Modulated Calcium Homeostasis and Release Events Under Atrial Fibrillation and Its Risk Factors: A Meta-Analysis
 Sarah Pei Ting Fong
Sarah Pei Ting Fong Shaleka Agrawal
Shaleka Agrawal Mengqi Gong
Mengqi Gong Jichao Zhao
Jichao Zhao- 1Auckland Bioengineering Institute, The University of Auckland, Auckland, New Zealand
- 2Department of Cardiology, The First Affiliated Hospital of Anhui Medical University, Hefei, China
Background: Atrial fibrillation (AF) is associated with calcium (Ca2+) handling remodeling and increased spontaneous calcium release events (SCaEs). Nevertheless, its exact mechanism remains unclear, resulting in suboptimal primary and secondary preventative strategies.
Methods: We searched the PubMed database for studies that investigated the relationship between SCaEs and AF and/or its risk factors. Meta-analysis was used to examine the Ca2+ mechanisms involved in the primary and secondary AF preventative groups.
Results: We included a total of 74 studies, out of the identified 446 publications from inception (1982) until March 31, 2020. Forty-five were primary and 29 were secondary prevention studies for AF. The main Ca2+ release events, calcium transient (standardized mean difference (SMD) = 0.49; I2 = 35%; confidence interval (CI) = 0.33–0.66; p < 0.0001), and spark amplitude (SMD = 0.48; I2 = 0%; CI = −0.98–1.93; p = 0.054) were enhanced in the primary diseased group, while calcium transient frequency was increased in the secondary group. Calcium spark frequency was elevated in both the primary diseased and secondary AF groups. One of the key cardiac currents, the L-type calcium current (ICaL) was significantly downregulated in primary diseased (SMD = −1.07; I2 = 88%; CI = −1.94 to −0.20; p < 0.0001) and secondary AF groups (SMD = −1.28; I2 = 91%; CI = −2.04 to −0.52; p < 0.0001). Furthermore, the sodium–calcium exchanger (INCX) and NCX1 protein expression were significantly enhanced in the primary diseased group, while only NCX1 protein expression was shown to increase in the secondary AF studies. The phosphorylation of the ryanodine receptor at S2808 (pRyR-S2808) was significantly elevated in both the primary and secondary groups. It was increased in the primary diseased and proarrhythmic subgroups (SMD = 0.95; I2 = 64%; CI = 0.12–1.79; p = 0.074) and secondary AF group (SMD = 0.66; I2 = 63%; CI = 0.01–1.31; p < 0.0001). Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) expression was elevated in the primary diseased and proarrhythmic drug subgroups but substantially reduced in the secondary paroxysmal AF subgroup.
Conclusions: Our study identified that ICaL is reduced in both the primary and secondary diseased groups. Furthermore, pRyR-S2808 and NCX1 protein expression are enhanced. The remodeling leads to elevated Ca2+ functional activities, such as increased frequencies or amplitude of Ca2+ spark and Ca2+ transient. The main difference identified between the primary and secondary diseased groups is SERCA expression, which is elevated in the primary diseased group and substantially reduced in the secondary paroxysmal AF subgroup. We believe our study will add new evidence to AF mechanisms and treatment targets.
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia, with markedly increasing prevalence (1, 2). It is associated with significant mortality and morbidity and becomes more challenging to treat as it advances (3–5). AF is mainly managed by primary and/or secondary preventative therapies. Primary prevention includes early detection and intervention on risk factors before AF develops, while secondary prevention involves diagnosing and treating AF (4). However, current pharmacological strategies are often associated with limited efficacy and adverse consequences, mainly due to an incomplete understanding of underlying cellular mechanisms related to AF (4, 6). In particular, calcium (Ca2+) is one of the most crucial ions for cardiac excitation–contraction coupling and Ca2+-dependent signaling pathways for maintaining cardiac function (7, 8).
Intracellular Ca2+ release events are exclusively investigated in myocardial physiology and pathophysiology, as they hold the key to understanding how cardiomyocyte Ca2+ signaling is regulated by ionic channels and Ca2+ proteins (7, 8). In a single cardiac cycle, the L-type calcium channels (LTCCs) localized on the sarcolemma and tubules are first activated (9). The opening of LTCCs results in the movement of Ca2+ into the cytosol, which induces the cardiac type 2 ryanodine receptors (RyR2) located on the junctional sarcoplasmic reticulum (SR) to release Ca2+ from its stores into the cytosol (9–11). This elementary Ca2+ release event is observed as a form of a Ca2+ spark, and the process is known as calcium-induced calcium release (12). Increases in highly localized, short-lived Ca2+ signals raise intracellular Ca2+ [Ca2+]i, which contributes to global Ca2+ waves or transients that propagate through the cell (10, 13). [Ca2+]i then binds to troponin to allow myosin adenosine triphosphatase (ATPase) to bind to actin in the sarcomere to initiate cardiac contraction (9, 14). Ca2+ is mainly recycled back into the SR via the SR Ca2+-ATPase (SERCA2a) pump or extruded across the cell membrane through the cardiac sodium–calcium exchanger (NCX1) (15). The reduction in [Ca2+]i causes Ca2+ to dissociate from troponin and terminate myofilament cross-bridge cycling for cardiac relaxation (9, 16). SERCA2a activity is directly modulated by phospholamban (PLN). In its unphosphorylated state, PLN acts as an inhibitor to SERCA2a. When phosphorylated by protein kinase A (PKA), PLN dislodges from SERCA2a to enable the reuptake of Ca2+ (9). Another important signaling protein besides PKA is the Ca2+/calmodulin-dependent protein kinase II (CAMKII), which is responsible for transducing cytosolic Ca2+, and calmodulin, a Ca2+-binding messenger protein that modulates RyR activity and transduces Ca2+ signals to other protein kinases or phosphatases (17–19).
In diseased states, spontaneous Ca2+ release events (SCaEs) are observed as spontaneous Ca2+ sparks or arrhythmogenic Ca2+ waves are substantially enhanced (20, 21). Such defective Ca2+ homeostasis often results from remodeled Ca2+-handling proteins (22–24). However, current studies reported conflicting results on how these Ca2+-handling proteins were remodeled in AF and its risk factors, which hinders the development of effective AF treatment and prevention. In this study, we aim to illustrate the precise mechanisms and targeted therapies for AF and AF prevention by investigating the pathophysiological role of Ca2+ and its arrhythmogenicity. This systematic review has compared the different Ca2+ mechanisms between the primary and secondary AF preventative groups in the existing studies to date.
Methods
This systematic review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines (refer to the PRISMA 2009 checklist in Supplementary Table 1) (25).
Search Strategy and Eligibility
The systematic electronic search was performed using the terms “atrial fibrillation” AND “calcium wave”/“calcium transient”/“calcium spark” in all fields to identify articles in PubMed from inception through March 31, 2020. Based on their titles and abstracts, the searched articles were screened manually for inclusion. Screening criteria included publications that mapped SCaEs in atrial cardiomyocytes in sinus rhythm and/or AF. Publications that did not conduct any experimental studies on atrial cells, such as mathematical modeling, population-based or organ-level studies, review papers, and editorial reports, were excluded. We also excluded papers that focused on genes and/or miRNA, signaling pathways, tissue, and organelle calcium experimental studies. Two co-authors then reviewed the screened articles in full text for eligibility, and those that met the criteria were selected. Any discrepancies were resolved by a third author through discussion and consensus. Full details of the search terms were presented in Supplementary Table 1. The quality of the included studies was assessed according to the Newcastle–Ottawa Scale (Supplementary Table 2) (26). A study with a score of 5 and above was considered satisfactory.
Data Extraction
The atrial cellular activities were extracted from selected studies. It included SCaEs [Ca2+ spark frequency (CaSpF), Ca2+ transient frequency (CaTF), Ca2+ spark amplitude (CaSpA), and Ca2+ transient amplitude (CaTA)], and Ca2+ load and leak. It also included atrial current densities, such as L-type calcium current (ICaL), sodium–calcium exchanger current (INCX), late sodium current (INa−Late), and potassium current (IK), and protein expressions, such as L-type alpha 1C subunit voltage-dependent calcium current (Cav1.2), NCX1, RyR2, phosphorylated ryanodine receptor 2 (pRyR2), SERCA2a, PLN, phosphorylated phospholamban (pPLN), CAMKII, phosphorylated Ca2+/calmodulin-dependent protein kinase II (pCAMKII), and PKA. We further categorized the above results into two main groups: primary and secondary preventative therapies for AF. Primary prevention was divided into three subclasses: the diseased group (risk factors for AF), the application of proarrhythmogenic agents or antiarrhythmogenic agents. Secondary prevention was classified into either the paroxysmal or chronic AF group. The analysis was conducted using R.
Data Synthesis and Statistical Analysis
Statistical analyses were performed using R (27). Dichotomous values were used to calculate 95% confidence interval (CI) of relative risk ratios, and continuous values to quantify standardized mean difference (SMD). Each study was given a weighting factor to determine its importance in the meta-analysis, which was represented by gray boxes in forest plots. When the boundaries of the CI were within the box, a white horizontal line was plotted; otherwise, it was illustrated by a black horizontal line. Studies with the CI not crossing zero were deemed to be statistically significant.
The overall SMD was interpreted using Cohen's guidelines (28), where a value of 0.2–0.49 was deemed to be small, 0.5–0.79 represented medium, and 0.8 and above was large. Statistical heterogeneity was calculated using I2 for all studies (29). In general, heterogeneity was classified into three main categories, low, medium, and high, when I2 values were ≤ 25%, between 25 and 50%, and ≥75%, respectively. Statistical significance was measured with p-values. We considered a result to be statistically significant when p-value was ≤ 0.05. We also employed influence analysis and graphic display of heterogeneity (GOSH) plots to detect influential studies and remove outliers (27).
Results
Study Characteristics
Our literature search identified a total of 446 publications from inception (1982) to March 31, 2020 (Figure 1). When screening the titles and abstracts, a total of 372 papers were excluded (Supplementary Table 3): 25 articles focused on other diseases instead of AF, 101 included AF but did not conduct experiments on SCaEs, 13 papers mentioned signaling pathways but not Ca2+, 23 were tissue or organelle studies, another 24 studied mRNA and genes, 27 indicated mathematical models, 23 articles were controlled trials or case reports, and 136 were review articles. Eventually, a total of 74 studies (12, 24, 30–100), consisting of 45 primary and 29 secondary prevention studies for AF, were eligible and included for this systematic review.
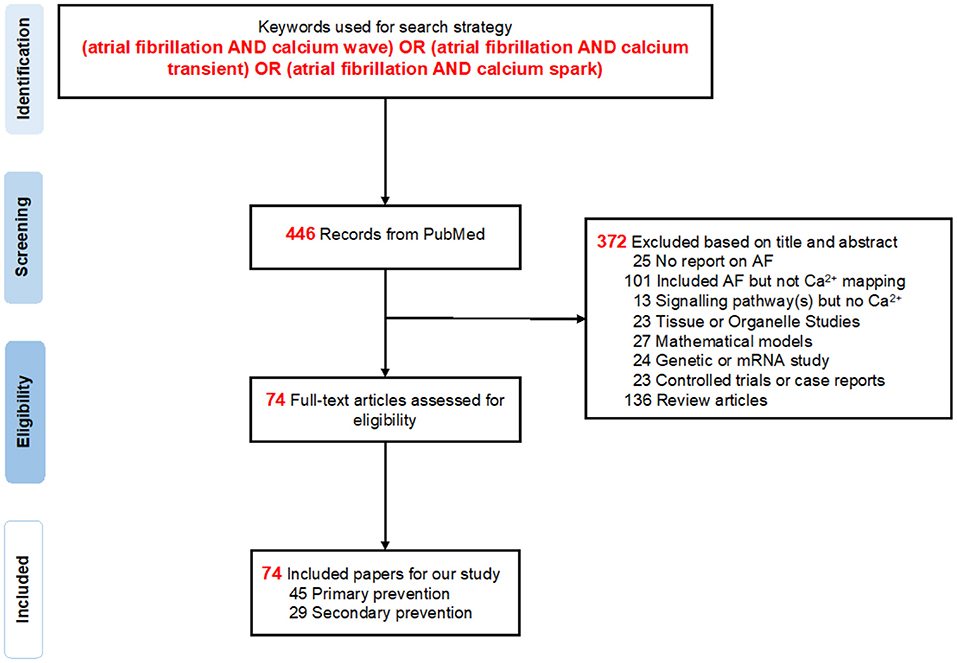
Figure 1. PRISMA flow diagram. A total of 446 records were found in PubMed, from which 74 studies were selected for further analysis in this systematic review.
Based on the 74 selected studies, pharmacological targets were grouped by their mechanism of action on the ionic channel(s) or protein(s). We discovered that ICaL was the most widely studied current in both primary and secondary pharmacological therapy for AF, followed by INCX and RyR2 channels (Figure 2). This coincides with the present targeted drug therapies available for AF, where LTCC antagonists are one of the most frequently prescribed drugs for the treatment of hypertension and AF. Figure 2 aids us in understanding and exploring other potential pathways for therapeutic drug discovery, such as the INCX and RyR2. It is noteworthy that the late sodium current was only commonly studied for primary prevention.
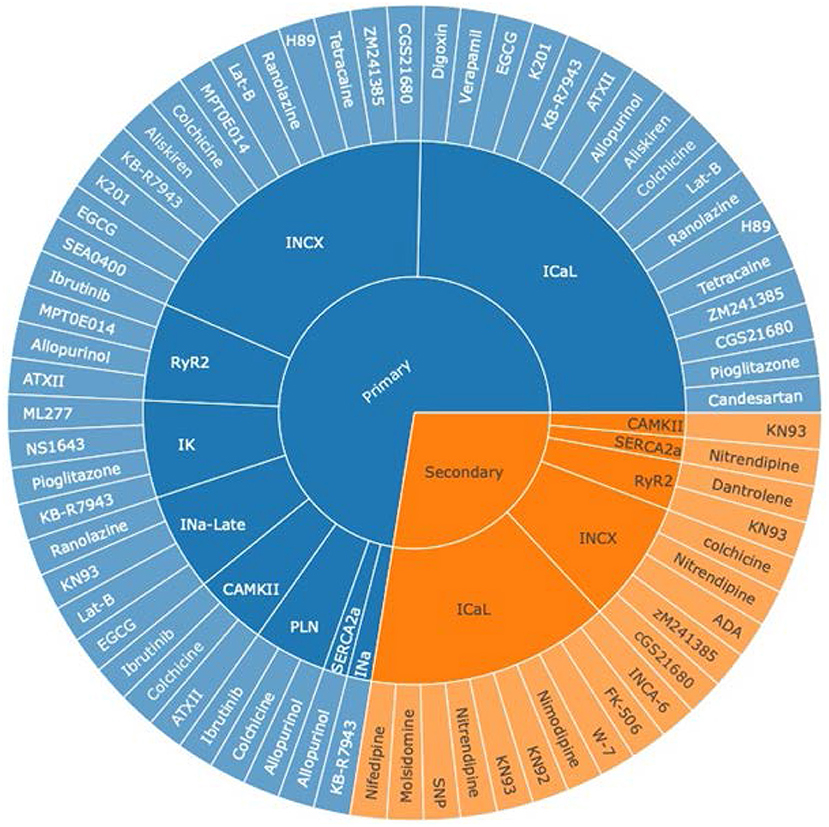
Figure 2. An overview of pharmacologically targeted ionic channels or proteins for AF treatment, grouped by primary (blue) and secondary (orange) prevention. The sunburst plot's core was divided into two groups, primary and secondary prevention for AF, where the middle section represented ion channels, and the outer segment symbolized various drug therapies. INCX stands for the sodium–calcium exchanger current; ICaL, the L-type calcium current; RyR2, the ryanodine receptor 2; IK, the potassium current; INa−Late, the late sodium current; CAMKII, the Ca2+/calmodulin-dependent protein kinase II; PLN, phospholamban; SERCA2a, the sarco/endoplasmic reticulum Ca2+-ATPase 2a pump; INa, the sodium current.
Spontaneous Calcium Release Events
The evolution of cardiac Ca2+ waves is influenced by local elevations of [Ca2+]i, seen as Ca2+ sparks. The properties (frequency and amplitude) of these Ca2+ release events are key determinants to the arrhythmogenicity of the cardiomyocytes. In our study, key calcium-handling remodeling including ionic currents, calcium release events, and protein expressions was summarized for the primary prevention and secondary AF groups in Figure 3. In the primary prevention group, CaSpF was significantly enhanced in the diseased subgroup (SMD = 0.6; I2 = 0%; CI = 0.30–0.89; p = 0.6601) and proarrhythmic drug subgroup (SMD = 0.89; I2 = 79%; CI = 0.48–1.30; p < 0.0001) (Supplementary Figures 1A,B). When these results were combined from both subgroups, they displayed a similar result (SMD = 0.81; I2 = 71%; CI = 0.54–1.09; p < 0.0001) (Figure 3A). The addition of antiarrhythmic drugs significantly decreased CaSpF (SMD = −0.80; I2 = 54%; CI = −0.97 to −0.62; p = 0.0054) (Figure 3B). A similar trend was observed for CaTA. CaTA was increased in the diseased subgroup (SMD = 0.49; I2 = 35%; CI = 0.33–0.66; p < 0.0001) (Figure 3C) and reduced by antiarrhythmic drugs (SMD = −0.79; I2 = 53%; CI = −1.00 to −0.58; p = 0.0002) (Figure 3D). In addition, CaSpA was enhanced in the diseased subgroup (Supplementary Figure 4).
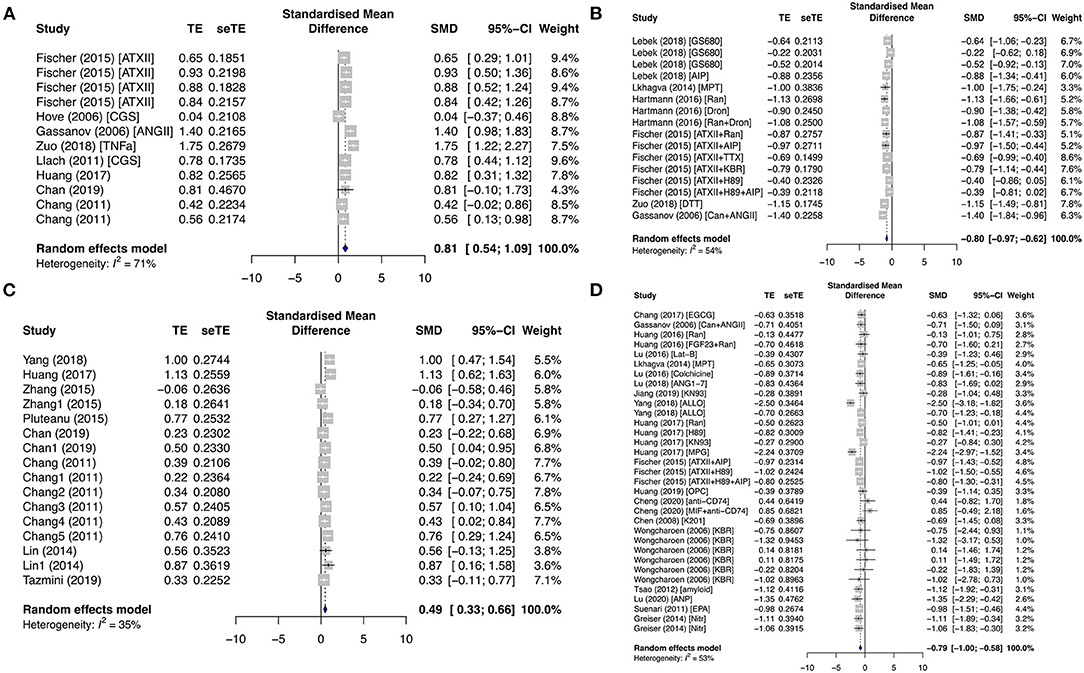
Figure 3. Calcium-release profiles in the primary prevention group. Calcium spark frequency (CaSpF) was represented in the (A) diseased and proarrhythmic subgroups and the (B) antiarrhythmic subgroup. Calcium transient amplitude (CaTA) was presented in the (C) diseased and (D) antiarrhythmic subgroups. TE, estimated treatment effect; seTE, standard error of treatment estimate; SMD, standard mean difference; 95% CI, 95% confidence interval; ATXII, anemonia viridis toxin 2; ANGII, angiotensin II; CGS21680, adenosine 2A agonist; TNFα, tumor necrosis factor-alpha; Ran, ranolazine; Dan, dantrolene; KN92, an inactive analog of KN93; KN93, calcium/calmodulin-dependent protein kinase II inhibitor; FGF23, fibroblast growth factor 23; Aldo, aldosterone; EGCG, epigallocatechin gallate; Can, candesartan; MPT0E014, histone deacetylase inhibitor; ANG1–7, angiotensin 1–7; ALLO, allopurinol; H89, protein kinase inhibitor; MPG, N-[2-mercaptopropionyl]glycine; OPC21286, arginine vasopressin antagonists; AIP, autocamide-2-related inhibitory peptide; MIF, macrophage inhibitory factor; K201, 1,4-benzothiazepine derivative; KB-R7943, reverse-mode sodium/calcium exchanger inhibitor; ANP, atrial natriuretic peptide; EPA, eicosapentaenoic acid; Nitr, nitrendipine.
In contrast, the CaSpF and calcium transient frequency (CaTF) were significantly elevated in both the secondary paroxysmal and chronic AF subgroups, with respective SMD = 0.81; I2 = 96%; CI = −0.14–1.76; p < 0.0001 (Figure 4A), and SMD = 0.85; I2 = 92%; CI = 0.12–1.57; p < 0.0001 (Figure 4B), and high heterogeneities. However, the change in CaSpA was almost negligible in both subgroups (SMD = 0.06; I2 = 55%; CI = 0.27–0.39; p < 0.0386) (Supplementary Figure 4G). Surprisingly, CaTA was unaltered in both paroxysmal (SMD = −0.07; I2 = 66%; CI = −0.34–0.20; p < 0.0001) and permanent AF (SMD = −0.06; I2 = 79%; CI = −0.49–0.38; p < 0.0001) (Supplementary Figures 2C,D).
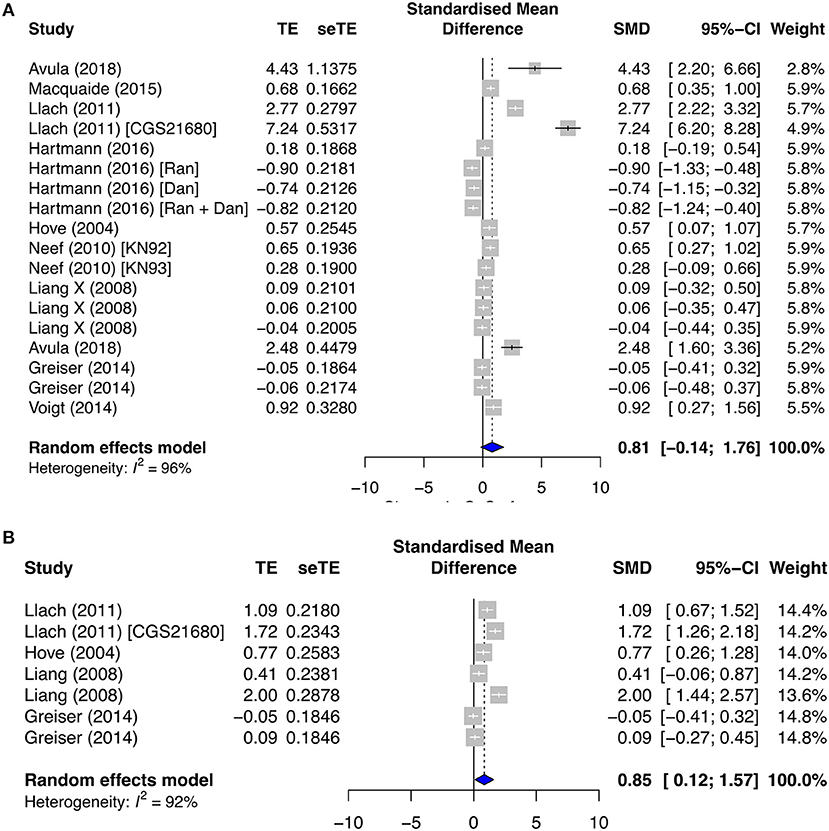
Figure 4. Calcium release profiles in the secondary prevention group. (A) Calcium spark frequency (CaSpF) and (B) calcium transient frequency (CaTF). TE, estimated treatment effect; seTE, standard error of treatment estimate; SMD, standard mean difference; 95% CI, 95% confidence interval; CGS21680, adenosine 2A agonist; Ran, ranolazine; Dan, dantrolene; KN92, an inactive analog of KN93; and KN93, calcium/calmodulin-dependent protein kinase II inhibitor.
SR Ca2+ Leak–Load Relationship
SR Ca2+ release is affected by the opening of RyR channels from its stores. In particular, SR Ca2+ leak is a major contributor to cardiac arrhythmia. Ca2+ load remained relatively unchanged in both the primary and secondary subgroups, except when antiarrhythmic drugs were applied in the primary group (SMD = −0.40; I2 = 59%; CI = −0.62 to −0.17; p < 0.0001) (Figure 5A). No change in Ca2+ leak was observed in the secondary prevention group (Supplementary Figure 5), but it was significantly affected by pro- and antiarrhythmic drugs in the primary subgroups. Ca2+ leak was raised by proarrhythmic agents (SMD = 0.81; I2 = 0%; CI = 0.54–1.09; p = 0.7583) (Figure 5B) and antagonized by antiarrhythmic agents (SMD = −0.66; I2 = 33%; CI = −0.81 to −0.50; p = 0.0932) (Figure 5C).
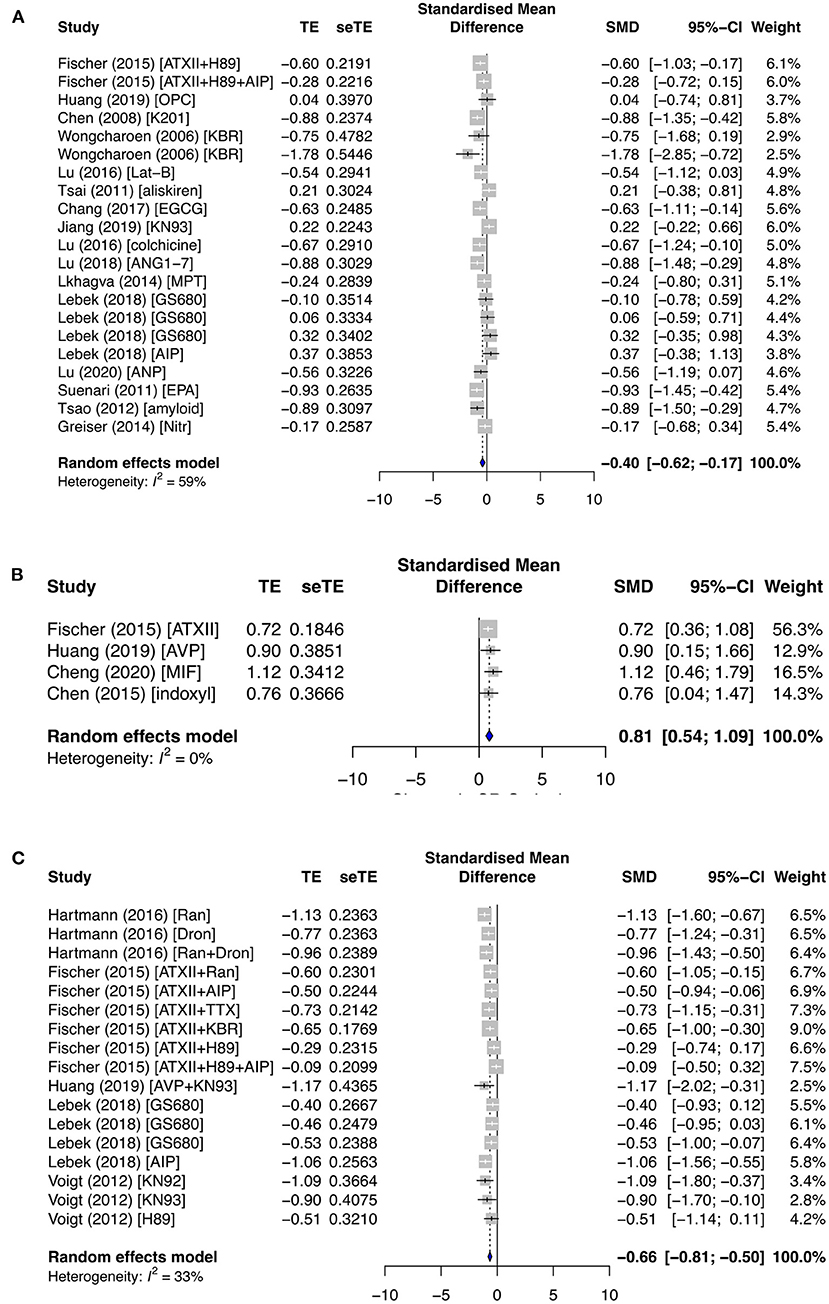
Figure 5. The effect of primary groups on SR calcium leak–load relationship. SR calcium load was affected by (A) antiarrhythmic drugs, and SR calcium leak was influenced by (B) proarrhythmic and (C) antiarrhythmic agents. TE, estimated treatment effect; seTE, standard error of treatment estimate; SMD, standard mean difference; 95% CI, 95% confidence interval; ATXII, anemonia viridis toxin 2; H89, protein kinase inhibitor; AIP, autocamide-2-related inhibitory peptide; OPC21286, arginine vasopressin antagonists; MIF, macrophage inhibitory factor; K201, 1,4-benzothiazepine derivative; KB-R7943, reverse-mode sodium/calcium exchanger inhibitor; Lat-B, latrunculin-B; EGCG, epigallocatechin gallate; ANG1–7, angiotensin 1–7; MPT0E014, histone deacetylase inhibitor; GS680, calcium/calmodulin-dependent protein kinase II inhibitor; EPA, eicosapentaenoic acid; Nitr, nitrendipine; AVP, arginine vasopressin; Ran, ranolazine; Dan, dantrolene; and TTX implies tetrodotoxin.
Ionic Mechanisms of Atrial Remodeling
One of the most important currents for atrial cardiac action potential generation is ICaL. ICaL was significantly downregulated in the primary diseased subgroup (SMD = −1.07; I2 = 88%; CI = −1.94 to −0.20; p < 0.0001) (Figure 6A), antiarrhythmic drug subgroup (SMD = −0.96; I2 = 61%; CI = −1.31 to −0.61; p < 0.0001) (Figure 6B), and secondary AF subgroups (SMD = −1.28; I2 = 91%; CI = −2.04 to −0.52; p < 0.0001) (Figure 6C). These results were consistent with Cav1.2 protein expression in the primary antiarrhythmic subgroup (SMD = −0.70; I2 = 30%; CI = −1.25 to −0.16; p = 0.2027) (Figure 6D) and secondary permanent AF group (SMD = −1.69; I2 = 0%; CI = −7.05–3.67; p < 0.0001) (Supplementary Figure 6F).
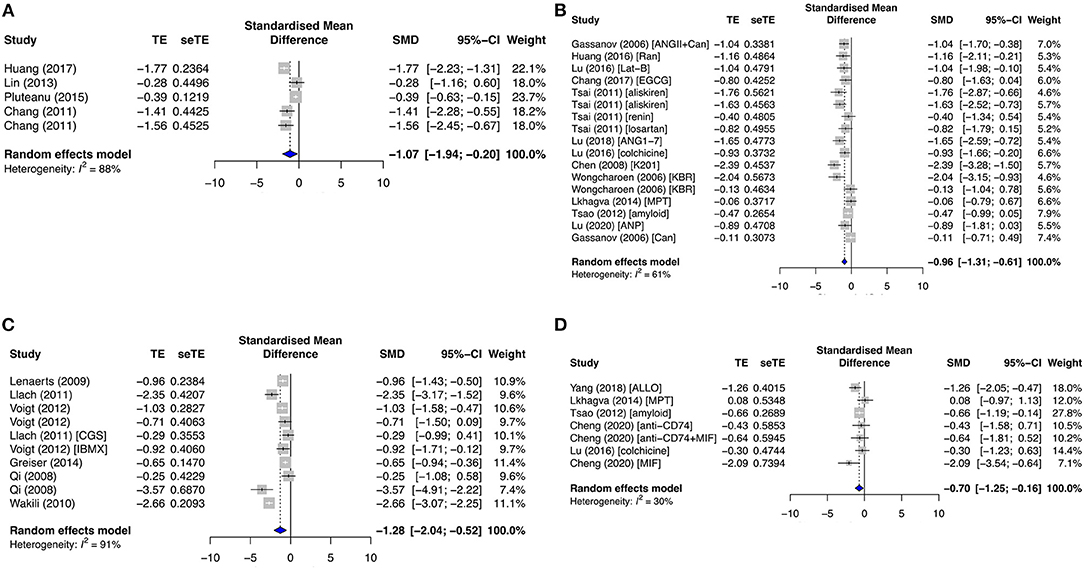
Figure 6. Measured L-type calcium current density (ICaL) and its protein expression (Cav1.2) in the primary prevention group. (A) Diseased, (B) antiarrhythmic drug, and (C) secondary prevention group with AF studies, and (D) Cav1.2 expression in the primary antiarrhythmic subgroup. TE, estimated treatment effect; seTE, standard error of treatment estimate; SMD, standard mean difference; 95% CI, 95% confidence interval; ANGII, angiotensin II; Can, candesartan; Ran, ranolazine; Lat-B, latrunculin-B; EGCG, epigallocatechin gallate; ANG1–7, angiotensin 1–7; K201, 1,4-benzothiazepine derivative; KB-R7943, reverse-mode sodium/calcium exchanger inhibitor; MPT0E014, histone deacetylase inhibitor; ANP, atrial natriuretic peptide; CGS21680, adenosine 2A agonist; IBMX, 3-isobutyl-1-methylxanthine; ALLO, allopurinol; MIF, macrophage inhibitory factor.
The extrusion of [Ca2+]i for Ca2+ recycling is via the cardiac NCX. INCX was significantly enhanced in both the primary diseased and proarrhythmic subgroups (SMD = 0.68; I2 = 89%; CI = 0.01–1.35; p < 0.0001) (Figure 7A) and reduced in the primary antiarrhythmic drug group (SMD = −1.03; I2 = 76%; CI = −1.51 to −0.55; p < 0.0001) (Figure 7B). Likewise, NCX1 protein expression was upregulated in the primary diseased and proarrhythmic subgroup (SMD = 0.43; I2 = 50%; CI = −0.25–1.10; p = 0.0638) (Supplementary Figure 7E) and significantly inhibited in the primary antiarrhythmic subgroup (SMD = −0.82; I2 = 0%; CI = −1.31 to −0.33; p < 0.0001) (Figure 7D). As opposed to the primary group, AF studies demonstrated that INCX and NCX1 protein expression had mixed results [SMD = 0.14; I2 = 74%; CI = −0.26–0.54; p < 0.0001 (Figure 7C), and SMD = 0.62; I2 = 61%; CI = −0.29–1.54; p = 0.0638 (Supplementary Figure 7H), respectively].
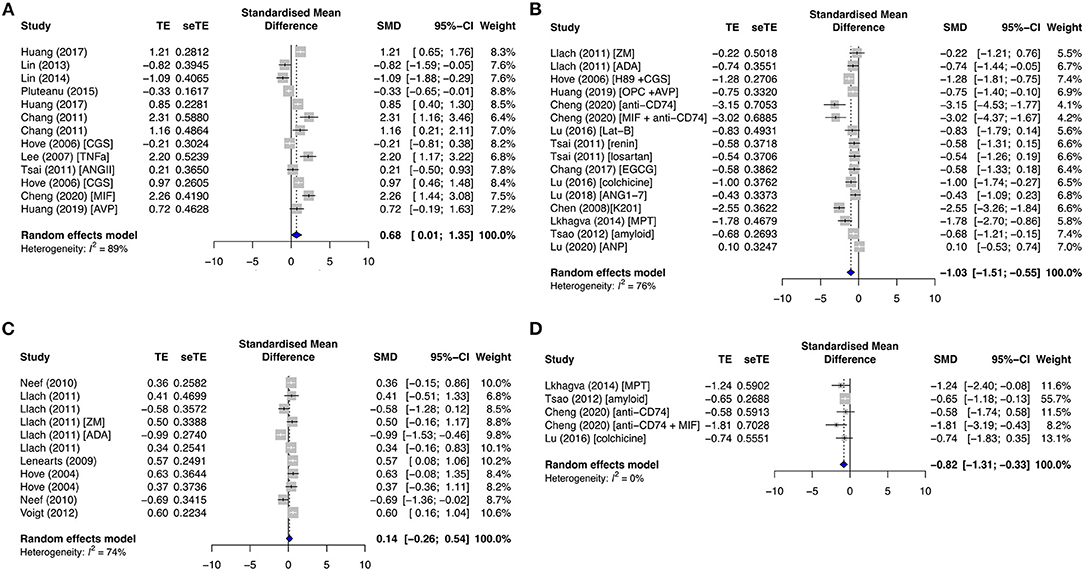
Figure 7. Sodium–calcium current density (INCX) and its protein expression (NCX1), INCX, in the (A) primary diseased and proarrhythmic groups, (B) antiarrhythmic group, and (C) secondary group, and (D) NCX1 expression in the primary antiarrhythmic group. TE, estimated treatment effect; seTE, standard error of treatment estimate; SMD, standard mean difference; 95% CI, 95% confidence interval; CGS21680, adenosine 2A agonist; TNFα, tumor necrosis factor-alpha; ANGII, angiotensin II; MIF, macrophage inhibitory factor; AVP, arginine vasopressin; ZM241385, adenosine A2A receptor antagonist; ADA, adenosine deaminase; MPT0E014, histone deacetylase inhibitor.
RyR is a major cardiac channel and mediator of the myocardial excitation–contraction coupling. It contains two key phosphorylation sites, serine S2808 (pRyR-S2808) and S2814 (pRyR-S2814). S2808 is phosphorylated by PKA, while S2814 is modulated by CAMKII. Only pRyR-S2808 was significantly affected in both primary and secondary groups, while total RyR (tRyR) and pRyR-S2814 showed no significant changes. pRyR-S2808 expression was increased in the primary diseased and proarrhythmic subgroups (SMD = 0.95; I2 = 64%; CI = 0.12–1.79; p = 0.074) (Figure 8A) and the secondary AF group (SMD = 0.66; I2 = 63%; CI = 0.01–1.31; p < 0.0001) (Figure 8C) but was inhibited by antiarrhythmic drugs (SMD = −1.45; I2 = 57%; CI = −2.56 to −0.34; p = 0.0315) (Figure 8B).
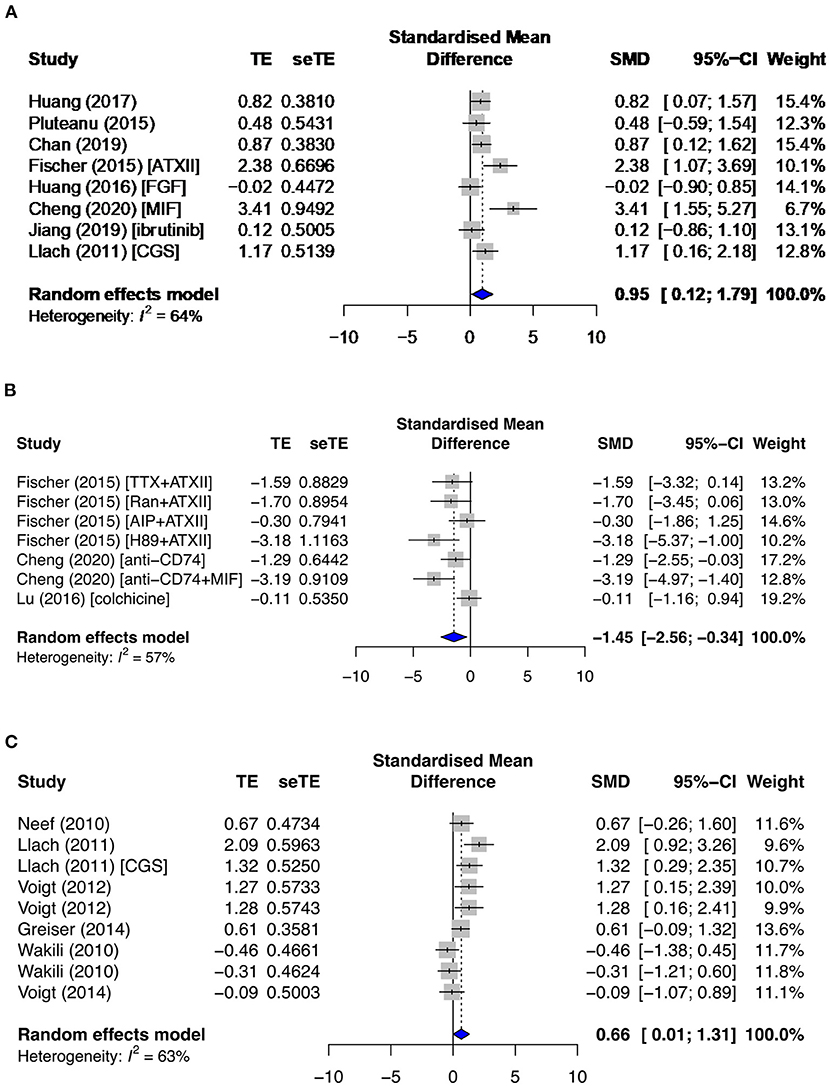
Figure 8. Phosphorylated ryanodine receptor at serine 2808 (pRyR-S2808) protein expression in the (A) diseased and proarrhythmic subgroups, (B) antiarrhythmic subgroups, and (C) secondary AF group. TE, estimated treatment effect; seTE, standard error of treatment estimate; SMD, standard mean difference; 95% CI, 95% confidence interval; ATXII, anemonia viridis toxin 2; FGF23, fibroblast growth factor 23; MIF, macrophage inhibitory factor; CGS21680, adenosine 2A agonist; TTX, tetrodotoxin; Ran, ranolazine; AIP, autocamide-2-related inhibitory peptide; H89, protein kinase inhibitor.
Another pathway for Ca2+ recycling is via the SERCA2a pump, in which activity is directly controlled by PLN. Few studies reported on SERCA and PLN expression, resulting in inconsistent and non-significant results. SERCA expression was elevated in the primary diseased and proarrhythmic subgroups but substantially reduced in the secondary paroxysmal AF subgroup. Total PLN (tPLN) remained relatively constant in all primary subgroups but was similarly decreased in paroxysmal AF. The results for phosphorylated PLN at sites serine 16 (pPLN-S16) and threonine 17 (pPLN-T17) in both groups were markedly diverse.
The signaling proteins, CAMKII and PKA, pCAMKII in particular, were affected by proarrhythmic drugs (SMD = 1.58; I2 = 60%; CI = 0.77–2.40; p = 0.0192) (Figure 9A) and antiarrhythmic therapies (SMD = −0.88; I2 = 66%; CI = −2.04–0.28; p = 0.0015), and total CAMKII (t CAMKII) by AF groups (SMD = 1.89; I2 = 63%; CI = 0.47–3.32; p = 0.023) (Figure 9B). Phosphorylated CAMKII had a greater activity than tCAMKII in most subgroups. Only primary studies reported on the sodium current (INa), INa−Late, and IK current densities. However, all data extracted were non-significant and/or inconsistent except for INa−Late. INa−Late was moderately enhanced in diseased (SMD = 0.74; I2 = 29%; CI = 0.33–1.15; p = 0.2206) (Figure 9C) and proarrhythmic drugs (SMD = 0.64; I2 = 0%; CI = 0.36–0.92; p = 0.7447) (Figure 9D) and significantly antagonized by antiarrhythmic agents (SMD = −1.00; I2 = 11%; CI = −1.29 to −0.70; p = 0.3408) (Figure 9E), with low heterogeneities across all three primary subgroups.
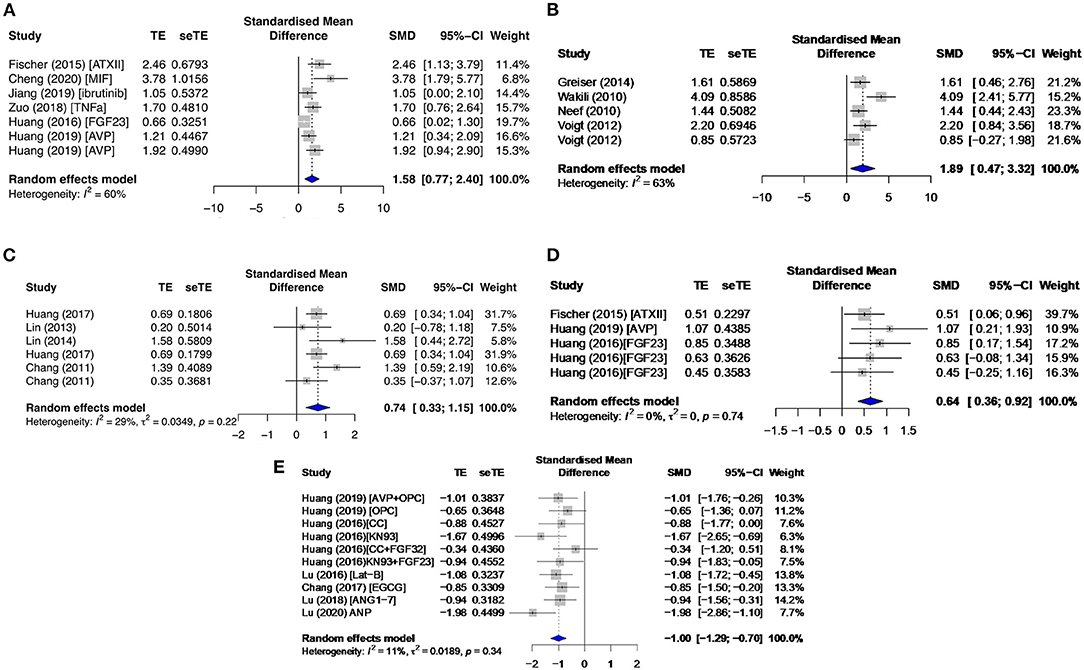
Figure 9. Phosphorylated calcium/calmodulin-dependent protein kinase II at threonine 286 (pCAMKII) and total CAMKII (tCAMKII) protein expressions and late sodium current density (INa−Late) in the primary group. This includes pCAMKII in the (A) proarrhythmic subgroup, tCAMKII in the (B) AF group, and INa−Late in the (C) diseased, (D) proarrhythmic drugs, and (E) antiarrhythmic agents. TE, estimated treatment effect; seTE, standard error of treatment estimate; SMD, standard mean difference; 95% CI, 95% confidence interval; ATXII, anemonia viridis toxin 2; FGF23, fibroblast growth factor 23; MIF, macrophage inhibitory factor; AVP, arginine vasopressin; OPC21286, arginine vasopressin antagonists; CC, chelerythrine chloride; Lat-B, latrunculin-B; EGCG, epigallocatechin gallate; ANG1–7, angiotensin 1–7; ANP, atrial natriuretic peptide.
The action of various drugs or reagents in the different experimental models were summarized in Supplementary Tables 4, 5. Additionally, the renin–angiotensin system, targeted by reagents such as angiotensin (ANG) and renin, was most commonly reported. The renin–angiotensin system, which is currently a drug target for hypertension, could also be a potential pharmacological discovery for the treatment and prevention of AF.
Discussion
Main Findings
This preclinical systematic review study analyzed 74 articles identified from 446 searched primary and secondary AF prevention articles. Forty-five publications were classified as primary AF prevention studies and 29 others as secondary prevention. To our knowledge, this is the largest study of this kind to explore the association between modulated calcium homeostasis and release events for primary and secondary prevention of AF. Our principal findings are summarized as follows (Table 1).
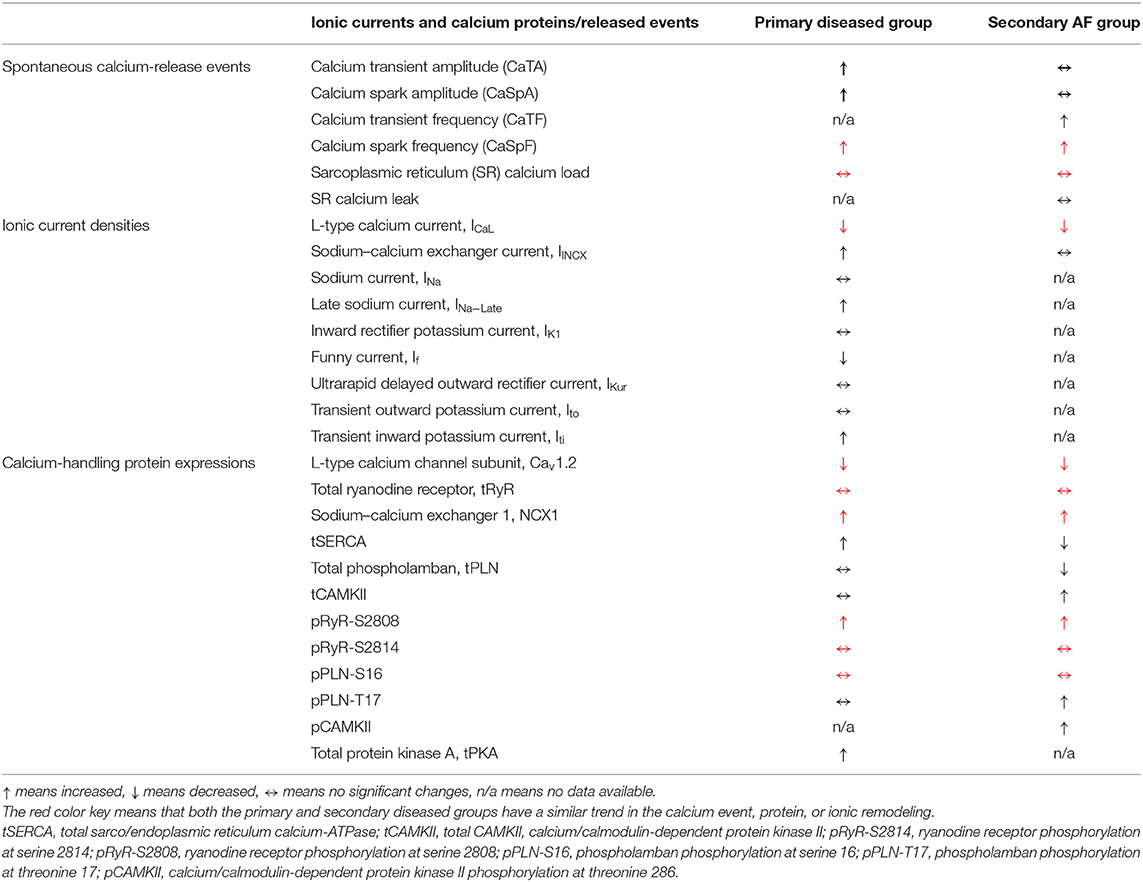
Table 1. The key calcium handling remodeling in the primary prevention group and the secondary diseased group.
With regard to the key Ca2+ channels/proteins/mediators, our study found that ICaL was the most widely studied current in both primary and secondary AF prevention, followed by INCX and RyR2 channels.
• We showed that ICaL was significantly downregulated in primary and secondary diseased groups, which were largely consistent with our results for Cav1.2 protein expression. Antiarrhythmic drugs in the primary group further reduced ICaL significantly.
• Furthermore, the NCX1 protein expression was significantly enhanced in both the primary and secondary diseased groups, but INCX was only elevated in the primary diseased group.
• In addition, our study demonstrated that the key phosphorylation expression for RyR was enhanced at serine 2808 in both the primary diseased and secondary AF groups, and inhibited in the primary antiarrhythmic drug subgroup. On the other hand, the other key RyR phosphorylation expression at serine 2814 showed no significant changes in both the primary and secondary diseased groups.
• SERCA expression was elevated in the primary diseased and proarrhythmic drug subgroups but substantially reduced in the secondary paroxysmal AF subgroup. tPLN remained relatively constant in all primary subgroups but was decreased in paroxysmal AF.
• Finally, the Ca2+ signaling mediator CAMKII was increased in the secondary AF group. With its phosphorylation activity at threonine 286, pCAMII-T286 was significantly raised by proarrhythmic drugs and significantly reduced by antiarrhythmic therapies.
It is noteworthy that there is a growing surge of interest for the late sodium current INa−Late and its direct effect on arrhythmia. Our study identified many primary preventative publications that showed that INa−Late was moderately enhanced in the primary diseased and proarrhythmic drug subgroups but significantly antagonized by antiarrhythmic agents, with low heterogeneities across all three subgroups.
As a result of atrial remodeling in the ionic channels and protein/signaling expressions in diseased and AF conditions, we observed changed Ca2+ functional activities, i.e., Ca2+ spark, Ca2+ transient, and Ca2+ load/leak. In the primary prevention group, CaSpF, CaTA, and CaSpA were significantly enhanced in the diseased subgroup and decreased by antiarrhythmic drug agents. On the other hand, CaTF and CaSpF were significantly elevated in both the secondary paroxysmal and chronic AF subgroups. Interestingly, we discovered that SR Ca2+ load and Ca2+ leak remained relatively constant in the primary and secondary subgroups, except when SR Ca2+ load was reduced when antiarrhythmic drugs were applied in the primary group. Furthermore, we found that Ca2+ leak was raised by proarrhythmic agents and antagonized by antiarrhythmic agents in the primary group.
Potential Mechanisms for Primary AF Diseases
The pathophysiological mechanism that causes spontaneous sarcoplasmic calcium release in the primary group involves the downregulation of ICaL and dysfunction of the Ca2+-handling proteins, in particular pRyR at S2808. The enhanced pRyR-S2808 activity may increase the frequency of the Ca2+ spark due to calcium-induced calcium releases. This would eventually lead to a high Ca2+ level in the cytosol and enhanced trigger activities via the forward mode of INCX, which was demonstrated in this review as the elevation of INCX activity. Furthermore, the enhancement of INa−Late was shown to potentially play a more significant role in the generation of arrhythmia in our review and recent studies (95–97). The reduction in ICaL was predicted to result in reduced SR load and diminished spontaneous activity. On the contrary, the downregulation of Cav1.2 current and protein expression has resulted in SCaEs, which could possibly be due to cell compensation (98, 99). Overall, this interplay led to an overload of Ca2+ in the cell, potentially causing AF.
Abnormal Ca2+ Activity in AF
The electrophysiological remodeling induced in the fibrillating atria and its molecular basis were extensively reviewed. Recent and past AF studies (95–97) have suggested that ICaL density was downregulated, together with the reduction of its protein expression, Cav1.2. Strangely, reduced ICaL density did not diminish the SR load; it remained unchanged (96). In contrast to the primary AF disease, consistently reduced SERCA levels were identified, reducing the releasable SR Ca2+ in the cytosol (98, 99).
On the other hand, some studies observed that the SR Ca2+ leak and activity of RyR were consistently upregulated (97, 100). Their observation justified the increase in SCaEs. The NCX expression was also increased, in contrast to what we saw earlier in the primary mechanism (100). The increased NCX expression could also account for the increase in frequencies of both the Ca2+ sparks and Ca2+ transients. The overextrusion of Ca2+ explains the unchanged CaSpA and CaTA.
We have shown that ICaL and NCX1 protein are the primary remodeling targets identified, and this leads to spontaneous calcium activity due to its interrelationship with the SR proteins. The conclusion of the secondary group meta-analysis aligns perfectly with the AF mechanism provided by Madsen et al. (34). On the other hand, the proposed primary mechanism is the best agglomeration of the mechanisms acquired from individual modifiable/non-modifiable risk factors associated with AF.
Limitations
This review aims to provide a better understanding of the mechanisms involved in the [Ca2+]i homeostasis within atrial cardiomyocytes and compare their activities among the primary and secondary AF subgroups. Although this review has comprehensively compiled the Ca2+ activity from inception to date, it still presents several limitations. The activity of SERCA and PLN appears to be unclear. It is certain that ICaL was reduced in the primary diseased and secondary AF groups. Surprisingly, the SR Ca2+ load–leak relationship was unaltered in the primary diseased and secondary AF groups with high heterogeneities. This could be influenced by a variety of non-controllable factors, such as the variability in the animal and human studies at various stages of AF or diseased states, the type and strength of pharmacological agents applied, and the different experimental settings and methodologies (101, 102).
Conclusion
Our study identified that ICaL is reduced in both primary and secondary diseased groups. Furthermore, pRyR-S2808 and NCX1 protein expression are enhanced. The remodeling leads to elevated Ca2+ functional activities, such as the frequencies or amplitude of Ca2+ spark and Ca2+ transient. The main difference identified between primary and secondary diseased groups is the SERCA expression, which is elevated in the primary diseased group and substantially reduced in the secondary paroxysmal AF subgroup. We believe our study will add new evidence to AF mechanisms and treatment targets.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
SF and SA undertook data extraction, post-processing, and analysis, as well as drafting the manuscript. MG assisted in assessing the quality of the included studies. JZ guided the project and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Health Research Council of New Zealand (16/385) and National Heart Foundation of New Zealand.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Kevin Jamart and Joseph Ashby for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.662914/full#supplementary-material
References
1. Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. (2014) 129:837–47. doi: 10.1161/CIRCULATIONAHA.113.005119
2. Hajhosseiny R, Matthews GK, Lip GY. Metabolic syndrome, atrial fibrillation, and stroke: tackling an emerging epidemic. Heart Rhythm. (2015) 12:2332–43. doi: 10.1016/j.hrthm.2015.06.038
3. Hersi A, Mitchell LB, Wyse DG. Atrial fibrillation: challenges and opportunities. Can J Cardiol. (2006) 22(Suppl.C):21C−6C. doi: 10.1016/S0828-282X(06)70998-9
4. Lau DH, Nattel S, Kalman JM, Sanders P. Modifiable risk factors and atrial fibrillation. Circulation. (2017) 136:583–96. doi: 10.1161/CIRCULATIONAHA.116.023163
5. Xu J, Luc JG, Phan K. Atrial fibrillation: review of current treatment strategies. J Thorac Dis. (2016) 8:E886–900. doi: 10.21037/jtd.2016.09.13
6. Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. (2016) 37:2893–962. doi: 10.15829/1560-4071-2017-7-7-86
7. Sutanto H, Heijman J. The role of calcium in the human heart: with great power comes great responsibility. Front Young Minds. (2019) 7:65. doi: 10.3389/frym.2019.00065
8. Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and Excitation-Contraction Coupling in the Heart. Circ Res. (2017) 121:181–95. doi: 10.1161/CIRCRESAHA.117.310230
9. Shkryl VM, Blatter LA. Ca(2+) release events in cardiac myocytes up close: insights from fast confocal imaging. PLoS ONE. (2013) 8:e61525. doi: 10.1371/journal.pone.0061525
10. Lipscombe D. L-type calcium channels: highs and new lows. Circ Res. (2002) 90:933–5. doi: 10.1161/01.RES.0000019740.52306.92
11. Bootman MD, Smyrnias I, Thul R, Coombes S, Roderick HL. Atrial cardiomyocyte calcium signalling. Biochim Biophys Acta. (2011) 1813:922–34. doi: 10.1016/j.bbamcr.2011.01.030
12. Pasqualin C, Yu A, Malécot CO, Gannier F, Cognard C, Godin-Ribuot D, et al. Structural heterogeneity of the rat pulmonary vein myocardium: consequences on intracellular calcium dynamics and arrhythmogenic potential. Sci Rep. (2018) 8:3244. doi: 10.1038/s41598-018-21671-9
13. Bootman MD, Higazi DR, Coombes S, Roderick HL. Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. J Cell Sci. (2006) 119:3915–25. doi: 10.1242/jcs.03223
14. Bers DM. Cardiac excitation-contraction coupling. Nature. (2002) 415:198–205. doi: 10.1038/415198a
15. Ono K, Iijima T. Cardiac T-type Ca(2+) channels in the heart. J Mol Cell Cardiol. (2010) 48:65–70. doi: 10.1016/j.yjmcc.2009.08.021
16. Boron, Walter F, Boulpaep EL. Medical Physiology: A Cellular and Molecular Approach. 2nd ed. Philadelphia, PA: Saunders/Elsevier; International ed. (2009).
17. Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. (2009) 54:180–7. doi: 10.1097/FJC.0b013e3181a25078
18. Dobrev D, Wehrens XH. Role of RyR2 phosphorylation in heart failure and arrhythmias: controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. (2014) 114:1311–9. doi: 10.1161/CIRCRESAHA.114.300568
19. Grandi E, Edwards AG, Herren AW, Bers DM. CaMKII comes of age in cardiac health and disease. Front Pharmacol. (2014) 5:154. doi: 10.3389/fphar.2014.00154
20. Zhang JC, Wu HL, Chen Q, Xie XT, Zou T, Zhu C, et al. Calcium-mediated oscillation in membrane potentials and atrial-triggered activity in atrial cells of Casq2R33Q/R33Q mutation mice. Front Physiol. (2018) 9:1447. doi: 10.3389/fphys.2018.01447
21. Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. (2008) 88:1491–545. doi: 10.1152/physrev.00030.2007
22. Harbord RM, Egger M, Sterne JA A. modified test for small-study effects in meta-analyses of controlled trials with binary endpoints. Stat Med. (2006) 25:3443–57. doi: 10.1002/sim.2380
23. Peters JL, Sutton AJ, Jones DR, Abrams KR, Rushton L. Comparison of two methods to detect publication bias in meta-analysis. J Am Med Assoc. (2006) 295:676–80. doi: 10.1001/jama.295.6.676
24. Chiang DY, Li N, Wang Q, Alsina KM, Quick AP, Reynolds JO, et al. Impaired local regulation of ryanodine receptor type 2 by protein phosphatase 1 promotes atrial fibrillation. Cardiovasc Res. (2014) 103:178–87. doi: 10.1093/cvr/cvu123
25. Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. (2009) 62:e1–34. doi: 10.1016/j.jclinepi.2009.06.006
26. Luchini C, Stubbs B, Solmi M, Veronese N. Assessing the quality of studies in meta-analyses: advantages and limitations of the Newcastle Ottawa Scale. World J Meta-Anal. (2017) 5:80–4. doi: 10.13105/wjma.v5.i4.80
27. Brian S. Everitt and Torsten Hothorn. A Handbook of Statistical Analyses Using R. ISBN: 1584885394. Chapman & Hall/CRC (2006).
28. Faraone SV. Interpreting estimates of treatment effects: implications for managed care. P T. (2008) 33:700–11.
29. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. (2003) 327:557–60. doi: 10.1136/bmj.327.7414.557
30. Coutu P, Chartier D, Nattel S. Comparison of Ca2+-handling properties of canine pulmonary vein and left atrial cardiomyocytes. Am J Physiol Heart Circ Physiol. (2006) 291:H2290–300. doi: 10.1152/ajpheart.00730.2005
31. Zhou WP Li F, Wu JJ, Lu YN, Qian YJ. Calcium-handling abnormalities underlying atrial arrhythmogenesis in a Fontan operation canine model. World J Pediatr. (2018) 14:576–84. doi: 10.1007/s12519-018-0177-z
32. Hartmann N, Mason FE, Braun I, Pabel S, Voigt N, Schotola H, et al. The combined effects of ranolazine and dronedarone on human atrial and ventricular electrophysiology. J Mol Cell Cardiol. (2016) 94:95–106. doi: 10.1016/j.yjmcc.2016.03.012
33. Hove-Madsen L, Prat-Vidal C, Llach A, Ciruela F, Casadó V, Lluis C, et al. Adenosine A2A receptors are expressed in human atrial myocytes and modulate spontaneous sarcoplasmic reticulum calcium release. Cardiovasc Res. (2006) 72:292–302. doi: 10.1016/j.cardiores.2006.07.020
34. Hove-Madsen L, Llach A, Bayes-Genís A, Roura S, Rodriguez Font E, Arís A, et al. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. (2004) 110:1358–63. doi: 10.1161/01.CIR.0000141296.59876.87
35. Huang JH, Chen YC, Lu YY, Lin YK, Chen SA, Chen YJ. Arginine vasopressin modulates electrical activity and calcium homeostasis in pulmonary vein cardiomyocytes. J Biomed Sci. (2019) 26:71. doi: 10.1186/s12929-019-0564-3
36. Kanaporis G, Blatter LA. Membrane potential determines calcium alternans through modulation of SR Ca2+ load and L-type Ca2+ current. J Mol Cell Cardiol. (2017) 105:49–58. doi: 10.1016/j.yjmcc.2017.02.004
37. Kanaporis G, Kalik ZM, Blatter LA. Action potential shortening rescues atrial calcium alternans. J Physiol. (2019) 597:723–40. doi: 10.1113/JP277188
38. Huang SY, Chen YC, Kao YH, Hsieh MH, Lin YK, Chung CC, et al. Fibroblast growth factor 23 dysregulates late sodium current and calcium homeostasis with enhanced arrhythmogenesis in pulmonary vein cardiomyocytes. Oncotarget. (2016) 7:69231–42. doi: 10.18632/oncotarget.12470
39. Lu YY, Lin YK, Wen ZH, Chen YC, Chen SA, Chen YJ. Latrunculin B modulates electrophysiological characteristics and arrhythmogenesis in pulmonary vein cardiomyocytes. Clin Sci. (2016) 130:721–32. doi: 10.1042/CS20150593
40. Liang X, Xie H, Zhu PH, Hu J, Zhao Q, Wang CS, et al. Ryanodine receptor-mediated Ca2+ events in atrial myocytes of patients with atrial fibrillation. Cardiology. (2008) 111:102–10. doi: 10.1159/000119697
41. Tazmini K, Frisk M, Lewalle A, Laasmaa M, Morotti S, Lipsett DB, et al. Hypokalemia promotes arrhythmia by distinct mechanisms in atrial and ventricular myocytes. Circ Res. (2020) 126:889–906. doi: 10.1161/CIRCRESAHA.119.315641
42. Llach A, Molina CE, Prat-Vidal C, Fernandes J, Casadó V, Ciruela F, et al. Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine A2A receptors. Eur Heart J. (2011) 32:721–9. doi: 10.1093/eurheartj/ehq464
43. Cheng WL, Kao YH, Chen YC, Lin YK, Chen SA, Chen YJ. Macrophage migration inhibitory factor increases atrial arrhythmogenesis through CD74 signaling. Transl Res. (2020) 216:43–56. doi: 10.1016/j.trsl.2019.10.002
44. Lee SH, Chen YC, Chen YJ, Chang SL, Tai CT, Wongcharoen W, et al. Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. (2007) 80:1806–15. doi: 10.1016/j.lfs.2007.02.029
45. Henry AD, MacQuaide N, Burton FL, Rankin AC, Rowan EG, Drummond RM. Spontaneous Ca2+ transients in rat pulmonary vein cardiomyocytes are increased in frequency and become more synchronous following electrical stimulation. Cell Calcium. (2018) 76:36–47. doi: 10.1016/j.ceca.2018.09.001
46. Musa H, Kaur K, O'Connell R, Klos M, Guerrero-Serna G, Avula UM, et al. Inhibition of platelet-derived growth factor-AB signaling prevents electromechanical remodeling of adult atrial myocytes that contact myofibroblasts. Heart Rhythm. (2013) 10:1044–51. doi: 10.1016/j.hrthm.2013.03.014
47. Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N. et al. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP126 knockout mice. Circ Res. (2012) 110:465–70. doi: 10.1161/CIRCRESAHA.111.253229
48. Mancarella S, Yue Y, Karnabi E, Qu Y, El-Sherif N, Boutjdir M. Impaired Ca2+ homeostasis is associated with atrial fibrillation in the alpha1D L-type Ca2+ channel KO mouse. Am J Physiol Heart Circ Physiol. (2008) 295:H2017–24. doi: 10.1152/ajpheart.00537.2008
49. Zhang YH, Wu HJ, Che H, Sun HY, Cheng LC, Li X, et al. Functional transient receptor potential canonical type 1 channels in human atrial myocytes. Pflugers Arch. (2013) 465:1439–49. doi: 10.1007/s00424-013-1291-3
50. Jones SA, Yamamoto M, Tellez JO, Billeter R, Boyett MR, Honjo H, et al. Distinguishing properties of cells from the myocardial sleeves of the pulmonary veins: a comparison of normal and abnormal pacemakers. Circ Arrhythm Electrophysiol. (2008) 1:39–48. doi: 10.1161/CIRCEP.107.748467
51. Tsai CF, Chen YC, Lin YK, Chen SA, Chen YJ. Electromechanical effects of the direct renin inhibitor (aliskiren) on the pulmonary vein and atrium. Basic Res Cardiol. (2011) 106:979–93. doi: 10.1007/s00395-011-0206-8
52. Logantha SJ, Cruickshank SF, Rowan EG, Drummond RM. Spontaneous and electrically evoked Ca2+ transients in cardiomyocytes of the rat pulmonary vein. Cell Calcium. (2010) 48:150–60. doi: 10.1016/j.ceca.2010.08.002
53. Zhang H, Cannell MB, Kim SJ, Watson JJ, Norman R, Calaghan SC, et al. Cellular hypertrophy and increased susceptibility to spontaneous calcium-release of rat left atrial myocytes due to elevated afterload. PLoS ONE. (2015) 10:e0144309. doi: 10.1371/journal.pone.0144309
54. Zhang Y, Fraser JA, Schwiening C, Zhang Y, Killeen MJ, Grace AA, et al. Acute atrial arrhythmogenesis in murine hearts following enhanced extracellular Ca(2+) entry depends on intracellular Ca(2+) stores. Acta Physiol. (2010) 198:143–58. doi: 10.1111/j.1748-1716.2009.02055.x
55. Wang T, Wang QX, Wu PY, Chen YT, Yang SS, Huang Y, et al. Effects and mechanism of digoxin on atrium electrical remodeling and susceptibility of atrial fibrillation in aged rabbits. Zhonghua Xin Xue Guan Bing Za Zhi. (2019) 47:608–13. doi: 10.3760/cma.j.issn.0253.3758.2019.08.004
56. Jiang L, Li L, Ruan Y, Zuo S, Wu X, Zhao Q, et al. Ibrutinib promotes atrial fibrillation by inducing structural remodeling and calcium dysregulation in the atrium. Heart Rhythm. (2019) 16:1374–82. doi: 10.1016/j.hrthm.2019.04.008
57. Tsai WC, Chen YC, Kao YH, Lu YY, Chen SA, Chen YJ. Distinctive sodium and calcium regulation associated with sex differences in atrial electrophysiology of rabbits. Int J Cardiol. (2013) 168:4658–66. doi: 10.1016/j.ijcard.2013.07.183
58. Chang JH, Chang SL, Hong PD, Chen PN, Hsu CH, Lu YY, et al. Epigallocatechin-3-gallate modulates arrhythmogenic activity and calcium homeostasis of left atrium. Int J Cardiol. (2017) 236:174–80. doi: 10.1016/j.ijcard.2017.01.090
59. Shafaattalab S, Lin E, Christidi E, Huang H, Nartiss Y, Garcia A, et al. Ibrutinib displays atrial-specific toxicity in human stem cell-derived cardiomyocytes. Stem Cell Reports. (2019) 12:996–1006. doi: 10.1016/j.stemcr.2019.03.011
60. Lu YY, Chen YC, Kao YH, Lin YK, Yeh YH, Chen SA, et al. Colchicine modulates calcium homeostasis and electrical property of HL-1 cells. J Cell Mol Med. (2016) 20:1182–90. doi: 10.1111/jcmm.12818
61. Lin YK, Chen YC, Kao YH, Tsai CF, Yeh YH, Huang JL, et al. A monounsaturated fatty acid (oleic acid) modulates electrical activity in atrial myocytes with calcium and sodium dysregulation. Int J Cardiol. (2014) 176:191–8. doi: 10.1016/j.ijcard.2014.07.004
62. Suenari K, Chen YC, Kao YH, Cheng CC, Lin YK, Chen YJ, et al. Discrepant electrophysiological characteristics and calcium homeostasis of left atrial anterior and posterior myocytes. Basic Res Cardiol. (2011) 106:65–74. doi: 10.1007/s00395-010-0132-1
63. Opel A, Nobles M, Montaigne D, Finlay M, Anderson N, Breckenridge R, et al. Absence of the regulator of G-protein signaling, RGS4, predisposes to atrial fibrillation and is associated with abnormal calcium handling. J Biol Chem. (2015) 290:19233–44. doi: 10.1074/jbc.M115.666719
64. Fischer TH, Herting J, Mason FE, Hartmann N, Watanabe S, Nikolaev VO, et al. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc Res. (2015) 107:184–96. doi: 10.1093/cvr/cvv153
65. Pluteanu F, Heß J, Plackic J, Nikonova Y, Preisenberger J, Bukowska A, et al. Early subcellular Ca2+ remodelling and increased propensity for Ca2+ alternans in left atrial myocytes from hypertensive rats. Cardiovasc Res. (2015) 106:87–97. doi: 10.1093/cvr/cvv045
66. Lin YK, Chen YC, Huang JH, Lin YJ, Huang SS, Chen SA, et al. Leptin modulates electrophysiological characteristics and isoproterenol-induced arrhythmogenesis in atrial myocytes. J Biomed Sci. (2013) 20:94. doi: 10.1186/1423-0127-20-94
67. Chen YJ, Chen YC, Wongcharoen W, Lin CI, Chen SA. Effect of K201, a novel antiarrhythmic drug on calcium handling and arrhythmogenic activity of pulmonary vein cardiomyocytes. Br J Pharmacol. (2008) 153:915–25. doi: 10.1038/sj.bjp.0707564
68. Voigt N, Zhou XB, Dobrev D. Isolation of human atrial myocytes for simultaneous measurements of Ca2+ transients and membrane currents. J Vis Exp. (2013) 77:e50235. doi: 10.3791/50235
69. Ríos-Pérez EB, García-Castañeda M, Monsalvo-Villegas A, Avila G. Chronic atrial ionic remodeling by aldosterone: potentiation of L-type Ca2+ channels and its arrhythmogenic significance. Pflugers Arch. (2016) 468:1823–35. doi: 10.1007/s00424-016-1876-8
70. Avula UMR, Hernandez JJ, Yamazaki M, Valdivia CR, Chu A, Rojas-Pena A, et al. Atrial infarction-induced spontaneous focal discharges and atrial fibrillation in sheep: role of dantrolene-sensitive aberrant ryanodine receptor calcium release. Circ Arrhythm Electrophysiol. (2018) 11:e005659. doi: 10.1161/CIRCEP.117.005659
71. Tsai CT, Chiang FT, Chen WP, Hwang JJ, Tseng CD, Wu CK, et al. Angiotensin II induces complex fractionated electrogram in a cultured atrial myocyte monolayer mediated by calcium and sodium-calcium exchanger. Cell Calcium. (2011) 49:1–11. doi: 10.1016/j.ceca.2010.10.005
72. Wongcharoen W, Chen YC, Chen YJ, Chang CM, Yeh HI, Lin CI, et al. Effects of a Na+/Ca2+ exchanger inhibitor on pulmonary vein electrical activity and ouabain-induced arrhythmogenicity. Cardiovasc Res. (2006) 70:497–508. doi: 10.1016/j.cardiores.2006.02.026
73. Wakili R, Yeh YH, Yan Qi X, Greiser M, Chartier D, Nishida K, et al. Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ Arrhythm Electrophysiol. (2010) 3:530–41. doi: 10.1161/CIRCEP.109.933036
74. Gassanov N, Brandt MC, Michels G, Lindner M, Er F, Hoppe UC. Angiotensin II-induced changes of calcium sparks and ionic currents in human atrial myocytes: potential role for early remodeling in atrial fibrillation. Cell Calcium. (2006) 39:175–86. doi: 10.1016/j.ceca.2005.10.008
75. Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chartier D, et al. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res. (2008) 103:845–54. doi: 10.1161/CIRCRESAHA.108.175463
76. Lkhagva B, Chang SL, Chen YC, Kao YH, Lin YK, Chiu CT, et al. Histone deacetylase inhibition reduces pulmonary vein arrhythmogenesis through calcium regulation. Int J Cardiol. (2014) 177:982–9. doi: 10.1016/j.ijcard.2014.09.175
77. Lebek S, Plößl A, Baier M, Mustroph J, Tarnowski D, Lücht CM, et al. The novel CaMKII inhibitor GS-680 reduces diastolic SR Ca leak and prevents CaMKII-dependent pro-arrhythmic activity. J Mol Cell Cardiol. (2018) 118:159–68. doi: 10.1016/j.yjmcc.2018.03.020
78. Chan YH, Chang GJ, Lai YJ, Chen WJ, Chang SH, Hung LM, et al. Atrial fibrillation and its arrhythmogenesis associated with insulin resistance. Cardiovasc Diabetol. (2019) 18:125. doi: 10.1186/s12933-019-0928-8
79. Tsao HM, Weerateerangkul P, Chen YC, Kao YH, Lin YK, Huang JH, et al. Amyloid peptide regulates calcium homoeostasis and arrhythmogenesis in pulmonary vein cardiomyocytes. Eur J Clin Invest. (2012) 42:589–98. doi: 10.1111/j.1365-2362.2011.02618.x
80. Lin YK, Chen YC, Chen JH, Chen SA, Chen YJ. Adipocytes modulate the electrophysiology of atrial myocytes: implications in obesity-induced atrial fibrillation. Basic Res Cardiol. (2012) 107:293. doi: 10.1007/s00395-012-0293-1
81. Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res. (2001) 52:226–35. doi: 10.1016/S0008-6363(01)00380-7
82. Lenaerts I, Holemans P, Pokreisz P, Sipido KR, Janssens S, Heidbüchel H, et al. Nitric oxide delays atrial tachycardia-induced electrical remodelling in a sheep model. Europace. (2011) 13:747–54. doi: 10.1093/europace/eur021
83. Greer-Short A, Musa H, Alsina KM, Ni L, Word TA, Reynolds JO, et al. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm. (2020) 17:503–11. doi: 10.1016/j.hrthm.2019.10.016
84. Huang SY, Chen YC, Kao YH, Hsieh MH, Lin YK, Chen SA, et al. Redox and activation of protein kinase A dysregulates calcium homeostasis in pulmonary vein cardiomyocytes of chronic kidney disease. J Am Heart Assoc. (2017) 6:e005701. doi: 10.1161/JAHA.117.005701
85. Hartmann N, Pabel S, Herting J, Schatter F, Renner A, Gummert J, et al. Antiarrhythmic effects of dantrolene in human diseased cardiomyocytes. Heart Rhythm. (2017) 14:412–9. doi: 10.1016/j.hrthm.2016.09.014
86. Herraiz-Martínez A, Álvarez-García J, Llach A, Molina CE, Fernandes J, Ferrero-Gregori A, et al. Ageing is associated with deterioration of calcium homeostasis in isolated human right atrial myocytes. Cardiovasc Res. (2015) 106:76–86. doi: 10.1093/cvr/cvv046
87. Chen WT, Chen YC, Hsieh MH, Huang SY, Kao YH, Chen YA, et al. The uremic toxin indoxyl sulfate increases pulmonary vein and atrial arrhythmogenesis. J Cardiovasc Electrophysiol. (2015) 26:203–10. doi: 10.1111/jce.12554
88. Chang SL, Chen YC, Yeh YH, Lin YK, Wu TJ, Lin CI, et al. Heart failure enhanced pulmonary vein arrhythmogenesis and dysregulated sodium and calcium homeostasis with increased calcium sparks. J Cardiovasc Electrophysiol. (2011) 22:1378–86. doi: 10.1111/j.1540-8167.2011.02126.x
89. Yan J, Zhao W, Thomson JK, Gao X, DeMarco DM, Carrillo E, et al. Stress signaling JNK2 crosstalk with CaMKII underlies enhanced atrial arrhythmogenesis. Circ Res. (2018) 122:821–35. doi: 10.1161/CIRCRESAHA.117.312536
90. Zuo S, Li LL, Ruan YF, Jiang L, Li X, Li SN, et al. Acute administration of tumour necrosis factor-α induces spontaneous calcium release via the reactive oxygen species pathway in atrial myocytes. Europace. (2018) 20:1367–74. doi: 10.1093/europace/eux271
91. Yang Y, Zhao J, Qiu J, Li J, Liang X, Zhang Z, et al. Xanthine oxidase inhibitor allopurinol prevents oxidative stress-mediated atrial remodeling in alloxan-induced diabetes mellitus rabbits. J Am Heart Assoc. (2018) 7:e008807. doi: 10.1161/JAHA.118.008807
92. Lu YY, Cheng CC, Wu HJ, Lin YK, Chen YC, Chen SA, et al. Effects of ANP on pulmonary vein electrophysiology, Ca2+ homeostasis and adrenergic arrhythmogenesis via PKA. Clin Exp Pharmacol Physiol. (2020) 47:247–54. doi: 10.1111/1440-1681.13199
93. Suenari K, Chen YC, Kao YH, Cheng CC, Lin YK, Kihara Y, et al. Eicosapentaenoic acid reduces the pulmonary vein arrhythmias through nitric oxide. Life Sci. (2011) 89:129–36. doi: 10.1016/j.lfs.2011.05.013
94. Gu J, Liu X, Wang QX, Guo M, Liu F, Song ZP, et al. Beneficial effects of pioglitazone on atrial structural and electrical remodeling in vitro cellular models. J Mol Cell Cardiol. (2013) 65:1–8. doi: 10.1016/j.yjmcc.2013.09.016
95. Macquaide N, Tuan HT, Hotta J, Sempels W, Lenaerts I, Holemans P, et al. Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc Res. (2015) 108:387–98. doi: 10.1093/cvr/cvv231
96. Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P. D'hooge J, et al. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. (2009) 105:876–85. doi: 10.1161/CIRCRESAHA.109.206276
97. Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. (2012) 125:2059–70. doi: 10.1161/CIRCULATIONAHA.111.067306
98. Greiser M, Kerfant BG, Williams GS, Voigt N, Harks E, Dibb KM, et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest. (2014) 124:4759–72. doi: 10.1172/JCI70102
99. Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. (2014) 129:145–56. doi: 10.1161/CIRCULATIONAHA.113.006641
100. Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, et al. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. (2010) 106:1134–44. doi: 10.1161/CIRCRESAHA.109.203836
101. Lo ACY, Bai J, Gladding PA, Fedorov VV, Zhao J. Afterdepolarizations and abnormal calcium handling in atrial myocytes with modulated SERCA uptake: a sensitivity analysis of calcium handling channels. Philos Trans A Math Phys Eng Sci. (2020) 378:20190557. doi: 10.1098/rsta.2019.0557
Keywords: atrial fibrillation, calcium handling, calcium release events, Ca2+ sparks, primary AF prevention, secondary AF prevention
Citation: Fong SPT, Agrawal S, Gong M and Zhao J (2021) Modulated Calcium Homeostasis and Release Events Under Atrial Fibrillation and Its Risk Factors: A Meta-Analysis. Front. Cardiovasc. Med. 8:662914. doi: 10.3389/fcvm.2021.662914
Received: 01 February 2021; Accepted: 11 June 2021;
Published: 20 July 2021.
Edited by:
Gary Tse, Second Hospital of Tianjin Medical University, ChinaReviewed by:
Osmar Antonio Centurion, National University of Asunción, ParaguayQiongling Wang, University of Missouri, United States
Copyright © 2021 Fong, Agrawal, Gong and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jichao Zhao, j.zhao@auckland.ac.nz
†These authors share first authorship