 Francesca Marini1,2
Francesca Marini1,2 Francesca Giusti1Gaia Palmini1Giuliano Perigli3Roberto Santoro3
Francesca Giusti1Gaia Palmini1Giuliano Perigli3Roberto Santoro3 Maria Luisa Brandi2*
Maria Luisa Brandi2*- 1Department of Experimental and Clinical Biomedical Sciences, University of Florence, Florence, Italy
- 2Fondazione Italiana per la Ricerca sulle Malattie dell'Osso (F.I.R.M.O.) Italian Foundation for the Research on Bone Diseases, Florence, Italy
- 3Department of Experimental and Clinical Medicine, University of Florence, Azienda Ospedaliero-Universitaria (AOU)-Careggi, Florence, Italy
Parathyroid carcinoma (PC) is an extremely rare malignancy, accounting less than 1% of all parathyroid neoplasms, and an uncommon cause of primary hyperparathyroidism (PHPT), characterized by an excessive secretion of parathyroid hormone (PTH) and severe hypercalcemia. As opposed to parathyroid hyperplasia and adenomas, PC is associated with a poor prognosis, due to a commonly unmanageable hypercalcemia, which accounts for death in the majority of cases, and an overall survival rate of 78-85% and 49-70% at 5 and 10 years after diagnosis, respectively. No definitively effective therapies for PC are currently available. The mainly employed treatment for PC is the surgical removal of tumoral gland(s). Post-surgical persistent or recurrent disease manifest in about 50% of patients. The comprehension of genetic and epigenetic bases and molecular pathways that characterize parathyroid carcinogenesis is important to distinguish malignant PCs from benign adenomas, and to identify specific targets for novel therapies. Germline heterozygote inactivating mutations of the CDC73 tumor suppressor gene, with somatic loss of heterozygosity at 1q31.2 locus, account for about 50-75% of familial cases; over 75% of sporadic PCs harbor biallelic somatic inactivation/loss of CDC73. Recurrent mutations of the PRUNE2 gene, a recurrent mutation in the ADCK1 gene, genetic amplification of the CCND1 gene, alterations of the PI3K/AKT/mTOR signaling pathway, and modifications of microRNA expression profile and gene promoter methylation pattern have all been detected in PC. Here, we review the current knowledge on gene mutations and epigenetic changes that have been associated with the development of PC, in both familial and sporadic forms of this malignancy.
Parathyroid Carcinoma
Parathyroid tumors are rare endocrine neoplasms affecting 0.1-0.3% of the general population (1); less than 1% of are malignant parathyroid carcinomas (PCs). Age at diagnosis of PCs is commonly in the mid fifth decade of life, usually 10 years earlier than benign parathyroid adenomas (PAs), with no gender prevalence. PC most commonly occurs as a sporadic single-gland disease, without a familial history, with only a minority of cases manifesting as isolated inherited cancer or as part of a complex hereditary syndrome.
The great majority of PCs are functioning tumors over-secreting parathyroid hormone (PTH), responsible for primary hyperparathyroidism (PHPT) and elevated serum levels of calcium; only 2% of PCs are non-functioning variants characterized by normal serum calcium and PTH values, manifesting through compressive signs and symptoms of local invasion (1). Contrary to parathyroid hyperplasia and adenomas, PCs are associated with poor prognosis due to more severely elevated PTH, and higher, commonly unmanageable, hypercalcemia, which accounts for death in a majority of cases, with an overall survival rate of 78-85% and 49-70% at 5 and 10 years after diagnosis, respectively (2). Moreover, 10-30% patients with PC already have metastatic disease at diagnosis, most commonly in the lungs, bone, or liver (3).
The absence of distinctive growth and histological characteristics, as well as of idiosyncratic serum concentration values of calcium ion and intact PTH, distinguishing malignant PCs from PAs, makes a clear pre-operative histopathological or biochemical diagnosis of PCs extremely difficult. A diagnosis of malignancy usually occurs during surgery, through an intraoperative assessment of a marked local invasion of adjacent structures and surrounding lymph nodes, and/or confirmed during a prolonged post-operative follow-up in presence of local relapses or distant metastases. Since PCs require a more radical surgical approach than benign PAs, with en bloc gland resection and tumor safety margins, often including the excision of the adjacent ipsilateral thyroid lobe and thymus, a misdiagnosis may lead to inadequate surgical resection and a high recurrence rate and risk of distant metastases. Currently, up to 50% of PC patients manifest metastases following an initial wrong diagnosis of benign disease, with a consequent severe impairment of the prognosis and reduction of overall survival (4).
In this light, the identification of distinctive genetic bases, epigenetic changes, and molecular signatures characterizing parathyroid malignancies is fundamental to identify potential diagnostic biomarkers able to clearly distinguish PCs from benign parathyroid tumors, as well to provide potential therapeutic targets for these malignant neoplasms.
Genetics of Parathyroid Carcinoma
PCs occur either as sporadic non-hereditary single-gland disease, or as isolated inherited malignancy in a percentage of patients with familial isolated primary hyperparathyroidism (FIPH), or in the context of hyperparathyroidism-jaw tumor (HPT-JT) syndrome, affecting about 15% of HPT-JT patients. Studies on hereditary and sporadic forms of PCs revealed specific germline and somatic genetic alterations underlying PC development (Table 1).
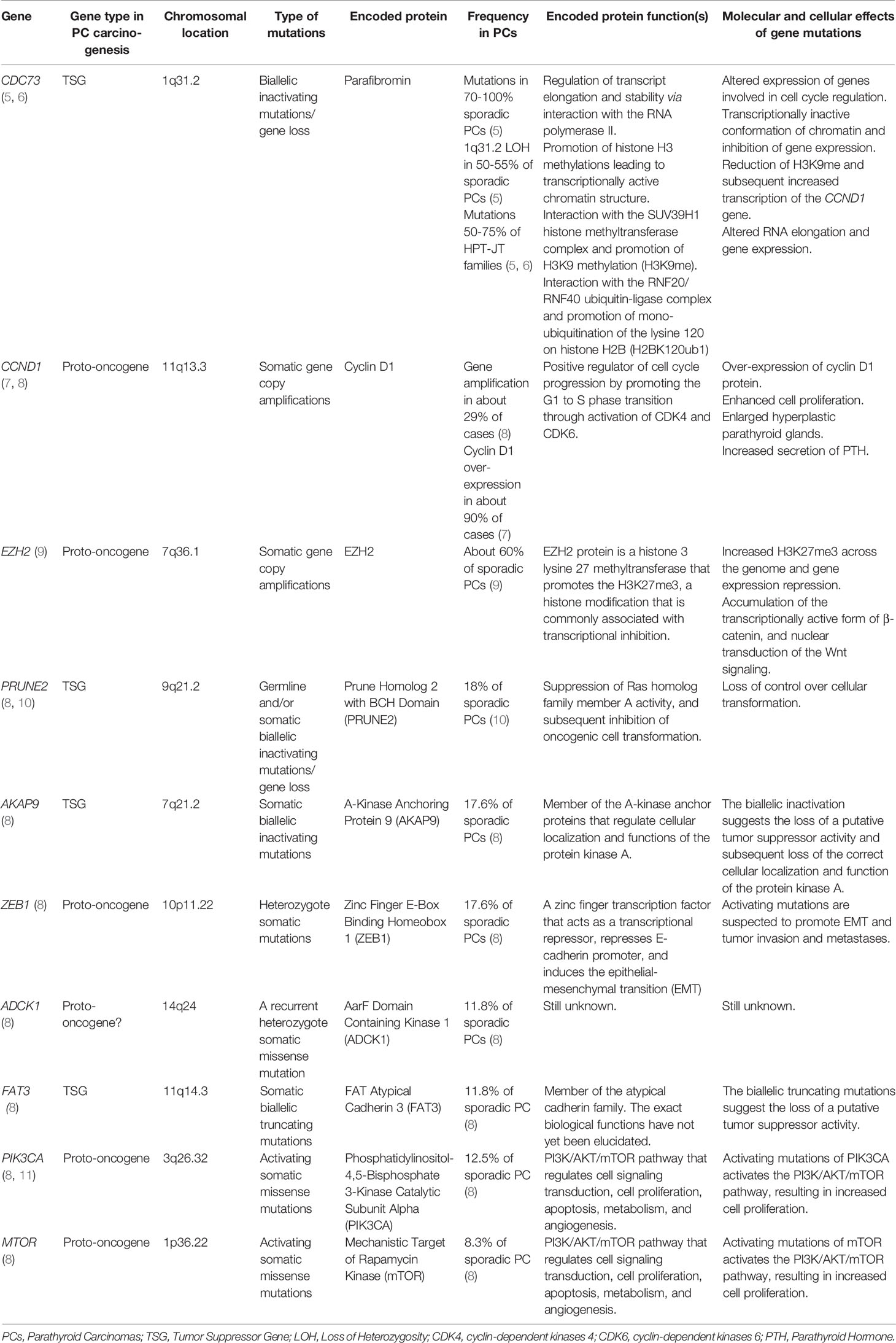
Table 1 Main genetic determinants in parathyroid carcinomas.
Extremely rare cases of malignant PCs have been reported in Multiple Endocrine Neoplasia type 1 (MEN1) syndrome (less than 15 cases) and in Multiple Endocrine Neoplasia type 2A (MEN2A) syndrome (only two cases), as a consequence of germline heterozygote inactivating mutations of the MEN1 tumor suppressor gene and germline heterozygote activating mutations of the RET proto-oncogene, respectively (5). No genotype-phenotype correlation has been found to explain why these few individuals develop PCs instead of PAs, which typically affect patients with MEN1 and MEN2A.
CDC73 Gene
Biallelic loss-of-function mutations/inactivation of the Cell Division Cycle 73 (CDC73) tumor suppressor gene are the major driver genetic defects in the etiology of PCs and, since they are found in less than 1% of benign PAs (12), they can be considered the main genetic hallmark of parathyroid malignancy risk.
In the hereditary forms (HPT-JT- and FIPH-related cancers), CDC73 mutations comprise one germline mutation and one somatic inactivation/loss of the gene, the first inherited from the affected parent, and the second occurring in the parathyroid cell, often as chromosomal loss at 1q31.2. Germline inactivating mutations of the CDC73 gene are detected in 50-75% of HPT-JT families (5, 6), and in about 8% of FIPH pedigrees (13). Over 85% of germline CDC73 mutations in hereditary PCs are frameshift and nonsense variants that create a premature stop codon, predicting a truncated protein, while the second somatic hit involves loss of heterozygosity (LOH) at the 1q31.2 locus in a great majority of cases.
In sporadic non-inherited PCs, both the CDC73 loss-of-function mutations occur at somatic level in the parathyroid cell. Somatic CDC73 mutations have been reported in 40-100% of sporadic PCs (5). Interestingly, germline CDC73 mutations are identified in 20-40% of patients with apparently sporadic PC (5), suggesting them as probable inherited diseases in those cases where the familial history is unknown, or cases of mosaicism with a germline mutation that originated de novo at the embryonic level (14). LOH at the CDC73 locus is reported in 50-55% of sporadic PCs (5). Hewitt et al. (14) showed hypermethylation of the CDC73 gene promoter in 18% (2/11) of sporadic PCs and 17% (1/6) of PAs from HPT-JT patients, all without mutation of the CDC73 gene, but not in 37 sporadic PAs, suggesting this epigenetic gene-silencing tool as an additional mechanism by which loss of the CDC73 gene expression may give rise to PCs.
The CDC73 gene is located on chromosome 1q31.2 and consists of 17 exons encoding parafibromin, a ubiquitously expressed 60 KDa protein that is an essential component of the Polymerase-Associated Factor 1 (PAF1) complex, whose main biological roles are resumed in Figure 1.
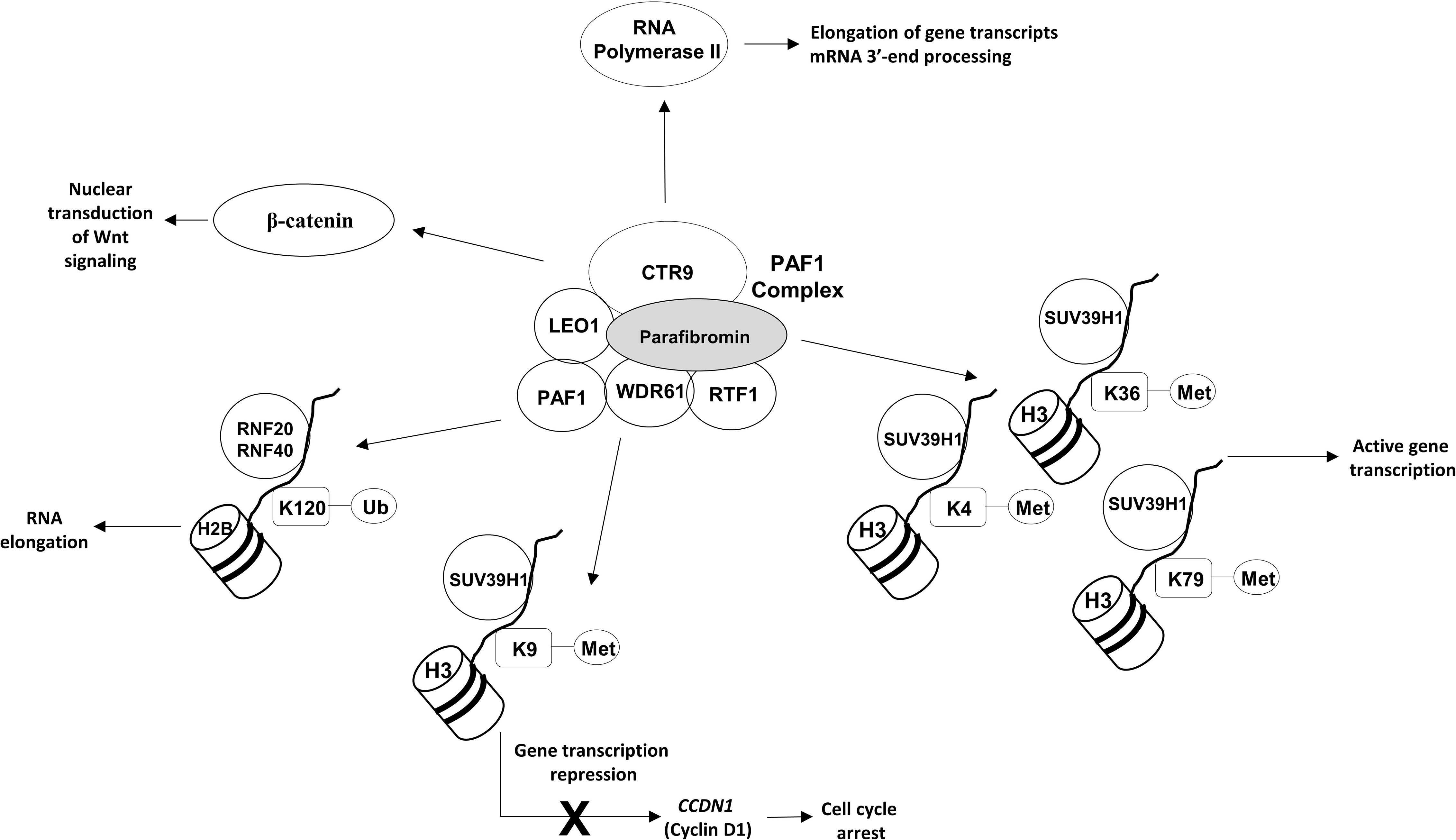
Figure 1 Main roles of parafibromin.
The PAF1 complex directly interacts with both the non-phosphorylated and the Ser2- and Ser5-phosphorylated forms of the RNA polymerase II large subunit, coordinating the correct recruitment of transcript elongation and RNA processing machineries (15). Moreover, the PAF1 complex is involved in coupling transcriptional and post-transcriptional events, in modulating the transition of gene transcripts from initiation to elongation steps, and in the mRNA 3’-end processing through modulation of the levels of Ser2 phosphorylation in the carboxy-terminal domain (CTD) of the RNA polymerase II (15). The interaction between RNA polymerase II and PAF1 depends on the C-terminal domain of parafibromin, which is deleted in over 80% of clinically relevant mutations of the CDC73 gene. Loss of parafibromin results in a significant reduction in PAF1 complex binding to RNA polymerase II, in a reduction of Ser2 phosphorylation of the RNA polymerase II CTD, and in shortened poly(A) tails on most cellular transcripts, altering the correct expression of several genes, including genes involved in cell cycle regulation, protein synthesis, and lipid and nucleic acid metabolism (16).
In the absence of parafibromin, replication-dependent histone mRNAs are not cleaved and contain poly(A) tails, being more stable. In sporadic PCs, loss of parafibromin has been associated with over-expression of some members of the replication-dependent histones family, H1.2, H2A and H2B, whose genes belong to a histone micro-cluster on chromosome 6p22.2-p21.3 (17). H1.2 inhibits transcription of growth suppressive genes via modulation of chromatin structure (18), thus, the H1.2 increased expression may result in a promotion of cell proliferation.
The PAF1 complex is also involved in multiple aspects of histone modifications (Figure 1). It promotes histone 3 methylations on lysine residues 4, 36 and 79 (H3K4me, H3K36me and H3K79me), which are generally associated with transcriptionally active chromatin (16). PAF1 has also been demonstrated to promote histone 3 methylation on lysine 9 (H3K9me) (19) through a direct interaction of the central region of parafibromin (amino acids 128–250) with SUV39H1 histone methyltransferase complex. Amino acids 128-227 have been demonstrated to be the minimal necessary for functional interaction with SUV39H1 (19). PAF1-induced H3K9me results in a transcriptional repression of the CCDN1 gene, encoding cyclin D1, and in a subsequent cell cycle arrest at the G1 to S phase transition. Moreover, PAF1 promotes the histone H2B mono-ubiquitination on the lysine residue 120 (H2BK120ub1), a histone modification that is positively involved in RNA elongation through a direct interaction with the RNF20/RNF40 ubiquitin-ligase complex (20).
The PAF1 complex has been demonstrated to be an essential regulator of the nuclear transduction of the Wnt signal via a direct protein-protein interaction between parafibromin, LEO1, and β-catenin. Parafibromin has been shown to directly bind to the C-terminal region of the β-catenin through the evolutionarily conserved central region of the parafibromin protein (amino acids 218-263), which itself has been found to be sufficient for potent β-catenin binding (21). The exact role of parafibromin in Wnt signaling has not been clearly established yet, and it might vary by cell type.
Parafibromin is located prevalently in the nucleus, thanks to a nuclear localization signal (amino acids 125-136) and a nucleolar localization signal (amino acids 76-92); loss of one or both of these domains, following CDC73 truncating mutations, prevents the nuclear localization of parafibromin and its main functions, and it has been associated with HPT-JT syndrome (22, 23). Loss of parafibromin nuclear immunostaining has been found in PCs and appears to be a distinctive histological feature characterizing malignant tumors from benign parathyroid tumors (24). Both sporadic and HPT-JT-related PCs with biallelic mutation/loss of the CDC73 gene have shown loss of nuclear localization and activity of parafibromin that were strongly associated with tumor malignant behavior, with a younger age of patients and larger tumor size with respect to other CDC73-positive PCs (12). Moreover, PCs negative for parafibromin nuclear immunostaining have shown distinctive morphological (i.e., eosinophilic cytoplasm, very frequent presence of perinuclear cytoplasmic clearing, nuclear atypia, and nuclear enlargement with distinctive coarse chromatin) and proliferative (extensive sheet-like growth rather than acinar architecture) features, which suggest parafibromin-deficient parathyroid malignancies to be a distinct histological and clinical subtype of PCs (12).
CCND1 Gene
Cyclin D1 is a positive regulator of cell cycle progression that specifically promotes the G1 to S phase transition of the cell cycle through the activation of the cyclin-dependent kinases 4 and 6 (CDK4 and CDK6). Cyclin D1 is encoded by the CCND1 proto-oncogene, located on chromosome 11q13.3. The over-expression of cyclin D1 protein is an extremely common event in parathyroid carcinogenesis, characterizing about 90% of PCs (7).
A direct pathogenic role of cyclin D1 in parathyroid tumorigenesis has been demonstrated in transgenic mouse models containing a chromosomal rearrangement of the human CCND1 locus, mimicking the one found in two human parathyroid tumors, and over-expressing cyclin D1. As a consequence of cyclin D1 over-expression, mutated mice developed enlarged hyperplastic parathyroid glands, characterized by increased cell proliferation and enhanced PTH secretion, and manifested a chronic PHPT with hypercalcemia and bone abnormalities (25). In these mice, the cyclin D1-induced abnormal parathyroid proliferation temporarily preceded the dysregulation of the calcium-PTH axis, confirming the direct action of up-regulated cyclin D1 on enhancement of parathyroid cell growth, and indicating that the disturbed parathyroid proliferation is the crucial primary initiator leading to the PHPT phenotype (26).
The exact mechanisms responsible for cyclin D1 over-expression in parathyroid carcinogenesis are still largely unclear. As for other types of human cancers, over-expression of cyclin D1 in parathyroid tumor cells presumably derives from gene copy number alterations or chromosomal rearrangements and trans-acting altered regulation of gene expression, rather than activating mutations of the CCDN1 gene. A heterozygote p15-q13 pericentromeric inversion in chromosome 11 was reported in two unrelated cases of benign PAs, positioning the 5’-regulatory element of the PTH gene from the 11p15 locus directly upstream of the CCDN1 gene on the 11q13 locus, inducing over-expression of cyclin D1 protein, and, at the same time, maintaining one intact copy of the PTH gene and, thus, the capability to express PTH (27). However, this chromosomal translocation has not been described in PCs. A whole-genome study on sporadic PCs showed somatic amplification of the genomic region containing the CCDN1 gene in 29% of analyzed cases, 80% of them being mutually exclusive with cases harboring CDC73 somatic mutations (8), suggesting that CCDN1 amplification is an alternative genetic mechanism to CDC73 inactivation to up-regulate cyclin D1 expression. Since cyclin D1 up-regulation is a frequent event also in benign sporadic PAs (20-40% of cases), the over-expression of this protein cannot be used as a hallmark to distinguish benign from malignant tumors.
Over-expression of cyclin D1 in mouse fibroblasts has been associated with an enhanced and more rapid cell cycle progression, due to a shortened duration of the G1 phase, and with the induction of anchorage-independent growth and enhanced invasion ability (28). At the molecular level, up-regulated cyclin D1 led to the activation of the pRB/E2F pathway, a pathway that has been shown to be deregulated in a large majority of human tumors, and the consequent increased expression of Fibroblast Growth Factor Receptor 1 (FGFR1), both at the mRNA and protein levels, which presumably acts as a direct mediator of the increased cell invasion of the matrigel in response to treatment with basic Fibroblast Growth Factor (bFGF). These data suggest that the pro-oncogenic action of up-regulated cyclin D1 can be mediated, at least in part, by potentiating the stimulatory effects of bFGF, a pro-proliferation factor that is often produced by stromal cells, on the growth and invasiveness of adjacent tumor cells (28), a mechanism that can also be suspected in parathyroid cancer.
PRUNE Gene
The Prune Homolog 2 with BCH Domain (PRUNE2) gene was recently identified as a novel PC-associated gene (10). The protein encoded by PRUNE2 gene belongs to the B-cell CLL/lymphoma 2 and adenovirus E1B 19 kDa interacting family, whose members play roles in many cellular processes, including apoptosis and cell transformation. PRUNE2 protein is a tumor suppressor exerting various biological functions, including the suppression of Ras homolog family member A activity, which results in inhibition of oncogenic cellular transformation.
A whole-exome sequencing analysis on PCs showed recurrent mutations of the PRUNE2 gene in 18% of cases (10). No mutation was detected in the 40 PA validation set, suggesting PRUNE2 gene as a novel candidate gene in malignant carcinogenesis and a possible genetic determinant to diagnose PCs. A germline missense mutation (p.Val452Met) has been identified in a PC sample in absence of CDC73 mutation and with a somatic LOH at the chromosome 9, containing the PRUNE2 gene. Two somatic nonsense mutations (c.Glu474X and p.Glu537X), presumably inactivating both the two gene alleles, were found in a PC sample bearing a concomitant CDC73 mutation, suggesting a possible synergic effect of these two mutated tumor suppressor genes in determining malignant cancer development. No data were available on the clinical characteristic of that PC, indicating a more severe cancer phenotype as a consequence of the presence of double-gene mutations. These two stop-gain mutations were confirmed as possible causes of PC development by another genome-sequencing study, and both were found at somatic levels in one single PC sample, but not in the corresponding normal parathyroid tissue (8). Two somatic missense mutations (p.Gly455Asp and p.Ser450Asn) were identified in two PC cases, both without CDC73 and MEN1 mutations.
All three identified missense mutations were predicted to be probably damaging and likely pathogenic by disrupting the function of the PRUNE2 protein. The two nonsense mutations were predicted to produce a truncated PRUNE2 protein lacking the BCH domain and, thus, presumably losing the overall tumor suppressor functions, leading to loss of control over cellular transformation. A study by Yu et al. (10) performed a whole-gene sequencing on 8 PCs included in the discovery cohort, localizing all the three identified mutations of the PRUNE2 gene in the exon 8, which appeared to be a possible mutational hot spot. As a consequence, the additional 13 PC samples of the validation cohort were sequenced only for exon 8, presumably leading to an underestimation of the real PRUNE2 mutation rate in PCs. Whole PRUNE2 gene sequencing on larger and different series of PC patients is needed to confirm PRUNE2 mutations as a genetic cause of malignancy of parathyroid glands and to identify a possible occurrence of mutations in other regions of the gene.
EZH2 Gene
The Enhancer of Zeste Homolog 2 (EZH2) gene encodes a histone 3 lysine 27 methyltransferase that is a member of the Polycomb Repressive 2 (PCR2) complex. EZH2 gene amplification at 7q36.1 locus is common in PCs (about 60% of cases), acting as an oncogene in parathyroid carcinogenesis and showing a significant over-expression of EZH2 mRNA and protein in malignant carcinomas compared with adenomas and hyperplastic parathyroid glands (9). Conversely, no EZH2 gene mutations were found in sporadic PCs (29), while a recurrent p.Tyr641Asp missense mutation was identified in unrelated PAs (30, 31). Therefore, different types of genetic aberrations of the EZH2 gene appear to differentially characterize PCs and PAs.
Increased expression and enzymatic activity of EZH2 protein result in an elevated histone trimethylation on lysine 27 (H3K27me3) across the genome, a histone modification that is commonly associated with transcriptional inhibition. A suspected target gene, repressed by EZH2-mediated H3K27me3, is the HIC1 tumor suppressor gene, which is normally involved in control of parathyroid growth; an aberrant down-regulation of HIC1 has been observed in both benign and malignant parathyroid, presumably due to the repressive action of H3K27me3 on gene expression (32) (Figure 2).
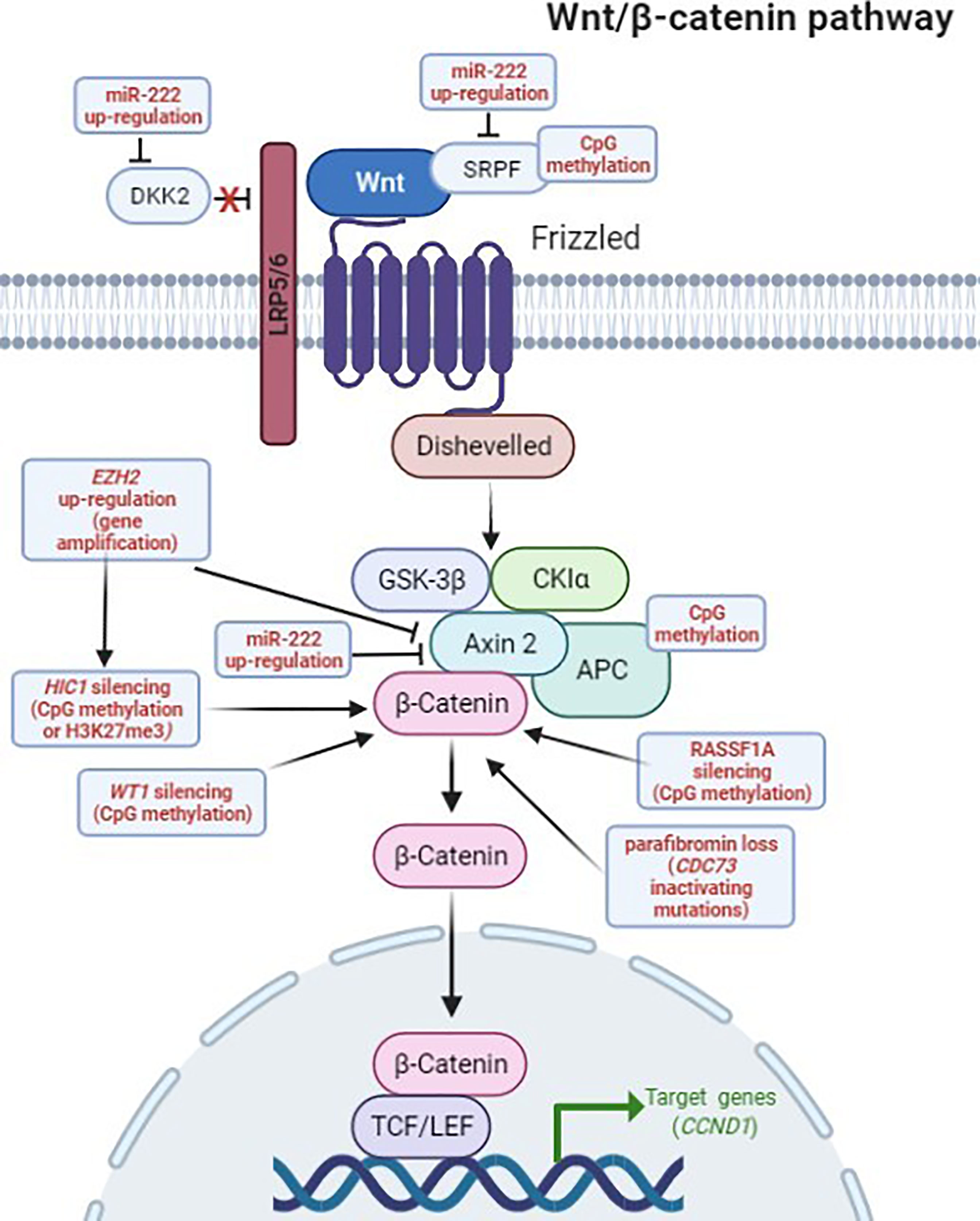
Figure 2 Deregulation of the Wnt/β-catenin signaling in PCs. Genetic and epigenetic aberrations detected in PC, altering the Wnt/β-catenin pathway, are highlighted in red. In the canonical Wnt/β-catenin pathway, the Wnt ligands initiate signaling by interacting, on the cell surface, with the Frizzled (FZD) receptor and the low density lipoprotein receptor-related protein 5/6 (LRP5/6) coreceptor. The ligand-activated FZD-LRP5/6 complex recruits, respectively, Dishevelled and AXIN2 intracellular proteins, preventing the constitutive destruction of cytosolic β-catenin by disassembling the β-catenin destruction complex, consisting of AXIN2, adenomatous polyposis coli (APC), glycogen synthase kinase3β (GSK3β) and casein kinase 1 (CK1), and ultimately leading to active β-catenin accumulation and nuclear translocation. In the nucleus, β-catenin binds and activates transcription factors of the lymphoid enhancer-binding factor (LEF)/T-cell factor (TCF) family, leading to the transcription of Wnt target genes (i.e. CCND1). DKK2 and SFRP proteins are natural extracellular inhibitors of the Wnt signaling, by respectively binding, and inactivating, the LRP5/6 coreceptor and the Wnt ligands. Figure created in BioRender.com, accessed on 10 January 2022.
Normally, EZH2 mediates the epigenetic gene transcription repression of several Wnt antagonists, including the growth-suppressive AXIN2 protein, a negative regulator of cytoplasmatic levels and stability of β-catenin in the absence of Wnt ligand. As a consequence, an excessive expression of EZH2 represses AXIN2, and it has been demonstrated to significantly contribute to an aberrant accumulation of the transcriptionally active (non-phosphorylated) form of β-catenin in the cytoplasm and the consequent nuclear translocation, resulting in the nuclear transduction of Wnt signaling that leads to increased cyclin D1 expression and enhanced cell proliferation (9) (Figure 2).
ADCK1 Gene
In a whole-genome sequencing analysis, a recurrent missense mutation p.Ile482Met in the ADCK1 gene, encoding the AarF Domain-Containing Kinase 1 (ADCK1), was found in 2 of the 17 analyzed PC cases (11.8%) (8). The role of the ADCK1 protein is not yet clear; it is not known if it has a protein kinase activity or which amino acid residue it would phosphorylate (Ser, Thr or Tyr). The monoallelic somatic ADCK1 mutation suggests a dominant-negative effect of the mutated protein and the ADCK1 gene as a candidate oncogene in PC development.
AKAP9 Gene
A whole-genome sequencing analysis on 17 sporadic PC samples has identified the AKAP9 gene as recurrently mutated (3/17; 17.6% of analyzed cases) (8). The AKAP9 gene encodes the A-Kinase Anchoring Protein 9 (AKAP9), a member of the A-kinase anchor proteins, that regulates cellular localization and function of the protein kinase A. Two missense and one nonsense somatic mutations were identified in 3 cases, two of them showing the biallelic inactivation of the gene, thus suggesting a tumor suppressor activity of AKAP9 in parathyroids. Further functional investigations are needed to assess the potential role of AKAP9 loss/inactivation as oncogenic driver in PC development.
ZEB1 Gene
The ZEB1 gene encodes the Zinc Finger E-Box Binding Homeobox 1 (ZEB1) protein, a zinc finger transcription factor that acts as a transcriptional repressor, represses E-cadherin promoter and induces the epithelial-to-mesenchymal transition (EMT), promoting tumor invasion and metastases.
Somatic heterozygote mutations of the ZEB1 gene have been recurrently identified in PC samples (17.6% of cases analyzed by a whole-genome sequencing) (8). The molecular and cellular effects of the identified ZEB1mutations in PCs have not yet been evaluated, but activating mutations are suspected to promote EMT and consequent tumor invasivity and metastatic capability.
FAT3 Gene
The FAT3 gene has recently been recognized as a possible candidate gene in sporadic PC development by whole-genome profiling (8). This gene encodes the FAT Atypical Cadherin 3 (FAT3) protein, whose biological role has not yet been clearly defined. FAT3 belongs to a family of proteins that includes two known tumor suppressors, FAT1 and FAT4.
Two different somatic nonsense mutations were identified in 2/17 (11.8%) analyzed cases, both with the biallelic inactivation of the gene and creating a premature stop codon near to the N-terminal region of the FAT3 protein, suggesting the FAT3 gene as a putative tumor suppressor gene in parathyroid carcinogenesis, possibly regulating the Wnt signaling.
PI3K/AKT/mTOR Signaling Pathway
The PI3K/AKT/mTOR pathway is an intracellular signaling pathway important for regulating cell signal transduction and biological processes, such as cell proliferation, apoptosis, metabolism, and angiogenesis. Several studies have shown that the deregulation of this signaling pathway is involved in the occurrence and development of different types of human malignancies (33). A genomic profiling of 24 PCs revealed the presence of somatic activating missense variants in genes encoding components of this signaling pathway in 21% (5/24) of cases: p.Lys111Glu, p.Glu545Ala, and p.His1047Arg in the PIK3CA gene, and p.Leu1460Pro and p.Gln2524Leu in the MTOR gene (8). Results from this study suggest the altered PI3K/AKT/mTOR pathway as a major oncogenic factor in parathyroid carcinogenesis.
The somatic activating missense p.Glu545Lys variant of the PIK3CA gene was identified, through a complete genome landscape mutational analysis of tumor specimens, in a primary sporadic PC, but not in the 7 PC recurrences, suggesting a role for mutated PIK3CA protein in tumor initiation, but not in tumor progression and tissue invasion (11).
YAP1 Gene
The Yes Associated Protein 1 (YAP1) gene encodes a downstream nuclear effector of the Hippo signaling pathway, which is known to play a role in the development and progression of multiple cancers, by acting as a transcriptional regulator of this signaling pathway. Recently Tavanti et al. (34) found a remarkable loss of YAP1 protein nuclear staining in formalin-fixed paraffin-embedded PC specimens, compared with normal parathyroid and PA samples that were both characterized by a positive nuclear expression of YAP1. Loss of YAP1 nuclear localization was independent of CDC73 and MEN1 gene status. No data were reported about YAP1 gene status at both germline and somatic levels in PCs, and the molecular mechanism responsible for loss of YAP1 nuclear localization in PC remains to be fully elucidated.
FLNA Gene
The filamin A (FLNA) gene encodes the homonym filamin A protein, a non-muscle actin filament cross-linking scaffold protein that influences the intracellular localization of numerous proteins. A very recent study by Storvall et al. (35) found that PCs were characterized by significantly increased cytoplasmic FLNA expression, compared to both PAs and atypical parathyroid adenomas (aPAs), suggesting that the increased cytoplasmic, and membranous, FLNA expression could be a marker of malignancy, and a potential prognostic predictor of aggressive behavior in parathyroid neoplasms, mostly in combination with parafibromin expression. However, a previous study (36) showed opposite results, reporting a decreased expression of full-length FLNA in the cytoplasm of tumoral parathyroid epithelial cells, both for PAs (50% to 10% of FLNA-positive cells) and PCs (less than 10% or totally absent FLNA-positive cells), with respect to the normal parathyroid counterpart.
Therefore, the role of FLNA expression levels in parathyroid carcinogenesis needs to be clearly elucidated by further studies.
TBX1 Gene
A greater than two-fold reduced expression of the T-Box Transcription Factor 1 (TBX1) gene, with respect to normal parathyroids, was found in 71% (10/14) of human PCs at mRNA level, and confirmed, at the protein level, by in a significant decrease of TBX1-positive immunostained cells (37). Interestingly, in all the 10 PC samples characterized by a reduced number of TBX1-expressing cells, this molecular signature was associated with both the downregulation of TBX1 transcript and the loss of parafibromin immunostaining, while the 4 PC samples conserving parafibromin expression had TBX1 mRNA expression levels similar to those of normal parathyroid, suggesting a regulatory relationship between TBX1 and parafibromin, involved in parathyroid carcinogenesis.
Epigenetics of Parathyroid Carcinoma
Epigenetic processes are one of the most important regulators of gene expression and one of the most often deregulated mechanisms in human tumors. Epigenetic mechanisms are heritable but reversible, and they include DNA methylation, histone modifications, and small and long non-coding RNAs.
DNA Methylation in Parathyroid Carcinoma
DNA methylation consists of the covalent addition of a methyl group at the 5’-position of a cytosine, usually followed by a guanine (CpG dinucleotide). DNA methylation is catalyzed by DNA methyltransferases (DNMTs), while DNA demethylation is promoted by ten-eleven translocation (TET) methylcytosine dioxygenases. CpG dinucleotides are spread out across the genome, being commonly heavily methylated, with the exception of CpG islands, which are prevalently located in the promoter regions of genes. Global CpG methylation in intergenic regions is a fundamental mechanism to repress the expression of potentially harmful genetic sequences, such as transportable and retroviral elements. The majority of human gene promoters (roughly 70%) reside within CpG islands; the hypermethylation of these CpG islands is a reversible mechanism of gene silencing, responsible for regulating tissue-specific gene expression.
Human cancers are generally characterized by a global DNA hypomethylation, with respect to their normal counterparts. In contrast, both benign and malignant parathyroid tumors show a global methylation pattern similar to normal parathyroid glands (38, 39).
A genome-wide analysis of CpG island methylation identified 367 gene promoters significantly differentially methylated between PAs and normal parathyroids, 175 between PCs and normal parathyroids and 263 between PAs and PCs (40). In-depth analysis of the top 100 differentially methylated CpG islands showed, in PC samples, the presence of hypermethylation of all the examined sites (40).
Promoter hypermethylations of Adenomatous Polyposis Coli (APC) and Ras-association Domain Family Member 1A (RASSF1A) tumor suppressor genes are two common epigenetic changes in parathyroid tumors (38–41), which have been interestingly found in the 100% of analyzed PC samples (39, 41) (Table 2). Both silencing of APC and RASSF1A genes lead to the accumulation of the active form of β-catenin and to the subsequent activation of the canonical Wnt signaling, resulting in transcription of TCF/LEF-responsive target genes, including CCND1.
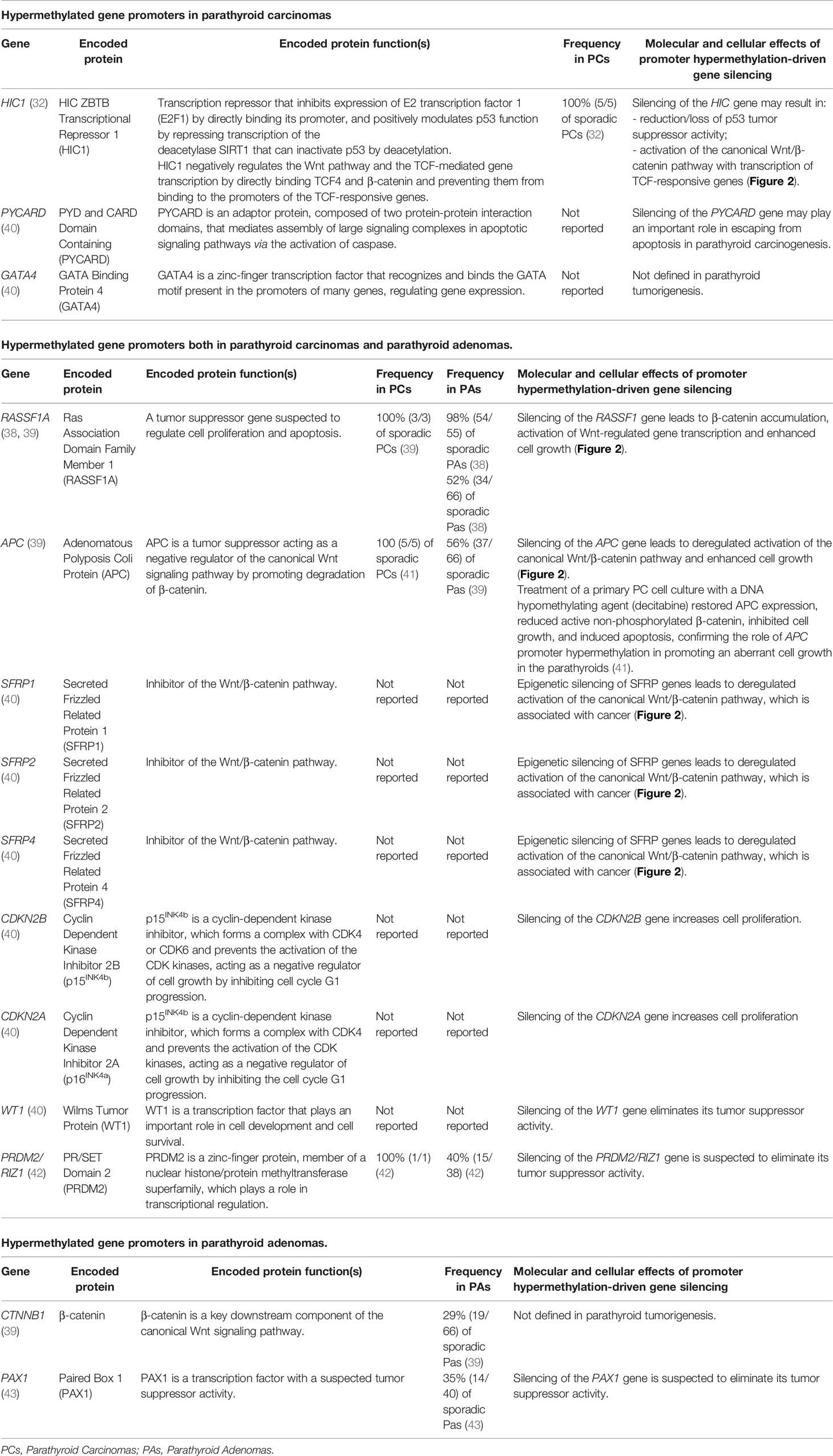
Table 2 Hypermethylated gene promoters in parathyroid tumors compared to normal parathyroid tissue.
Hypermethylation of promoters of HIC1, PYCARD, and GATA4 gene has specifically found to characterize PCs, with respect to both normal parathyroid glands and PAs (32, 40).
Main hypermethylated gene promoters identified in parathyroid tumors are reported in Table 2.
Histone Modifications in Parathyroid Carcinoma
Histones are the protein component of chromosomes, exerting a key role in the packing of chromatin and the regulation of gene expression. These proteins undergo post translational modifications, regulated by specific histone-modifying enzymes, consisting in the addition or removal of one or more methyl, acetyl, phosphate, or ubiquitin groups to specific amino acid residues. Histone modifications are reversible epigenetic mechanisms that alter chromatin state, modifying the accessibility of transcription factors to the promoters of target genes, thus positively or negatively regulating transcriptional activity.
As described above, one of the most commonly lost tumor suppressors in sporadic and syndromic PCs, the parafibromin, is a key regulator of various histone modifications (Figure 1). Normally, this nuclear tumor repressor interacts with the SUV39H1 histone methyltransferase complex, promoting H3K4me and H3K79me, and with the RNF20/RNF40 ubiquitine ligase complex, promoting H2BK120ub1.
Two other genes, EZH2 (9) and PRDM2/RIZ1 (42), whose expression has been reported to be deregulated in human PCs, respectively by gene amplification and promoter hypermethylation, encode methyl transferases, indicating a role of deregulated histone methylation in parathyroid carcinogenesis.
Deregulated MicroRNAs in Parathyroid Carcinoma
MicroRNAs (miRNAs) are single-stranded non-coding small RNAs (19-25 nucleotides in length), that negatively regulate the expression of target mRNAs, through a base-pairing mechanism, which leads to transcript endonucleolytic cleavage and degradation, or to transcriptional repression. Over 2,500 different mature human miRNAs have been identified thus far (www.mirbase.org), encoded by about 1,800 genes, which in more than 50% of cases are assembled in genomic clusters, rather than being casually distributed along the genome.
Deregulation of miRNA expression/activity has been found in many types of human cancers, acting as oncogene (oncomiR) or tumor suppressors; tumor cells show a larger alteration of miRNA expression compared to their normal counterparts.
Interestingly, deregulated signatures in miRNA expression appear to specifically characterize PCs, compared to both PAs and normal parathyroid glands, suggesting the possibility of using these epigenetic changes as markers to distinguish different parathyroid tumor types. PCs show a miRNA global down-regulation (about 80%) with respect to normal parathyroids. The most significantly down-regulated miRNAs in PCs were miR-26b (44), miR-30b (44), miR-126-5p (44), and miR-296-5p (45), while significantly up-regulated miRNAs were miR-222 (45), miR-517c (46), and miR-503 (45). Conversely, miR-139-3p resulted similarly expressed in PCs and PAs, and significantly down-regulated in both parathyroid tumor types compared to healthy glands (45).
Deregulated miRNAs specifically characterizing PC, and their possible role in carcinogenesis, are reported in Table 3.
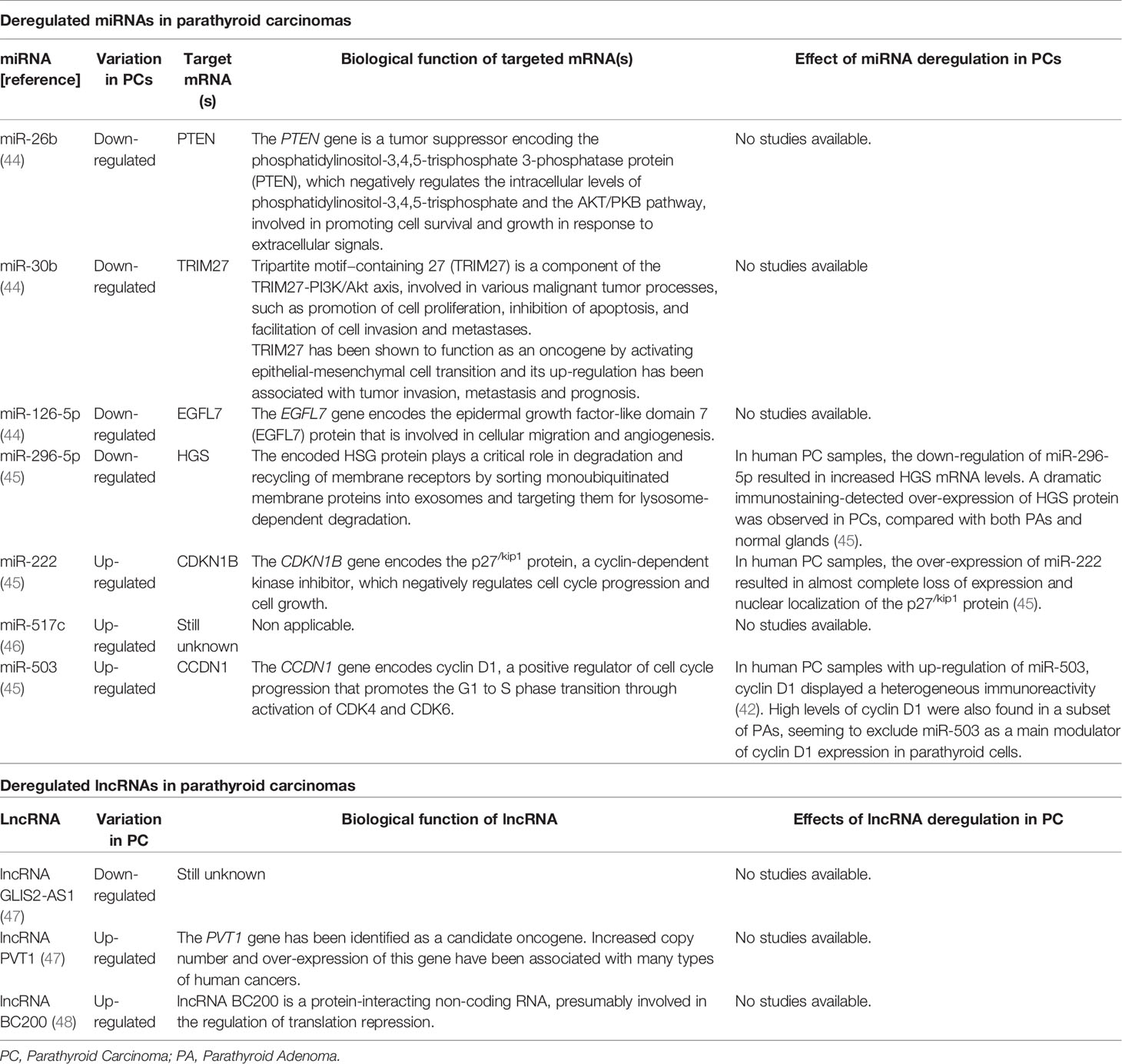
Table 3 Deregulated miRNAs and lncRNAs in parathyroid carcinomas.
Among deregulated miRNAs, miR-517c, located within the C19MC miRNA cluster at 19q13.42, showed the most significant expression difference between PC and PA, and its expression in PA was similar to that observed in normal parathyroids, strongly suggesting this miRNA as a potential diagnostic biomarker of PC, and potentially involved in determining the malignant phenotype. In PC samples, the up-regulation of miR-517c significantly correlated with over-expression of miR-371 and miR-372, two miRNAs of the miR-371-373 cluster, closely distal to C19MC. Deregulated expression profiles of miR-517c, miR-371, and miR-372, observed in primary PC, were conserved in their matched metastases (46). Considering clinical features of PC patients, expression levels of miR-517c in PC samples positively correlated with serum values of calcium and PTH, and with tumor weight (46). MiR-517c showed a tumor suppressor activity in human glioblastoma (49) and hepatocellular carcinoma cells (50), respectively through the inhibition of the epithelial-to-mesenchymal-like transition phenotype by targeting KPNA2 mRNA and, thus, disrupting the TP53 nuclear translocation, and through the inhibition of cell proliferation by targeting PTK2B/Pyk2 mRNA. Conversely, in PC, miR-517c appears to act as a proto-oncogene, although the pro-oncogenic effect of miR-517c up-regulation and the target mRNA(s) in parathyroid glands are not yet known.
Down-regulation of miR-296-5p showed a valuable predictive value in distinguishing PCs from healthy parathyroids (45); this miRNA was also down-regulated in a lung metastasis from a primary PC (46). MiR-296-5p is located at the imprinted complex locus GNAS (encoding the alpha subunit S of the guanine nucleotide-binding protein) on chromosome 20q13 and is expressed from the paternally-inherited allele (GNAS antisense RNA1; GNAS-AS1), but not from the maternally-derived allele (51). Interestingly, down-regulation of miR-296-5p seems to be restricted to PC, and it has not yet been reported in other human malignancies, indicating this miRNA as a possible parathyroid tissue-specific tumor suppressor. The putative mRNA target of miR-296-5p, the Hepatocyte Growth Factor-Regulated Tyrosine Kinase Substrate (HGS), which has been found to be over-expressed in PC samples with down-regulated miR-296-5p, is involved in the Wnt pathway, which is often altered in parathyroid tumors, suggesting a pro-oncogenic role of HGS up-regulation in parathyroid glands via Wnt signaling.
Interestingly, miR-222 was shown to suppress the expression of multiple negative regulators of the Wnt/β-catenin pathway, including DKK2, WIF2, SFRP2 and AXIN2 (Figure 2) (52). This miRNA is over-expressed in the majority of epithelial tumors, where it decreases the expression of epithelial-specific genes and increases the expression of mesenchymal-specific genes, promoting the epithelial-to-mesenchymal transition, and cell migration.
Deregulated Long Non-Coding RNAs in Parathyroid Carcinoma
Long non-coding RNAs (lncRNAs) are non-coding transcripts, longer than 200 nucleotides, acting as epigenetic regulators of gene expression, mainly in a tissue-specific fashion. A role of these molecules in regulating chromatin modifications and transcriptional and post-transcriptional gene expression has been demonstrated (53), as well as the fact that lncRNA deregulation is strongly associated with human tumorigenesis and cancer progression, through the activation of pro-oncogenic pathways and crosstalk with other RNA subtypes and epigenetic mechanisms (48). Alterations in lncRNA expression have been associated with endocrine diseases (54) and cancers (55, 56), including endocrine malignancies (57).
Up-regulation of two lncRNAs, [lncRNA PVT1 (47) and lncRNA BC200 (48)], and down-regulation of lncRNA GLIS2-AS1 (55), were significantly associated with PCs compared to healthy tissue and PAs. LncRNA BC200 has also been shown to discriminate PCs from aPAs, appearing to be a specific molecular signature of malignant tumors (48). Interestingly, lncRNA BC200 resulted significantly upregulated in the group of PCs with CDC73 mutations compared to that of PCs with wild type parafibromin, in association with more aggressive clinical features, such as higher levels of PTH and calcium ion (48). Over-expression of lncRNA BC200 has been reported in a broad spectrum of tumor cells, being responsible for cell viability, invasion, and migration (58).
lncRNA PVT1 has been shown to promote cell proliferation of non-small cell lung cancer through the epigenetic regulation of the large tumor suppressor kinase 2 (LATS2) expression (59), by directly binding to EZH2, a transcriptional repression factor that has been shown to act as a pro-oncogenic factor in parathyroid carcinogenesis (9).
To date, there are no reports in cancer or other human diseases regarding lncRNA GLIS2-AS1, and the biological function of this lncRNA is still unknown. A possible parathyroid tissue-specific tumor suppressor activity could be considered and remains to be investigated.
Circular RNA Expression Profile in Parathyroid Carcinoma
Circular RNAs (circRNAs) are a recently identified class of single-strand non-coding RNAs, consisting in a covalently closed loop generated by the 3’ and 5’ ends of the RNA molecule that have been joined together, which modulate miRNA activity by acting as miRNA sponges, regulate alternative splicing, and are suspected to have a role in human diseases and tumor progression. A microarray-based analysis of circRNA expression profile has been recently performed, for the first time, in human PAs, PCs and healthy parathyroids (60). Selected differentially expressed circRNAs and their corresponding mRNAs were validated by RT-qPCR, showing that 4 circRNAs (circRNA_0035563, circRNA_0017545, circRNA_0001687 and circRNA_0075005) and 4 mRNAs (MYC, FSCN1, ANXA2 and AKR1C3) were all significantly up-regulated in PCs than PAs, with circRNA_0075005 appearing the most promising diagnostic marker for distinguishing PC from PA. Expression of circ_0035563 and circ_0075005 was furtherly validated in formalin-fixed paraffin-embedded samples of PC and PA by fluorescence in situ hybridization, confirming both these molecules to be significantly higher in PC than in benign lesions. In addition, the study found circ_0035563 as significantly related to CDC73 mutations and malignant recurrence, suggesting this parameter as a suitable marker of prognosis in PC patients. Results of this pivotal study open to the possibility of using circRNA expression as marker for the differential diagnosis of PC and PA; the effective clinical value of these molecules needs to be confirmed in further studies on larger sample size from multiple centers, to be analyzed in association with histopathological and clinical features of tumors, and to be functionally studied on PC cell lines.
Gene Expression Profile and Proteomic Analysis in Parathyroid Carcinoma
Given the lack of specific PC biomarkers in clinical practice, two recent studies have investigated: 1) gene expression profile (61), and 2) global proteomic expression pattern (62), as possible molecular signatures to distinguish PC from PA.
The first study (61) compared the expression of 740 genes, known to be involved in main cancer progression processes [angiogenesis, extracellular matrix (ECM) remodeling, EMT, metastasis], between PAs, non-metastatic PCs, and metastatic PCs, identifying differentially expressed groups of genes able to discriminate PAs from PCs, and also differentiate non-metastatic PCs from metastatic PCs. Two specific signatures of 87 gene expressions (43 up-regulated and 44 down-regulated) and of 103 gene expressions (65 up-regulated and 38 down-regulated) characterized non-metastatic PCs and metastatic PCs, respectively, with respect to PAs. Among genes showing up-regulation in both metastatic and non-metastatic PCs vs PAs, there were genes encoding components of the ECM (COL1A1, COL1A2, COL3A1, COL5A2, FN1, LUM, LOX, THBS1, THBS2, BGN, and SFRP2) and genes encoding enzymes involved in proteolysis, cross-linking and assembly processes of proteins of the ECM (FAP, LOX, SULF1 and PXDN), suggesting a key role of ECM deregulation in carcinogenesis, presumably altering cell proliferation (61). Metastatic PCs showed a significant up-regulation of MMP9, SOX2, CD24, BMP7, ANGPTL4, and FGFR1 genes, and a down-regulation of ERBB3, TBX1, RAB25, and FBP1 genes, compared to both PAs and non-metastatic PCs, suggesting a role of this gene expression signature in tumor progression and metastatic spread (61).
The second study (62) compared the global proteomic profile between a PC and a PA, co-existing in the same patient, by using two-dimensional differential gel electrophoresis coupled with mass spectrometry. The possibility to compare the two different types of tumors from the same patient allowed the identification of distinctive PC protein features without any external confounding influences, such as age, sex, germline background, lifestyle, and environmental factors. PC showed the specific over-expression of 23 proteins and the specific under-expression of 10 proteins, mainly belonging to specific biological processes, such as cell metabolism, cell signaling, and protein ubiquitination. In particular, the Ubiquitin C-terminal Hydrolase L1 (UCH-L1), a member of the deubiquitinase family involved in processing ubiquitin precursors and in regulating angiogenesis (63), and whose up-regulation has previously been demonstrated to enhance cell invasion/metastasis (64, 65), and increase multidrug resistance in breast cancer (66, 67), resulted highly over-expressed in PC, suggesting an oncogenic role of this protein in the malignant carcinogenesis of parathyroid tissue and, at the same time, indicating UCH-L1 as a possible distinctive immunohistochemical signature of PC with respect to benign parathyroid tumors. Other promising protein biomarkers, all over-expressed in PCs, were: Malate Dehydrogenase 1 (MDH1), an enzyme that catalyzes the NAD/NADH-dependent reversible oxidation of malate to oxaloacetate in many metabolic pathways and was showed to be up-regulated in cancer (68); Chloride Intracellular Channel Protein 1 (CLIC1), a protein involved in regulating cell proliferation and differentiation, and metastasis (69, 70), and whose up-regulation has previously been associated with human malignancies; and Superoxide Dismutase 2 (SOD2), which is a key player of the antioxidant system of the cell and whose increased expression appears correlated to the stage of tumor progression (71).
Concluding Remarks
● Pre-operative diagnosis of PCs is complicated. There are no specific histological and molecular features helping to pre-operatively distinguish PAs, aPAs, and PCs.
● Loss-of-function mutations and/or loss of the CDC73 tumor suppressor gene are the main genetic defects in PCs, both in the sporadic form and in the context of FIPH or HPT-JT syndrome. However, the absence of nuclear staining for parafibromin in tumor samples is not sufficient, alone, for a histological diagnosis of PCs.
● Hypermethylation of specific gene promoters has been specifically associated with PC, leading to gene silencing and presumably triggering parathyroid carcinogenesis. Theoretically, treatment with inhibitors of DNA methyltransferase would be expected to reduce the methylation density at hypermethylated promoters, restoring the normal expression of silenced genes. However, the anti-oncogenic beneficial of these molecules is controversial, since they exert a non-specific cytosine demethylation action that cannot be specifically directed on the target hypermethylated promoters. This may lead to a global DNA hypomethylation, increasing the risk of DNA damages.
● Deregulation of expression of specific miRNAs showed a promising capability to discriminate parathyroid tumors from healthy glands, as well as among different types of parathyroid tumors. MiRNA signatures specifically characterizing PCs have been identified.
● Down-regulation of miR-296-5p and miR-126-5p and up-regulation of miR-517c appear to be valuable molecular discriminators between PCs and PAs. These molecules might be, thus, valuable molecular targets for epigenetic therapy of PCs.
● Target inhibition of up-regulated miRNAs having an oncogenic activity (oncomiR) may be a valuable approach for epigenetic therapy of PC, which can be easily obtained by small molecule miRNA inhibitors, miRNA sponges, miRNA masks, or synthetic, modified antisense anti-miRNAs.
● The deregulated expression of specific lncRNAs (lncRNA PVT1, lncRNA GLIS2-SA1, and lncRNA BC200) also appears to characterize molecular signatures of human PCs, distinguishing between benign and malignant parathyroid tumors. However, the real contribution of lncRNAs to parathyroid carcinogenesis remains to be fully elucidated.
● The interplay between different epigenetic mechanisms, and their possible synergic action in promoting parathyroid carcinogenesis, should be studied as well.
● Epigenetic deregulation, rewiring the cell translational network, is thought to initiate PC tumorigenesis, by inducing a dedifferentiated tumor phenotype, characterized by an “embryonic-like” gene expression signature. Mutated oncogenes and tumor suppressor genes are suspected to contribute to parathyroid cancer development, presumably not only by inducing an increased cell proliferation, but also because of their role in deregulating the cell epigenome.
● Further investigations, to identify and/or confirm deregulated epigenetic factors specifically characterizing PCs, are needed, in association with functional in vitro studies, to understand their role in PC initiation, development and progression. The high-throughput “omic” technologies (genomics, transcriptomics, proteomics, metabolomics) will allow collective quantification and characterization of wide numbers of biological molecules, at different molecular levels, helping in deciphering the regulatory networks, whose deregulation underlies the development and progression of parathyroid cancer, and in identifying novel therapeutic targets.
Author Contributions
FM wrote the manuscript and drew tables and figure. FG and GPa performed the review of published literature and contributed to drawing tables and figure. GPe, RS, and MB critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Authors are grateful to the F.I.R.M.O. Foundation for the support.
References
1. Cetani F, Marcocci C, Torregrossa L, Pardi E. Atypical Parathyroid Adenomas: Challenging Lesions in the Differential Diagnosis of Endocrine Tumors. Endocr Relat Cancer (2019) 26(7):R441–64. doi: 10.1530/ERC-19-0135
2. Long KL, Sippel RS. Current and Future Treatments for Parathyroid Carcinoma. Int J Endo Oncol (2018) 5(1):IJE06. doi: 10.2217/ije-2017-0011
3. Fingeret AL. Contemporary Evaluation and Management of Parathyroid Carcinoma. JCO Oncol Pract (2021) 17(1):17–21. doi: 10.1200/JOP.19.00540
4. Wang P, Xue S, Wang S, Lv Z, Meng X, Wang G, et al. Clinical Characteristics and Treatment Outcomes of Parathyroid Carcinoma: A Retrospective Review of 234 Cases. Oncol Lett (2017) 14(6):7276–82. doi: 10.3892/ol.2017.7076
5. Cardoso L, Stevenson M, Thakker RV. Molecular Genetics of Syndromic and Non-Syndromic Forms of Parathyroid Carcinoma. Hum Mut (2017) 38:1621–48. doi: 10.1002/humu.23337
6. Hendy GN, Cole DEC. Genetic Defects Associated With Familial and Sporadic Hyperparathyroidism. Front Horm Res (2013) 41:149–65. doi: 10.1159/000345675
7. Alvelos MI, Mendes M, Soares P. Molecular Alterations in Sporadic Primary Hyperparathyroidism. Genet Res Int (2011) 2011:275802. doi: 10.4061/2011/275802
8. Pandya C, Uzilov AV, Bellizzi J, Lau CY, Moe AS, Strahl M, et al. Genomic Profiling Reveals Mutational Landscape in Parathyroid Carcinomas. JCI Insight (2017) 2(6):e92061. doi: 10.1172/jci.insight.92061
9. Svedlund J, Barazeghi E, Stålberg P, Hellman P, Åkerström G, Björklund P, et al. The Histone Methyltransferase EZH2, an Oncogene Common to Benign and Malignant Parathyroid Tumors. Endocr Relat Cancer (2014) 21(2):231–9. doi: 10.1530/ERC-13-0497
10. Yu W, McPherson JR, Stevenson M, van Eijk R, Heng HL, Newey P, et al. Whole-Exome Sequencing Studies of Parathyroid Carcinomas Reveal Novel PRUNE2 Mutations, Distinctive Mutational Spectra Related to APOBEC-Catalyzed DNA Mutagenesis and Mutational Enrichment in Kinases Associated With Cell Migration and Invasion. J Clin Endocrinol Metab (2015) 100(2):E360–4. doi: 10.1210/jc.2014-3238
11. Kasaian K, Wiseman SM, Thiessen N, Mungall KL, Corbett RD, Qian JQ, et al. Complete Genomic Landscape of a Recurring Sporadic Parathyroid Carcinoma. J Pathol (2013) 230(3):249–60. doi: 10.1002/path.4203
12. Gill AJ, Lim G, Cheung VKY, Andrici J, Perry-Keene JL, Paik J, et al. Parafibromin-Deficient (HPT-JT Type, CDC73 Mutated) Parathyroid Tumors Demonstrate Distinctive Morphologic Features. Am J Surg Pathol (2019) 43(1):35–46. doi: 10.1097/PAS.0000000000001017
13. Pontikides N, Karras S, Kaprara A, Anagnostis P, Mintziori G, Goulis DG, et al. Genetic Basis of Familial Isolated Hyperparathyroidism: A Case Series and a Narrative Review of the Literature. J Bone Miner Metab (2014) 32(4):351–66. doi: 10.1007/s00774-013-0551-9
14. Hewitt KM, Sharma PK, Samowitz W, Hobbs M. Aberrant Methylation of the HRPT2 Gene in Parathyroid Carcinoma. Ann Otol Rhinol Laryngol (2007) 116(12):928–33. doi: 10.1177/000348940711601210
15. Yart A, Gstaiger M, Wirbelauer C, Pecnik M, Anastasiou D, Hess D, et al. The HRPT2 Tumor Suppressor Gene Product Parafibromin Associates With Human PAF1 and RNA Polymerase II. Mol Cell Biol (2005) 25(12):5052–60. doi: 10.1128/MCB.25.12.5052-5060.2005
16. Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, Copeland TD, Guszczynski T, et al. The Parafibromin Tumor Suppressor Protein Is Part of a Human Paf1 Complex. Mol Cell Biol (2005) 25(2):612–20. doi: 10.1128/MCB.25.2.612-620.2005
17. Haven CJ, Howell VM, Eilers PH, Dunne R, Takahashi M, van Puijenbroek M, et al. Gene Expression of Parathyroid Tumors: Molecular Subclassification and Identification of the Potential Malignant Phenotype. Cancer Res (2004) 64:e7405–11. doi: 10.1158/0008-5472.CAN-04-2063
18. Kim JM, Kim K, Punj V, Liang G, Ulmer TS, Lu W, et al. Linker Histone H1.2 Establishes Chromatin Compact and Gene Silencing Through Recognition of H3k27me3. Sci Rep (2015) 5:16714. doi: 10.1038/srep16714
19. Yang YJ, Han JW, Youn HD, Cho EJ. The Tumor Suppressor, Parafibromin, Mediates Histone H3 K9 Methylation for Cyclin D1 Repression. Nucleic Acids Res (2010) 38(2):382–90. doi: 10.1093/nar/gkp991
20. Ng HH, Dole S, Struhl K. The Rtf1 Component of the Paf1 Transcriptional Elongation Complex Is Required for Ubiquitination of Histone H2B. J Biol Chem (2003) 278(36):33625–8. doi: 10.1074/jbc.C300270200
21. Mosimann C, Hausmann G, Basler K. Parafibromin/Hyrax Activates Wnt/Wg Target Gene Transcription by Direct Association With Beta-Catenin/Armadillo. Cell (2006) 125(2):327–41. doi: 10.1016/j.cell.2006.01.053
22. Lin L, Czapiga M, Nini L, Zhang JH, Simonds WF. Nuclear Localization of the Parafibromin Tumor Suppressor Protein Implicated in the Hyperparathyroidism-Jaw Tumor Syndrome Enhances Its Proapoptotic Function. Mol Cancer Res (2007) 5(2):183–93. doi: 10.1158/1541-7786.MCR-06-0129
23. Panicker LM, Zhang JH, Dagur PK, Gastinger MJ, Simonds WF. Defective Nucleolar Localization and Dominant Interfering Properties of a Parafibromin L95P Missense Mutant Causing the Hyperparathyroidism-Jaw Tumor Syndrome. Endocr Relat Cancer (2010) 17(2):513–24. doi: 10.1677/ERC-09-0272
24. Tan MH, Morrison C, Wang P, Yang X, Haven CJ, Zhang C, et al. Loss of Parafibromin Immunoreactivity Is a Distinguishing Feature of Parathyroid Carcinoma. Clin Cancer Res (2004) 10(19):6629–37. doi: 10.1158/1078-0432.CCR-04-0493
25. Imanishi Y, Hosokawa Y, Yoshimoto K, Schipani E, Mallya S, Papanikolaou A, et al. Primary Hyperparathyroidism Caused by Parathyroid-Targeted Overexpression of Cyclin D1 in Transgenic Mice. J Clin Invest (2001) 107(9):1093–102. doi: 10.1172/JCI10523
26. Mallya SM, Gallagher JJ, Wild YK, Kifor O, Costa-Guda J, Saucier K, et al. Abnormal Parathyroid Cell Proliferation Precedes Biochemical Abnormalities in a Mouse Model of Primary Hyperparathyroidism. Mol Endocrinol (2005) 19(10):2603–9. doi: 10.1210/me.2005-0116
27. Arnold A, Kim HG, Gaz RD, Eddy RL, Fukushima Y, Byers MG, et al. Molecular Cloning and Chromosomal Mapping of DNA Rearranged With the Parathyroid Hormone Gene in a Parathyroid Adenoma. J Clin Invest (1989) 83(6):2034–40. doi: 10.1172/JCI114114
28. Tashiro E, Maruki H, Minato Y, Doki Y, Weinstein IB, Imoto M. Overexpression of Cyclin D1 Contributes to Malignancy by Up-Regulation of Fibroblast Growth Factor Receptor 1 via the pRB/E2F Pathway. Cancer Res (2003) 63(2):424–31.
29. Sanpaolo E, Miroballo M, Corbetta S, Verdelli C, Baorda F, Balsamo T, et al. EZH2 and ZFX Oncogenes in Malignant Behaviour of Parathyroid Neoplasms. Endocrine (2016) 54(1):55–9. doi: 10.1007/s12020-016-0892-y
30. Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP, et al. Identification of Somatic Mutations in Parathyroid Tumors Using Whole-Exome Sequencing. J Clin Endocrinol Metab (2012) 97(9):E1774–81. doi: 10.1210/jc.2012-1743
31. Wei Z, Sun B, Wang ZP, He JW, Fu WZ, Fan, et al. Whole-Exome Sequencing Identifies Novel Recurrent Somatic Mutations in Sporadic Parathyroid Adenomas. Endocrinology (2018) 159(8):3061–8. doi: 10.1210/en.2018-00246
32. Svedlund J, Koskinen Edblom S, Marquez VE, Åkerström G, Björklund P, Westin G. Hypermethylated in Cancer 1 (HIC1), a Tumor Suppressor Gene Epigenetically Deregulated in Hyperparathyroid Tumors by Histone H3 Lysine Modification. J Clin Endocrinol Metab (2012) 97(7):E1307–15. doi: 10.1210/jc.2011-3136
33. Rascio F, Spadaccino F, Rocchetti MT, Castellano G, Stallone G, Netti GS, et al. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers (Basel) (2021) 13(16):3949. doi: 10.3390/cancers13163949
34. Tavanti GS, Verdelli C, Morotti A, Maroni P, Guarnieri V, Scillitani A, et al. Yes-Associated Protein 1 Is a Novel Calcium Sensing Receptor Target in Human Parathyroid Tumors. Int J Mol Sci (2021) 22(4):2016. doi: 10.3390/ijms22042016
35. Storvall S, Leijon H, Ryhänen EM, Vesterinen T, Heiskanen I, Schalin-Jäntti C, et al. Filamin A and Parafibromin Expression in Parathyroid Carcinoma. Eur J Endocrinol (2021) 185(6):803–12. doi: 10.1530/EJE-21-0668
36. Mingione A, Verdelli C, Ferrero S, Vaira V, Guarnieri V, Scillitani A, et al. Filamin A Is Reduced and Contributes to the CASR Sensitivity in Human Parathyroid Tumors. J Mol Endocrinol (2017) 58(2):91–103. doi: 10.1530/JME-16-0184
37. Verdelli C, Avagliano L, Guarnieri V, Cetani F, Ferrero S, Vicentini L, et al. Expression, Function, and Regulation of the Embryonic Transcription Factor TBX1 in Parathyroid Tumors. Lab Invest (2017) 97(12):1488–99. doi: 10.1038/labinvest.2017.88
38. Juhlin CC, Kiss NB, Villablanca A, Haglund F, Nordenstrom J, Hoog A, et al. Frequent Promoter Hypermethylation of the APC and RASSF1A Tumour Suppressors in Parathyroid Tumours. PloS One (2010) 5:e9472. doi: 10.1371/journal.pone.0009472
39. Sulaiman L, Juhlin CC, Nilsson IL, Fotouhi O, Larsson C, Hashemi J. Global and Gene-Specific Promoter Methylation Analysis in Primary Hyperparathyroidism. Epigenetics (2013) 8:646e655. doi: 10.4161/epi.24823
40. Starker LF, Svedlund J, Udelsman R, Dralle H, Akerström G, Westin G, et al. The DNA Methylome of Benign and Malignant Parathyroid Tumors. Genes Chromosomes Cancer (2011) 50:735–45. doi: 10.1002/gcc.20895
41. Svedlund J, Aurén M, Sundström M, Dralle H, Akerström G, Björklund P, et al. Aberrant WNT/beta-Catenin Signaling in Parathyroid Carcinoma. Mol Cancer (2010) 9:294. doi: 10.1186/1476-4598-9-294
42. Carling T, Du Y, Fang W, Correa P, Huang S. Intragenic Allelic Loss and Promoter Hypermethylation of the RIZ1 Tumor Suppressor Gene in Parathyroid Tumors and Pheochromocytomas. Surgery (2003) 134(6):932–939; discussion 939-940. doi: 10.1016/s0039-6060(03)00422-7
43. Singh P, Bhadada SK, Arya AK, Saikia UN, Sachdeva N, Dahiya D, et al. Aberrant Epigenetic Alteration of PAX1 Expression Contributes to Parathyroid Tumorigenesis. J Clin Endocrinol Metab (2021) 107(2):e783–92. doi: 10.1210/clinem/dgab626
44. Rahbari R, Holloway AK, He M, Khanafshar E, Clark OH, Kebebew E. Identification of Differentially Expressed microRNA in Parathyroid Tumors. Ann Surg Oncol (2011) 18:1158–65. doi: 10.1245/s10434-010-1359-7
45. Corbetta S, Vaira V, Guarnieri V, Scillitani A, Eller-Vainicher C, Ferrero S, et al. Differential Expression of microRNAs in Human Parathyroid Carcinomas Compared With Normal Parathyroid Tissue. Endocr Relat Cancer (2010) 17(1):135–46. doi: 10.1677/ERC-09-0134
46. Vaira V, Elli F, Forno I, Guarnieri V, Verdelli C, Ferrero S, et al. The microRNA Cluster C19MC Is Deregulated in Parathyroid Tumours. J Mol Endocrinol (2012) 49(2):115–24. doi: 10.1530/JME-11-0189
47. Zhang X, Hu Y, Wang M, Zhang R, Wang P, Cui M, et al. Profiling Analysis of Long Non-Coding RNA and mRNA in Parathyroid Carcinoma. Endocr Relat Cancer (2019) 26(2):163–76. doi: 10.1530/ERC-18-0480
48. Morotti A, Forno I, Verdelli C, Guarnieri V, Cetani F, Terrasi A, et al. The Oncosuppressors MEN1 and CDC73 Are Involved in lncRNA Deregulation in Human Parathyroid Tumors. J Bone Miner Res (2020) 35(12):2423–31. doi: 10.1002/jbmr.4154
49. Lu Y, Xiao L, Liu Y, Wang H, Li H, Zhou Q, et al. MIR517C Inhibits Autophagy and the Epithelial-to-Mesenchymal (-Like) Transition Phenotype in Human Glioblastoma Through KPNA2-Dependent Disruption of TP53 Nuclear Translocation. Autophagy (2015) 11(12):2213–32. doi: 10.1080/15548627.2015.1108507
50. Liu RF, Xu X, Huang J, Fei QL, Chen F, Li YD, et al. Down-Regulation of miR-517a and miR-517c Promotes Proliferation of Hepatocellular Carcinoma Cells via Targeting Pyk2. Cancer Lett (2013) 329(2):164–73. doi: 10.1016/j.canlet.2012.10.027
51. Robson JE, Eaton SA, Underhill P, Williams D, Peters J. MicroRNAs 296 and 298 Are Imprinted and Part of the GNAS/Gnas Cluster and miR-296 Targets IKBKE and Tmed9. RNA (2012) 18(1):135–44. doi: 10.1261/rna.029561.111
52. Liu S, Wang Z, Liu Z, Shi S, Zhang Z, Zhang J, et al. miR-221/222 Activate the Wnt/beta-Catenin Signaling to Promote Triple-Negative Breast Cancer. J Mol Cell Biol (2018) 10(4):302–15. doi: 10.1093/jmcb/mjy041
53. Wang KC, Chang HY. Molecular Mechanisms of Long Noncoding RNAs. Mol Cell (2011) 43:904–14. doi: 10.1016/j.molcel2011.08.018
54. Knoll M, Lodish HF, Sun L. Long Non-Coding RNAs as Regulators of the Endocrine System. Nat Rev Endocrinol (2015) 11(3):151–60. doi: 10.1038/nrendo.2014.229
55. Bartonicek N, Maag JL, Dinger ME. Long Noncoding RNAs in Cancer: Mechanisms of Action and Technological Advancements. Mol Cancer (2016) 15(1):43. doi: 10.1186/s12943-016-0530-6
56. Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer Cell (2016) 29(4):452–63. doi: 10.1016/j.ccell.2016.03.010
57. Murugan AK, Munirajan AK, Alzahrani AS. Long Noncoding RNAs: Emerging Players in Thyroid Cancer Pathogenesis. Endocr Relat Cancer (2018) 25(2):R59–82. doi: 10.1530/ERC-17-0188
58. Samson J, Cronin S, Dean K. BC200 (BCYRN1) - The Shortest, Long, Non-Coding RNA Associated With Cancer. Noncoding RNA Res (2018) 3(3):131–43. doi: 10.1016/j.ncrna.2018.05.003
59. Wan L, Sun M, Liu GJ, Wei CC, Zhang EB, Kong R, et al. Long Noncoding RNA PVT1 Promotes Non-Small Cell Lung Cancer Cell Proliferation Through Epigenetically Regulating LATS2 Expression. Mol Cancer Ther (2016) 15(5):1082–94. doi: 10.1158/1535-7163.MCT-15-0707
60. Hu Y, Zhang X, Cui M, Wang M, Su Z, Liao Q, et al. Circular RNA Profile of Parathyroid Neoplasms: Analysis of Co-Expression Networks of Circular RNAs and mRNAs. RNA Biol (2019) 16(9):1228–36. doi: 10.1080/15476286.2019.1622962
61. Condello V, Cetani F, Denaro M, Torregrossa L, Pardi E, Piaggi P, et al. Gene Expression Profile in Metastatic and Non-Metastatic Parathyroid Carcinoma. Endocr Relat Cancer (2021) 28(2):111–34. doi: 10.1530/ERC-20-0450
62. Ciregia F, Cetani F, Pardi E, Soggiu A, Piras C, Zallocco L, et al. Parathyroid Carcinoma and Adenoma Co-Existing in One Patient: Case Report and Comparative Proteomic Analysis. Cancer Genomics Proteomics (2021) 18(6):781–96. doi: 10.21873/cgp.20297
63. Song IK, Kim HJ, Magesh V, Lee KJ. Ubiquitin C-Terminal Hydrolase-L1 Plays a Key Role in Angiogenesis by Regulating Hydrogen Peroxide Generated by NADPH Oxidase 4. Biochem Biophys Res Commun (2018) 495(1):1567–72. doi: 10.1016/j.bbrc.2017.11.051
64. Kim HJ, Magesh V, Lee JJ, Kim S, Knaus UG, Lee KJ. Ubiquitin C-Terminal Hydrolase-L1 Increases Cancer Cell Invasion by Modulating Hydrogen Peroxide Generated via NADPH Oxidase 4. Oncotarget (2015) 6(18):16287–162303. doi: 10.18632/oncotarget.3843
65. Luo Y, He J, Yang C, Orange M, Ren X, Blair N, et al. UCH-L1 Promotes Invasion of Breast Cancer Cells Through Activating Akt Signaling Pathway. J Cell Biochem (2018) 119(1):691–700. doi: 10.1002/jcb.26232
66. Jin Y, Zhang W, Xu J, Wang H, Zhang Z, Chu C, et al. UCH-L1 Involved in Regulating the Degradation of EGFR and Promoting Malignant Properties in Drug-Resistant Breast Cancer. Int J Clin Exp Pathol (2015) 8(10):12500–8.
67. Wang W, Zou L, Zhou D, Zhou Z, Tang F, Xu Z, et al. Overexpression of Ubiquitin Carboxyl Terminal Hydrolase-L1 Enhances Multidrug Resistance and Invasion/Metastasis in Breast Cancer by Activating the MAPK/Erk Signaling Pathway. Mol Carcinog (2016) 55(9):1329–42. doi: 10.1002/mc.22376
68. Wang YP, Zhou W, Wang J, Huang X, Zuo Y, Wang TS, et al. Arginine Methylation of MDH1 by CARM1 Inhibits Glutamine Metabolism and Suppresses Pancreatic Cancer. Mol Cell (2016) 64(4):673–87. doi: 10.1016/j.molcel.2016.09.028
69. Peng JM, Lin SH, Yu MC, Hsieh SY. CLIC1 Recruits PIP5K1A/C to Induce Cell-Matrix Adhesions for Tumor Metastasis. J Clin Invest (2021) 131(1):e133525. doi: 10.1172/JCI133525
70. Qiu Y, Mao YT, Zhu JH, Zhao K, Wang JF, Huang JM, et al. CLIC1 Knockout Inhibits Invasion and Migration of Gastric Cancer by Upregulating AMOT-P130 Expression. Clin Transl Oncol (2021) 223(3):514–25. doi: 10.1007/s12094-020-02445-0
Keywords: parathyroid carcinoma, gene mutation, tumor suppressor genes, molecular signatures, epigenetic changes, DNA methylation, microRNAs, long non-coding RNAs
Citation: Marini F, Giusti F, Palmini G, Perigli G, Santoro R and Brandi ML (2022) Genetics and Epigenetics of Parathyroid Carcinoma. Front. Endocrinol. 13:834362. doi: 10.3389/fendo.2022.834362
Received: 13 December 2021; Accepted: 28 January 2022;
Published: 24 February 2022.
Edited by:
Filomena Cetani, University of Pisa, ItalyReviewed by:
Sabrina Corbetta, University of Milan, ItalyJessica Costa-Guda, University of Connecticut, United States
Copyright © 2022 Marini, Giusti, Palmini, Perigli, Santoro and Brandi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Luisa Brandi, marialuisa.brandi@unifi.it