The Role of the TGF-β Superfamily in Myocardial Infarction
 Anis Hanna
Anis Hanna Nikolaos G. Frangogiannis
Nikolaos G. Frangogiannis- Department of Medicine (Cardiology), The Wilf Family Cardiovascular Research Institute, Albert Einstein College of Medicine, Bronx, NY, United States
The members of the transforming growth factor β (TGF-β) superfamily are essential regulators of cell differentiation, phenotype and function, and have been implicated in the pathogenesis of many diseases. Myocardial infarction is associated with induction of several members of the superfamily, including TGF-β1, TGF-β2, TGF-β3, bone morphogenetic protein (BMP)-2, BMP-4, BMP-10, growth differentiation factor (GDF)-8, GDF-11 and activin A. This manuscript reviews our current knowledge on the patterns and mechanisms of regulation and activation of TGF-β superfamily members in the infarcted heart, and discusses their cellular actions and downstream signaling mechanisms. In the infarcted heart, TGF-β isoforms modulate cardiomyocyte survival and hypertrophic responses, critically regulate immune cell function, activate fibroblasts, and stimulate a matrix-preserving program. BMP subfamily members have been suggested to exert both pro- and anti-inflammatory actions and may regulate fibrosis. Members of the GDF subfamily may also modulate survival and hypertrophy of cardiomyocytes and regulate inflammation. Important actions of TGF-β superfamily members may be mediated through activation of Smad-dependent or non-Smad pathways. The critical role of TGF-β signaling cascades in cardiac repair, remodeling, fibrosis, and regeneration may suggest attractive therapeutic targets for myocardial infarction patients. However, the pleiotropic, cell-specific, and context-dependent actions of TGF-β superfamily members pose major challenges in therapeutic translation.
Introduction
In human patients, myocardial infarction is typically caused by thrombotic occlusion of a coronary artery, leading to prolonged cessation of blood flow in the territory subserved by the vessel. Ischemia-induced disruption of aerobic metabolism in cardiomyocytes immediately perturbs their contractile function and ultimately results in catastrophic structural changes (1). After 20–30 min of ischemia, subendocardial cardiomyocytes in the area at risk exhibit irreversible changes. Longer ischemic intervals trigger a “wavefront” of cardiomyocyte death that ultimately involves the entire thickness of the ventricular wall supplied by the occluded coronary (2). Because the adult mammalian heart has negligible regenerative capacity, repair of the infarcted myocardium is dependent on formation of a collagen-based scar, and requires activation of a superbly orchestrated inflammatory/reparative response (3). Inflammatory chemokines and cytokines are critically involved in the reparative process, mediating activation of macrophages that clear the wound from dead cells and matrix debris, and triggering expansion of reparative cells (including fibroblasts and vascular cells) that form granulation tissue and secrete a collagen-based matrix network. Although scar formation protects the heart from catastrophic early complications (such as cardiac rupture), infarct healing is closely intertwined with adverse remodeling, a constellation of cellular events that involve both infarcted and non-infarcted myocardium, and lead to chamber dilation, exacerbating dysfunction.
The members of the TGF-β superfamily play important roles in cell survival, differentiation, proliferation, and function and have been implicated in regulation of inflammatory and reparative responses. Following myocardial infarction, induction, and activation of endogenous TGF-β signaling pathways has been suggested to modulate injury, regulate inflammation, and orchestrate repair (4). This review manuscript discusses our current knowledge on the role of TGF-β superfamily members in injury, repair, and remodeling of the infarcted heart.
The Members of the TGF-β Superfamily
In humans, the TGF-β superfamily is comprised of 33 members that can be subclassified into several subfamilies, including the TGF-βs (TGF-β1, -β2, and -β3), the bone morphogenetic proteins (BMPs), the Growth differentiation factors (GDFs), the activins, the inhibins, nodal, and anti-Mullerian hormone proteins. Although TGF-β1 remains the best-studied member of the superfamily in myocardial infarction, a growing body of evidence implicates several other members in regulation of cardiac repair and post-infarction remodeling.
The TGF-βs
TGF-β Expression in Normal Myocardium
High constitutive expression of TGF-β is noted in both embryonic and adult mammalian hearts, and is predominantly localized in cardiomyocytes, or bound to the extracellular matrix (5). TGF-βs play an important role in cardiac development; loss of TGF-β isoforms in neonatal mice is associated with abnormal cardiac development and defective valve formation (6, 7). In normal adult mammalian hearts, TGF-β is stored in a latent form; whether low-level constitutive TGF-β activity is required to support homeostatic cardiac function remains unknown. In vivo, targeting Smad4 (the common intracellular effector of responses triggered by many members of the TGF-β superfamily) in adult cardiomyocytes was associated with impaired ventricular function and with perturbations in ion channel gene expression (8). Moreover, in neonatal rat cardiomyocytes cultured on fibroblast matrix under serum-free conditions, exogenous TGF-β sustains spontaneous rhythmic beating (9). However, the in vivo relevance of this observation is unclear.
Induction and Cellular Origin of TGF-βs in the Infarcted Myocardium
Upregulation of TGF-βs is well-documented in both mouse and large animal models of myocardial infarction (5, 10–15). In a mouse model of reperfused myocardial infarction, TGF-β1 and TGF-β2 mRNA levels peak early after 6–72 h of reperfusion; in contrast, TGF-β3 exhibits a prolonged time course and is persistently upregulated after 7 days of reperfusion (10). The distinct time course of TGF-β3 may be due to cell-specific expression patterns, or may reflect isoform-specific effects of stimuli inducing transcription of TGF-βs.
Although most myocardial cell types are capable of synthesizing and releasing significant amounts of TGF-βs; their relative contribution remains poorly defined. In a porcine model of coronary occlusion, cardiomyocytes were a major source of TGF-β (11). On the other hand, studies in mouse models of myocardial infarction suggest that infarct macrophages may be key contributors to the TGF-β response (16). Genetic disruption of the chemokine monocyte chemoattractant protein (MCP)-1/CCL2, a mediator with a crucial role in recruitment of monocytes/macrophages in inflamed tissues, was associated with reduced TGF-β2 and TGF-β3 mRNA expression following myocardial infarction, consistent with an important role for infiltrating mononuclear cells in TGF-β synthesis in the infarcted heart (17). TGF-β synthesis may mark specific subsets of infarct macrophages that respond to cytokine stimulation in the pro-inflammatory environment of the infarct (18). In addition to cardiomyocytes and macrophages, several other cell types may also secrete TGF-βs in the infarcted heart; however, robust documentation of their contribution is lacking. Platelets constitutively express significant amounts of growth factors and have been suggested to be an important source of TGF-β1 in the pressure-overloaded myocardium (19). Abundant platelets infiltrate the infarct during the early stages of repair; however, their relative role as a source of TGF-βs has not been investigated. Activated fibroblasts, vascular cells, mast cells and lymphocyte subsets can produce and secrete TGF-βs, and are found in significant numbers in infarcted hearts (20–23). However, whether they are a significant source of TGF-β isoforms following infarction remains unknown.
TGF-β Activation in the Infarcted Myocardium
In the infarcted myocardium, activated cells enrich the existing stores of latent TGF-β through de novo synthesis of all 3 isoforms. However, this is not sufficient for activation of TGF-β cascades. Transduction of TGF-β signals in sites of injury requires liberation of the active TGF-β dimer from the latent complexes. TGF-βs are secreted in a latent form that is comprised of the TGF-β dimer, the latency-associated peptide (LAP), which confers latency to TGF-β (24, 25), and a latent TGF-β-binding protein (LTBP), which serves to sequester the complex into the extracellular matrix (26). Extensive data from animal models suggest that myocardial infarction is associated with rapid activation of TGF-β (27), followed by stimulation of downstream Smad-dependent signaling cascades (28). However, the molecular signals that trigger TGF-β activation following infarction remain poorly understood. Several mechanisms may be involved. First, cell surface integrins may interact with the LAP, thus releasing the TGF-β dimer (29). Although in vitro studies have implicated αvβ5 and αvβ3 integrins in latent TGF-β activation and in subsequent myofibroblast conversion (30); the in vivo significance of these interactions in the infarcted myocardium remains unknown. Second, a wide range of proteases (including serine proteases, cathepsins, matrix metalloproteinases, and cysteine proteases) have been implicated in TGF-β activation following injury. Proteases are rapidly activated following myocardial infarction (31–33); however, their potential role in activation of TGF-β in infarcted hearts remains poorly documented. Third, reactive oxygen species (ROS) are rapidly generated in ischemic hearts and may be involved in activation of TGF-β activation in the infarcted myocardium. Fourth, specialized extracellular matrix proteins (including matricellular proteins, such as thrombospondin-1), are markedly upregulated in the infarcted heart (34–36) and may play an important role in TGF-β activation. Thrombospondin-1 has been suggested to interact with the LAP, promoting release of the active TGF-β dimer from the latent complex (37). In the infarcted myocardium, TSP-1 upregulation is associated with activation of TGF-β signaling in the infarct border zone in both mouse and canine infarcts (34). Finally, an acidic environment has been suggested to promote TGF-β activation (38). Generation of lactic acid in the ischemic myocardium may reduce the pH, contributing to activation of latent TGF-β stores following infarction.
Cellular Actions of TGF-β in the Infarcted Myocardium (Figure 1)
TGF-β1, β2, and β3 are activated in the infarcted myocardium and may regulate phenotype and function of all cell types involved in cardiac injury and repair. Although the 3 TGF-β isoforms exhibit distinct patterns of regulation following infarction, little is known regarding isoform-specific actions in the infarcted heart. Most in vitro studies have investigated TGF-β1-mediated actions. In vivo experiments on the other hand, have focused on the role of TGF-β receptor-activated signaling, exploring pathways common to all three isoforms and to other members of the TGF-β superfamily.
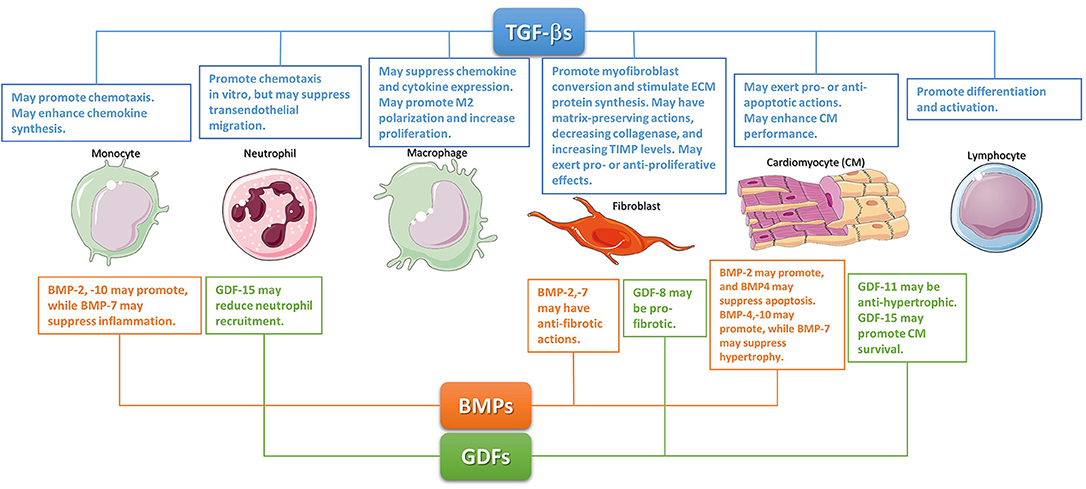
Figure 1. The diverse cellular actions of the TGF-βs, BMPs, and GDFs in the infarcted myocardium. During the inflammatory phase of infarct healing, TGF-βs may regulate cardiomyocyte survival, promote monocyte chemotaxis, and modulate lymphocyte differentiation and activation. TGF-βs may be critical in modulating macrophage phenotype toward an anti-inflammatory M2 phenotype. These effects may act as a switch from inflammation to repair. The effects of BMPs and GDFs have not been systematically studied; however, several members of these subfamilies may regulate inflammation. During the reparative phase, TGF-βs are critical regulators of fibroblast activation. Although studies in cardiac fibroblasts suggested TGF-βs can have either pro- or anti- proliferative effects, TGF-βs have consistent effects on fibroblast activation, mediating myofibroblast conversion and stimulating exctracellular matrix protein synthesis. Moreover, TGF-βs also promote a matrix-preserving program by decreasing collagenase synthesis, and by upregulating tissue inhibitors of metalloproteinases (TIMPs). Members of the GDF and BMP subfamilies may also regulate phenotype and function of reparative cells. The cartoon was designed using Servier Medical Art (https://smart.servier.com/).
Effects of TGF-β on Ischemic Cardiomyocytes
Early studies in isolated perfused hearts undergoing brief myocardial ischemia followed by reperfusion, suggested protective effects of TGF-β, mediated through attenuation of oxidative stress and reduced release of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α (39). Ex vivo and in vitro experiments examining the effects of TGF-β on cardiomyocyte survival have produced conflicting results. In a model of reperfused feline myocardial infarction, TGF-β1 administration reduced cardiomyocyte death, and attenuated neutrophil recruitment in the infarcted myocardium (40). It is unclear whether TGF-β-mediated protection in these studies was due to activation of direct pro-survival effects in cardiomyocytes, or reflected suppression of injurious inflammatory signaling. TGF-β1 infusion in isolated perfused hearts, protected cardiomyocytes from apoptosis through actions involving p42/p44 mitogen-activated protein kinase (MAPK) signaling during early reperfusion (41). In contrast, in rat cardiomyocytes, the pro-apoptotic effects of angiotensin II were attributed to activation of TGF-β signaling (42). Moreover, in vitro studies have suggested direct actions of TGF-β on cardiomyocyte function, mediated through upregulation of the laminin receptor 37/67 (43). Considering its notoriously pleiotropic and context-dependent actions, the effects of exogenous TGF-β administration are likely dose- and time-dependent, and are affected by the experimental conditions.
In vivo, mouse models of cardiomyocyte-specific TGF–β receptor disruption have been used to investigate endogenous effects of TGF–βs on cardiomyocytes (44), In a model of non-reperfused myocardial infarction, mice with cardiomyocyte-specific loss of the type 1 or type 2 TGF-β receptor were protected from death due to cardiac rupture, exhibiting marked reduction in neutrophil infiltration. The mechanism of protection remains poorly defined. It was suggested that TGF-β signaling in cardiomyocytes may suppress synthesis of cardioprotective genes, such as interleukin (IL)-33, GDF-15, and TSP-4.
TGF-β and the Post-Infarction Inflammatory Reaction
TGF-βs are critical modulators of immune cell phenotype and function (45). In vitro, TGF-βs can exert pro- or anti-inflammatory depending on the phenotype and the state of differentiation of the cells, and the presence or absence of other mediators. TGF-β is a potent neutrophil (46) and monocyte (47) chemoattractant; these effects may be important for recruitment of leukocytes in inflamed tissues. Despite exhibiting strong chemotactic actions for monocytes and neutrophils in single-cell assays, TGF-β may attenuate transendothelial leukocyte migration by reducing surface expression of adhesion molecules (48). Moreover, in monocytes picomolar concentrations of TGF-β stimulate synthesis of pro-inflammatory cytokines and chemokines (47, 49). In contrast to its pro-inflammatory actions in monocytes, TGF-β is known to deactivate macrophages, suppressing expression of chemokines (such as MCP-1/CCL2) and cytokines, (including IL-1β and TNF-α) (50–52). Whether TGF–β acts as a pro- or anti-inflammatory mediator in vivo is dependent on the context: the cell types involved, their phenotypic profile, and the cytokine environment. Thus, attribution of pro- or anti-inflammatory properties to TGF-βs should be based on documentation of robust in vivo effects, rather than on in vitro assays.
In vivo, TGF-β1 plays an essential role in immune cell homeostasis by preventing spontaneous inflammation in mammalian tissues. 50% of TGF-β1 null mice develop massive multi-organ inflammation 2–4 weeks after birth, mainly affecting the heart and lungs (53). The anti-inflammatory effects of TGF-β in normal tissues may involve suppressive actions on T cell inflammatory activity (54).
In the healing infarct, timely recruitment of inflammatory leukocytes, and subsequent activation of reparative macrophages play a crucial role in repair, but may also mediate fibrotic responses that promote adverse remodeling (3, 55, 56). TGF–β may critically regulate inflammatory leukocyte recruitment and function following myocardial infarction. Ex vivo studies using cardiac lymph collected following reperfused infarction as a window to the cardiac microenvironment suggested that TGF-β activity may be implicated in recruitment of inflammatory monocytes in the healing infarct (27). In vivo experiments have suggested anti-inflammatory actions of TGF-β following infarction. Early systemic inhibition of TGF-β signaling through transfection with the extracellular domain of TβRII increased mortality in a model of non-reperfused infarction, accentuating neutrophil recruitment, and inducing expression of cytokines and chemokines (57). The specific effects of TGF-β signaling on the phenotype of immune cell subpopulations infiltrating the infarcted myocardium have not been systematically studied. TGF-β may be a critical modulator of macrophage phenotype, promoting M2 polarization (58) and suppressing expression of inflammatory cytokines and chemokines (58, 59), enhancing macrophage-colony stimulating factor (M-CSF)-induced proliferation (60), inhibiting nitrite release (61), and reducing cytotoxic activity (62). TGF-β also acts as a central mediator in T lymphocyte differentiation and activation, critically regulating phenotype and function of all subpopulations (63–65). CD4+ T cells (66) and foxp3+ regulatory T cells (Tregs) (20–22), have been implicated in regulation of repair and remodeling following myocardial infarction Whether TGF-β signaling cascades play a crucial role in differentiation and phenotypic modulation of lymphocytes in the infarcted heart remains unknown.
Effects of TGF-β on Fibroblast Phenotype and Function
During the reparative phase of cardiac repair, resident cardiac fibroblast populations become activated (67, 68), undergoing myofibroblast conversion, and secreting large amounts of structural extracellular matrix proteins (69–73). Activated infarct fibroblasts also produce cytokines, growth factors, and matricellular proteins, and participate in matrix metabolism through secretion of MMPs. TGF-β plays a crucial role in expansion of the myofibroblast population in the healing infarct and stimulates expression of extracellular matrix proteins. TGF-β potently induces expression of α-smooth muscle actin in cardiac fibroblasts, the hallmark of myofibroblast conversion (74, 75). Moreover, TGF-β significantly and consistently stimulates synthesis and secretion of extracellular matrix proteins (such as collagen I, collagen III, and fibronectin) (76, 77), and promotes a matrix-preserving phenotype by suppressing collagenase expression, and by accentuating synthesis of tissue inhibitor of metalloproteinases (TIMPs) (28, 78, 79). Whether TGF-β stimulates fibroblast proliferation in the infarcted heart is less clear. In vitro studies have demonstrated both proliferative and anti-proliferative effects of TGF-β on cardiac fibroblasts (77, 80). Differences in TGF–β concentration, distinct phenotypic profiles, or states of differentiation of fibroblast populations, and differences in the growth factor and extracellular matrix environment may account for the conflicting observations.
Although genetic interventions to study the in vivo effects of fibroblast-specific TGF-β signaling in infarcted hearts have not been performed, a large body of evidence suggests that TGF-β may play a critical role in regulation of fibroblast phenotype in healing infarcts. In a model of non-reperfused infarction, TGF-β inhibition through administration of a neutralizing antibody had detrimental effects, accentuating chamber dilation, increasing myocardial MMP expression, and reducing collagen synthesis (81). Moreover, two independent investigations inhibiting TGF-β through gene transfer of the extracellular domain of TβRII suggested that TGF-β may play a crucial role in fibrosis of the infarcted heart (57, 82). TGF-β inhibition after the inflammatory phase of cardiac repair attenuated deposition of fibrous tissue in the infarcted region (82). The observed effects may reflect, at least in part, loss of TGF-β signaling in cardiac fibroblasts.
Effects of TGF-β on Endothelial Cell Phenotype and on Infarct Angiogenesis
Endothelial cells, the most abundant non-cardiomyocytes in the adult mammalian myocardium (83), play an important role in regulation of the post-infarction inflammatory response and in repair of the infarcted heart. During the inflammatory phase of infarct healing, endothelial cells undergo activation, producing large amounts of chemokines (84, 85), and expressing adhesion molecules. Stimulated endothelial cells interact with activated circulating leukocytes promoting their recruitment in the healing infarct (86). During the proliferative phase, angiogenic activation of endothelial cells (87) may be important for generation of neovessels that supply the healing tissue with oxygen and nutrients. Finally, during the maturation phase, endothelial cells may recruit mural cells, by producing platelet-derived growth factor-BB (88, 89); thus restraining angiogenesis, suppressing inflammation and contributing to formation of a mature scar (90).
Although both endothelial cells and pericytes are highly responsive to TGF-β (91), the role of TGF-β-mediated actions on the infarct microvasculature remains unknown. It has been suggested that suppressive effects of TGF-β on endothelial cell chemokine synthesis may contribute to suppression and resolution of the post-infarction inflammatory response (92). TGF-β may also be implicated in regulation of infarct angiogenesis and may contribute to vascular maturation by modulating interactions between endothelial cells and mural cells. It should be emphasized that TGF-β actions on endothelial cells may be either angiogenic, or angiostatic, depending on contextual factors (91, 93).
TGF-β and Cardiac Regeneration
The adult mammalian myocardium has extremely limited regenerative capacity that is overwhelmed by the massive loss of cardiomyocytes following myocardial infarction. Accentuation of the regenerative capacity of the heart in order to remuscularize the infarcted myocardium is a major visionary goal in cardiovascular research. Experimental evidence suggests a broad and context-dependent role for TGF-β in cardiac regeneration. TGF-β may regulate cardiomyocyte differentiation in vitro (94), may be involved in endogenous remuscularization in regenerative models of cardiac injury (95), and may modulate regenerative responses to cell therapy following myocardial infarction (96). An in vitro study showed that TGF-β stimulation increases expression of cardiac transcription factors in murine embryonic stem cells driving cardiomyocyte differentiation (97). In contrast, in other in vitro experiments, a highly selective TGF-β inhibitor that promotes proteasomal degradation of TβRII enhanced differentiation of uncommitted mesoderm to cardiomyocytes (98). In vivo studies in fish models have supported the involvement of TGF-β in regulation of endogenous cardiac regeneration. In zebrafish, pharmacologic inhibition of TβRI abolished heart regeneration following cryoinjury (95). In contrast, a pharmacological inhibitor of the type I TGFβ receptor was found to enhance proliferation of Nkx2.5+ cardiomyoblasts (99), and improved endogenous cardiomyoblast-mediated regeneration in mice (96). TGF-β has also been found to modulate the effects of cell therapy. In a mouse model of myocardial infarction, intramyocardial implantation of TGF-β pre-programmed CD117+ stem cells was reported to promote regeneration and stimulate angiogenesis (100). The conflicting findings from various studies likely reflect the differences between experimental models and the context-dependent actions of TGF-β that typically depend on the state of differentiation of target cells. Considering that high levels of active TGF-β in the infarcted mammalian myocardium are not associated with any significant regenerative activity, it is highly unlikely that stimulation of the TGF-β cascade may be sufficient to promote remuscularization following cardiac injury. However, through its broad and pronounced effects on differentiation of many cell types, TGF-β may modulate phenotype and function of myocardial progenitor cells, thus critically regulating their regenerative capacity.
The BMP/GDF Subfamily
BMPs
BMPs are a group of evolutionarily conserved, structurally related secreted cytokines with an essential role in development and in the pathogenesis of many pathologic conditions (101). Although the name reflects the well-documented osteo-inductive properties of many members of the group, BMPs are pleiotropic and multifunctional, exerting a wide range of actions in many different cell types (102). Development of genetic loss of function models has documented the role of several BMPs in cardiac development (103, 104). Unfortunately, the potential involvement of BMPs in repair, remodeling and fibrosis of the infarcted heart remains poorly investigated.
BMP2
In vitro, BMP2 induces differentiation of cardiac progenitor cells into mature cardiomyocytes, stimulating expression of cardiac-specific genes (105). Knockout models in mice have demonstrated that BMP2 is critically involved in cardiac development (106), coordinating atrioventricular canal morphogenesis (107). Whether BMP2 plays a role in homeostatic function of the adult heart remains unclear. In vitro, BMP2 was found to stimulate contraction in adult rat cardiomyocytes, through activation of PI-3K (108).
Following myocardial infarction, BMP2 shows an early time course of upregulation. In a model of non-reperfused myocardial infarction, transient early induction of BMP2 was followed by increased expression of other BMPs (109). BMP2 expression in infarcted hearts was localized in both cardiomyocytes and interstitial fibroblasts (110, 111); however, the mechanism of upregulation remains unknown. Very little is known regarding the function of endogenous BMP-2 in myocardial infarction. Accentuation of post-infarction inflammation in mice lacking the BMP inhibitor gremlin (109) may suggest important pro-inflammatory actions of BMPs. Whether neutrophils, macrophages and lymphocytes, essential effector cells of post-infarction inflammation, are major targets of BMP2 is unknown. Moreover, BMP2 may exert cytoprotective actions on cardiomyocytes. In vitro, BMP2 inhibits apoptosis of serum-deprived cardiomyocytes (112). In mice undergoing permanent coronary occlusion protocols, intravenous BMP-2 injection attenuated apoptosis of infarct border zone cardiomyocytes, reducing the size of the infarct (113). Although inhibitory effects of BMP-2 on fibroblast function have been reported (114), the significance of such actions in repair following myocardial infarction and in cardiac fibrosis remains unclear. Angiogenic effects of BMP2 have been documented in vitro and in vivo (115); however, the potential role of BMP2 as a regulator of neovessel formation in healing myocardial infarction has not been investigated (Figure 1).
BMP4
Much like BMP2, BMP4 also promotes cardiomyocyte differentiation of cardiac progenitor cells (116, 117) Defective atrioventricular septation in BMP4 KO mice suggests a critical role for BMP4 in cardiac development (118). Adult human and mouse hearts exhibit constitutive BMP4 expression that increases following myocardial infarction (109, 119). The molecular mechanisms and predominant cellular sources responsible for BMP4 upregulation following infarction remain unknown. Limited information is available on the role of BMP4 following myocardial infarction. In vitro BMP4 has been suggested to promote apoptosis in neonatal rat cardiomyocytes (120). In vivo, in a model of ischemia/reperfusion, heterozygous BMP4 +/– null mice had reduced infarct size, and attenuated cardiomyocyte apoptosis (121). BMP4 may also stimulate a hypertrophic response in cardiomyocytes of the infarct border zone or the remote remodeling myocardial areas (122). In addition to effects on cardiomyocytes, BMP4 may also modulate inflammatory, fibrogenic and angiogenic responses. In vitro studies using endothelial cells and macrophages have suggested both pro- and anti-inflammatory actions of BMP4 (123, 124). BMP4 also stimulates angiogenic activation of endothelial cells through a Smad1/5-mediated pathway (125). Unfortunately, experimental studies exploring the role of these BMP4-mediated actions in myocardial infarction have not been performed (Figure 1).
BMP6
BMP6 is critically involved in iron homeostasis. Targeted disruption of BMP6 induces rapid and massive deposition of iron in many organs, including the heart (126). Iron overload in BMP6 null mice is associated with marked reduction of hepcidin (126), a key regulator of iron transport. BMP6 is expressed in developing hearts and mice with combined BMP6 and BMP7 loss exhibit a delay in formation of the outflow tract endocardial cushions (127). Although a clinical study showed increased circulating levels of BMP6 in patients with advanced heart failure (128), information on the regulation and role of BMP6 in the infarcted myocardium is lacking.
BMP7
Although BMP7 is highly expressed in the developing myocardium (129), BMP7 loss does not affect cardiac development, and defects in BMP7 null mice are confined to the kidney and the eye (130). A systematic study of myocardial BMP expression in non-reperfused myocardial infarcts found no significant upregulation of BMP7 mRNA expression 1–21 days following coronary occlusion, but suggested trends toward reduced levels at early timepoints (109). BMP7 exerts important actions on fibroblasts, cardiomyocytes and macrophages. Anti-fibrotic effects of BMP7 have been reported in many systems and may be mediated, at least in part, through suppression of collagen synthesis by cardiac fibroblasts (131), and through inhibition of endothelial to mesenchymal transition (EndMT) (132). In cardiomyocytes, BMP7 attenuates hypertrophy by inhibiting TGF-β responses (131). In macrophages, BMP7 has been reported to promote M2 polarization (133). Considering the absence of endogenous BMP7 induction in infarcted and remodeling hearts, administration of exogenous BMP7 has been suggested as a potential strategy to attenuate fibrosis and adverse remodeling. In a rat model of non-reperfused infarction systemic administration of BMP7 was reported to reduce mortality and attenuate ventricular dysfunction (134). Although the protective effects of exogenous BMP7 were attributed to anti-fibrotic actions; dissection of the cellular targets and molecular mechanisms was not performed.
BMP10
In the developing heart, BMP10 is regulated by myocardin and plays an important role in cardiomyocyte proliferation and chamber maturation (135, 136). In the postnatal heart, BMP10 expression seems to be restricted to the right atrium (137). Experiments in a non-reperfused mouse model of myocardial infarction showed a 2-fold increase in BMP10 levels 21 days after coronary occlusion. The cellular source of BMP10 in the infarcted heart, and the basis for the delayed pattern of induction remain unknown. Moreover, the effects of endogenous BMP10 in repair and remodeling of the infarcted heart have not been investigated. In vitro assays demonstrated that BMP10 enhances TNF-α-driven monocyte recruitment to the vascular endothelium promoting inflammation (138). However, considering the late time course of induction in the healing infarct, pro-inflammatory effects of BMP10 are unlikely to play an important role in the infarcted heart. BMP10 has been also reported to exert pro-hypertrophic actions in cardiomyocytes through an interaction with titin-cap (Tcap) (139). A study in a rat model of myocardial infarction suggested that exogenous BMP10 administration may stimulate cardiomyocyte cell cycle reentry (140). Considering the negligible proliferative activity of cardiomyocytes in infarcted and remodeling hearts, despite increased BMP10 levels and the challenges in robust documentation of proliferating cardiomyocytes, the significance of these findings in cardiac repair is unclear.
GDFs
Growth/differentiation factors (GDF1 to 15) are produced as inactive precursor proteins, which are then cleaved and assembled into active secreted homodimers. GDF dimers are disulfide-linked ligands that bind to a diverse array of receptors. (141, 142).
GDF8
GDF8 (also known as myostatin) is a critical negative regulator of growth and mass of skeletal and cardiac muscle, and plays an important role in energy homeostasis in adult cardiomyocytes. Cardiomyocyte-specific deletion of GDF8 in adult mice is associated with cardiomyocyte hypertrophy and heart failure, by promoting AMP-activated kinase activation (141). Following infarction, rapid upregulation of GDF8 has been reported within minutes after coronary ischemia (143). Surviving cardiomyocytes in the border zone of the infarct have been suggested as the main cellular source of GDF8 in infarcted and remodeling hearts (142). The functional role and cellular targets of GDF8 in the infarcted heart remain poorly understood. A study using a global loss-of-function model GDF8 loss may protect the infarcted heart, attenuating late dysfunction. The protective effects of GDF8 loss were attributed to attenuated fibrosis (144). The findings of this study are difficult to interpret, considering the baseline effects of myostatin loss on the myocardium, and the challenges in identifying cell-specific mechanisms. Pro-fibrotic actions of GDF8 in the myocardium may involve direct activation of fibroblasts (145–147), or effects on other cell types such as cardiomyocytes and macrophages.
GDF11
Publication of a high impact study suggesting that GDF11 levels decline with aging and that GDF11 administration may reverse age-related hypertrophy (148) (Figure 1) generated great interest in the potential role of this approach as therapy to reverse aging in many tissues (149). Subsequent studies have challenged the notion that GDF11 can rejuvenate the aging heart (150). Thus, additional work is necessary to settle this controversy. Although effects of GDF11 on hypertrophic responses have been suggested in models of left ventricular pressure overload (151), the regulation, cellular effects and actions of endogenous GDF11 in the infarcted myocardium have not been systematically investigated. Exogenous delivery of GDF11 was found to be protective in mouse and rat models of myocardial ischemia/reperfusion. (152, 153) Beneficial effects of GDF11 were attributed to improved angiogenesis and homing of endothelial progenitors (152), or to attenuated oxidative stress (153).
GDF15
GDF-15 is an inflammation-associated hormone with an important protective role in a wide range of infectious and inflammatory conditions (154, 155). In the mammalian myocardium, GDF15 is secreted by cardiomyocytes and may regulate body growth. In pediatric patients with heart disease, increased secretion of GDF15 negatively regulates body growth through effects on liver-derived growth hormone signaling (156). GDF15 expression is rapidly increased following myocardial ischemia and is predominantly localized in cardiomyocytes (157). Extensive experimental evidence suggests a crucial role for GDF15 in regulation of cardiomyocyte survival, post-infarction inflammation, repair, and remodeling of the infarcted heart. In a model of reperfused myocardial infarction, GDF15 exerted pro-survival actions on cardiomyocytes, reducing the size of the infarct (157). Moreover, in a model of non-reperfused infarction, GDF15 protected from cardiac rupture, restraining neutrophil recruitment through suppression of leukocyte integrin activation (155). Clinical studies have suggested that serum GDF15 levels may improve risk stratification in both STEMI and non-STEMI patients, providing important prognostic information beyond established clinical and biochemical markers (158–160) (Figure 1).
The Inhibin/Activin Subfamily and the Role of the Follistatins
Activins are dimeric polypeptides composed of two β subunits of inhibin linked by a disulfide bridge (161). Five types of β subunits have been described (βA, βB, βC, βD, and βE); activin nomenclature is defined by the type of subunits that form the dimer. In mammals, activins A (βA/βA), B (βB/βB), AB (βA/βB), C (βC/βC), and E (βE/βE) have been isolated; however, only activins A, B and AB have been studied and are known to exhibit biological activity (162).
In a rat model of non-reperfused myocardial infarction activin βA was markedly upregulated after 2 days of coronary occlusion and remained elevated for at least 28 days. Activin A protein was predominantly localized in surviving cardiomyocytes (163). A very rapid upregulation of activin A was reported in a model of reperfused myocardial infarction, occurring as early as 30 min after coronary ischemia. Early induction of activin A was dependent on TLR4 signaling (164). Overexpression experiments in vivo and in vitro suggested that activin A may protect ischemic cardiomyocytes from apoptosis (165). Activin A may also activate fibroblasts, promoting cell proliferation and collagen synthesis through p38 and Erk MAPK pathways (166).
The activin system is regulated by follistatin and follistatin-like 3 (Fstl3), extracellular proteins that bind activins with high affinity to form inactive complexes. In a rat model of myocardial infarction, administration of exogenous follistatin was reported to attenuate inflammation (167). Endogenous Flstl3 is upregulated following myocardial infarction and attenuates the cardioprotective effects of activin A (165). Cardiomyocyte-derived Flstl3 may also exert hypertrophic actions (168) and may stimulate fibroblasts in a paracrine manner, promoting a fibrogenic phenotype (169). It should be emphasized that Flstl3 actions in vivo may be, at least in part, independent of activin inhibition. Activin actions may also be relevant in human ischemic heart disease. Patients with both ischemic and non-ischemic heart failure had elevated circulating levels of activin A (163).
Signaling Pathways Activated by the TGF-β Superfamily in the Infarcted Myocardium (Figure 2)
The members of the TGF-β superfamily signal through binding to dual specificity kinase receptors (that have characteristics of both serine/threonine and tyrosine kinases) on the cell surface. Binding of a TGF-β family member assembles a heterotetrameric complex of 2 type 1 and 2 type II TGF-β receptors (170, 171). In humans, there are 7 type I receptors, termed activin-like receptor kinase (ALK)1-7, and 5 type II receptors; each member of the TGF-β superfamily signals by binding to characteristic combinations of these receptors. TGF-β isoforms transduce signaling through a single type II receptor (TβRII); in contrast, most BMPs are more promiscuous binding to three different type II receptors (ActRII, ActRIIB, and BMPRII). Binding of TGF-βs superfamily members to the type II receptor promotes oligomerization and transphoshorylates a type I receptor kinase. Activation of specific type I receptors accounts for part of the cellular specificity of TGF-β actions. For example, in most cell types TGF-βs activate the ubiquitously expressed type I receptor ALK5 (172). In endothelial cells, TGF-βs exerts distinct effects through another type I TGF-β receptor, ALK1 (173). Subsequently, activated type I receptors phosphorylate intracellular transcriptional effectors, the receptor-activated Smads (R-Smads). TGF-β-induced ALK5 activation, or activin-mediated ALK4 stimulation typically trigger phosphorylation of Smad2 and Smad3. TGF-β-induced ALK1 activation or BMP signaling generally phosphorylate Smad1, Smad5, and Smad8. Activated R-Smads form complexes with the common Smad, Smad4, and translocate to the nucleus, where they recruit co-activators or co-repression of transcriptional complexes and modulate gene transcription (174). In addition to their effects mediated through activation of Smads, the members of the TGF-β superfamily also signal through non-Smad pathways, by activating mitogen-activated protein kinase (MAPK) family responses, PI3K/Akt, focal adhesion kinase (FAK), and Rho GTPase pathways (175, 176). The multiple interactions between signaling responses of TGF-β superfamily members and other cascades (such as Wnt and Notch signaling) (177) generate additional layers of complexity that may explain, at least in part, the context-dependent actions of TGF-βs.
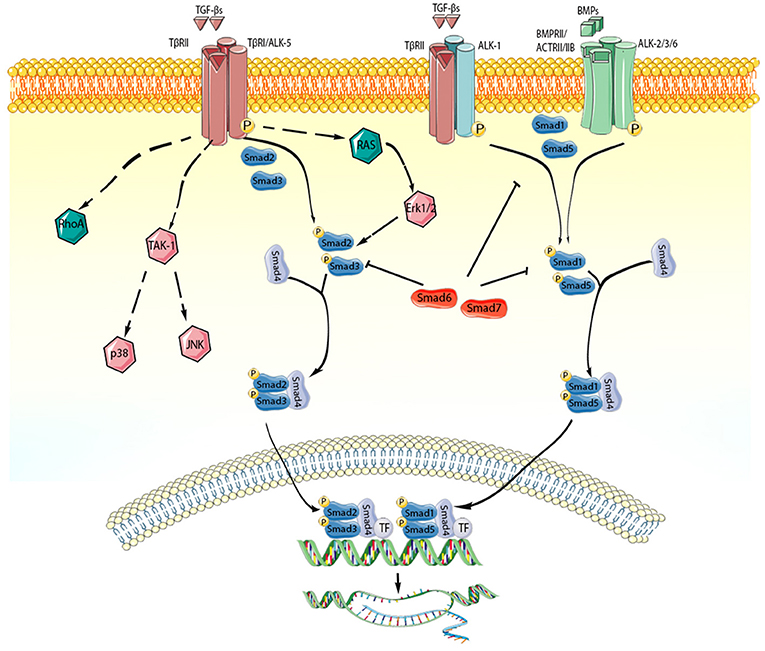
Figure 2. TGF-β superfamily members act through activation of Smad-dependent and Smad-independent signaling pathways. Each member of the TGF-β superfamily signals by binding to distinct combinations of type II, and type I receptors. TGF-β dimers bind TβRII inducing transphosphorylation of the GS segments in the type I receptor (ALK-5). Activated ALK-5 phosphorylates the receptor-activated Smads (R-Smads) Smad2 and Smad3, which then form a heterotrimeric complex with the common Smad, Smad4, promoting the translocation of the Smad complex to the nucleus. Interactions between the Smad and transcriptional activators or repressors regulate transcription of target genes. In some cell types (such as endothelial cells), TGF-βs may act through the type I receptor ALK1, stimulating Smad1/5/8 signaling. BMP dimers on the other hand bind their specific type II receptors, subsequently activating type I receptors (ALK-2,-3,-6) and phosphorylating Smad1/5. Smad1 and Smad5 then bind to Smad4 and translocate to the nucleus regulating transcription. In addition, TGF-β superfamily members signal through non-canonical Smad-independent cascades. The inhibitory Smads (Smad6 and Smad7) negatively regulate TGF-β superfamily cascades. The cartoon was designed using Servier Medical Art (https://smart.servier.com/).
Smad-Dependent Signaling in Myocardial Infarction
The ALK5-Smad2/3 Cascade in the Infarcted Heart
Activation of Smad2 and Smad3 signaling is consistently noted in infarcted hearts, and is localized in border zone cardiomyocytes, fibroblasts, and immune cells (13, 28, 178–180). ALK5 inhibition attenuated Smad2 activation in healing infarcts (181). However, the relative contribution of various TGF-β superfamily members in activating Smad2 and Smad3 remains unclear. In vivo experiments using cell-specific knockout strategies suggested that Smad3 signaling exerts important actions in regulating cardiomyocyte, fibroblast, and macrophage function following myocardial infarction. In cardiomyocytes, Smad3 activation increased apoptosis, enhancing nitrosative stress, and increasing expression of MMPs in the remodeling myocardium (178). On the other hand, activation of Smad3 in myofibroblasts protected the infarcted heart from late rupture, and played a major role in repair of the infarct, promoting formation of organized arrays of reparative myofibroblasts in the border zone, through activation of an integrin-ROS axis (178). Macrophage-specific Smad3 activated a phagocytic program and played a major role in transition of macrophages to an anti-inflammatory phenotype (179). In contrast, Smad2 activation may have more subtle and limited effects in cardiac repair. Myofibroblast-specific loss of Smad2 was associated with a transient improvement in post-infarction remodeling, without affecting mortality, scar organization, or long-term outcome (180).
The Role of the Smad1/5/8 Cascade in Myocardial Infarction
Although the Smad1 cascade is also activated in the ischemic and infarcted heart (182–184) relatively little is known regarding its role in regulation of phenotype and function of surviving cardiomyocytes, immune cells, fibroblasts, and vascular cells. Whether Smad1 activation in infarcts is mediated predominantly through BMPs, or reflects effects of TGF-βs on cells expressing ALK1 is unknown. Cardiomyocyte-specific overexpression of Smad1was found to be protective, reducing infarct size in a model of ischemia/reperfusion, by attenuating activation of a pro-apoptotic program (182). However, the potential role of endogenous Smad1 activation in regulation of cardiac injury, repair, and remodeling has not been investigated.
Activation of Smad-Independent Pathways in the Infarcted Heart
Although the members of the TGF-β superfamily are known to activate a wide range of non-canonical cascades, (including MAPK, TAK-1, and Rho GTPase) (177, 185, 186), the relative contribution of these pathways in myocardial infarction remains unknown. In vitro experiments and associative data have suggested that activation of a TAK-1/p38 MAPK axis by TGF-β may be involved in hypertrophic remodeling of surviving myocardium in the infarcted heart (187). In cardiac fibroblasts, stimulation of p38 MAPK may play an important role in fibrogenic activation (188) and may mediate hypertrophy (189). However, considering the broad range of mediators that signal through MAPKs, the role of TGF-β-driven activation of p38 in the infarcted heart is unclear.
Targeting the TGF-β Superfamily in Myocardial Infarction
Considering the critical role of TGF-β signaling cascades in tissue repair, remodeling, fibrosis and regeneration, the members of the TGF-β superfamily have been considered attractive therapeutic targets for patients with myocardial infarction (190). Unfortunately, the pleiotropic and context-dependent actions of TGF-βs, the unique and complex biology of TGF-β activation, and the complexity of the signaling cascades activated by the members of the superfamily pose major challenges in therapeutic translation. Clearly, activation of TGF-β signaling affects phenotype and function of all cells involved in cardiac repair, exerting both protective and detrimental effects. Our current knowledge of the cell-specific actions of TGF-βs in vivo is not sufficient to design therapy. Moreover, the distinct actions of TGF-β-mediated Smad-dependent and non-Smad pathways add a layer of complexity that further complicates therapeutic translation. The pathophysiologic heterogeneity of myocardial infarction represents another major challenge. In the clinic, patients surviving acute myocardial infarction may exhibit very different responses, which are not always dependent on the extent of initial injury. Some patients tend to develop dilative remodeling that may be due to accentuated or unrestrained inflammatory activation. Other patients may have extensive fibrosis, associated with diastolic heart failure (191). Patient-specific TGF-β responses may be critical in determining the outcome in myocardial infarction. Thus, identification of patient subpopulations with overactive TGF-β signaling through the use of carefully validated biomarkers, or imaging studies, will be essential in order to design personalized treatment strategies. In any case, targeting the TGF-β system carries significant risks, because of its critical role in homeostasis and repair and its broad effects on many different cell types (192). For example, extensive evidence suggests that disruption of the TGF-β/Smad3 axis may promote aortic aneurysm formation and rupture (193–195). Thus, attempts for clinical translation of TGF-β/Smad inhibition strategies following infarction should focus on brief therapeutic interventions, and may need to exclude vulnerable patients.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Dr. Frangogiannis' laboratory was supported by NIH R01 grants HL76246 and HL85440, and by Department of Defense grants PR151029, PR151134, and PR181464.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. (2015) 5:1841–75. doi: 10.1002/cphy.c150006
2. Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation. (1977) 56:786–94. doi: 10.1161/01.CIR.56.5.786
3. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. (2012) 110:159–73. doi: 10.1161/CIRCRESAHA.111.243162
4. Frangogiannis NG. The role of transforming growth factor (TGF)-beta in the infarcted myocardium. J Thorac Dis. (2017) 9:S52–63. doi: 10.21037/jtd.2016.11.19
5. Thompson NL, Flanders KC, Smith JM, Ellingsworth LR, Roberts AB, Sporn MB. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J Cell Biol. (1989) 108:661–9. doi: 10.1083/jcb.108.2.661
6. Heine U, Munoz EF, Flanders KC, Ellingsworth LR, Lam HY, Thompson NL, et al. Role of transforming growth factor-beta in the development of the mouse embryo. J Cell Biol. (1987) 105:2861–76. doi: 10.1083/jcb.105.6.2861
7. Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, et al. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-β(2)-knockout mice. Circulation. (2001) 103:2745–52. doi: 10.1161/01.CIR.103.22.2745
8. Umbarkar P, Singh AP, Gupte M, Verma VK, Galindo CL, Guo Y, et al. Cardiomyocyte SMAD4-dependent TGF-β signaling is essential to maintain adult heart homeostasis. JACC Basic Transl Sci. (2019) 4:41–53. doi: 10.1016/j.jacbts.2018.10.003
9. Roberts AB, Roche NS, Winokur TS, Burmester JK, Sporn MB. Role of transforming growth factor-beta in maintenance of function of cultured neonatal cardiac myocytes. Autocrine action and reversal of damaging effects of interleukin-1. J Clin Invest. (1992) 90:2056–62. doi: 10.1172/JCI116087
10. Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. (2004) 164:665–77. doi: 10.1016/S0002-9440(10)63154-9
11. Wunsch M, Sharma HS, Markert T, Bernotat-Danielowski S, Schott RJ, Kremer P, et al. In situ localization of transforming growth factor beta 1 in porcine heart: enhanced expression after chronic coronary artery constriction. J Mol Cell Cardiol. (1991) 23:1051–62. doi: 10.1016/0022-2828(91)91640-D
12. Vilahur G, Juan-Babot O, Pena E, Onate B, Casani L, Badimon L. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J Mol Cell Cardiol. (2011) 50:522–33. doi: 10.1016/j.yjmcc.2010.12.021
13. Hao J, Ju H, Zhao S, Junaid A, Scammell-La Fleur T, Dixon IM. Elevation of expression of Smads 2, 3, and 4, decorin and TGF-β in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol. (1999) 31:667–78. doi: 10.1006/jmcc.1998.0902
14. Deten A, Holzl A, Leicht M, Barth W, Zimmer HG. Changes in extracellular matrix and in transforming growth factor beta isoforms after coronary artery ligation in rats. J Mol Cell Cardiol. (2001) 33:1191–207. doi: 10.1006/jmcc.2001.1383
15. Bujak M, Frangogiannis NG. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. (2007) 74:184–95. doi: 10.1016/j.cardiores.2006.10.002
16. van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. (2007) 170:818–29. doi: 10.2353/ajpath.2007.060547
17. Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. (2005) 96:881–9. doi: 10.1161/01.RES.0000163017.13772.3a
18. Jia D, Jiang H, Weng X, Wu J, Bai P, Yang W, et al. Interleukin-35 promotes macrophage survival and improves wound healing after myocardial infarction in mice. Circ Res. (2019) 124:1323–36. doi: 10.1161/CIRCRESAHA.118.314569
19. Meyer A, Wang W, Qu JX, Croft L, Degen JL, Coller BS, et al. Platelet TGF-β 1 contributions to plasma TGF-β 1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. (2012) 119:1064–74. doi: 10.1182/blood-2011-09-377648
20. Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol. (2014) 307:H1233–42. doi: 10.1152/ajpheart.00328.2014
21. Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. (2010) 176:2177–87. doi: 10.2353/ajpath.2010.090759
22. Weirather J, Hofmann U, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. (2014) 115:55–67. doi: 10.1161/CIRCRESAHA.115.303895
23. Frangogiannis NG, Perrard JL, Mendoza LH, Burns AR, Lindsey ML, Ballantyne CM, et al. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation. (1998) 98:687–98. doi: 10.1161/01.CIR.98.7.687
24. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. (2003) 116:217–24. doi: 10.1242/jcs.00229
25. Gentry LE, Nash BW. The pro domain of pre-pro-transforming growth factor beta 1 when independently expressed is a functional binding protein for the mature growth factor. Biochemistry. (1990) 29:6851–7. doi: 10.1021/bi00481a014
26. Robertson IB, Rifkin DB. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb Perspect Biol. (2016) 8:a021907. doi: 10.1101/cshperspect.a021907
27. Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, MacKay CR, et al. Complement C5a, TGF-β 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation. (1997) 95:684–92. doi: 10.1161/01.CIR.95.3.684
28. Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, et al. Essential role of smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. (2007) 116:2127–38. doi: 10.1161/CIRCULATIONAHA.107.704197
29. Nishimura SL. Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol. (2009) 175:1362–70. doi: 10.2353/ajpath.2009.090393
30. Sarrazy V, Koehler A, Chow ML, Zimina E, Li CX, Kato H, et al. Integrins αvβ5 and αvβ3 promote latent TGF-β 1 activation by human cardiac fibroblast contraction. Cardiovasc Res. (2014) 102:407–17. doi: 10.1093/cvr/cvu053
31. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. (2007) 87:1285–342. doi: 10.1152/physrev.00012.2007
32. Villarreal F, Omens J, Dillmann W, Risteli J, Nguyen J, Covell J. Early degradation and serum appearance of type I collagen fragments after myocardial infarction. J Mol Cell Cardiol. (2004) 36:597–601. doi: 10.1016/j.yjmcc.2004.01.004
33. Liu CL, Guo J, Zhang X, Sukhova GK, Libby P, Shi GP. Cysteine protease cathepsins in cardiovascular disease: from basic research to clinical trials. Nat Rev Cardiol. (2018) 15:351–70. doi: 10.1038/s41569-018-0002-3
34. Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, et al. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. (2005) 111:2935–42. doi: 10.1161/CIRCULATIONAHA.104.510354
35. Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. (2012) 92:635–88. doi: 10.1152/physrev.00008.2011
36. Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res. (2004) 64:24–31. doi: 10.1016/j.cardiores.2004.06.006
37. Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-β by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. (2000) 11:59–69. doi: 10.1016/S1359-6101(99)00029-5
38. Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med. (2012) 186:740–51. doi: 10.1164/rccm.201201-0084OC
39. Lefer AM, Tsao P, Aoki N, Palladino MA Jr. Mediation of cardioprotection by transforming growth factor-beta. Science. (1990) 249:61–4. doi: 10.1126/science.2164258
40. Lefer AM, Ma XL, Weyrich AS, Scalia R. Mechanism of the cardioprotective effect of transforming growth factor beta 1 in feline myocardial ischemia and reperfusion. Proc Natl Acad Sci USA. (1993) 90:1018–22. doi: 10.1073/pnas.90.3.1018
41. Baxter GF, Mocanu MM, Brar BK, Latchman DS, Yellon DM. Cardioprotective effects of transforming growth factor-beta1 during early reoxygenation or reperfusion are mediated by p42/p44 MAPK. J Cardiovasc Pharmacol. (2001) 38:930–9. doi: 10.1097/00005344-200112000-00015
42. Schroder D, Heger J, Piper HM, Euler G. Angiotensin II stimulates apoptosis via TGF-β 1 signaling in ventricular cardiomyocytes of rat. J Mol Med. (2006) 84:975–83. doi: 10.1007/s00109-006-0090-0
43. Wenzel S, Henning K, Habbig A, Forst S, Schreckenberg R, Heger J, et al. TGF-β 1 improves cardiac performance via up-regulation of laminin receptor 37/67 in adult ventricular cardiomyocytes. Basic Res Cardiol. (2010) 105:621–9. doi: 10.1007/s00395-010-0108-1
44. Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, et al. Cardiomyocyte-specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res. (2014) 114:1246–57. doi: 10.1161/CIRCRESAHA.114.302653
45. Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol. (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
46. Fava RA, Olsen NJ, Postlethwaite AE, Broadley KN, Davidson JM, Nanney LB, et al. Transforming growth factor beta 1 (TGF-β 1) induced neutrophil recruitment to synovial tissues: implications for TGF-β -driven synovial inflammation and hyperplasia. J Exp Med. (1991) 173:1121–32. doi: 10.1084/jem.173.5.1121
47. Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, et al. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci USA. (1987) 84:5788–92. doi: 10.1073/pnas.84.16.5788
48. Smith WB, Noack L, Khew-Goodall Y, Isenmann S, Vadas MA, Gamble JR. Transforming growth factor-beta 1 inhibits the production of IL-8 and the transmigration of neutrophils through activated endothelium. J Immunol. (1996) 157:360–8.
49. Letterio JJ, Roberts AB. Regulation of immune responses by TGF-β. Annu Rev Immunol. (1998) 16:137–61. doi: 10.1146/annurev.immunol.16.1.137
50. Werner F, Jain MK, Feinberg MW, Sibinga NE, Pellacani A, Wiesel P, et al. Transforming growth factor-beta 1 inhibition of macrophage activation is mediated via Smad3. J Biol Chem. (2000) 275:36653–8. doi: 10.1074/jbc.M004536200
51. Feinberg MW, Shimizu K, Lebedeva M, Haspel R, Takayama K, Chen Z, et al. Essential role for Smad3 in regulating MCP-1 expression and vascular inflammation. Circ Res. (2004) 94:601–8. doi: 10.1161/01.RES.0000119170.70818.4F
52. Kitamura M, Suto T, Yokoo T, Shimizu F, Fine LG. Transforming growth factor-beta 1 is the predominant paracrine inhibitor of macrophage cytokine synthesis produced by glomerular mesangial cells. J Immunol. (1996) 156:2964–71.
53. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. (1993) 90:770–4. doi: 10.1073/pnas.90.2.770
54. Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. (2000) 12:171–81. doi: 10.1016/S1074-7613(00)80170-3
55. Dutta P, Nahrendorf M. Monocytes in myocardial infarction. Arterioscler Thromb Vasc Biol. (2015) 35:1066–70. doi: 10.1161/ATVBAHA.114.304652
56. Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. (2016) 126:2151–66. doi: 10.1172/JCI85782
57. Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, et al. Inhibition of TGF-β signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. (2004) 64:526–35. doi: 10.1016/j.cardiores.2004.07.017
58. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
59. Xiao YQ, Freire-de-Lima CG, Janssen WJ, Morimoto K, Lyu D, Bratton DL, et al. Oxidants selectively reverse TGF-β suppression of proinflammatory mediator production. J Immunol. (2006) 176:1209–17. doi: 10.4049/jimmunol.176.2.1209
60. Celada A, Maki RA. Transforming growth factor-beta enhances the M-CSF and GM-CSF-stimulated proliferation of macrophages. J Immunol. (1992) 148:1102–5.
61. Ding A, Nathan CF, Graycar J, Derynck R, Stuehr DJ, Srimal S. Macrophage deactivating factor and transforming growth factors-beta 1 -beta 2 and -beta 3 inhibit induction of macrophage nitrogen oxide synthesis by IFN-gamma. J Immunol. (1990) 145:940–4.
62. Nelson BJ, Ralph P, Green SJ, Nacy CA. Differential susceptibility of activated macrophage cytotoxic effector reactions to the suppressive effects of transforming growth factor-beta 1. J Immunol. (1991) 146:1849–57.
63. Oh SA, Li MO. TGF-β: guardian of T cell function. J Immunol. (2013) 191:3973–9. doi: 10.4049/jimmunol.1301843
64. Chen W, Konkel JE. Development of thymic Foxp3(+) regulatory T cells: TGF-β matters. Eur J Immunol. (2015) 45:958–65. doi: 10.1002/eji.201444999
65. Wahl SM, Swisher J, McCartney-Francis N, Chen W. TGF-β: the perpetrator of immune suppression by regulatory T cells and suicidal T cells. J Leukoc Biol. (2004) 76:15–24. doi: 10.1189/jlb.1103539
66. Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. (2012) 125:1652–63. doi: 10.1161/CIRCULATIONAHA.111.044164
67. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. (2016) 7:12260. doi: 10.1038/ncomms12260
68. Ruiz-Villalba A, Simon AM, Pogontke C, Castillo MI, Abizanda G, Pelacho B, et al. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J Am Coll Cardiol. (2015) 65:2057–66. doi: 10.1016/j.jacc.2015.03.520
69. Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc Res. (2000) 48:89–100. doi: 10.1016/S0008-6363(00)00158-9
70. Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. (2014) 70C:74–82. doi: 10.1016/j.yjmcc.2013.11.015
71. Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. (1995) 147:325–38.
72. Shinde AV, Frangogiannis NG. Mechanisms of fibroblast activation in the remodeling myocardium. Curr Pathobiol Rep. (2017) 5:145–52. doi: 10.1007/s40139-017-0132-z
73. Humeres C, Frangogiannis NG. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl Sci. (2019) 4:449–67. doi: 10.1016/j.jacbts.2019.02.006
74. Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. (2005) 97:900–7. doi: 10.1161/01.RES.0000187457.24338.3D
75. Shinde AV, Humeres C, Frangogiannis NG. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta. (2017) 1863:298–309. doi: 10.1016/j.bbadis.2016.11.006
76. Eghbali M, Tomek R, Sukhatme VP, Woods C, Bhambi B. Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts. Regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res. (1991) 69:483–90. doi: 10.1161/01.RES.69.2.483
77. Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. (2010) 107:418–28. doi: 10.1161/CIRCRESAHA.109.216101
78. Chua CC, Chua BH, Zhao ZY, Krebs C, Diglio C, Perrin E. Effect of growth factors on collagen metabolism in cultured human heart fibroblasts. Connect Tissue Res. (1991) 26:271–81. doi: 10.3109/03008209109152444
79. Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, et al. Protective effects of activated myofibroblasts in the pressure-overloaded myocardium are mediated through smad-dependent activation of a matrix-preserving program. Circ Res. (2019) 124:1214–27. doi: 10.1161/CIRCRESAHA.118.314438
80. Yi X, Li X, Zhou Y, Ren S, Wan W, Feng G, Jiang X. Hepatocyte growth factor regulates the TGF-β 1-induced proliferation, differentiation and secretory function of cardiac fibroblasts. Int J Mol Med. (2014) 34:381–90. doi: 10.3892/ijmm.2014.1782
81. Frantz S, Hu K, Adamek A, Wolf J, Sallam A, Maier SK, et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. (2008) 103:485–92. doi: 10.1007/s00395-008-0739-7
82. Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. (2005) 111:2430–7. doi: 10.1161/01.CIR.0000165066.71481.8E
83. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res. (2016) 118:400–9. doi: 10.1161/CIRCRESAHA.115.307778
84. Frangogiannis NAG. Chemokines in ischemia and reperfusion. Thromb Haemost. (2007) 97:738–47. doi: 10.1160/TH07-01-0022
85. Kumar AG, Ballantyne CM, Michael LH, Kukielka GL, Youker KA, Lindsey ML, et al. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation. (1997) 95:693–700. doi: 10.1161/01.CIR.95.3.693
86. Frangogiannis NAG. The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities. J Cardiovasc Pharmacol. (2014) 63:185–95. doi: 10.1097/FJC.0000000000000003
87. Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. (2003) 163:2433–40. doi: 10.1016/S0002-9440(10)63598-5
88. Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. (2002) 50:71–9. doi: 10.1177/002215540205000108
89. Dobaczewski M, Akrivakis S, Nasser K, Michael LH, Entman ML, Frangogiannis NG. Vascular mural cells in healing canine myocardial infarcts. J Histochem Cytochem. (2004) 52:1019–29. doi: 10.1369/jhc.3A6210.2004
90. Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, et al. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. (2006) 48:2315–23. doi: 10.1016/j.jacc.2006.07.060
91. Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. (1997) 8:21–43. doi: 10.1016/S1359-6101(96)00048-2
92. Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. (2001) 15:1428–30. doi: 10.1096/fj.00-0745fje
93. Pardali E, ten Dijke P. Transforming growth factor-beta signaling and tumor angiogenesis. Front Biosci. (2009) 14:4848–61. doi: 10.2741/3573
94. Massague J, Xi Q. TGF-β control of stem cell differentiation genes. FEBS Lett. (2012) 586:1953–8. doi: 10.1016/j.febslet.2012.03.023
95. Chablais F, Jazwinska A. The regenerative capacity of the zebrafish heart is dependent on TGFβ signaling. Development. (2012) 139:1921–30. doi: 10.1242/dev.078543
96. Chen WP, Liu YH, Ho YJ, Wu SM. Pharmacological inhibition of TGFβ receptor improves Nkx2.5 cardiomyoblast-mediated regeneration. Cardiovasc Res. (2015) 105:44–54. doi: 10.1093/cvr/cvu229
97. Behfar A, Zingman LV, Hodgson DM, Rauzier JM, Kane GC, Terzic A, et al. Stem cell differentiation requires a paracrine pathway in the heart. Faseb J. (2002) 16:1558–66. doi: 10.1096/fj.02-0072com
98. Willems E, Cabral-Teixeira J, Schade D, Cai W, Reeves P, Bushway PJ, et al. Small molecule-mediated TGF-β type II receptor degradation promotes cardiomyogenesis in embryonic stem cells. Cell Stem Cell. (2012) 11:242–52. doi: 10.1016/j.stem.2012.04.025
99. Chen WP, Wu SM. Small molecule regulators of postnatal Nkx2.5 cardiomyoblast proliferation and differentiation. J Cell Mol Med. (2012) 16:961–5. doi: 10.1111/j.1582-4934.2011.01513.x
100. Li TS, Hayashi M, Ito H, Furutani A, Murata T, Matsuzaki M, et al. Regeneration of infarcted myocardium by intramyocardial implantation of ex vivo transforming growth factor-beta-preprogrammed bone marrow stem cells. Circulation. (2005) 111:2438–45. doi: 10.1161/01.CIR.0000167553.49133.81
101. Wu DH, Hatzopoulos AK. Bone morphogenetic protein signaling in inflammation. Exp Biol Med. (2019) 244:147–156. doi: 10.1177/1535370219828694
102. Mueller TD, Nickel J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. (2012) 586:1846–59. doi: 10.1016/j.febslet.2012.02.043
103. Morrell NW, Bloch DB, ten Dijke P, Goumans MJ, Hata A, Smith J, et al. Targeting BMP signalling in cardiovascular disease and anaemia. Nat Rev Cardiol. (2016) 13:106–20. doi: 10.1038/nrcardio.2015.156
104. Noseda M, Peterkin T, Simoes FC, Patient R, Schneider MD. Cardiopoietic factors: extracellular signals for cardiac lineage commitment. Circ Res. (2011) 108:129–52. doi: 10.1161/CIRCRESAHA.110.223792
105. Monzen K, Shiojima I, Hiroi Y, Kudoh S, Oka T, Takimoto E, et al. Bone morphogenetic proteins induce cardiomyocyte differentiation through the mitogen-activated protein kinase kinase kinase TAK1 and cardiac transcription factors Csx/Nkx-2.5 and GATA-4. Mol Cell Biol. (1999) 19:7096–105. doi: 10.1128/MCB.19.10.7096
106. Zhang H, Bradley A. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development. (1996) 122:2977–86.
107. Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. (2005) 132:5601–11. doi: 10.1242/dev.02156
108. Ghosh-Choudhury N, Abboud SL, Chandrasekar B, Ghosh Choudhury G. BMP-2 regulates cardiomyocyte contractility in a phosphatidylinositol 3 kinase-dependent manner. FEBS Lett. (2003) 544:181–4. doi: 10.1016/S0014-5793(03)00507-6
109. Sanders LN, Schoenhard JA, Saleh MA, Mukherjee A, Ryzhov S, McMaster WG Jr, et al. BMP Antagonist Gremlin 2 Limits Inflammation After Myocardial Infarction. Circ Res. (2016) 119:434–49. doi: 10.1161/CIRCRESAHA.116.308700
110. Rutkovskiy A, Sagave J, Czibik G, Baysa A, Enayati KZ, Hillestad V, et al. Connective tissue growth factor and bone morphogenetic protein 2 are induced following myocardial ischemia in mice and humans. Scand J Clin Lab Invest. (2017) 77:321–31. doi: 10.1080/00365513.2017.1318447
111. Chang SA, Lee EJ, Kang HJ, Zhang SY, Kim JH, Li L, et al. Impact of myocardial infarct proteins and oscillating pressure on the differentiation of mesenchymal stem cells: effect of acute myocardial infarction on stem cell differentiation. Stem Cells. (2008) 26:1901–12. doi: 10.1634/stemcells.2007-0708
112. Izumi M, Fujio Y, Kunisada K, Negoro S, Tone E, Funamoto M, et al. Bone morphogenetic protein-2 inhibits serum deprivation-induced apoptosis of neonatal cardiac myocytes through activation of the Smad1 pathway. J Biol Chem. (2001) 276:31133–41. doi: 10.1074/jbc.M101463200
113. Ebelt H, Hillebrand I, Arlt S, Zhang Y, Kostin S, Neuhaus H, et al. Treatment with bone morphogenetic protein 2 limits infarct size after myocardial infarction in mice. Shock. (2013) 39:353–60. doi: 10.1097/SHK.0b013e318289728a
114. Yang YL, Liu YS, Chuang LY, Guh JY, Lee TC, Liao TN, et al. Bone morphogenetic protein-2 antagonizes renal interstitial fibrosis by promoting catabolism of type I transforming growth factor-beta receptors. Endocrinology. (2009) 150:727–40. doi: 10.1210/en.2008-0090
115. Benn A, Hiepen C, Osterland M, Schutte C, Zwijsen A, Knaus P. Role of bone morphogenetic proteins in sprouting angiogenesis: differential BMP receptor-dependent signaling pathways balance stalk vs. tip cell competence. FASEB J. (2017) 31:4720–33. doi: 10.1096/fj.201700193RR
116. Cagavi E, Bartulos O, Suh CY, Sun B, Yue Z, Jiang Z, et al. Functional cardiomyocytes derived from Isl1 cardiac progenitors via Bmp4 stimulation. PLoS ONE. (2014) 9:e110752. doi: 10.1371/journal.pone.0110752
117. Takei S, Ichikawa H, Johkura K, Mogi A, No H, Yoshie S, et al. Bone morphogenetic protein-4 promotes induction of cardiomyocytes from human embryonic stem cells in serum-based embryoid body development. Am J Physiol Heart Circ Physiol. (2009) 296:H1793–803. doi: 10.1152/ajpheart.01288.2008
118. Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, et al. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. (2003) 17:2362–7. doi: 10.1101/gad.1124803
119. Wu X, Sagave J, Rutkovskiy A, Haugen F, Baysa A, Nygard S, et al. Expression of bone morphogenetic protein 4 and its receptors in the remodeling heart. Life Sci. (2014) 97:145–54. doi: 10.1016/j.lfs.2013.12.030
120. Lu J, Sun B, Huo R, Wang YC, Yang D, Xing Y, et al. Bone morphogenetic protein-2 antagonizes bone morphogenetic protein-4 induced cardiomyocyte hypertrophy and apoptosis. J Cell Physiol. (2014) 229:1503–10. doi: 10.1002/jcp.24592
121. Pachori AS, Custer L, Hansen D, Clapp S, Kemppa E, Klingensmith J. Bone morphogenetic protein 4 mediates myocardial ischemic injury through JNK-dependent signaling pathway. J Mol Cell Cardiol. (2010) 48:1255–65. doi: 10.1016/j.yjmcc.2010.01.010
122. Sun B, Huo R, Sheng Y, Li Y, Xie X, Chen C, et al. Bone morphogenetic protein-4 mediates cardiac hypertrophy, apoptosis, and fibrosis in experimentally pathological cardiac hypertrophy. Hypertension. (2013) 61:352–60. doi: 10.1161/HYPERTENSIONAHA.111.00562
123. Li Z, Wang J, Wang Y, Jiang H, Xu X, Zhang C, et al. Bone morphogenetic protein 4 inhibits liposaccharide-induced inflammation in the airway. Eur J Immunol. (2014) 44:3283–94. doi: 10.1002/eji.201344287
124. Helbing T, Arnold L, Wiltgen G, Hirschbihl E, Gabelmann V, Hornstein A, et al. Endothelial BMP4 regulates leukocyte diapedesis and promotes inflammation. Inflammation. (2017) 40:1862–74. doi: 10.1007/s10753-017-0627-0
125. Heinke J, Wehofsits L, Zhou Q, Zoeller C, Baar KM, Helbing T, et al. BMPER is an endothelial cell regulator and controls bone morphogenetic protein-4-dependent angiogenesis. Circ Res. (2008) 103:804–12. doi: 10.1161/CIRCRESAHA.108.178434
126. Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. (2009) 41:478–81. doi: 10.1038/ng.320
127. Kim RY, Robertson EJ, Solloway MJ. Bmp6 and Bmp7 are required for cushion formation and septation in the developing mouse heart. Dev Biol. (2001) 235:449–66. doi: 10.1006/dbio.2001.0284
128. Banach J, Gilewski W, Slomka A, Buszko K, Blazejewski J, Karasek D, et al. Bone morphogenetic protein 6-a possible new player in pathophysiology of heart failure. Clin Exp Pharmacol Physiol. (2016) 43:1247–50. doi: 10.1111/1440-1681.12665
129. Helder MN, Ozkaynak E, Sampath KT, Luyten FP, Latin V, Oppermann H, et al. Expression pattern of osteogenic protein-1 (bone morphogenetic protein-7) in human and mouse development. J Histochem Cytochem. (1995) 43:1035–44. doi: 10.1177/43.10.7560881
130. Dudley AT, Robertson EJ. Overlapping expression domains of bone morphogenetic protein family members potentially account for limited tissue defects in BMP7 deficient embryos. Dev Dyn. (1997) 208:349–62.
131. Merino D, Villar AV, Garcia R, Tramullas M, Ruiz L, Ribas C, et al. BMP-7 attenuates left ventricular remodelling under pressure overload and facilitates reverse remodelling and functional recovery. Cardiovasc Res. (2016) 110:331–45. doi: 10.1093/cvr/cvw076
132. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. (2007) 13:952–61. doi: 10.1038/nm1613
133. Urbina P, Singla DK. BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2014) 307:H762–72. doi: 10.1152/ajpheart.00367.2014
134. Jin Y, Cheng X, Lu J, Li X. Exogenous BMP-7 facilitates the recovery of cardiac function after acute myocardial infarction through counteracting TGF-β 1 signaling pathway. Tohoku J Exp Med. (2018) 244:1–6. doi: 10.1620/tjem.244.1
135. Huang J, Elicker J, Bowens N, Liu X, Cheng L, Cappola TP, et al. Myocardin regulates BMP10 expression and is required for heart development. J Clin Invest. (2012) 122:3678–91. doi: 10.1172/JCI63635
136. Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. (2004) 131:2219–31. doi: 10.1242/dev.01094
137. Susan-Resiga D, Essalmani R, Hamelin J, Asselin MC, Benjannet S, Chamberland A, et al. Furin is the major processing enzyme of the cardiac-specific growth factor bone morphogenetic protein 10. J Biol Chem. (2011) 286:22785–94. doi: 10.1074/jbc.M111.233577
138. Mitrofan CG, Appleby SL, Nash GB, Mallat Z, Chilvers ER, Upton PD, et al. Bone morphogenetic protein 9 (BMP9) and BMP10 enhance tumor necrosis factor-alpha-induced monocyte recruitment to the vascular endothelium mainly via activin receptor-like kinase 2. J Biol Chem. (2017) 292:13714–26. doi: 10.1074/jbc.M117.778506
139. Nakano N, Hori H, Abe M, Shibata H, Arimura T, Sasaoka T, et al. Interaction of BMP10 with Tcap may modulate the course of hypertensive cardiac hypertrophy. Am J Physiol Heart Circ Physiol. (2007) 293:H3396–403. doi: 10.1152/ajpheart.00311.2007
140. Sun L, Yu J, Qi S, Hao Y, Liu Y, Li Z. Bone morphogenetic protein-10 induces cardiomyocyte proliferation and improves cardiac function after myocardial infarction. J Cell Biochem. (2014) 115:1868–76. doi: 10.1002/jcb.24856
141. Biesemann N, Mendler L, Wietelmann A, Hermann S, Schafers M, Kruger M, et al. Myostatin regulates energy homeostasis in the heart and prevents heart failure. Circ Res. (2014) 115:296–310. doi: 10.1161/CIRCRESAHA.115.304185
142. Sharma M, Kambadur R, Matthews KG, Somers WG, Devlin GP, Conaglen JV, et al. Myostatin, a transforming growth factor-beta superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J Cell Physiol. (1999) 180:1–9. doi: 10.1002/(SICI)1097-4652(199907)180:1<1::AID-JCP1>3.3.CO;2-M
143. Castillero E, Akashi H, Wang C, Najjar M, Ji R, Kennel PJ, et al. Cardiac myostatin upregulation occurs immediately after myocardial ischemia and is involved in skeletal muscle activation of atrophy. Biochem Biophys Res Commun. (2015) 457:106–11. doi: 10.1016/j.bbrc.2014.12.057
144. Lim S, McMahon CD, Matthews KG, Devlin GP, Elston MS, Conaglen JV. Absence of myostatin improves cardiac function following myocardial infarction. Heart Lung Circ. (2018) 27:693–701. doi: 10.1016/j.hlc.2017.05.138
145. Li ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. (2008) 283:19371–8. doi: 10.1074/jbc.M802585200
146. Biesemann N, Mendler L, Kostin S, Wietelmann A, Borchardt T, Braun T. Myostatin induces interstitial fibrosis in the heart via TAK1 and p38. Cell Tissue Res. (2015) 361:779–87. doi: 10.1007/s00441-015-2139-2
147. Artaza JN, Singh R, Ferrini MG, Braga M, Tsao J, Gonzalez-Cadavid NF. Myostatin promotes a fibrotic phenotypic switch in multipotent C3H 10T1/2 cells without affecting their differentiation into myofibroblasts. J Endocrinol. (2008) 196:235–49. doi: 10.1677/JOE-07-0408
148. Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. (2013) 153:828–39. doi: 10.1016/j.cell.2013.04.015
149. Walker RG, Poggioli T, Katsimpardi L, Buchanan SM, Oh J, Wattrus S, et al. Biochemistry and biology of GDF11 and myostatin: similarities, differences, and questions for future investigation. Circ Res. (2016) 118:1125–41. doi: 10.1161/CIRCRESAHA.116.308391
150. Smith SC, Zhang X, Zhang X, Gross P, Starosta T, Mohsin S, et al. GDF11 does not rescue aging-related pathological hypertrophy. Circ Res. (2015) 117:926–32. doi: 10.1161/CIRCRESAHA.115.307527
151. Harper SC, Johnson J, Borghetti G, Zhao H, Wang T, Wallner M, et al. GDF11 decreases pressure overload-induced hypertrophy, but can cause severe cachexia and premature death. Circ Res. (2018) 123:1220–31. doi: 10.1161/CIRCRESAHA.118.312955
152. Du GQ, Shao ZB, Wu J, Yin WJ, Li SH, Wu J, et al. Targeted myocardial delivery of GDF11 gene rejuvenates the aged mouse heart and enhances myocardial regeneration after ischemia-reperfusion injury. Basic Res Cardiol. (2017) 112:7. doi: 10.1007/s00395-016-0593-y
153. Su HH, Liao JM, Wang YH, Chen KM, Lin CW, Lee IH, et al. Exogenous GDF11 attenuates non-canonical TGF-β signaling to protect the heart from acute myocardial ischemia-reperfusion injury. Basic Res Cardiol. (2019) 114:20. doi: 10.1007/s00395-019-0728-z
154. Luan H, Wang A, Hilliard B, Carvalho F, Rosen C, Ahasic A, et al. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell. (2019) 178:1231–44. doi: 10.1016/j.cell.2019.07.033
155. Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. (2011) 17:581–8. doi: 10.1038/nm.2354
156. Wang T, Liu J, McDonald C, Lupino K, Zhai X, Wilkins BJ, et al. GDF15 is a heart-derived hormone that regulates body growth. EMBO Mol Med. (2017) 9:1150–64. doi: 10.15252/emmm.201707604
157. Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res. (2006) 98:351–60. doi: 10.1161/01.RES.0000202805.73038.48
158. Kempf T, Bjorklund E, Olofsson S, Lindahl B, Allhoff T, Peter T, et al. Growth-differentiation factor-15 improves risk stratification in ST-segment elevation myocardial infarction. Eur Heart J. (2007) 28:2858–65. doi: 10.1093/eurheartj/ehm465
159. Wollert KC, Kempf T, Lagerqvist B, Lindahl B, Olofsson S, Allhoff T, et al. Growth differentiation factor 15 for risk stratification and selection of an invasive treatment strategy in non ST-elevation acute coronary syndrome. Circulation. (2007) 116:1540–8. doi: 10.1161/CIRCULATIONAHA.107.697714
160. Wollert KC, Kempf T, Peter T, Olofsson S, James S, Johnston N, et al. Prognostic value of growth-differentiation factor-15 in patients with non-ST-elevation acute coronary syndrome. Circulation. (2007) 115:962–71. doi: 10.1161/CIRCULATIONAHA.106.650846
161. Bloise E, Ciarmela P, Dela Cruz C, Luisi S, Petraglia F, Reis FM. Activin A in mammalian physiology. Physiol Rev. (2019) 99:739–80. doi: 10.1152/physrev.00002.2018
162. Walton KL, Makanji Y, Harrison CA. New insights into the mechanisms of activin action and inhibition. Mol Cell Endocrinol. (2012) 359:2–12. doi: 10.1016/j.mce.2011.06.030
163. Yndestad A, Ueland T, Oie E, Florholmen G, Halvorsen B, Attramadal H, et al. Elevated levels of activin A in heart failure: potential role in myocardial remodeling. Circulation. (2004) 109:1379–85. doi: 10.1161/01.CIR.0000120704.97934.41
164. Chen Y, Rothnie C, Spring D, Verrier E, Venardos K, Kaye D, et al. Regulation and actions of activin A and follistatin in myocardial ischaemia-reperfusion injury. Cytokine. (2014) 69:255–62. doi: 10.1016/j.cyto.2014.06.017
165. Oshima Y, Ouchi N, Shimano M, Pimentel DR, Papanicolaou KN, Panse KD, et al. Activin A and follistatin-like 3 determine the susceptibility of heart to ischemic injury. Circulation. (2009) 120:1606–15. doi: 10.1161/CIRCULATIONAHA.109.872200
166. Hu J, Wang X, Wei SM, Tang YH, Zhou Q, Huang CX. Activin A stimulates the proliferation and differentiation of cardiac fibroblasts via the ERK1/2 and p38-MAPK pathways. Eur J Pharmacol. (2016) 789:319–27. doi: 10.1016/j.ejphar.2016.07.053
167. Hu J, Wang X, Tang YH, Shan YG, Zou Q, Wang ZQ, et al. Activin A inhibition attenuates sympathetic neural remodeling following myocardial infarction in rats. Mol Med Rep. (2018) 17:5074–80. doi: 10.3892/mmr.2018.8496
168. Shimano M, Ouchi N, Nakamura K, Oshima Y, Higuchi A, Pimentel DR, et al. Cardiac myocyte-specific ablation of follistatin-like 3 attenuates stress-induced myocardial hypertrophy. J Biol Chem. (2011) 286:9840–8. doi: 10.1074/jbc.M110.197079
169. Panse KD, Felkin LE, Lopez-Olaneta MM, Gomez-Salinero J, Villalba M, Munoz L, et al. Follistatin-like 3 mediates paracrine fibroblast activation by cardiomyocytes. J Cardiovasc Transl Res. (2012) 5:814–26. doi: 10.1007/s12265-012-9400-9
170. Massague J. How cells read TGF-β signals. Nat Rev Mol Cell Biol. (2000) 1:169–78. doi: 10.1038/35043051
171. Heldin CH, Moustakas A. Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol. (2016) 8:a022053. doi: 10.1101/cshperspect.a022053
172. Rahimi RA, Leof EB. TGF-β signaling: a tale of two responses. J Cell Biochem. (2007) 102:593–608. doi: 10.1002/jcb.21501
173. Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. Embo J. (2002) 21:1743–53. doi: 10.1093/emboj/21.7.1743
174. Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through smads. Annu Rev Cell Dev Biol. (2005) 21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018
175. Funaba M, Zimmerman CM, Mathews LS. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J Biol Chem. (2002) 277:41361–8. doi: 10.1074/jbc.M204597200
176. Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. (1999) 13:804–16. doi: 10.1101/gad.13.7.804
177. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
178. Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, et al. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation. (2018) 137:707–24. doi: 10.1161/CIRCULATIONAHA.117.029622
179. Chen B, Huang S, Su Y, Wu YJ, Hanna A, Brickshawana A, et al. Macrophage Smad3 protects the infarcted heart, stimulating phagocytosis and regulating inflammation. Circ Res. (2019) 125:55–70. doi: 10.1161/CIRCRESAHA.119.315069
180. Huang S, Chen B, Su Y, Alex L, Humeres C, Shinde AV, et al. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J Mol Cell Cardiol. (2019) 132:84–97. doi: 10.1016/j.yjmcc.2019.05.006
181. Tan SM, Zhang Y, Connelly KA, Gilbert RE, Kelly DJ. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. (2010) 298:H1415–25. doi: 10.1152/ajpheart.01048.2009
182. Masaki M, Izumi M, Oshima Y, Nakaoka Y, Kuroda T, Kimura R, et al. Smad1 protects cardiomyocytes from ischemia-reperfusion injury. Circulation. (2005) 111:2752–9. doi: 10.1161/CIRCULATIONAHA.104.490946
183. Ogura Y, Ouchi N, Ohashi K, Shibata R, Kataoka Y, Kambara T, et al. Therapeutic impact of follistatin-like 1 on myocardial ischemic injury in preclinical models. Circulation. (2012) 126:1728–38. doi: 10.1161/CIRCULATIONAHA.112.115089
184. Kumagai S, Nakayama H, Fujimoto M, Honda H, Serada S, Ishibashi-Ueda H, et al. Myeloid cell-derived LRG attenuates adverse cardiac remodelling after myocardial infarction. Cardiovasc Res. (2016) 109:272–82. doi: 10.1093/cvr/cvv273
185. Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. (1999) 274:37413–20. doi: 10.1074/jbc.274.52.37413
186. Yu L, Hebert MC, Zhang YE. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. Embo J. (2002) 21:3749–59. doi: 10.1093/emboj/cdf366
187. Matsumoto-Ida M, Takimoto Y, Aoyama T, Akao M, Takeda T, Kita T. Activation of TGF-β 1-TAK1-p38 MAPK pathway in spared cardiomyocytes is involved in left ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. (2006) 290:H709–15. doi: 10.1152/ajpheart.00186.2005
188. Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, et al. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. (2017) 136:549–61. doi: 10.1161/CIRCULATIONAHA.116.026238
189. Bageghni SA, Hemmings KE, Zava N, Denton CP, Porter KE, Ainscough JFX, et al. Cardiac fibroblast-specific p38alpha MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J. (2018) 32:4941–54. doi: 10.1096/fj.201701455RR
190. Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. (2010) 106:1675–80. doi: 10.1161/CIRCRESAHA.110.217737
191. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. (2014) 11:255–65. doi: 10.1038/nrcardio.2014.28
192. Frangogiannis NG. Targeting the transforming growth factor (TGF)-β cascade in the remodeling heart: benefits and perils. J Mol Cell Cardiol. (2014) 76:169–71. doi: 10.1016/j.yjmcc.2014.09.001
193. Tan CK, Tan EH, Luo B, Huang CL, Loo JS, Choong C, et al. SMAD3 deficiency promotes inflammatory aortic aneurysms in angiotensin II-infused mice via activation of iNOS. J Am Heart Assoc. (2013) 2:e000269. doi: 10.1161/JAHA.113.000269
194. Biernacka A, Cavalera M, Wang J, Russo I, Shinde A, Kong P, et al. Smad3 signaling promotes fibrosis while preserving cardiac and aortic geometry in obese diabetic mice. Circ Heart Fail. (2015) 8:788–98. doi: 10.1161/CIRCHEARTFAILURE.114.001963
195. Regalado ES, Guo DC, Villamizar C, Avidan N, Gilchrist D, McGillivray B, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. (2011) 109:680–6. doi: 10.1161/CIRCRESAHA.111.248161
Keywords: TGF-β, myocardial infarction, Smad, BMP, GDF, fibrosis, inflammation
Citation: Hanna A and Frangogiannis NG (2019) The Role of the TGF-β Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 6:140. doi: 10.3389/fcvm.2019.00140
Received: 13 June 2019; Accepted: 03 September 2019;
Published: 18 September 2019.
Edited by:
JeanSébastien Silvestre, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Clement Cochain, Universitätsklinikum Würzburg, GermanyElisa Avolio, University of Bristol, United Kingdom
Copyright © 2019 Hanna and Frangogiannis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolaos G. Frangogiannis, nikolaos.frangogiannis@einstein.yu.edu