). Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J. Virol. 79, 12273–12279. doi: 10.1128/JVI.79.19.12273-12279.2005
Heyl, K. A., Klassert, T. E., Heinrich, A., Muller, M. M., Klaile, E., Dienemann, H., et al. (2014). Dectin-1 is expressed in human lung and mediates the proinflammatory immune response to nontypeable haemophilus influenzae. mBio 5:e01492–4. doi: 10.1128/mBio.01492-14
Hirota, J. A., Hirota, S. A., Warner, S. M., Stefanowicz, D., Shaheen, F., Beck, P. L., et al. (2012). The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J. Allergy Clin. Immunol. 129, 1116–1125. e1116. doi: 10.1016/j.jaci.2011.11.033
Hong, J. Y., Bentley, J. K., Chung, Y., Lei, J., Steenrod, J. M., Chen, Q., et al. (2014). Neonatal rhinovirus induces mucous metaplasia and airways hyperresponsiveness through IL-25 and type 2 innate lymphoid cells. J. Allergy Clin. Immunol. 134, 429–439. doi: 10.1016/j.jaci.2014.04.020
Hudy, M. H., and Proud, D. (2013). Cigarette smoke enhances human rhinovirus-induced CXCL8 production via HuR-mediated mRNA stabilization in human airway epithelial cells. Respir. Res. 14:88. doi: 10.1186/1465-9921-14-88
Jackson, W. T., Giddings, T. H. Jr., Taylor, M. P., Mulinyawe, S., Rabinovitch, M., Kopito, R. R., et al. (2005). Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. doi: 10.1371/journal.pbio.0030156
Jakiela, B., Brockman-Schneider, R., Amineva, S., Lee, W. M., and Gern, J. E. (2008). Basal cells of differentiated bronchial epithelium are more susceptible to rhinovirus infection. Am. J. Respir. Cell Mol. Biol. 38, 517–523. doi: 10.1165/rcmb. 2007-0050OC
Jamieson, K. C., Traves, S. L., Kooi, C., Wiehler, S., Dumonceaux, C. J., Maciejewski, B. A., et al. (2019). Rhinovirus and bacteria synergistically induce IL-17C release from human airway epithelial cells to promote neutrophil recruitment. J. Immunol. 202, 160–170. doi: 10.4049/jimmunol.1800547
Jayaraman, A., Jackson, D. J., Message, S. D., Pearson, R. M., Aniscenko, J., Caramori, G., et al. (2014). IL-15 complexes induce NK- and T-cell responses independent of type I IFN signaling during rhinovirus infection. Mucosal. Immunol. 7, 1151–1164. doi: 10.1038/mi.2014.2
Jheng, J. R., Lau, K. S., Tang, W. F., Wu, M. S., and Horng, J. T. (2010). Endoplasmic reticulum stress is induced and modulated by enterovirus 71. Cell Microbiol. 12, 796–813. doi: 10.1111/j.1462-5822.2010.01434.x
Jing, Y., Gimenes, J. A., Mishra, R., Pham, D., Comstock, A. T., Yu, D., et al. (2019). NOTCH3 contributes to rhinovirus-induced goblet cell hyperplasia in COPD airway epithelial cells. Thorax 74, 18–32. doi: 10.1136/thoraxjnl-2017-210593
Kato, A., Favoreto, S. Jr., Avila, P. C., and Schleimer, R. P. (2007). TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 179, 1080–1087. doi: 10.4049/jimmunol.179.2.1080
Kaufman, R. J. (1999). Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes. Dev. 13, 1211–1233. doi: 10.1101/gad.13.10.1211
Kaul, P., Biagioli, M. C., Singh, I., and Turner, R. B. (2000). Rhinovirus-induced oxidative stress and interleukin-8 elaboration involves p47-phox but is independent of attachment to intercellular adhesion molecule-1 and viral replication. J. Infect. Dis. 181, 1885–1890. doi: 10.1086/315504
Kelsen, S. G. (2016). The unfolded protein response in chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 13(Suppl. 2), S138–S145. doi: 10.2174/156652412801318764
Kesimer, M., Ehre, C., Burns, K. A., Davis, C. W., Sheehan, J. K., and Pickles, R. J. (2013). Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal. Immunol. 6, 379–392. doi: 10.1038/mi.2012.81
Kim, J., Myers, A. C., Chen, L., Pardoll, D. M., Truong-Tran, Q. A., Lane, A. P., et al. (2005). Constitutive and inducible expression of b7 family of ligand s by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 33, 280–289. doi: 10.1165/rcmb.2004-0129OC
Klein, K. A., and Jackson, W. T. (2011). Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J. Virol. 85, 9651–9654. doi: 10.1128/JVI.00316-11
Knowles, M. R., and Boucher, R. C. (2002). Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 109, 571–577. doi: 10.1172/JCI0215217
Koch, S., and Nusrat, A. (2009). Dynamic regulation of epithelial cell fate and barrier function by intercellular junctions. Ann. N.Y. Acad. Sci. 1165, 220–227. doi: 10.1111/j.1749-6632.2009.04025.x
Koetzler, R., Zaheer, R. S., Wiehler, S., Holden, N. S., Giembycz, M. A., and Proud, D. (2009). Nitric oxide inhibits human rhinovirus-induced transcriptional activation of CXCL10 in airway epithelial cells. J. Allergy Clin. Immunol. 123, 201–208.e209. doi: 10.1016/j.jaci.2008.09.041
Kolatkar, P. R., Bella, J., Olson, N. H., Bator, C. M., Baker, T. S., and Rossmann, M. G. (1999). Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 18, 6249–6259. doi: 10.1093/emboj/18.22.6249
Korolchuk, V. I., Menzies, F. M., and Rubinsztein, D. C. (2010). Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398. doi: 10.1016/j.febslet.2009.12.047
Koumenis, C. (2006). ER stress, hypoxia tolerance and tumor progression. Curr. Mol. Med. 6, 55–69. doi: 10.2174/156652406775574604
Lamborn, I. T., Jing, H., Zhang, Y., Drutman, S. B., Abbott, J. K., Munir, S., et al. (2017). Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J. Exp. Med. 214, 1949–1972. doi: 10.1084/jem.20161759
Lee, W. M., Lemanske, R. F. Jr., Evans, M. D., Vang, F., Pappas, T., Gangnon, R., et al. (2012). Human rhinovirus species and season of infection determine illness severity. Am J. Respir. Crit. Care. Med. 186, 886–891. doi: 10.1164/rccm.201202-0330OC
Levine, S. J., Wu, T., and Shelhamer, J. H. (1997). Extracellular release of the type I intracellular IL-1 receptor antagonist from human airway epithelial cells: differential effects of IL-4, IL-13, IFN-gamma, and corticosteroids. J. Immunol. 158, 5949–5957.
Levy, B. D., and Serhan, C. N. (2014). Resolution of acute inflammation in the lung. Annu. Rev. Physiol. 76, 467–492. doi: 10.1146/annurev-physiol-021113-170408
Liu, K., Gualano, R. C., Hibbs, M. L., Anderson, G. P., and Bozinovski, S. (2008). Epidermal growth factor receptor signaling to Erk1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. J. Biol. Chem. 283, 9977–9985. doi: 10.1074/jbc.M710257200
Liu, Y., Bochkov, Y. A., Eickhoff, J. C., Hu, T., Zumwalde, N. A., Tan, J. W., et al. (2020). Orosomucoid like 3 (ORMDL3) supports rhinovirus replication in human epithelial cells. Am. J. Respir. Cell Mol. Biol. 62, 783–792. doi: 10.1165/rcmb.2019-0237OC
Liu, Y., Hill, M. G., Klose, T., Chen, Z., Watters, K., Bochkov, Y. A., et al. (2016). Atomic structure of a rhinovirus C, a virus species linked to severe childhood asthma. Proc. Natl. Acad. Sci. U.S.A. 113, 8997–9002. doi: 10.1073/pnas.1606595113
Lopez-Souza, N., Favoreto, S., Wong, H., Ward, T., Yagi, S., Schnurr, D., et al. (2009). In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J. Allergy Clin. Immunol. 123, 1384–1390.e1382. doi: 10.1016/j.jaci.2009.03.010
Makela, M. J., Puhakka, T., Ruuskanen, O., Leinonen, M., Saikku, P., Kimpimaki, M., et al. (1998). Viruses and bacteria in the etiology of the common cold. J. Clin. Microbiol. 36, 539–542. doi: 10.1128/JCM.36.2.539-542.1998
Mammucari, C., Schiaffino, S., and Sand ri, M. (2008). Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 4, 524–526. doi: 10.4161/auto.5905
Manley, G. C. A., Stokes, C. A., Marsh, E. K., Sabroe, I., and Parker, L. C. (2019). DUSP10 negatively regulates the inflammatory response to rhinovirus through interleukin-1beta signaling. J. Virol. 93:e01659–18. doi: 10.1128/JVI.01659-18
Matsumura, M., Inoue, H., Matsumoto, T., Nakano, T., Fukuyama, S., Matsumoto, K., et al. (2007). Endogenous metalloprotease solubilizes IL-13 receptor alpha2 in airway epithelial cells. Biochem. Biophys. Res. Commun. 360, 464–469. doi: 10.1016/j.bbrc.2007.06.076
Mayer, A. K., Bartz, H., Fey, F., Schmidt, L. M., and Dalpke, A. H. (2008). Airway epithelial cells modify immune responses by inducing an anti-inflammatory microenvironment. Eur. J. Immunol. 38, 1689–1699. doi: 10.1002/eji.200737936
Mayer, A. K., Muehmer, M., Mages, J., Gueinzius, K., Hess, C., Heeg, K., et al. (2007). Differential recognition of TLR-dependent microbial ligand s in human bronchial epithelial cells. J. Immunol. 178, 3134–3142. doi: 10.4049/jimmunol.178.5.3134
Mehta, A. K., Duan, W., Doerner, A. M., Traves, S. L., Broide, D. H., Proud, D., et al. (2016). Rhinovirus infection interferes with induction of tolerance to aeroantigens through OX40 ligand, thymic stromal lymphopoietin, and IL-33. J. Allergy Clin. Immunol. 137, 278–288.e276. doi: 10.1016/j.jaci.2015.05.007
Menzel, M., Ramu, S., Calven, J., Olejnicka, B., Sverrild, A., Porsbjerg, C., et al. (2019). Oxidative stress attenuates TLR3 responsiveness and impairs anti-viral mechanisms in bronchial epithelial cells from COPD and asthma patients. Front Immunol. 10:2765. doi: 10.3389/fimmu.2019.02765
Mihaylova, V. T., Kong, Y., Fedorova, O., Sharma, L., Dela Cruz, C. S., Pyle, A. M., et al. (2018). Regional differences in airway epithelial cells reveal tradeoff between defense against oxidative stress and defense against rhinovirus. Cell Rep. 24, 3000–3007.e3003. doi: 10.1016/j.celrep.2018.08.033
Mijaljica, D., Prescott, M., and Devenish, R. J. (2011). Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7, 673–682. doi: 10.4161/auto.7.7.14733
Moffatt, M. F., Kabesch, M., Liang, L., Dixon, A. L., Strachan, D., Heath, S., et al. (2007). Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448, 470–473. doi: 10.1038/nature06014
Mordstein, M., Kochs, G., Dumoutier, L., Renauld, J. C., Paludan, S. R., Klucher, K., et al. (2008). Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4:e1000151. doi: 10.1371/journal.ppat.1000151
Mordstein, M., Neugebauer, E., Ditt, V., Jessen, B., Rieger, T., Falcone, V., et al. (2010). Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 84, 5670–5677. doi: 10.1128/JVI.00272-10
Newcomb, D. C., Sajjan, U., Nanua, S., Jia, Y., Goldsmith, A. M., Bentley, J. K., et al. (2005). Phosphatidylinositol 3-kinase is required for rhinovirus-induced airway epithelial cell interleukin-8 expression. J. Biol. Chem. 280, 36952–36961. doi: 10.1074/jbc.M502449200
Niwa, M., Fujisawa, T., Mori, K., Yamanaka, K., Yasui, H., Suzuki, Y., et al. (2018). IL-17A Attenuates IFN-lambda expression by inducing suppressor of cytokine signaling expression in airway epithelium. J. Immunol. 201, 2392–2402. doi: 10.4049/jimmunol.1800147
O'Grady, S. M. (2019). Oxidative stress, autophagy and airway ion transport. Am. J. Physiol. Cell Physiol. 316, C16–C32. doi: 10.1152/ajpcell.00341.2018
Okuda, K., Chen, G., Subramani, D. B., Wolf, M., Gilmore, R. C., Kato, T., et al. (2019). Localization of secretory mucins MUC5AC and MUC5B in normal/healthy human airways. Am J. Respir. Crit. Care Med. 199, 715–727. doi: 10.1164/rccm.201804-0734OC
Orvedahl, A., MacPherson, S., Sumpter, R. Jr., Talloczy, Z., Zou, Z., and Levine, B. (2010). Autophagy protects against sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127. doi: 10.1016/j.chom.2010.01.007
Ostedgaard, L. S., Moninger, T. O., McMenimen, J. D., Sawin, N. M., Parker, C. P., Thornell, I. M., et al. (2017). Gel-forming mucins form distinct morphologic structures in airways. Proc. Natl. Acad. Sci. U.S.A. 114, 6842–6847. doi: 10.1073/pnas.1703228114
Palmenberg, A. C., and Gern, J. E. (2015). Classification and evolution of human rhinoviruses. Methods Mol. Biol. 1221, 1–10. doi: 10.1007/978-1-4939-1571-2_1
Pohl, C., Hermanns, M. I., Uboldi, C., Bock, M., Fuchs, S., Dei-Anang, J., et al. (2009). Barrier functions and paracellular integrity in human cell culture models of the proximal respiratory unit. Eur. J. Pharm. Biopharm. 72, 339–349. doi: 10.1016/j.ejpb.2008.07.012
Proud, D., Hudy, M. H., Wiehler, S., Zaheer, R. S., Amin, M. A., Pelikan, J. B., et al. (2012). Cigarette smoke modulates expression of human rhinovirus-induced airway epithelial host defense genes. PLoS ONE 7:e40762. doi: 10.1371/journal.pone.0040762
Proud, D., Turner, R. B., Winther, B., Wiehler, S., Tiesman, J. P., Reichling, T. D., et al. (2008). Gene expression profiles during in vivo human rhinovirus infection: insights into the host response. Am. J. Respir. Crit. Care Med. 178, 962–968. doi: 10.1164/rccm.200805-670OC
Rahmatpanah, F., Agrawal, S., Jaiswal, N., Nguyen, H. M., McClelland, M., and Agrawal, A. (2019). Airway epithelial cells prime plasmacytoid dendritic cells to respond to pathogens via secretion of growth factors. Mucosal. Immunol. 12, 77–84. doi: 10.1038/s41385-018-0097-1
Ravi, A., Koster, J., Dijkhuis, A., Bal, S. M., Sabogal Pineros, Y. S., Bonta, P. I., et al. (2019). Interferon-induced epithelial response to rhinovirus 16 in asthma relates to inflammation and FEV1. J. Allergy Clin. Immunol. 143, 442–447.e410. doi: 10.1016/j.jaci.2018.09.016
Rock, J. R., Onaitis, M. W., Rawlins, E. L., Lu, Y., Clark, C. P., Xue, Y., et al. (2009). Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. U.S.A. 106, 12771–12775. doi: 10.1073/pnas.0906850106
Rock, J. R., Randell, S. H., and Hogan, B. L. (2010). Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis. Models Mech. 3, 545–556. doi: 10.1242/dmm.006031
Sajjan, U., Wang, Q., Zhao, Y., Gruenert, D. C., and Hershenson, M. B. (2008). Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 178, 1271–1281. doi: 10.1164/rccm.200801-136OC
Sajjan, U. S., Jia, Y., Newcomb, D. C., Bentley, J. K., Lukacs, N. W., LiPuma, J. J., et al. (2006). H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 20, 2121–2123. doi: 10.1096/fj.06-5806fje
Sanders, S. P., Proud, D., Permutt, S., Siekierski, E. S., Yachechko, R., and Liu, M. C. (2004). Role of nasal nitric oxide in the resolution of experimental rhinovirus infection. J. Allergy Clin. Immunol. 113, 697–702. doi: 10.1016/j.jaci.2004.01.755
Sanders, S. P., Siekierski, E. S., Richards, S. M., Porter, J. D., Imani, F., and Proud, D. (2001). Rhinovirus infection induces expression of type 2 nitric oxide synthase in human respiratory epithelial cells in vitro and in vivo. J. Allergy Clin. Immunol. 107, 235–243. doi: 10.1067/mai.2001.112028
Schmeisser, H., Fey, S. B., Horowitz, J., Fischer, E. R., Balinsky, C. A., Miyake, K., et al. (2013). Type I interferons induce autophagy in certain human cancer cell lines. Autophagy 9, 683–696. doi: 10.4161/auto.23921
Schneeberger, E. E., and Lynch, R. D. (1992). Structure, function, and regulation of cellular tight junctions. Am. J. Physiol. 262:L647–661. doi: 10.1152/ajplung.1992.262.6.L647
Schneider, D., Ganesan, S., Comstock, A. T., Meldrum, C. A., Mahidhara, R., Goldsmith, A. M., et al. (2010). Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 182, 332–340. doi: 10.1164/rccm.200911-1673OC
Senft, D., and Ronai, Z. A. (2015). UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40, 141–148. doi: 10.1016/j.tibs.2015.01.002
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 3, 99–111. doi: 10.1016/S1534-5807(02)00203-4
Shi, J., Wong, J., Piesik, P., Fung, G., Zhang, J., Jagdeo, J., et al. (2013). Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy 9, 1591–1603. doi: 10.4161/auto.26059
Shin, K., Fogg, V. C., and Margolis, B. (2006). Tight junctions and cell polarity. Annu. Rev. Cell Dev. Biol. 22, 207–235. doi: 10.1146/annurev.cellbio.22.010305.104219
Singh, P. K., Tack, B. F., McCray, P. B. Jr., and Welsh, M. J. (2000). Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am. J. Physiol. Lung Cell Mol. Physiol. 279, L799–L805. doi: 10.1152/ajplung.2000.279.5.L799
Slater, L., Bartlett, N. W., Haas, J. J., Zhu, J., Message, S. D., Walton, R. P., et al. (2010). Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 6:e1001178. doi: 10.1371/journal.ppat.1001178
Slevogt, H., Seybold, J., Tiwari, K. N., Hocke, A. C., Jonatat, C., Dietel, S., et al. (2007). Moraxella catarrhalis is internalized in respiratory epithelial cells by a trigger-like mechanism and initiates a TLR2- and partly NOD1-dependent inflammatory immune response. Cell Microbiol. 9, 694–707. doi: 10.1111/j.1462-5822.2006.00821.x
Sommereyns, C., Paul, S., Staeheli, P., and Michiels, T. (2008). IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 4:e1000017. doi: 10.1371/journal.ppat.1000017
Song, J., Chi, M., Luo, X., Song, Q., Xia, D., Shi, B., et al. (2019). Non-structural protein 2B of human rhinovirus 16 activates both PERK and ATF6 rather than IRE1 to trigger ER stress. Viruses 11:133. doi: 10.3390/v11020133
Spurrell, J. C., Wiehler, S., Zaheer, R. S., Sand ers, S. P., and Proud, D. (2005). Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am. J. Physiol. Lung Cell Mol Physiol. 289, L85–L95. doi: 10.1152/ajplung.00397.2004
Staring, J., von Castelmur, E., Blomen, V. A., van den Hengel, L. G., Brockmann, M., Baggen, J., et al. (2017). PLA2G16 represents a switch between entry and clearance of picornaviridae. Nature 541, 412–416. doi: 10.1038/nature21032
Stolz, D., Papakonstantinou, E., Grize, L., Schilter, D., Strobel, W., Louis, R., et al. (2019). Time-course of upper respiratory tract viral infection and COPD exacerbation. Eur. Respir. J. 54:1900407. doi: 10.1183/13993003.00407-2019
Su, H. L., Liao, C. L., and Lin, Y. L. (2002). Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 76, 4162–4171. doi: 10.1128/JVI.76.9.4162-4171.2002
Tan, K. S., Ong, H. H., Yan, Y., Liu, J., Li, C., Ong, Y. K., et al. (2018). In vitro model of fully differentiated human nasal epithelial cells infected with rhinovirus reveals epithelium-initiated immune responses. J. Infect Dis. 217, 906–915. doi: 10.1093/infdis/jix640
Tang, F. S., Hansbro, P. M., Burgess, J. K., Ammit, A. J., Baines, K. J., and Oliver, B. G. (2016). A novel immunomodulatory function of neutrophils on rhinovirus-activated monocytes in vitro. Thorax 71, 1039–1049. doi: 10.1136/thoraxjnl-2015-207781
Tardif, K. D., Mori, K., Kaufman, R. J., and Siddiqui, A. (2004). Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279, 17158–17164. doi: 10.1074/jbc.M312144200
Telcian, A. G., Laza-Stanca, V., Edwards, M. R., Harker, J. A., Wang, H., Bartlett, N. W., et al. (2011). RSV-induced bronchial epithelial cell PD-L1 expression inhibits CD8+ T cell nonspecific antiviral activity. J. Infect. Dis. 203, 85–94. doi: 10.1093/infdis/jiq020
Triantafilou, K., Vakakis, E., Richer, E. A., Evans, G. L., Villiers, J. P., and Triantafilou, M. (2011). Human rhinovirus recognition in non-immune cells is mediated by Toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence 2, 22–29. doi: 10.4161/viru.2.1.13807
Uller, L., Leino, M., Bedke, N., Sammut, D., Green, B., Lau, L., et al. (2010). Double-strand ed RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-beta in bronchial epithelial cells from donors with asthma. Thorax 65, 626–632. doi: 10.1136/thx.2009.125930
Unger, B. L., Faris, A. N., Ganesan, S., Comstock, A. T., Hershenson, M. B., and Sajjan, U. S. (2012). Rhinovirus attenuates non-typeable Hemophilus influenzae-stimulated IL-8 responses via TLR2-dependent degradation of IRAK-1. PLoS Pathog. 8:e1002969. doi: 10.1371/journal.ppat.1002969
Unger, B. L., Ganesan, S., Comstock, A. T., Faris, A. N., Hershenson, M. B., and Sajjan, U. S. (2014). Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J. Virol. 88, 3705–3718. doi: 10.1128/JVI.03039-13
Vareille, M., Kieninger, E., Alves, M. P., Kopf, B. S., Moller, A., Geiser, T., et al. (2012). Impaired type I and type III interferon induction and rhinovirus control in human cystic fibrosis airway epithelial cells. Thorax 67, 517–525. doi: 10.1136/thoraxjnl-2011-200405
Vareille, M., Kieninger, E., Edwards, M. R., and Regamey, N. (2011). The airway epithelium: soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 24, 210–229. doi: 10.1128/CMR.00014-10
Wang, Q., Nagarkar, D. R., Bowman, E. R., Schneider, D., Gosangi, B., Lei, J., et al. (2009). Role of double-strand ed RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol. 183, 6989–6997. doi: 10.4049/jimmunol.0901386
Wark, P. A., Johnston, S. L., Bucchieri, F., Powell, R., Puddicombe, S., Laza-Stanca, V., et al. (2005). Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 201, 937–947. doi: 10.1084/jem.20041901
Warner, S. M., Wiehler, S., Michi, A. N., and Proud, D. (2019). Rhinovirus replication and innate immunity in highly differentiated human airway epithelial cells. Respir. Res. 20:150. doi: 10.1186/s12931-019-1120-0
Watson, A., Spalluto, C. M., McCrae, C., Cellura, D., Burke, H., Cunoosamy, D., et al. (2020). Dynamics of IFN-β responses during respiratory viral infection. Insights for therapeutic strategies. Am. J. Respir. Crit. Care Med. 201, 83–94. doi: 10.1164/rccm.201901-0214OC
Watters, K., and Palmenberg, A. C. (2018). CDHR3 extracellular domains EC1-3 mediate rhinovirus C interaction with cells and as recombinant derivatives, are inhibitory to virus infection. PLoS Pathog. 14:e1007477. doi: 10.1371/journal.ppat.1007477
Xander, N., Reddy Vari, H., Eskandar, R., Li, W., Bolla, S., Marchetti, N., et al. (2019). Rhinovirus-induced SIRT-1 via TLR2 regulates subsequent type I and type III IFN responses in airway epithelial cells. J. Immunol. 203, 2508–2519. doi: 10.4049/jimmunol.1900165
Yang, J., Tian, B., Sun, H., Garofalo, R. P., and Brasier, A. R. (2017). Epigenetic silencing of IRF1 dysregulates type III interferon responses to respiratory virus infection in epithelial to mesenchymal transition. Nat. Microbiol. 2:17086. doi: 10.1038/nmicrobiol.2017.86
Yorimitsu, T., and Klionsky, D. J. (2005). Autophagy: molecular machinery for self-eating. Cell Death Differ. 12(Suppl. 2), 1542–1552. doi: 10.1038/sj.cdd.4401765
Zachari, M., and Ganley, I. G. (2017). The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 61, 585–596. doi: 10.1042/EBC20170021
Zaheer, R. S., Koetzler, R., Holden, N. S., Wiehler, S., and Proud, D. (2009). Selective transcriptional down-regulation of human rhinovirus-induced production of CXCL10 from airway epithelial cells via the MEK1 pathway. J. Immunol. 182, 4854–4864. doi: 10.4049/jimmunol.0802401
Zaheer, R. S., and Proud, D. (2010). Human rhinovirus-induced epithelial production of CXCL10 is dependent upon IFN regulatory factor-1. Am. J. Respir. Cell Mol. Biol. 43, 413–421. doi: 10.1165/rcmb.2009-0203OC
Zaheer, R. S., Wiehler, S., Hudy, M. H., Traves, S. L., Pelikan, J. B., Leigh, R., et al. (2014). Human rhinovirus-induced ISG15 selectively modulates epithelial antiviral immunity. Mucosal. Immunol. 7, 1127–1138. doi: 10.1038/mi.2013.128
Zasloff, M. (2002). Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. doi: 10.1038/415389a
Keywords: dsRNA, rhinovirus, antiviral responses, ER stress, autophagy, COPD, asthma
Citation: Ganjian H, Rajput C, Elzoheiry M and Sajjan U (2020) Rhinovirus and Innate Immune Function of Airway Epithelium. Front. Cell. Infect. Microbiol. 10:277. doi: 10.3389/fcimb.2020.00277
Received: 18 March 2020; Accepted: 12 May 2020;
Published: 19 June 2020.
Edited by:
David Proud, University of Calgary, CanadaReviewed by:
John W. Upham, The University of Queensland, AustraliaTeresa de los Santos, Agricultural Research Service (USDA), United States
Copyright © 2020 Ganjian, Rajput, Elzoheiry and Sajjan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Umadevi Sajjan, uma.sajjan@temple.edu