Dynamic Macrophages: Understanding Mechanisms of Activation as Guide to Therapy for Atherosclerotic Vascular Disease
 Julius L. Decano1
Julius L. Decano1  Masanori Aikawa1,2,3*
Masanori Aikawa1,2,3*- 1Center for Interdisciplinary Cardiovascular Sciences, Division of Cardiovascular Medicine, Department of Medicine, Brigham Women's Hospital, Harvard Medical School, Boston, MA, United States
- 2Center for Excellence in Vascular Biology, Division of Cardiovascular Medicine, Department of Medicine, Brigham Women's Hospital, Harvard Medical School, Boston, MA, United States
- 3Channing Division of Network Medicine, Department of Medicine, Brigham Women's Hospital, Harvard Medical School, Boston, MA, United States
An emerging theory is that macrophages are heterogenous; an attribute that allows them to change behavior and execute specific functions in disease processes. This review aims to describe the current understanding on factors that govern their phenotypic changes, and provide insights for intervention beyond managing classical risk factors. Evidence suggests that metabolic reprogramming of macrophages triggers either a pro-inflammatory, anti-inflammatory or pro-resolving behavior. Dynamic changes in bioenergetics, metabolome or influence from bioactive lipids may promote resolution or aggravation of inflammation. Direct cell-to-cell interactions with other immune cells can also influence macrophage activation. Both paracrine signaling and intercellular molecular interactions either co-stimulate or co-inhibit activation of macrophages as well as their paired immune cell collaborator. More pathways of activation can even be uncovered by inspecting macrophages in the single cell level, since differential expression in key gene regulators can be screened in higher resolution compared to conventional averaged gene expression readouts. All these emerging macrophage activation mechanisms may be further explored and consolidated by using approaches in network biology. Integrating these insights can unravel novel and safer drug targets through better understanding of the pro-inflammatory activation circuitry.
Introduction
In the last few decades, accumulating evidence has supported modulation of inflammatory signals, and regulation of immune cell to cell interactions in atherosclerosis are key therapeutic strategies for atherothrombotic disease (1–3) The recent Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS) trial, involving over 10,000 patients, show conclusive proof that reduction of inflammation, specifically targeting the interleukin-1β (IL-1β) pathway activation, independent of LDL cholesterol lowering, can significantly lower coronary artery disease (CAD) morbidity and mortality. Antagonizing the IL-1β signaling resulted in marked reduction of plasma high-sensitivity C-reactive protein (hs-CRP) levels among patients with elevated hs-CRP levels and history of myocardial infarction, which eventually led to decreased major adverse cardiac/CV events (MACE and MACE+) (4) hs-CRP is a predictive marker of the severity of atherosclerosis and extent of future cardiovascular events (5, 6). Success of anti-inflammatory drug trials are reliant on rigorous basic science research, integrating a plurality of approaches. These include analyses of pathologic specimens, tightly controlled in vitro experiments and extensive use of pre-clinical small animal models to gather basic mechanistic information about the disease. In the context of chronic inflammation in cardiovascular disease, basic science research in macrophage biology has undoubtedly been the guiding compass for pursuing this anti-inflammation focus of atherosclerosis therapy.
Macrophage Activation in Vascular Inflammatory Disease
Both acute and chronic forms of vascular inflammation are typified by the multitude of vasculitides and atherothrombotic pathologies. CAD, peripheral artery disease, vein graft failure, and arterio-venous fistula failure have seen various macrophage subtypes playing crucial roles. They either drive disease progression or cessation, or promote vessel repair and healing (7). Understanding the various phenotypes that allow macrophages to be categorized into subclasses with stereotyped behavior and function is crucial. This helps design strategies to precisely modulate immune signaling in vascular inflammation (1–3, 8). By limiting cellular subpopulations promoting plaque development, intimal cell proliferation, and tissue damage may be mitigated. This may also spare the subpopulation deemed beneficial for achieving disease control and resolution to allow return to homeostasis. In addition, understanding the profound adaptability, and plasticity of macrophages is key to knowing how to trigger phenotypic and functional changes within these cells and how far they can be reprogrammed.
What we have learned from the past is that both in vitro modeling of human and mouse primary macrophages complemented by experiments on small animal models of vascular disease have been important in elucidating mechanisms of macrophage activation and their role in the progression of the atherothrombotic lesions in CAD (9, 10). It is known that majority of the release of matrix metalloproteinases, MMPs, in human atherosclerotic plaques may derive from macrophages and foam cells, and to a lesser extent from smooth muscle cells (SMCs) and endothelial cells (ECs) (11). Excessive activation of proteases in the lesion lead to increased degradation of fibrillar collagen. This determines plaque integrity, leading to friable and unstable lesions. This may also lead to adverse remodeling prompting rupture and embolism of plaque debris, often seen in plaques (12, 13). Our preclinical studies used genetically-altered mouse strains to demonstrate that MMP-collagenases, major macrophage products, indeed promote the paucity of plaque collagen (14, 15). In vein grafts, MMP-2 and MMP-9 may play important roles in degrading the basement membrane which leads to enhanced infiltration of pro-inflammatory monocyte and macrophage populations (16, 17). These unstable plaque features are also most prominent among CAD patients with elevated low density lipoprotein (LDL) cholesterol levels (18), elevated lipoprotein (a) [Lp(a)] (19) and other metabolic derangements, reiterating a close interplay between inflammation and dysregulation of lipid handling (and other metabolic syndromes). LDL modifications, LDL cholesterol efflux and reverse cholesterol efflux all contribute to how cholesterol crystals instigate initial stages of atherosclerosis.
The process begins with recruiting monocytes from the circulating blood, followed by several processes including their differentiation into macrophages, foam cell formation and activation of the NOD-like receptor-pyrin domain (PYD)- containing-3 (NLRP3) inflammasome complex. Cholesterol crystals may cause phagolysosomal damage in macrophages priming them to activate NLRP3. Activation of NF-κB induces macrophages to produce pro-IL-1β and a pro-form of NLRP3. Upon activation of NLRP3, activated caspase-1 cleaves the pro-IL-1β releasing IL-1β, which amplifies the cascade of inflammatory signals (20, 21) including IL-6, tumor necrosis alpha (TNF-α) and pro-thrombotic initiators such as tissue factor (coagulation factor III). Clinical relevance for this is reflected in patients with typically high serum LDL cholesterol levels having increased incidence and severity of CAD and subsequent MACE sequelae. This paved way for the extensive use of statins in aggressively lowering elevated LDL cholesterol levels. Eventually, aggressive lipid lowering therapy indeed improved survival rates, as well as MACE/MACE+ outcomes as shown in clinical studies (22–25). However, certain patient populations did not benefit much due to having a baseline coronary atheroma predominantly dictating their prognosis (MACE) (26, 27). This leads to the burgeoning field of statin related research focused on elucidating mechanisms of statins that mitigate inflammation in cardiovascular disease, independent of their cholesterol lowering action.
For recalcitrant cases, LDL cholesterol levels can be further lowered by increasing availability of hepatic clearance via LDL receptors. Here, reduction of proprotein convertase subtilisin/kexin type 9 (PCSK9) activity and circulating levels have been quite effective, as seen in the success of drugs like evolucumab (Repatha) (28) and alirocumab (Praluent) (21, 29). Whether these inhibiting antibodies can reduce macrophage activation in coronary lesions remains to be proven. Interestingly, with successful reduction of LDL cholesterol levels in the at-risk population, physicians have inevitably selected for and identified a subpopulation of patients with considerable CAD morbidity despite sufficiently lowered LDL cholesterol levels. Since then, multiple points of evidence recognize inflammation, beyond hyperlipidemia, as a key regulatory hub by which CAD risk factors, co-morbid metabolic disease, and cardiovascular adverse events intersect (5, 30).
Mechanisms for Macrophage Activation
Traditional Thoughts on Macrophage Inflammatory Pathways in CAD
For years, many thought of resting macrophages as being activated in disease states into polarized subclasses that are diametrically opposed. Either they are “classically-activated” or pro-inflammatory, designated as “M1” and “alternatively-activated” or non/anti-inflammatory/pro-resolving “M2.” However, with much utilization of techniques allowing for multiple parametric assessments like single cell assays (FACS, etc), -OMICs profiling, and networks medicine approach, a more recent multi-dimensional model has emerged (31–35). Unsurprisingly, many authors have shied away from the M1 and M2 designations. Our recent study used single cell gene expression analysis to reveal that interferon gamma (INFγ)-induced “classically-activated” human primary macrophages remain largely heterogeneous, which is not consistent with a traditional “polarization” theory (36).
Acknowledging that macrophage subclasses fall into a spectrum of activation that is beyond bi-directional is the currently accepted paradigm. However, it may be still helpful, from a drug development and therapeutic research stand point, to think of macrophages in CVD to be either pro-inflammatory state(s) or any state(s) that is otherwise, most likely either immunosuppressive or pro-resolving (2, 37), (Figure 1A) because cause-effect relations are clear in these systems. So-called M1 macrophages are the ones that generate pro-inflammatory cytokines like TNF-α, inducible nitric oxide synthase (iNOS), and IL-6 (38). INFγ or lipopolysaccharide (LPS) is used to promote the pro-inflammatory phenotype in vitro (32) by triggering toll-like receptor (TLR)4 signaling associated pathways NF-κB, Notch, and INFγ/STAT-1. The NF-κB and NLRP3 inflammasome pathways are enhanced in pro-inflammatory macrophages. As stated above, experiments in small animal models reveal microscopic cholesterol crystals that activate the NLRP3 inflammasome resulting in the secretion of interleukin 1 family of cytokines (20). In another pathway, Delta-like 4 ligand (Dll4)-triggered Notch signaling can activate the NF-κB pathway to induce pro-inflammatory mediators such as iNOS, IL-1β, and CCL2/MCP-1, while Dll4 suppression reduce expression of pro-inflammatory factors of so-called M1 macrophages (8, 39–41). More recently, we demonstrated that the interplay between ADP-rbosylation enzymes PARP9 and PARP14 regulates the balance of pro- vs. anti-inflammatory macrophages (36).
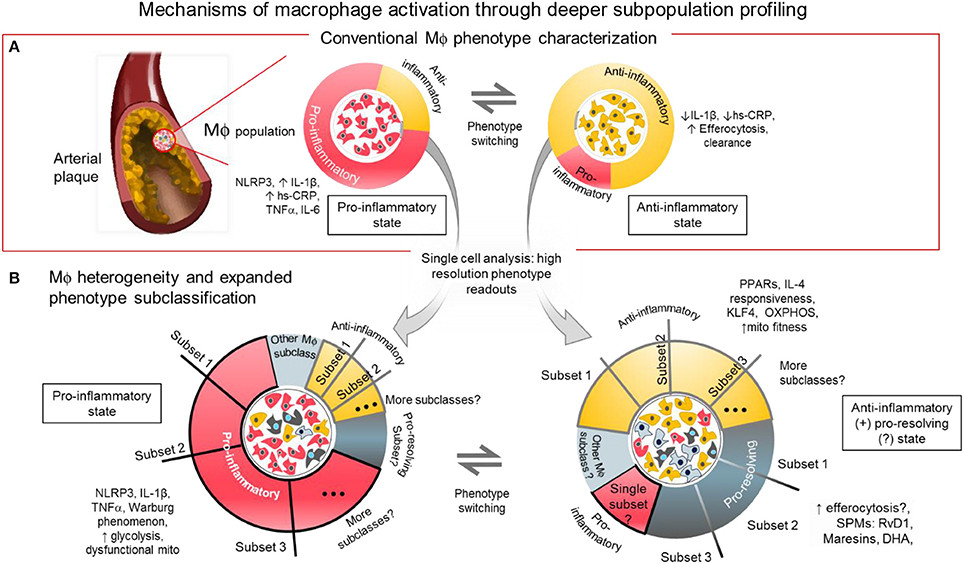
Figure 1. Mechanisms of macrophage activation through deeper subpopulation profiling. (A) Conventional characterization of macrophages. Macrophages and foam cells in atherosclerotic lesions comprise a mixture of phenotypes and different activation states. The balance of proinflammatory, and anti-inflammatory/pro-resolving states may determine the fate of the plaque whether it will be stable or prone to rupture. The balance of inflammatory signaling molecules, metabolic states and presence of SPMs, among others, may influence the level of macrophage activation in distinct local regions within the atherosclerotic plaque. (B) Macrophage heterogeneity and expanded subclassification. The balance of factors within the macrophages may depend on the number and size of subpopulations with distinct functional phenotypes as revealed by single cell analysis. One or more subset populations within a specifically activated macrophage population and a disproportional balance of any one subset may drive the outcome of disease progression. Identifying the driver population(s) may be the key to identifying regulators of the disease.
The loosely characterized anti-inflammatory or alternatively activated type of macrophages (traditionally called M2-like cells) is a general designation to refer to different subclasses that generate molecules that either suppress activity of the M1-like cells, initiate efferocytosis or promote resolution of inflammation (31) (Figure 1A). In mouse models, unlike in humans, these M2-like cells are identified with markers including arginase 1, mannose receptor C type 1, Ym1 and Fizz1 (38, 42). Although these markers do not identify M2-like macrophages in humans, information gleaned from using these murine M2-like cells may yield genes and proteins that are present and crucial in human macrophages for controlling inflammation. To produce M2-like phenotype in vitro, resting or activated macrophages need to be stimulated with any of the following chemokines: IL-4, IL-13, and IL-10, (42, 43) in contrast to the LPS or IFNγ stimulated M1-like cells. To avoid confusion, recent guidelines for macrophage subclass nomenclature propose to call these in vitro stimulated cells M(LPS), M(IFNγ), M(IL-4) or M(IL-10) (32).
IL-4 polarization of macrophages involve the Krüppel-like factor-4 (KLF-4) pathway and the IL-4/STAT-6 pathway. Paucity of KLF-4 in macrophages produce elevated expression of pro-inflammatory genes iNOS, and TNFα (44). MCP-1-induced protein (MCPIP) generated by KLF4 inhibits M1 activation. Likewise, it also promotes an M2-like phenotype. Furthermore, IL-4 triggered STAT6 induction promotes KLF4 expression, which mediates M2 activation through MCPIP activity as stated above (45). Peroxisome proliferator-activated receptors (PPARs) are also considered to be promoters of an anti-inflammatory phenotype (46). IL-4 and STAT-6 mediate transcription of several metabolism-related genes and regulators including PPARγ (47). Furthermore, absence of PPARγ in macrophages fail to induce oxidative metabolism and are also unable to exhibit M2-like phenotype (48). Independent from IL-4, IL-10 has recently been identified to promote mitochondrial fitness during pro-inflammatory activation of macrophages by mitigating the mitochondrial damage caused by reactive oxygen species (ROS) after iNOS activation, essentially paving the way for control, and resolution of inflammation (43).
Position along the macrophage phenotype/activation spectrum may simply be a function of the balance or imbalance of pro-inflammatory vs. anti-inflammatory factors present in every macrophage. Macrophages modeled in vitro are inherently heterogenous (Figure 1B). This means that although most of every single macrophage may be sitting at or near the extremes of the M1/M2 spectrum, some will remain near or at the median of the spectrum. The state of activation for groups of macrophages accumulating in local areas of the chronically inflamed tissue may be a function of the combined effect of the activation states of each macrophage single cell state. It is tempting to simplify that attenuating any and all M1-like macrophage populations will attenuate inflammatory burden. Disease state can therefore be reverted back to homeostasis. However, reducing pro-inflammatory activation without balancing the pro-resolving macrophage population and/or leaving a “patrolling” subpopulation might foreseeably lead to a more vulnerable immune system (8). That is why aggressive anti-inflammatory therapy targeting only macrophage accumulation and activation may increase incidence of severe infections. Calibrating the overall balance may lead to the development of efficient and safe therapies with minimal risk for unfavorable immunologic consequences. For example, inhibiting a specific signaling mechanism such as the Dll4-Notch pathway may suppress damaging macrophage products while promoting protective factors (39–41, 49).
Mechanisms Through Metabolic Reprogramming
Glycolytic Energy Preference of the Activated Macrophage
Evidence suggests that metabolic pathways are vital regulators of macrophage activation (50). As early as 1963, researchers recognized how metabolism affects monocyte and macrophage physiology. Based on experimental findings, alveolar macrophages, and circulating monocytes tend to have better physiologic function when energy is being fueled by aerobic respiration. Moreover, the macrophages were noted as capable of aerobic respiration in greater magnitudes than neutrophils and monocytes, and that inhibiting part of oxidative phosphorylation (OXPHOS) had a depressive impact on phagocytic activity (51). Later, G.C Hard showed that peritoneal macrophages from immune mice (activated) challenged by injecting a virulent strain of C. ovis intraperitoneally, had higher production of lactic acid compared to peritoneal macrophages from non-immune mice (resting macrophages). It was not clearly distinguished whether the peritoneal macrophages from the non-immune mice were truly differentiated and not an admixture of macrophages and patrolling monocytes in the peritoneal cavity. Still, his findings are among the first to show enhanced glycolysis through increases in lactate production along with decreased O2 uptake in immune activated primary macrophages (52). After more than a decade, experiments with thioglycolate-elicited mouse peritoneal macrophages show surprisingly high amounts of hexokinase, glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase implying further glycolytic induction in activated macrophages. In contrast, resting macrophages from saline injected mice showed less than 10% activity of 6-phosphofructokinase, complementing the prior observation that glycolytic rate is increased dramatically during phagocytosis or increased secretory activity (53). These early studies into macrophage metabolism research have broaden an entire field for mining biological mechanisms that allow us to appreciate immune activation as a reflection of metabolic state. Pivotal insights on macrophage metabolic reprograming came when it was reported that pro-inflammatory type polarization of macrophages with either LPS or INFγ could switch bioenergetic preferences of macrophages from OXPHOS to the glycolytic route (54, 55), a phenomena many observed to be reminiscent of the Warburg phenomenon happening in cancer cells (56–58). Other evidence supporting this, demonstrates that stimulation of TLRs by LPS increase PFKFB3 (6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3) expression, resulting in an increase of the key glycolytic allosteric regulator fructose 2,6-bisphosphate and a glycolytic flux (59).
Role of Metabolites in Classical Activation of Macrophages
Afterwards, work done in the lab of Luke O'Neill showed how metabolite key players in the glycolytic pathway, tri-carboxylic acid cycle (TCA) and OXPHOS play important roles in determining inflammatory activation states of immune cells like macrophages (56). They confirm that LPS causes macrophages to switch their core metabolism from OXPHOS to glycolysis. Inhibiting glycolysis could also suppress LPS mediated IL-1β secretion, albeit not TNFα, in mouse macrophages. While LPS does decrease expression of mitochondrial genes, there is a concomitant increase on levels of the TCA intermediate succinate. Their findings show that succinate: (1) stabilizes HIF-1α which support IL-1β release; (2) increases succinylation of several proteins; and (3) reduces desuccinylation of Sirt5 (60), a known epigenetic regulator of metabolism and inflammation. This Warburg-like phenomenon in macrophages implicate reprogramming at the epigenetic level through changing levels of acetyl-CoA potentially affecting increased acetylation and decreased deacetylations of histone proteins (61) Pro-inflammatory macrophages also have increased glucose transporter 1 (GLUT1) expression and availability to drive glucose uptake. This causes both hyper inflammatory proteome and transcriptome seen in RAW264.7 cells overexpressing GLUT1, leading to elevated secretion of inflammatory mediators, increase in reactive oxygen species (ROS) production and oxidative stress intracellularly (62) LPS-stimulated TLR4 increases mammalian target of rapamycin (mTOR) signaling, which induces the expression of lactate dehydrogenase and hypoxia induced genes (63, 64). Both events shunt acetyl-CoA away from TCA consumption. Moreover, this activation also leads up to elevated levels of an isoform of PFKFB3 favoring glycolysis (63–65).
Metabolic Changes in Anti-inflammatory Type of Macrophages
On the other hand, anti-inflammatory conditioning of macrophages have an opposite effect of promoting OXPHOS while reducing preference for glycolysis (66). IL-4 for instance promotes OXPHOS resulting in elevated oxygen consumption rate (OCR) in RAW264.7 cells. Extracellular acidification rate (ECAR), a measure of lactate production (anaerobic glycolysis) is also lower compared to pro-inflammatory LPS stimulated RAW264.7 cells (67). But as a caveat, LPS and/or IFNγ-polarized macrophages may not repolarize into an anti-inflammatory phenotype (IL-4 re-stimulation) if there is too much mitochondrial dysfunction brought out by LPS-induced hyper-glycolysis and nitric oxide production (68). This results in inhibition of OXPHOS in the mitochondria (66). This also prevents plasticity of LPS+IFNγ conditioned macrophages to convert into an anti-inflammatory phenotype (66) underlying the importance of OXPHOS in the resolution of inflammation or the direction of macrophage activation. In this case of proinflammatory macrophages resistant to phenotype switching, IL-10 may be the key signaling molecule that may aid IL-4 and accomplish the phenotype switch.
Arguably considered an anti-inflammatory chemokine, IL-10 could increase clearance of dysfunctional mitochondria through mitophagy. IL-10 activates STAT3 signaling which in turn activates DDIT4 transcription factor leading to inhibition of the NLRP3 inflammasome activated mTORC1 pathway, thereby releasing the restraint against autophagy. DDIT4 itself promotes clearance of ROS-damaged mitochondria by mitophagy while IL-10 maintains membrane gradient potential of mitochondria of primary macrophage promoting organelle integrity and fitness during macrophage activation. This allows for rapid resolution of inflammation (43). All these changes in bioenergetics during macrophage polarization may be accompanied by a changing metabolome and specifically, a shift in the lipidome as well.
Lipidome Changes in Proinflammatory Macrophages and the Pro-Resolving Factors
E. Dennis' group did important work on macrophage lipidomics having contributed to the LIPID MAPS consortium to develop quantitative methods for evaluating the composition, biosynthesis, and function of all macrophage lipids. In one of their findings, they identified endotoxin Kdo2-Lipid A (KLA, a defined form of LPS) of E.coli activates macrophages via TLR4 similar to “regular” LPS. Prostaglandin D2 (PGD2) is the predominant eicosanoid produced after KLA—proinflammatory stimulation of RAW264.7 cells (> 120 ng/106 cells), with almost nil 11-HETE production in contrast. PGE2 and PGD2 increase in a dose dependent manner with both LPS and KLA stimulation. PGE2 and PGD2 increase in a more potent but similar fashion to TNFα chemokine after LPS or KLA stimulation (69). Using RAW264.7 cells, their laboratory again assessed mouse macrophage lipidome changes with KLA treatment vs. drug perturbations like statins which are clinically relevant as statins inhibit cholesterol biosynthesis pathway and are able to reduce further inflammation in CAD of patients with high LDL profile (70). KLA tended to increase almost all categories of sphingolipid analyzed, cholesterol esters, and some glycrophospholipids. Statins like mevastatin (or compactin), a parent compound of pravastatin promote lipid changes intracellularly in these macrophages. Expectedly, statins blocked KLA induced increases in desmosterol and other components of the sterol biosynthetic pathway yet had no effect on actual intracellular cholesterol levels per se (71).
Still, there is more to uncover regarding metabolome and lipidome changes during the phenotypic differentiation by activated macrophages. With large metabolomic/lipidomic datasets, a systems approach to analysis may be the key to uncover comprehensive understanding of the dynamics of lipid and non-lipid metabolite pathways in the macrophage. This initial characterization in the mouse system is a first step to finding desirable therapeutic targets (71). However, there is still need to further elevate these studies to clinical relevance by using human samples. To this end, Tabas et al. further advanced the utility of mass-spectrometry lipidomic profiling (6) for atherosclerosis therapeutics by demonstrating specialized pro-resolving lipid mediators (SPMs) in atherosclerotic plaques. SPMs are generally essential fatty acids-derived autacoids (2) They found that bioactive lipid derivative resolvin D1 (RvD1) levels were low relative to pro-inflammatory lipid leukotriene B4 (LTB4) in vulnerable plaques of the human carotid artery. This was further confirmed in hyperlipidemic mouse models by showing that administration of RvD1 increases plaque stability, lowers oxidative stress and necrosis, and thickens fibrous caps. Their findings support a–omics assisted mechanistic rationale for SPM therapy in CAD to mitigate plaque vulnerability (37).
Another set of SPMs, the maresins which are studied extensively by the Serhan group, are macrophage derived and are produced via 14-lipoxygenation of docosahexaenoic acid (DHA) that is either converted by enzymes into mediators with two-OH groups or into autacoids that are peptide-lipid conjugates, called maresin conjugates. These SPMs promote the uptake and clearance of apoptotic cells by macrophages. Maresins also regulate portions of tissue repair (2) therefore, resolution of the inflammatory damage. SPMs can potentially influence switching of macrophage function.
Macrophage Crosstalk With Other Immune Cells
Not only are macrophages known for their plasticity, these cells have the ability to influence and be influenced by other immune cells like T cells that results in a similar macrophage phenotype switching as above. The reciprocity of T cells and macrophages through either paracrine signaling or molecular interactions may dictate the direction of inflammation. It may either drive well into a vicious cycle of unmitigated chronic pro-inflammatory atherosclerotic events or toward inflammation resolution (72). Several reports have already shown that these macrophages communicate with other immune cells via specified protein pairs on their cell surfaces. Pairs can be co-stimulatory or co-inhibitory, whereby molecular interaction between these pairs triggers downstream biomolecular cascades that may promote or limit macrophage activation in atherosclerosis (73). Oncology researchers took advantage of this T cell-macrophage crosstalk, in order to combat cancer cells (74). Immunologists have coined the term immune checkpoints to identify these pairs of proteins that interact to either promote a pro-inflammatory activation or an anti-inflammatory one (75).
One of these pairs of co-stimulatory proteins are CD40 and CD40L (ligand) which are expressed in macrophages among other cell types found in atherosclerotic vessels (76). Both CD40 and CD40L are expressed highly in atherosclerotic lesions (77) and plasma which may predict patients with features of high-risk atherosclerotic lesions corroborated with MRI (78). Abrogating CD40L activity effectively reduces release of pro-inflammatory factors together with reducing activation of macrophages by activated T cells in vivo using mouse models of atherosclerosis. Deletion of either CD40 or CD40L has atheroprotective effects by mitigating macrophage activation (73). CD80/B7-1 and CD86/B7-2 are a pair of recognized M1-like markers for macrophages are also expressed in dendritic cells (DCs) in atherosclerotic plaques. They co-stimulate and bind to CD28 on T cells, B cells, and other macrophages (79). CD80 and CD86 double deficiency in hyperlipidemic LDL receptor-deficient (LDLr−/−) mice results in lesser atherosclerotic burden (80) Other co-stimulatory immune checkpoint proteins found in human atherosclerotic lesions and in pre-clinical models that tend to skew macrophages and DCs to the pro-inflammatory end of the spectrum are: OX40-OX40L, CD137-CD137L, and CD30-CD30L (81) Immune checkpoint and co-inhibitory proteins PD-1 and PD-L1/2 inhibit T cell immune response resulting in a beneficial atheroprotective effect. Stimulation of PD-L1 expression in vitro could attenuate the stimulatory ability on allogeneic T cell proliferation and its cytokine production, including IFNγ (82). Such effect, however, is detrimental to immune clearance of tumor cells in cancer, hence the success of the anti-PD-1 immunotherapy oncologic drug pembrolizumab (83). CD27-CD70 is a another possible co-inhibitory pair since CD27 paucity on mice models show markedly increased atherosclerotic burden. In addition, CD27 is demonstrated to be essential in maintain a healthy pool of regulatory T cells (Tregs), preventing increased apoptosis of Tregs (84). CD70 promotes macrophage function and viability, and is important for effective efferocytosis and extrusion of oxLDL. CD70 deficiency results in more advanced atheroma (85). These are just a few of the immune checkpoints that affect macrophage behavior. Other proteins include, though not limited to, CTLA-4, ICOS-ICOSL, GITR-GITRL, and TIM (81). Other potential macrophage checkpoints are CD47 and signal regulatory protein alpha, SIRPα. In cancer, activation of SIRPα by CD47 on macrophages suppress both phagocytosis and respiratory burst (74). Therefore, as expected in mouse atherosclerotic models, blockade of CD47 exerts anti-atherosclerotic effects, halting lesion progression and preventing plaque rupture and restores phagocytosis/efferocytosis (86). A more recently studied immune regulator is the V-domain containing Ig Suppressor of T cell Activation (VISTA, aka PD-1H, DD1α; gene name DIES1). VISTA is both a receptor and a ligand with immunosuppressive effects on IFNγ, and TNFα with T cells and macrophages (74). In fact, majority of VISTA+ macrophages have the anti-inflammatory M2-like phenotype (87). Other proteins that can potentially act as immune checkpoints by control macrophage behavior via T cell and other immune cell 2-way interaction include: T cell immunoglobulin and ITIM domain (TIGIT) and indoleamine-2,3-dioxygenase (IDO). More studies are, however, required to clarify their roles in the macrophage inflammatory phenotype spectrum (74).
A summary comparison of these biomolecular signatures of atherosclerosis, as evidenced by findings in studies of mouse vs. human macrophages is enumerated in Table 1.
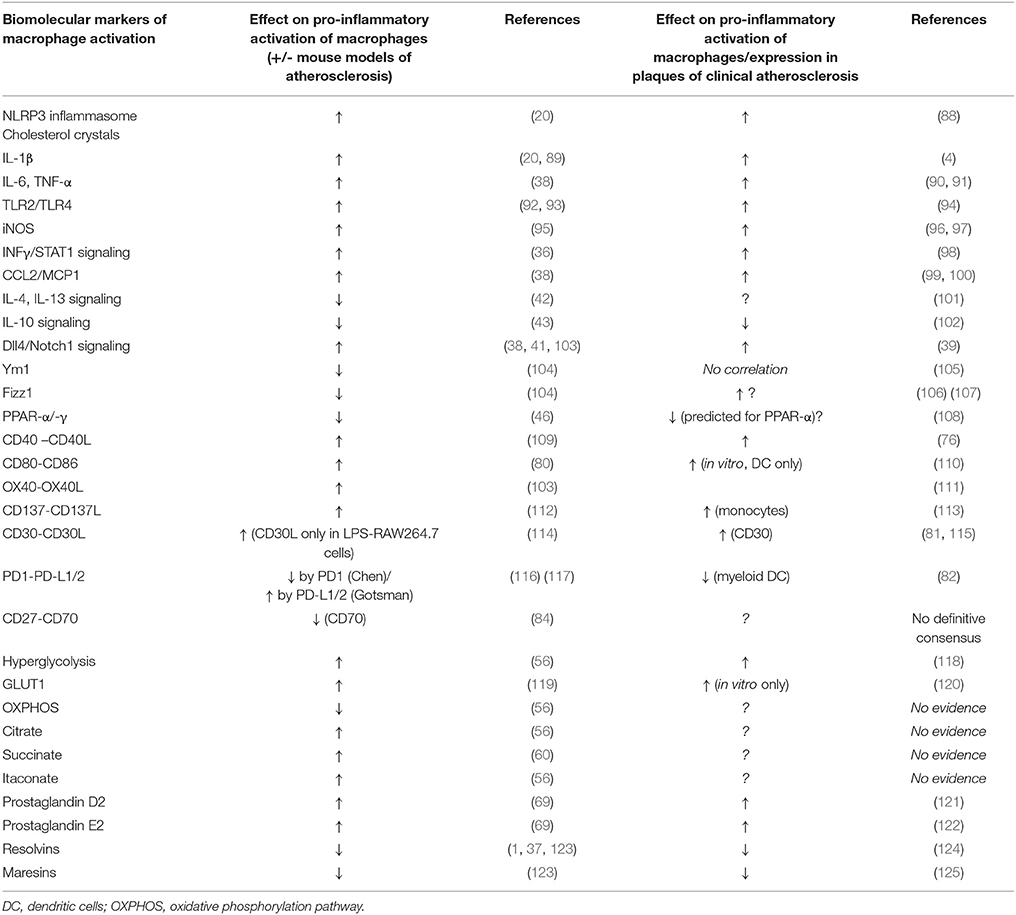
Table 1. Comparison of the effects of some biomolecular markers of macrophage activation as seen in mouse in vitro & pre-clinical models of atherosclerosis vs. human atherosclerosis (including in vitro studies).
Macrophage Heterogeneity in Atherosclerosis
Advances in single cell analysis provided insights of how heterogenous human primary macrophages are. Single cell qPCR reveals how heterogenous resting macrophages, M0 or M(-), are. Yet after a pro-inflammatory polarization by IFNγ activation, M(IFNγ) macrophages even have a further heterogenous response showing subpopulations that are more responsive to pro-inflammatory signals, even as some populations remain resistant and reticent, maintaining an unstimulated phenotype (35, 36). As cited earlier, identifying these macrophages and contrasting them vs. the highly responsive ones may help filter out by enrichment of key regulatory genes. These genes may promote phenotype switching thereby unraveling them to be desirable therapeutic targets. These targets or pathways may be otherwise hidden when examining an average transcriptomic readout from “bulk” macrophage populations processed from conventional qPCR or mRNAseq assays instead of the high resolution readouts of single cell analysis.
Future Perspectives: New Paradigms of Discovery Science and Drug Development in Vascular inFlammation
With the multitude of mechanistic perspectives that make up the macrophage behavior in atherosclerosis, there are many aspects to consider when designing effective therapies with the highest potential of leaping from the laboratory bench to clinical translation. Consolidating these various angles intelligently, in order to arrive at a viable drug target in the fastest possible way, is an attractive goal for the medical science community. However, due to limited resources, insufficient funding, and lack of expertise, many academic investigators may fail to develop and deliver their target discovery beyond the laboratory space (126, 127). Despite the evident strengths of pharmaceutical industry in drug development, its recent tendency to avoid investing in early, high-risk projects appears to have enhanced this gap in translation of academic target discovery into the clinic (128, 129). These roadblocks often pre-maturely remove the possibility of an otherwise promising target from being transferred to pharmaceutical development and eventually becoming a novel, even first-in-class, drug. (Figure 2A). Arriving at the most cost effective strategy for pharmaceutical translation requires a balancing act of carefully prioritizing the targets discovered with very rational and discreet management of resources for target(s) validation while still within the hands of the academic researcher (130).
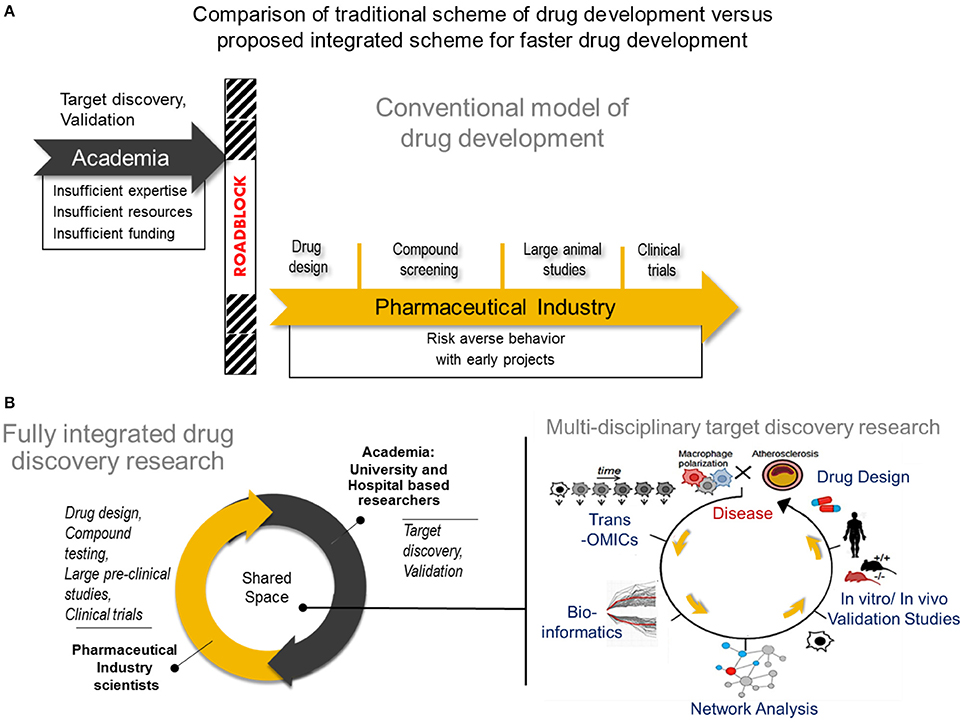
Figure 2. Comparison of tradional drug development vs. proposed integrated drug discovery research (A) The conventional model of drug development Target discovery and validation studies are often done by academic researchers who usually have insufficient expertise and resources to conduct such studies resulting years of development. After characterizing a potential drug target, many academic investigators struggle to translate their finding into the pharmaceutical space due to a natural disconnect between academia and industry. In rare instances, academic investigators may manage to cross the roadblock and transfer their breakthroughs to industry, followed by a lengthy process of drug design, compound testing and animal studies before the drug will be considered for human studies. (B) A new paradigm for drug development Center) A fully integrated drug discovery research in our laboratory involves close collaboration between academic and pharmaceutical industry scientists. Right) Use of multi-omics approach to disease characterization with systems approach analysis for faster target discovery and prioritization and drug design. A right panel was reproduced from Iwata et al. (36). Trans-OMICs: genomics, transcriptomics, proteomics, epigenomics, metabolomics, lipidomics, etc.
Close collaboration between academia and industry will foster faster exchange of expertise and resources that could lead to mutually beneficial research outputs (130, 131). In such a model established in our laboratory (Figure 2B, center panel), drug development is facilitated when effectively combining the exploratory nature of innovative academic research and the extensive expertise in drug design and the rigor and resource intensive validation from pharmaceutical industry. Much advancement in computational biology and networks medicine, as well as steadily declining costs and time required for—omics experiments and single cell experiments also help to speed the transition of academic target discovery into drug development. It is easier now to consolidate various biological mechanisms that define macrophage activation, and use an intergrated approach to arrive at viable targets in a more comprehensive and unbiased manner. A typical workflow of target discovery research in our laboratory is demonstrated in Figure 2B (right panel). We expect that paired with an ever-expanding knowledge base and expertise in performing these–omics experiments, as well as use of machine learning approaches, a more solid understanding of pathways that drive macrophage activation will emerge. Novel pathways of activation may be uncovered when we actively use network science to incorporate comprehensive readouts from–omics and single cell experiments. With all these innovative large-scale approaches in biological research and data analyses, pairing with synergistic efforts from academic and industry scientists and cell and computational biologists, more effective and better defined drugs may arrive to the market sooner. Better understanding of biological mechanisms for macrophage activation and heterogeneity through big data, particularly of clinical samples, and integrated analysis, may also lead to safer drugs that target specific subsets of populations as opposed to a more generalized approach.
Author Contributions
JD and MA both contributed to the concept and key contents of this manuscript. JD drafted the manuscript and MA critically reviewed and edited it.
Funding
This work was supported in part by research grants from the National Heart, Lung, and Blood Institute (R01HL107550 and R01HL126901 to MA) and Kowa Company, Ltd., (Nagoya, Japan to MA).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Tabas I, Lichtman AH. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 47:621–34. doi: 10.1016/j.immuni.2017.09.008
2. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nature Rev Immunol. (2013) 13:709–21. doi: 10.1038/nri3520
3. Koltsova EK, Hedrick CC, Ley K. Myeloid cells in atherosclerosis: a delicate balance of anti-inflammatory and proinflammatory mechanisms. Curr Opin lipidol. (2013) 24:371–80. doi: 10.1097/MOL.0b013e328363d298
4. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N E J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
5. Sabatine MS, Morrow DA, Jablonski KA, Rice MM, Warnica JW, Domanski MJ, et al. Prognostic significance of the centers for disease control/american heart association high-sensitivity C-reactive protein cut points for cardiovascular and other outcomes in patients with stable coronary artery disease. Circulation (2007) 115:1528–36. doi: 10.1161/CIRCULATIONAHA.106.649939
6. Ridker PM. A test in context: high-sensitivity C-reactive protein. J Am. College Cardiol. (2016) 67:712–23. doi: 10.1016/j.jacc.2015.11.037
7. Shirai T, Hilhorst M, Harrison DG, Goronzy JJ, Weyand CM. Macrophages in vascular inflammation–From atherosclerosis to vasculitis. Autoimmunity (2015) 48:139–51. doi: 10.3109/08916934.2015.1027815
8. Nakano T, Fukuda D, Koga J, Aikawa M. Delta-Like Ligand 4-Notch Signaling in Macrophage Activation. Arterioscler Thromb Vasc Biol. (2016) 36:2038–47. doi: 10.1161/ATVBAHA.116.306926
9. Nakashima Y, Plump AS, Raines EW, Breslow J.L, Ross, R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arteriosc Thromb Vasc Biol. (1994) 14:133. doi: 10.1161/01.ATV.14.1.133
10. Bisgaard LS, Mogensen CK, Rosendahl A, Cucak H, Nielsen LB, Rasmussen SE, Pedersen TX. Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression - implications for atherosclerosis research. Sci Rep. (2016) 6:35234. doi: 10.1038/srep35234
11. Shah P.K. Mechanisms of plaque vulnerability and rupture. J Am College Cardiol. (2003) 41:S15–22. doi: 10.1016/S0735-1097(02)02834-6
12. Molloy KJ, Thompson MM, Jones JL, Schwalbe EC, Bell PRF, Naylor AR, Loftus IM. Unstable carotid plaques exhibit raised matrix metalloproteinase-8 activity. Circulation (2004) 110:337. doi: 10.1161/01.CIR.0000135588.65188.14
13. Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med. (2002) 8:1257. doi: 10.1038/nm1102-1257
14. Fukumoto Y, Deguchi J, Libby P, Rabkin-Aikawa E, Sakata Y, Chin MT, et al. Genetically determined resistance to collagenase action augments interstitial collagen accumulation in atherosclerotic plaques. Circulation (2004) 110:1953–9. doi: 10.1161/01.CIR.0000143174.41810.10
15. Deguchi J, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, Whittaker P, Aikawa M. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation (2005) 112:2708–15. doi: 10.1161/CIRCULATIONAHA.105.562041
16. Southgate KM, Mehta D, Izzat MB, Newby AC, Angelini GD. Increased secretion of basement membrane–degrading metalloproteinases in pig saphenous vein into carotid artery interposition grafts. Arteriosc Thromb Vascul Biol. (1999) 19:1640. doi: 10.1161/01.ATV.19.7.1640
17. George SJ, Zaltsman AB, Newby AC. Surgical preparative injury and neointima formation increase MMP-9 expression and MMP-2 activation in human saphenous vein. Cardiovasc Res (1997) 33:447–59. doi: 10.1016/S0008-6363(96)00211-8
18. Yamada K, Yoshimura S, Kawasaki M, Enomoto Y, Asano T, Minatoguchi S, Iwama T. Effects of atorvastatin on carotid atherosclerotic plaques: a randomized trial for quantitative tissue characterization of carotid atherosclerotic plaques with integrated backscatter ultrasound. Cerebrovasc Dis. (2009) 28:417–24. doi: 10.1159/000235746
19. Zampoulakis JD, Kyriakousi AA, Poralis KA, Karaminas NT, Palermos ID, Chimonas ET, Cokkino SDV. Lipoprotein(a) is related to the extent of lesions in the coronary vasculature and to unstable coronary syndromes. Clin Cardiol. (2009) 23:895–900. doi: 10.1002/clc.4960231208
20. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
21. Gross O, Thomas Christina J, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. (2011) 243:136–51. doi: 10.1111/j.1600-065X.2011.01046.x
22. Post coronary artery bypass graft trial investigators. The effect of aggressive lowering of low-density lipoprotein cholesterol levels and low-dose anticoagulation on obstructive changes in saphenous-vein coronary-artery bypass grafts. N Engl J Med. (1997) 336:153–63.
23. Pitt B, Waters D, Brown WV, van Boven AJ, Schwartz L, Title LM, Eisenberg D, Shurzinske L, McCormick LS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med. (1999) 341:70–6. doi: 10.1056/NEJM199907083410202
24. Kinlay S, Schwartz GG, Olsson AG, Rifai N, Leslie SJ, Sasiela WJ, et al. High-dose atorvastatin enhances the decline in inflammatory markers in patients with acute coronary syndromes in the MIRACL study. Circulation (2003) 108:1560–6. doi: 10.1161/01.CIR.0000091404.09558.AF
25. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. (2008) 359:2195–207. doi: 10.1056/NEJMoa0807646
26. Puri R, Nissen SE, Shao M, Ballantyne CM, Barter PJ, Chapman MJ, et al. Coronary atheroma volume and cardiovascular events during maximally intensive statin therapy. Eur Heart J. (2013) 34:3182–90. doi: 10.1093/eurheartj/eht260
27. Ridker PM. How common is residual inflammatory risk? Circ Res. (2017) 120:617–9. doi: 10.1161/CIRCRESAHA.116.310527
28. Toth PP, Worthy G, Gandra SR, Sattar N, Bray S, Cheng LI, et al. Systematic review and network meta-analysis on the efficacy of evolocumab and other therapies for the management of lipid levels in hyperlipidemia. J Am Heart Assoc. (2017) 6:e005367. doi: 10.1161/JAHA.116.005367
29. Cannon CP, Cariou B, Blom D, McKenney JM, Lorenzato C, Pordy R, Chaudhari U, Colhoun HM. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J. (2015) 36:1186–94. doi: 10.1093/eurheartj/ehv028
30. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. (1997) 336:973–79. doi: 10.1056/NEJM199704033361401
31. Martinez FO, Gordon S. The evolution of our understanding of macrophages and translation of findings toward the clinic. Exp Rev Clin Immunol. (2015) 11:5–13. doi: 10.1586/1744666X.2015.985658
32. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
33. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
34. Beyer M, Mallmann MR, Xue J, Staratschek-Jox A, Vorholt D, Krebs W, et al. High-resolution transcriptome of human macrophages. PloS ONE (2012) 7:e45466. doi: 10.1371/journal.pone.0045466
35. Nahrendorf M, Swirski FK. Abandoning M1/M2 for a network model of macrophage function. Circ Res. (2016) 119:414–7. doi: 10.1161/CIRCRESAHA.116.309194
36. Iwata H, Goettsch C, Sharma A, Ricchiuto P, Goh WWB, Halu A, et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat Commun. (2016) 7:12849. doi: 10.1038/ncomms12849
37. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. (2016) 7:12859. doi: 10.1038/ncomms12859
38. Fukuda D, Aikawa M. The expanding role of delta-like 4 mediated notch signaling in cardiovascular and metabolic diseases. Circ J. (2013) 77:2462–8. doi: 10.1253/circj.CJ-13-0873
39. Fung E, Tang SM, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, Carlesso N, Aster J.C, Aikawa M. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation (2007) 115:2948–56. doi: 10.1161/CIRCULATIONAHA.106.675462
40. Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, Gorgun CZ, et al. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci U S A. (2012) 109:E1868–77. doi: 10.1073/pnas.1116889109
41. Koga J, Nakano T, Dahlman JE, Figueiredo JL, Zhang H, Decano J, et al. Macrophage notch ligand delta-like 4 promotes vein graft lesion development: implications for the treatment of vein graft failure. Arterioscler Thromb Vasc Biol (2015) 35:2343–53. doi: 10.1161/ATVBAHA.115.305516
42. Ng Kuet Leong N, Brombacher F, Dalpke A.H, Weitnauer M. Crosstalk between glucocorticoids and IL-4 modulates Ym1 expression in alternatively activated myeloid cells. Immunobiology (2017) 222:759–67. doi: 10.1016/j.imbio.2017.02.003
43. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science (2017) 356:513. doi: 10.1126/science.aal3535
44. Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest. (2011) 121:2736–49. doi: 10.1172/JCI45444.
45. Kapoor N, Niu J, Saad Y, Kumar S, Sirakova T, Becerra E, Li X, Kolattukudy PE. Transcription factors STAT6 and KLF4 implement macrophage polarization via the dual catalytic powers of MCPIP. J Immunol (2015) 194:6011–023. doi: 10.4049/jimmunol.1402797
46. Van Ginderachter JA, Movahedi K, Van den Bossche J, De Baetselier P. Macrophages, PPARs, and Cancer. PPAR Res. (2008) 2008:169414. doi: 10.1155/2008/169414
47. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metabol. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
48. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447:1116–20. doi: 10.1038/nature05894
49. Sweet DR, Fan L, Hsieh PN, Jain MK. Kruppel-like factors in vascular inflammation: mechanistic insights and therapeutic potential. Front Cardiovasc Med. (2018) 5:6. doi: 10.3389/fcvm.2018.00006
50. Galvan-Pena S, O'Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. (2014) 5:420. doi: 10.3389/fimmu.2014.00420
51. Oren R, Farnham AE, Saito K, Milofsky E, Karnovsky ML. Metabolic patterns in three types of phagocytizing cells. J Cell Biol. (1963) 17:487–501. doi: 10.1083/jcb.17.3.487
53. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J. (1986) 239:121–5. doi: 10.1042/bj2390121
54. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
55. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. (2014) 12:e1001759. doi: 10.1371/journal.pbio.1001759
56. O'Neill LA. A critical role for citrate metabolism in LPS signalling. in Biochem J. (2011) 438 e5–6. doi: 10.1042/BJ20111386
57. Ponizovskiy MR. Cancer metabolism and the warburg effect as anabolic process outcomes of oncogene operation. Crit Rev Eukaryot Gene Expr. (2010) 20:325–39. doi: 10.1615/CritRevEukarGeneExpr.v20.i4.40·
58. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519. doi: 10.1085/jgp.8.6.519
59. Ruiz-Garcia A, Monsalve E, Novellasdemunt L, Navarro-Sabate A, Manzano A, Rivero S, et al. Cooperation of adenosine with macrophage Toll-4 receptor agonists leads to increased glycolytic flux through the enhanced expression of PFKFB3 gene. J Biol Chem. (2011) 286:19247–58. doi: 10.1074/jbc.M110.190298
60. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496:238–42. doi: 10.1038/nature11986
61. Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. (2015) 33:125–31. doi: 10.1016/j.ceb.2015.02.003
62. Johnson AR, Qin Y, Cozzo AJ, Freemerman AJ, Huang MJ, Zhao L, et al. Metabolic reprogramming through fatty acid transport protein 1 (FATP1) regulates macrophage inflammatory potential and adipose inflammation. Mol Metab. (2016) 5:506–26. doi: 10.1016/j.molmet.2016.04.005
63. Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
64. Semenza GL, Jiang B, Leung SW, Passantino R, Concordet J, Maire P, Giallongo A. Hypoxia Response elements in the aldolase a, enolase 1, and lactate dehydrogenase a gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. (1996) 271:32529–37. doi: 10.1074/jbc.271.51.32529
65. Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, Bosca L. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. (2010) 185:605–14. doi: 10.4049/jimmunol.0901698
66. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
67. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. (2012) 15:813–26. doi: 10.1016/j.cmet.2012.04.023
68. Albina JE, Mastrofrancesco B. Modulation of glucose metabolism in macrophages by products of nitric oxide synthase. Am J Physiol Cell Physiol. (1993) 264:C1594–9. doi: 10.1152/ajpcell.1993.264.6.C1594
69. Raetz CR, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC Jr, et al. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J Lipid Res. (2006) 47:1097–111. doi: 10.1194/jlr.M600027-JLR200
70. Schoenhagen P, Tuzcu EM, Apperson-Hansen C, Wang C, Wolski K, Lin S, et al. Determinants of arterial wall remodeling during lipid-lowering therapy: serial intravascular ultrasound observations from the Reversal of Atherosclerosis with Aggressive Lipid Lowering Therapy (REVERSAL) trial. Circulation (2006) 113:2826–34. doi: 10.1161/CIRCULATIONAHA.105.585703
71. Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, et al. A mouse macrophage lipidome. J Biol Chem. (2010) 285:39976–85. doi: 10.1074/jbc.M110.182915
72. Gotsman I, Sharpe AH, Lichtman AH. T-cell costimulation and coinhibition in atherosclerosis. Circ Res. (2008) 103:1220–31. doi: 10.1161/CIRCRESAHA.108.182428
73. Lutgens E, Lievens D, Beckers L, Donners M, Daemen M. CD40 and Its Ligand in Atherosclerosis. Trends Cardiovasc Med. (2007) 17:118–23. doi: 10.1016/j.tcm.2007.02.004
74. Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. (2017) doi: 10.1016/j.semcancer.2017.10.001 [Epub ahead of print].
75. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
76. Hakkinen T, Karkola K, Yla-Herttuala S. Macrophages, smooth muscle cells, endothelial cells, and T-cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions. Colocalization with epitopes of oxidized low-density lipoprotein, scavenger receptor, and CD16 (Fc gammaRIII). Virchows Arch. (2000) 437:396–405. doi: 10.1007/s004280000239
77. Schonbeck U. Oxidized low-density lipoprotein augments and 3-Hydroxy-3-Methylglutaryl coenzyme a reductase inhibitors limit CD40 and CD40L expression in human vascular cells. Circulation (2002) 106:2888–93. doi: 10.1161/01.CIR.0000043029.52803.7B
78. Blake GJ. Soluble CD40 ligand levels indicate lipid accumulation in carotid atheroma: an in vivo study with high-resolution MRI. Arteriosc Thromb Vascular Biol. (2002) 23:11e−14. doi: 10.1161/01.ATV.0000050143.22910.62
79. Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. (2002) 2:116–26. doi: 10.1038/nri727
80. Buono C, Pang H, Uchida Y, Libby P, Sharpe AH, Lichtman AH. B7-1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation (2004) 109:2009–15. doi: 10.1161/01.CIR.0000127121.16815.F1
81. Foks AC, Kuiper J. Immune checkpoint proteins: exploring their therapeutic potential to regulate atherosclerosis. Br J Pharmacol. (2017) 174:3940–55. doi: 10.1111/bph.13802
82. Lee J, Zhuang Y, Wei X, Shang F, Wang J, Zhang Y, et al. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J Mol Cell Cardiol. (2009) 46:169–76. doi: 10.1016/j.yjmcc.2008.10.028
83. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. (2015) 372:2018–28. doi: 10.1056/NEJMoa1501824
84. Winkels H, Meiler S, Lievens D, Engel D, Spitz C, Bürger C, et al. CD27 co-stimulation increases the abundance of regulatory T cells and reduces atherosclerosis in hyperlipidaemic mice. Eur Heart J. (2017) 38:3590–9. doi: 10.1093/eurheartj/ehx517
85. Winkels H, Meiler S, Smeets E, Lievens D, Engel D, Spitz C, et al. CD70 limits atherosclerosis and promotes macrophage function. Thromb Haemost. (2017) 117:164–75. doi: 10.1160/TH16-04-0318
86. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature (2016) 536:86–90. doi: 10.1038/nature18935
87. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. (2017) 23:551. doi: 10.1038/nm.4308
88. Paramel Varghese G, Folkersen L, Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J Am Heart Assoc. (2016) 5:e003031. doi: 10.1161/JAHA.115.003031
89. Netea MG, Demacker PN, Kullberg BJ, Boerman OC, Verschueren I, Stalenhoef AF, Van Der Meer JW. Increased interleukin-1alpha and interleukin-1beta production by macrophages of low-density lipoprotein receptor knock-out mice stimulated with lipopolysaccharide is CD11c/CD18-receptor mediated. Immunology (1998) 95:466–72. doi: 10.1046/j.1365-2567.1998.00598.x
90. Plutzky J. Inflammatory pathways in atherosclerosis and acute coronary syndromes. The Am J Cardiol. (2001) 88:10–5. doi: 10.1016/S0002-9149(01)01924-5
91. Simon TG, Trejo MEP, McClelland R, Bradley R, Blaha MJ, Zeb I, Corey KE, Budoff MJ, Chung RT. Circulating Interleukin-6 is a biomarker for coronary atherosclerosis in nonalcoholic fatty liver disease: results from the Multi-Ethnic Study of Atherosclerosis. Int J Cardiol. (2018) 259:198–204. doi: 10.1016/j.ijcard.2018.01.046
92. Wang J.G, Aikawa M. Toll-like receptors and Src-family kinases in atherosclerosis – focus on macrophages. Circ J. (2015) 79:2332–4. doi: 10.1253/circj.CJ-15-1039
93. Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, Witztum JL, Bae YS, Miller YI. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ Res. (2009) 104:1355–63. doi: 10.1161/CIRCRESAHA.108.192880
94. Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation (2001) 104:3103–8. doi: 10.1161/hc5001.100631
95. Huang H, Koelle P, Fendler M, Schrottle A, Czihal M, Hoffmann U, Conrad M, Kuhlencordt P.J. Induction of inducible nitric oxide synthase (iNOS) expression by oxLDL inhibits macrophage derived foam cell migration. Atherosclerosis (2014) 235:213–22. doi: 10.1016/j.atherosclerosis.2014.04.020
96. van Dijk RA, Rijs K, Wezel A, Hamming JF, Kolodgie FD, Virmani R, Schaapherder AF, Lindeman JH. Systematic evaluation of the cellular innate immune response during the process of human atherosclerosis. J Am Heart Assoc. (2016) 5:e002860. doi: 10.1161/JAHA.115.002860
97. Perrotta I, Brunelli E, Sciangula A, Conforti F, Perrotta E, Tripepi S, Donato G, Cassese M. iNOS induction and PARP-1 activation in human atherosclerotic lesions: an immunohistochemical and ultrastructural approach. Cardiovas Pathol. (2011) 20:195–203. doi: 10.1016/j.carpath.2010.06.002
98. Li N, McLaren JE, Michael DR, Clement M, Fielding CA, Ramji DP. ERK is integral to the IFN-gamma-mediated activation of STAT1, the expression of key genes implicated in atherosclerosis, and the uptake of modified lipoproteins by human macrophages. J Immunol (2010) 185:3041–8. doi: 10.4049/jimmunol.1000993
99. McDermott DH, Yang Q, Kathiresan S, Cupples LA, Massaro JM, Keaney JF Jr, et al. CCL2 polymorphisms are associated with serum monocyte chemoattractant protein-1 levels and myocardial infarction in the Framingham Heart Study. Circulation (2005) 112:1113–20. doi: 10.1161/CIRCULATIONAHA.105.543579
100. Domschke G, Gleissner CA. CXCL4-induced macrophages in human atherosclerosis. Cytokine (2017). doi: 10.1016/j.cyto.2017.08.021. [Epub ahead of print].
101. Boles U, Johansson A, Wiklund U, Sharif Z, David S, McGrory S, Henein MY. Cytokine disturbances in coronary artery ectasia do not support atherosclerosis pathogenesis. Int J Mol Sci. (2018) 19:E260. doi: 10.3390/ijms19010260
102. Han X, Boisvert WA. Interleukin-10 protects against atherosclerosis by modulating multiple atherogenic macrophage function. Thromb Haemost. (2015) 113:505–12. doi: 10.1160/TH14-06-0509
103. Nakano M, Fukumoto Y, Satoh K, Ito Y, Kagaya Y, Ishii N, Sugamura K, Shimokawa H. OX40 ligand plays an important role in the development of atherosclerosis through vasa vasorum neovascularization. Cardiovasc Res. (2010) 88:539–46. doi: 10.1093/cvr/cvq211
104. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev. Cardiology (2015) 12:10–7. doi: 10.1038/nrcardio.2014.173
105. Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang MJ, He CH, et al. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Ann Rev Physiol. (2011) 73:10.1146. doi: 10.1146/annurev-physiol-012110-142250
106. Schwartz DR, Lazar MA. Human resistin: found in translation from mouse to man. Trends Endocrinol Metabol. (2011) 22:259–65. doi: 10.1016/j.tem.2011.03.005
107. Lehrke M, Reilly MP, Millington SC, Iqbal N, Rader DJ, Lazar MA. An inflammatory cascade leading to hyperresistinemia in humans. PLoS Med. (2004) 1:e45. doi: 10.1371/journal.pmed.0010045
108. Han L, Shen WJ, Bittner S, Kraemer FB, Azhar S. PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α. Fut Cardiol. (2017) 13:259–78. doi: 10.2217/fca-2016-0059
109. Lievens D, Eijgelaar WJ, Biessen EA, Daemen MJ, Lutgens E. The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb Haemost. (2009) 102:206–14. doi: 10.1160/TH09-01-0029
110. Dopheide FJ, Sester JU, Schlitt JA, Horstick JG, Rupprecht JH, Münzel JT, Blankenberg JS. Monocyte-derived dendritic cells of patients with coronary artery disease show an increased expression of costimulatory molecules CD40, CD80 and CD86 in vitro. Coron Art Dis. (2007) 18:523–31. doi: 10.1097/MCA.0b013e3282eff1ad
111. Yang J.H, Ren F. Clinical implications of tenascin-C and OX40 ligand in patients with acute coronary syndrome. Biomed Rep. (2014) 2:132–6. doi: 10.3892/br.2013.195
112. Jeon HJ, Choi JH, Jung IH, Park JG, Lee MR, Lee MN, et al. CD137 (4-1BB) deficiency reduces atherosclerosis in hyperlipidemic mice. Circulation (2010) 121:1124–33. doi: 10.1161/CIRCULATIONAHA.109.882704
113. Yu Y, He Y, Yang TT, Jiang H, Xiang YJ, Fang LB, et al. Elevated plasma levels and monocyte-associated expression of CD137 ligand in patients with acute atherothrombotic stroke. Eur Rev Med Pharmacol Sci. (2014) 18:1525–32. Available online at: https://www.europeanreview.org/article/7415
114. Goldie-Cregan LC, Croager EJ, Abraham LJ. Characterization of the murine CD30 ligand (CD153) gene: gene structure and expression. Tissue Antig. (2002) 60:139–46. doi: 10.1034/j.1399-0039.2002.600204.x
115. Boyle JJ. Association of coronary plaque rupture and atherosclerotic inflammation. J Pathol (1997) 181:93–9. doi: 10.1002/(SICI)1096-9896(199701)181:1<93::AID-PATH696>3.0.CO;2-H
116. Chen W, Wang J, Jia L, Liu J, Tian Y. Attenuation of the programmed cell death-1 pathway increases the M1 polarization of macrophages induced by zymosan. Cell Death Dis. (2016) 7:e2115. doi: 10.1038/cddis.2016.33
117. Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. (2007) 117:2974–82. doi: 10.1172/JCI31344
118. Bekkering S, van den Munckhof I, Nielen T, Lamfers E, Dinarello C, Rutten J, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis (2016) 254:228–36. doi: 10.1016/j.atherosclerosis.2016.10.019
119. Sarrazy V, Viaud M, Westerterp M, Ivanov S, Giorgetti-Peraldi S, Guinamard R, et al. Disruption of glut1 in hematopoietic stem cells prevents myelopoiesis and enhanced glucose flux in atheromatous plaques of ApoE(-/-) Mice. Circ Res. (2016) 118:1062–77. doi: 10.1161/CIRCRESAHA.115.307599
120. Lee SJ, Thien Quach CH, Jung KH, Paik JY, Lee JH, Park JW, et al. Oxidized low-density lipoprotein stimulates macrophage 18F-FDG uptake via hypoxia-inducible factor-1alpha activation through Nox2-dependent reactive oxygen species generation. J Nuc Med. (2014) 55:1699–705. doi: 10.2967/jnumed.114.139428
121. Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. 15-Deoxy-Δ12,14-prostaglandin J2 : a prostaglandin d2 metabolite generated during inflammatory processes. J Biol Chem. (2002) 277:10459–66. doi: 10.1074/jbc.M110314200
122. Gomez-Hernandez A, Sanchez-Galan E, Martin-Ventura JL, Vidal C, Blanco-Colio LM, Ortego M, et al. Atorvastatin reduces the expression of prostaglandin E2 receptors in human carotid atherosclerotic plaques and monocytic cells: potential implications for plaque stabilization. J Cardiovasc Pharmacol. (2006) 47:60–9. doi: 10.1097/01.fjc.0000194252.38683.68
123. Akagi D, Chen M, Toy R, Chatterjee A, Conte MS. Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J. (2015) 29:2504-2513. doi: 10.1096/fj.14-265363
124. Laguna-Fernandez A, Checa A, Carracedo M, Artiach G, Petri MH, Baumgartner R, et al. ERV1/ChemR23 signaling protects from atherosclerosis by modifying oxLDL Uptake and Phagocytosis in Macrophages. Circulation (2018). doi: 10.1161/CIRCULATIONAHA.117.032801. [Epub ahead of print].
125. Deng B, Wang CW, Arnardottir HH, Li Y, Cheng CY, Dalli J, Serhan C.N. Maresin biosynthesis and identification of maresin 2, a new anti-inflammatory and pro-resolving mediator from human macrophages. PloS One (2014) 9:e102362. doi: 10.1371/journal.pone.0102362
126. Bjerrum Ole J. New safe medicines faster: a proposal for a key action within the european union's 6th framework programme. Pharmacol Toxicol. (2003) 86:23–6. doi: 10.1034/j.1600-0773.86.s1.7.x
127. Petsko GA. When failure should be the option. BMC Biol. (2010) 8:61–1. doi: 10.1186/1741-7007-8-61
128. Munro TP, Mahler SM, Huang EP, Chin DY, Gray PP. Bridging the gap: facilities and technologies for development of early stage therapeutic mAb candidates. mAbs (2011) 3:440–52. doi: 10.4161/mabs.3.5.16968
129. Gaspar R, Aksu B, Cuine A, Danhof M, Takac MJ, Linden HH, et al. Towards a European strategy for medicines research (2014-2020): the EUFEPS position paper on Horizon 2020. Eur J Pharmaceut Sci. (2012) 47:979–87. doi: 10.1016/j.ejps.2012.09.020
130. Schuhmacher A, Gassmann O, Hinder M. Changing R&D models in research-based pharmaceutical companies. J Trans Med. (2016) 14:105. doi: 10.1186/s12967-016-0838-4
Keywords: macrophages, cardiovascular diseases, drug development, inflammation, cell metabolism
Citation: Decano JL and Aikawa M (2018) Dynamic Macrophages: Understanding Mechanisms of Activation as Guide to Therapy for Atherosclerotic Vascular Disease. Front. Cardiovasc. Med. 5:97. doi: 10.3389/fcvm.2018.00097
Received: 27 December 2017; Accepted: 02 July 2018;
Published: 03 August 2018.
Edited by:
Andrew Newby, University of Bristol, United KingdomReviewed by:
Cinzia Perrino, Università degli Studi di Napoli Federico II, ItalyJian Xu, University of Oklahoma Health Sciences Center, United States
Copyright © 2018 Decano and Aikawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masanori Aikawa, maikawa@bwh.harvard.edu