Therapeutic SERPINs: Improving on Nature
 Coen Maas
Coen Maas Steven de Maat
Steven de Maat- CDL Research, University Medical Center Utrecht, Utrecht, Netherlands
Serine proteases drive important physiological processes such as coagulation, fibrinolysis, inflammation and angiogenesis. These proteases are controlled by serine protease inhibitors (SERPINs) that neutralize their activity. Currently, over 1,500 SERPINs are known in nature, but only 37 SERPINs are found in humans. Thirty of these are functional protease inhibitors. The inhibitory potential of SERPINs is in perfect balance with the proteolytic activities of its targets to enable physiological protease activity. Hence, SERPIN deficiency (either qualitative or quantitative) can lead to disease. Several SERPIN resupplementation strategies have been developed to treat SERPIN deficiencies, including concentrates derived from plasma and recombinant SERPINs. SERPINs usually inhibit multiple proteases, but only in their active state. Over the past decades, considerable insights have been acquired in the identification of SERPIN biological functions, their inhibitory mechanisms and specificity determinants. This paves the way for the development of therapeutic SERPINs. Through rational design, the inhibitory properties (selectivity and inhibitory potential) of SERPINs can be reformed and optimized. This review explores the current state of SERPIN engineering with a focus on reactive center loop modifications and backbone stabilization. We will discuss the lessons learned from these recombinant SERPINs and explore novel techniques and strategies that will be essential for the creation and application of the future generation of therapeutic SERPINs.
Introduction
Approximately one third of all proteases belong to the superfamily of serine proteases, which can be found throughout all kingdoms of life. In humans, ~180 serine proteases govern essential physiological processes such as vascular hemostasis (1), inflammation (2), tissue remodeling (3) or angiogenesis (4). Many of these processes are regulated by chymotrypsin-like serine proteases, which are the most abundant class of serine proteases. These have a highly conserved proteolytic mechanism [reviewed in ((5))] and operate in “sequential activation” cascade mechanisms (e.g., coagulation or complement). The activity of serine proteases needs to be controlled, as excessive activity causes disease. This is where serine protease inhibitors (SERPINs) are of high importance.
SERPINs in Human Physiology
The superfamily of SERPINs consists of ~1,500 identified members (6). There is evidence for the existence of 37 human SERPINs at protein level. Thirty of these have proven inhibitory function, where they act as suicide substrate inhibitors. Loss of SERPIN function can have severe pathological consequences. For example, patients with α1-antitrypsin (α1AT) deficiency develop pulmonary emphysema due to uncontrolled activity of neutrophil elastase (7). C1-esterase inhibitor (C1INH) deficiency leads to attacks of angioedema, due to excessive bradykinin formation by the plasma contact system (8), whereas patients with low levels of antithrombin (ATIII) have an increased risk of ischemic stroke, deep vein thrombosis or pulmonary embolism due to increased activity of the coagulation system (9). Currently, the majority of SERPIN therapeutics are meant as supplementation therapy to overcome these defects.
SERPINs Mode of Action
SERPINS have a generally well-conserved secondary structure consisting of three β-sheets (A, B and C; highlighted in green in Figure 1) and nine α-helices (6, 10). Additionally, SERPINs contain an exposed reactive center loop (RCL; highlighted in red in Figure 1), which is a flexible loop structure on top of the SERPIN backbone. The RCL serves as a bait sequence for target proteases. The tertiary structure of native SERPINs is metastable, which can shift into a hyperstable conformation. This process is critical for the SERPIN function. While this shift can occur spontaneously, it becomes actively triggered when the RCL is cleaved by a protease (11). When the protease cleaves the RCL at the P1-P1'scissile bond, the serine (or a cysteine in case of cysteine proteases) of the catalytic triad of the protease attacks the carbonyl of the RCL, forming a tetrahedral intermediate (12). Hereafter, two situations may occur: (I) The SERPIN assumes its hyperstable state, pulling the protease to the opposite side of the SERPIN (13). Meanwhile, the N-terminal remainder of the RCL becomes inserted next to the five strands of β-sheet A, effectively making it the sixth strand (11, 14). During this process, the protease active site becomes distorted, and it can no longer hydrolyze the tetrahedral intermediate (15). When this occurs in the extracellular space, the SERPIN-protease complex will be cleared via scavenger receptors. II) The protease hydrolyzes the tetrahedral intermediate and releases it before active site disruption. The speed by which the C-terminal loop of the RCL is inserted into β-sheet A is critical and determines whether the SERPIN becomes a substrate or an inhibitor. Nonetheless, the SERPIN still folds into its hyperstable state (because its RCL has been successfully cleaved, which is irreversible) and will be rapidly cleared, while the regenerated protease remains active.
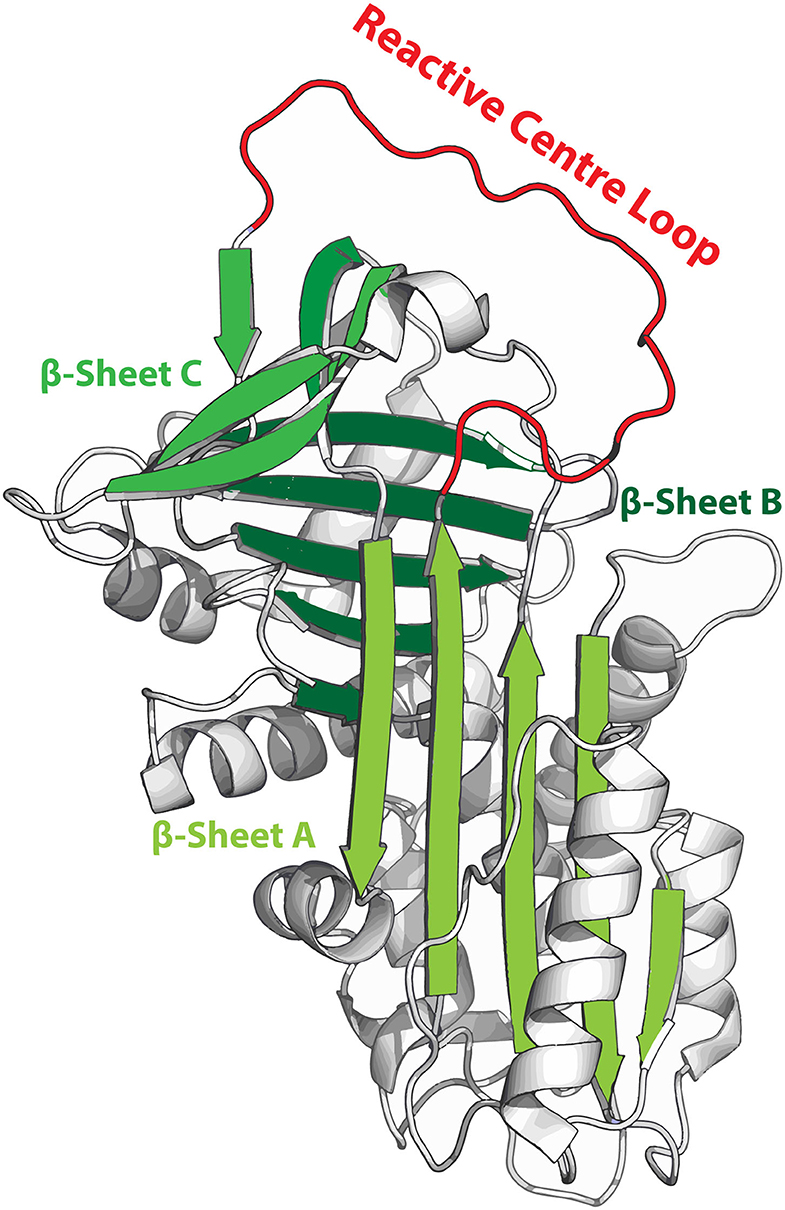
Figure 1. Metastable structure of α1-antitrypsin. β-sheets are highlighted in green and the reactive center loop (RCL) is highlighted in red. When the RCL becomes cleaved it integrates into β-sheet A, effectively becoming the 6th strand of this sheet.
The ability of SERPINs to interact with multiple targets offers a unique opportunity for the therapeutic management of pathological enzyme systems. This review will explore the current state of SERPIN engineering, with a special focus on stabilizing SERPIN function and altering SERPIN specificity.
Supercharging SERPINs
To supercharge SERPINs, co-factors can be administered to patients. Glycosaminoglycans can modulate the activity of several SERPINs by enhancing SERPIN functionality and therefore complex formation (16–21). For example, heparin (amongst others) enhances the efficiency of thrombin inhibition by endogenous antithrombin (from 7.2*103 to 1.3*107 M−1 · s−1 respectively) (22). More recently, polyphosphate was identified as a novel cofactor in the regulation of the complement system by enhancing the interaction between C1s and C1INH to an extent, similar to that of heparin (23). However, with the rise of recombinant protein technology, the doorway has been opened to change SERPINS for the treatment of disease states beyond SERPIN deficiency and can be used to control new therapeutic target proteases.
Stability
Ideally, therapeutics are stable and in the case of life-long diseases should have a long half-life time in the human circulation. SERPINs are dependent upon their ability to shift from a metastable to a hyperstable conformation for function. This shift becomes problematic if it occurs spontaneously (i.e., without cleavage of the RCL by a target protease). This results in an inert SERPIN with the propensity to polymerize into pathogenic Z and S variants. Such behavior results in intracellular SERPIN accumulation and leads to tissue damage in the form of liver cirrhosis in the case of α1AT deficiency (24). Also for C1INH, intracellular accumulation has been reported for some mutations (25). However, there is little evidence to suggest that this is accompanied by liver cirrhosis. The differences in expression levels between both SERPINs are a logical explanation for this unwishful clinical phenotype. In order to avoid SERPIN polymerization during drug development, efforts are ongoing to achieve stabilization of the SERPIN backbone without it losing its inhibitory potential.
Lessons From Antitrypsin
Kwon et al. increased the thermostability of α1AT 13-fold by using a single mutation (F51C) without harming its inhibitory activity (26). Similarly, mutation F51L increased thermostability, but reduced the misfolding and polymerization of the pathogenic α1AT Z variant (27). Interestingly, the naturally-occurring (non-pathogenic) mutation F51S leads to α1AT retention in CHO-cells and reduces its stability (28). All the above mutations are at the same position: residue 51, demonstrating that subtle differences have a large impact on backbone stabilization (28). Further mutagenesis of the α1AT-F51L backbone identified six additional mutations (T59A, T68A. A70G, M374I, S381A) that improve α1AT stability without influencing inhibitory activity (29, 30). Other stabilizing mutations influenced the inhibitory capacity and are therefore less interesting for the development of therapeutic SERPINs (31).
Lessons From PAI-1
Not all SERPINs have a similar half-life and stability. Compared to other SERPINs, plasminogen activator I (PAI-1) has a short stability half-life of 1–2 h at 37°C and is considered relatively unstable (32). The binding of vitronectin to PAI-1 increases its stability 2–3 fold, but this complex would be considered unfavorable for therapeutic purposes. A single mutation (I91L) increases PAI-1 stability by nine-fold (32), whereas the combination of four mutations (N150H, K154T, Q319L and M354I) was able to increases the half-life 72-fold to 145 h (33). A set of 10 mutations (T50A, Q56R, A61V, G70D, T94A, N150D, D222G, I223V, G264D, and S331G) increases the stability even further to 540 h (34). Remarkably, the introduction of additional disulfide bridge (Cys 197–Cys 355) in the original PAI-1 backbone increases the stability to 700 h (35).
A Uniform Serpin Backbone
These results from work on α1AT and PAI-1 show the potential of engineering SERPIN stability, however these results cannot be directly extrapolated to other SERPIN molecules. To expedite the development of therapeutic SERPINs, attempts have been undertaken to create uniform SERPIN backbones that have been optimized for stability. Hereto, the group of Porebski et al. aligned the sequence of various SERPINs to identify their consensus sequence (36). The resulting molecule “Conserpin,” is stable up to temperatures of 110°C and shows resistance against polymerization. Furthermore, Conserpin is able to reversibly fold in response to chemical denaturation. Unfortunately, the inhibitory activity of Conserpin was found to be poor as it is unable to form stable covalent serpin-protease complexes (36, 37). While this behavior is improved by replacing nine amino acids of the Conserpin RCL (P7-P2') by that of α1AT, it still underperforms in comparison to wild type α1AT. As such, further insight in to SERPIN-protease interaction is required to allow therapeutic SERPINs to be optimized for stability without it affecting their efficacy. Although the RCL is very important, there are other motifs present in SERPINs that are important for target engagement such as exosites.
Circulatory Half-Life
In humans, the circulatory half-life time between extracellular SERPINs differs quite significantly. For example, plasma-derived α1AT has a circulatory half-life of 4.5–8.7 days (38, 39). By comparison, its recombinant counterpart has a six-fold decrease in circulatory half-life (39, 40), which is thought to be the result of lacking, wrong or incomplete glycosylation. Although α1AT has been expressed in almost every host, the lack of proper glycosylation and circulatory half-life has been a major hurdle for any recombinant form of α1AT from reaching the market. Similar to α1AT, plasma-derived C1INH has a circulatory half-life of 22–56 h, but its recombinant variant (isolated from rabbit milk) only has a half-life of 2.4–3 h (41, 42). It is remarkable that prophylactic treatment with this molecule has therapeutic value (43), suggesting that it has biological properties unlike its natural counterpart which may facilitate alternative bio-distribution or cellular uptake which are beneficial to its therapeutic properties.
Enhanced Glycosylation
These obstacles have motivated efforts to optimize recombinant SERPIN glycosylation. Introduction of an additional N-glycosylation site (introduced via the Q9N mutation) was able to further increase the circulatory half-life of α1AT in rats (44). Recently, a modified CHO cell line was presented that delivered full humanized N-glycosylation profiles for both α1AT and the C1INH (45). Here, ten genes were knocked out to prevent glycosylation errors by the CHO cell line. Furthermore, the α-2,6-sialyltransferase enzyme (ST6GAL1) was overexpressed to improve capping of the N-glycans with alpha-2,6-linked sialic acid. While these recombinant variants of α1AT and C1INH exactly match their plasma derived counterparts when it comes to N-glycosylation profiles, their circulatory half-life times remain to be investigated.
Pegylation
To overcome the short circulation half-life of recombinant SERPINs, strategies have focused shielding the SERPIN via PEGylation (46–48). PEGylation of therapeutic proteins generally increases their biological stability and decreases their immunogenicity. Furthermore, PEGylation via a cysteine residue with an exposed thiol group (naturally present on certain SERPINs, including α1AT) is relatively straightforward and inexpensive. For recombinant α1AT, PEGylation did not influence its inhibitory potential in vitro, while pegylated α1AT variants (with 20 or 40 kDa PEG chains) showed increased circulation half-life, matching plasma-derived α1AT (48). Finally, in an in vivo elastase-mediated lung damage model, the PEGylated recombinant α1AT variants even outperformed its plasma derived counterpart.
Fusion Proteins
As an alternative approach to increase the circulation half-life, SERPINs have been fused to the Fc domains from IgG. This results in a homo-dimeric protein that should increase both efficacy and extend circulatory half-life (49). Currently, a phase I trial with α1AT-FC fusion protein (INBRX-101) is ongoing (https://clinicaltrials.gov/ct2/show/NCT03815396).
In vivo Expression
Trials with recombinant SERPINS are proven to be successful, patients would require weekly life-long therapy with injectables. As an attractive alternative, in vivo expression of SERPINs via gene therapy has been considered. For α1AT, both a phase I and II trial have been undertaken using a recombinant adeno-associated virus (AAV) vector which was administered to α1AT-deficient patients via intramuscular injection (50, 51). Patients tolerated the treatment and showed long term expression of α1AT. All subjects developed anti-AAV antibodies, but none developed antibodies against α1AT. While these studies confirmed the feasibility, patients only produced 20 μg/ml (0.38 μM) of α1AT in plasma serum, where therapeutic levels have to be at least 600 μg/ml (11.54 μM), where as normal levels are ~1.5 mg/ml (28.85 μM). While improved delivery of the gene therapy and improved SERPIN expression might help to overcome this problem, increasing the inhibitory activity of α1AT through mutagenesis might help to lower the levels that are required for therapy.
Tailoring SERPIN Efficacy and Specificity
The RCL together with exosites are the major regulators of SERPIN activity. Exosites can directly improve the SERPIN-protease interaction, whereas the RCL sequence determines the SERPIN specificity by controlling which proteases active sites can cleave it. Even a single amino acid mutation in the RCL can have functional consequences. For example, wild-type α1AT is a potent inhibitor of neutrophil elastase, trypsin, chymotrypsin, tissue kallikrein 7 & 14, cathepsin G, neutrophil proteinase 3 and pancreatic elastase (Table 1), but not of coagulation proteases. Contrastingly, the RCL P1 mutation M358R (α1AT-Pittsburgh) converts it into a potent inhibitor of thrombin, activated protein C (APC), plasmin, factor XIa, factor Xa, plasma kallikrein and factor XIIa (Table 1). As a net result, patients with α1AT-Pittsburgh suffer from a life-long bleeding disorder (66, 67). This experiment of nature shows the impact of small RCL modifications.
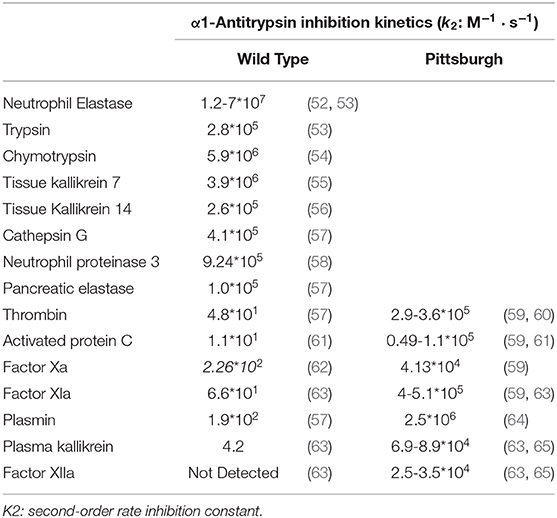
Table 1. The Pittsburgh (M358R) mutation dramatically alters α1-antitrypsin specificity.
Despite its pathological nature, α1AT-Pittsburgh has been investigated as treatment for coagulopathy and mortality in sepsis. While α1AT-Pittsburgh treatment decreased mortality and coagulopathy was reported in a piglet sepsis model (68), a baboon model was unable to confirm these results and even showed signs of increased coagulopathy (69). The overall consensus was that the inhibition of APC and plasmin in this setting were unfavorable.
Redesigning RCL Specificity
Various groups have attempted to refine the specificity of α1AT-Pittsburgh. Initial redesign of SERPIN specificity started as an “exchange program” by grafting RCL sequences onto different SERPINs backbones. This led to some success (70, 71); but was limited by the inhibitory behavior of the initial donor sequences. Although APC inhibition is considered unfavorable in the treatment of sepsis-related coagulopathy, Polderdijk et al. recently demonstrated that a refined α1AT variant (357KRK359), which selectively inhibits APC, has therapeutic value for the treatment of hemophilia A- and B (59). This molecule is currently in clinical development (https://www.clinicaltrials.gov/ct2/show/NCT04073498).
To unlock the true potential of SERPIN engineering for diverse diseases, further mutagenesis of the RCL is warranted. Yet, with each position that is mutagenized, the amount of total possibilities rises exponentially. Indeed, to fully mutagenize a sequence of eight amino acids (stretching the P4-P4' region) and use all 20 naturally occurring amino acids for, leads to a total of 2.56*1010 RCL sequences variants. Specific RCL positions have been thoroughly researched, which provides valuable information. For example, Schapira et al. showed that a single mutation helps to refine the inhibitory potential of α1AT-Pittsburgh (60). By altering the P2 position from a proline to an alanine (P357A) in α1AT-Pittsburgh, the inhibition of thrombin was diminished to the extent that it had no effects on the ex vivo thrombin time in plasma of Wistar rats. Interestingly, this 357AR358 mutation left the inhibition of FXIIa and PKa intact, protecting the rats in a model of bradykinin induced hypotensia. In 2002, Sulikowski et al. changed the RCL of α1AT-Pittsburgh into 356LGR358 or 356PFR358 to create a SERPIN to inhibit FXIIa, PKa and C1s (72). Where the 356LGR358mutant inhibited its designated targets, it also potently inhibited APC. By comparison, 356PFR358 showed an increase in specificity toward PKa. More recently, our group attempted to further improve the inhibition of the bradykinin producing proteases FXIIa and PKa. Based upon naturally occurring sequences and data from substrate peptide libraries, we created 18 α1AT variants. We found that only two new variants SMRT/V and SLLR/V (/ indicates RCL cleavage site) with a potent ability to inhibit FXIIa, PKa and FXIa, while showing negligible inhibition of thrombin, FXa and APC. These variants were effective in inflammatory models of carrageenan induced-paw swelling (driven by bradykinin) and dextran sulfate sodium-induced colitis as well as an injury-driven model of arterial thrombosis (73).
Peptide Libraries
While data from synthetic substrate peptide libraries (74) can be used to guide selection of lead RCL sequences, we experienced that data from these libraries unfortunately poorly translates into the wanted inhibitory behavior of full-length SERPINS (73). This probably relates to the non-linear structure of the RCL loop. To overcome this obstacle, others have performed high throughput SERPIN screening studies with the T7 phage display system (75, 76). While this method allowed to find thrombin inhibitors that are twice more potent that a1AT-Pittsburgh, their specificity toward other proteases remains to be investigated.
Viral SERPINs
Like humans, viruses also express SERPINs to inhibit targets in their respective hosts (77). Examples of viruses that express SERPINs are the Orthopoxviruses, Myxoma viruses, Cowpox virus, Baculovirus and the Swinepox virus. These “cross-class” SERPINs enhance infection and suppress host inflammatory responses. Deletion of these SERPINs dramatically reduces the lethality rates, showing that these SERPINs act as virulence factors (78, 79). The Myxovirus expresses the SERPIN Serp-1, which inhibits urokinase plasminogen activator (uPA), tissue plasminogen activator, factor Xa, plasmin, and thrombin (in the presence of heparin). Serp-1 requires the uPA receptor to function in vivo (80–82). Interestingly, Serp-1 effectively suppresses arterial inflammation and plaque growth (83–85). In addition, a peptide mimicking the Serp-1 RCL showed therapeutic benefits in a MHV68 virus-induced vasculitis mouse model (86). However, the activity and stability of this peptide was different from full-length Serp-1. Protein modeling studies were performed to improve the inhibitory (and antiplaque) activity of Serp-1-based peptides. The resulting peptides indeed displayed increased inhibitory activity and were able to increase the survival rate of the mice in a MHV68 infection model of IFNγR KO mice (87). This work demonstrates the power of protein-modeling and shows its value for SERPIN design.
Discussion
Over the past years, new molecular insights have been rapidly acquired that help recombinant SERPINs to fulfill their therapeutic promise. While the first recombinant SERPIN variants (the α1AT-FC fusion protein and the APC inhibiting α1AT variant) are moving into clinical development, the design of new SERPIN variants is still a very specialized and labor-intensive exercise. Improvements in molecular cloning strategies combined with protein modeling approaches will be of great importance to efficiently unlock the potential of SERPINs as therapeutic agents.
Author Contributions
CM and SM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the students of BMW2023 group 9, Biomedical Sciences at Utrecht University Medical Center for carefully reading the manuscript and providing constructive feedback. CM gratefully acknowledges the Landsteiner Foundation for Blood Transfusion Research and the Netherlands Thrombosis Foundation. SM gratefully acknowledges the TTW section of the Netherlands Organization for Scientific Research (NWO, 2019/TTW/00704802).
References
1. Bianchini EP, Auditeau C, Razanakolona M, Vasse M, Borgel D. Serpins in hemostasis as therapeutic targets for bleeding or thrombotic disorders. Front Cardiovasc Med. (2020) 7:622778. doi: 10.3389/fcvm.2020.622778
2. Bao J, Pan G, Poncz M, Wei J, Ran M, Zhou Z. Serpin functions in host-pathogen interactions. PeerJ. (2018) 6:e4557. doi: 10.7717/peerj.4557
3. Francois D, Venisse L, Marchal-Somme J, Jandrot-Perrus M, Crestani B, Arocas V, et al. Increased expression of protease nexin-1 in fibroblasts during idiopathic pulmonary fibrosis regulates thrombin activity and fibronectin expression. Lab Invest. (2014) 94:1237–46. doi: 10.1038/labinvest.2014.111
4. Aimes RT, Zijlstra A, Hooper JD, Ogbourne SM, Sit ML, Fuchs S, et al. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb Haemost. (2003) 89:561–572. doi: 10.1055/s-0037-1613388
5. Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, Gettins PG, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. (2001) 276:33293–6. doi: 10.1074/jbc.R100016200
6. Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, et al. An overview of the serpin superfamily. Genome Biol. (2006) 7:216. doi: 10.1186/gb-2006-7-5-216
7. Gooptu B, Ekeowa UI, Lomas DA. Mechanisms of emphysema in alpha1-antitrypsin deficiency: molecular and cellular insights. Eur Respir J. (2009) 34:475–88. doi: 10.1183/09031936.00096508
8. De Maat S, Hofman ZLM, Maas C. Hereditary angioedema: the plasma contact system out of control. J Thromb Haemost. (2018) 16:1674–85. doi: 10.1111/jth.14209
9. Marciniak E, Farley CH, DeSimone PA. Familial thrombosis due to antithrombin 3 deficiency. Blood. (1974) 43:219–31.
10. Elliott PR, Abrahams JP, Lomas DA. Wild-type alpha 1-antitrypsin is in the canonical inhibitory conformation. J Mol Biol. (1998) 275:419–25. doi: 10.1006/jmbi.1997.1458
11. Loebermann H, Tokuoka R, Deisenhofer J, Huber R. Human alpha 1-proteinase inhibitor. Crystal structure analysis of two crystal modifications, molecular model and preliminary analysis of the implications for function. J Mol Biol. (1984) 177:531–57.
12. Lawrence DA, Ginsburg D, Day DE, Berkenpas MB, Verhamme IM, Kvassman JO, et al. Serpin-protease complexes are trapped as stable acyl-enzyme intermediates. J Biol Chem. (1995) 270:25309–12. doi: 10.1074/jbc.270.43.25309
13. Stratikos, Gettins EPG. Formation of the covalent serpin-proteinase complex involves translocation of the proteinase by more than 70 A and full insertion of the reactive center loop into beta-sheet A. Proc Natl Acad Sci USA. (1999) 96:4808–13. doi: 10.1073/pnas.96.9.4808
14. Huntington JA, Read RJ, Carrell RW. (2000) Structure of a serpin-protease complex shows inhibition by deformation. Nature. (1999) 407:923–26. doi: 10.1038/35038119
15. Kaslik G, Kardos J, Szabo E, Szilagyi L, Zavodszky P, Westler WM, et al. Effects of serpin binding on the target proteinase: global stabilization, localized increased structural flexibility and conserved hydrogen bonding at the active site. Biochemistry. (1997) 36:5455–64. doi: 10.1021/bi962931m
16. Li, Huntington WJA. The heparin binding site of protein C inhibitor is protease-dependent. J Biol Chem. (2008) 283:36039–45. doi: 10.1074/jbc.M805974200
17. Ehrlich HJ, Keijer J, Preissner KT, Gebbink RK, Pannekoek H. Functional interaction of plasminogen activator inhibitor type 1 (PAI-1) and heparin. Biochemistry. (1991) 30:1021–1028. doi: 10.1021/bi00218a020
18. Cunningham DD, Wagner SL, Farrell D. H. Regulation of protease nexin-1 activity by heparin and heparan sulfate. Adv Exp Med Biol. (1992) 313:297–306. doi: 10.1007/978-1-4899-2444-5_29
19. Huang X, Rezaie AR, Broze GJ Jr. Olson S. T. Heparin is a major activator of the anticoagulant serpin, protein Z-dependent protease inhibitor. J Biol Chem. (2011) 286:8740–8751. doi: 10.1074/jbc.M110.188375
20. Olson ST, Bjork I. Predominant contribution of surface approximation to the mechanism of heparin acceleration of the antithrombin-thrombin reaction. Elucidation from salt concentration effects. J Biol Chem. (1991) 266:6353–64.
21. Verhamme IM, Bock PE, Jackson C. M. The preferred pathway of glycosaminoglycan-accelerated inactivation of thrombin by heparin cofactor II. J Biol Chem. (2004) 279:9785–95. doi: 10.1074/jbc.M313962200
22. Beeler D, Rosenberg R, Jordan R. Fractionation of low molecular weight heparin species and their interaction with antithrombin. J Biol Chem. (1979) 254:2902–13.
23. Wijeyewickrema LC, Lameignere E, Hor L, Duncan RC, Shiba T, Travers RJ, et al. Polyphosphate is a novel cofactor for regulation of complement by a serpin, C1 inhibitor. Blood. (2016) 128:1766–76. doi: 10.1182/blood-2016-02-699561
24. Behrens MA, Sendall TJ, Pedersen JS, Kjeldgaard M, Huntington JA, Jensen JK. The shapes of Z-alpha1-antitrypsin polymers in solution support the C-terminal domain-swap mechanism of polymerization. Biophys J. (2014) 107:1905–12. doi: 10.1016/j.bpj.2014.08.030
25. Haslund D, Ryo LB, Seidelin Majidi S, Rose I, Skipper KA, Fryland T, et al. Dominant-negative SERPING1 variants cause intracellular retention of C1 inhibitor in hereditary angioedema. J Clin Invest. (2019) 129:388–405. doi: 10.1172/JCI98869
26. Kwon KS, Kim J, Shin HS, Yu MH. Single amino acid substitutions of alpha 1-antitrypsin that confer enhancement in thermal stability. J Biol Chem. (1994) 269:9627–31.
27. Kim J, Lee KN, Yi GS, Yu MH. A thermostable mutation located at the hydrophobic core of alpha 1-antitrypsin suppresses the folding defect of the Z-type variant. J Biol Chem. (1995) 270:8597–601. doi: 10.1074/jbc.270.15.8597
28. Trinh HN, Jang SH, Lee C. Functional characterization of a SNP (F51S) found in human alpha 1-antitrypsin. Mol Genet Genomic Med. (2019) 7:e819. doi: 10.1002/mgg3.819
29. Lee KN, Park SD, Yu MH. Probing the native strain iin alpha1-antitrypsin. Nat Struct Biol. (1996) 3:497–500. doi: 10.1038/nsb0696-497
30. Lee KN, Im H, Kang SW, Yu MH. Characterization of a human alpha1-antitrypsin variant that is as stable as ovalbumin. J Biol Chem. (1998) 273:2509–16. doi: 10.1074/jbc.273.5.2509
31. Seo EJ, Im H, Maeng JS, Kim KE, Yu MH. Distribution of the native strain in human alpha 1-antitrypsin and its association with protease inhibitor function. J Biol Chem. (2000) 275:16904–09. doi: 10.1074/jbc.M001006200
32. Berkenpas MB, Lawrence DA, Ginsburg D. Molecular evolution of plasminogen activator inhibitor-1 functional stability. EMBO J. (1995) 14:2969–77.
33. Vleugels N, Gils A, Mannaerts S, Knockaert I, Declerck PJ. (1998) Evaluation of the mechanism of inactivation of plasminogen activator inhibitor-1 by monoclonal antibodies using a stable variant. Fibrinolysis Proteolysis. (1998) 12:277–82. doi: 10.1016/S0268-9499(98)80020-9
34. Stoop AA, Eldering E, Dafforn TR, Read RJ, Pannekoek H. Different structural requirements for plasminogen activator inhibitor 1 (PAI-1) during latency transition and proteinase inhibition as evidenced by phage-displayed hypermutated PAI-1 libraries. J Mol Biol. (2001) 305:773–83. doi: 10.1006/jmbi.2000.4356
35. Chorostowska-Wynimko J, Swiercz R, Skrzypczak-Jankun E, Wojtowicz A, Selman SH, Jankun J. A novel form of the plasminogen activator inhibitor created by cysteine mutations extends its half-life: relevance to cancer and angiogenesis. Mol Cancer Ther. (2003) 2:19–28.
36. Porebski BT, Keleher S, Hollins JJ, Nickson AA, Marijanovic EM, Borg NA, et al. Smoothing a rugged protein folding landscape by sequence-based redesign. Sci Rep. (2016) 6:33958. doi: 10.1038/srep33958
37. Yang L, Irving JA, Dai W, Aguilar MI, Bottomley SP. Probing the folding pathway of a consensus serpin using single tryptophan mutants. Sci Rep. (2018) 8:2121. doi: 10.1038/s41598-018-19567-9
38. Vidal Pla R, Padulles Zamora N, Sala Pinol F, Jardi Margaleff R, Rodriguez Frias F, Montoro Ronsano JB. [Pharmacokinetics of alpha1-antitrypsin replacement therapy in severe congenital emphysema]. Arch Bronconeumol. (2006) 42:553–6.
39. Wewers MD, Casolaro MA, Sellers SE, Swayze SC, McPhaul KM, Wittes JT, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. (1987) 316:1055–62. doi: 10.1056/NEJM198704233161704
40. Arjmand S, Bidram E, Lotfi AS, Shamsara M, Mowla SJ. Expression and purification of functionally active recombinant human alpha 1-antitrypsin in methylotrophic yeast pichia pastoris. Avicenna J Med Biotechnol. (2011) 3:127–34.
41. Bernstein JA, Ritchie B, Levy RJ, Wasserman RL, Bewtra AK, Hurewitz DS, et al. Population pharmacokinetics of plasma-derived C1 esterase inhibitor concentrate used to treat acute hereditary angioedema attacks. Ann Allergy Asthma Immunol. (2010) 105:149–54. doi: 10.1016/j.anai.2010.06.005
42. Farrell C, Hayes S, Relan A, van Amersfoort ES, Pijpstra R, Hack CE. Population pharmacokinetics of recombinant human C1 inhibitor in patients with hereditary angioedema. Br J Clin Pharmacol. (2013) 76:897–907. doi: 10.1111/bcp.12132
43. Riedl MA, Grivcheva-Panovska V, Moldovan D, Baker J, Yang WH, Giannetti BM, et al. Recombinant human C1 esterase inhibitor for prophylaxis of hereditary angio-oedema: a phase 2, multicentre, randomised, double-blind, placebo-controlled crossover trial. Lancet. (2017) 390:1595–602. doi: 10.1016/S0140-6736(17)31963-3
44. Chung HS, Kim JS, Lee SM, Park SJ. Additional N-glycosylation in the N-terminal region of recombinant human alpha-1 antitrypsin enhances the circulatory half-life in Sprague-Dawley rats. Glycoconj J. (2016) 33:201–8. doi: 10.1007/s10719-016-9657-3
45. Amann T, Hansen AH, Kol S, Hansen HG, Arnsdorf J, Nallapareddy S, et al. Glyco-engineered CHO cell lines producing alpha-1-antitrypsin and C1 esterase inhibitor with fully humanized N-glycosylation profiles. Metab Eng. (2019) 52:143–52. doi: 10.1016/j.ymben.2018.11.014
46. Mast AE, Salvesen G, Brucato FH, Schnebli HP. Pizzo SV. Polyethylene glycol modification of serpins improves therapeutic potential. Biol Chem Hoppe Seyler. (1990) 371:101–9.
47. Mast AE, Salvesen G, Schnebli HP, Pizzo SV. Evaluation of the rapid plasma elimination of recombinant alpha 1-proteinase inhibitor: synthesis of polyethylene glycol conjugates with improved therapeutic potential. J Lab Clin Med. (1990) 116:58–65.
48. Cantin AM, Woods DE, Cloutier D, Dufour EK, Leduc R. Polyethylene glycol conjugation at Cys232 prolongs the half-life of alpha1 proteinase inhibitor. Am J Respir Cell Mol Biol. (2002) 27:659–65. doi: 10.1165/rcmb.4866
49. Lee S, Lee Y, Hong K, Hong J, Bae S, Choi J, et al. Effect of recombinant alpha1-antitrypsin Fc-fused (AAT-Fc)protein on the inhibition of inflammatory cytokine production and streptozotocin-induced diabetes. Mol Med. (2013) 19:65–71. doi: 10.2119/molmed.2012.00308
50. Brantly ML, Spencer LT, Humphries M, Conlon TJ, Spencer CT, Poirier A, et al. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 alphal-antitrypsin (AAT) vector in AAT-deficient adults. Hum Gene Ther. (2006) 17:1177–86. doi: 10.1089/hum.2006.17.1177
51. Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo RF, et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: interim results. Hum Gene Ther. (2011) 22:1239–47. doi: 10.1089/hum.2011.053
52. Hopkins PC, Carrell RW, Stone SR. Effects of mutations in the hinge region of serpins. Biochemistry. (1993) 32:7650–57. doi: 10.1021/bi00081a008
53. Avron A, Reeve FH, Lickorish JM, Carrell RW. Effect of alanine insertion (P'5) on the reactive centre of alpha 1-antitrypsin. FEBS Lett. (1991) 280:41–3. doi: 10.1016/0014-5793(91)80199-d
54. Djie MZ, Stone SR, Le Bonniec BF. Intrinsic specificity of the reactive site loop of alpha1-antitrypsin, alpha1-antichymotrypsin, antithrombin III and protease nexin I. J Biol Chem. (1997) 272:16268–73. doi: 10.1074/jbc.272.26.16268
55. Luo LY, Jiang W. Inhibition profiles of human tissue kallikreins by serine protease inhibitors. Biol Chem. (2006) 387:813–6. doi: 10.1515/BC.2006.103
56. Felber LM, Kundig C, Borgono CA, Chagas JR, Tasinato A, Jichlinski P, et al. Mutant recombinant serpins as highly specific inhibitors of human kallikrein 14. FEBS J. (2006) 273:2505–14. doi: 10.1111/j.1742-4658.2006.05257.x
57. Beatty K, Bieth J, Travis J. Kinetics of association of serine proteinases with native and oxidized alpha-1-proteinase inhibitor and alpha-1-antichymotrypsin. J Biol Chem. (1980) 255:3931–34.
58. Sinden NJ, Baker MJ, Smith DJ, Kreft JU, Dafforn TR, Stockley RA. alpha-1-antitrypsin variants and the proteinase/antiproteinase imbalance in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L179–L190. doi: 10.1152/ajplung.00179.2014
59. Polderdijk SG, Adams TE, Ivanciu L, Camire RM, Baglin TP, Huntington JA. Design and characterization of an APC-specific serpin for the treatment of hemophilia. Blood. (2017) 129:105–13. doi: 10.1182/blood-2016-05-718635
60. Schapira M, Ramus MA, Waeber B, Brunner HR, Jallat SD, et al. Protection by recombinant alpha 1-antitrypsin Ala357 Arg358 against arterial hypotension induced by factor XII fragment. J Clin Invest. (1987) 80:582–5. doi: 10.1172/JCI113108
61. Heeb MJ, Bischoff R, Courtney M, Griffin JH. Inhibition of activated protein C by recombinant alpha 1-antitrypsin variants with substitution of arginine or leucine for methionine358. J Biol Chem. (1990) 265:2365–69.
62. Ellis V, Scully M, MacGregor I, Kakkar V. Inhibition of human factor Xa by various plasma protease inhibitors. Biochim Biophys Acta. (1982) 701:24–31.
63. Scott CF, Carrell RW, Glaser CB, Kueppers F, Lewis JH, Colman RW. Alpha-1-antitrypsin-Pittsburgh. A potent inhibitor of human plasma factor XIa, kallikrein and factor XIIf. J Clin Invest. (1986) 77:631–4. doi: 10.1172/JCI112346
64. Patston PA, Roodi N, Schifferli JA, Bischoff R, Courtney M, Schapira M. Reactivity of alpha 1-antitrypsin mutants against proteolytic enzymes of the kallikrein-kinin, complement and fibrinolytic systems. J Biol Chem. (1990) 265:10786–91.
65. Schapira M, Ramus MA, Jallat S, Carvallo D, Courtney M. Recombinant alpha 1-antitrypsin Pittsburgh (Met 358—-Arg) is a potent inhibitor of plasma kallikrein and activated factor XII fragment. J Clin Invest. (1986) 77:635–7. doi: 10.1172/JCI112347
66. Lewis JH, Iammarino RM, Spero JA, Hasiba U. Antithrombin Pittsburgh: an alpha1-antitrypsin variant causing hemorrhagic disease. Blood. (1978) 51:129–37.
67. Owen MC, Brennan SO, Lewis JH, Carrell RW. Mutation of antitrypsin to antithrombin. alpha 1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N Engl J Med. (1983) 309:694–8. doi: 10.1056/NEJM198309223091203
68. Colman RW, Flores DN, De La Cadena RA, Scott CF, Cousens L, Barr PJ, et al. Recombinant alpha 1-antitrypsin Pittsburgh attenuates experimental gram-negative septicemia. Am J Pathol. (1988) 130:418–26.
69. Harper PL, Taylor FB, DeLa Cadena RA, Courtney M, Colman RW, Carrell RW. Recombinant antitrypsin Pittsburgh undergoes proteolytic cleavage during E.coli sepsis and fails to prevent the associated coagulopathy in a primate model. Thromb Haemost. (1998) 80:816–21.
70. Hopkins PC, Crowther DC, Carrell RW, Stone SR. Development of a novel recombinant serpin with potential antithrombotic properties. J Biol Chem. (1995) 270:11866–71. doi: 10.1074/jbc.270.20.11866
71. Hopkins PC, Pike RN, Stone SR. Evolution of serpin specificity: cooperative interactions in the reactive-site loop sequence of antithrombin specifically restrict the inhibition of activated protein C. J Mol Evol. (2000) 51:507–15. doi: 10.1007/s002390010114
72. Sulikowski T, Bauer BA, Patston PA. alpha(1)-Proteinase inhibitor mutants with specificity for plasma kallikrein and C1s but not C1. Protein Sci. (2002) 11:2230–6. doi: 10.1110/ps.0207302
73. de Maat S, Sanrattana W, Mailer RK, Parr NMJ, Hessing M, Koetsier RM, et al. Design and characterization of alpha1-antitrypsin variants for treatment of contact system-driven thromboinflammation. Blood. (2019) 134:1658–69. doi: 10.1182/blood.2019000481
74. Gosalia DN, Salisbury CM, Ellman JA, Diamond SL. High throughput substrate specificity profiling of serine and cysteine proteases using solution-phase fluorogenic peptide microarrays. Mol Cell Proteomics. (2005) 4:626–36. doi: 10.1074/mcp.M500004-MCP200
75. Scott BM, Matochko WL, Gierczak RF, Bhakta V, Derda R, Sheffield WP. Phage display of the serpin alpha-1 proteinase inhibitor randomized at consecutive residues in the reactive centre loop and biopanned with or without thrombin. PLoS ONE. (2014) 9:e84491. doi: 10.1371/journal.pone.0084491
76. de Souza LR, Scott BM, Bhakta V, Donkor DA, Perruzza DL, Sheffield WP. Serpin phage display: the use of a T7 system to probe reactive center loop libraries with different serine proteinases. Methods Mol Biol. (2018) 1826:41–64. doi: 10.1007/978-1-4939-8645-3_3
77. Yaron JR, Zhang L, Guo Q, Burgin M, Schutz LN, Awo E, et al. Deriving immune modulating drugs from viruses-a new class of biologics. J Clin Med. (2020) 9:972. doi: 10.3390/jcm9040972
78. Upton C, Macen JL, Wishart DS, McFadden G. Myxoma virus and malignant rabbit fibroma virus encode a serpin-like protein important for virus virulence. Virology. (1990) 179:618–31. doi: 10.1016/0042-6822(90)90129-f
79. Lomas DA, Evans DL, Upton C, McFadden G, Carrell RW. Inhibition of plasmin, urokinase, tissue plasminogen activator and C1S by a myxoma virus serine proteinase inhibitor. J Biol Chem. (1993) 268:516–21.
80. Nash P, Whitty A, Handwerker J, Macen J, McFadden G. Inhibitory specificity of the anti-inflammatory myxoma virus serpin, SERP-1. J Biol Chem. (1998) 273:20982–91. doi: 10.1074/jbc.273.33.20982
81. Li X, Schneider H, Peters A, Macaulay C, King E, Sun Y, et al. Heparin alters viral serpin, serp-1, anti-thrombolytic activity to anti-thrombotic activity. Open Biochem J. (2008)2:6–15. doi: 10.2174/1874091X00802010006
82. Viswanathan K, Richardson J, Togonu-Bickersteth B, Dai E, Liu L, Vatsya P, et al. Myxoma viral serpin, Serp-1, inhibits human monocyte adhesion through regulation of actin-binding protein filamin B. J Leukoc Biol. (2008) 85:418–26. doi: 10.1189/jlb.0808506
83. Dai E, Viswanathan K, Sun Y, Li M, Liu X, Togonu-Bickersteth LY, et al. Identification of myxomaviral serpin reactive site loop sequences that regulate innate immune responses. J Biol Chem. (2006) 281:8041–50. doi: 10.1074/jbc.M509454200
84. Bedard EL, Jiang J, Arp J, Qian H, Wang H, Guan H, et al. Prevention of chronic renal allograft rejection by SERP-1 protein. Transplantation. (2006) 81:908–14. doi: 10.1097/01.tp.0000203141.02725.8a
85. Jiang J, Arp J, Kubelik D, Zassoko R, Liu W, Wise Y, et al. Induction of indefinite cardiac allograft survival correlates with toll-like receptor 2 and 4 downregulation after serine protease inhibitor-1 (Serp-1) treatment. Transplantation. (2007) 84:1158–67. doi: 10.1097/01.tp.0000286099.50532.b0
86. Ambadapadi S, Munuswamy-Ramanujam G, Zheng D, Sullivan C, Dai E, Morshed S, et al. Reactive center loop (RCL) peptides derived from serpins display independent coagulation and immune modulating activities. J Biol Chem. (2016) 291:2874–87. doi: 10.1074/jbc.M115.704841
87. Mahon BP, Ambadapadi S, Yaron JR, Lomelino CL, Pinard MA, Keinan S, et al. Crystal structure of cleaved Serp-1, a myxomavirus-derived immune modulating serpin: structural design of serpin reactive center loop peptides with improved therapeutic function. Biochemistry. (2018) 57:1096–107. doi: 10.1021/acs.biochem.7b01171
Keywords: SERPIN, α1-antitrypsin, C1 esterase inhibitor, reactive center loop, therapy
Citation: Maas C and de Maat S (2021) Therapeutic SERPINs: Improving on Nature. Front. Cardiovasc. Med. 8:648349. doi: 10.3389/fcvm.2021.648349
Received: 31 December 2020; Accepted: 10 March 2021;
Published: 31 March 2021.
Edited by:
Marie-Christine Bouton, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Margarethe Geiger, Medical University of Vienna, AustriaLakshmi Carmel Wijeyewickrema, La Trobe University, Australia
Copyright © 2021 Maas and de Maat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven de Maat, s.demaat@umcutrecht.nl