Extracellular Endothelial Cell-Derived Vesicles: Emerging Role in Cardiac and Vascular Remodeling in Heart Failure
 Alexander E. Berezin
Alexander E. Berezin Alexander A. Berezin
Alexander A. Berezin- 1Internal Medicine Department, State Medical University, Ministry of Health of Ukraine, Zaporozhye, Ukraine
- 2Internal Medicine Department, Medical Academy of Post-graduate Education, Ministry of Health of Ukraine, Zaporozhye, Ukraine
Extracellular vesicles play a pivotal role in numerous physiological (immune response, cell-to-cell cooperation, angiogenesis) and pathological (reparation, inflammation, thrombosis/coagulation, atherosclerosis, endothelial dysfunction) processes. The development of heart failure is strongly associated with endothelial dysfunction, microvascular inflammation, alteration in tissue repair, and cardiac and vascular remodeling. It has been postulated that activated endothelial cell-derived vesicles are not just transfer forms of several active molecules (such as regulatory peptides, coagulation factors, growth factors, active molecules, hormones that are embedded onto angiogenesis, tissue reparation, proliferation, and even prevention from ischemia/hypoxia), but are instead involved in direct myocardial and vascular damage due to regulation of epigenetic responses of the tissue. These responses are controlled by several factors, such as micro-RNAs, that are transferred inside extracellular vesicles from mother cells to acceptor cells and are transductors of epigenetic signals. Finally, it is not a uniform opinion whether different phenotypes of heart failure are the result of altered cardiac and vascular reparation due to certain epigenetic responses, which are yielded by co-morbidities, such as diabetes mellitus and obesity. The aim of the review is to summarize knowledge regarding the role of various types of extracellular endothelial cell-derived vesicles in the regulation of cardiac and vascular remodeling in heart failure.
Introduction
Heart failure (HF) is a complex condition which is often accompanied by co-morbidities and a high prevalence in the general population, and is a final stage of various cardiovascular (CV) diseases (1). Despite sufficient improvements in diagnosis, prevention, and treatment of HF, new incidences of HF with reduced ejection fraction (HFrEF) and mid-range ejection fraction (HFmrEF) continue to occur due to a poor prognosis and need for mechanical support devices and heart transplantation (2, 3). The nature of the evolution of HF is tightly associated with substantial structural cardiac and vascular remodeling that is controlled by both genetic and epigenetic factors (4). Previous preclinical and clinical studies have revealed that epigenetic mechanisms, including chromatin modifications and non-coding RNAs, have emerged as molecular transducers of age, etiology triggers and co-existing metabolic factors, environmental stimuli, and inflammatory and neurohumoral regulatory molecules to control gene expression (5, 6). In fact, pre- and post-ischemic conditioning, post-ischemic injury, oxidative stress and hypertrophic remodeling, endothelial dysfunction, accelerating atherosclerosis, plaque rapture, microvascular inflammation and occlusion, thrombosis and sub-intimal lipids' modification, extracellular matrix accumulation and cardiac/vessel fibrosis are the processes which may be potentially regulated by underlying altered chromatin modifications and non-coding RNAs dyshomeostasis in HF (7–9).
Extracellular vesicles (EVs) are a wide range of particles that are released from the most viable cells and transfer active molecules, such as hormones, regulatory peptides, growth factors, and chromatin, and play a pivotal role in cell-to-cell cooperation, immunity, inflammation, apoptosis, and repairs (10). Developing HF adds to EVs' formation from the numerous types of cells including cardiac myocytes, fibroblasts, mononuclear cells, platelets, endothelial cell, progenitor cells, and even stem cells (11). Endothelial cell-derived EVs are a secretome of the progenitor and mature endothelial cells and are involved in functional and structural repairs of myocardium, endothelium, and vascular vasculature (12). Therefore, chromatin materials are able to be transferred as a cargo with EVs from cell to cell due to cell activation or apoptosis and thereby influence target cells acting as epigenetic factors (13). Finally, the epigenetic changes may influence many intercellular communication signaling systems, including the nitric oxide, angiotensin, and endothelin-1 signaling systems, which are embedded onto pathogenesis of cardiac and vascular remodeling (14, 15). The aim of the review is to summarize knowledge regarding the role of various types of extracellular endothelial cell-derived vesicles in the regulation of cardiac and vascular remodeling in HF.
Extracellular Vesicles: Definition and Nomenclature
Previously secreted membrane-enclosed particles, which are collectively called extracellular vesicles (EVs), include exosomes, ectosomes, microvesicles, small size microvesicles, microparticles, nano particles, apoptotic bodies, and other EVs. Some of them (ectosomes and microparticles) were not determined as distinct from each other, and several classification approaches (sedimentation speed-derived criteria, immune phenotype, origin, mechanism of release, and size) were applied to EVs' subsets to qualify them in some classes. According to the Executive Committee of the International Society for Extracellular Vesicles, EVs are defined as mixture particles ranging from 30 to 2,000 nm in diameter, which are released by various types of viable cells in several different mechanisms (blebbing and budding of endosomal or plasma membranes) and they include exosomes, microvesicles, and apoptotic bodies (16). Table 1 reports nomenclature and basic characteristics of several subtypes of EVs.
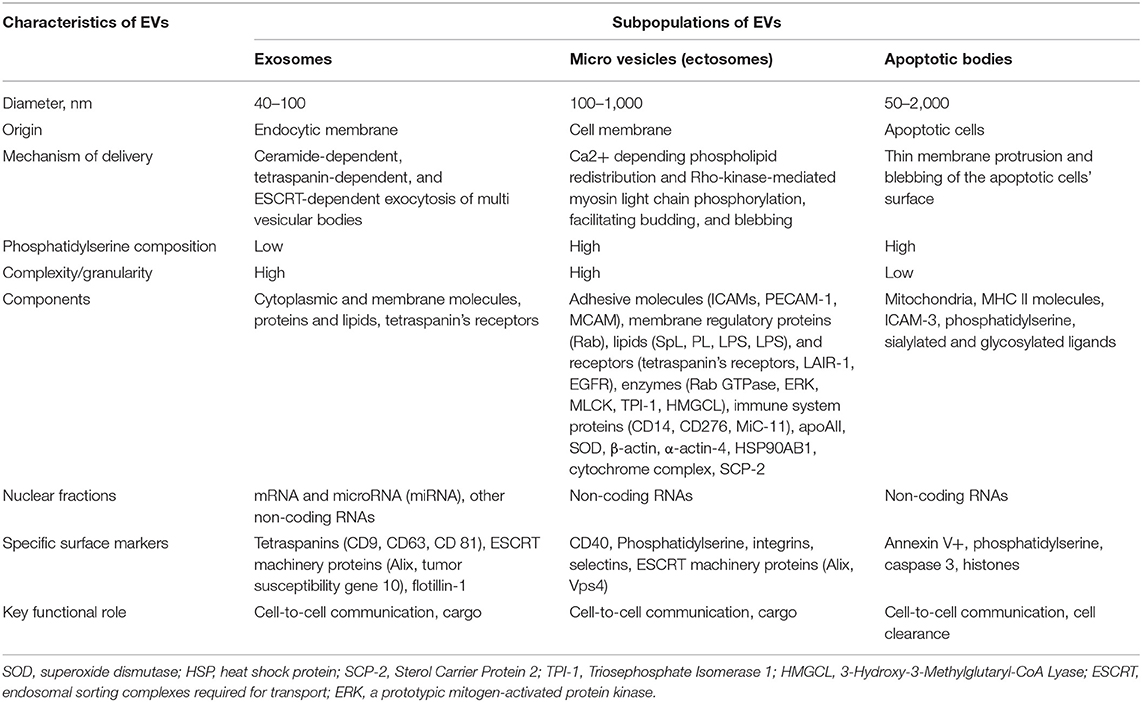
Table 1. Nomenclature and basic characteristics of several subtypes of EVs.
Exosomes
Exosomes are derivates of the endocytic membrane that have an average diameter of 40–100 nm and are released from several types of cells after exocytosis and the shaping of multivesicular bodies (MVBs) (17, 18). MVBs move along intracellular tubules, fuse with plasmatic membranes, and release exosomes onto extracellular space. Exosomes have various cellular components including cytoplasmic and membrane molecules, proteins, hormones (aldosterone), growth factors (vascular endothelial growth factor, transforming growth factor), cytokines (interleukin [IL]-1β, IL-6, IL-8), and lipids, and may also contain fragments of chromatin, such as non-coding RNAs and several inactive forms of micro-RNAs (17, 18). There are a common set of membranes and cytosolic proteins, which are embedded onto exosomes originated from distinct types of cells (19). The specific surface markers that ensure recognition of the exosomes are tetraspanins (CD9, CD63, CD 81), ESCRT (endosomal sorting complexes required for transport), machinery proteins (Alix, tumor susceptibility gene 10), and flotillin-1 (20).
Microvesicles
Microvesicles (equally known as microparticles or ectosomes) typically have a range from 100 to 1,000 nm in diameter and are shaped as a result of budding of the cell membrane (21). Microvesicles are heavily enriched in phospholipids, such as phosphatidylserine and phosphatidylcholine, and numerous membrane-depended structures (receptors, CD markers) originated from the parent cells (22). Proteomics and lipidomics arrangement of microvesicles is extremely variable and includes membrane regulatory (Rab, Sterol Carrier Protein 2) and structure (β-actin, α-actin-4) proteins, heat shock protein HSP90AB1, adhesive molecules (ICAMs, PECAM-1, MCAM), lipids (SpL, PL, LPS, LPS) and receptors (tetraspanin's receptors, LAIR-1, EGFR), enzymes (superoxide dismutase, Rab GTPase, cytochrome complex, Akt/ ERK, triosephosphate isomerase−1, 3-Hydroxy-3-Methylglutaryl-CoA Lyase), immune system proteins (CD14, CD276, MiC-11), and apo-lipoproteins (apo-A-II) (23–25). Therefore, microvesicles may yield several non-coding RNAs and chromatin fragments coupled with the complexity of the other components (26).
Apoptotic Cell-Derived Extracellular Vesicles
Apoptotic cell-derived EVs include two types of apoptotic bodies: large membrane-bound vesicles (large apoptotic bodies [ABs] with diameter ≥1,000 nm) and small apoptotic microvesicles (small ABs with diameter <1000 nm) (27). Apoptotic bodies (ABs) are particles generally larger in size in comparison to both exosomes and microvesicles, while ABs have a variable diameter that fluctuates around 1,000 nm (from 1,000 to 2000 nm) (28). Both subpopulations of ABs result in blebbing of the surface of the apoptotic cells and contain regulatory specific proteins, numerous cell organelles, and chromatin fractions, like non-coding nucleus or nucleolus RNAs (29). The process of ABs' generation is precisely controlled by several distinct morphological steps (i.e., membrane permeability and blebs, membrane protrusion, and cell fragmentation), which are consequently regulated by several molecular factors including the Rho-associated protein kinase and the plasma membrane channel pannexin-1 (Figure 1).
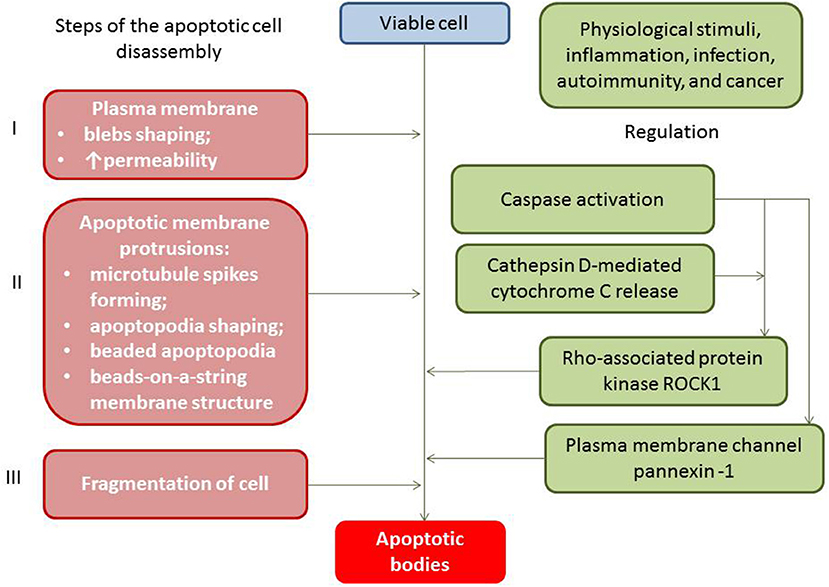
Figure 1. Apoptotic bodies generation and regulation.
ABs contain mitochondria, MHC II molecules, ICAM-3, phosphatidylserine, sialylated and glycosylated ligands, fragments of chromatin granules, DNAs, and non-coding RNAs. It has to be noted that the packaging of chromatin content (DNAs and non-coding RNAs) into the structure of the ABs is regulated during apoptosis and there are ABs that have no fragments of chromatin or remarkably low amounts of DNAs (30). ABs are classified depending on their origin from the mother cells including antigen-presenting cells, mononuclears, endothelial cells, fibroblasts, cardiac myocytes, and epithelial cells (31). The clearance of ABs has been ensured by phagocytes (32). To accurately differentiate ABs from other particles, such as cells and debris, there are several specific surface markers, such as Annexin V+/ phosphatidylserine (33).
Biological Function and Pathological Role of Extracellular Vesicles
The key biological functions of EVs typically originate from various viable cells that use cell-to-cell communication and transfer materials called secretome. Acting as cargo for numerous molecules (Heat shock proteins [HSP-90, HSP-70], ILs, tumor necrosis factor-alpha, active molecules, enzymes, peptides, growth factors), EVs are recognized by target cells through specific antigens' presentation, bind to target cells, fuse with them, and abundantly supply the packaged materials to the cells. Therefore, exosomes and microvesicles naturally have a wide range of pleiotropic biological functions including immune response, antigen presentation, and the transfer of RNA and DNAs (28, 34). The full spectrum of pleiotropic effects of circulating EVs is reported Figure 2.
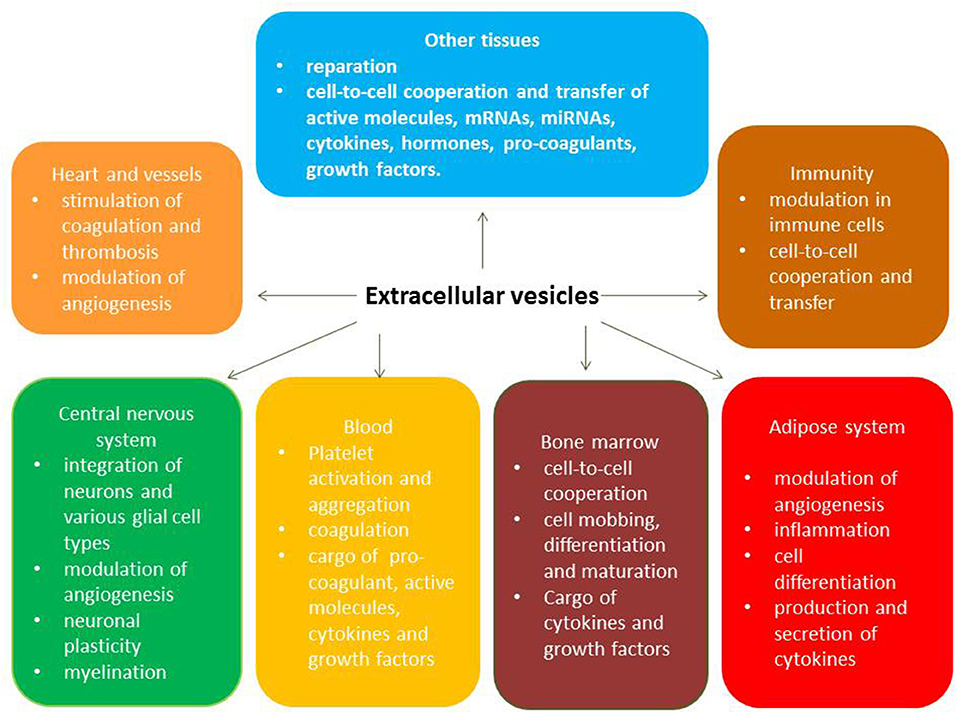
Figure 2. Pleotropic effects of circulating extracellular vesicles.
Recent studies have revealed that EVs may contain inactive forms of non-coding RNAs, which can be properly transferred to another cell and be functional in that new microenvironment (35, 36). Although 585 microRNAs were found to be up-regulated in HF patients, and 4,623 microRNAs were found to be down-regulated, most of them are circulating extracellular microRNAs, but a much smaller portion is transported using EVs (10, 26, 35). Indeed, under ischemia/hypoxic conditions, STEMI, HF, the up-regulated myocardial expression of pro-fibrotic (transforming growth factor [TGF]-β, growth differentiation factor 11 [GDF-11] and Rho-associated coiled-coil containing kinase-2 [ROCK-2]), and pro-inflammatory (inducible NO synthase, nuclear factor-kB, IL-2, IL-8, CCL5, STAT1, VEGF, TNF-alpha) genes and down-regulated gene expression of the matrix metalloproteinases (MMP-1, MMP-3, MMP-9) and their tissue inhibitors were found (35–37). In fact, EVs-related transfer microRNAs that have demonstrated abilities to up- (microRNA-210. microRNA-132) and down- (microRNA-17-3p, microRNA-222) regulate these genes through several intracellular signaling mechanisms (extracellular signal–regulated kinases 1/2 [ERK1/2], heat shock protein 27 (HSP27) signaling).
There is strong evidence that hypoxia and ischemia are triggers for mononuclear-depending production of pro-inflammatory cytokines including IL-2 and TNF-alpha, while supply of these cytokines to the target cells mediates through the package into EVs (37). On the contrary, HSPs, growth factors, non-coding RNAs, and active molecules, which are transferred by EVs, are involved in the regulation of reparative response, immunity reaction, and mediating cytoprotection (38, 39). However, a wide spectrum of biological active molecules that are transported by EVs from mother cells to the target cells yielded the ability to regulate endogenous repair system activity including proliferation, differentiation and mobbing of progenitor cells, and angiogenesis (40, 41). Through appropriate receptor-ligand (integrin αvβ3, CD40 ligand, neuregulin 1, VE-cadherin and beta-catenin) interactions and content cargo, EVs are able to regulate intracellular signaling pathways ensuring the activation of endothelial cells and the attraction and internalization of various circulating blood cells (platelets, mononuclears, macrophages, lymphocytes) to the endothelial cell surface (41). Moreover, vascular growth, restoring vascular integrity and function, and the recruitment of inflammatory cells may be directly related to up-regulated expression of the neuregulin 1 in the endothelial cells in results of EV-depended stimulation, because circulating EVs can be a source of variety of pro-angiogenic mRNAs including mRNA neuregulin 1 (42). Additionally, EVs may naturally induce a cytoskeleton-junction response of endothelial cells that is properly characterized by myosin light chain phosphorylation, contractile fiber reorganization, VE-cadherin phosphorylation, and adherent junction dissociation. This process is a key mechanism in the permeability of the vascular wall, release of neutrophil extracellular traps containing citrullinated histones and myeloperoxidase, and development of senescence and accelerating atherosclerosis (43–45). Proteome of EVs contains pro-coagulant components, such as tissue factor and phospholipids, which play a pivotal role in coagulation and the triggering of vasoocclusions in several diseases (46, 47).
Extracellular Vesicles and Nature Evolution of Heart Failure
There is evidence that various cells in the failing heart and vasculature including cardiomyocyte progenitor cells, cardiac fibroblasts, circulating blood cells, and mature and progenitor endothelial cells, are largely mediated by the paracrine release of EVs conveying the reparative potency. Although transcriptomics and proteinomics of these cells have been widely investigated, the role of paracrine factors, such as EVs, in the regulation of cardiac and vascular remodeling in HF has not been fully understood.
Cardiomyocyte Progenitor Cell-Derived EVs
Previously, cardiomyocyte progenitor cell (CPC)-derived EVs have shown beneficial effects on cardiac function and remodeling throughout the enhancement of the differentiation of cardiac progenitor cells into cardiac cells (48). CPC-derived EVs strongly inhibit lymphocyte and monocyte proliferation, suppressed inflammation, and prevented extracellular matrix accumulation (49). Indeed, CPC-derived EVs have significantly lowered the levels of pro-inflammatory cytokines, such as IgG1, IgG4, IgM, IL-1α, IL-2, IL-6, and TNF-alpha, among end-stage HF patients (48, 50). Therefore, CPC-derived EVs have reduced the number of pro-inflammatory Ly6Chigh monocytes, M1 macrophages, and suppressed NK cell degranulation in myocardium, while increasing the number of anti-inflammatory M2 macrophages (50). In fact, corresponding changes in the transcriptomic signature of the cardiac myocytes, CPC-derived EVs have demonstrated an ability to decrease tissue stiffness and BNP release and exhibited beneficial effects with regard to post-STEMI remodeling (49). Additionally, CPC-derived EVs contain a distinct repertoire of biologically active miRNAs, such as microRNA-373 and microRNA-21, that have strongly yielded anti-fibrotic effects and ameliorated fibrosis in the infarcted area targeting key pro-fibrogenic genes, i.e., TGF-β, GDF-11, and ROCK-2 (51, 52). Interestingly, EVs significantly inhibited microRNA-21 degradation and thereby mediate the anti-apoptotic effect in cardiac myocytes and endothelial cells (53). It has been demonstrated that the paracrine inhibitory impact of CPC on both cardiac fibroblast activation and collagen synthesis continues through cross-talk between cardiac fibroblasts and CPC-derived EVs (54). Thus, CPC-derived EVs ensure cardiac protection through paracrine output regarding cardiac myocytes that is attributable to decreased production of pro-healing cytokines and increased anti-inflammatory and anti-fibrotic microRNAs (55).
Circulating Blood Cells-Derived EVs
Previous clinical studies have shown that there were no significant differences in the circulating number of EVs derived from platelets (CD41a+), neutrophils (CD66b+), erythrocytes (CD235a+), monocytes (CD14+), T lymphocytes (CD3+), and B lymphocytes (CD19+) between healthy volunteers and HF patients (56). In contrast, a decreased number of circulating endothelial cells (CD31+CD41a-) EVs was found in HF patients (57). However, the total number of EVs enriched phosphatidylserines was significantly increased in HF patients compared with healthy volunteers (56). In fact, an increased number of phosphatidylserines EVs derived from various cells, including platelets and erythrocytes, was associated with hypercoagulability of HF and mostly related to atrial fibrillation and reduced LVEF (58, 59). However, EVs derived from circulating blood cells other than endothelial cells are unlikely to play a significant role in the pathogenesis of HF, but several co-morbidities (diabetes, atrial fibrillation, chronic kidney disease, chronic obstructive pulmonary disease) may have a direct effect on EV releasing from blood cells and, thereby, exacerbate clinical evolution of the HF via pro-inflammatory and pro-coagulative potencies.
Extracellular Endothelial Cell-Derived Vesicles
Extracellular endothelial cell-derived vesicles are released in both progenitor and mature endothelial cells after activation or apoptosis. The main triggers for EVs' synthesis and secretion vary depending on the presentation of various co-morbidities, the stage of HF evolution, medication use, as well as the implementation of mechanical support devices.
Innate molecular mechanisms of cardiac and vascular remodeling in HF has been investigated from several directions, such as myocardial hypertrophy and fibrosis, myocardial and microvascular inflammation, and myocardial mitochondrial dysfunction, as well as autophagy, apoptosis, and reparation. In fact, EVs play a pivotal role in various stages of the nature evolution of HF and mediate the pathological processes mentioned above (Table 2).
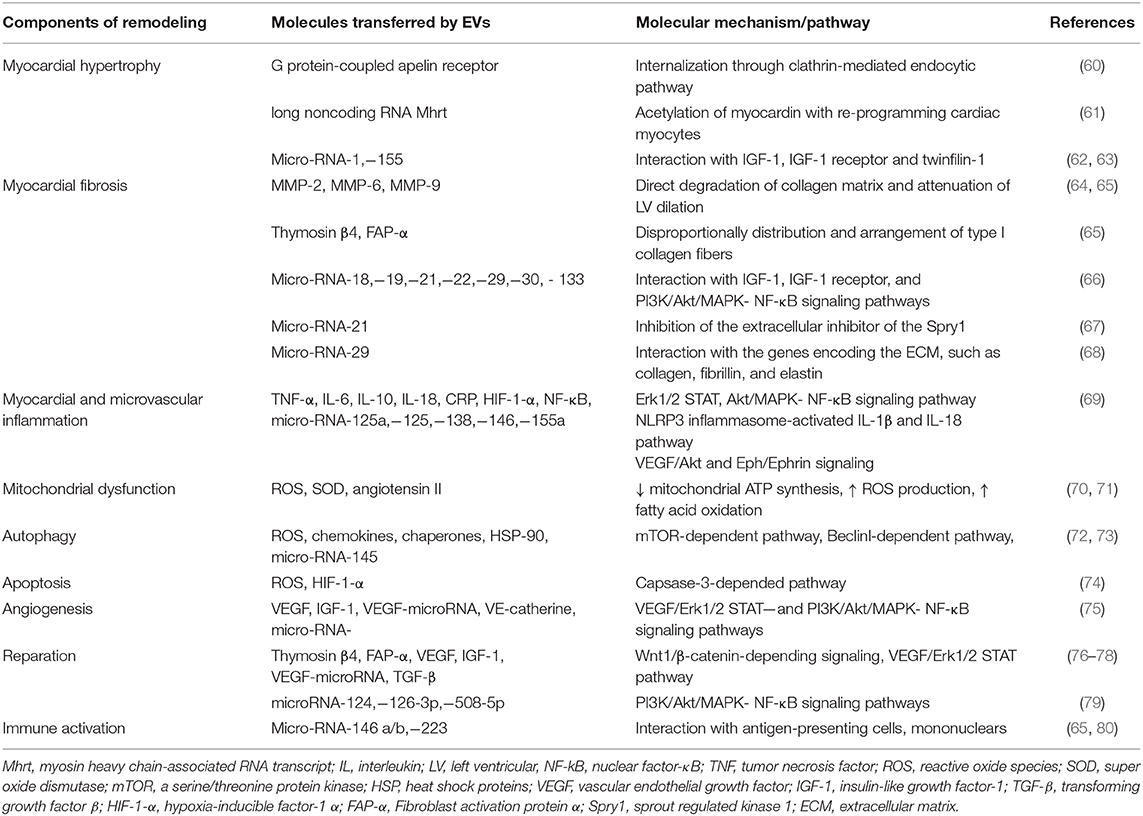
Table 2. EV-related pathways to regulate cardiac and vascular remodeling.
In fact, at early stages of nature evolution of HF, the circulating levels of EVs derived from activated endothelial cells were higher when compared with healthy volunteers, while the levels of apoptotic endothelial cell-derived EVs were similar in stage A HF patients and healthy volunteers (81, 82). Therefore, numerous metabolic risk factors, such as resistance to insulin, hyperglycemia, abdominal obesity, and hyperuricemia, are considered to be early triggers for the mobilization of endothelial progenitor cells from bone marrow and peripheral tissue. These factors can also influence the transformation of several cells, such as fibroblasts and smooth muscle cells of vasculature into cells with endothelial cells' phenotype (83–86). This process is under strong epigenetic control and circulating EVs originated from activated and apoptotic endothelial cells and their precursors are able to regulate the repair of tissues such as endothelium and vasculature myocardium through attraction of cells with high innate ability to post-natal transformation (87, 88). Finally, increased levels of extracellular activated endothelial cell-derived vesicles characterize a tendency in endogenous repair systems to restore the integrity and function of target organs including the endothelium, myocardium, kidney, and brain (89).
Previous clinical studies have shown that the number of circulating EVs produced by progenitor precursors of endothelial cells or mature endothelial cells declines depending on the severity of HF, and patients with HFrEF had significantly lowered levels of EVs when compared with patients with HFpEF (90–92). In contrast, the advance of HF was associated with a steady increase in the circulating levels of apoptotic endothelial cell-derived EVs and gradual development of deficiencies in the pool of activated endothelial cell-derived EVs (93). However, lowered number of circulating EVs originated from activated endothelial cells was determined to be a marker of endothelial dysfunction with possible discriminative value to all-cause mortality, cardiovascular mortality, a risk of acute HF and acute decompensated HF onset, and an admission due to HF (94). Some evidence suggests that the ratio between the number of EVs derived from activated and apoptotic endothelial cells may yield a pronouncedly higher predictive potency for clinical outcomes intimately related to HF than a simple amount of EVs originated from several cell subpopulations (95).
Thus, clinical data received from numerous investigators have indicated that the deficiency of the circulating activated endothelial cell-derived EVs and/or increased number of apoptotic endothelial cell-derived EVs might have a discriminative capability in HF with different phenotypes. This fact can be met with several difficulties, while the principal scheme regarding the role of activated and apoptotic endothelial cell-derived EVs in HF is reported in Figure 3. It has been suggested that organ protective effect is ensured by activated endothelial cell-derived EVs rather than apoptotic endothelial cell-derived EVs. Perhaps, proteinomics (β1 integrin, vascular endothelial growth factor, fibroblast growth factor-2, platelet-derived growth factor, enzymatic activity of matrix metalloproteinase [MMP]-2, MMP-6 and MMP-9), lipidomics (sphingosine-1-phosphate), oxidative stress components and enzymes (oxidized lipids, superoxide dismutase), non-coding RNA (micro-RNA [miRNA] 126-3p, mi-RNA-214, mi-RNA-125a, mi-RNA-150) profiles, and chromatin fragments are sufficiently distinguished in both subsets of EVs. There are several molecular mechanisms, which mediate the protective and deteriorating impact of endothelial cell-derived EVs on target tissues (Figure 4). In fact, endothelial cell-derived EVs are able to promote the protective effect that is associated with angiogenesis, tissue reparation, and pre- and post-conditioning due to VEGF/Erk 1/2 pSTAT- depending signaling pathway, whereas stimulation of Fyn kinases results in the internationalization of EV tissue factors with β-integrin, degradation of MMPs including neprilysin and C-reactive protein-embarked EVs provoke oxidative stress, cell injury, coagulation, and increase in vascular permeability, respectively (96, 97). Yet, EVs enriched Nox2-NADPH oxidase micro-RNA and insulin growth factor-1 (IGF-1) are involved into the regulation of oxidative stress and cell injury (97). Therefore, MMPs (MMP-2, MMP-6) transferred by endothelial cell-derived EVs translates the angiogenic impact of endothelial cells and promotes vascular integrity through VEGF/Erk 1/2 signaling pathway (98). It has been suggested that endothelial cell-derived EVs that are released in response to IL-3 stimulation contain angiopoetic factors, such as micro-RNA-124,−126-3p. Additionally, there are indirect angiopoetic effects that relate to post-ischemic formation of capillary-like structures and collateral vessel formation as a result in delta-like 4/Notch signaling, as well as from the cooperation of EVs with β1 integrin leading to Ras-related C3 botulinum toxin substrate 1-extracellular signal-related kinase 1 and 2-avian erythroblastosis virus E26 homolog-1 signaling and secretion of the CCL2 (99). Moreover, the activation of plasminogen into plasmin at the surface of endothelial cell-derived EVs mediates angiogenic properties of endothelial progenitor cells (100). Finally, support of endothelial structure integrity by EV cargo materials leads to improved endothelial function and a reduction of fibrosis in vasculature and myocardium (101). Previous studies have demonstrated that endothelial cell-derived EVs may promote vascular mineralization after the release of various specific mineralization-promoting cargos (tissue non-specific alkaline phosphatase, annexin-II and annexin-VI) (102, 103). Interestingly, it has identified a specific trafficking protein called sortilin, which was an initial trigger to shape EVs from progenitor endothelial cells, vascular smooth muscle cells, and mononuclears (103). In fact, the secretion of calcifying EVs is under the control of pro-inflammatory cytokines and is probably regulated epigenetically (104). However, the hypothesis regarding that the endothelial cell-derived EVs are embedded onto epigenetic regulation of endogenous repair system mediating tissue protective effects requires further investigation to be clearly understood.
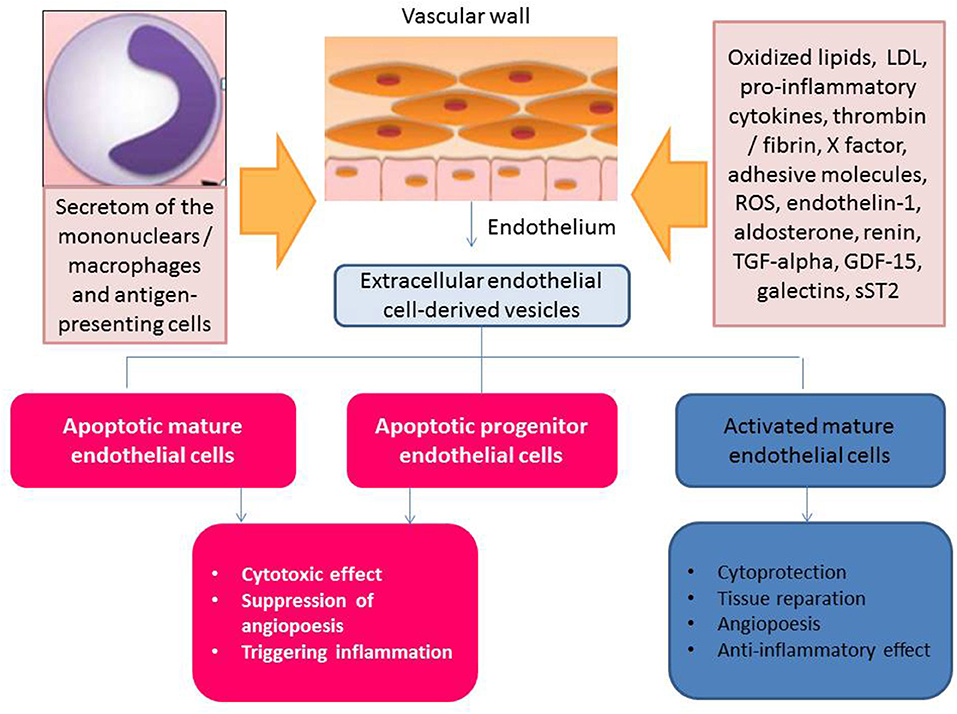
Figure 3. Apoptotic endothelial cell-derived and activated endothelial cell-derived extracellular vesicles: the role in HF pathogenesis.
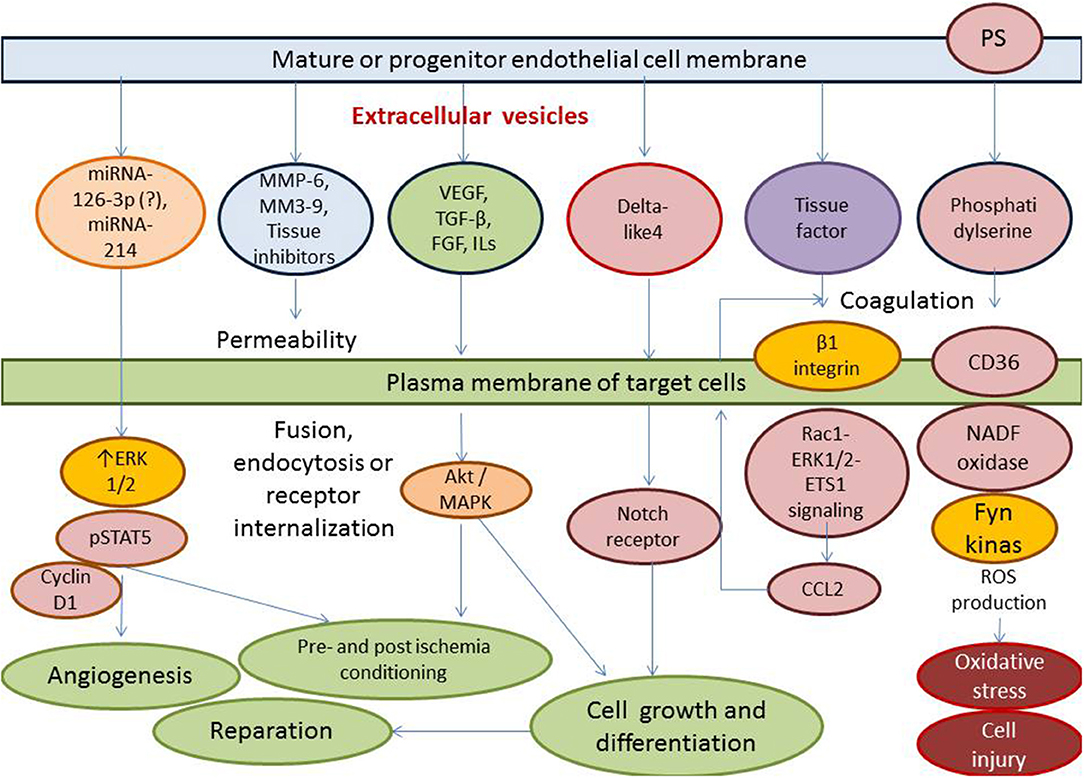
Figure 4. Molecular mechanisms ensuring the protective and deteriorating impact of endothelial cell-derived EVs on the target tissues (endothelium, vasculature, and myocardium). ROS, reactive oxide species; TGF, transforming growth factor; VEGF, vascular endothelial growth factor; MMP, matrix metalloproteinase; miRNA, micro ribonucleic acid; MAPK, mitogen-activated protein kinase; PS, phosphatidylserine; NADF, nicotinamide dinucleotide phosphate; CCL2, chemokine ligand−2; Rac1, Ras-related C3 botulinum toxin substrate 1; ERK1/2, extracellular signal-related kinase 1 and 2.
EV-Derived Non-Coding RNAs in Cardiac and Vascular Remodeling in Heart Failure
There are four epigenetic mechanisms: histone acetylation, histone methylation, DNA hyper- and hypo-methylation, and non-coding RNA regulation. Multiple pre-clinical and clinical studies have shown that non-coding RNAs transferred by EVs are the most important epigenetic regulators of cell differentiation, proliferation, survival, development, regeneration, and neovascularization (52, 105, 106). Interestingly, some subsets of free cell non-coding RNAs, such as mi-RNAs, are normally derived to the target cells by high-density lipoproteins (107), however, the majority of long non-coding RNAs and short chains of mi-RNAs are enriched and stable in EVs and can be delivered by EVs acting as gene regulators (108). Several characteristics of various progenitors cells, which are embedded onto cardiac and vascular remodeling and are expected to carry benefits to the failing heart and vasculature, such as trans-differentiation, paracrine output, migration, survival, are able to be potentially regulated by non-coding RNAs disembarked from endothelial cell-derived EVs (90, 109). For instance, endothelial cell-derived EVs through a transfer of long noncoding RNA Mhrt have exhibited the ability to cause acetylation of myocardin, which plays a pivotal role in re-programming cardiac myocytes (50, 51). There is strong evidence that micro-RNA-1 and mi-RNA−155 via interaction with free fatty acids cardiac binding protein FABP3, insulin-like growth factor-1 (IGF-1), IGF-1 receptor, and twinfilin-1 regulate cardiac myocyte free fatty acids uptake, provide proliferative response, and mediate myocardial hypertrophy (62, 63). Moreover, the spectrum of mi-RNAs that cooperate with impaired insulin sensitivity, insulin signaling, ATP production, ketone bodies, free fatty acids, and amino acids utilization, and thereby impact on cardiac relaxation, contractile function and remodeling, is wide. For instance, mi-RNA-26a,−103, -and 107 have been shown to predominantly be regulators for insulin receptor function and free fatty acid metabolism (53, 110–116). Additionally, recent pre-clinical studies have revealed that mi-RNA-378 and mi-RNA- 451 may play a crucial role in energy metabolism control through interacting with carnitine O-acetyltransferase, the peroxisome proliferator-activated receptor γ coactivator 1β, and LKB1/AMPK-signaling (117–119). However, there is no strong evidence showing that the endothelial cell-derived EVs were cargo for these molecules and this area remains largely unexplored.
Ischemia/hypoxia are triggers for endothelial cells to derive EVs in which were found 66 up-regulated microRNAs for VEGF/Akt and Eph/Ephrin signaling, as well as NO-depending pathway and 119 down-regulated microRNAs for TGF-beta receptor complex and endogenous sterols' synthesis (90, 110). It has been noted that TGF-beta receptor complex pathway, SMAD, and endogenous sterols' synthesis play crucial roles in initiating reperfusion-induced pathological events and fibrotic response (111). Additionally, EVs accumulate in the ischemic myocardium and regulate local inflammatory responses and vascular function through Erk1/2 STAT, Akt/MAPK- NF-κB signaling pathway (69, 112). Therefore, NLRP3 inflammasomes and endothelial cell-derived EVs act as cargo for a wide spectrum of active molecules, including inflammatory cytokines (TNF-α, IL-6, IL-10, IL-18, CRP, HIF-1-α), regulatory peptides (NF-κB), mi-RNAs (-125a,−125,−138,−146,−155a) act IL-1β and IL-18 pathway (69).
There is evidence showing that mi-RNA-21, after a delivery into cardiac myocytes and endothelial cells, have reduced apoptosis through decreases in Programmed Cell Death gene-4 expression, inhibition of the extracellular inhibitor of the sprout regulated kinase 1 (Spry1), and stimulation of the expression of VEGF (53, 67, 113). In animal models, the protective effect of microRNA-21 against ischemia-induced myocardial damage was confirmed by diminished cell apoptosis around the infarcted areas after treatment with antibody vs. miRNA-21 (114). Therefore, mi-RNA-29 has interacted with the genes encoding the extracellular matrix components, such as collagen, fibrillin, and elastin, and thereby reduces the risk of early rupture of the cardiac wall after myocardial infarction (68). In fact, several mi-RNAs were found to be involved in the provision of the myocardial fibrosis and vascular elastosis through interplay with IGF-1/IGF-1 receptor and PI3K/Akt/MAPK- NF-κB signaling pathways that lead to disproportionate distribution and exaggerated arrangement of type I collagen fibers in the extracellular matrix (66). Mi-RNA-378 also had a critical role in the regulation of cardiac fibrosis and the effects of biomechanical stress on cardiac remodeling (120–123). It has been reported that mi-RNA-378 inhibited cardiac fibrosis in EVs-dependent secretory manner, partially via its role as regulator of p38 MAP kinase phosphorylation by targeting MKK6 in cardiac fibroblasts (120).
Interestingly, there are some mi-RNAs (-146a,−155) that were associated with various metabolic comorbidities (type 2 diabetes mellitus, abdominal obesity, resistance to insulin) among patients with HF and adverse cardiac remodeling (70, 108), but the role of endothelial cell-derived EVs in transportation of these molecules still needs to be confirmed further. In contrast, micro-RNA-126 being a component of endothelial cell-derived EVs mediates protein kinase G activity, VCAM-1 expression on the surface of endothelial cells, and increases monocyte recruitment and differentiation (53, 90, 109–119). Several specific mi-RNAs (-92a,−126, and−133) were determined as regulators of microvascular coronary endothelial function and blood coagulation (120, 121), and mi-RNA-138 and−155 were negatively associated with NO production and cell-cell communication, respectively (122, 123). Animal study has revealed that mi-RNA-17-3p-dependent inhibition of TIMP3 can increase cardiac proliferation and endothelial cell survival (124–131). Additionally, mi-RNA-124 and mi-RNA−126-3p were determined to be key epigenetic regulators of PI3K/Akt/MAPK- NF-κB signaling pathways in progenitor endothelial cells, which are a core element of endogenous repair systems (79). The number, activity, and survival of progenitor endothelial cells were found to be significantly reduced in HF and corresponded to poor clinical outcomes (132); consequently, the role of several epigenetic regulators could be investigated in the direction of creating new biomarker predictive models.
There are data that confirm the idea regarding the ability of endothelial cell-derived EVs to be a driver for hypercoagulable phenotypes at the acute phase of decompensated HF in contrast with the well-known platelet-dependent pro-thrombotic state that occurs in HF (133). Probably, endothelial cell-derived EVs may ensure a control for neutrophil extracellular trap formation and pro-thrombotic profile (protein C, thrombin generation, tissue factor supply). However, the impact of these findings on the clinical outcomes among patients with different HF phenotypes and with/without sinus rhythm is not fully understood.
EVs-Derived Micro-RNAs as Predictive Biomarkers in HF
It has been suggested that exosomal micro-RNAs can be used as predictive biomarkers among HF patients (134). There is evidence that circulating levels of exosomal mi-RNAs (92b-5p,−192-5p, and−320a) in acute decompensated HF patients were significantly higher than in healthy volunteers and that the levels of exosomal mi-RNAs correlated positively with age and cardiac cavities enlargement, and inversely with LVEF and LV fraction shortening. Interestingly, the signature of circulating cell-free mi-RNAs (-423-5p,−320,−22, and−92b) was previously determined as a predictor of HF in patients after dilated cardiomyopathy and myocardial infarction (135–137). Additionally, mi-RNA-126 and mi-RNA-199a, which were contained in EVs, were related to cardiovascular clinical outcomes, whereas the levels of circulating free-RNAs were not associated with HF-related events (138). However, there are several controversies between the data received from different investigators in this issue. For instance, there was no significant difference between HFrEF patients and healthy volunteers in the expression of circulating mi-RNAs between EVs and unfractionated serum (139). In contrast, mi-RNA-192-5p expression was significantly elevated in patients who developed HFpEF within 1 year after acute myocardial infarction compared with healthy volunteers (140). Thus, the discriminative ability of exosomal micro-RNAs remains uncertain and requires further evaluation. Finally, it is not clear whether different phenotypes of HF (HFrEF and HFpEF) are the result of altered cardiac and vascular repair due to certain epigenetic responses, which are yielded by co-morbidities, such as type 2 diabetes mellitus and abdominal obesity (141). In this context, the role of endothelial cell-derived EVs that transfer several biological active molecules, including non-coding RNAs, is not fully understood and should be studied further.
Future Directions and Challenges in EV Research
The transcriptomics of EVs, including signature of EV-derived microRNAs and RNA-derived fragments, is disputed as a promising source of biomarkers in liquid biopsies (142). Future studies that could clearly explain the potency of EVs as biomarkers for personalized care of HF are required. Probably brand new technological solutions, such as an integrated microfluidic exosome analysis platform, will become powerful non-invasive diagnostic tools for easy screening and monitoring of the EV-based Liquid biopsy (143). There are expectations that EVs will be a promising tool for transfer of the drugs and vector signals to the target cells to regulate many processes involved in myocardium and vasculature reparation, endothelial homoeostasis, and adaptations to myocardial injury (144). These advances have made EV-based point-of-care applications possible and promising, while new devices for use in liquid biopsy need to be developed in the future.
Conclusion
Endothelial cell-derived EVs have been identified as enveloped particles that are very heterogeneous in size, composition, and biogenesis that play a pivotal role in the evolution of HF, including cardiac and vascular remodeling. Several co-morbidities, such as type 2 diabetes mellitus, insulin resistance, and abdominal obesity, have been found to be closely related to the deterioration of repairs, and an increase in ischemia, inflammation, fibrosis, cardiac hypertrophy, accelerate atherosclerosis, and thereby to mediate shaping of HFpEF or HFrEF. EVs produced by progenitor and mature endothelial cells are co-regulators of these responses influencing HF nature evolution and probably having predictive potency to clinical outcomes. Large pre-clinical and clinical studies are needed to further understand the role of endothelial cell-derived EVs in the pathogenesis of HFrEF/HFpEF and prediction of HF-related events.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ABs, apoptotic bodies; CCL2, chemokine ligand−2; CV, cardiovascular; ECM, extracellular matrix; ERK1/2, extracellular signal-related kinase 1 and 2; EVs, extracellular vesicles; FAP-α, Fibroblast activation protein α; HF, heart failure; HFpEF, HF with preserved ejection fraction; HFrEF, HF with reduced ejection fraction; HIF-1-α, hypoxia-inducible factor-1 α; HSP, heat shock proteins; GDF-11, growth differentiation factor 11; IGF-1, insulin-like growth factor-1; IL, interleukin; LV, left ventricular; MAPK, mitogen-activated protein kinase; Mhrt, myosin heavy chain-associated RNA transcript; miRNA, micro ribonucleic acid; MMP, matrix metalloproteinase; MVBs, multi vesicular bodies; mTOR, a serine/threonine protein kinase; NADF, nicotinamide dinucleotide phosphate; NF-kB, nuclear factor-κB; PS, phosphatidylserine; Rac1, Ras-related C3 botulinum toxin substrate 1; ROCK-2, Rho-associated coiled-coil containing kinase-2; ROS, reactive oxide species; SOD, super oxide dismutase; Spry1, sprout regulated kinase 1; TGF-β, transforming growth factor β; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
References
1. Carter HE, Schofield D, Shrestha R. Productivity costs of cardiovascular disease mortality across disease types and socioeconomic groups. Open Heart. (2019) 6:e000939. doi: 10.1136/openhrt-2018-000939
2. Sato Y, Yoshihisa A, Oikawa M, Nagai T, Yoshikawa T, Saito Y, et al. Prognostic impact of worsening renal function in hospitalized heart failure patients with preserved ejection fraction -A report from the JASPER Registry. J Card Fail. (2019) 25:631–42. doi: 10.1016/j.cardfail.2019.04.009
3. Patel KV, Mauricio R, Grodin JL, Ayers C, Fonarow GC, Berry JD, et al. Identifying a low-flow phenotype in heart failure with preserved ejection fraction: a secondary analysis of the RELAX trial. ESC Heart Fail. (2019) 6:613–20. doi: 10.1002/ehf2.12431
4. Kim SY, Morales CR, Gillette TG, Hill JA. Epigenetic regulation in heart failure. Curr Opin Cardiol. (2016) 31:255–65. doi: 10.1097/HCO.0000000000000276
5. Wang W, Zhang Y, Wang R, Shrestha Y, Xu Y, Peng L, et al. Risk factors and epigenetic markers of left ventricular diastolic dysfunction with preserved ejection fraction in a community-based elderly chinese population. Clin Interv Aging. (2019) 14:1719–28. doi: 10.2147/CIA.S219748
6. Huotari J, Helenius A. Endosome maturation. EMBO J. (2011) 30:3481–500. doi: 10.1038/emboj.2011.286
7. Forini F, Nicolini G1, Pitto L1, Iervasi G1. Novel insight into the epigenetic and post-transcriptional control of cardiac gene expression by thyroid hormone. Front Endocrinol. (2019) 10:601. doi: 10.3389/fendo.2019.00601
8. Huynh DTN, Heo KS. Therapeutic targets for endothelial dysfunction in vascular diseases. Arch Pharm Res. (2019) 42:848–61. doi: 10.1007/s12272-019-01180-7
9. Brandt MM, Nguyen ITN, Krebber MM, van de Wouw J, Mokry M, Cramer MJ, et al. Limited synergy of obesity and hypertension, prevalent risk factors in onset and progression of heart failure with preserved ejection fraction. J Cell Mol Med. (2019) 23:6666–78. doi: 10.1111/jcmm.14542
10. Berezin AE. Microparticles in chronic heart failure. Adv Clin Chem. (2017) 81:1–41. doi: 10.1016/bs.acc.2017.01.001
11. Fujita J, Tohyama S, Kishino Y, Okada M, Morita Y. Concise review: genetic and epigenetic regulation of cardiac differentiation from human pluripotent stem cells. Stem Cells. (2019) 37:992–1002. doi: 10.1002/stem.3027
12. Berezin AE, Kremzer AA, Berezina TA, Martovitskaya YV. The signature of circulating microparticles in heart failure patients with metabolic syndrome. J Circul Biomarkers. (2016) 5:1–10. doi: 10.1177/1849454416663659
13. Pepin ME, Drakos S, Ha CM, Tristani-Firouzi M, Selzman CH, Fang JC, et al. DNA methylation reprograms cardiac metabolic gene expression in end-stage human heart failure. Am J Physiol Heart Circ Physiol. (2019) 317:H674–84. doi: 10.1152/ajpheart.00016.2019
14. Segers VFM, Gevaert AB, Boen JRA, Van Craenenbroeck EM, De Keulenaer GW. Epigenetic regulation of intercellular communication in the heart. Am J Physiol Heart Circ Physiol. (2019) 316:H1417–25. doi: 10.1152/ajpheart.00038.2019
15. McKinsey TA, Vondriska TM, Wang Y. Epigenomic regulation of heart failure: integrating histone marks, long noncoding RNAs, and chromatin architecture. F1000Res. (2018) 7:1713. doi: 10.12688/f1000research.15797.1
16. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. (2014) 3:26913. doi: 10.3402/jev.v3.26913
17. Clayton A, Boilard E, Buzas EI, Cheng L, Falcón-Perez JM, Gardiner C, et al. Considerations towards a roadmap for collection, handling and storage of blood extracellular vesicles. J Extracell Vesicles. (2019) 8:1647027. doi: 10.1080/20013078.2019.1647027
18. Hill AF, Pegtel DM, Lambertz U, Leonardi T, O'Driscoll L, Pluchino S, et al. ISEV position paper: extracellular vesicle RNA analysis and bioinformatics. J Extracell Vesicles. (2014) 2:22859. doi: 10.3402/jev.v2i0.22859
19. Raimondo F, Morosi L, Chinello C, Magni F, Pitto M. Advances in membranous vesicle and exosome proteomics improving biological understanding and biomarker discovery. Proteomics. (2011) 11:709–20. doi: 10.1002/pmic.201000422
20. Corrado C, Raimondo S, Chiesi A, Ciccia F, De Leo G, Alessandro R. Exosomes as intercellular signaling organelles involved in health and disease: basic science and clinical applications. Int J Mol Sci. (2013) 14:5338–66. doi: 10.3390/ijms14035338
21. Tual-Chalot S, Leonetti D, Andriantsitohaina R, Martinez MC. Microvesicles: Intercellular vectors of biological messages. Mol Intervent. (2011) 11:88–94. doi: 10.1124/mi.11.2.5
22. Soo CY, Song YQ, Zheng Y, Campbell EC, Riches AC, Gunn-Moore F, et al. Nanoparticle tracking analysis monitors microvesicle and exosome secretion from immune cells. Immunology. (2012) 136:192–7. doi: 10.1111/j.1365-2567.2012.03569.x
23. Huang XY, Yuan TZ, Tschannen M, Sun ZF, Jacob H, Du MJ, et al. Characterization of human plasma-derived exosomal rnas by deep sequencing. BMC Genomics. (2013) 14:14. doi: 10.1186/1471-2164-14-319
24. de Jong OG, Verhaar MC, Chen Y, Vader P, Gremmels H, Posthuma G, et al. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J Extracell Vesicles. (2012) 1:1. doi: 10.3402/jev.v1i0.18396
25. Waldenstrom A, Genneback N, Hellman U, Ronquist G. Cardiomyocyte microvesicles contain dna/rna and convey biological messages to target cells. PLoS ONE. (2012) 7:e34653. doi: 10.1371/journal.pone.0034653
26. Camussi G, Deregibus MC, Bruno S, Grange C, Fonsato V, Tetta C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res. (2011) 1:98–110.
27. Caruso S, Poon IKH. Apoptotic cell-derived extracellular vesicles: more than just debris. Front Immunol. (2018) 9:1486. doi: 10.3389/fimmu.2018.01486
28. Boon RA, Vickers KC. Intercellular transport of micrornas. Arterioscl Thromb Vasc Biol. (2013) 33:186–92. doi: 10.1161/ATVBAHA.112.300139
29. Noguchi M, Hirata N, Edamura T, Ishigaki S, Suizu F. Intersection of apoptosis and autophagy cell death pathway. Austin J Mol Cell Biol. (2015) 2:1004.
30. Jiang L, Paone S, Caruso S, Atkin-Smith GK, Phan TK, Hulett MD, et al. Determining the contents and cell origins of apoptotic bodies by flow cytometry. Sci Rep. (2017) 7:14444. doi: 10.1038/s41598-017-14305-z
31. Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol. (2013) 113:1–11. doi: 10.1007/s11060-013-1084-8
32. Orlando KA, Stone NL, Pittman RN. Rho kinase regulates fragmentation and phagocytosis of apoptotic cells. Exp Cell Res. (2006) 312:5–15. doi: 10.1016/j.yexcr.2005.09.012
33. Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, et al. Methods for assessing autophagy and autophagic cell death. Autophagosome Phagosome. (2008) 445:29–76. doi: 10.1007/978-1-59745-157-4_3
34. Simpson RJ, Lim JW, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics. (2009) 6:267–83. doi: 10.1586/epr.09.17
35. Mause SF, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res. (2010) 107:1047–57. doi: 10.1161/CIRCRESAHA.110.226456
36. El Andaloussi S, Maeger I, Breakefield XO, Wood MJA. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat Rev Drug Discov. (2013) 12:348–58. doi: 10.1038/nrd3978
37. Yu X, Deng LY, Wang D, Li N, Chen X, Cheng X, et al. Mechanism of TNF-alpha autocrine effects in hypoxic cardiomyocytes: Initiated by hypoxia inducible factor 1 alpha, presented by exosomes. J Mol Cell Cardiol. (2012) 53:848–57. doi: 10.1016/j.yjmcc.2012.10.002
38. Tian J, Guo X, Liu XM, Liu L, Weng QF, Dong SJ, et al. Extracellular hsp60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovasc Res. (2013) 98:391–401. doi: 10.1093/cvr/cvt047
39. Hoyer FF, Nickenig G, Werner N. Microparticles - messengers of biological information. J Cell Mol Med. (2010) 14:2250–6. doi: 10.1111/j.1582-4934.2010.01114.x
40. Todorova D, Simoncini S, Lacroix R, Sabatier F, Dignat-George F. Extracellular vesicles in angiogenesis. Circ Res. (2017) 120:1658–73. doi: 10.1161/CIRCRESAHA.117.309681
41. Bagi Z, Couch Y, Broskova Z, Perez-Balderas F, Yeo T, Davis S, et al. Extracellular vesicle integrins act as a nexus for platelet adhesion in cerebral microvessels. Sci Rep. (2019) 9:15847. doi: 10.1038/s41598-019-52127-3
42. Figliolini F, Ranghino A, Grange C, Cedrino M, Tapparo M, Cavallari C, et al. Extracellular vesicles from adipose stem cells prevent muscle damage and inflammation in a mouse model of hind limb ischemia: role of Neuregulin-1. Arterioscler Thromb Vasc Biol. (2019) 40:239–54. doi: 10.1161/ATVBAHA.119.313506
43. Chatterjee V, Yang X, Ma Y, Cha B, Meegan JE, Wu M, et al. Endothelial microvesicles carrying Src-rich cargo impair adherens junction integrity and cytoskeleton homeostasis. Cardiovasc Res. (2019) cvz238. doi: 10.1093/cvr/cvz238. [Epub ahead of print].
44. Berezin A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab Syndr. (2019) 13:3017–23. doi: 10.1016/j.dsx.2018.07.010
45. Dalli J, Montero-Melendez T, Norling LV, Yin X, Hinds C, Haskard D, et al. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol Cell Proteomics. (2013) 12:2205–19. doi: 10.1074/mcp.M113.028589
46. Banfi C, Brioschi M, Wait R, Begum S, Gianazza E, Pirillo A, et al. Proteome of endothelial cell-derived procoagulant microparticles. Proteomics. (2005) 5:4443–55. doi: 10.1002/pmic.200402017
47. Camus SM, De Moraes JA, Bonnin P, Abbyad P, Le Jeune S, Lionnet F, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vaso occlusions in sickle cell disease. Blood. (2015) 125:3805–14. doi: 10.1182/blood-2014-07-589283
48. van den Hoogen P, de Jager SCA, Mol EA, Schoneveld AS, Huibers MMH, Vink A, et al. Potential of mesenchymal- and cardiac progenitor cells for therapeutic targeting of B-cells and antibody responses in end-stage heart failure. PLoS ONE. (2019) 14:e0227283. doi: 10.1371/journal.pone.0227283
49. Mastikhina O, Moon BU, Williams K, Hatkar R, Gustafson D, Mourad O, et al. Human cardiac fibrosis-on-a-chip model recapitulates disease hallmarks and can serve as a platform for drug testing. Biomaterials. (2020) 233:119741. doi: 10.1016/j.biomaterials.2019.119741
50. Harane NE, Correa BL, Gomez I, Hocine HR, Vilar J, Desgres M, et al. Extracellular vesicles from human cardiovascular progenitors trigger a reparative immune response in infarcted hearts. Cardiovasc Res. (2020) cvaa028. doi: 10.1093/cvr/cvaa028. [Epub ahead of print].
51. Xuan W, Wang L, Xu M, Weintraub NL, Ashraf M. miRNAs in extracellular vesicles from ips-derived cardiac progenitor cells effectively reduce fibrosis and promote angiogenesis in infarcted heart. Stem Cells Int. (2019) 2019:3726392. doi: 10.1155/2019/3726392
52. Peters MMC, Sampaio-Pinto V, da Costa Martins PA. Non-coding RNAs in endothelial cell signalling and hypoxia during cardiac regeneration. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118515. doi: 10.1016/j.bbamcr.2019.07.010
53. Song Y, Zhang C, Zhang J, Jiao Z, Dong N, Wang G, et al. Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics. (2019) 9:2346–60. doi: 10.7150/thno.29945
54. Bracco Gartner TCL, Deddens JC, Mol EA, Magin Ferrer M, van Laake LW, Bouten CVC, et al. Anti-fibrotic effects of cardiac progenitor cells in a 3D-model of human cardiac fibrosis. Front Cardiovasc Med. (2019) 6:52. doi: 10.3389/fcvm.2019.00052
55. Ge X, Meng Q, Zhuang R, Yuan D, Liu J, Lin F, et al. Circular RNA expression alterations in extracellular vesicles isolated from murine heart post ischemia/reperfusion injury. Int J Cardiol. (2019) 296:136–40. doi: 10.1016/j.ijcard.2019.08.024
56. Kou Y, Zou L, Liu R, Zhao X, Wang Y, Zhang C, et al. Intravascular cells and circulating microparticles induce procoagulant activity via phosphatidylserine exposure in heart failure. J Thromb Thrombolysis. (2019) 48:187–94. doi: 10.1007/s11239-019-01889-8
57. Berezin AE, Berezin AA. Platelet-derived vesicles: diagnostic and predictive value in cardiovascular diseases. J Unexpl Med Data. (2019) 4:4. doi: 10.20517/2572-8180.2019.05
58. Wernly B, Mirna M, Rezar R, Prodinger C, Jung C, Podesser BK, et al. Regenerative cardiovascular therapies: stem cells and beyond. Int J Mol Sci. (2019) 20:E1420. doi: 10.3390/ijms20061420
59. Berezin A. Pattern of micro vesicles in heart failure: novel biomarker of endothelial dysfunction and vascular reparation. Biomark J. (2018) 4:14–26. doi: 10.21767/2472-1646.100050
60. He L, Chen L, Li L. The mechanosensitive APJ internalization via clathrin-mediated endocytosis: a new molecular mechanism of cardiachypertrophy. Med Hypotheses. (2016) 90:6–10. doi: 10.1016/j.mehy.2016.02.017
61. Luo Y, Xu Y, Liang C, Xing W, Zhang T. The mechanism of myocardial hypertrophy regulated by the interaction between Mhrt andmyocardin. Cell Signal. (2018) 43:11–20. doi: 10.1016/j.cellsig.2017.11.007
62. Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M, et al. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. (2007) 100:416–24. doi: 10.1161/01.RES.0000257913.42552.23
63. Heymans S, Corsten MF, Verhesen W, Carai P, van Leeuwen RE, Custers K, et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation. (2013) 128:1420–32. doi: 10.1161/CIRCULATIONAHA.112.001357
64. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology (Bethesda). (2013) 28:391–403. doi: 10.1152/physiol.00029.2013
65. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis TissueRepair. (2011) 5:15. doi: 10.1186/1755-1536-5-15
66. Limongelli G, Caiazza M, Masarone D. Are microRNA useful to predict prognosis in acute heart failure? J Lab Precis Med. (2018) 3:14. doi: 10.21037/jlpm.2018.01.11
67. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. (2008) 456:980–4. doi: 10.1038/nature07511
68. Thum T, Catalucci D, Bauersachs J. MicroRNAs: novel regulators in cardiac development and disease. Cardiovasc Res. (2008) 79:562–70. doi: 10.1093/cvr/cvn137
69. Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. (2018) 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001
70. Schirone L, Forte M, Palmerio S, Yee D3, Nocella C, Angelini F, et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid Med Cell Longev. (2017) 2017:1–16. doi: 10.1155/2017/3920195
71. Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. (2008) 80:30–9. doi: 10.1093/cvr/cvn184
72. Nishida K, Otsu K. Autophagy during cardiac remodeling. J Mol Cell Cardiol. (2016) 95:11–8. doi: 10.1016/j.yjmcc.2015.12.003
73. Higashi K, Yamada Y, Minatoguchi S, Baba S, Iwasa M, Kanamori H, et al. MicroRNA-145 repairs infarcted myocardium by accelerating cardiomyocyte autophagy. Am J Physiol Heart Circ Physiol. (2015) 309:H1813–26. doi: 10.1152/ajpheart.00709.2014
74. Teringova E, Tousek P. Apoptosis in ischemic heart disease. J Transl Med. (2017) 15:87. doi: 10.1186/s12967-017-1191-y
75. Xiao N, Qi XY, Tang LN, Tan LL, Chen YQ, Zhao HM. VEGF promotes cardiac stem cells differentiation into vascular endothelial cells via the PI3K/Akt signaling pathway. Artif Cells Nanomed Biotechnol. (2014) 42:400–5. doi: 10.3109/21691401.2013.837473
76. Shrivastava S, Srivastava D, Olson EN, DiMaio JM, Bock-Marquette I. Thymosin beta4 and cardiac repair. Ann NY Acad Sci. (2010) 1194:87–96. doi: 10.1111/j.1749-6632.2010.05468.x
77. Cerrada I, Ruiz-Sauri A, Carrero R, Trigueros C, Dorronsoro A, Sanchez-Puelles JM, et al. Hypoxia-inducible factor 1 alpha contributes to cardiac healing in mesenchymal stem cells-mediated cardiac repair. Stem Cells Dev. (2013) 22:501–11. doi: 10.1089/scd.2012.0340
78. Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. (2014) 70:74–82. doi: 10.1016/j.yjmcc.2013.11.015
79. Qiang L, Hong L, Ningfu W, Huaihong C, Jing W, et al. Expression of miR-126 and miR-508-5p in endothelial progenitor cells is associated with the prognosis of chronic heart failure patients. Int J Cardiol. (2013) 168:2082–8. doi: 10.1016/j.ijcard.2013.01.160
80. Essandoh K, Li Y, Huo J, Fan GC. MiRNA-Mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock. (2016) 46:122–31. doi: 10.1097/SHK.0000000000000604
81. Lawson C, Vicencio JM, Yellon DM, Davidson SM. Microvesicles and exosomes: new players in metabolic and cardiovascular disease. J Endocrinol. (2016) 228:R57–71. doi: 10.1530/JOE-15-0201
82. Berezin AE, Kremzer AA, Samura TA, Berezina TA, Kruzliak P. Impaired immune phenotype of circulating endothelial-derived microparticles in patients with metabolic syndrome and diabetes mellitus. J Endocrinol Invest. (2015) 38:865–74. doi: 10.1007/s40618-015-0273-z
83. de la Cuesta F, Passalacqua I, Rodor J, Bhushan R, Denby L, Baker AH. Extracellular vesicle cross-talk between pulmonary artery smooth muscle cells and endothelium during excessive TGF-β signalling: implications for PAH vascular remodelling. Cell Commun Signal. (2019) 17:143. doi: 10.1186/s12964-019-0449-9
84. Akbar N, Azzimato V, Choudhury RP, Aouadi M. Extracellular vesicles in metabolic disease. Diabetologia. (2019) 62:2179–87. doi: 10.1007/s00125-019-05014-5
85. Berezin AE, Kremzer AA Relationship between circulating endothelial progenitor cells and insulin resistance in non-diabetic patients with ischemic chronic heart failure. Diab Metab Syndr Clin Res Rev. (2014) 8:138–44. doi: 10.1016/j.dsx.2014.07.001
86. Puhm F, Afonyushkin T, Resch U, Obermayer G, Rohde M, Penz T, et al. Mitochondria are a subset of extracellular vesicles released by activated monocytes and induce type I IFN and TNF responses in endothelial cells. Circ Res. (2019) 125:43–52. doi: 10.1161/CIRCRESAHA.118.314601
87. Berezin A. Epigenetics in heart failure phenotypes. BBA Clin. (2016) 6:31–7. doi: 10.1016/j.bbacli.2016.05.005
88. Wong PF, Tong KL, Jamal J, Khor ES, Lai SL, Mustafa MR. Senescent HUVECs-secreted exosomes trigger endothelial barrier dysfunction in young endothelial cells. EXCLI J. (2019) 18:764–76. doi: 10.17179/excli2019-1505
89. Berezin A. Endogenous vascular repair system in cardiovascular disease: The role of endothelial progenitor cells. AMJ. (2019) 12:42–8. doi: 10.21767/AMJ.2018.3464
90. Schüttler D, Clauss S, Weckbach LT, Brunner S. Molecular mechanisms of cardiac remodeling and regeneration in physical exercise. Cells. (2019) 8:E1128. doi: 10.3390/cells8101128
91. Yang J, Xue FT, Li YY, Liu W, Zhang S. Exosomal piRNA sequencing reveals differences between heart failure and healthy patients. Eur Rev Med Pharmacol Sci. (2018) 22:7952–61. doi: 10.26355/eurrev_201811_16423
92. Gohar A, de Kleijn DPV, Hoes AW, Rutten FH, Hilfiker-Kleiner D, Ferdinandy P, Sluijter JPG, den Ruijter HM. Vascular extracellular vesicles in comorbidities of heart failure with preserved ejection fraction in men and women: the hidden players. A mini review. Vascul Pharmacol. (2018) 111:1–6. doi: 10.1016/j.vph.2018.05.006
93. Berezin AE, Kremzer AA, Martovitskaya YV, Berezina TA, Gromenko EA. Pattern of endothelial progenitor cells and apoptotic endothelial cell-derived microparticles in chronic heart failure patients with preserved and reduced left ventricular ejection fraction. EBioMedicine. (2016) 4:86–94. doi: 10.1016/j.ebiom.2016.01.018
94. McKinsey TA. Therapeutic potential for hdac inhibitors in the heart. Ann Rev Pharmacol Toxicol. (2012) 52:303–19. doi: 10.1146/annurev-pharmtox-010611-134712
95. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Failure. (2010) 3:420–30. doi: 10.1161/CIRCHEARTFAILURE.109.888479
96. Turu MM, Slevin M, Matou S, West D, Rodríguez C, Luque A, et al. C-reactive protein exerts angiogenic effects on vascular endothelial cells and modulates associated signalling pathways and gene expression. BMC Cell Biol. (2008) 9:47. doi: 10.1186/1471-2121-9-47
97. Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol. (2002) 160:673–80. doi: 10.1016/S0002-9440(10)64887-0
98. Tang H, He Y, Li L, Mao W, Chen X, Ni H, et al. Exosomal MMP2 derived from mature osteoblasts promotes angiogenesis of endothelial cells via VEGF/Erk1/2 signaling pathway. Exp Cell Res. (2019) 383:111541. doi: 10.1016/j.yexcr.2019.111541
99. Arderiu G, Peña E, Badimon L. Angiogenic microvascular endothelial cells release microparticles rich in tissue factor that promotes postischemic collateral vessel formation. Arterioscler Thromb Vasc Biol. (2015) 35:348–57. doi: 10.1161/ATVBAHA.114.303927
100. Lacroix R, Sabatier F, Mialhe A, Basire A, Pannell R, Borghi H, et al. Activation of plasminogen into plasmin at the surface of endothelial microparticles: a mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood. (2007) 110:2432–9. doi: 10.1182/blood-2007-02-069997
101. Leroyer AS, Rautou PE, Silvestre JS, Castier Y, Lesèche G, Devue C, et al. CD40 ligand+ microparticles from human atherosclerotic plaques stimulate endothelial proliferation and angiogenesis a potential mechanism for intraplaque neovascularization. J Am Coll Cardiol. (2008) 52:1302–11. doi: 10.1016/j.jacc.2008.07.032
102. Kurabayashi M. Molecular mechanism of vascular calcification. Clin Calcium. (2019) 29:157–63. doi: 10.20837/4201902157
103. Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. (2016) 126:1323–36. doi: 10.1172/JCI80851
104. Krohn JB, Hutcheson JD, Martínez-Martínez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol. (2016) 594:2895–903. doi: 10.1113/JP271338
105. Chung JJ, Han J, Wang LL, Arisi MF, Zaman S, Gordon J, et al. Delayed delivery of endothelial progenitor cell-derived extracellular vesicles via shear thinning gel improves postinfarct hemodynamics. J Thorac Cardiovasc Surg. (2019). doi: 10.1016/j.jtcvs.2019.06.017. [Epub ahead of print].
106. Ragvin A, Valvatne H, Erdal S, Arskog V, Tufteland KR, Breen K, et al. : Nucleosome binding by the bromodomain and PHD finger of the transcriptional cofactor p300. J Mol Biol. (2004) 337:773–88. 10.1016/j.jmb.2004.01.051. doi: 10.1016/j.jmb.2004.01.051
107. Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. (2011) 13:423–33. doi: 10.1038/ncb2210
108. El Harane N, Kervadec A, Bellamy V, Pidial L, Neametalla HJ, Perier MC, et al. Acellular therapeutic approach for heart failure: in vitro production of extracellular vesicles from human cardiovascular progenitors. Eur Heart J. (2018) 39:1835–47. doi: 10.1093/eurheartj/ehy012
109. Mount S, Kanda P, Parent S, Khan S, Michie C, Davila L, et al. Physiologic expansion of human heart-derived cells enhances therapeutic repair of injured myocardium. Stem Cell Res Ther. (2019) 10:316. doi: 10.1186/s13287-019-1418-3
110. Coulthard MG, Morgan M, Woodruff TM, Arumugam TV, Taylor SM, Carpenter TC, et al. Eph/Ephrin signaling in injury and inflammation. Am J Pathol. (2012). 181:1493e1503. doi: 10.1016/j.ajpath.2012.06.043
111. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblastspecific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. (2017) 127:3770–83. doi: 10.1172/JCI94753
112. Loyer X, Zlatanova I, Devue C, Yin M, Howangyin KY, Klaihmon P, et al. Intra-cardiac release of extracellular vesicles shapes inflammation following myocardial infarction. Circ Res. (2018) 123:100–6. doi: 10.1161/CIRCRESAHA.117.311326
113. Gu H, Liu Z, Li Y, Xie Y, Yao J, Zhu Y, et al. Serum-derived extracellular vesicles protect against acute myocardial infarction by regulating miR-21/PDCD4 signaling pathway. Front Physiol. (2018) 9:348. doi: 10.3389/fphys.2018.00348
114. Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, et al. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. (2009) 284:29514–25. doi: 10.1074/jbc.M109.027896
115. Dong C, Ma A, Shang L. Nanoparticles for postinfarct ventricular remodeling. Nanomedicine. (2018) 13:3037–50. doi: 10.2217/nnm-2018-0264
116. Feng B, Chen S, Gordon AD, Chakrabarti S. miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J Mol Cell Cardiol. (2017) 105:70–6. doi: 10.1016/j.yjmcc.2017.03.002
117. van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. (2012) 126:830–9. doi: 10.1161/CIRCULATIONAHA.111.076075
118. Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res. (2010) 107:810–7. doi: 10.1161/CIRCRESAHA.110.226357
119. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. (2008) 15:261–71. doi: 10.1016/j.devcel.2008.07.002
120. Witkowski M, Weithauser A, Tabaraie T, Steffens D, Krankel N, Witkowski M, et al. Micro-RNA-126 reduces the blood thrombogenicity in diabetes mellitus via targeting of tissue factor. Arterioscler Thromb Vasc Biol. (2016) 36:1263–71. doi: 10.1161/ATVBAHA.115.306094
121. Widmer RJ, Chung WY, Herrmann J, Jordan KL, Lerman LO, Lerman A. The association between circulating microRNA levels and coronary endothelial function. PLoS ONE. (2014) 9:e109650. doi: 10.1371/journal.pone.0109650
122. Sen A, Most P, Peppel K. Induction of microRNA-138 by pro-inflammatory cytokines causes endothelial cell dysfunction. FEBS Lett. (2014) 588:906–14. doi: 10.1016/j.febslet.2014.01.033
123. Lopez-Ramirez MA, Wu D, Pryce G, Simpson JE, Reijerkerk A, King-Robson J, et al. MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J. (2014) 28:2551–65. doi: 10.1096/fj.13-248880
124. Fu X, Dong B, Tian Y, Lefebvre P, Meng Z, Wang X, et al. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J Clin Invest. (2015) 125:2497–509. doi: 10.1172/JCI75438
125. Bork-Jensen J, Thuesen AC, Bang-Bertelsen CH, Grunnet LG, Pociot F, Beck-Nielsen H, et al. Genetic versus non-genetic regulation of miR-103, miR-143 and miR-483-3p expression in adipose tissue and their metabolic implications-A twin study. Genes. (2014) 5:508–17. doi: 10.3390/genes5030508
126. Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. (2011) 474:649–53. doi: 10.1038/nature10112
127. Carrer M, Liu N, Grueter CE, Williams AH, Frisard MI, Hulver MW, et al. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc Natl Acad Sci USA. (2012) 109:15330–5. doi: 10.1073/pnas.1207605109
128. Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res. (2015) 116:279–88. doi: 10.1161/CIRCRESAHA.116.304707
129. Pinti MV, Hathaway QA, Hollander JM. Role of microRNA in metabolic shift during heart failure. Am J Physiol Heart Circ Physiol. (2017) 312:H33–45. doi: 10.1152/ajpheart.00341.2016
130. Yuan J, Liu H, Gao W, Zhang L, Ye Y, Yuan L, et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics. (2018) 8:2565–82. doi: 10.7150/thno.22878
131. Liu Z, Zhang Z, Yao J, Xie Y, Dai Q, Zhang Y, et al. Serum extracellular vesicles promote proliferation of H9C2 cardiomyocytes by increasing miR-17-3p. Biochem Biophys Res Commun. (2018) 499:441–6. doi: 10.1016/j.bbrc.2018.03.157
132. Berezin AE, Kremzer AA, Samura TA, Martovitskaya YV. Apoptotic microparticles to progenitor mononuclear cells Ratio in heart failure: relevance of clinical status and outcomes. J Cardiovasc Dis. (2014) 2:50–7. Available online at: http://www.researchpub.org/journal/jcvd/jcvd.html
133. Popovic B, Zannad F, Louis H, Clerc-Urmès I, Lakomy C, Gibot S, et al. Endothelial-driven increase in plasma thrombin generation characterising a new hypercoagulable phenotype in acute heart failure. Int J Cardiol. (2019) 274:195–201. doi: 10.1016/j.ijcard.2018.07.130
134. Wu T, Chen Y, Du Y, Tao J, Zhou Z, Yang Z. Serum Exosomal MiR-92b-5p as a potential biomarker for acute heart failure caused by dilated cardiomyopathy. Cell Physiol Biochem. (2018) 46:1939–50. doi: 10.1159/000489383
135. Gupta MK, Halley C, Duan ZH, Lappe J, Viterna J, Jana S, et al. miRNA-548c: a specific signature in circulating PBMCs from dilated cardiomyopathy patients. J Mol Cell Cardiol. (2013) 62:131–41. doi: 10.1016/j.yjmcc.2013.05.011
136. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. (2008) 105:13027–32. doi: 10.1073/pnas.0805038105
137. Molina-Navarro MM, Rosello-Lleti E, Ortega A, Tarazon E, Otero M, Martinez-Dolz L, et al. Differential gene expression of cardiac ion channels in human dilated cardiomyopathy. PLoS ONE. (2013) 8:e79792. doi: 10.1371/journal.pone.0079792
138. Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, et al. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. (2009) 119:2357–66. doi: 10.1161/CIRCULATIONAHA.108.814145
139. Goren Y, Kushnir M, Zafrir B, Tabak S, Lewis BS, Amir O. Serum levels of microRNAs in patients with heart failure. Eur J Heart Fail. (2012). 14:147–54. doi: 10.1093/eurjhf/hfr155
140. Matsumoto S, Sakata Y, Suna S, Nakatani D, Usami M, Hara M, et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ Res. (2013) 113:322–6. doi: 10.1161/CIRCRESAHA.113.301209
141. Cesselli D, Parisse P, Aleksova A, Veneziano C, Cervellin C, Zanello A, et al. Extracellular vesicles: how drug and pathology interfere with their biogenesis and function. Front Physiol. (2018) 9:1394. doi: 10.3389/fphys.2018.01394
142. Tosar JP, Cayota A. Extracellular tRNAs and tRNA-derived fragments. RNA Biol. (2020) 1–19. doi: 10.1080/15476286.2020.1729584. [Epub ahead of print].
143. Li G, Tang W, Yang F. Cancer liquid biopsy using integrated microfluidic exosome analysis platforms. Biotechnol J. (2020) e1900225. doi: 10.1002/biot.201900225. [Epub ahead of print].
Keywords: extracellular vesicles, cardiac and vascular remodeling, heart failure, epigenetics, co-morbidities
Citation: Berezin AE and Berezin AA (2020) Extracellular Endothelial Cell-Derived Vesicles: Emerging Role in Cardiac and Vascular Remodeling in Heart Failure. Front. Cardiovasc. Med. 7:47. doi: 10.3389/fcvm.2020.00047
Received: 11 December 2019; Accepted: 10 March 2020;
Published: 15 April 2020.
Edited by:
Susumu Minamisawa, Jikei University School of Medicine, JapanReviewed by:
Andrea Caporali, University of Edinburgh, United KingdomTetsuo Sasano, Tokyo Medical and Dental University, Japan
Copyright © 2020 Berezin and Berezin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander E. Berezin, aeberezin@gmail.com
†ORCID: Alexander E. Berezin orcid.org/0000-0002-0446-3999