Loss of Metabolic Flexibility in the Failing Heart
 Qutuba G. Karwi
Qutuba G. Karwi Golam M. Uddin
Golam M. Uddin Kim L. Ho
Kim L. Ho Gary D. Lopaschuk
Gary D. Lopaschuk- Cardiovascular Research Centre, University of Alberta, Edmonton, AB, Canada
To maintain its high energy demand the heart is equipped with a highly complex and efficient enzymatic machinery that orchestrates ATP production using multiple energy substrates, namely fatty acids, carbohydrates (glucose and lactate), ketones and amino acids. The contribution of these individual substrates to ATP production can dramatically change, depending on such variables as substrate availability, hormonal status and energy demand. This “metabolic flexibility” is a remarkable virtue of the heart, which allows utilization of different energy substrates at different rates to maintain contractile function. In heart failure, cardiac function is reduced, which is accompanied by discernible energy metabolism perturbations and impaired metabolic flexibility. While it is generally agreed that overall mitochondrial ATP production is impaired in the failing heart, there is less consensus as to what actual switches in energy substrate preference occur. The failing heart shift toward a greater reliance on glycolysis and ketone body oxidation as a source of energy, with a decrease in the contribution of glucose oxidation to mitochondrial oxidative metabolism. The heart also becomes insulin resistant. However, there is less consensus as to what happens to fatty acid oxidation in heart failure. While it is generally believed that fatty acid oxidation decreases, a number of clinical and experimental studies suggest that fatty acid oxidation is either not changed or is increased in heart failure. Of importance, is that any metabolic shift that does occur has the potential to aggravate cardiac dysfunction and the progression of the heart failure. An increasing body of evidence shows that increasing cardiac ATP production and/or modulating cardiac energy substrate preference positively correlates with heart function and can lead to better outcomes. This includes increasing glucose and ketone oxidation and decreasing fatty acid oxidation. In this review we present the physiology of the energy metabolism pathways in the heart and the changes that occur in these pathways in heart failure. We also look at the interventions which are aimed at manipulating the myocardial metabolic pathways toward more efficient substrate utilization which will eventually improve cardiac performance.
Introduction
Heart failure is a major cause of death and disability, with more than 26 million people diagnosed with heart failure worldwide (1). The mortality rate of heart failure is approximately 30% within 5 years following diagnosis (2). Thus, heart failure presents a tremendous burden on society, the health care system and the economy (3, 4). While pharmacological management, primary prevention and earlier diagnosis have dramatically improved in the last 20 years, there is still a high morbidity and mortality associated with heart failure.
Different energy substrates, namely fatty acids, carbohydrates (glucose and lactate), ketones and amino acid, contribute differently to meet the high energy demand of the heart. Fatty acid oxidation is the biggest contributor to ATP production (~40–60%) while carbohydrates metabolism generates the remainder (~20–40%). The heart has a “metabolic flexibility,” a virtue which allows it to switch between these energy substrates according to the workload, availability of the substrates, and the hormonal status. There is a general consensus that this metabolic flexibility is impaired in heart failure which affects ATP production and, consequently, cardiac contractility. Another important dimension of the metabolic flexibility is the effect of inotropic agents on energy substrates mobilization and use which occurs in response to an increase in heart work. It has been demonstrated that adrenergic stimulation, using epinephrine, enhances glycogenolysis and increases glucose oxidation in the normal rat heart (5, 6). Nevertheless, whether this form of metabolic flexibility is also impaired in the failing heart has yet to be characterized. As a disease, the complexity of heart failure is represented by differences in etiology, severity and the presence of comorbidities, such as hypertension, diabetes and obesity. Despite its diverse nature, the failing heart is characterized by its energy deficient state, as evidenced by a 30–40% reduction in ATP production and reduced phosphocreatine/ATP (PCr/ATP) ratio (7–10). This energy-starved state may be due to mitochondrial dysfunction as a result of reactive oxygen species generation, compromised bioenergetics and impaired mitochondrial fission and fusion [see De Jong et al. (11) and Neubauer (10) for reviews]. Furthermore, mitochondrial dysfunction also directly affects myocardial energy substrate metabolism further aggravating the energy crisis. Alterations in myocardial energy substrate metabolism are discernible in the failing heart as a result of compromised mitochondrial oxidative phosphorylation [see Lopaschuk et al. (12) for review]. This results in an increased reliance on glycolysis and decreased glucose oxidation to produce the required energy (13, 14). While ketone oxidation increases in the failing heart (15–17), fatty acid oxidation has been suggested to decrease (10, 18–20), or not change (13, 21). Regardless, these metabolic shifts in heart failure can negatively affect cardiac contractility and myocardial energy management, leading to decreased metabolic flexibility at a time when the heart needs it most. While the pathological consequences of changes to fatty acid and glucose metabolism have been extensively studied, the alterations and implications of altered ketone body metabolism during heart failure remain unclear even though the heart uses the most ketone bodies per unit mass (22, 23). Lastly, there is an increasing amount of evidence suggesting that restoring metabolic flexibility by enhancing glucose oxidation directly (24) or indirectly through inhibition of fatty acid oxidation (25) could improve myocardial ATP production, improve cardiac function and limit cardiac remodeling (12, 13, 26, 27).
This review will focus on the alterations in fatty acid oxidation, carbohydrate metabolism and ketone metabolism that occur in the heart in the setting of ischemic heart failure. Amino acid metabolism is out of the scope of this review and the reader is referred to other reviews about the subject (28, 29). In addition, we will illustrate how energy metabolism in ischemic heart failure is controlled by the interplay of transcriptional regulation, post-translational modifications, and cytosolic/mitochondrial signaling kinases. We will also critically appraise some of the published data and discuss the controversies surrounding energy substrate preference in the failing heart. Lastly, we will briefly discuss the advances in pharmacological interventions which target myocardial energy metabolism as an approach to treat heart failure.
Cardiac Metabolism in the Healthy Heart
Cardiomyocytes have a virtue of metabolic adaptability as they can omnivorously utilize different energy substrates, including fatty acids, carbohydrates (glucose and lactate), ketones and amino acids, to meet the high energy demand of the heart and to sustain contractile function. Due to this metabolic flexibility, the heart's substrate preference can rapidly change based on the availability of energy substrates supplied to the heart, hormonal status and the changing workload of the heart. Mitochondrial oxidative phosphorylation produces the majority of the high-energy phosphates needed to sustain contractile function (Figure 1). Of this, fatty acid oxidation is the biggest contributor to mitochondrial oxidative metabolism, providing approximately 40–60% of the total energy produced, with the oxidation of glucose, lactate, ketone and amino acids providing the remaining 20–40%. To better understand the use of each substrate, first, we will discuss glucose, fatty acid, and ketone metabolism in the normal healthy heart.
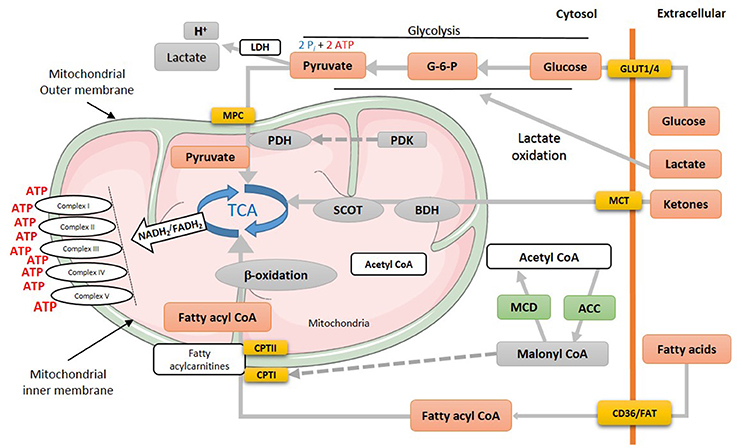
Figure 1. Energy metabolism in normal heart. Various metabolic pathways contribute to mitochondrial ATP production in the heart. Mitochondrial ATP production uses mostly fatty acids, glucose and ketones as a fuel source. The production of acetyl CoA by fatty acid ß-oxidation first requires the mitochondrial uptake of fatty acids via a carnitine carrier system. Oxidation of glucose involves the production of pyruvate via glycolysis, which produces acetyl CoA for the TCA cycle via PDH. Acetyl CoA production by ketone body oxidation is facilitated by BDH and SCOT. MPC, mitochondrial pyruvate career; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; TCA, tricarboxylic acid cycle; SCOT, succinyl-CoA-3-oxaloacid CoA transferase; BDH, β-hydroxybutyrate dehydrogenase; CPT, Carnitine palmitoyltransferase; MCD, malonyl CoA dehydrogenase; ACC, acetyl CoA carboxylase; MCT, monocarboxylate transporter; CD36, cluster of differentiation; FAT, fatty acid translocase; GLUT, glucose transporter type (1 or 4).
Glucose Metabolism
Locke and Rosenheim in 1907 were the first to study myocardial glucose uptake in an isolated rabbit heart model (30). Thirty years later, Albert Szent-Gyorgyi, Hans A. Krebs and William A. Johnson reported the conversion of pyruvate to succinate, unearthing the Krebs cycle (31). The uptake of glucose into the cardiomyocyte occurs though insulin-independent (GLUT1) and insulin-dependent (GLUT4) transporters. Transported glucose is first phosphorylated by hexokinase to glucose-6-phosphate (G-6-P), which can then undergo different metabolic pathways. G-6-P can be used as a substrate for glycolysis to produce pyruvate, NADH and 2 ATP (Figure 1) or it can enter the hexosamine biosynthesis pathway. In addition, G-6-P can be used for glycogen synthesis or shuttled to the pentose phosphate pathway (32). Glycolysis-derived pyruvate can either be converted to lactate by lactate dehydrogenase (LDH) or alternatively transferred to the mitochondria matrix by the mitochondrial pyruvate carrier (MPC) (33) (Figure 1). In the mitochondrial matrix, pyruvate dehydrogenase (PDH), the rate limiting enzyme of glucose oxidation, catalyzes the conversion of pyruvate to acetyl CoA which feeds into the tricarboxylic acid (TCA) cycle. Mitochondrial pyruvate can also supply the TCA cycle with essential intermediates, namely oxaloacetate and malate, via carboxylation. The enzymatic activity of PDH can be inhibited through phosphorylation by PDH kinase (PDK) and be reactivated by PDH phosphatase (34).
In terms of energy producing efficiency, oxidation of each glucose molecule requires 6 O2 molecules and produces 31 high-energy phosphate (ATP) molecules. Accordingly, glucose's phosphate/oxygen (P/O) ratio is 2.58, making glucose the most efficient energy substrate. Glucose metabolism also produces two high energy phosphate (Pi) molecules by converting glucose to pyruvate through glycolysis (Figure 1).
Fatty Acid Oxidation
Beta-oxidation of fatty acids was first proposed by a work of Knoop (35) which was later verified by a following study by Dakin (36). After a half century, Richard Bing and colleagues discovered that the human heart prefers fatty acids for respiration (37). Later in 1963, Philip Randle and his group (38) proposed that glucose and fatty acids are working in a concert to meeting the high energy demand of the heart, which is later known as the Randle cycle. The source of fatty acids for mitochondrial oxidative metabolism originates from circulating free fatty acids (FFAs) bound to albumin and/or released from triacylglycerols (TAG) contained in chylomicrons or very-low-density lipoproteins (VLDL) (39–41). Some fatty acids also originate from intracellular TAG stores (42). Extracellular fatty acids are transported to the cardiomyocytes through passive diffusion or via the fatty acid transport protein (FATP) or fatty acid translocase (FAT, CD36) (39, 43, 44). Once FFAs are in the cytosol, they are esterified to long-chain fatty acyl CoA via an ATP-dependent pathway initiated by a family of fatty acyl-CoA synthase enzymes (45). The majority (90%) of long-chain fatty acyl CoAs are shuttled directly to the mitochondria, facilitated by carnitine palmitoyltransferase isomers (CPTI and CPTII), with the remainder stored in the myocardial TAG reservoir. CPTI converts long chain fatty acyl CoAs to long chain acylcarnitine in the outer mitochondrial membrane which can then be converted back to long chain fatty acyl CoA by CPTII in the inner mitochondrial membrane (27, 46–48). Flux of fatty acids through CPT1 is regulated by malonyl CoA, a potent inhibitor of CPT1 (49–51). Malonyl CoA levels in turn are primarily dependent on a balance between acetyl CoA synthesis by acetyl CoA carboxylase (ACC) (52–54) and degradation by malonyl CoA decarboxylase (MCD) (27, 55, 56) (Figure 1). It is worth mentioning that via a carnitine shuttle system, mitochondrial acetyl CoA can be shuttled back to the cytosol to further affect malonyl CoA levels (57). Additionally, citrates produced from the TCA cycle can also translocate to the cytosolic compartment where it can activate ACC1, produce more malonyl CoA, and send a feedback response to decrease fatty acid uptake into the mitochondria (27).
In terms of ATP production efficiency, the oxidation of a representative fatty acid molecule, namely palmitate, consumes 23 molecules of O2 and produces 105 high-energy phosphate (ATP) molecules. This means that fatty acids are the least efficient energy substrate with a P/O ratio of 2.33 compared to glucose oxidation (2.58). Moreover, esterification of fatty acid to fatty acyl CoA requires 2 ATP-derived Pi. Similarly, cytosolic processing of fatty acid to fatty acyl CoA or TAG as well as reconverting TAG to fatty acid also consumes 2 Pi. This further reduces the total energy profit from fatty acid oxidation.
Ketone Body Oxidation
Ketone bodies are produced during times of fasting and starvation via hepatic ketogenesis, producing three types of ketone bodies: acetone, β-hydroxybutyrate (βOHB) and acetoacetate. Acetone, present in low abundance, is excreted by exhalation while βOHB and acetoacetate are the main ketone bodies circulating in our blood (58, 59). The predominant amount of ketogenesis occurs in the liver and the heart itself cannot produce ketones. Therefore, once circulating ketones reach the heart, ketones can enter the cardiomyocytes and be transported to the mitochondrial matrix where βOHB is oxidized into acetoacetate by βOHB dehydrogenase (BDH1), after which acetoacetate is activated by succinyl-CoA to acetoacetyl-CoA by the rate limiting enzyme, succinyl-CoA:3-oxoacid-CoA-transferase (SCOT) which is encoded by the gene Oxct1. Lastly, acetyl-CoA acetyltransferase converts acetoacetyl-CoA into acetyl-CoA (60, 61). Acetyl-CoA can then enter the tricarboxylic acid cycle and electron transport chain to generate ATP. Regulation of ketone oxidation enzymes in the heart remains poorly understood although hepatic BDH1 has been shown to be post-translationally modified by SIRT5 (62) and cardiac SCOT may be regulated by residue-specific nitration in the settings of diabetes (63, 64) and aging (65).
Ketone body as a fuel for the heart has long been recognized (37, 66). Barnes and Waters along with their associates (67, 68) first demonstrated that the heart can use ßOHB in 1938. In 1965, Rudolph et al. (69) showed that ßOHB and acetoacetate accounted for 2.6% ± 0.3% and 6.0% ± 1.0%, respectively, of the heart's total oxidative metabolism. Despite minor contribution to the total ATP production compared to fatty acids or glucose (12), myocardial oxidation of ketones can increase significantly in response to changes in the arterial concentration (69). Therefore, blood ketone levels are a key factor in determining myocardial ketone body oxidation rates and influencing the heart's metabolic flexibility. Lastly, it is important to consider, since the P/O ratio of ßOHB, is 2.50, this would make ketones less efficient than glucose but more efficient than palmitate.
Cardiac Metabolism in the Failing Heart
In heart failure, cardiac energy metabolism is compromised due to impaired cardiac ATP production, metabolic flexibility, mitochondrial TCA cycle activity and overall oxidative metabolism (9, 70, 71). More specifically, impaired mitochondrial function and oxidative capacity (10, 19, 72) results in reduced ATP production by up to 40% compared to the normal heart. In humans, the phosphocreatine/ATP ratio has been shown to decrease in heart failure patients (73–75), alongside impaired mitochondrial electron transport chain activity (19, 76). However, the exact energy metabolic profile of heart failure remains controversial as there is still discrepancy regarding substrate preferences. For example, both myocardial fatty acid, glucose, and ketone body oxidation rates have been shown to vary depending on the heart failure model used and the duration of heart failure. Other molecular changes may also contribute to the diverse nature of heart failure energy metabolic pathology. This includes changes to transcriptional control, post-translational modifications, and mitochondrial biogenesis, all of which occur in response to heart failure's metabolic inflexibility.
Glucose Metabolism in the Failing Heart
As discussed, the ischemic failing heart has a considerable energy deficit (30–40%) due to impaired mitochondrial oxidative metabolism, as well as due to the fact that there is a shortage in oxygen transport and an increased reliance on less efficient energy substrates which have a low O2/ATP ratio (9, 26). Importantly, other forms of heart failure, including non-ischemic failing heart, might not have the same oxygen deficit as was shown in animals (77) and human (78). This could potentially have implications on substrates metabolism, but it is out of the scope of this review. While there is a general consensus that the heart switches from fatty acid oxidation to glucose metabolism to produce energy in heart failure, we believe it is more plausible to state that the failing heart switches from mitochondrial oxidative phosphorylation to glycolysis as a main source of energy.
Glycolysis
Glycolysis is anaerobic metabolic process which its rates increase in the failing heart (18) (Figure 2). Direct measurements of myocardial glycolytic rates showed a marked increase in both abdominal aorta constriction (ACC)- and transverse aortic-constriction (TAC)-induced heart failure animal models (21, 79–81). In patients with heart failure with reduced ejection fraction (HFrEF), there is a marked increase in glucose uptake and glycolysis that is associated with an increased lactate and pyruvate accumulation (82). This increase in glycolysis and the accumulation of glycolysis by-products, namely lactate, and proton, is also seen in heart failure in Dahl salt-sensitive rats fed a high salt diet (83), and in patients with heart failure (82). While glycolysis provides an anaerobic source of ATP, the accumulation of glycolysis-derived protons in the cytosol can contribute to acidosis which can reduce cardiac contractility, as it desensitizes contractile proteins to Ca2+ and inhibits the slow Ca2+ current (27, 84). It also leads to Na+ and Ca2+ overload as cardiomyocytes attempt to extrude excess proton to the extracellular space. It can also lead to an ionic imbalance which can aggravate the energy deficit seen in heart failure, as the already limited cardiac ATP supply is shifted toward re-establishing ionic homeostasis.
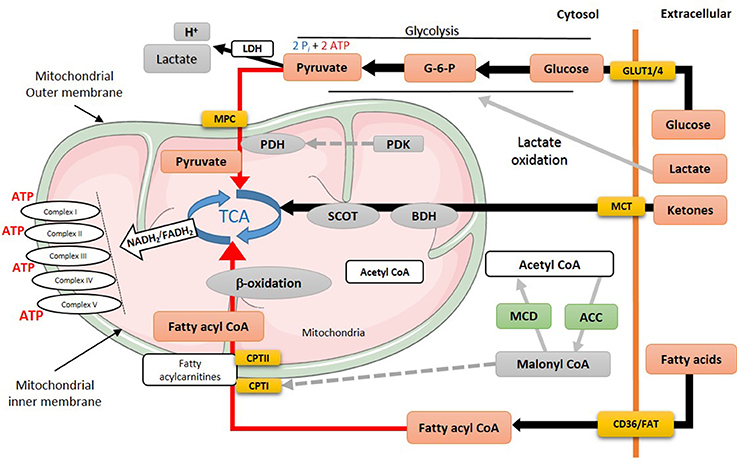
Figure 2. Energy metabolism in heart failure. During heart failure, overall mitochondrial oxidative metabolism and electron transport chain activity is compromised. Increased flux is indicated by black lines, while red lines indicate impaired utilization of different substrates. MPC, mitochondrial pyruvate career; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; TCA, tricarboxylic acid cycle; SCOT, succinyl-CoA-3-oxaloacid CoA transferase; BDH, β-hydroxybutyrate dehydrogenase; CPT, carnitine palmitoyltransferase; MCD, malonyl CoA dehydrogenase; ACC, acetyl CoA carboxylase; MCT, monocarboxylate transporter; CD36, cluster of differentiation; FAT, fatty acid translocase; GLUT, glucose transporter type (1 or 4).
An important point to consider when measuring myocardial glycolytic rates is the animal model used. Employing murine hearts to address myocardial energy metabolism has increased dramatically due to the flexibility mice provide for knockdown or overexpression of target genes. The murine model, however, may not be ideal in cases where myocardial glycolytic measurements are required, especially if glycolytic rates are differentially affected due to a pathological disease. For example, heart failure-induced up-regulation in glycolysis rate is not detectable if the mouse heart is used as the model to study heart failure (13, 14, 83). This is probably due to the fact that the mouse heart is highly glycolytic compared to other animals models (85) and this high glycolysis is already saturating its contribution to overall ATP production (~20%). Therefore, differences in energy substrates utilization between different animal models need to be considered (86, 87). Nevertheless, other studies have also reported a marked increase in glycolysis rate in other animal models of heart failure (80, 88–90).
One of the characteristic changes in heart failure is the development of insulin resistance which has been shown in vivo (13, 14, 91–95) and in human (96–98). Insulin regulates glucose uptake by enhancing GLUT4 translocation (99, 100) and increases glycolysis (101–103). In insulin resistance in heart failure, the heart switches to GLUT1 to take up glucose. Despite this impaired insulin signaling, glycolysis is increased in the failing heart.
Glucose Oxidation
We and others have reported that impairment of glucose oxidation is a metabolic marker that precedes the development of cardiac dysfunction in different animal models of heart failure (14, 94). Although glycolysis rates increase in heart failure, this does not necessarily translate into an increase in glucose oxidation since glycolysis and glucose oxidation are differentially regulated in the heart (104). The majority of studies directly examining the failing heart's glucose oxidation rates in humans and animals show a marked decrease in glucose oxidation in the failing heart, and a reduced contribution of glucose oxidation to overall ATP production (13, 14, 91, 92, 94, 96, 98, 105). A study by Diakos et al. (82) also demonstrated that the increase in cardiac glycolysis seen in severe heart failure patients was not accompanied by an increase in lactate and pyruvate accumulation, suggesting that the increase in glycolysis is not matched by an increase in glucose oxidation. In support of this, Paolisso et al. (96) reported an abrogated rate of glucose oxidation in patients with congestive heart failure. Furthermore, impairment of pyruvate oxidation in transgenic mice is associated with the development of left ventricular hypertrophy (89), emphasizing the relationship between maintained glucose oxidation and normal cardiac function. In support of this, Kato et al. (106) showed that in Dahl salt sensitive rats with heart failure (which have high cardiac glucose uptake and glycolysis), stimulating PDH with dichloroacetate improved heart function and decreased lactate production (presumably due to an increase in glucose oxidation). Combined, these studies suggest an important role of cardiac metabolic inflexibility, which occurs in heart failure, with regards to glucose oxidation in mediating heart failure severity.
While the majority of studies suggest a decrease in glucose oxidation in the failing heart, not all studies are consistent with this finding. The extent of reduction in cardiac glucose oxidation in heart failure varies according to the severity of heart failure, as well as the experimental model of heart failure used and the availability of other energy substrates. In a rat model of transverse aortic constriction (TAC), for instance, Doenst et al. (107) showed that glucose oxidation rates remained unchanged in a rat model of compensated heart failure (due to mild TAC) glucose oxidation was only reduced after systolic dysfunction occurred. Whether the slow development of diastolic dysfunction over a relatively long period of time in animal models has an impact on energy metabolism changes needs further investigation. In support of this, Zhang et al. (14) also reported that glucose oxidation rate was only decreased as an early sign of cardiac dysfunction in another mild heart failure model induced by AAC. As the severity of heart failure increases, this will have a direct impact on glucose oxidation rate. This was shown in a number of studies where a mouse model of pressure-overload-heart failure with severe cardiac dysfunction showed a marked impaired glucose oxidation rates (93–95). However, Osorio et al. (108) reported that glucose oxidation rate was increased in a canine cardiac pacing model of heart failure which, however, contrasts the decrease in glucose oxidation seen in pacing-induced heart failure seen in pigs (88). Unfortunately, cardiac glycolytic rates were not measured in the study by Osorio et al. (108). In a TAC model of murine heart failure, an increase in the proportion of glucose oxidized by the hearts was also seen (109). The reason for these discrepant findings is unclear. However, it should be acknowledged that high pyruvate supply from increased glycolysis has the potential to augment glucose oxidation (110). Indeed, the study by Kolwicz et al. (109), which suggests that glucose oxidation was increased in TAC hearts from mice, also observed a marked increase in lactate and alanine production, indicative of increased glycolysis rates. Furthermore, while pacing-induced heart failure in dogs showed an increase in glucose oxidation (111), cardiac function was markedly low in these dogs [left ventricular work (LVW) was 148 ± 28 Hg*mm while normal LVW is 800–900 Hg*mm]. In severe heart failure, it is possible that mitochondrial calcium control is compromised, potentially leading to mitochondrial calcium accumulation, and activation of the PDH complex due to calcium activation of PDH phosphatase (112).
The reduction in glucose oxidation that occurs in the failing heart is due, in part, to an overall deterioration in mitochondrial oxidative capacity, as well as due to an impaired activity of PDH, the rate-limiting enzyme for glucose oxidation (13, 14, 92, 98). There is also some evidence that the machinery of glucose oxidation could be impaired in the failing heart (104). Gupte et al. (113) demonstrated that the mRNA expression of PDH, MCT1, and pyruvate/alanine aminotransferase, were decreased in heart failure, suggesting impairment in pyruvate metabolism. In line, Dodd et al. (89) demonstrated, using hyperpolarised 13C-magnetic resonance spectroscopy, that PDH complex activity is impaired in a rat model of myocardial infarction-induced heart failure. Of importance, is that PDH complex impairment was progressive and proportional to the degree of cardiac dysfunction.
As it was discussed earlier (see Glycolysis section), it is generally accepted that insulin-induced stimulation of glucose oxidation is markedly attenuated in obesity and diabetes (114, 115), contributing to myocardial metabolic inflexibility (116). Impairment in insulin signaling and development of insulin resistant myocardium precedes cardiac dysfunction in heart failure and it is a major determine of its progression (14, 94, 117). Rutter et al. (118) showed that increased insulin resistance, which is also associated with obesity, was accompanied with worsening cardiac remodeling. In consistence, Peterson et al. (119) also, in consistence, demonstrated that insulin resistance in obese women was associated with deterioration in cardiac efficiency. Taken together, it seems plausible to suggest that insulin resistance is, at least in part, responsible for the reduction in glucose oxidation in heart failure. In a rat model of streptozotocin-induced diabetic cardiomyopathy, insulin resistance is associated with a marked decrease in PDH flux and diastolic function (120). Of interest, is that dichloroacetate (DCA), a PDK inhibitor that enhances PDH activity, treatment reversed insulin resistance, increase PDH flux and improving cardiac function (120). This further emphasizes the potential therapeutic applications of improving glucose oxidation to mitigate cardiac dysfunction in heart failure. In the same context, cardiac-specific PDHA1(−/−) knockout mice showed impaired insulin signaling which was associated with diastolic dysfunction (121). Consistent with this, Sankaralingam et al. (115) demonstrated that cardiac dysfunction and insulin resistance were both worsened by high fat diet-induced obesity in abdominal aortic constriction-induced heart failure mice model. Collectively, this further emphasizes the crucial role of insulin resistance and high circulating FFAs in the pathogenesis of heart failure.
Another mechanism by which glucose oxidation could be attenuated in heart failure is through heart failure-induced mitochondrial hyperacetylation. Hyperacetylation occurs in the failing heart (122) and hyperacetylation of PDH has previously been shown to have an inhibitory effect on its activity (123, 124). Thus, heart-failure induced hyperacetylation could inhibit PDH activity and decrease glucose oxidation rates. It has also been reported that hyperacetylation can increase the activity of fatty acid oxidation enzymes, such as LCAD and β-HAD (125). As such, enhancing the activity of these ß-oxidation enzymes could negatively feedback to inhibit glucose oxidation through the Randle cycle (38, 126).
As discussed, the failing heart has a considerable energy deficit (30–40%) due to impaired mitochondrial oxidative metabolism, as well as due to the fact that there is a shortage in oxygen transport and an increased reliance on less efficient energy substrates which have a low O2/ATP ratio (9, 26). While there is a general consensus that the heart switches from fatty acid oxidation to glucose metabolism to produce energy in heart failure, we believe it is more plausible to state that the failing heart switches from mitochondrial oxidative phosphorylation to glycolysis as a main source of energy.
Fatty Acid Oxidation in the Failing Heart
While it is generally agreed that the failing heart has reduced cardiac energetics, mitochondrial TCA cycle activity and overall oxidative metabolism (9, 70, 71), it is less clear whether myocardial fatty acid oxidation rates are also decreased. It is generally assumed that cardiac fatty acid oxidation is decreased in heart failure (10, 127–129), which is supported by decreased transcription of a number of enzymes involved in fatty acid oxidation (10, 128–132). However, direct measurements of fatty acid oxidation rates in both human and experimental models of heart failure do not always support this assumption.
Heart failure can be associated with an increase in circulating FFA levels due to high lipolysis rates (96, 133, 134). Of importance, is that increased level of circulating FFAs in failing heart is an important determinant of fatty acid oxidation rates in the heart. For example, decompensated heart failure patients show an increase in circulating FFAs levels, which is accompanied by enhanced myocardial fatty acid uptake and fatty acid oxidation (135, 136). In support of this, Taylor et al. (137) also demonstrated that fatty acid uptake rates are up-regulated in patients with severe heart failure (ejection fraction ~24%), using a positron emission tomography (PET) imaging technique. In contrast, Dávila-Román et al. (73), who also using (PET) imaging, showed a decrease in circulating FFAs level in patients with non-ischaemic heart failure. Neglia et al. (75) also showed a decreased fatty acid oxidation in patients with idiopathic dilated cardiomyopathy.
Animal studies also show differing results as to what happens to fatty acid oxidation in the failing heart. Studies in mice in which heart failure was produced secondary to pressure overload or a myocardial infarction have shown that cardiac fatty acid oxidation rates are unchanged (94, 95, 110). Mori et al. (91) also showed that fatty acid oxidation rate was not changed in Ang II-induced heart failure. However, it should be recognized that these maintained rates were seen despite a decrease in cardiac function, suggesting that fatty acid oxidation per unit work may actually increase in heart failure. In addition, these studies showed that the contribution of fatty acid oxidation to total ATP production increased, due primarily to a decreased contribution of glucose oxidation to ATP production. Others have also shown that with compensatory heart failure, fatty acid oxidation enzymes are preserved (21, 79). In contrast, Byrne et al. (94) and Sung et al. (93) have shown that cardiac fatty acid oxidation rates are impaired in TAC-induced severe heart failure in mice. In rats subjected to TAC, Doenst et al. (107) also demonstrated that fatty acid oxidation rates decreased in parallel with an overall decrease in mitochondrial oxidative phosphorylation (107). Moreover, a reduction in fatty acid oxidation rates was also seen in canine models of severe heart failure (138, 139).
The issue of what happens to fatty acid oxidation in the failing heart becomes more complex in the presence of obesity and/or diabetes is present. Even in the absence of heart failure, fatty acid oxidation rates are elevated under these conditions (119, 140–142). These high cardiac fatty acid oxidation rates persist if evidence of heart failure is seen in obesity and diabetes (115, 141, 143). Furthermore a strong link between reduced cardiac efficiency and excessive reliance on fatty acid oxidation has been shown in ob/ob mice (141) and obese humans (119).
The reasons for the confusion as to what is happening to fatty acid oxidation in heart failure, may be related to alterations in the control of fatty acid oxidation at multiple levels, including changes in fatty acid supply to the heart, alterations in allosteric control of fatty acid oxidation, alterations in transcriptional control of fatty acid oxidation, and alterations in post-translational control of fatty acid oxidation. Increased fatty acid supply to the heart will increase fatty acid oxidation, as will the presence of insulin resistance (12). Indeed, Tuunanen et al. (144) showed that while cardiac fatty acid oxidation rates were decreased in patients with idiopathic dilated cardiomyopathy, as cardiac function deteriorated insulin resistance occurred with a subsequent increase in fatty acid oxidation rates. As discussed, in heart failure a marked cardiac insulin resistance occurs (13, 14, 92). This includes a decreased ability of insulin to inhibit fatty acid oxidation (14, 92). In the presence of obesity and/or diabetes, this cardiac insulin resistance is even more dramatic (115). As result, an increased cardiac insulin resistance in heart failure may contribute to maintaining fatty acid oxidation rates. Heart failure is often associated with impairment in insulin signaling which could have a marked impact on energy metabolism in the heart. Insulin has an inhibitory effect on fatty acid oxidation through enhancing the activity of acetyl CoA carboxylase which increases the tissue level of malonyl CoA thereby decreasing mitochondrial fatty acid uptake. Insulin-induced inhibition of fatty acid oxidation is impaired in the failing heart leading to an increase in fatty acid contribution to the total ATP production, despite being inefficient substrate during heart failure. Furthermore, inactivation of the carnitine shuttle system increases cytosolic fatty acyl CoA levels (or long chain acyl CoA) and, in addition to the accumulation of TAG and diacylglycerol (DAG), can have a negative impact on insulin signaling (14, 145, 146). For instance, excess lipid metabolites can phosphorylate serine residues on IRS-1 by activating IKK-NF-κB, JNK-AP-1, and the PKC pathway all of which can reduce glucose uptake by decreasing Akt and PI3K activity (14, 147).
Changes at the transcriptional level of genes involved in fatty acid oxidation are often cited as a key reason why cardiac fatty acid oxidation rates may be decreased in heart failure (148, 149). A down regulated gene expression of fatty acid oxidative enzyme (LCAD, MCAD) has been observed in heart failure patients, as well as during the progression of heart failure in animal models (130). Three distinct isoforms of peroxisome proliferator activated receptor [PPARα (cardiac abundant), PPARβ/δ, and PPARγ] are responsible for the transcriptional changes of fatty acid metabolic genes (150, 151). PPAR and the retinoid X receptor (RXR) complex are transferred to the nucleus to bind with a specific PPAR response element (PPRE), which is located in the target gene's promoter. An inducible PPARγ coactivator-1α (PGC-1α) is also correlated with the transcriptional activity of PPAR superfamily (149, 152). Fatty acids are the endogenous ligand of the PPARα and may activate the PPARα/PGC1α pathway for the transcriptional regulations. PPARα/PGC1α transcriptional activity has also been shown to regulate pyruvate dehydrogenease kinase 4 (PDK4), which can reduce glucose oxidation by inactivation of PDH activity (153, 154), but not glycolysis. Again in heart failure patients, cardiac PPARa expression was shown to down regulated (113, 155). The expression of PGC-1α, important for mitochondrial biogenesis, is also down regulated in heart failure [(20, 21, 48, 156)]. In pressure-overload-induced heart failure, abnormal mitochondrial morphology and reduced mitochondrial density is seen, which is associated with altered electron transport chain proteins expression (20). Patients with heart failure also show a reduction in mitochondrial DNA contents which was accompanied by down regulation of PGC-1α-associated proteins (157). Furthermore, based on DNA microarray analysis, it has been shown that a subset of downstream gene targets of PGC-1a are also down-regulated in the failing heart, which is correlated with the reduced left ventricular ejection fraction (158). However, it is still not clear whether this attenuation in the role of PGC-1α in heart failure is enough to manipulate mitochondrial biogenesis. However, a reduction in the number of mitochondria could contribute to the changes in fatty acid oxidation observed in heart failure.
Post-translational modifications may also alter fatty acid oxidation in the failing heart This includes mitochondrial lysine acetylation, in which an acetyl group is transferred to a lysine residues or mitochondrial proteins. Acetylation can be mediated through histone and non-histone acetyl-transferase (159). Furthermore, mitochondrial acetyltransferase, namely GCN5L, has also been shown to promote acetylation (160). On the reverse reaction, sirtuins (SIRTs) act as deacetylases to reverse the effect of acetylation (161, 162). Acetylation controls the activity of number of metabolic enzymes (163). Hyperacetylation of LCAD and ßHAD results in an increase in fatty acid oxidation rates (125). In obese mice with heart failure, an elevated GCN5L expression in abdominal aortic constriction-induced heart failure is associated with increased in LCAD acetylation and an increase in fatty acid oxidation (115). Furthermore, switching to a low fat diet in obese mice showed the opposite effect on the post-translational modification associated with reduced fatty acid oxidation (115). Moreover, we also showed glucose oxidation could be inhibited through hyperacetylation in heart failure (92). Taken together, post-translational modifications may be another factor to be considered which might to explain the metabolic inflexibility during heart failure.
Insulin Resistance and Heart Failure
Glucose and fatty acid metabolism are tightly controlled by insulin signaling in the heart. Evidence from clinical studies have shown a strong association between insulin resistance and cardiac dysfunction (118, 164). Moreover, patients with insulin resistance have high rates of lipolysis in adipose tissue with increases in TAG hydrolysis (164–166). Of importance, insulin resistance-induced shifts in favor of fatty acid oxidation and is associated with attenuation of glucose uptake by the heart [see review by (116)]. This change in metabolic preference was also observed in an experimental setting (13, 14, 91, 167) and clinical studies of heart failure (82, 88). Of interest, insulin resistance precedes any changes in cardiac energy metabolism in mice subjected to abdominal aortic constriction (14). Impairment in insulin signaling primarily has a direct inhibitory effect on fatty acid oxidation by increasing the malonyl CoA levels and secondarily inhibiting glucose oxidation through a negative feedback effect of the Randle cycle (38). Moreover, increasing fatty acid oxidation rates could also indirectly cause attenuation in glucose oxidation by triggering the activity of PDK and limiting the activity of the PDH complex. In a model of angiotensin II-induced heart failure with preserved ejection fraction (HFpEF), glucose oxidation rates were reduced by 45% as PDK activity was enhanced and PDH complex activity was attenuated (91). These results are further supported as PDK deletion improved the HFpEF-induced reduction in glucose oxidation (92).
Ketone Oxidation in the Failing Heart
Circulating Ketone Body Concentrations in Heart Failure
A major determinant of ketone oxidation rates in the heart are the levels of circulating ketones. Earlier studies have suggested that blood ketone levels are elevated in congestive heart failure (with reduced ejection fraction) patients proportional to the severity of cardiac dysfunction (168, 169). These results were recently challenged by Melenovsky et al. (170) who found that the plasma level of ßOHB in heart failure patients were similar to healthy subjects. However, in support of the earlier observations by Lommi et al. (168) and Lommi et al. (169), recent metabolomics studies have found increased blood ketone levels in HFrEF patients (171) and HFpEF patients (172). It is interesting to note however that Zordoky and colleagues also found that HFpEF patients had significantly higher blood ketone levels than HFrEF patients while HFrEF patients had lower ketone levels than healthy controls (172). The discrepancy in reported levels of circulating ketones in heart failure patients may be due to multiple reasons including differences in severity, duration and type of heart failure. This is especially the case in severe heart failure where insulin resistance-induced increases in hepatic ketogenesis could be inevitably contributing to increases in circulating ketones (173). In addition, it is important to note that cardiac ketone levels are dynamic. In general, the circulating levels and cellular uptake of ketones is proportional to its contribution to ATP production (174). In that regard, it is difficult to pin point cardiac ketone levels without concurrently considering pathological circulating serum levels, uptake, oxidative rates and secretion rates of ketone (175). For example, it is still not clear whether HFrEF patients, with lower blood ketone levels than HFpEF patients (172), have a greater reliance on ketone bodies and increased myocardial ketone body oxidation. Alternatively, it is possible that HFpEF patients with high circulating ketone body levels could be associated with increased muscle uptake and oxidation of ketone bodies. Therefore, changes in circulating ketone body levels are temporal, indefinite, and direct measurements of flux through myocardial ketone body oxidation rates are required.
Ketone Body Oxidation in Heart Failure
Arterio-venous measurements for ßOHB in HFrEF patients, a surrogate for myocardial ketone body utilization, reported no differences compared to healthy controls (176), and a slight increase, however not significant, in HFrEF patients (98). However, recent studies have suggested that myocardial ketone body oxidation is increased in heart failure. In a mouse model of compensated and decompensated pressure overload cardiac hypertrophy, proteomics data demonstrated that a key enzyme involved in ketone body oxidation, namely ß-hydroxybutyrate dehydrogenase (BDH1), was up-regulated 2 to 3-fold (15). Moreover, the myocardial metabolite profile of mice with heart failure was comparable to mice fed a 4-week ketogenic diet. In parallel, Bedi et al. (16) also observed an increased ratio of serum to myocardial ketone bodies with up-regulated expression of BDH1, BDH2, and succinyl-CoA:3-ketoacid CoA transferase (SCOT) in human heart failure patients. We have also seen an increase in myocardial ketone body oxidation rates in the ex vivo isolated failing murine heart (unpublished data) (17). Taken together, these studies suggest that the failing heart has an increased reliance on ketone body oxidation. Nevertheless, Nagao et al. (177) found that βOHB levels were elevated in mice with ascending aortic banding-induced heart failure. In vitro, subjecting cardiomyocytes to oxidative stress also resulted in elevated levels of βOHB, increased levels of anti-oxidative factors, and concurrent down-regulation of the rate limiting enzyme in ketone body oxidation, SCOT (177). These findings would suggest that ketone body oxidation decreases during heart failure to maintain elevated levels of βOHB as a compensatory response to protect the heart against oxidative stress. The reason for the contradictory results could be due to the severity and duration of heart failure in these studies (15, 177). Since heart failure is a chronic condition, it seems plausible to suggest that in the early stage of heart failure, βOHB may have an antioxidant role and only become an adaptive fuel source in the end-stage heart failure (177). It is worth mentioning that βOHB has previously been shown to be an HDAC inhibitor and protects against oxidative stress (178). However, with this uncertainty comes the question of whether ketone body oxidation in heart failure is adaptive or maladaptive (23, 173)?
To address whether ketones are adaptive or maladaptive in failing hearts, several recent studies have investigated this. Schugar et al. (179) reported that mice with a cardiac-specific knockout of Oxct1 (or SCOT, the rate limiting enzyme in ketone body oxidation) subjected to TAC-induced heart failure was associated with increased rates of anaplerosis, mitochondrial ultrastructure abnormality and accelerated pathological cardiac remodeling. Similarly, overexpression of cardiac BDH1, the first enzyme in the ketone body oxidation pathway, mitigated oxidative stress and attenuated cardiac remodeling following pressure overload-induced hypertrophy (180). Together, these studies suggest that heart failure-induced increases in ketone body oxidation are adaptive for a failing heart. However, there are still several aspects to consider in light of the preliminary suggestion that ketone body oxidation is adaptive in the setting of heart failure. One factor to consider is the change in cardiac ketone levels, a dynamic concentration that would decrease if heart failure is characterized by elevated myocardial ketone body oxidation rates. Keeping this in mind, the up-regulation of cardiokine/myokine follistatin-like protein 1 (FSTL1), having been shown to be cardioprotective in heart failure, is accompanied by a reduction in cardiac ketone body uptake in a canine model of tachypacing-induced heart failure (181). In such a scenario, high cardiac ketone levels would be suggested to be undesirable and accelerating myocardial ketone body oxidation would be adaptive in heart failure. Alternatively, since ßOHB has been shown to be an HDAC inhibitor (178) and recent work has demonstrated that HDAC inhibitors can enhance myofibril relaxation kinetics and improve diastolic function (182), maintaining ketone levels may indeed be beneficial in the setting of HFpEF as opposed to HFrEF. Second, another factor to consider are post-translational modifications which may be responsible for myocardial energy metabolic derangements that contribute to the progression of heart failure (122). In this case, increasing myocardial ketone body oxidation and increasing the myocardial acetyl CoA pool has been suggested to provide more substrate for lysine acetylation, ultimately contributing to the failing heart's hyperacetylated state (122). This may or may not be desirable since hyperacetylation of glucose and fatty acid oxidation enzymes (183), and its effects on enzyme activity, require further investigation in the setting of heart failure.
Ketones' Effects on Glucose and Fatty Acid Oxidation
Ketones have the potential to suppress glucose oxidation and vice versa (37, 69, 184) as they both compete for available oxygen and as a source of TCA cycle acetyl CoA. Williamson and Krebs (184) observed that in the presence of insulin, acetoacetate decreased glucose oxidation by half in the perfused rat heart. This may be explained by the ability of ketones to increase the mitochondrial acetyl-CoA to CoA ratio and consequently inhibit the activity and flux through PDH, the rate limiting enzyme of glucose oxidation (185–187). Of importance is whether ketones add a new dimension of complexity to the Randle cycle (38). Furthermore, the influence of ketone levels and its inhibitory effect on glucose oxidation also needs to be characterized to understand whether it is beneficial to enhance either of these pathways. Recently, ketone bodies were found to decrease myocardial glucose uptake and increase myocardial blood flow in a PET study in healthy humans (188). In connection with this displaced glucose uptake, leucine metabolism into ketone bodies has also been shown to inhibit GLUT4 translocation in cardiomyocytes due to an increase in lysine acetylation, ultimately hampering cardiac glucose uptake (189). Since heart failure is characterized by an increase in acetylation (122), the failing heart's hyperacetylated state could be potentiating leucine to ketone-mediated GLUT 4 inhibition and partially conferring insulin resistance. However, this is in contrast to a study that showed that administration of ßOHB and acetoacetate were able to recapitulate insulin-induced improvements in an isolated perfused rat heart's ex vivo cardiac efficiency (190). Since the failing heart is insulin resistant, ketones may be a viable substrate to improve cardiac efficiency in the failing heart (190). However, more studies measuring ketone body oxidation flux in the presence and absence of insulin are required.
It has also been reported that ketones can inhibit myocardial fatty acid oxidation (191, 192). For example, intravenous infusion of ßOHB suppressed myocardial fatty acid oxidation independent of changes in malonyl-CoA levels or the ratio of acetyl-CoA to CoA in pigs (193). Since fatty acid oxidation rates in heart failure remains controversial, it is unclear whether the inhibitory effects of ketones on myocardial fatty acid oxidation are beneficial or detrimental for the failing heart. This, however, further underlines the crucial role of ketone bodies in myocardial energy metabolism and implicates ketones as an important role-player that is currently neglected from the Randle cycle.
Therapeutic Approaches to Address the Failing Heart's Metabolic Profile
Stimulating Glucose Oxidation
Targeting glucose metabolism has been shown to be an effective approach to mitigate cardiac remodeling and improve heart function. Ikegami et al. (117) and Liao et al. (194) both reported that enhancing glucose uptake by GLUT1 and GLUT4 improved cardiac function and attenuated pressure-overload-induced hypertrophy in mice. Dichloroacetate (DCA) is a direct PDK inhibitor which increases glucose oxidation via enhancing PDH complex activity in the setting of heart failure. In the isolated working rat heart DCA enhances post-ischemic cardiac function and efficiency which is associated with improved coupling between glycolysis and glucose oxidation (195). DCA-induced improvement in coupling between glycolysis and glucose oxidation was also later demonstrated in suprarenal abdominal aortic constriction in rat (196). Similarly, Kato et al. (106) demonstrated that DCA administration to Dahl-salt sensitive rats increased cardiac energy reserve, reduced oxidative stress and slowed the transition from compensated heart failure to failing heart. In line with experimental studies, small clinical studies, although few, have shown promising improvement in cardiac contractility with DCA treatment in patients with coronary artery disease (197) and heart failure (198). Clinical data were not all consistent as DCA infusion in patients with congestive heart failure did not show significant beneficial effects (199).
Inhibiting Fatty Acid Oxidation
There are number of pharmacological approaches which are shown to successfully reduce fatty acid oxidation. Two molecules, namely etomoxir and perhexiline, is shown to inhibit CPT1 (Figure 3) and limit fatty acid oxidation with parallel increase in glucose oxidation in mouse and rat models of heart failure (200, 201). In humans, etomoxir showed improvement in ejection fraction and cardiac output (202, 203). Perhexiline also improves cardiac function and symptoms of heart failure (204) However, clinical trials to validate the preliminary encouraging finding with these two molecules were terminated due to the hepatotoxicity (204).
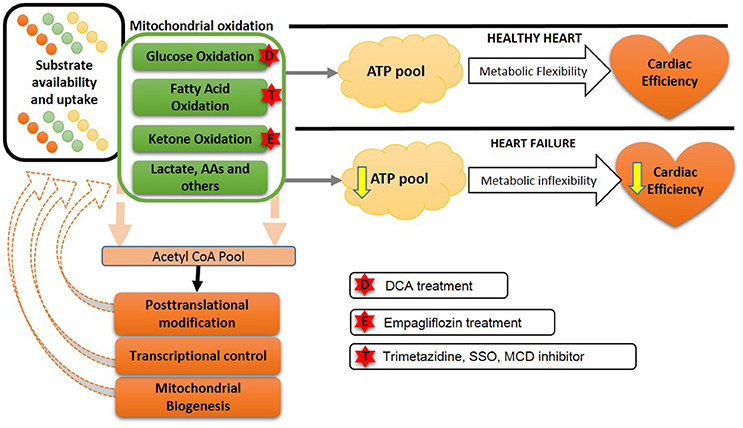
Figure 3. Future approaches to overcome the metabolic balance or inflexibility: In the healthy heart, a variety of energy substrates produce ATP to maintain metabolic flexibility and cardiac efficiency. However, in heart failure a reduced ATP production occurs due to a decreased metabolic inflexibility and a less efficient heart. Transcriptional changes and altered mitochondrial biogenesis also contribute to this metabolic inflexibility in heart failure. Possible approaches to improve metabolic flexibility are shown by stars. DCA, dichloroacetate; SSO, sulfo-N-succinimidyl-oleate; MCD, malonyl CoA dehydrogenase; AA, amino acids.
Furthermore, sulfo-N-succinimidyl-oleate (SSO) is an inhibitor of CD36 and has been shown to decrease fatty acid oxidation followed by an indirect increase in glucose oxidation (205). While these approaches showed beneficial effects in experimental studies involving heart failure, clinical studies with these agents have yet to be performed.
Another approach to inhibiting fatty acid oxidation is to inhibit the last enzyme of fatty acid ß-oxidation, 3-keotacyl CoA thiolase, with trimetazidine (206). Clinically trimetazidine has shown beneficial effects by increasing cardiac efficiency, where there is a shift in myocardial substrate utilization from fatty acid oxidation to glucose oxidation. It has been using as an antianginal agent in more than 100 countries (207). A combination of chronic trimetazidine treatment along with the other conventional therapy in heart failure patients improves cardiac function in humans (208). Treatment with trimetazidine in heart failure patients with idiopathic dilated cardiomyopathy shows a decrease in myocardial fatty acid oxidation rates, as well as improved left ventricular function and insulin sensitivity (208). A meta-analysis of clinical trials with trimetazidine in heart failure, showed a beneficial effect of trimetazidine on left ventricular systolic function, clinical symptoms for patients with chronic heart failure and importantly may result in decreasing all-cause mortality (209). We have also shown that trimetazidine prevents obesity-related reductions in cardiac function in obese mice with heart failure (210, 211).
Another potential intervention to decrease fatty acid oxidation in heart failure is with ranolazine. Ranolazine has been clinically used as anti-anginal agent since 2006 (212, 213). While considered to be an inhibitor of the late Na+ current, ranolazine is also a fatty acid oxidation inhibitor, which is capable of activating PDH a rate limiting enzyme for glucose oxidation (214). However, ranolazines efficacy in treating heart failure has not been extensively studied.
Enhancing Ketone Body Oxidation
In diabetic cardiomyopathy, ketones have been popularized as a thrifty fuel substrate for the heart (215, 216). The role of myocardial ketone metabolism has attracted huge attention since empagliflozin, a sodium glucose co-transporter-2 inhibitor used to treat type 2 diabetes, has shown cardioprotective effects in type 2 diabetic patients. This cardioprotection was associated with increased plasma ketone levels which led to the proposed cardioprotective role of increased ketone body oxidation in the diabetic failing heart (215–217). Furthermore, a recent study found that non-diabetic mice with experimental TAC-induced heart failure was protected against heart failure-induced decreases in in vivo and ex vivo function following a 2-week treatment with empagliflozin (218). However, despite the promising and exciting findings, it is still not clear whether empagliflozin increases ketone oxidation in the heart, or whether empagliflozin-mediated cardioprotection is through a ketone-independent mechanism (219). Therefore, future studies are required to elucidate whether empagliflozin's cardiovascular benefits are mediated by changes in myocardial ketone oxidation.
Future Directions
There is a growing recognition and understanding of the importance of metabolic flexibility of the heart and how metabolic inflexibility in heart failure could contribute or even cause deterioration of cardiac contractility and affect the disease progression. Of importance, is that metabolic inflexibility could also be influenced by other comorbidities such as diabetes, obesity, hyperlipidemia and hypertension. Taken together, aiming to re-establish metabolic flexibility in the failing heart is shown to be an effective approach to improve cardiac function and therapeutic outcome. In addition, it is important to recognize the need for a “tailored therapy” for different categories of patients with heart failure based on the type and severity of heart failure as well as the comorbidities which co-exist.
Here, we will discuss some of the recent pharmacological interventions which could have clinical value for failing heart patients.
Glucose Oxidation
The shift toward utilizing a more oxygen-efficient substrate, namely glucose, could potentially have favorable effects in terms of the energy production and cardiac function of the ischemic failing heart which is oxygen deficient. This approach would also improve the coupling between glycolysis and glucose oxidation and produce more ATP per mole of glucose oxidized. It would also limit glycolysis-induced acidosis and its consequent inhibitory effect on cardiac contractility. As discussed earlier, DCA is shown to increase the contribution of glucose to the total ATP production through increasing the glucose oxidation rate with a secondary reduction in fatty acid oxidation in different experimental models and in pilot human studies. However, it is important to note that DCA has a poor pharmacokinetic profile (short half-life) and a low potency. Therefore, future investigations using DCA treatment should potentially consider continuous infusion to administer DCA to maintain effective concentration.
Fatty Acid Oxidation
In the same context of utilizing oxygen efficient energy substrate in a failing heart which is under oxygen deficit, enhancing cardiac function could be achieved by reducing the reliance of the heart on fatty acid oxidation. While a considerable number of approaches exist that can directly or indirectly inhibit fatty acid oxidation, clinical trials targeting a reduction of fatty acid uptake or enzymatic activity are either limited with the confounding factors or underpowered. Therefore, framing animal models along with potential drug targeting fatty acid oxidation to optimize its effective dose, length and other possible side effects, under different diseases states (i.e. obesity, diabetes), different age, and sex should be a prime consideration prior to design future heart failure clinical studies. Unlike etomoxir and perhexiline (CPT1 inhibitors), it is not known if SSO cause hepatotoxicity. Another possible therapeutic approach is using malonyl CoA decarboxylase (MCD) inhibitors. It has been shown that by increasing malonyl CoA levels, fatty acid oxidation is reduced with a compensatory increase in glucose oxidation (25, 220). Inhibition of MCD, using the novel compound CBM-301106, increases cardiac malonyl CoA levels and decreases fatty acid oxidation (221). However, MCD inhibitors have yet to be tested in the clinical
Furthermore, inhibition of beta-oxidation enzymes, such as 3-keotacyl CoA thiolase, also has potential in reducing fatty acids oxidation l. Based on the promising outcomes in animal and human studies (described in Therapeutic Approaches section), modulation of fatty acid oxidation using trimetazidine is a potential approach to treating heart failure. Again, large randomized clinical trials are still needed to confirm this.
Of importance, is that modulating a particular energy substrate use byu the mitochondria to enhance the overall oxidative phosphorylation could be hindered by a decrease in the number and quality of the mitochondria in the myocardium. Targeting PGC1α to enhance mitochondrial biogenesis and improve the transcriptional changes in the failing heart could potentially have a therapeutic application in heart failure. Furthermore, a combination of the potential therapeutic components (trimetazidine, CD36 inhibitors, MCD inhibitors, PDK inhibitors), targeting both fatty acid and glucose oxidation during heart failure could potentially restore metabolic inflexibility and improve cardiac function in heart failure.
Ketone Oxidation
Stimulating ketone oxidation has been proposed as a potential approach for improving cardiac function in the failing heart (180). Increasing myocardial ketone oxidation has also been indirectly implies to be beneficial in the context of diabetic cardiomyopathy through empagliflozin's “thrifty fuel hypothesis.” However, there are presently no cardiac-specific drugs that can specifically modulate myocardial ketone body metabolism. While ketogenic diets are available, the extraneous systemic effects and cardiovascular risk factors associated with these high-fat diets need to be assessed as it may not be appropriate for heart failure patients. Furthermore, in light of recent work suggesting that increased ketone body oxidation is adaptive in the setting of heart failure (179, 180), increasing myocardial ketone body oxidation may be desirable, assuming it is not doing so at the cost of displacing glucose uptake, glucose oxidation or fatty acid oxidation. This would be undesirable in the context of an already depressed glucose oxidation (see section “Glucose Metabolism in Heart Failure”). Therefore, future studies characterized by normalizing ketone body oxidation in the setting of heart failure need to be conducted with measurements of the effects on other substrates and its overall cardiac energy metabolic consequences.
Targeting the Mitochondria
Recognising the central role of the mitochondria in energy metabolism and how impaired oxidative phosphorylation influences the progression of heart failure, targeting the mitochondria is another approach to regain metabolic flexibility and to improve cardiac function. Preclinical studies using mitochondrial-targeted antioxidants, such as AP39 and elamipretide, can preserve mitochondrial integrity through a marked reduction in mitochondrial ROS generation, which is associated with an improved post-infarction cardiac function in vivo (222, 223). Consistent with this, chronic treatment with elamipretide mitigates cardiac dysfunction in an advance heart failure model in dog, induced by a serial intracoronary microembolizations, which is accompanied with enhanced mitochondrial respiration and ATP production (224). Very recently, a double-blind, placebo-controlled clinical trial using elamipretifde in patients with HPrEF (ejection fraction ≤ 35%) showed a good tolerability and safety profile of the employed dosing range (225), encouraging future studies to characterize long-term safety and efficacy.
Targeting Cardiac Contractility
As contraction and metabolism are so inextricably linked, another approach to improve cardiac metabolism is through “rationalization of ATP usage.” This approach could involve more reliance on less ATP-consuming processes to maintain ionic homeostasis and efficient utilization of the limited ATP to provide more ATP for cardiac contraction. For example, cytoplasmic Ca2+ handling which is mainly governed by the sarcoplasmic reticulum (SR) ATPase (SERCA2a) activity, which requires ATP to transfer Ca2+ into the SR. Therefore, enhancing the Ca2+ sensitivity of the myofibrils, which is impaired due to glycolysis-induced acidosis, could not only improve cardiac contractility but also reduce energy cost of contractility through reducing the amount of ATP used for Ca2+ homeostasis. Levosimendan is a Ca2+ sensitizer and it is shown to improve cardiac contractility in animal (226) and human (227), through enhancing Ca2+ sensitivity of troponin C without affecting Ca2+-influx. Following small clinical trials have shown that levosimendan infusion improved left ventricle contractility in different types of heart failure including congestive (228), decompensated (229) advanced/end-stage (230–232) heart failure. Future large clinical trials are warranted to validate the promising effect of Levosimendan in failing heart patients.
Conclusions
Due to the heart's constant high energy demand, a fine balance between energy substrate utilization is crucial in maintaining metabolic flexibility. The metabolic profile of the failing heart is not simply a shift from “fatty acids to glucose.” Rather, the failing heart can be considered to have increased rates of glycolysis, depressed glucose oxidation rates and increased ketone body oxidation rates. With regards to the controversial nature of fatty acid oxidation, while the genes involved in fatty acid oxidation are down-regulated, direct measurements of rates have presented conflicting results. Thus, future studies that consider the transcriptional regulation, post-translational modifications (acetylation), absolute metabolic rates, and mitochondrial biogenesis are all required to fully understand the way in which fatty acid oxidation is perturbed in heart failure. Finally, definitively characterizing the metabolic profile of the failing heart will help direct future pharmacological therapies that can combine approaches to harmonize and normalize the metabolic flexibility of the failing heart.
Author Contributions
QK, GU, KH, and GL designed the literature search strategies and contributed to the critical analysis and interpretation of the published data. QK, GU, and KH carried out the literature search, collected the data and wrote the manuscript which was edited by GL and approved my all authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was funded by a Foundation Grant from the Canadian Institutes of Health Research to GL.
References
1. Savarese G, Lund LH. Global public health burden of heart failure. Cardiac Fail Rev. (2017) 3:7–11. doi: 10.15420/cfr.2016:25:2
2. Roger VL, Weston SA, Redfield MM, Hellermann-Homan JP, Killian J, Yawn BP, et al. Trends in heart failure incidence and survival in a community-based population. JAMA (2004) 292:344–50. doi: 10.1001/jama.292.3.344
3. Wang G, Zhang Z, Ayala C, Wall HK, Fang J. Costs of heart failure-related hospitalizations in patients aged 18 to 64 years. Am J Manag Care (2010) 16:769–76.
4. Benjamin EJ, Virani SS, Callaway CW, Chang AR, Cheng S, Chiuve SE, et al. Heart disease and stroke statistics - 2017 update: a report from the american heart association. Circulation (2017) 135:e146–603. doi: 10.1161/CIR.0000000000000485
5. Collins-Nakai RL, Noseworthy D, Lopaschuk GD. Epinephrine increases ATP production in hearts by preferentially increasing glucose metabolism. Am J Physiol Heart Circ Physiol. (1994) 267:H1862–71. doi: 10.1152/ajpheart.1994.267.5.H1862
6. Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. (1998) 273:29530–9. doi: 10.1074/jbc.273.45.29530
7. Hearse DJ. Oxygen deprivation and early myocardial contractile failure: a reassessment of the possible role of adenosine triphosphate. Am J Cardiol. (1979) 44:1115–21. doi: 10.1016/0002-9149(79)90177-2
8. Krahe T, Schindler R, Neubauer S, Ertl G, Horn M, Lackner K. 31P-Kardio-MR-Spektroskopie bei Myokardinsuffizienz. Fortschr Röntgenstr. (1993) 159:64–70. doi: 10.1055/s-2008-1032723
9. Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation (1997) 96:2190–6. doi: 10.1161/01.CIR.96.7.2190
10. Neubauer S. The failing heart — an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
11. De Jong KA, Lopaschuk GD. Complex energy metabolic changes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. Can J Cardiol. (2017) 33:860–71. doi: 10.1016/j.cjca.2017.03.009
12. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
13. Zhabyeyev P, Gandhi M, Mori J, Basu R, Kassiri Z, Clanachan A, et al. Pressure-overload-induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc Res. (2013) 97:676–85. doi: 10.1093/cvr/cvs424
14. Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, et al. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circulation (2013) 6:1039–48. doi: 10.1161/CIRCHEARTFAILURE.112.000228
15. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
16. Bedi KC, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
17. Ho K, Wagg C, Zhang L, Ussher J, Lopaschuk G. The contribution of fatty acid and ketone body oxidation to energy production increases in the failing heart and is associated with a decrease in cardiac efficiency. J Mol Cell Cardiol. (2017) 112:143. doi: 10.1016/j.yjmcc.2017.07.041
18. Allard M, Schonekess B, Henning S, English D, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol Heart Circ Physiol. (1994) 267:H742–50. doi: 10.1152/ajpheart.1994.267.2.H742
19. Casademont J, Miró Ò. Electron transport chain defects in heart failure. Heart Fail Rev. (2002) 7:131–9. doi: 10.1023/A:1015372407647
20. Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc Res. (2009) 85:376–84. doi: 10.1093/cvr/cvp344
21. Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, et al. PGC-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res. (2011) 111:243964. doi: 10.1161/CIRCRESAHA.111.243964
22. Cahill GF Jr, Veech RL. Ketoacids? Good medicine? Trans Am Clin Climatol Assoc. (2003) 114:143–9.
23. Taegtmeyer H. Failing heart and starving brain. Circulation (2016) 134:265–6. doi: 10.1161/CIRCULATIONAHA.116.022141
24. Wambolt RB, Lopaschuk GD, Brownsey RW, Allard MF. Dichloroacetate improves postischemic function of hypertrophied rat hearts. J Am Coll Cardiol. (2000) 36:1378–85. doi: 10.1016/S0735-1097(00)00856-1
25. Dyck JRB, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, et al. Absence of malonyl coenzyme a decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation (2006) 114:1721–8. doi: 10.1161/CIRCULATIONAHA.106.642009
26. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. (2005) 85:1093–129. doi: 10.1152/physrev.00006.2004
27. Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation—a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta Mol Cell Res. (2011) 1813:1333–50. doi: 10.1016/j.bbamcr.2011.01.015
28. Huang Y, Zhou M, Sun H, Wang Y. Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res. (2011) 90:220–3. doi: 10.1093/cvr/cvr070
29. Sun H, Wang Y. Branched chain amino acid metabolic reprogramming in heart failure. Biochim Biophys Acta Mol Basis Dis. (2016) 1862:2270–5. doi: 10.1016/j.bbadis.2016.09.009
30. Locke FS, Rosenheim O. Contributions to the physiology of the isolated heart. J Physiol. (1907) 36:205–20. doi: 10.1113/jphysiol.1907.sp001229
31. Kornberg H. Krebs and his trinity of cycles. Nat Rev Mol Cell Biol. (2000) 1:225–8. doi: 10.1038/35043073
32. Lehninger AL, Nelson DL, Cox MM. Lehninger Principles of Biochemistry. New York, NY: Worth Publishers (2000).
33. Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science (2012) 337:93–6. doi: 10.1126/science.1218530
34. Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. (2003) 31:1143–51. doi: 10.1042/bst0311143
35. Knoop F. Der Abbau Aromatischer Fettsäuren im Tierkörper. Freiburg im Breisgau: Kuttruff (1904).
36. Dakin HD. The mode of oxidation in the animal organism of phenyl derivatives of fatty acids. Part V: studies on the fate of phenylvaleric acid and its derivatives. J Biol Chem. (1909) 6:221–33.
37. Bing RJ, Siegel A, Ungar I, Gilbert M. Metabolism of the human heart. II. Studies on fat, ketone and amino acid metabolism. Am J Med. (1954) 16:504–15. doi: 10.1016/0002-9343(54)90365-4
38. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet (1963) 281:785–9. doi: 10.1016/S0140-6736(63)91500-9
39. Van Der Vusse GJ, Van Bilsen M, Glatz JF. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res. (2000) 45:279–93. doi: 10.1016/S0008-6363(99)00263-1
40. Augustus AS, Kako Y, Yagyu H, Goldberg IJ. Routes of FA delivery to cardiac muscle: modulation of lipoprotein lipolysis alters uptake of TG-derived FA. Am J Physiol Endocrinol Metab. (2003) 284:E331–9. doi: 10.1152/ajpendo.00298.2002
41. Niu YG, Hauton D, Evans RD. Utilization of triacylglycerol-rich lipoproteins by the working rat heart: routes of uptake and metabolic fates. J Physiol. (2004) 558:225–37. doi: 10.1113/jphysiol.2004.061473
42. Saddik M, Gamble J, Witters L, Lopaschuk G. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J Biol Chem. (1993) 268:25836–45.
43. Glatz JF, Luiken JJ, Bonen A. Involvement of membrane-associated proteins in the acute regulation of cellular fatty acid uptake. J Mol Neurosci. (2001) 16:123–32. doi: 10.1385/JMN:16:2-3:123
44. Schwenk RW, Luiken JJ, Bonen A, Glatz JF. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc Res. (2008) 79:249–58. doi: 10.1093/cvr/cvn116
45. Chiu H-C, Kovacs A, Ford DA, Hsu F-F, Garcia R, Herrero P, et al. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. (2001) 107:813–22. doi: 10.1172/JCI10947
46. Murthy M, Pande S. Mechanism of carnitine acylcarnitine translocase-catalyzed import of acylcarnitines into mitochondria. J Biol Chem. (1984) 259:9082–9.
47. Mcgarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system—from concept to molecular analysis. FEBS J. (1997) 244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x
48. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
49. Mcgarry JD, Mannaerts G, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest. (1977) 60:265–70. doi: 10.1172/JCI108764
50. Mcgarry JD, Leatherman GF, Foster DW. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J Biol Chem. (1978) 253:4128–36.
51. Mcgarry J, Foster D. Regulation of hepatic fatty acid oxidation and ketone body production. Ann Rev Biochem. (1980) 49:395–420. doi: 10.1146/annurev.bi.49.070180.002143
52. Savage DB, Choi CS, Samuel VT, Liu Z-X, Zhang D, Wang A, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. (2006) 116:817–24. doi: 10.1172/JCI27300
53. Wakil SJ, Abu-Elheiga LA. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res. (2009) 50:S138–43. doi: 10.1194/jlr.R800079-JLR200
55. Dyck JR, Lopaschuk GD. Malonyl CoA control of fatty acid oxidation in the ischemic heart. J Mol Cell Cardiol. (2002) 34:1099–109. doi: 10.1006/jmcc.2002.2060
56. Ussher JR, Lopaschuk GD. The malonyl CoA axis as a potential target for treating ischaemic heart disease. Cardiovasc Res. (2008) 79:259–68. doi: 10.1093/cvr/cvn130
57. Reszko AE, Kasumov T, David F, Thomas KR, Jobbins KA, Cheng J-F, et al. Regulation of malonyl-CoA concentration and turnover in the normal heart. J Biol Chem. (2004) 279:34298–301. doi: 10.1074/jbc.M405488200
58. Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Phys Heart Circ Physiol. (2013) 304:H1060–76. doi: 10.1152/ajpheart.00646.2012
59. Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. (2017) 25:262–84. doi: 10.1016/j.cmet.2016.12.022
60. Williamson DH, Hems R. Metabolism and function of ketone bodies. Essays Cell Metab. (1970) 257–81.
61. Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostagl Leukot Essent Fatty Acids (2004) 70:243–51. doi: 10.1016/j.plefa.2003.11.001
62. Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. (2013) 18:920–33. doi: 10.1016/j.cmet.2013.11.013
63. Turko IV, Marcondes S, Murad F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am J Physiol Heart Circ Physiol. (2001) 281:H2289–94. doi: 10.1152/ajpheart.2001.281.6.H2289
64. Wang Y, Peng F, Tong W, Sun H, Xu N, Liu S. The nitrated proteome in heart mitochondria of the db/db mouse model: characterization of nitrated tyrosine residues in SCOT. J Prot Res. (2010) 9:4254–63. doi: 10.1021/pr100349g
65. Rebrin I, Brégère C, Kamzalov S, Gallaher TK, Sohal RS. Nitration of tryptophan 372 in succinyl-CoA:3-ketoacid CoA transferase during aging in rat heart mitochondria. Biochemistry (2007) 46:10130–44. doi: 10.1021/bi7001482
67. Barnes RH, Mackay EM, Moe GK, Visscher MB. The utilization of ß hydroxybutyric acid by the isolated mammalian heart and lungs. Am J Physiol Legacy Cont. (1938) 123:272–9. doi: 10.1152/ajplegacy.1938.123.1.272
68. Waters ET, Fletcher JP, Mirsky IA. The relation between carbohydrate and ß-hydroxybutyric acid utilization by the heart-lung preparation. Am J Physiol Legacy Cont. (1938) 122:542–6. doi: 10.1152/ajplegacy.1938.122.2.542
69. Rudolph W, Maas D, Richter J, Hasinger F, Hofmann H, Dohrn P. [On the significance of acetoacetate and beta-hydroxybutyrate in human myocardial metabolism]. Klin Wochenschr. (1965) 43:445–51. doi: 10.1007/BF01483852
70. Chandler MP, Kerner J, Huang H, Vazquez E, Reszko A, Martini WZ, et al. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am J Physiol Heart Circ Physiol. (2004) 287:H1538–43. doi: 10.1152/ajpheart.00281.2004
71. O'donnell JM, Fields AD, Sorokina N, Lewandowski ED. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J Mol Cell Cardiol. (2008) 44:315–22. doi: 10.1016/j.yjmcc.2007.11.006
72. Aubert G, Vega RB, Kelly DP. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim Biophys Acta Mol Cell Res. (2013) 1833:840–7. doi: 10.1016/j.bbamcr.2012.08.015
73. Dávila-Román VG, Vedala G, Herrero P, De Las Fuentes L, Rogers JG, Kelly DP, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. (2002) 40:271–7. doi: 10.1016/S0735-1097(02)01967-8
74. Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci USA. (2005) 102:808–13. doi: 10.1073/pnas.0408962102
75. Neglia D, De Caterina A, Marraccini P, Natali A, Ciardetti M, Vecoli C, et al. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. (2007) 293:H3270–8. doi: 10.1152/ajpheart.00887.2007
76. Fukushima A, Milner K, Gupta A, Lopaschuk GD. Myocardial energy substrate metabolism in heart failure: from pathways to therapeutic targets. Curr Pharmac Design (2015) 21:3654–64. doi: 10.2174/1381612821666150710150445
77. Jameel MN, Hu Q, Zhang J. Myocytes oxygenation and high energy phosphate levels during hypoxia. PLoS ONE (2014) 9:e101317. doi: 10.1371/journal.pone.0101317
78. Dass S, Holloway CJ, Cochlin LE, Rider OJ, Mahmod M, Robson M, et al. No evidence of myocardial oxygen deprivation in nonischemic heart failure. Circ Heart Fail. (2015) 8:1088–93. doi: 10.1161/CIRCHEARTFAILURE.114.002169
79. Degens H, De Brouwer KF, Gilde AJ, Lindhout M, Willemsen PH, Janssen BJ, et al. Cardiac fatty acid metabolism is preserved in the compensated hypertrophic rat heart. Basic Res Cardiol. (2006) 101:17–26. doi: 10.1007/s00395-005-0549-0
80. Akki A, Smith K, Seymour A-ML. Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol. Cell. Biochem. (2008) 311:215–24. doi: 10.1007/s11010-008-9711-y
81. Symons JD, Abel ED. Lipotoxicity contributes to endothelial dysfunction: a focus on the contribution from ceramide. Rev Endocr Metab Dis. (2013) 14:59–68. doi: 10.1007/s11154-012-9235-3
82. Diakos NA, Navankasattusas S, Abel ED, Rutter J, Mccreath L, Ferrin P, et al. Evidence of glycolysis up-regulation and pyruvate mitochondrial oxidation mismatch during mechanical unloading of the failing human heart: implications for cardiac reloading and conditioning. JACC Basic Transl Sci. (2016) 1:432–44. doi: 10.1016/j.jacbts.2016.06.009
83. Fillmore N, Levasseur JL, Fukushima A, Wagg CS, Wang W, Dyck JRB, et al. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol Med. (2018) 24:3. doi: 10.1186/s10020-018-0005-x
84. Fiolet JW, Baartscheer A. Cellular calcium homeostasis during ischemia; a thermodynamic approach. Cardiovasc Res. (2000) 45:100–6. doi: 10.1016/S0008-6363(99)00294-1
85. Aasum E, Hafstad AD, Larsen TS. Changes in substrate metabolism in isolated mouse hearts following ischemia-reperfusion. Mol Cell Biochem. (2003) 249:97–103. doi: 10.1023/A:1024734605562
86. Barrick CJ, Dong A, Waikel R, Corn D, Yang F, Threadgill DW, et al. Parent-of-origin effects on cardiac response to pressure overload in mice. Am J Phys Heart Circ Physiol. (2009) 297:H1003–9. doi: 10.1152/ajpheart.00896.2008
87. Garcia-Menendez L, Karamanlidis G, Kolwicz S, Tian R. Substrain specific response to cardiac pressure overload in C57BL/6 mice. Am J Physiol Heart Circ Physiol. (2013) 305:H397–402. doi: 10.1152/ajpheart.00088.2013
88. Schroeder MA, Lau AZ, Chen AP, Gu Y, Nagendran J, Barry J, et al. Hyperpolarized (13)C magnetic resonance reveals early- and late-onset changes to in vivo pyruvate metabolism in the failing heart. Eur J Heart Fail. (2013) 15:130–40. doi: 10.1093/eurjhf/hfs192
89. Dodd MS, Atherton HJ, Carr CA, Stuckey DJ, West JA, Griffin JL, et al. Impaired in vivo mitochondrial Krebs cycle activity after myocardial infarction assessed using hyperpolarized magnetic resonance spectroscopy. Circ Cardiovasc Imaging (2014) 7:895–904. doi: 10.1161/CIRCIMAGING.114.001857
90. Seymour AM, Giles L, Ball V, Miller JJ, Clarke K, Carr CA, et al. In vivo assessment of cardiac metabolism and function in the abdominal aortic banding model of compensated cardiac hypertrophy. Cardiovasc Res. (2015) 106:249–60. doi: 10.1093/cvr/cvv101
91. Mori J, Basu R, Mclean BA, Das SK, Zhang L, Patel VB, et al. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ Heart Fail. (2012) 5:493–503. doi: 10.1161/CIRCHEARTFAILURE.112.966705
92. Mori J, Alrob OA, Wagg CS, Harris RA, Lopaschuk GD, Oudit GY. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. Am J Physiol Heart Circ Physiol. (2013) 304:H1103–13. doi: 10.1152/ajpheart.00636.2012
93. Sung MM, Das SK, Levasseur J, Byrne NJ, Fung D, Kim TT, et al. Resveratrol treatment of mice with pressure-overload-induced heart failure improves diastolic function and cardiac energy metabolism. Circ Heart Fail. (2015) 8:128–37. doi: 10.1161/CIRCHEARTFAILURE.114.001677
94. Byrne NJ, Levasseur J, Sung MM, Masson G, Boisvenue J, Young ME, et al. Normalization of cardiac substrate utilization and left ventricular hypertrophy precede functional recovery in heart failure regression. Cardiovasc Res. (2016) 110:249–57. doi: 10.1093/cvr/cvw051
95. Sung MM, Byrne NJ, Robertson IM, Kim TT, Samokhvalov V, Levasseur J, et al. Resveratrol improves exercise performance and skeletal muscle oxidative capacity in heart failure. Am J Physiol Heart Circ Physiol. (2017) 312:H842–53. doi: 10.1152/ajpheart.00455.2016
96. Paolisso G, Gambardella A, Galzerano D, D'amore A, Rubino P, Verza M, et al. Total-body and myocardial substrate oxidation in congestive heart failure. Metabolism (1994) 43:174–9. doi: 10.1016/0026-0495(94)90241-0
97. Swan JW, Anker SD, Walton C, Godsland IF, Clark AL, Leyva F, et al. Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol. (1997) 30:527–32. doi: 10.1016/S0735-1097(97)00185-X
98. Funada J, Betts TR, Hodson L, Humphreys SM, Timperley J, Frayn KN, et al. Substrate utilization by the failing human heart by direct quantification using arterio-venous blood sampling. PLoS ONE (2009) 4:e7533. doi: 10.1371/journal.pone.0007533
99. Slot JW, Geuze HJ, Gigengack S, James DE, Lienhard GE. Translocation of the glucose transporter GLUT4 in cardiac myocytes of the rat. Proc Natl Acad Sci USA. (1991) 88:7815–9. doi: 10.1073/pnas.88.17.7815
100. Zorzano A, Sevilla L, Camps M, Becker C, Meyer J, Kammermeier H, et al. Regulation of glucose transport, and glucose transporters expression and trafficking in the heart: studies in cardiac myocytes. Am J Cardiol. (1997) 80:65A–76. doi: 10.1016/S0002-9149(97)00459-1
101. Lefebvre V, Méchin M-C, Louckx MP, Rider MH, Hue L. Signaling pathway involved in the activation of heart 6-phosphofructo-2-kinase by insulin. J Biol Chem. (1996) 271:22289–92. doi: 10.1074/jbc.271.37.22289
102. Hue L, Beauloye C, Marsin A-S, Bertrand L, Horman S, Rider MH. Insulin and ischemia stimulate glycolysis by acting on the same targets through different and opposing signaling pathways. J Mol Cell Cardiol. (2002) 34:1091–7. doi: 10.1006/jmcc.2002.2063
103. Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. (2004) 381:561–79. doi: 10.1042/BJ20040752
104. Lopaschuk GD, Wambolt RB, Barr RL. An imbalance between glycolysis and glucose oxidation is a possible explanation for the detrimental effects of high levels of fatty acids during aerobic reperfusion of ischemic hearts. J Pharmacol Exp Ther. (1993) 264:135–44.
105. Moravec J, El Alaoui-Talibi Z, Moravec M, Guendouz A. Control of oxidative metabolism in volume-overloaded rat hearts: effect of pretreatment with propionyl-L-carnitine. Adv Exp Med Biol. (1996) 388:205–12. doi: 10.1007/978-1-4613-0333-6_25
106. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. (2010) 3:420–30. doi: 10.1161/CIRCHEARTFAILURE.109.888479
107. Doenst T, Pytel G, Schrepper A, Amorim P, Färber G, Shingu Y, et al. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res. (2010) 86:461–70. doi: 10.1093/cvr/cvp414
108. Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, et al. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation (2002) 106:606–12. doi: 10.1161/01.CIR.0000023531.22727.C1
109. Kolwicz SC Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. (2012) 111:728–38. doi: 10.1161/CIRCRESAHA.112.268128
110. Masoud WGT, Ussher JR, Wang W, Jaswal JS, Wagg CS, Dyck JR, et al. Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc Res. (2014) 101:30–8. doi: 10.1093/cvr/cvt216
111. Recchia FA, Mcconnell PI, Bernstein RD, Vogel TR, Xu X, Hintze TH. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res. (1998) 83:969–79. doi: 10.1161/01.RES.83.10.969
112. Bround MJ, Wambolt R, Cen H, Asghari P, Albu RF, Han J, et al. Cardiac ryanodine receptor (Ryr2)-mediated calcium signals specifically promote glucose oxidation via pyruvate dehydrogenase. J Biol Chem. (2016) 291:23490–505. doi: 10.1074/jbc.M116.756973
113. Gupte AA, Hamilton DJ, Cordero-Reyes AM, Youker KA, Yin Z, Estep JD, et al. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet. (2014) 7:266–76. doi: 10.1161/CIRCGENETICS.113.000404
114. Ussher JR, Koves TR, Jaswal JS, Zhang L, Ilkayeva O, Dyck JRB, et al. Insulin-stimulated cardiac glucose oxidation is increased in high-fat diet–induced obese mice lacking malonyl CoA decarboxylase. Diabetes (2009) 58:1766–75. doi: 10.2337/db09-0011
115. Sankaralingam S, Abo Alrob O, Zhang L, Jaswal JS, Wagg CS, Fukushima A, et al. Lowering body weight in obese mice with diastolic heart failure improves cardiac insulin sensitivity and function: implications for the obesity paradox. Diabetes (2015) 64:1643–57. doi: 10.2337/db14-1050
117. Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, et al. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest. (1999) 104:1703–14. doi: 10.1172/JCI7605
118. Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, et al. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation (2003) 107:448–54. doi: 10.1161/01.CIR.0000045671.62860.98
119. Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation (2004) 109:2191–6. doi: 10.1161/01.CIR.0000127959.28627.F8
120. Le Page LM, Rider OJ, Lewis AJ, Ball V, Clarke K, Johansson E, et al. Increasing pyruvate dehydrogenase flux as a treatment for diabetic cardiomyopathy: a combined 13C hyperpolarized magnetic resonance and echocardiography study. Diabetes (2015) 64:2735–43. doi: 10.2337/db14-1560
121. Gopal K, Almutairi M, Al Batran R, Eaton F, Gandhi M, Ussher JR. Cardiac-specific deletion of pyruvate dehydrogenase impairs glucose oxidation rates and induces diastolic dysfunction. Front Cardiovasc Med. (2018) 5:17. doi: 10.3389/fcvm.2018.00017
122. Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight (2016) 1:e84897. doi: 10.1172/jci.insight.84897
123. Ozden O, Park SH, Wagner BA, Song HY, Zhu Y, Vassilopoulos A, et al. SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radic Biol Med. (2014) 76:163–72. doi: 10.1016/j.freeradbiomed.2014.08.001
124. Zhang X, Ji R, Liao X, Deng LY, Castillero E, Givens R, et al. Abstract 18978: lysine acetylation of pyruvate dehydrogenase reduces enzymatic activity and contributes to impaired substrate metabolism in the failing myocardium. Circulation (2014) 130:A18978.
125. Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc. Res. (2014) 103:485–97. doi: 10.1093/cvr/cvu156
126. Khan D, Sarikhani M, Dasgupta S, Maniyadath B, Pandit AS, Mishra S, et al. SIRT6 deacetylase transcriptionally regulates glucose metabolism in heart. J Cell Physiol. (2018) 233:5478–89. doi: 10.1002/jcp.26434
127. Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation (2004) 110:894–6. doi: 10.1161/01.CIR.0000139340.88769.D5
128. Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. (2005) 115:547–55. doi: 10.1172/JCI24405
129. Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. (2008) 81:412–9. doi: 10.1093/cvr/cvn301
130. Sack MN, Rader TA, Park S, Bastin J, Mccune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation (1996) 94:2837–42. doi: 10.1161/01.CIR.94.11.2837
131. Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci. (1997) 94:6438–43. doi: 10.1073/pnas.94.12.6438
132. Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. (2007) 12:331–43. doi: 10.1007/s10741-007-9034-1
133. Lommi J, Kupari M, Yki-Järvinen H. Free fatty acid kinetics and oxidation in congestive heart failure. Am J Cardiol. (1998) 81:45–50. doi: 10.1016/S0002-9149(97)00804-7
134. Nørrelund H, Wiggers H, Halbirk M, Frystyk J, Flyvbjerg A, Bøtker HE, et al. Abnormalities of whole body protein turnover, muscle metabolism and levels of metabolic hormones in patients with chronic heart failure. J Int Med. (2006) 260:11–21. doi: 10.1111/j.1365-2796.2006.01663.x
135. Tuunanen H, Engblom E, Naum A, Scheinin M, Någren K, Airaksinen J, et al. Decreased myocardial free fatty acid uptake in patients with idiopathic dilated cardiomyopathy: evidence of relationship with insulin resistance and left ventricular dysfunction. J Card Fail. (2006) 12:644–52. doi: 10.1016/j.cardfail.2006.06.005
136. Tuunanen H, Ukkonen H, Knuuti J. Myocardial fatty acid metabolism and cardiac performance in heart failure. Curr Cardiol Rep. (2008) 10:142–8. doi: 10.1007/s11886-008-0024-2
137. Taylor M, Wallhaus TR, Degrado TR, Russell DC, Stanko P, Nickles RJ, et al. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F] fluoro-6-thia-heptadecanoic acid and [18F] FDG in patients with congestive heart failure. J Nucl Med. (2001) 42:55–62.
138. Lei B, Lionetti V, Young ME, Chandler MP, D'agostino C, Kang E, et al. Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J Mol Cell Cardiol. (2004) 36:567–76. doi: 10.1016/j.yjmcc.2004.02.004
139. Qanud K, Mamdani M, Pepe M, Khairallah RJ, Gravel J, Lei B, et al. Reverse changes in cardiac substrate oxidation in dogs recovering from heart failure. Am J Phys Heart Circ Physiol. (2008) 295:H2098–105. doi: 10.1152/ajpheart.00471.2008
140. Lopaschuk GD, Tsang H. Metabolism of palmitate in isolated working hearts from spontaneously diabetic “BB” Wistar rats. Circ Res. (1987) 61:853–8. doi: 10.1161/01.RES.61.6.853
141. Mazumder PK, O'neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes (2004) 53:2366–74. doi: 10.2337/diabetes.53.9.2366
142. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology (2005) 146:5341–9. doi: 10.1210/en.2005-0938
143. Lin CH, Kurup S, Herrero P, Schechtman KB, Eagon JC, Klein S, et al. Myocardial oxygen consumption change predicts left ventricular relaxation improvement in obese humans after weight loss. Obesity (2011) 19:1804–12. doi: 10.1038/oby.2011.186
144. Tuunanen H, Engblom E, Naum A, Nagren K, Hesse B, Airaksinen KE, et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation (2006) 114:2130–7. doi: 10.1161/CIRCULATIONAHA.106.645184
146. Fukushima A, Lopaschuk GD. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim Biophys Acta Mol Cell Biol Lipids (2016) 1861:1525–34. doi: 10.1016/j.bbalip.2016.03.020
147. Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes (2002) 51:2005–11. doi: 10.2337/diabetes.51.7.2005
148. Finck BN, Kelly DP. Peroxisome proliferator–activated receptor γ coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation (2007) 115:2540–8. doi: 10.1161/CIRCULATIONAHA.107.670588
149. Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. (2008) 44:968–75. doi: 10.1016/j.yjmcc.2008.03.021
150. Kanda H, Nohara R, Hasegawa K, Kishimoto C, Sasayama S. A nuclear complex containing PPARα/RXRα is markedly downregulated in the hypertrophied rat left ventricular myocardium with normal systolic function. Heart Vessels (2000) 15:191–6. doi: 10.1007/s003800070022
151. Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. (2004) 95:568–78. doi: 10.1161/01.RES.0000141774.29937.e3
152. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. (2006) 116:615–22. doi: 10.1172/JCI27794
153. Hopkins TA, Sugden MC, Holness MJ, Kozak R, Dyck JR, Lopaschuk GD. Control of cardiac pyruvate dehydrogenase activity in peroxisome proliferator-activated receptor-α transgenic mice. Am J Physiol Heart Circ Physiol. (2003) 285:H270–6. doi: 10.1152/ajpheart.00852.2002
154. Schummer CM, Werner U, Tennagels N, Schmoll D, Haschke G, Juretschke HP, et al. Dysregulated pyruvate dehydrogenase complex in Zucker diabetic fatty rats. Am J Physiol Endocrinol Metab. (2008) 294:E88–96. doi: 10.1152/ajpendo.00178.2007
155. Karbowska J, Kochan Z, Smolenski RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett. (2003) 8:49–54.
156. Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proc Natl Acad Sci USA. (2006) 103:10086–91. doi: 10.1073/pnas.0603615103
157. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, Del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. (2010) 106:1541–8. doi: 10.1161/CIRCRESAHA.109.212753
158. Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. PGC-1α and ERRα target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. (2009) 46:201–12. doi: 10.1016/j.yjmcc.2008.10.025
159. Xiong Y, Guan K-L. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol. (2012) 198:155–64. doi: 10.1083/jcb.201202056
160. Webster BR, Scott I, Han K, Li JH, Lu Z, Stevens MV, et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J Cell Sci. (2013) 126:4843–9. doi: 10.1242/jcs.131300
161. Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. (2008) 7:104–12. doi: 10.1016/j.cmet.2007.11.006
162. Newman JC, He W, Verdin E. Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease. J Biol Chem. (2012) 287:42436–43. doi: 10.1074/jbc.R112.404863
163. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science (2010) 327:1000–4. doi: 10.1126/science.1179689
164. Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet (2004) 364:1786–8. doi: 10.1016/S0140-6736(04)17402-3
165. Kim G, Jo K, Kim KJ, Lee Y-H, Han E, Yoon H-J, et al. Visceral adiposity is associated with altered myocardial glucose uptake measured by 18FDG-PET in 346 subjects with normal glucose tolerance, prediabetes, and type 2 diabetes. Cardiovasc Diabetol. (2015) 14:148. doi: 10.1186/s12933-015-0310-4
166. Li C, Lu C, Zhao X, Chen X. Comparison between myocardial infarction and diabetes mellitus damage caused angiogenesis or energy metabolism. Int J Clin Exp Med. (2015) 8:22371–6.
167. Murray AJ, Cole MA, Lygate CA, Carr CA, Stuckey DJ, Little SE, et al. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J Mol Cell Cardiol. (2008) 44:694–700. doi: 10.1016/j.yjmcc.2008.01.008
168. Lommi J, Kupari M, Koskinen P, Näveri H, Leinonen H, Pulkki K, et al. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. (1996) 28:665–72. doi: 10.1016/0735-1097(96)00214-8
169. Lommi J, Koskinen P, Näveri H, Härkönen M, Kupari M. Heart failure ketosis. J Int Med. (1997) 242:231–8. doi: 10.1046/j.1365-2796.1997.00187.x
170. Melenovsky V, Kotrc M, Polak J, Pelikanova T, Bendlova B, Cahova M, et al. Availability of energetic substrates and exercise performance in heart failure with or without diabetes. Eur J Heart Fail. (2014) 14:754–63. doi: 10.1093/eurjhf/hfs080
171. Du Z, Shen A, Huang Y, Su L, Lai W, Wang P, et al. 1H-NMR-based metabolic analysis of human serum reveals novel markers of myocardial energy expenditure in heart failure patients. PLoS ONE (2014) 9:e88102. doi: 10.1371/journal.pone.0088102
172. Zordoky BN, Sung MM, Ezekowitz J, Mandal R, Han B, Bjorndahl TC, et al. Metabolomic fingerprint of heart failure with preserved ejection fraction. PLoS ONE (2015) 10:e0124844. doi: 10.1371/journal.pone.0124844
173. Kolwicz SC, Airhart S, Tian R. Ketones step to the plate: a game changer for metabolic remodeling in heart failure? Circulation (2016) 133:689–91. doi: 10.1161/CIRCULATIONAHA.116.021230
174. Ikegami R, Shimizu I, Yoshida Y, Minamino T. Metabolomic analysis in heart failure. Circ J. (2017) 82:10–6. doi: 10.1253/circj.CJ-17-1184
175. Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. (1980) 60:143–87. doi: 10.1152/physrev.1980.60.1.143
176. Janardhan A, Chen J, Crawford PA. Altered systemic ketone body metabolism in advanced heart failure. Tex Heart Inst J. (2011) 38:533–8.
177. Nagao M, Toh R, Irino Y, Mori T, Nakajima H, Hara T, et al. β-Hydroxybutyrate elevation as a compensatory response against oxidative stress in cardiomyocytes. Biochem Biophys Res Commun. (2016) 475:322–8. doi: 10.1016/j.bbrc.2016.05.097
178. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science (2013) 339:211–4. doi: 10.1126/science.1227166
179. Schugar RC, Moll AR, André D'avignon D, Weinheimer CJ, Kovacs A, Crawford PA. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab. (2014) 3:754–69. doi: 10.1016/j.molmet.2014.07.010
180. Uchihashi M, Hoshino A, Okawa Y, Ariyoshi M, Kaimoto S, Tateishi S, et al. Cardiac-specific Bdh1 overexpression ameliorates oxidative stress and cardiac remodeling in pressure overload-induced heart failure. Circ Heart Fail. (2017) 10:e004417. doi: 10.1161/CIRCHEARTFAILURE.117.004417
181. Seki M, Powers JC, Maruyama S, Zuriaga MA, Wu CL, Kurishima C, et al. Acute and chronic increases of circulating FSTL1 normalize energy substrate metabolism in pacing-induced heart failure. Circ Heart Fail. (2018) 11:e004486. doi: 10.1161/CIRCHEARTFAILURE.117.004486
182. Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med. (2018) 10:eaao0144. doi: 10.1126/scitranslmed.aao0144
183. Al Batran R, Ussher JR. Revisiting protein acetylation and myocardial fatty acid oxidation. Am J Physiol Heart Circ Physiol. (2017) 313:H617–9. doi: 10.1152/ajpheart.00303.2017
184. Williamson JR, Krebs HA. Acetoacetate as fuel of respiration in the perfused rat heart. Biochem J. (1961) 80:540–7. doi: 10.1042/bj0800540
185. Wieland O, Funcke HV, Löffler G. Interconversion of pyruvate dehydrogenase in rat heart muscle upon perfusion with fatty acids or ketone bodies. FEBS Lett. (1971) 15:295–8. doi: 10.1016/0014-5793(71)80641-5
186. Hiltunen JK, Hassinen IE. Energy-linked regulation of glucose and pyruvate oxidation in isolated perfused rat heart. Role of pyruvate dehydrogenase. Biochim Biophys Acta Bioenerget. (1976) 440:377–90. doi: 10.1016/0005-2728(76)90072-4
187. Kerbey AL, Randle PJ, Cooper RH, Whitehouse S, Pask HT, Denton RM. Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes: role of coenzyme A, acetyl-coenzyme A and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem J. (1976) 154:327–48. doi: 10.1042/bj1540327
188. Gormsen LC, Svart M, Thomsen HH, Sondergaard E, Vendelbo MH, Christensen N, et al. Ketone body infusion with 3-hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: a positron emission tomography study. J Am Heart Assoc. (2017) 6:e005066. doi: 10.1161/JAHA.116.005066
189. Renguet E, Ginion A, Gelinas R, Bultot L, Auquier J, Robillard Frayne I, et al. Metabolism and acetylation contribute to leucine-mediated inhibition of cardiac glucose uptake. Am J Physiol Heart Circ Physiol. (2017) 313:H432–45. doi: 10.1152/ajpheart.00738.2016
190. Kashiwaya Y, King MT, Veech RL. Substrate signaling by insulin: a ketone bodies ratio mimics insulin action in heart. Am J Cardiol. (1997) 80:50A–64. doi: 10.1016/S0002-9149(97)00458-X
191. Chen V, Wagner G, Spitzer JJ. Regulation of substrate oxidation in isolated myocardial cells by beta-hydroxybutyrate. Horm Metab Res. (1984) 16:243–7. doi: 10.1055/s-2007-1014756
192. Forsey RG, Reid K, Brosnan JT. Competition between fatty acids and carbohydrate or ketone bodies as metabolic fuels for the isolated perfused heart. Can J Physiol Pharmacol. (1987) 65:401–6. doi: 10.1139/y87-067
193. Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD. beta-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am J Physiol Heart Circ Physiol. (2003) 285:H1626–31. doi: 10.1152/ajpheart.00332.2003
194. Liao R, Jain M, Cui L, D'agostino J, Aiello F, Luptak I, et al. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation (2002) 106:2125–31. doi: 10.1161/01.CIR.0000034049.61181.F3
195. Liu B, Clanachan AS, Schulz R, Lopaschuk GD. Cardiac efficiency is improved after ischemia by altering both the source and fate of protons. Circ Res. (1996) 79:940–8. doi: 10.1161/01.RES.79.5.940
196. Lydell CP, Chan A, Wambolt RB, Sambandam N, Parsons H, Bondy GP, et al. Pyruvate dehydrogenase and the regulation of glucose oxidation in hypertrophied rat hearts. Cardiovasc Res. (2002) 53:841–51. doi: 10.1016/S0008-6363(01)00560-0
197. Wargovich TJ, Macdonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Myocardial metabolic and hemodynamic effects of dichloroacetate in coronary artery disease. Am J Cardiol. (1988) 61:65–70. doi: 10.1016/0002-9149(88)91306-9
198. Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, et al. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol. (1994) 23:1617–24. doi: 10.1016/0735-1097(94)90665-3
199. Lewis JF, Dacosta M, Wargowich T, Stacpoole P. Effects of dichloroacetate in patients with congestive heart failure. Clin Cardiol. (1998) 21:888–92. doi: 10.1002/clc.4960211206
200. Lopaschuk GD, Wall SR, Olley PM, Davies NJ. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res. (1988) 63:1036–43. doi: 10.1161/01.RES.63.6.1036
201. Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest. (2002) 109:1381–9. doi: 10.1172/JCI0214596
202. Schmidt-Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci. (2000) 99:27–35. doi: 10.1042/cs0990027
203. Holubarsch CJ, Rohrbach M, Karrasch M, Boehm E, Polonski L, Ponikowski P, et al. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci. (2007) 113:205–12. doi: 10.1042/CS20060307
204. Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, et al. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation (2005) 112:3280–8. doi: 10.1161/CIRCULATIONAHA.105.551457
205. Coort SL, Willems J, Coumans WA, Van Der Vusse GJ, Bonen A, Glatz JF, et al. Sulfo-N-succinimidyl esters of long chain fatty acids specifically inhibit fatty acid translocase (FAT/CD36)-mediated cellular fatty acid uptake. Mol Cell Biochem. (2002) 239:213–9. doi: 10.1023/A:1020539932353
206. Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. (2000) 86:580–8. doi: 10.1161/01.RES.86.5.580
207. Peng S, Zhao M, Wan J, Fang Q, Fang D, Li K. The efficacy of trimetazidine on stable angina pectoris: a meta-analysis of randomized clinical trials. Int J Cardiol. (2014) 177:780–5. doi: 10.1016/j.ijcard.2014.10.149
208. Fragasso G, Palloshi A, Puccetti P, Silipigni C, Rossodivita A, Pala M, et al. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol. (2006) 48:992–8. doi: 10.1016/j.jacc.2006.03.060
209. Gao D, Ning N, Niu X, Hao G, Meng Z. Trimetazidine: a meta-analysis of randomised controlled trials in heart failure. Heart (2011) 97:278–86. doi: 10.1136/hrt.2010.208751
210. Ussher JR, Fillmore N, Keung W, Mori J, Beker DL, Wagg CS, et al. Trimetazidine therapy prevents obesity-induced cardiomyopathy in mice. Canad J Cardiol. (2014) 30:940–4. doi: 10.1016/j.cjca.2014.04.023
211. Ussher JR, Keung W, Fillmore N, Koves TR, Mori J, Zhang L, et al. Treatment with the 3-ketoacyl-CoA thiolase inhibitor trimetazidine does not exacerbate whole-body insulin resistance in obese mice. J Pharmacol Exp Ther. (2014) 349:487–96. doi: 10.1124/jpet.114.214197
212. Chaitman BR, Pepine CJ, Parker JO, Skopal J, Chumakova G, Kuch J, et al. Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercise tolerance and angina frequency in patients with severe chronic angina: a randomized controlled trial. JAMA (2004) 291:309–16. doi: 10.1001/jama.291.3.309
213. Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non–ST-segment–elevation acute coronary syndrome: results from the Metabolic Efficiency with Ranolazine for Less Ischemia in Non–ST-Elevation Acute Coronary Syndrome–Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation (2007) 116:1647–52. doi: 10.1161/CIRCULATIONAHA.107.724880
214. Mccormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation (1996) 93:135–42. doi: 10.1161/01.CIR.93.1.135
215. Ferrannini E, Mark M, Mayoux E. CV Protection in the EMPA-REG OUTCOME trial: a “Thrifty Substrate” hypothesis. Diabetes Care (2016) 39:1108–14. doi: 10.2337/dc16-0330
216. Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis. Diabetes Care (2016) 39:1115–22. doi: 10.2337/dc16-0542
217. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, Cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
218. Byrne NJ, Parajuli N, Levasseur JL, Boisvenue J, Beker D, Masson G, et al. Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload-induced heart failure. JACC Basic Transl Sci. (2017) 2:347–54 doi: 10.1016/j.jacbts.2017.07.003
219. Lopaschuk GD, Verma S. Empagliflozin's fuel hypothesis: not so soon. Cell Metabolism (2016) 24:200–2. doi: 10.1016/j.cmet.2016.07.018
220. Stanley WC, Morgan EE, Huang H, Mcelfresh TA, Sterk JP, Okere IC, et al. Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation and reduces lactate production during demand-induced ischemia. Am J Physiol Heart Circ Physiol. (2005) 289:H2304–9. doi: 10.1152/ajpheart.00599.2005
221. Dyck JRB, Cheng J-F, Stanley WC, Barr R, Chandler MP, Brown S, et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. (2004) 94:e78–84. doi: 10.1161/01.RES.0000129255.19569.8f
222. Dai W, Shi J, Gupta RC, Sabbah HN, Hale SL, Kloner RA. Bendavia, a mitochondria-targeting peptide, improves postinfarction cardiac function, prevents adverse left ventricular remodeling, and restores mitochondria-related gene expression in rats. J Cardiovasc Pharmacol. (2014) 64:543–53. doi: 10.1097/FJC.0000000000000155
223. Karwi QG, Bornbaum J, Boengler K, Torregrossa R, Whiteman M, Wood ME, et al. AP39, a mitochondria-targeting hydrogen sulfide (H2S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br J Pharmacol. (2017) 174:287–301. doi: 10.1111/bph.13688
224. Sabbah HN, Gupta RC, Kohli S, Wang M, Hachem S, Zhang K. Chronic Therapy With Elamipretide (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circ Heart Fail. (2016) 9:e002206. doi: 10.1161/CIRCHEARTFAILURE.115.002206
225. Daubert MA, Yow E, Dunn G, Marchev S, Barnhart H, Douglas PS, et al. Novel mitochondria-targeting peptide in heart failure treatment. A randomized, placebo-controlled trial of elamipretide Circ Heart Fail. (2017) 10:e004389. doi: 10.1161/CIRCHEARTFAILURE.117.004389
226. Edes I, Kiss E, Kitada Y, Powers FM, Papp JG, Kranias EG, et al. Effects of levosimendan, a cardiotonic agent targeted to troponin C, on cardiac function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart. Circ Res. (1995) 77:107–13.
227. Hasenfuss G, Pieske B, Castell M, Kretschmann B, Maier LS, Just H. Influence of the novel inotropic agent levosimendan on isometric tension and calcium cycling in failing human myocardium. Circulation (1998) 98:2141–7. doi: 10.1161/01.CIR.98.20.2141
228. Heikki U, Markku S, Juha A, Juhani K, Meri K, Hidehiro I, et al. Myocardial efficiency during levosimendan infusion in congestive heart failure. Clin Pharmacol Ther. (2000) 68:522–31. doi: 10.1067/mcp.2000.110972
229. Duman D, Palit F, Simsek E, Bilgehan K, Sacide A. Effects of levosimendan versus dobutamine on left atrial function in decompensated heart failure. Canad J Cardiol. (2009) 25:e353–6. doi: 10.1016/S0828-282X(09)70721-4
230. Feola M, Lombardo E, Taglieri C, Vallauri P, Piccolo S, Valle R. Effects of levosimendan/furosemide infusion on Plasma Brain Natriuretic Peptide, echocardiographic parameters and cardiac output in end-stage heart failure patients. Med Sci Monit (2011) 17:PI7–13. doi: 10.12659/MSM.881433
231. Malfatto G, Della Rosa F, Villani A, Rella V, Branzi G, Facchini M, et al. Intermittent levosimendan infusions in advanced heart failure: favourable effects on left ventricular function, neurohormonal balance, and one-year survival. J Cardiov Pharmacol. (2012) 60:450–5. doi: 10.1097/FJC.0b013e31826b86aa
Keywords: fatty acid oxidation, glucose oxidation, ketone oxidation, cardiac metabolism, heart failure, insulin resistant
Citation: Karwi QG, Uddin GM, Ho KL and Lopaschuk GD (2018) Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 5:68. doi: 10.3389/fcvm.2018.00068
Received: 30 March 2018; Accepted: 18 May 2018;
Published: 06 June 2018.
Edited by:
Thomas Pulinilkunnil, Dalhousie University, CanadaReviewed by:
Heinrich Taegtmeyer, University of Texas Health Science Center at Houston, United StatesLuc Bertrand, Fonds National de la Recherche Scientifique, Belgium
Copyright © 2018 Karwi, Uddin, Ho and Lopaschuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary D. Lopaschuk, gary.lopaschuk@ualberta.ca
†These authors have contributed equally to this work.