Mitochondrial Dysfunction: An Emerging Link in the Pathophysiology of Cardiorenal Syndrome
 Shuqing Shi1†
Shuqing Shi1†  Bingxuan Zhang1†
Bingxuan Zhang1†  Yumeng Li1†
Yumeng Li1†  Xia Xu1 Jiayu Lv1
Xia Xu1 Jiayu Lv1  Qiulei Jia2 Ruoning Chai3 Wenjing Xue2 Yuan Li4 Yajiao Wang1 Huaqin Wu3* Qingqiao Song1* Yuanhui Hu3*
Qiulei Jia2 Ruoning Chai3 Wenjing Xue2 Yuan Li4 Yajiao Wang1 Huaqin Wu3* Qingqiao Song1* Yuanhui Hu3*- 1Department of Internal Medicine, Guang'anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 2Beijing University of Chinese Medicine, Beijing, China
- 3Department of Cardiovascular, Guang'anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 4Reproductive and Genetic Center, Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan, China
The crosstalk between the heart and kidney is carried out through various bidirectional pathways. Cardiorenal syndrome (CRS) is a pathological condition in which acute or chronic dysfunction in the heart or kidneys induces acute or chronic dysfunction of the other organ. Complex hemodynamic factors and biochemical and hormonal pathways contribute to the development of CRS. In addition to playing a critical role in generating metabolic energy in eukaryotic cells and serving as signaling hubs during several vital processes, mitochondria rapidly sense and respond to a wide range of stress stimuli in the external environment. Impaired adaptive responses ultimately lead to mitochondrial dysfunction, inducing cell death and tissue damage. Subsequently, these changes result in organ failure and trigger a vicious cycle. In vitro and animal studies have identified an important role of mitochondrial dysfunction in heart failure (HF) and chronic kidney disease (CKD). Maintaining mitochondrial homeostasis may be a promising therapeutic strategy to interrupt the vicious cycle between HF and acute kidney injury (AKI)/CKD. In this review, we hypothesize that mitochondrial dysfunction may also play a central role in the development and progression of CRS. We first focus on the role of mitochondrial dysfunction in the pathophysiology of HF and AKI/CKD, then discuss the current research evidence supporting that mitochondrial dysfunction is involved in various types of CRS.
Introduction
The crosstalk between the heart and kidney plays an important role in regulating fluid balance, metabolite excretion, and neuroendocrine function to maintain homeostasis (1), and there are common pathological risk factors between these two organs. Cardiorenal syndrome (CRS) is broadly defined as “a disorder of the heart and kidney whereby acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other” (2, 3). In 2010, the Acute Dialysis Quality Initiative (ADQI) classified CRS into five types based on the organ leading to the syndrome and the time course of disease progression (i.e., acute or chronic) (2). Epidemiological studies have shown that 25–63% of patients with heart failure (HF) suffered from a form of CRS defined by the ADQI (4). According to the Global Burden of Disease Study, the global prevalence of chronic kidney disease (CKD) in 2015 was estimated to be approximately 323 million, and 10–47% of CKD patients had cardiovascular disease, which was the leading cause of death in the CKD population (5). Furthermore, as renal function declined, the risk of all-cause mortality increased from 20% in stage 3a to nearly 500% in stage 5. Moreover, 25–50% of patients with HF had CKD, including an estimated 5.7 million people in the United States. Mortality increased by 56% in HF patients with concomitant CKD and further increased to 131% in patients with moderate or severe renal impairment (6, 7).
Despite decades of basic and clinical research, CRS remains a significant public health burden due to its unclear pathogenesis and lack of effective treatment (8). Both in acute and chronic settings, the pathophysiological pathways that exacerbate the cardiac or renal injury, such as persistent activation of the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS), chronic inflammation, oxidative stress, and fibrosis, play a vital role in CRS (9, 10) (Figure 1). Although the above theoretical mechanisms explain the pathological changes in CRS, there is no single-factor hypothesis to describe how damage to an organ triggers distal organ dysfunction or failure.
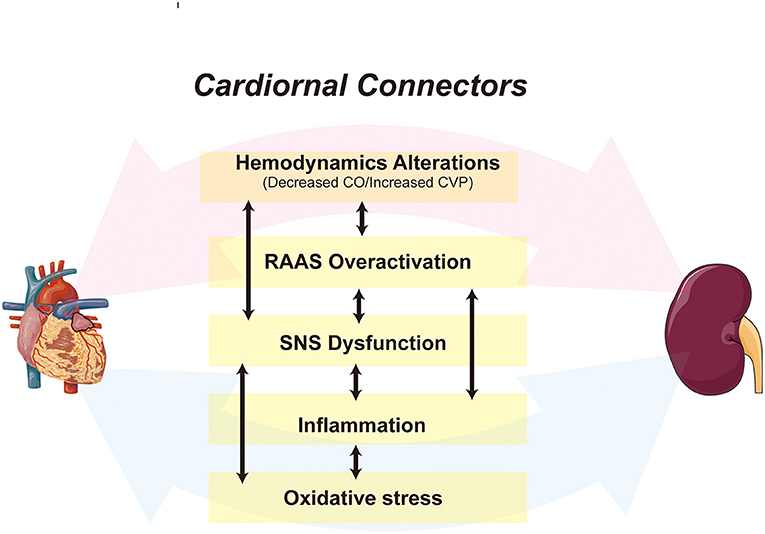
Figure 1. A schematic depiction of the cardiorenal connectors in CRS. The cardiorenal connectors, including hemodynamic factors, RAAS, SNS, inflammation, and oxidative stress are core mechanisms of CRS, all of which synergize and activate each other, leading to further deterioration of cardiac and renal function. CO, Cardiac Output; CVP, central venous pressure.
It is widely known that the heart contains the highest amounts of mitochondria in the human body. Given the high energy consumption of cardiomyocytes, more than 90% of the ATP is produced by mitochondria, which account for approximately one-third of the cardiomyocyte volume (11). At the renal level, owing to the high energy demands for solute reabsorption (12), the kidney, especially the cells in proximal tubules and medullary thick ascending limb, contains abundant mitochondria (13). A wide range of stress stimuli, such as ischemia, hypoxia or toxic injury, primarily target the cardiomyocytes as well as tubular epithelial cells especially the highly metabolically active proximal tubular segment (14, 15). In fact, mitochondria rapidly sense and respond to the insults to maintain their homeostasis through morphological alterations, bioenergetics adaptations, and enhanced self-renewal/degradation (12, 16). Impaired adaptive responses are closely associated with a decline in cardiac/renal function through various mechanisms affecting metabolism, oxidative stress, inflammation, calcium dynamics and mitophagy, and it is widely recognized that mitochondrial malfunction is an early and prominent signature of organic depression. Therefore, disrupted mitochondrial homeostasis is considered not only as a consequence of myocardial injury but also as a possible cause of HF (17). Meanwhile, Plotnikov and colleagues concluded that mitochondrial dysfunction was an independent risk factor for CKD regardless of the underlying etiology (18), and it was closely associated with the progression of CKD (19). Maintaining mitochondrial function significantly preserves the structural integrity of the left ventricular myocardium and renal parenchyma and the function of the heart and kidney (20, 21).
Furthermore, all the above-mentioned pathological factors in CRS damage the mitochondria in distal organs through circulatory effects (21–23), suggesting that mitochondria play a connecting role in cardiorenal interaction, and may be a core link in CRS progression (Figure 2). However, the specific mechanism and evidence have not been well summarized. In this article, we first outline the unique characteristics of heart and kidney mitochondria and their important role in organ injury, then summarize the current evidence of mitochondrial involvement in the pathogenesis of various types of CRS, including the potential of mitochondria-targeted CRS therapy.
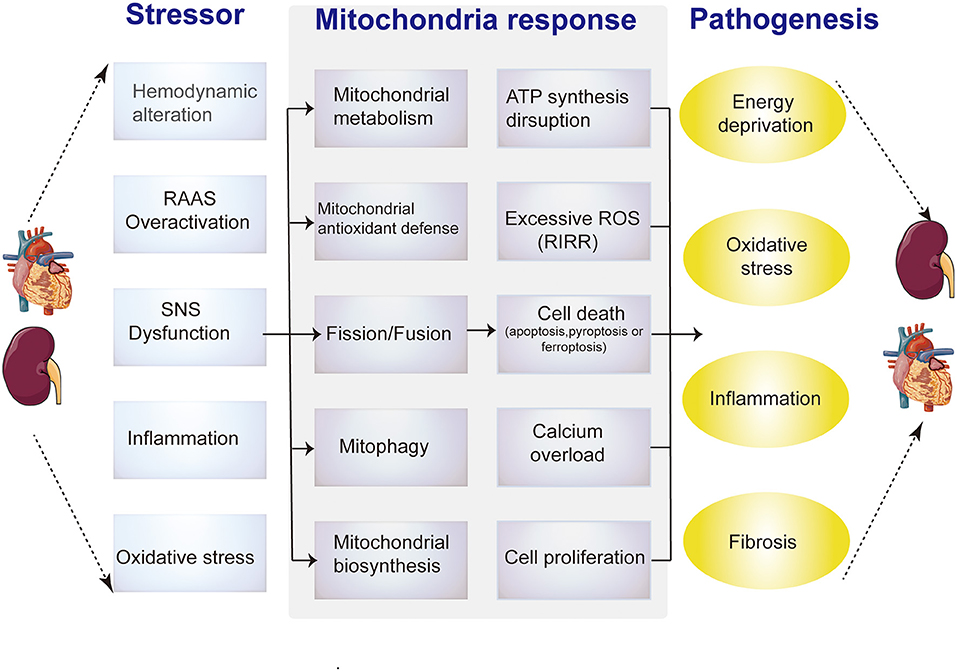
Figure 2. Diagrammatic representation shows the relationship between pathological alterations in CRS and mitochondrial dysfunction mechanisms. Intrinsic and extrinsic stress signals, such as hemodynamic alteration, RAAS overactivation, SNS dysfunction, inflammation or oxidative stress, can activate mitochondrial responses, including mitochondrial metabolism, antioxidant defense imbalance, abnormal dynamics, mitophagy and biogenesis. Impaired adaptive responses ultimately lead to mitochondrial dysfunction, include ATP synthesis reduction, excessive ROS, aberrant calcium signaling, (de) differentiation or cell death (apoptosis or necrosis). These changes in cardiomyocytes or renal tubular epithelial cells induce energy deprivation, oxidative stress, inflammation and fibrosis.
Impaired Mitochondrial Metabolism
Under physiological conditions, mitochondria from adult cardiomyocytes and renal proximal tubular cells (PTCs) preferentially use fatty acyl-CoA, the primary substrate for mitochondrial fatty acid β-oxidation (FAO), rather than pyruvate to generate ATP (24, 25). It has been estimated that fatty acids supply ~60–90% of the energy used to synthesize ATP in cardiomyocytes, whereas 10–40% of ATP is derived from glucose metabolism (26). PTCs almost exclusively depend on FAO and subsequent mitochondrial oxidative phosphorylation (OXPHOS) as their sole energy source (25). When faced with various biological stresses, such as temporary hypoxia, FAO in most hypermetabolic cells shuts down for a period of time, allowing the flexible conversion of metabolic substrates from FAs to glucose (i.e., the Randle cycle, proposed in 1963) (27, 28). This switch compensates for impaired energy in a short period and exerts a partial organ protective effect. However, the conversion affects the activity of electron transport chain (ETC)-related proteins, leading to impaired OXPHOS and restricted ATP production (29, 30). The hallmarks of metabolic remodeling are FAO downregulation and increased glucose utilization, which are observed in both early-stage heart and kidney injury (31, 32) (Figure 3).
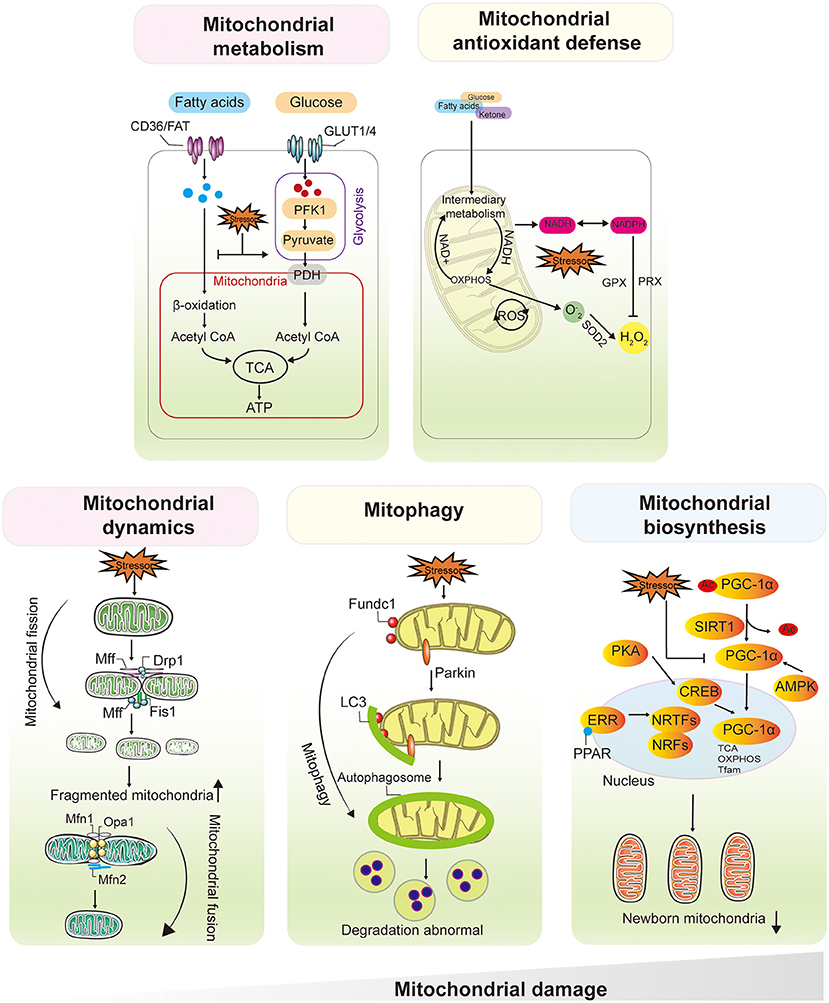
Figure 3. Mitochondrial Dysfunction Plays a Central and Multifaceted Role in HF and AKI/CKD. Mitochondria from cardiomyocytes and renal proximal tubular cells preferentially use fatty acyl-CoA, the primary substrate for mitochondrial FAO, rather than pyruvate to generate ATP. The hallmarks of metabolic remodeling are FAO downregulation and increased glucose utilization, which are observed in both early-stage heart and kidney injury. ROS were initially thought to be by-products of mitochondrial OXPHOS, the balance between mtROS production and scavenging is critical for maintaining mitochondrial function and cell viability. Mitochondrial dynamics contribute to the functional integrity of mitochondria. Fusion allows mixing of contents within the mitochondrial network and protects the mitochondria from stress. Damaged mitochondria undergo selective mitochondrial autophagy via Parkin, Fundc1, through autophagosome formation and lysosome-mediated degradation. Upregulation of PGC1α and activation of NRF1/2 initiate mitochondrial biogenesis, followed by mtDNA amplification and synthesis of nuclear-encoded mitochondrial proteins. After severe mitochondrial damage, increased fission leads to mitochondrial fragmentation, depolarization of the mitochondrial membrane potential inhibits fusion, and frequent abnormal fission events will affect mitochondrial autophagy, leading to abnormal degradation of damaged mitochondria, along with a decrease in the number and quality of nascent mitochondria mediated by mitochondrial biosynthesis. NRFs, nuclear respiratory factors, PGC1α, PPARγ coactivator-1α. FAO, fatty acid β-oxidation, OXPHOS, oxidative phosphorylation.
Role in Cardiac Injury
As a central mechanism of energy deprivation in the failing heart, impaired mitochondrial metabolism has inspired decades of research to date. In vitro studies have shown that ATP is reduced in failing cardiomyocytes, particularly in mitochondria (33, 34). Both animal models and clinical trials have demonstrated that ATP flux is reduced in advanced HF, and ATP supplementation improves cardiac function and even reverses the subsequent structural remodeling of ventricles (34–36). The utilization of myocardial FAs, which are metabolized via non-oxidative pathways to produce lipotoxic ceramides and diacylglycerols in rats with severe HF, leading to mitochondrial dysfunction and apoptosis and further aggravating the impaired energy metabolism (37). Previous studies confirmed that a greater dependence on glucose in the failing heart promotes glycolysis, leading to increased lactate production and anaplerosis (38). It also drives more glucose into collateral pathways, all of which reduce the efficiency of ATP synthesis and exacerbate pathological myocardial remodeling (38, 39).
Role in Kidney Injury
Experimental evidence indicates that FAO dysregulation profoundly affects the fate of PTCs by promoting inflammation, ultimately leading to mesenchymal fibrosis and epithelial-to-mesenchymal transition (40). High levels of albumin-bound long-chain saturated FAs promote the progression of renal tubular damage and interstitial fibrosis through the activation of pro-inflammatory pathways, including tumor necrosis factor-alpha (TNF-α), CC motif chemokine 2, and interleukin (IL)-6, and increase the production of ROS (40–42). Kang HM et al. reported that PTCs treated with the CPT-1 inhibitor etomoxir undergo morphologic and genomic changes, with increased expression of mesenchymal genes, such as ACTA2 (encoding α-smooth muscle actin), VIM (encoding vimentin), and COL1A1 and COL3A1 (encoding fibrillar collagens) (43). In addition, Lan R et al. found that the shift from FAO to glycolysis in PTCs undergoing renal ischemia/reperfusion (I/R) injury promoted the development of tubular atrophy and transition from AKI to CKD (44). This may be related to the increased expression of the key glycolytic enzyme glycolysis pyruvate kinase M2 and levels of lactate metabolites, cooperatively leading to hypoxic and acidic environments and eventually inhibiting the proliferation and differentiation of podocytes and aggravating fibrosis (45, 46).
Mitochondrial Antioxidant Defense Imbalance
ROS were initially thought to be by-products of mitochondrial OXPHOS. Electrons that leak out of the major site of complex I and complex III in the ETC and react with O2 generate superoxide anions (47), which are then converted to H2O2 by superoxide dismutase (48). Mitochondrial antioxidant systems, including catalase, glutathione peroxidases, and peroxiredoxin, further reduce H2O2 to water. Under physiological conditions, the mitochondrial antioxidant defense system maintains low levels of ROS in the organelle (49, 50). Under stress conditions, ROS accumulate when mitochondrial ROS (mtROS) production exceeds the mitochondrial antioxidant defense capacity or when the antioxidant defense system is impaired (51). Subsequently, mtROS react rapidly with nitric oxide to form a potent oxidant and nitrifying agent. As a result of mtROS accumulation, H2O2 release from mitochondria to the cytoplasm increases, exacerbating oxidative damage outside the mitochondria (52, 53). Therefore, the balance between mtROS production and scavenging is critical for maintaining mitochondrial function and cell viability (Figure 3).
Role in Cardiac Injury
In the failing heart, damage caused by excessive mtROS and innate antioxidant defense exhaustion are evident in human patients and animal models (54–56). The overproduction of mtROS is primarily due to increased expression and activity of NADPH oxidase through a variety of pathological stimuli, including mechanical stretch, angiotensin II, endothelin 1, and TNF-α (52). Moreover, experimental evidence suggests the activities of superoxide dismutase, catalase, and glutathione peroxidases are significantly reduced during HF (17). Mitochondria-targeted ROS scavenging (e.g., MitoQ, MitoTEMPO, or SS-31) has demonstrated benefit in animal models of HF (57–59). Interestingly, NAD+, a precursor for the phosphorylated dinucleotide pair NADP+/NADPH (plays a major role in the detoxification of ROS), was also significantly reduced in the myocardium of some HF mice (60). In addition, nicotinamide mononucleotide adenosyltransferase, an enzyme responsible for NAD+ production, was found to be substantially inhibited in both mouse HF models and patients (61, 62). Further studies indicate that increasing intracellular NAD+ levels by pharmacological or genetic approaches restore protein acetylation and improve cardiac function in mouse models of HF (63).
Role in Kidney Injury
Experimental evidence suggests that kidney injury is accompanied by an impaired mitochondrial antioxidant defense system. Increased production of mtROS is a common feature of AKI and CKD (64, 65). Enhanced mitochondrial antioxidant defenses by supplementation with mitochondria-targeted antioxidants have been shown to attenuate mitochondrial dysfunction and reduce kidney injury in animal models of IR-induced renal fibrosis, diabetic nephropathy, and unilateral ureteral obstruction (UUO)-induced CKD (66–68). Notably, a moderate increase in mtROS may regulate signaling pathways involved in renal injury and incomplete renal repair (69). For example, mtROS have been shown to activate hypoxia-inducible factor 1α in response to hypoxia; the NLRP3 pathway, which induces inflammation, cytokine production, and innate immune responses; and the transforming growth factor-β (TGFβ) pathway, which has a pro-fibrotic effect in disease conditions (70–72).
Noteworthily, excessive mtROS have been shown to affect a wide range of cellular functions in the context of HF and AKI/CKD. Mitochondrial damage caused by an initial increase in oxidative stress leads to a further increase in ROS production and more severe damage. This results in so-called ROS-induced ROS release in mitochondria, creating a vicious cycle and increasing the tendency for cell death (73). High levels of ROS damage mtDNA and induce mtDNA leakage, which can bind to Toll-like receptors or nucleotide-binding oligomerization domain-containing protein (NOD)-like receptors, leading to inflammation (74). Moreover, increased ROS also induce mitochondrial permeability transition pore opening, lead to cell death, and impair mitochondrial biogenesis (25, 75, 76). However, the specific role of these mechanisms in the development of damage remains to be fully elucidated.
Abnormal Mitochondrial Dynamics
Mitochondria are not static organelles. In fact, they continuously undergo fusion and fission, two processes that define mitochondrial dynamics (77). Through fission and fusion, mitochondrial size, distribution, shape, position, and mass are fine-tuned in response to changes in the metabolic state of the cell (77). Mitochondrial fission is a multistep process that allows a mitochondrion to split in two daughter mitochondria, and primarily mediated by dynamin-related protein 1 (Drp1), a large dynamin-related guanosine triphosphate hydrolase (GTPase) (78, 79). During the process, Drp1 is recruited to the outer mitochondrial membrane, and then GTP hydrolysis enhances membrane contraction, leading to the recruitment of dynamin 2 to terminate membrane cut-off (80). In contrast, mitochondrial fusion is driven by a two-step process induced by several GTPases, including mitofusin 1 (Mfn1) and Mfn2 involved in outer mitochondrial membrane fusion and optic atrophy 1 (Opa1) involved in inner membrane fusion (81, 82). The continuous alternation between fission and fusion maintains mitochondrial homeostasis and cellular function in response to physiological changes (83). However, the disruption of dynamic balance alters mitochondrial morphology and impairs mitochondrial function, leading to cell viability, which is closely associated with heart and kidney diseases (78, 84) (Figure 3).
Role in Cardiac Injury
Impaired mitochondrial fission may cause dilated cardiomyopathy, whereas impaired mitochondrial fusion leads to hypertrophy (85–87). Genetic ablation of genes associated with fission/fusion can lead to significant myocardial structural abnormalities, such as enlarged left ventricles, reduced cardiac output, and abnormal myocardial fibrosis (88, 89). Mice with a cardiomyocyte-specific conditional knockout of Drp1 die after 8–13 weeks and develop evidence of HF. At the cellular level, these hearts contain elevated mitochondrial counts, elongated mitochondria, and increased apoptosis (86). Similarly, myocardial Drp1 levels are elevated in HF patients through a mechanism potentially regulated by neurohormone norepinephrine-mediated myocardial hypertrophy. In contrast to mitochondrial fission, the mRNA transcription and protein expression of Mfn1/2 or Opa1 were enhanced in mouse cardiomyocytes (90), suggesting that under physiological conditions, mitochondrial fusion activity in cardiomyocytes is restricted by inhibitory mechanisms. Because of the redundant expression of Mfn1 and Mfn2 in cardiomyocytes, the ablation of either gene fails to significantly inhibit mitochondrial fusion in normal cardiomyocytes (91). At the subcellular level, most mitochondria in Mfn1/Mfn2 double-knockout cardiomyocytes are spherical and lack an elongated morphology, suggesting an imbalance between fission and fusion (92). Interestingly, in Drosophila and mice, Opa1 gene ablation induces mitochondrial fragmentation with mtROS production and respiratory dysfunction, which produces a cardiac phenotype different from that induced by Mfn1/Mfn2 ablation (93). Moreover, heart tube-specific Opa1 knockdown induces dilation of the heart tube, with severe contractile impairment (94). Conversely, Opa1 overexpression in vivo refines myocardial injury, and the underlying mechanism may be dependent on the stabilization of the mitochondrial cristae structure of Opa1, which increases mitochondrial respiratory function and prevents cytochrome c release, ROS production, and apoptosis (95).
Role in Kidney Injury
Mitochondrial fragmentation due to excessive fission and/or fusion inhibition is thought to be a key event in mitochondrial damage and renal tubular injury during AKI (12). In murine models of AKI induced by renal IR or cisplatin toxicity, mitochondrial fragmentation precedes tubular cell apoptosis, and the inhibition of fission attenuates tubular cell death and renal injury (96, 97). Consistent with this finding, proximal tubule-specific deletion of Drp1 protects mice from renal IR-induced tubular cell death, inflammation, and renal injury and accelerates renal recovery (98). In vitro studies have shown that Mfn2 deficiency enhances ATP depletion-induced cell injury and death (99). However, proximal tubule-specific Mfn2 knockout mice exhibit milder renal injury and higher survival rates compared with wild-type mice. After AKI in these mice, Mfn2 deficiency stimulates mitogen-activated protein kinase signaling pathway-dependent renal tubular cell proliferation, which may accelerate renal repair and thus overcome the detrimental effects of inhibiting mitochondrial fusion, leading to renal protection (99). Enhanced mitochondrial fragmentation in renal tubular cells and podocytes has been reported in experimental models of diabetic kidney disease (DKD) and renal biopsy samples from patients with DKD (100, 101). In addition, the knockdown of podocyte Drp1 blocked mitochondrial fragmentation, improved mitochondrial fitness, and protected mice from DKD progression (102). Consistent with these findings, pharmacological inhibition of Drp1 protected mice from DKD progression (101, 103). As mentioned above, mitochondrial fragmentation in renal tubular cells may reduce energy metabolism and increase ROS formation, thereby promoting tissue injury, inflammation, and maladaptive renal repair. The specific mechanisms underlying the deleterious role of mitochondrial fragmentation in renal repair need to be investigated in depth.
Abnormal Mitophagy
Mitophagy is a mechanism that selectively degrades excess and defective mitochondria (104, 105). Two major mechanisms for labeling mitochondria and transporting them to autophagosomes have been identified: one regulated by the serine/threonine-protein kinase PTEN-induced kinase 1 (PINK1), mitochondrial E3 ubiquitin-protein ligase parkin pathway and the other mediated by mitophagy receptors, including BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3-like (BNIP3L), FUN14 domain-containing 1 (FUNDC1), and E3 ubiquitin-protein ligase SMURF1 (106, 107). Among them, Parkin and Fundc1 appear to be inducers of protective mitophagy, whereas lethal mitophagy is more likely triggered by the upregulation of Bnip3 and Nix (106, 108). Specifically, although autophagy/mitophagy is considered by most researchers to be the “guardian” of mitochondrial function and cardiomyocyte homeostasis, different adaptors trigger mitophagy to varying degrees, promoting cell survival or cell death. Moderate mitophagy selectively removes impaired mitochondrial subsets, thereby increasing ATP production. In contrast, excessive mitophagy induces cell death by depleting cellular ATP reserves (109). It is worth noting that the degree of mitophagy activation also depends on the level and duration of cellular stress. However, the extent to which mitophagy activation contributes to mitochondrial dysfunction and cellular energy deficiency in HF or AKI/CKD remains unclear (Figure 3).
Role in Cardiac Injury
Impaired mitophagy in cardiomyocytes has been reported in experimental models of HF and cardiac biopsy samples from patients with HF. In a mouse model of HF induced by transverse aortic coarctation, PINK1 phosphorylation was reduced, accompanied by inhibited mitophagy and impaired mitochondrial function (110). Genetic ablation of Parkin disrupts the mitochondrial network, reduces ATP synthesis, and causes ROS overload in cardiomyocytes (111). AMPKα2 phosphorylates Ser495 of PINK1 to enhance pink/parkin pathway signaling, activate mitophagy, promote the elimination of damaged mitochondria, and improve mitochondrial function, thereby preventing the early progression of HF (110). Interestingly, the role of parkin-mediated mitophagy in the regulation of mitochondrial dynamics has also been reported. In parkin-deficient hearts and cardiomyocytes, mitochondria show fragmentation, which may be related to parkin-mediated inhibition of Mfn1/2 ubiquitination, leading to reduced mitochondrial fusion (110, 112). Furthermore, hearts with baseline genetic ablation of Fundc1 exhibit reduced early to late ventricular filling velocities, prolonged isovolumetric relaxation times, reduced ejection fractions, and decreased fractional shortening, suggesting that Fundc1 knockout mice have impaired cardiac function and are susceptible to HF (113). As a cardiomyocyte death factor, Nix regulates mitochondrial death and acts as a downstream effector of the stress-related hormone norepinephrine, which promotes cardiac fibrosis during ventricular remodeling. In addition, Nix overexpression promotes collagen and fibronectin expression (114).
Role in Kidney Injury
Increasing evidence indicates that mitophagy plays an essential role in kidney injury. In a mouse model of septic AKI induced by cecum ligation and puncture, renal tubular cells exhibited increased mitophagy in the early stages of septic AKI, followed by decreased mitophagy in the late stages of AKI (115). Increased mitochondrial loss and autophagosome formation have been reported in the regeneration of PTCs after renal IRI, and these abnormalities resolved in normally repaired tubules but persisted and progressively worsened in undifferentiated tubular cells, suggesting a role for mitophagy in renal repair after AKI (44). Furthermore, in the renal tubules of DKD mice, reduced expression of PINK1 and parkin in PTCs, decreased autophagosome formation, and optineurin overexpression enhanced mitophagy, thereby attenuating cellular senescence, mtROS accumulation, and NLRP3 inflammasome activation (116, 117). In addition to DKD, mitophagy has been associated with the development of non-diabetic CKD, with increased mitochondrial PINK1 and parkin formation and increased levels of autophagy in renal tubules and hypoxia-exposed PTCs in UUO mice, suggesting that mitophagy is activated in these settings. In contrast, another study reported reduced parkin expression and autophagy levels in kidney tissue from UUO mice (118, 119). These conflicting findings suggest the presence of context- and cell-type-specific alterations in mitophagy in CKD. However, both studies suggest that the deletion of PINK1 or Parkin exacerbates renal injury in UUO mice, supporting a protective role for mitophagy in CKD.
Abnormal Mitochondrial Biogenesis
In response to changing energy demands triggered by developmental signals and environmental pressures, cells initiate mitochondrial biogenesis, the process of generating new mitochondrial mass and mtDNA replication through the proliferation of existing mitochondria (120). PGC1α is considered to be a master regulator of mitochondrial biogenesis, and its expression is upregulated upon increased energy demands (eg, fasting or exercise) or stress conditions (eg, cold or hypoxia) (121, 122). Mechanistically, PGC-1a is activated by phosphorylation or deacetylation, which then stimulates the expression of a series of nuclear transcription factors, including nuclear respiratory factor 1, 2 (NRF1/2), and subsequently, the initiation of NRF binding to ETC genes and mitochondrial transcription factor family (Tfam) genes, which are responsible for mtDNA transcription and translation (122).The current data acknowledge that mitochondrial biosynthesis results in an increase in OXPHOS capacity, a decrease in pathological oxidative stress and the repair of mitochondria-related dysfunction (Figure 3).
Role in Cardiac Injury
Previous studies showed the essential role of mitochondrial biogenesis during adulthood, which involve conferring protection against long-term cardiac dysfunction, and in early embryonic cardiac development (123). Subepicardial biopsies from patients with HF exhibit decreased PGC1α expression (124). Consistent with the clinical finding, PGC1α ablation impairs mitochondrial energy metabolism and induces the development of HF in mice (125). Likewise, in vitro, dramatic reduction in mtDNA copy number in ischemic cardiomyocytes, which leads to reduced mitochondrial mass and increased mitochondrial fragmentation, while PGC1α overexpression attenuates these abnormalities (126). These findings suggest that downregulation of PGC1α facilitates the pathogenesis and progression of HF. However, the results of other studies sound a different tone. Hu's study showed no significant changes in PGC-1α gene expression in myocardial tissue of patients with congestive HF (127). Addition, in a transgenic mouse model, myocardial-specific PGC1α overexpression resulted in excessive mitochondrial proliferation and disruption of sarcoplasmic reticulum structure in cardiomyocytes, leading to cardiac enlargement with reduced myocardial contractile function (128), which indicate the overexpression of PGC1α does not improve mitochondrial function (129).This may be explained by the fact that the outcome of PGC1α overexpression and its effects on HF are tightly related to the activity and the interaction of mitochondrial biogenesis with other intracellular events. Altogether, the role of PGC1α in HF remains controversial, while the role of PGC1α in different types of HF needs to be further investigated.
Role in Kidney Injury
Mounting evidence supports a beneficial effect of mitochondrial biogenesis in kidney injury and repair after AKI. PGC1α was highly expressed in proximal tubules with abundant mitochondria (130). Decreased expression of PGC1α in the kidney was observed in both renal IRI- or cisplatin-induced AKI animal models and in renal biopsy samples from AKI patients compared with controls (65, 130). Further studies demonstrated that in mice models of septic AKI, the levels of PGC1α and downstream OXPHOS genes in the kidney were suppressed proportionally to the degree of kidney injury and were restored to normal levels during kidney recovery (131), suggesting a negative correlation between PGC1α expression in kidney and AKI severity. The pharmacological activation of PGC1α accelerated the recovery of renal function after IRI in mice supporting the mentioned findings. Notably, mouse podocyte-specific overexpression of PGC-1 resulted in altered mitochondrial properties, including formation of giant mitochondria, increased expression of ETC and mitochondrial fusion genes, and enhanced podocyte proliferation and dedifferentiation, leading to proteinuria and glomerulosclerosis (132).
Evidence for the Participation of Mitochondrial Dysfunction in the Pathogenesis of CRS
As previously mentioned, hemodynamic factors, RAAS, SNS, inflammation, and oxidative stress are core mechanisms of CRS, all of which synergize and activate each other, leading to further deterioration of cardiac and renal function (133, 134). In fact, all of the above factors cause mitochondrial damage in distal organs. First, mitochondria are the main consumers of oxygen in cells, and hypoxia directly or indirectly impairs mitochondrial dynamics, autophagy, and OXPHOS through the hypoxia-inducible factor pathway (56, 135). And one of the most deleterious effects of RAAS stimulation is the activation of NADPH oxidase, which results in increased mtROS in endothelial cells, renal tubular cells (136), and cardiomyocytes (137). Long-term SNS hyperactivity, on the one hand, promotes the growth of renal vascular wall and cultured cardiomyocytes through the production of mtROS (138, 139), on the other hand, it also promotes the activation of RAAS by directly stimulating the release of renin and plays a synergistic role in mitochondrial damage. In addition, as a hub of proinflammatory signaling, mitochondria are affected by elevated levels of multiple factors (such as C-reactive protein, IL-1β, IL-6, and TNF-a) in the chronic inflammatory environment of CRS (140, 141), further releasing inflammatory activators represented by mtDNA and ROS, triggering a vicious cycle of more severe inflammatory responses that predispose to fibrosis (142, 143) (Figure 4). Thus, mitochondrial dysfunction appears to play a key role in CRS. The role of mitochondrial dysfunction in different subtypes of CRS was summarized based on the available evidence (Table 1).
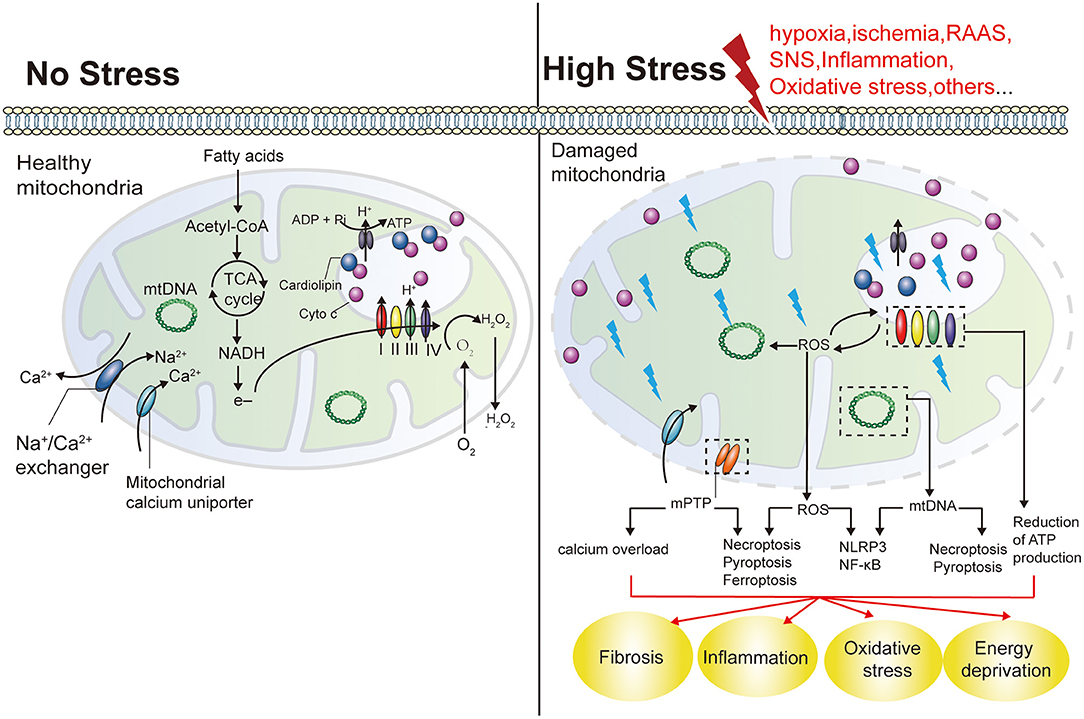
Figure 4. Mitochondrial functions and the effects of mitochondrial damage. Mitochondria play a key role in producing energy in the form of ATP. NADPH are formed by the oxidation of fatty acids and the cycle of TCA in the mitochondrial matrix, and their electrons are transferred to O2 through the electron transport chain (including COX I and IV). This results in the generation of a proton gradient across the IMM to produce ATP. Cyto C exists in free form in the IMS, or is anchored in the IMM by interaction with cardiolipin, acting as an electron carrier COX III and COX IV. Mitochondria are the main source of ROS. Mitochondria also play an important role in maintaining calcium balance in cells. Mitochondrial damage reduces ATP production and can result in the energetic failure of cells. An increase in mitochondrial ROS production by damaged mitochondria may also induce other forms of cell death, including necroptosis, pyroptosis and ferroptosis, as well as inflammation. NADH, nicotinamide adenine dinucleotides; TCA, tricarboxylic acids; IMM, mitochondrial intima; Cyto C, Cytochrome C; IMS, intermembrane space; IMM, intermembrane space; COX I, mitochondrial respiratory complex I; NLRs, nucleotide-binding and oligomerization domain-like receptors.

Table 1. Mechanisms and effects of mitochondrial dysfunction in different types of CRS.
Mitochondrial Dysfunction in CRS1
CRS1 is defined as a sudden deterioration of cardiac function, leading to AKI and/or dysfunction (2). It usually follows acute ischemic or non-ischemic heart disease (152, 153), most commonly acute decompensated heart failure (ADHF) (154). Although the above mechanisms are risk factors for renal mitochondrial, there are limited studies on the direct relationship between CRS1 and mitochondrial dysfunction. Levosimendan is an effective drug for the clinical treatment of ADHF. A recent study showed that it decreased the risk of AKI after cardiopulmonary resuscitation in rats by enhancing mitochondrial respiratory enzyme activity, promoting mitochondrial energy metabolism, regulating mitochondrial dynamics-related protein expression, improving mitochondrial dysfunction, and reducing the number of apoptotic cells caused by mitochondrial pathways (144).
Mitochondrial Dysfunction in CRS2
CRS2 is defined as chronic cardiac insufficiency, leading to progressive manifestations of kidney damage, which contributes to the progression of CKD (155, 156). Morphological studies in rat models of congestive HF-induced renal injury revealed mitochondrial swelling in renal tubular epithelial cells, possibly due to the release of Cyt-c, which mediated caspase 3 activation and nuclear transfer and triggered apoptosis, supporting the role of mitochondria-mediated apoptosis in CRS2 (145). Early empagliflozin treatment protects cardiac and renal function in CRS rats by reducing apoptin (mitochondrial-Bax/cleaved-caspase-3/cleaved-parp) and fibrosin (TGF-β/Smad3), reducing DNA/mitochondrial damage (γ-H2AX/cytoplasmic-Cyt-c), and maintaining mitochondrial function and integrity (146).
Mitochondrial Dysfunction in CRS3
CRS3 refers to a CRS subtype in which the onset of AKI leads to the progression of acute cardiac injury or dysfunction (9). Proteomic analysis of CRS3 rats showed alterations in myocardial pyruvate metabolism, glyoxylate and dicarboxylic acid metabolism, starch and sucrose metabolism, and amino acid biosynthesis, with 23 proteins enriched in signaling pathways related to mitochondrial function, suggesting that AKI may affect cardiomyocyte metabolism or mitochondrial bioenergy (23). Growth factor receptor-binding protein 2 (Grb2) is a regulator of AKI-related myocardial injury, and Grb2 activation promotes mitochondrial metabolic disorders in cardiomyocytes by inhibiting the Akt/mTOR signaling pathway. Additionally, the administration of Grb2-specific inhibitors reverses myocardial pathological changes in the context of AKI (23). Furthermore, the dysregulation of mitochondrial dynamics caused by increased Drp1 expression and cardiac apoptosis plays an important role in AKI-induced myocardial injury (147, 148). Renal ischemia-reperfusion injury induces mitochondrial calcium overload in cardiomyocytes through the inositol 1,4,5-trisphosphate receptor (IP3R)-mitochondrial calcium uniporter (MCU) signaling pathway, decreasing mitochondrial membrane potential and increasing pathological mitochondrial fission. In addition, melatonin attenuates myocardial injury caused by cytoplasmic and mitochondrial calcium overload by inhibiting IP3R phosphorylation and MCU expression (23). Thus, Doi K et al. proposed that CRS3-related studies should center on the new concept of “mitochondrial hormesis” (149).
Mitochondrial Dysfunction in CRS4
CRS4 is characterized by cardiovascular damage in patients with CKD in all stages (25). Pressure and fluid overload in CKD patients often lead to hypertrophy with histological changes, such as fibrosis (26). These changes are associated with inflammation and other cardiovascular factors, including hypertension, RAAS activation, or fluid overload, and are often accompanied by a decrease in GFR (157). Ang II is involved in CKD-induced myocardial interstitial fibrosis and cardiomyocyte hypertrophy by inducing mitochondrial structural damage and mitochondrial apoptosis. Ang II receptor blockers merely upregulate mitochondrial fusion-related protein levels, reduce mitochondrial swelling, and improve the spatial organization of cardiac mitochondria, suggesting that further identification of molecular pathways contributing to mitochondrial damage and appropriate intervention are essential in CRS4 (150). The drug Entresto protects cardiomyocytes and cardiac function in high-protein diet-fed CRS4 rats by regulating oxidative stress and Mfn2-mediated mitochondrial functional integrity (140). Hyperphosphatemia (HP) is a known serum hallmark of CKD. The transcription factor interferon regulatory factor 1 (IRF1) is a key molecule upregulated by HP through histone H3K9 acetylation, and it directly binds to the promoter region of HP-mediated PGC1α to play a role in its transcriptional repression. In contrast, the restoration of PGC1α expression or gene knockdown of IRF1 significantly attenuates HP-induced changes in vitro and in vivo. These findings suggest that IRF1-PGC1α axis-mediated myocardial mitochondrial biosynthesis plays a crucial role in the pathogenesis of CRS4 (22).
Mitochondrial Dysfunction in CRS5
CRS5 refers to systemic diseases (sepsis, drug toxicity, lupus, liver cirrhosis, or amyloidosis) resulting in simultaneous cardiac and renal damage and/or dysfunction (158). In sepsis-induced CRS5, the key pathway found to be altered is primary metabolism, which may affect local cycling through decreased ATP levels and mitochondrial dysfunction (151, 159). Tran et al. demonstrated that mitochondrial dysfunction, cell swelling, and marked acylglycerol accumulation in tubules led to reduced prostaglandin E2 and promoted medullary vasoconstriction in ischemic AKI (160). There is a clear correlation between the stage of mitochondrial dysfunction, disease severity, and prognosis of patients with septic myocardial injury (161). However, no studies have directly assessed cardiac and renal mitochondrial function in patients with sepsis.
Concluding Remarks and Perspectives
This article reviewed the pathophysiological mechanisms of CRS from the perspective of mitochondrial dysfunction. The elucidation of the mechanisms by which two organs are interconnected is an unmet medical need. Given that mitochondria play a vital role in renal and cardiovascular disease (Figure 2), this organelle may be an excellent candidate therapeutic target to disrupt the vicious cycle between HF and AKI/CKD. Accordingly, we propose a three-step mechanism that may explain the pathophysiology of CRS. First, damaged renal tissue (heart) releases pro-inflammatory factors and oxidative metabolites into the circulation, and changes in the neuroendocrine system lead to the secretion of several hormones into the blood. Second, kidney (cardiac)-derived biomolecules interact directly with receptors or adaptors on the surface of cardiomyocytes (renal tubular epithelial cells) and have the potential to exert indirect effects on cardiomyocytes (renal tubular epithelial cells) through other mechanisms. Finally, as one of the most sensitive intracellular organelles, mitochondria sense a wide range of stimuli in the extracellular environment and respond to cardiac (renal)-derived biomolecules by changing their morphology. Impaired mitochondrial adaptation leads to insufficient ATP synthesis, which further triggers oxidative stress, inflammation, apoptosis, and fibrosis, possibly due to the transition from adaptive organ dysfunction to maladaptive organ dysfunction (Figure 2). However, this assumption has several limitations. First, receptors or adaptors expressed on the surface of cardiomyocytes and renal tubular epithelial cells have not been demonstrated. Second, mitochondria are not the only determinant of cytopathological changes. Cardiomyocyte injury can also be caused by mechanical stress due to intracellular acidosis, disturbed calcium metabolism, and fluid overload. Third, as discussed above, despite exciting preclinical data, translation of mitochondria-targeted agents into clinical use in HF and AKI /CKD remains a big challenge, for example, the results obtained in antioxidants in patients with heart or kidney diseases have been very diverse, and not all therapies as effective as preclinical studies have shown. Therefore, further studies on the relationship between mitochondria and other intracellular stress responses are needed to understand the sequence of events contributing to organ damage.
Given the key role of mitochondrial dysfunction in CRS, specific interventions targeting mitochondrial homeostasis to prevent and treat CRS have emerged as promising therapeutic strategies. A variety of compounds targeting mitochondria have been shown to prevent kidney injury and/or accelerate kidney repair in patients with AKI and CKD. Additionally, “mitotherapy” is considered a potential strategy for HF treatment and should help assess the use of these compounds in CRS. Emerging evidence suggests that mitochondria-targeted therapies acting upstream of cellular damage may have advantages over therapies targeting downstream processes (inflammation and fibrosis). Therefore, from this point of view, mitochondrial dysfunction may be one of the molecular links between cardiac and renal in CRS and is an emerging link in the pathophysiology of the diseases.
Author Contributions
SS, BZ, and YL conceived and designed the study. XX and JL involved in database search and extracted the data. QJ, RC, and WX analyzed the data and wrote the manuscript. YL and YW polished the English. YW, HW, QS, and YH revised the manuscript. All authors listed approved it for publication.
Funding
This work was supported by the Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences (Nos. CI2021A03323 and C12021A01603) and National Natural Science Foundation of China (Grant No. 81573799).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Kang Ren, for editing the English text of a draft of this manuscript.
References
1. Virzì GM, Clementi A, Brocca A, de Cal M, Ronco C. Molecular and genetic mechanisms involved in the pathogenesis of cardiorenal cross talk. Pathobiology. (2016) 83:201–10. doi: 10.1159/000444502
2. Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, et al. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. (2010) 31:703–11. doi: 10.1093/eurheartj/ehp507
3. Melenovsky V, Cervenka L, Viklicky O, Franekova J, Havlenova T, Behounek M, et al. Kidney response to heart failure: proteomic analysis of cardiorenal syndrome. Kidney Blood Press Res. (2018) 43:1437–50. doi: 10.1159/000493657
4. Ronco C, Di Lullo L. Cardiorenal syndrome in western countries: epidemiology, diagnosis and management approaches. Kidney Dis (Basel). (2017) 2:151–63. doi: 10.1159/000448749
5. Hill NR, Fatoba ST, Oke JL, Hirst JA, O'Callaghan CA, Lasserson DS, et al. Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS ONE. (2016) 11:e158765. doi: 10.1371/journal.pone.0158765
6. Wheeler DC, Stefansson BV, Heerspink HJL, Wheeler David C, Stefánsson Bergur V. Jongs Niels, et al. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: a prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. (2021) 9:22–31. doi: 10.1016/S2213-8587(20)30369-7
7. Thompson S, James M, Wiebe N, Hemmelgarn B, Manns B, Klarenbach S, et al. Cause of death in patients with reduced kidney function. J Am Soc Nephrol. (2015) 26:2504–11. doi: 10.1681/ASN.2014070714
8. Lekawanvijit S, Krum H. Cardiorenal syndrome: acute kidney injury secondary to cardiovascular disease and role of protein-bound uraemic toxins. J Physiol. (2014) 592:3969–83. doi: 10.1113/jphysiol.2014.273078
9. Clementi A, Virzì GM, Brocca A, de Cal M, Pastori S, Clementi M, et al. Advances in the pathogenesis of cardiorenal syndrome type 3. Oxid Med Cell Longev. (2015) 2015:148082. doi: 10.1155/2015/148082
10. Di Lullo L, Bellasi A, Russo D, Cozzolino M, Ronco C. Cardiorenal acute kidney injury: Epidemiology, presentation, causes, pathophysiology and treatment. Int J Cardiol. (2017) 227:143–50. doi: 10.1016/j.ijcard.2016.11.156
11. Doenst T, Nguyen T D, Abel E D. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
12. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. (2009) 119:1275–85. doi: 10.1172/JCI37829
13. Takemura K, Nishi H, Inagi R. Mitochondrial dysfunction in kidney disease and uremic sarcopenia. Front Physiol. (2020) 11:565023. doi: 10.3389/fphys.2020.565023
14. Isaka Y, Kimura T, Takabatake Y. The protective role of autophagy against aging and acute ischemic injury in kidney proximal tubular cells. Autophagy. (2011) 7:1085–7. doi: 10.4161/auto.7.9.16465
15. Kaushal GP, Kaushal V, Herzog C, Yang C. Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy. (2008) 4:710–2. doi: 10.4161/auto.6309
16. Howell GM, Gomez H, Collage RD, Loughran P, Zhang X, Escobar DA, et al. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS One. (2013) 8:e69520. doi: 10.1371/journal.pone.0069520
17. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. (2018) 128:3716–26. doi: 10.1172/JCI120849
18. Plotnikov EY, Kazachenko AV, Vyssokikh MY, Vasileva AK, Tcvirkun DV, Isaev NK, et al. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. (2007) 72:1493–502. doi: 10.1038/sj.ki.5002568
19. Granata S, Zaza G, Simone S, Villani G, Latorre D, Pontrelli P, et al. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genomics. (2009) 10:388. doi: 10.1186/1471-2164-10-388
20. Yeh JN, Yue Y, Chu YC, Huang CR, Yang CC, Chiang JY, et al. Entresto protected the cardiomyocytes and preserved heart function in cardiorenal syndrome rat fed with high-protein diet through regulating the oxidative stress and Mfn2-mediated mitochondrial functional integrity. Biomed Pharmacother. (2021) 144:112244. doi: 10.1016/j.biopha.2021.112244
21. Wang J, Toan S, Li R, Zhou H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochem Pharmacol. (2020) 174:113832. doi: 10.1016/j.bcp.2020.113832
22. Huang Y, Wang S, Zhou J, Liu Y, Du C, Yang K, et al. IRF1-mediated downregulation of PGC1alpha contributes to cardiorenal syndrome type 4. Nat Commun. (2020) 11:4664. doi: 10.1038/s41467-020-18519-0
23. Wang J, Sun X, Wang X, Cui S, Liu R, Liu J, et al. Grb2 induces cardiorenal syndrome type 3: Roles of IL-6, cardiomyocyte bioenergetics, and Akt/mTOR pathway. Front Cell Dev Biol. (2021) 9:630412. doi: 10.3389/fcell.2021.630412
24. Raimundo N. Mitochondrial pathology: stress signals from the energy factory. Trends Mol Med. (2014) 20:282–92. doi: 10.1016/j.molmed.2014.01.005
25. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. (2019). 30:784–99.e5. doi: 10.1016/j.cmet.2019.08.003
26. Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation–a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta. (2011) 1813:1333–50. doi: 10.1016/j.bbamcr.2011.01.015
27. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. (2009) 297:E578–91. doi: 10.1152/ajpendo.00093.2009
28. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. (1963) 1:785–9. doi: 10.1016/S0140-6736(63)91500-9
29. Murphy SP, Ibrahim NE, Januzzi JL Jr. Heart failure with reduced ejection fraction: a review. JAMA. (2020). 324:488–504. doi: 10.1001/jama.2020.10262
30. Packer M, Metra M. Guideline-directed medical therapy for heart failure does not exist: a non-judgmental framework for describing the level of adherence to evidence-based drug treatments for patients with a reduced ejection fraction. Eur J Heart Fail. (2020) 22:1759–67. doi: 10.1002/ejhf.1857
31. Razeghi P, Young ME, Ying J, Depre C, Uray IP, Kolesar J, et al. Downregulation of metabolic gene expression in failing human heart before and after mechanical unloading. Cardiology. (2002) 97:203–9. doi: 10.1159/000063122
32. Koonen DP, Febbraio M, Bonnet S, Nagendran J, Young ME, Michelakis ED, et al. CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation. (2007) 116:2139–47. doi: 10.1161/CIRCULATIONAHA.107.712901
33. Hoes MF, Grote Beverborg N, Kijlstra JD, Kuipers J, Swinkels DW, Giepmans BNG, et al. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail. (2018) 20:910–9. doi: 10.1002/ejhf.1154
34. Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. (2013) 113:603–16. doi: 10.1161/CIRCRESAHA.113.302095
35. Hu D, Linders A, Yamak A, Correia C, Kijlstra JD, Garakani A, et al. Metabolic maturation of human pluripotent stem cell-derived cardiomyocytes by inhibition of HIF1α and LDHA. Circ Res. (2018) 123:1066–79. doi: 10.1161/CIRCRESAHA.118.313249
36. Ravassa S, Beaumont J, Huerta A, Barba J, Coma-Canella I, González A, et al. Association of low GLP-1 with oxidative stress is related to cardiac disease and outcome in patients with type 2 diabetes mellitus: a pilot study. Free Radic Biol Med. (2015) 81:1–12. doi: 10.1016/j.freeradbiomed.2015.01.002
37. Calvier L, Boucher P, Herz J, Hansmann G. LRP1 deficiency in vascular SMC leads to pulmonary arterial hypertension that is reversed by PPARγ activation. Circ Res. (2019) 124:1778–85. doi: 10.1161/CIRCRESAHA.119.315088
38. Pound KM, Sorokina N, Ballal K, Berkich DA, Fasano M, Lanoue KF, et al. Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res. (2009) 104:805–12. doi: 10.1161/CIRCRESAHA.108.189951
39. Lahey R, Carley AN, Wang X, Glass CE, Accola KD, Silvestry S, et al. Enhanced redox state and efficiency of glucose oxidation with miR based suppression of maladaptive NADPH-dependent malic enzyme 1 expression in hypertrophied hearts. Circ Res. (2018) 122:836–45. doi: 10.1161/CIRCRESAHA.118.312660
40. Simon N, Hertig A. Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front Med (Lausanne). (2015) 2:52. doi: 10.3389/fmed.2015.00052
41. Li Z, Lu S, Li X. The role of metabolic reprogramming in tubular epithelial cells during the progression of acute kidney injury. Cell Mol Life Sci. (2021) 78:5731–41. doi: 10.1007/s00018-021-03892-w
42. Chiba T, Peasley KD, Cargill KR, Maringer KV, Bharathi SS, Mukherjee E, et al. Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J Am Soc Nephrol. (2019) 30:2384–98. doi: 10.1681/ASN.2019020163
43. Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. (2015) 21:37–46. doi: 10.1038/nm.3762
44. Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol. (2016) 27:3356–67. doi: 10.1681/ASN.2015020177
45. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
46. Cheng Y, Feng Y, Xia Z, Li X, Rong J. ω-Alkynyl arachidonic acid promotes anti-inflammatory macrophage M2 polarization against acute myocardial infarction via regulating the cross-talk between PKM2, HIF-1α and iNOS. Biochim Biophys Acta Mol Cell Biol Lipids. (2017) 1862:1595–605. doi: 10.1016/j.bbalip.2017.09.009
47. Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. (2014) 171:2017–28. doi: 10.1111/bph.12468
48. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
49. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. (2003) 552:335–44. doi: 10.1113/jphysiol.2003.049478
50. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. (2020) 21:363–83. doi: 10.1038/s41580-020-0230-3
51. Bindoli A, Rigobello MP. Principles in redox signaling: from chemistry to functional significance. Antioxid Redox Signal. (2013) 18:1557–93. doi: 10.1089/ars.2012.4655
52. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. (2008) 102:488–96. doi: 10.1161/CIRCRESAHA.107.162800
53. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. (2021) 11:1845–63. doi: 10.7150/thno.50905
54. Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. (2011) 108:837–46. doi: 10.1161/CIRCRESAHA.110.232306
55. Goh KY, Qu J, Hong H, Liu T., Dell'Italia LJ, Wu Y, et al. Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc Res. (2016) 109:79–89. doi: 10.1093/cvr/cvv230
56. Fuhrmann DC, Brüne B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. (2017) 12:208–15. doi: 10.1016/j.redox.2017.02.012
57. Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation. (2015) 131:1082–97. doi: 10.1161/CIRCULATIONAHA.114.012725
58. Aramide Modupe Dosunmu-Ogunbi A, Galley JC, Yuan S, Schmidt HM, Wood KC, Straub AC. Redox switches controlling nitric oxide signaling in the resistance vasculature and implications for blood pressure regulation: mid-career award for research excellence (2020). Hypertension. (2021) 78:912–26. doi: 10.1161/HYPERTENSIONAHA.121.16493
59. Ning R, Li Y, Du Z, Li T, Sun Q, Lin L., et al. The mitochondria-targeted antioxidant MitoQ attenuated PM25-induced vascular fibrosis via regulating mitophagy. Redox Biol. (2021) 46:102113. doi: 10.1016/j.redox.2021.102113
60. Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. (2018) 137:2256–73. doi: 10.1161/CIRCULATIONAHA.116.026099
61. Byun J, Oka SI, Imai N, Huang CY, Ralda G, et al. Both gain and loss of Nampt function promote pressure overload-induced heart failure. Am J Physiol Heart Circ Physiol. (2019) 317:H711–25. doi: 10.1152/ajpheart.00222.2019
62. Zhou B, Wang DD, Qiu Y, Airhart S, Liu Y, Stempien-Otero A, et al. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J Clin Invest. (2020) 130:6054–63. doi: 10.1172/JCI138538
63. Walker MA., Tian R. Raising NAD in heart failure: time to translate? Circulation. (2018) 137:2274–7. doi: 10.1161/CIRCULATIONAHA.117.032626
64. Mapuskar KA, Wen H, Holanda DG, Rastogi P, Steinbach E, Han R, et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. (2019) 20:98–106. doi: 10.1016/j.redox.2018.09.020
65. Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. (2016) 531:528–32. doi: 10.1038/nature17184
66. Liu D, Jin F, Shu G, Xu X, Qi J, Kang X, et al. Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials. (2019) 211:57–67. doi: 10.1016/j.biomaterials.2019.04.034
67. Arulkumaran N, Pollen SJ, Tidswell R, Gaupp C, Peters VBM, Stanzani G, et al. Selective mitochondrial antioxidant MitoTEMPO reduces renal dysfunction and systemic inflammation in experimental sepsis in rats. Br J Anaesth. (2021) 127:577–86. doi: 10.1016/j.bja.2021.05.036
68. Kong MJ, Bak SH, Han KH, Kim JI, Park JW, et al. Fragmentation of kidney epithelial cell primary cilia occurs by cisplatin and these cilia fragments are excreted into the urine. Redox Biol. (2019) 20:38–45. doi: 10.1016/j.redox.2018.09.017
69. Angelova PR, Abramov AY. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic Biol Med. (2016) 100:81–5. doi: 10.1016/j.freeradbiomed.2016.06.005
70. Ding W, Guo H, Xu C, Wang B, Zhang M, Ding F. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget. (2016) 7:17479–91. doi: 10.18632/oncotarget.8243
71. Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, et al. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J Biol Chem. (2013) 288:770–7. doi: 10.1074/jbc.M112.431973
72. Kirkman DL, Muth BJ, Ramick MG, Townsend RR, Edwards DG. Role of mitochondria-derived reactive oxygen species in microvascular dysfunction in chronic kidney disease. Am J Physiol Renal Physiol. (2018) 314:F423–9. doi: 10.1152/ajprenal.00321.2017
73. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
74. Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res. (2018) 202:52–68. doi: 10.1016/j.trsl.2018.07.014
75. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–63.e3. doi: 10.1016/j.molcel.2018.10.042
76. Zhang Z, Shao X, Jiang N, Mou S, Gu L, Li S, et al. Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis. (2018) 9:983. doi: 10.1038/s41419-018-1023-x
77. Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. (2010) 88:16–29. doi: 10.1093/cvr/cvq237
78. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
79. Giacomello M, Pyakurel A, Glytsou C., Scorrano L.The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. (2020) 21:204–24. doi: 10.1038/s41580-020-0210-7
80. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. (2016) 27:105–17. doi: 10.1016/j.tem.2015.12.001
81. Gao S, Hu J. Mitochondrial fusion: the machineries in and out. Trends Cell Biol. (2021) 31:62–74. doi: 10.1016/j.tcb.2020.09.008
82. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. (2013) 5:a011072. doi: 10.1101/cshperspect.a011072
83. Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. (2016) 78:505–31. doi: 10.1146/annurev-physiol-021115-105011
84. Li T, Han J, Jia L, Hu X, Chen L, Wang Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell. (2019) 10:583–94. doi: 10.1007/s13238-019-0618-z
85. Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. (2011) 31:1309–28. doi: 10.1128/MCB.00911-10
86. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. (2015) 116:264–78. doi: 10.1161/CIRCRESAHA.116.303356
87. Hu L, Ding M, Tang D, Gao E, Li C, Wang K, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. (2019) 9:3687–706. doi: 10.7150/thno.33684
88. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. (2009) 11:958–66. doi: 10.1038/ncb1907
89. Chen Y, Liu Y, Dorn GW 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. (2011) 109:1327–31. doi: 10.1161/CIRCRESAHA.111.258723
90. Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. (2015) 21:273–86. doi: 10.1016/j.cmet.2014.12.011
91. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. (2003) 160:189–200. doi: 10.1083/jcb.200211046
92. Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM, et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. (2016). 7:e2238. doi: 10.1038/cddis.2016.139
93. Shahrestani P, Leung HT, Le PK, Pak WL, Tse S, Ocorr K, et al. Heterozygous mutation of Drosophila Opa1 causes the development of multiple organ abnormalities in an age-dependent and organ-specific manner. PLoS ONE. (2009) 4:e6867. doi: 10.1371/journal.pone.0006867
94. Dorn GW. 2nd, Clark CF, Eschenbacher WH, Kang MY, Engelhard JT, Warner SJ, et al. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ Res. (2011) 108:12–7. doi: 10.1161/CIRCRESAHA.110.236745
95. Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, et al. Down-regulation of OPA1 alters mouse mitochondrialmorphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. (2012) 94:408–17. doi: 10.1093/cvr/cvs117
96. Perry HM, Huang L, Wilson RJ, Bajwa A, Sesaki H, Yan Z, et al. Dynamin-related protein 1 deficiency promotes recovery from AKI. J Am Soc Nephrol. (2018) 29:194–206. doi: 10.1681/ASN.2017060659
97. Wei Q, Sun H, Song S, Liu Y, Liu P, Livingston MJ, et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J Clin Invest. (2018) 128:5448–64. doi: 10.1172/JCI121859
98. Liu Z, Li H, Su J, Xu S, Zhu F, Ai J, et al. Numb depletion promotes Drp1-mediated mitochondrial fission and exacerbates mitochondrial fragmentation and dysfunction in acute kidney injury. Antioxid Redox Signal. (2019) 30:1797–816. doi: 10.1089/ars.2017.7432
99. Gall JM, Wang Z, Bonegio RG, Havasi A, Liesa M, Vemula P, et al. Conditional knockout of proximal tubule mitofusin 2 accelerates recovery and improves survival after renal ischemia. J Am Soc Nephrol. (2015) 26:1092–102. doi: 10.1681/ASN.2014010126
100. Xiao L, Zhu X, Yang S, Liu F, Zhou Z, Zhan M, et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes. (2014) 63:1366–80. doi: 10.2337/db13-1412
101. Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol. (2015) 26:1304–21. doi: 10.1681/ASN.2014050457
102. Ayanga BA, Badal SS, Wang Y, Galvan DL, Chang BH, Schumacker PT, et al. Dynamin-related protein 1 deficiency improves mitochondrial fitness and protects against progression of diabetic nephropathy. J Am Soc Nephrol. (2016) 27:2733–47. doi: 10.1681/ASN.2015101096
103. Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan F, et al. Berberine Protects Glomerular Podocytes via Inhibiting Drp1-Mediated Mitochondrial Fission and Dysfunction. Theranostics. (2019) 9:1698–713. doi: 10.7150/thno.30640
104. Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos Science. (2011) 334:1141–4. doi: 10.1126/science.1210333
105. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. (2015) 524:309–14. doi: 10.1038/nature14893
106. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. (2018) 20:1013–22. doi: 10.1038/s41556-018-0176-2
107. Liu L, Sakakibara K, Chen Q., Okamoto K.Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. (2014) 24:787–95. doi: 10.1038/cr.2014.75
108. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. (2009) 16:939–46. doi: 10.1038/cdd.2009.16
109. Lenhausen AM, Wilkinson AS, Lewis EM, Dailey KM, Scott AJ, Khan S, et al. Apoptosis inducing factor binding protein PGAM5 triggers mitophagic cell death that is inhibited by the ubiquitin ligase activity of X-linked inhibitor of apoptosis. Biochemistry. (2016) 55:3285–302. doi: 10.1021/acs.biochem.6b00306
110. Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, et al. AMPKα2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ Res. (2018) 122:712–29. doi: 10.1161/CIRCRESAHA.117.312317
111. Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW 2nd. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res. (2014) 114:257–65. doi: 10.1161/CIRCRESAHA.114.302734
112. Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. (2013) 288:915–26. doi: 10.1074/jbc.M112.411363
113. Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. (2020) 30:101415. doi: 10.1016/j.redox.2019.101415
114. Liu W, Wang X, Gong J, Mei Z, Gao X, Zhao Y, et al. The stress-related hormone norepinephrine induced upregulation of Nix, contributing to ECM protein expression. Cell Stress Chaperones. (2014) 19:903–12. doi: 10.1007/s12192-014-0515-6
115. Liu JX, Yang C, Zhang WH, Su HY, Liu ZJ, Pan Q, et al. Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci. (2019) 235:116828. doi: 10.1016/j.lfs.2019.116828
116. Chen K, Dai H, Yuan J, Chen J, Lin L, Zhang W, et al. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. (2018) 9:105. doi: 10.1038/s41419-017-0127-z
117. Chen K, Feng L, Hu W, Chen J, Wang X, Wang L, et al. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. (2019) 33:4571–85. doi: 10.1096/fj.201801749RRR
118. Li S, Lin Q, Shao X, Zhu X, Wu J, Wu B, et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic Biol Med. (2020) 152:632–49. doi: 10.1016/j.freeradbiomed.2019.12.005
119. Bhatia D, Chung KP, Nakahira K, Patino E, Rice MC, Torres LK, et al. Mitophagy-dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight. (2019) 4:e132826. doi: 10.1172/jci.insight.132826
120. Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. (2008) 79:208–17. doi: 10.1093/cvr/cvn098
121. Karamanlidis G, Nascimben L, Couper GS, Shekar PS., del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. (2010) 106:1541–8. doi: 10.1161/CIRCRESAHA.109.212753
122. Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol. (2013) 5:a015008. doi: 10.1101/cshperspect.a015008
123. Pisano A, Cerbelli B, Perli E, Pelullo M, Bargelli V, Preziuso C, et al. Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end-stage ischemic heart failure. Cardiovasc Pathol. (2016) 25:103–12. doi: 10.1016/j.carpath.2015.09.009
124. Chaanine AH, Joyce LD, Stulak JM, Maltais S, Joyce DL, Dearani JA, et al. Mitochondrial morphology, dynamics, and function in human pressure overload or ischemic heart disease with preserved or reduced ejection fraction. Circ Heart Fail. (2019) 12:e005131. doi: 10.1161/CIRCHEARTFAILURE.118.005131
125. Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. (2006) 103:10086–91. doi: 10.1073/pnas.0603615103
126. Sun L, Zhao M, Yu XJ, Wang H, He X, Liu JK, et al. Cardioprotection by acetylcholine: a novel mechanism via mitochondrial biogenesis and function involving the PGC-1α pathway. J Cell Physiol. (2013) 228:1238–48. doi: 10.1002/jcp.24277
127. Hu X, Xu X, Lu Z, Zhang P, Fassett J, Zhang Y, et al. AMP activated protein kinase-α2 regulates expression of estrogen-related receptor-α, a metabolic transcription factor related to heart failure development. Hypertension. (2011) 58:696–703. doi: 10.1161/HYPERTENSIONAHA.111.174128
128. Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, et al. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. (2004) 94:525–33. doi: 10.1161/01.RES.0000117088.36577.EB
129. Pereira RO, Wende AR, Crum A, Hunter D, Olsen CD, Rawlings T, et al. Maintaining PGC-1α expression following pressure overload-induced cardiac hypertrophy preserves angiogenesis but not contractile or mitochondrial function. FASEB J. (2014) 28:3691–702. doi: 10.1096/fj.14-253823
130. Portilla D, Dai G, McClure T, Bates L, Kurten R, Megyesi J, et al. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. (2002) 62:1208–18. doi: 10.1111/j.1523-1755.2002.kid553.x
131. Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. (2011) 121:4003–14. doi: 10.1172/JCI58662
132. Li SY, Park J, Qiu C, Han SH, Palmer MB, Arany Z, et al. Increasing the level of peroxisome proliferator-activated receptor γ coactivator-1α in podocytes results in collapsing glomerulopathy. JCI Insight. (2017) 2:e92930. doi: 10.1172/jci.insight.92930
133. Colombo PC, Ganda A, Lin J, Onat D, Harxhi A, Iyasere JE, et al. Inflammatory activation: cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail Rev. (2012) 17:177–90. doi: 10.1007/s10741-011-9261-3
134. Bongartz LG, Braam B, Gaillard CA, Cramer MJ, Goldschmeding R, Verhaar MC, et al. Target organ cross talk in cardiorenal syndrome: animal models. Am J Physiol Renal Physiol. (2012) 303:F1253–63. doi: 10.1152/ajprenal.00392.2012
135. Li HS, Zhou YN Li L, Li SF, Long D, Chen XL, et al. HIF-1alpha protects against oxidative stress by directly targeting mitochondria. Redox Biol. (2019) 25:101109. doi: 10.1016/j.redox.2019.101109
136. Zafari AM, Ushio-Fukai M, Minieri CA, Akers M, Lassègue B, Griendling KK. Arachidonic acid metabolites mediate angiotensin II-induced NADH/NADPH oxidase activity and hypertrophy in vascular smooth muscle cells. Antioxid Redox Signal. (1999) 1:167–79. doi: 10.1089/ars.1999.1.2-167
137. Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. (2003) 41:2164–71. doi: 10.1016/S0735-1097(03)00471-6
138. Bleeke T, Zhang H, Madamanchi N, Patterson C, Faber JE. Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ Res. (2004) 94:37–45. doi: 10.1161/01.RES.0000109412.80157.7D
139. Amin JK, Xiao L, Pimental DR, Pagano PJ, Singh K, Sawyer DB, et al. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol. (2001) 33:131–9. doi: 10.1006/jmcc.2000.1285
140. Zebrack JS, Anderson JL, Beddhu S, Horne BD, Bair TL, Cheung A, et al. Do associations with C-reactive protein and extent of coronary artery disease account for the increased cardiovascular risk of renal insufficiency? J Am Coll Cardiol. (2003) 42:57–63. doi: 10.1016/S0735-1097(03)00564-3
141. Wang J, Sim AS, Wang XL, Salonikas C, Moriatis M, Naidoo D, et al. Relations between markers of renal function, coronary risk factors and the occurrence and severity of coronary artery disease. Atherosclerosis. (2008) 197:853–9. doi: 10.1016/j.atherosclerosis.2007.07.034
142. Angelini A, Castellani C, Virzì GM, Fedrigo M, Thiene G, Valente M, et al. The Role of Congestion in Cardiorenal Syndrome Type 2: New Pathophysiological Insights into an Experimental Model of Heart Failure. Cardiorenal Med. (2015) 6:61–72. doi: 10.1159/000440775
143. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med. (2011) 208:519–33. doi: 10.1084/jem.20102049
144. Zhao L, Tian L, Wang S, Yang W, Lu X, Zhu C. Levosimendan in rats decreases acute kidney injury after cardiopulmonary resuscitation by improving mitochondrial dysfunction. Transl Androl Urol. (2021) 10:3010–20. doi: 10.21037/tau-21-443
145. Aboryag NB, Mohamed DM, Dehe L, Shaqura M, Treskatsch S, Shakibaei M, et al. Histopathological Changes in the Kidney following Congestive Heart Failure by Volume Overload in Rats. Oxid Med Cell Longev. (2017) 2017:6894040. doi: 10.1155/2017/6894040
146. Yang CC, Chen YT, Wallace CG, Chen KH, Cheng BC, Sung PH, et al. Early administration of empagliflozin preserved heart function in cardiorenal syndrome in rat. Biomed Pharmacother. (2019) 109:658–70. doi: 10.1016/j.biopha.2018.10.095
147. Doi K. Kidney-heart interactions in acute kidney injury. Nephron. (2016) 134:141–4. doi: 10.1159/000447021
148. Sumida M, Doi K, Ogasawara E, Yamashita T, Hamasaki Y, Kariya T, et al. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J Am Soc Nephrol. (2015) 26:2378–87. doi: 10.1681/ASN.2014080750
149. Doi K, Noiri E. Mitochondrial dysfunction in cardiorenal syndrome. Antioxid Redox Signal. (2016) 25:200–7. doi: 10.1089/ars.2016.6654
150. Bigelman E, Cohen L, Aharon-Hananel G, Levy R, Rozenbaum Z, Saada A, et al. Pathological presentation of cardiac mitochondria in a rat model for chronic kidney disease. PLoS ONE. (2018) 13:e198196. doi: 10.1371/journal.pone.0198196
151. Bartz RR, Fu P, Suliman HB, Crowley SD, MacGarvey NC, Welty-Wolf K, et al. Staphylococcus aureus sepsis induces early renal mitochondrial DNA repair and mitochondrial biogenesis in mice. PLoS ONE. (2014) 9:e100912. doi: 10.1371/journal.pone.0100912
152. Heywood JT, Fonarow GC, Costanzo MR, Mathur VS, Wigneswaran JR, Wynne J, et al. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. (2007) 13:422–30. doi: 10.1016/j.cardfail.2007.03.011
153. Iglesias J, Ghetiya S, Ledesma KJ, Patel CS, Levine JS. Interactive and potentially independent roles of renin-angiotensin-aldosterone system blockade and the development of cardiorenal syndrome type 1 on in-hospital mortality among elderly patients admitted with acute decompensated congestive heart failure. Int J Nephrol Renovasc Dis. (2019) 12:33–48. doi: 10.2147/IJNRD.S185988
154. Ronco C, Cicoira M, McCullough P A. Cardiorenal syndrome type 1: pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J Am Coll Cardiol. (2012) 60:1031–42. doi: 10.1016/j.jacc.2012.01.077
155. Raina R, Nair N, Chakraborty R, Nemer L, Dasgupta R, Varian K. An Update on the Pathophysiology and Treatment of Cardiorenal Syndrome. Cardiol Res. (2020) 11:76–88. doi: 10.14740/cr955
156. Rangaswami J, Bhalla V, Blair JEA, Chang TI, Costa S, Lentine KL, et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation. (2019) 139:e840–78. doi: 10.1161/CIR.0000000000000664
157. Di Lullo L, Bellasi A, Barbera V, Russo D, Russo L, Di Iorio B, et al. Pathophysiology of the cardio-renal syndromes types 1-5: An uptodate. Indian Heart J. (2017) 69:255–65. doi: 10.1016/j.ihj.2017.01.005
158. Mehta RL, Rabb H, Shaw AD, Singbartl K, Ronco C, McCullough PA, et al. Cardiorenal syndrome type 5: clinical presentation, pathophysiology and management strategies from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol. (2013) 182:174–94. doi: 10.1159/000349970
159. Gomez H, Kellum J A, Ronco C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat Rev Nephrol. (2017) 13:143–51. doi: 10.1038/nrneph.2016.186
160. Collier J B, Schnellmann R G. Extracellular signal-regulated kinase 1/2 regulates NAD metabolism during acute kidney injury through microRNA-34a-mediated NAMPT expression. Cell Mol Life Sci. (2020) 77:3643–55. doi: 10.1007/s00018-019-03391-z
Keywords: cardiorenal syndrome, mitochondrial dysfunction, heart failure, kidney failure, oxidative stress, inflammation
Citation: Shi S, Zhang B, Li Y, Xu X, Lv J, Jia Q, Chai R, Xue W, Li Y, Wang Y, Wu H, Song Q and Hu Y (2022) Mitochondrial Dysfunction: An Emerging Link in the Pathophysiology of Cardiorenal Syndrome. Front. Cardiovasc. Med. 9:837270. doi: 10.3389/fcvm.2022.837270
Received: 16 December 2021; Accepted: 18 January 2022;
Published: 25 February 2022.
Edited by:
Ernesto Martinez-Martinez, Universidad Complutense de Madrid, SpainReviewed by:
Chiara Castellani, University of Padua, ItalyJaap Joles, Utrecht University, Netherlands
Copyright © 2022 Shi, Zhang, Li, Xu, Lv, Jia, Chai, Xue, Li, Wang, Wu, Song and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huaqin Wu, yuanshanyun2650@163.com; Qingqiao Song, Songqq985@126.com; Yuanhui Hu, huyhui55@sina.com
†These authors have contributed equally to this work and share first authorship