 Xiaoying Zhou1†
Xiaoying Zhou1† Chunli Wang
Chunli Wang Zhanjun Jia
Zhanjun Jia Bixia Zheng
Bixia Zheng- 1Department of Gastroenterology, Children’s Hospital of Nanjing Medical University, Nanjing, China
- 2Nanjing Key Laboratory of Pediatrics, Children’s Hospital of Nanjing Medical University, Nanjing, China
Next-generation sequencing is effective for the molecular diagnosis of genetic diseases. However, the identification of the clinical significance of synonymous variants remains a challenge. Our previous study showed that some synonymous variants in ATP7B gene produced splicing disruptions, leading to Wilson disease (WD). To test the hypothesis that synonymous variants of ATP7B cause abnormal splicing by disrupting authentic splice sites or splicing regulatory elements, we used computational tools and minigene assays to characterize 253 naturally occurring ATP7B gene synonymous variants in this study. Human Splicing Finder (HSF) and ESE Finder 3.0 were used to predict the impact of these rare synonymous variants on pre-mRNA splicing. Then, we cloned 14 different wild-type Minigene_ATP7B_ex constructs for in vitro minigene assay, including 16 exons of ATP7B gene. After computational prediction, 85 candidate variants were selected to be introduced into the corresponding Minigene_ATP7B_ex constructs for splicing assays. Using this two-step procedure, we demonstrated that 11 synonymous variants in ExAc database (c.1620C>T, c.3888C>T, c.1554C>T, c.1677C>T, c.1830G>A, c.1875T>A, c.2826C>A, c.4098G>A, c.2994C>T, c.3243G>A, and c.3747G>A) disrupted RNA splicing in vitro, and two (c.1620C>T and c.3243G>A) of these caused a complete exon skipping. The results not only provided a reliable experimental basis for the genetic diagnosis of WD patients but also offered some new insights into the pathogenicity of synonymous variants in genetic diseases.
Introduction
Wilson disease (OMIM, #277900, WD) is an autosomal, recessively inherited disorder characterized by the dramatic accumulation of intracellular hepatic copper with subsequent hepatic and neurologic abnormalities (Xie and Wu, 2017). The worldwide prevalence of Wilson disease is estimated to be on the order of 30 per one million, with a gene frequency of 0.56% and a carrier frequency of one in 90 (Sandahl et al., 2019). Loss-of-function mutations in ATPase copper-transporting beta gene (ATP7B) are responsible for WD. The ATP7B gene (OMIM, ∗606882) is located on chromosome 13q14.3 encoding copper-transporting ATPase 2 (ATP7B), acting as a plasma membrane copper-transport protein. Liver disease, which shows a high variability in patients, is the main clinical manifestation of WD, with or without neurological and/or psychological symptoms and Kayser–Fleischer corneal rings (Walshe, 2017). Due to the heterogeneity of the clinical features and the age of onset, WD is prone to be misdiagnosed or prolonged in the diagnosis cycle (Balashova et al., 2019). At present (Hedera, 2019), genetic screening of ATP7B gene is widely used in the diagnosis of WD, which can greatly benefit the clinical diagnosis and management of patients (Li et al., 2019). However, synonymous variants yield from mutation screening with uncertain clinical significance (VUS). Thus, the functional and clinical interpretations pose a challenge for genomic diagnosis of WD.
Up to now, a total of 995 mutations in ATP7B were annotated in the Human Gene Mutation Database (HGMD)1, in which 67 splice variants located in the non-coding region were included. No synonymous variants were reported in the HGMD. Synonymous variants are often considered to be neutral because they do not change the amino acid coding. However, recent studies have shown that synonymous/missense mutations can lead to abnormal splicing of mRNA by disrupting splicing regulatory elements (SREs), which act as a new, gradually recognized pathogenic mechanism in genetic diseases. This mechanism has been reported in Gitelman syndrome, progressive familial intrahepatic cholestasis, Netherton syndrome, and other genetic disorders (Byrne et al., 2009; Dal Mas et al., 2015; Takeuchi et al., 2015). Our previous study demonstrated that the synonymous variant (c.4014T>A) and missense variant (c.2755C>G/p.R919G) in WD patients resulted in exon skipping in pre-mRNA splicing, leading to protein truncation and WD (Wang et al., 2018).
The recognition of intron and exon boundaries by spliceosome is one of the key steps in the splicing process [9]. Exonic splicing enhancers (ESEs) and exonic splicing silencers (ESSs) are SREs located in the coding region. ESEs and ESSs are cis-acting elements and are composed of six to eight nucleotides. After binding to the ESEs, the SR protein can promote splicing by recruiting U1 snRNP to the donor splicing site or balancing the inhibitory effect of ESSs [10]. Human-derived SR proteins include SF2/ASF, SC35, SRp40, and SRp55, also known as SRSF1, SRSF2, SRSF5, and SRSF6, respectively (Claverie-Martin et al., 2015). ESE Finder 3.0 can recognize ESE motifs from the point of view of SR proteins, and this method can predict the disrupted SR binding protein by variation (Cartegni and Krainer, 2002). The hnRNP protein binds to ESSs, which can inhibit the binding of the spliceosomal complexes to the splicing site and block the combination of SR protein and ESEs (Cartegni et al., 2003). Donor or acceptor splice sites are the most conservative splicing sequence, which is recognized by different components of spliceosome in the process of splicing. Donor and acceptor splice sites were initially recognized by U1 snRNP and U2 auxiliary factor (U2AF), respectively, then formed complexes [13]. Synonymous mutations lead to changes in one or more of the above-mentioned splicing factors, which may lead to mRNA splicing abnormalities (Singh RnandSingh, 2018).
To test the hypothesis that synonymous variants of ATP7B cause abnormal splicing by disrupting authentic splice sites or splicing regulatory elements, we used computational tools and minigene assays to characterize 253 synonymous variants in the Exome Aggregation Consortium (ExAc) database and literature. There were 14 kinds of wild-type plasmids constructed by inserting exons into the splicing vector pSPL3, covering 16 exons (except exons 1, 2, 8, 9, and 21). A total of 85 candidate variants were investigated by minigene assays in vitro, 11 of which were shown to cause abnormal pre-mRNA splicing of ATP7B.
Materials and Methods
Variant Inclusion and Computational Tools
By searching the ExAc database2, which is incorporated into the gnomad database now3, 247 rare synonymous variations of ATP7B gene with allele frequencies less than 0.005 were included. The rare synonymous variations of ATP7B reported in the literature were also reviewed, and finally six more variants were also included. The list of the 253 synonymous variations is shown in Supplementary Table 1. Human Splicing Finder (HSF)4 was used to analyze the effect of these variants on the splicing of pre-mRNA. ESE Finder 3.05 was further applied to analyze variations that were predicted to affect ESEs (threshold is SRSF1: 1.956, SRSF2: 2.383, SRSF5: 2.67, SRSF6: 2.676).
Construction of Wild-Type Minigene_ATP7B_ex Plasmids
We used pSPL3 vector, a generous gift from Dr. Irene Bottillo (Sapienza University of Rome, Italy) and Dr. Leping Shao (Qingdao University, China). The exons and flanking introns of the ATP7B gene were amplified by Phanta® Super-Fidelity DNA Polymerase (Vazyme Biotech Co., Ltd., China) with the primers indicated in Supplementary Table 2. The primers used in this step were edited by Primer 5 software, and the corresponding viscous terminal sequence was added to the end. pSPL3 vector was cut with XhoI and BamHI (Invitrogen, United States). All of the indicated fragments were cloned into a pSPL3 vector with the XhoI and BamHI using ClonExpressTM II One Step Cloning Kit (Vazyme Biotech Co., Ltd., China). All constructs were sequenced to confirm by Sanger sequencing (TsingKe Co., Ltd., China), and the corresponding exon wild-type plasmids (Minigene ATP7B_ex) were screened. All clones were functionally checked in 293T cells (Cell Bank of the Chinese Academy of Sciences, China., Cat# GNHu17), and 14 kinds of wild-type plasmids were constructed by inserting exons into the splicing vector pSPL3, covering 16 exons (except exons 1, 2, 8, 9, and 21). The pattern for constructing the plasmids can be found in Supplementary Figure 1. The sequencing data of the RT-PCR products of the 14 kinds of wild-type plasmids which covered 16 exons of ATP7B are shown in Supplementary Figure 2.
Site-Directed Mutagenesis
Based on the construction of wild-type Minigene ATP7B_ex, various mutant Minigene ATP7B_ex capture plasmids were constructed by PCR-mediated in vitro site-directed mutagenesis using Mut Express MultiS Fast Mutagenesis Kit V2 (Vazyme Biotech Co., Ltd., China). The design of the 85 candidate DNA site-directed mutagenesis primers for synonymous mutations is shown in Supplementary Table 3. The primers used in this step were edited by Primer 5 software. The corresponding wild-type Minigene_ATP7B_ex was amplified by PCR, with the primers shown in Supplementary Table 3, to form a linear mutant plasmid and then reorganized. The gene sequences were confirmed by direct sequencing.
Transfection of Eukaryotic Cells and RT-PCR of Minigenes
293T cells were seeded in 12-well plates and grown in 1 ml Dulbecco’s Modified Eagle’s Medium (10% fetal bovine serum). After the cells were 50–70% confluent, 293T cells were transfected with 1 μg of DNA (containing either wild-type or mutant Minigene ATP7B_ex) using Lipofectamine 2000 (Invitrogen, America). At 24 h after transient transfection, total RNA was extracted from cells using TRIzol (Takara, Japan). The first cDNA strand was reversely transcribed using the Superscript III Reverse Transcriptase (Invitrogen, America) together with random hexanucleotide primers. Then, 2 μl of each cDNA was amplified by TaKaRa Ex Taq (Takara, Japan), with primers located in the two cassette exons of the pSPL3 vector. The primer sequences were as follows: forward SD 5′-TCTGAGTCACCTGGACA ACC-3′ and reverse SA 5′-ATCTCAGTGGTATTTGTGAGC-3′. The PCR products were analyzed by agarose gel electrophoresis and proven by sequencing of the extracted DNA. Gel containing 1.5% agarose in Tris-acetate-EDTA buffer was prepared and applied. The marker used in the agarose gels was DL2000 Plus DNA Marker (Vazyme, China). The ratio of normal and abnormal transcripts was analyzed by grayscale value, and the experiment was repeated four times in each group. The mean value was obtained.
Results
Computational Analysis of Synonymous Variations in ATP7B
A total of 253 rare synonymous variants that presented in ExAc database or were previously reported in literature were included in this study (El-Mougy et al., 2014; Dong et al., 2016; Qian et al., 2019). The allele frequencies of these variants are all less than 0.005 and not yet functionally investigated. To select possible spliceogenic variants, we firstly prioritized the variants using a computational algorithm HSF. Based on the predicted results of HSF, 46 variants were likely to create ESSs motifs, and 21 variants would affect donor or acceptor splice sites. What is more, 140 variants were likely to disrupt the ESEs. Previous studies showed that ESE variants that affected the binding of two or more SR proteins are more probable to produce splicing disruption (Takeuchi et al., 2015). Therefore, further prediction using ESE Finder 3.0 was carried out for these 140 variants, of which 18 variants that predicted to disrupt two or more SR proteins were screened in minigene assays (Supplementary Table 2). A flow chart in Figure 1 shows the two-step screening of all synonymous variants.
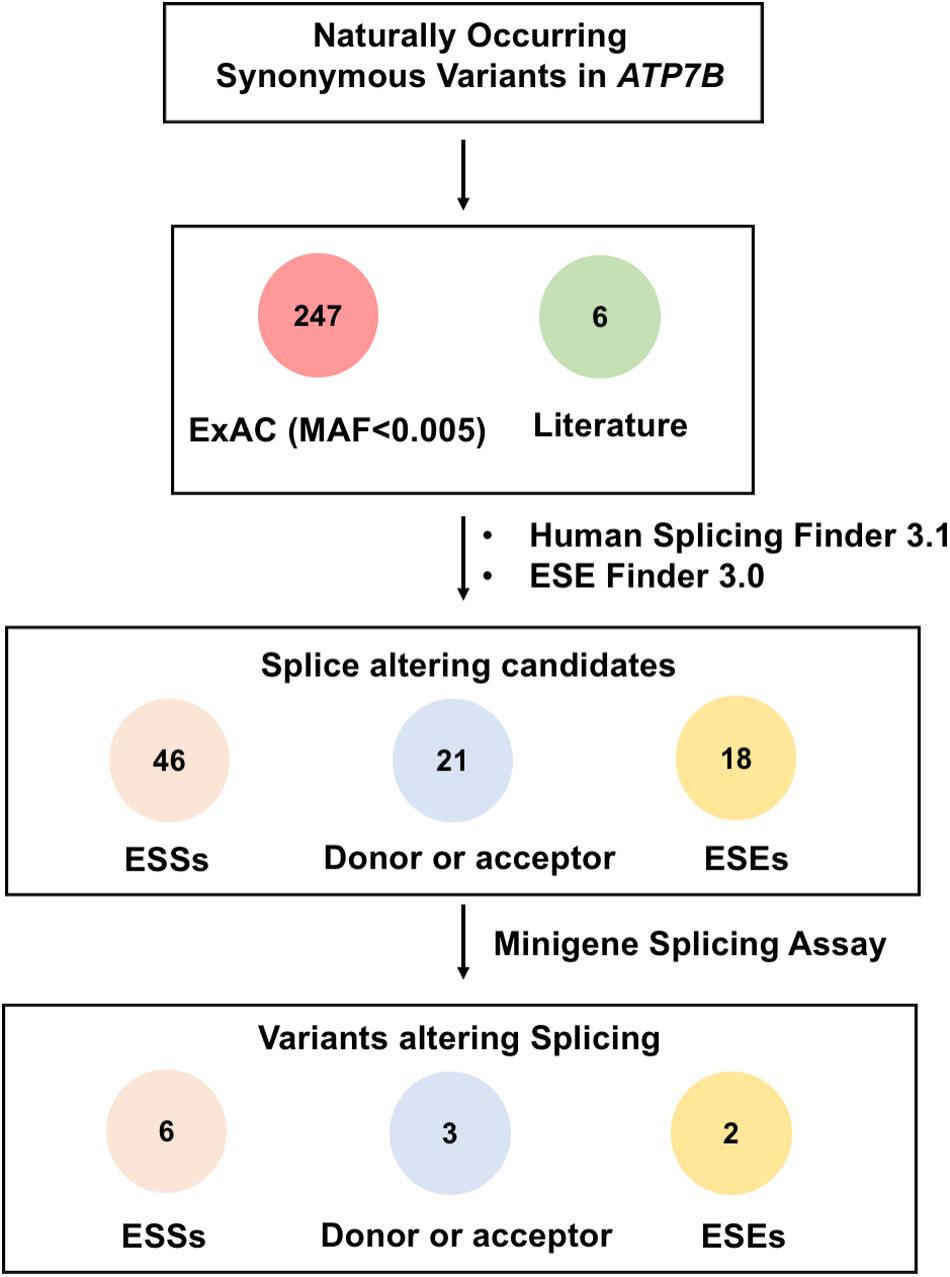
Figure 1. Schematic overview of prioritization and functional assessment of 253 naturally occurring synonymous variations in ATP7B gene. A total of 253 rare synonymous variants that presented in ExAc database or previously reported in literature were included in this study. Based on the predicted results of Human Splicing Finder 3.1 and ESE Finder 3.0, 46 variants were predicted to create ESS motifs, 21 variants would affect donor or acceptor splice sites, and 18 variants were likely to disrupt the binding of ESEs with two or more SR proteins. Then, all 85 candidate variants were screened by minigene splicing assays. As a result, 11 variants were confirmed to disrupt the splicing of pre-mRNA: Six variants were predicted to create ESS motifs, three variants would affect donor or acceptor splice sites, and two variants were predicted to disrupt ESE motifs.
Minigene Assays of 85 Synonymous Variants in ATP7B
We constructed 14 different wild-type Minigene_ATP7B_ex plasmids, including 16 exons of ATP7B gene. According to the results of computational calculation, we divided the 85 synonymous mutations into the following three groups for Minigene Assays: (1) 18 variants that may destroy a variety of ESEs, (2) 46 variants that may produce new ESSs, and (3) 21 variants that may affect donor or acceptor site splicing. Taken together, 85 variants were introduced into their corresponding Minigene_ATP7B_ex and assayed in 293T cells. As a result, 11 variants were confirmed to disrupt the splicing of pre-mRNA, which were divided into three groups as described below. The results of the computational calculation and minigene assays of the 11 variants are detailed in Table 1.
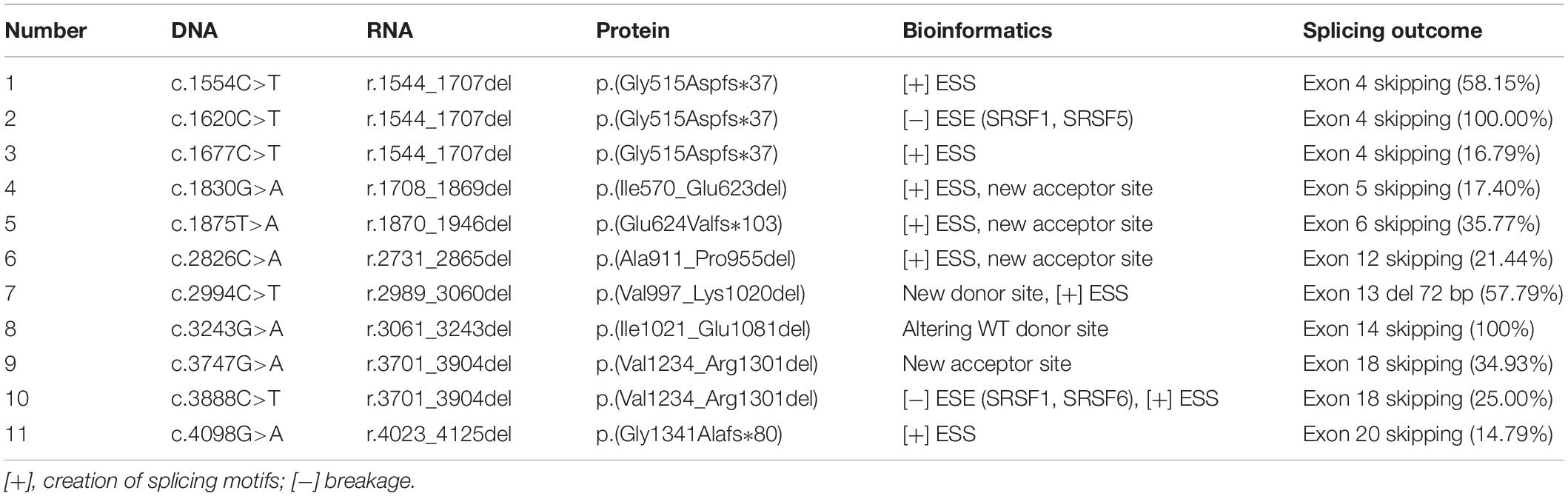
Table 1. Computational prediction and splicing outcome of 11 synonymous variants in the ATP7B gene.
Evaluation of 18 Synonymous Variants Predicted to Eliminate ESE Motif
There were 18 selected synonymous variations verified by in vitro minigene assays. Our results showed that c.1620C>T and c.3888C>T induced skipping of exons 4 (100%) and 18 (25%), respectively (Figure 2). Figure 2A shows the exact location of these two variants in the ATP7B gene. ESE Finder 3.0 predicted that the c.1620C>T would break ESEs binding to SRSF1 and SRSF5, and the c.3888C>T would disrupt the ESEs that bind to SRSF1 and SRSF6 (Figure 2B). Agarose electrophoresis and Sanger sequencing of RT-PCR products confirmed that the c.1620C>T produced a band consistent with a transcript that only contains vector exons SD and SA, and c.3888C>T produced the expected full-length transcript (656 nt) with exons 18 and 19 and a 453-nt transcript with exon 18 skipping (25%) (Figures 2C,D). In four repeated experiments, using grayscale value analysis, the c.1620C>T resulted in exon 4 (164 bp) being skipped completely, and the c.3888C>T increased the proportion of abnormal splicing by 25% compared with the wild type (Figure 2C).
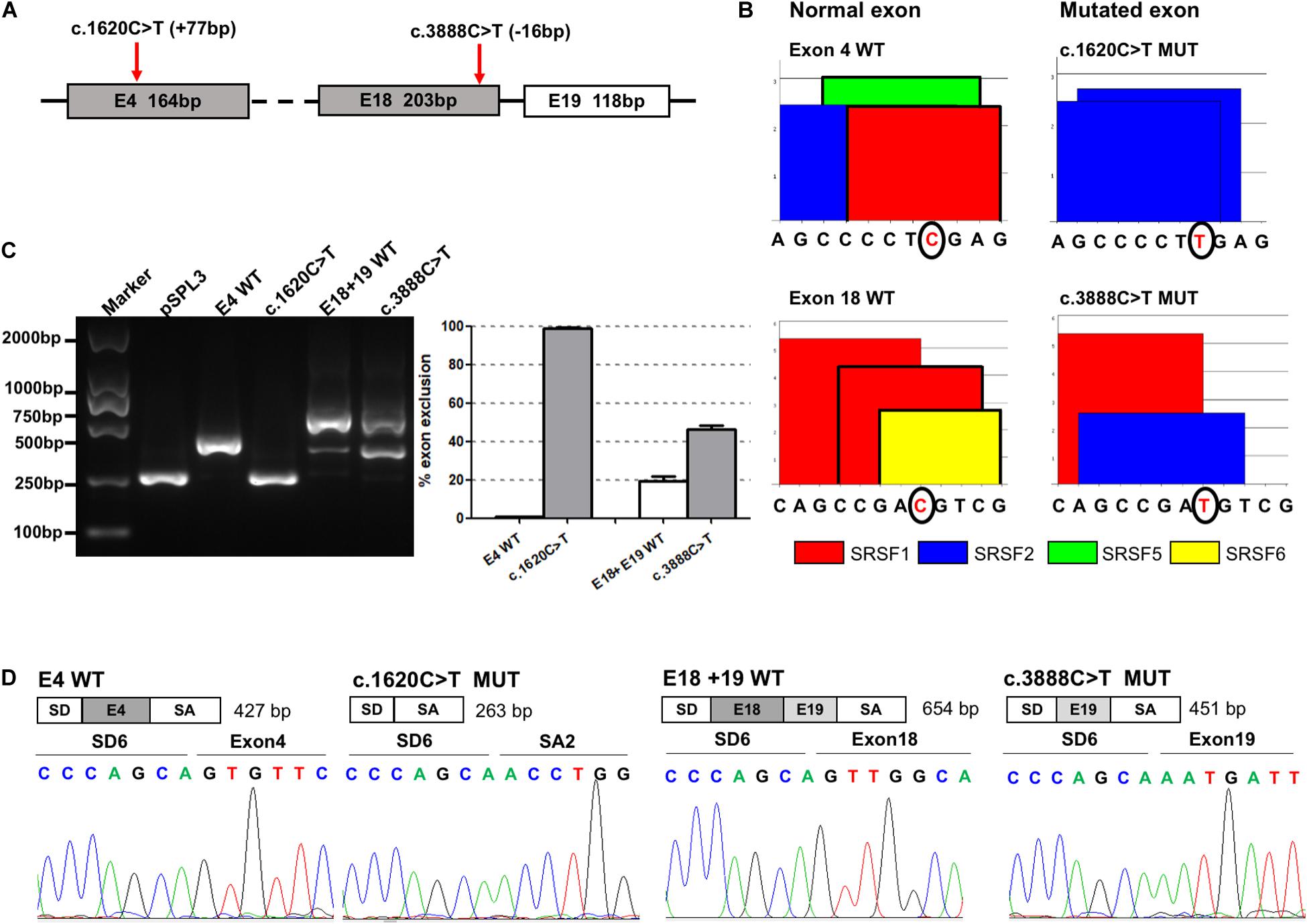
Figure 2. The effect of variants c.1620C>T and c.3888C>T on ATP7B pre-mRNA splicing. (A) The exonic localization of the c.1620C>T and c.3888C>T variants in the ATP7B gene. (B) ESE Finder 3.0 predicted that c.1620C>T would destroy ESEs binding to SRSF1 and SRSF5 and c.3888C>T would destroy ESEs binding to SRSF1 and SRSF6. (C) Agarose electrophoresis of RT-PCR products is shown on the left and the grayscale value analysis on the right. The RT-PCR results shows that the c.1620C>T mutant resulted in exon 4 (164 bp) being skipped completely, and the c.3888C>T mutant increased the proportion of abnormal splicing by 25% compared with the wild type. All data are represented as mean ± SEM (n = 4). Quantification of the aberrant splicing percentage in the graph was calculated by densitometric analyses of the gel-fractionated RT-PCR products as the percentage of exclusion (%) = [MUT band/(MUT band + WT band)] × 100. (D) Sequencing chromatograms of the RT-PCR products amplified from the wild type and mutants. WT, wild type; MUT, mutant.
Evaluation of 46 Synonymous Variants Predicted to Create ESS Motif
The 46 candidate variants in the second group were verified by in vitro minigene assays. The results indicated that six variants (c.1554C>T, c.1677C>T, c.1830G>A, c.1875T>A, c.2826C>A, and c.4098G>A) affected the pre-mRNA splicing of ATP7B (Figure 3). The specific locations of the six variants in the ATP7B gene are shown in Figure 3A, in which the c.1554C>T and c.1875T>A variants are located within 15 bp downstream of the exon and intron boundary. The electrophoretic results showed that these variants produced a mutant band with a transcript that only contains vector exons SD and SA (Figure 3B), which were all confirmed by Sanger sequencing of the RT-PCR products (Supplementary Figure 2). The grayscale value analysis showed that these variations produced more abnormal splicing compared with the wild type; c.1554C>T increased the proportion of abnormal splicing by 58.15%, c.1677C>T increased by 16.79%, c.1830G>A increased by 17.40%, c.1875T>A increased by 35.77%, c.2826C>A increased by 21.44%, and c.4098G>A increased by 14.79% (Figure 3C).
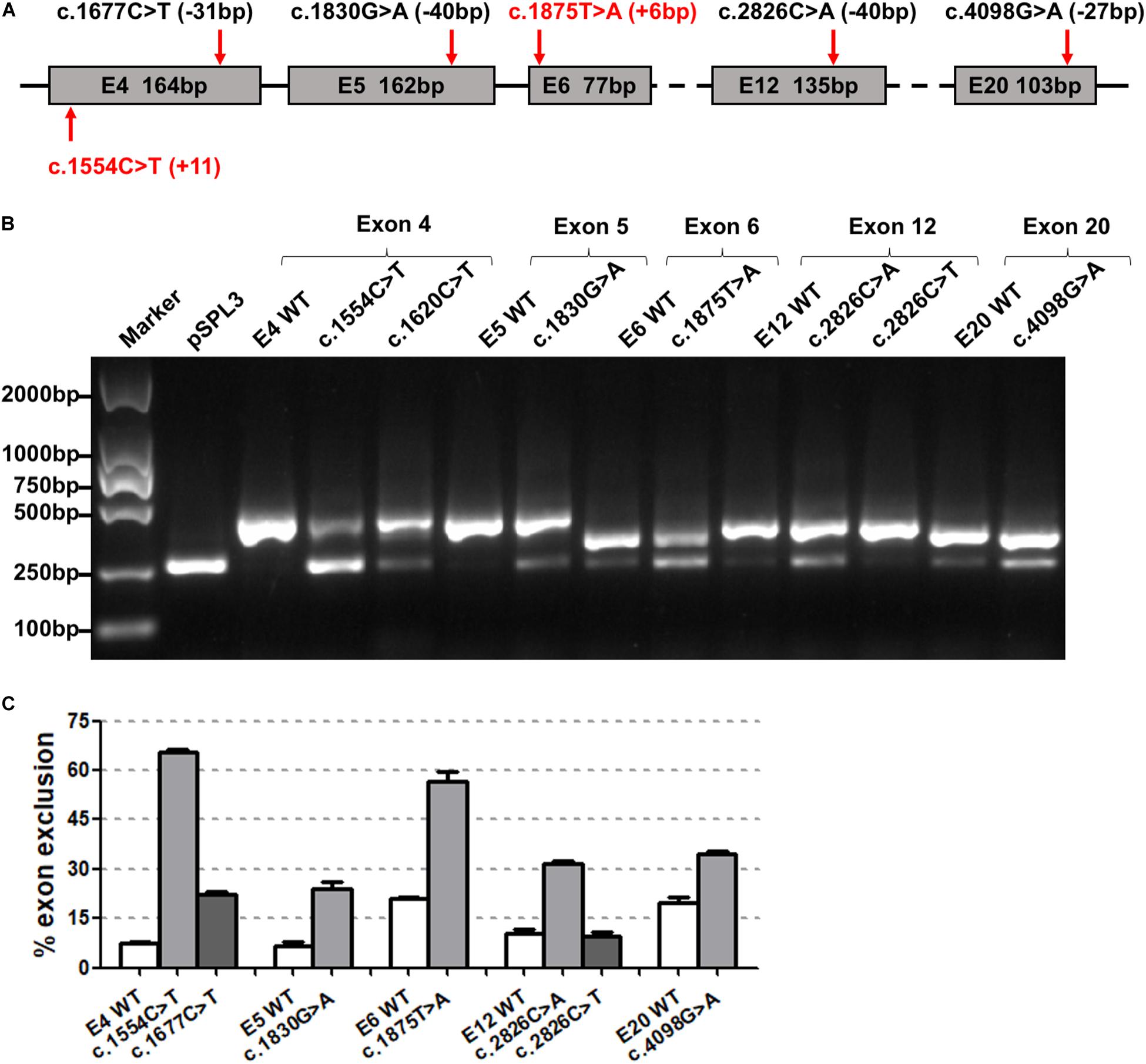
Figure 3. The effect of six synonymous variants predicted to create ESS motif on ATP7B pre-mRNA splicing. (A) The exonic localization of the six synonymous variants in the ATP7B gene. Variants marked in red are close to the junction of the intron and exon. (B) Agarose electrophoretic results of RT-PCR products of the six variants (c.1554C>T, c.1677C>T, c.1830G>A, c.1875T>A, c.2826C>A, and c.4098G>A) and corresponding wild types. (C) The grayscale value analysis shows that these six variations produced more abnormal splicing compared with the wild type: c.1554C>T increased the proportion of abnormal splicing by 58.15%, c.1677C>T increased by 16.79%, c.1830G>A increased by 17.40%, c.1875T>A increased by 35.77%, c.2826C>A increased by 21.44%, and c.4098G>A increased by 14.79%. All data are represented as mean ± SEM (n = 4). Quantification of the aberrant splicing percentage in the graph was calculated by densitometric analyses of the gel-fractionated RT-PCR products as the percentage of exclusion (%) = [MUT band/(MUT band + WT band)] × 100. WT, wild type; MUT, mutant.
The two variants (c.1554C>T and c.1677C>T) were both located in exon 4; the c.1554C>T nearby the boundary of the exon (+11 bp) and intron had a greater percentage of exon skipping. c.2826C>A and c.2826C>T were in the same position (c.2826), but abnormal splicing was observed only when cytosine (C) was mutated into adenine (A).
Evaluation of 21 Synonymous Variants Predicted to Affect Donor or Acceptor Sites
The minigene assays showed that three variants—c.2994C>T, c.3243G>A, and c.3747G>A—in the third group affected pre-mRNA splicing in vitro (Figure 4). The specific locations of the three variants in the ATP7B gene are detailed in Figure 4A. The c.2994C>T in exon 13 was predicted to activate an exonic cryptic donor site. The minigene assay showed that the c.2994C>T mutant produced an abnormal transcript which excluded 72 bp of exon 13 at the 3′ end, with a percentage of 57.79% to total transcripts (Figures 4B–D). c.3243G>A was located in a distinctive position, the last base of exon 14, and it was predicted probably to change canonical donor site (Figure 4A). The minigene verification showed that the variant led to the complete skipping of exon 14 (Figures 4B–D). c.3747G>A was selected because it predictably generated a new acceptor site, which unexpectedly showed exon 18 skipping in the minigene assay (Figures 4B–D). These results indicated that the c.3747G>A variant might disrupt the pre-mRNA splicing by altering some exonic splicing regulatory elements rather than creating a new acceptor site.
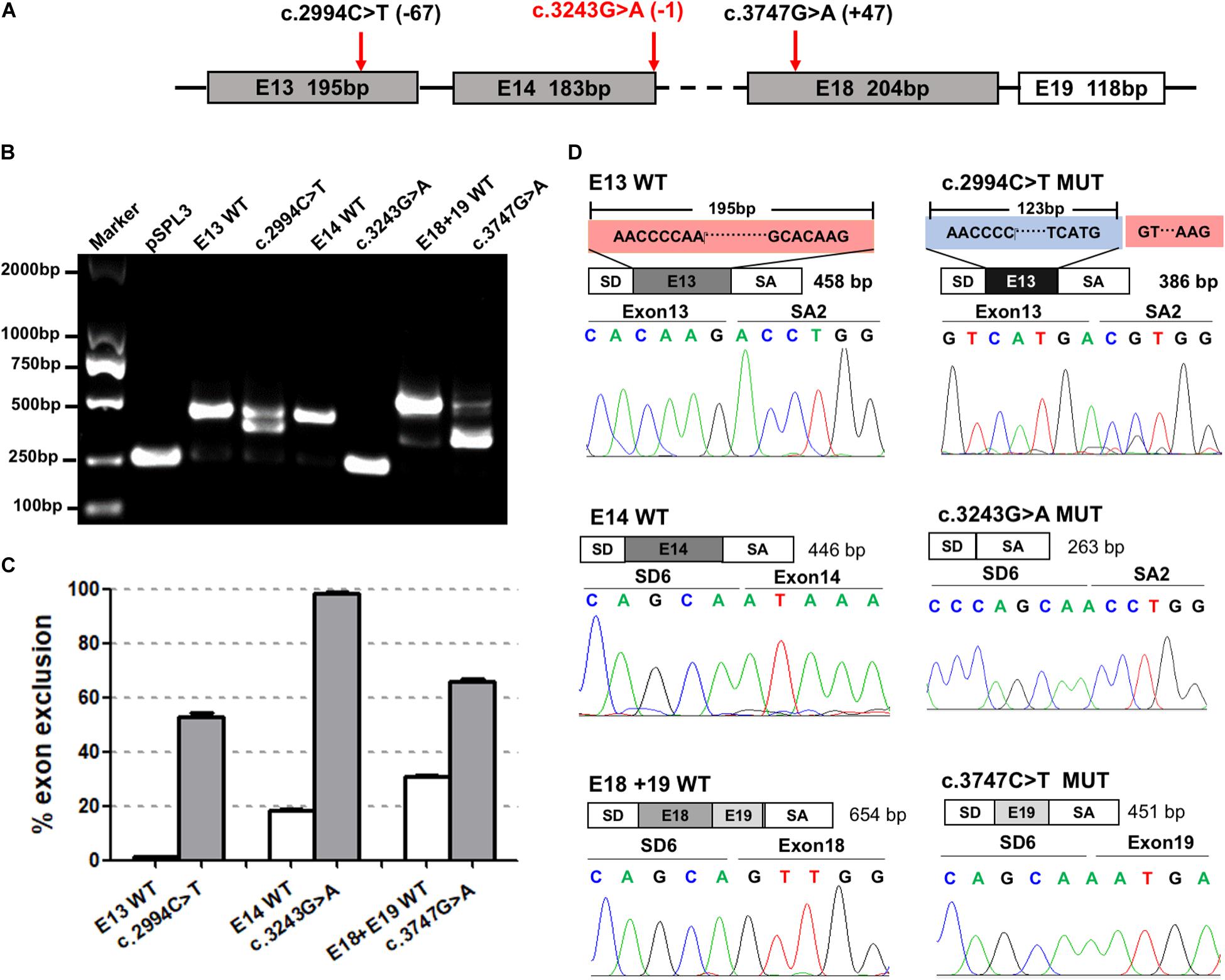
Figure 4. The effect of variants c.2994C>T, c.3243G>A, and c.3747G>A predicted to affect authentic donor or acceptor sites on ATP7B pre-mRNA splicing. (A) The exonic position of c.2994C>T, c.3243G>A, and c.3747G>A in the ATP7B gene. Variants marked in red are close to the junction of the intron and exon. (B) Agarose electrophoretic results of the RT-PCR products of c.2994C>T, c.3243G>A, c.3747G>A, and corresponding wild types. The RT-PCR band of the c.2994C>T mutant was ∼100 bp lower than the corresponding E13-WT band. The RT-PCR band of the c.3243G>A mutant was the same as the pSPL3 vector which only contains vector exons SD and SA. The RT-PCR band of the c.3747G>A WT produced the expected full-length transcript (656 bp) with exons 18 and 19, while the mutant showed one more minor band with a ∼450-bp length. (C) Grayscale value analysis shows that the c.2994C>T mutant produced an abnormal transcript with a percentage of 57.79% to total transcripts. The c.3243G>A mutant produced a complete exon 14 skipping (100%). The c.3747G>A mutant increased the exon 18 skipping by 34.93% compared with the wild type. All data are represented as mean ± SEM (n = 4). Quantification of the aberrant splicing percentage in the graph was calculated by densitometric analyses of the gel-fractionated RT-PCR products as the percentage of exclusion (%) = [MUT band/(MUT band + WT band)] × 100. (D) Sequencing chromatograms of the RT-PCR products amplified from the wild type and mutants. The sequencing of the c.2994C>T mutant band showed exon 13 was excluded (72 bp) compared with the 195-bp wildtype exon 13. The sequencing of the c.3243G>A and c.3747G>A mutants confirmed the skipping of exon 14 and exon 18, respectively. WT, wild type; MUT, mutant.
Discussion
Next-generation sequencing has become the most effective tool for the diagnosis of WD, and more and more variants with uncertain clinical significance have been detected. Evaluating the pathogenicity of these uncertain variants is a great challenge to genetic diagnosis. Particularly, it is more difficult to assess synonymous mutations that do not change amino acids (Tiulpakov et al., 2020). Our previous study showed that the synonymous mutation c. 4014T>A in WD patient partially caused exon 19 exclusion in ATP7B pre-mRNA splicing (Wang et al., 2018). Most synonymous mutations are non-pathogenic mutations, but synonymous changes that affect splicing could lead to severe clinical manifestations (Byrne et al., 2009; Diao et al., 2014). In this study, the effect of synonymous variants in the ATP7B gene on pre-mRNA splicing was comprehensively analyzed in large quantities for the first time. A total of 253 rare synonymous variants were predicted by bioinformatics, and 85 variants were screened for minigene assays. Finally, our results showed that 11 variants had a different degree of impact on the pre-mRNA maturation of ATP7B (Table 1). Especially the three variants [c.1620C>T (Dong et al., 2016), c.1875T>A (Qian et al., 2019), and c.3243G>A (Qian et al., 2019)] have been reported in WD patients, which had not been functionally investigated. Our experimental results show that these three variants hinder the normal pre-mRNA splicing, in which c.1620C>T and c.3243G>A mutations completely caused exon skipping, and c.1875T>A increased abnormal splicing by 35%. All the three variants yielded a deleterious prediction score on disrupting authentic splice sites by two algorithms (HSF and NN Splicing).
Alterations in pre-mRNA splicing levels play a significant role in the development of human genetic diseases, including monogenic diseases as well as complex diseases. Studies have shown that mutations that bring about splicing dysfunction may account for 50% of all known mutations (Baralle and Buratti, 2017). The splicing process is strictly regulated and controlled by a series of proteins and SREs. The identification of splice sites depends on the conservative sequences around splice sites in introns and exons. Specific genetic variations can alter sequences such as splice sites, branching sites, or SREs, generating abnormal splicing (Wheway et al., 2019). The occurrence of these mutations may result in the abnormal use of 3′ or 5′ splice sites and may also lead to the abnormal inclusion of intron or exon skipping, resulting in the translation of proteins with abnormal amounts of amino acids (Anna and Monika, 2018).
ATP7B contains domains common to all P-type ATPases, including the six copper binding domains, the transmembrane domain with eight membrane spanning helices, the actuator domain, the phosphorylation domain, and the nucleotide-binding domain (Lutsenko et al., 2007; Telianidis et al., 2013). Prediction based on these functional domains indicated that complete skipping of exons 4, 6, and 20 will lead to early termination of translation and destructive changes in the ATP7B protein. The skip of exons 5, 12, and 14 would lead to the loss of the copper binding domain, the transmembrane region 5, and the phosphorylation domain, respectively. The truncation of exon 13 caused by c.2994C>T would lead to the deletion of amino acids at positions 1,997 to 1,020, which is close to the sixth transmembrane helix (including the copper binding sites). Previously, Wan et al. (2010) showed that the biological activity of the ATP7B mutant lacking exon 12 was 20% lower than that of wild-type ATP7B in in vitro studies, which might be responsible for a relatively milder disease condition and later onset age in WD patients. Further functional studies are needed to elucidate the deleteriousness of the absence of these functional domains caused by splicing variants.
The accuracy of bioinformatics analysis is limited. The use of a variety of bioinformatics methods for assessment combined with in vitro experimental studies can effectively increase the accuracy. RT-PCR with the RNA of carriers can directly reflect the effects of variants on splicing, but it is difficult to obtain samples from target tissues regarding the genes with low expression in peripheral blood and some exons are also too long to be amplified by RT-PCR (Bryksin and Matsumura, 2010). A large number of studies have shown that a minigene (pSPL3) can simulate exon splicing in vitro and can be used to analyze the splicing effect (Byrne et al., 2009; Acedo et al., 2015; Dal Mas et al., 2015; Takeuchi et al., 2015; Wang et al., 2018). In our previous study, we demonstrated that the ATP7B RT-PCR results from the peripheral blood of affected individuals were consistent with those of minigene assays in vitro, suggesting that a minigene assay can be used to verify the splicing of the ATP7B gene (Wang et al., 2018). We resoundingly constructed 14 wild-type Minigene_ATP7B_ex in this study. The corresponding wild-type plasmids of the first and the last exon (exon 1 and exon 21) were not constructed. Moreover, exon 2 and exons 8 and 9 could not be simulated successfully in the pSPL3 system, which may be due to the existence of key splicing regulatory elements in the deep intron region (Garanto et al., 2019).
Several limitations of this study should be mentioned. Firstly, splicing is a very complex process, which can vary between different tissues and different cell types. Thus, human samples from the tissues of interest are always the best way to evaluate the splicing effect of variants. However, the ATP7B gene is mainly expressed in hepatocytes, and it is difficult to obtain liver tissue in vivo. What is more, the frameshift mutations could trigger nonsense-mediate decay (NMD)-driven degradation of the mutant mRNA (Zarraga et al., 2011; Gong and Zhou, 2018). NMD assay was not conducted in our study. Further studies are required to determine whether the abnormal mRNA produced by these synonymous variants in ATP7B is subject to NMD.
Conclusion
In conclusion, we prioritized 253 naturally occurring rare ATP7B synonymous variants by computational tools and subsequent functional assessment using a minigene splicing assay, which provides evidences that 11 variants alter pre-mRNA splicing in vitro. However, the specific mechanism underlying these splicing disruptions needs to be further studied. We expect that the application of these methodologies could provide reliable experimental basis for genetic diagnosis and genetic counseling of WD patients with these synonymous variations.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Author Contributions
XZ and WZ performed bioinformatics predictions and minigene assays and were major contributors in writing the manuscript. CW and LW screened the candidate variations. ZJ revised the manuscript. YJ, ZL, and BZ designed the study, guided the writing of the manuscript, and critically reviewed the manuscript. All the authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (nos. 81873542 and 81600405).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Irene Bottillo of Sapienza University of Rome and Dr. Leping Shao of Qingdao University for offering us the pSPL3 vector.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.592611/full#supplementary-material
Supplementary Figure 1 | A schematic representation of the pSPL3 vector.
Supplementary Figure 2 | Sanger sequencing results of the RT-PCR products of the 14 successfully constructed wild-type Minigene_ATP7B_ex plasmids.
Supplementary Table 1 | Basic information of 253 synonymous variants in the ATP7B gene.
Supplementary Table 2 | Primers for amplification and cloning of fragments containing individual exons.
Supplementary Table 3 | Primers for site-directed mutations.
Footnotes
- ^ http://www.hgmd.cf.ac.uk/ac/index.php
- ^ http://exac.broadinstitute.org
- ^ https://gnomad.broadinstitute.org/
- ^ http://www.umd.be/HSF/
- ^ http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi
References
Acedo, A., Hernández-Moro, C., Curiel-García, Á, Díez-Gómez, B., and Velasco, E. A. (2015). Functional classification of BRCA2 DNA variants by splicing assays in a large minigene with 9 exons. Hum. Mutat. 36, 210–221. doi: 10.1002/humu.22725
Anna, A., and Monika, G. (2018). Splicing mutations in human genetic disorders: examples, detection, and confirmation. J. Appl. Genet. 59, 253–268. doi: 10.1007/s13353-018-0444-7
Balashova, M. S., Tuluzanovskaya, I. G., Glotov, O. S., Glotov, A. S., Barbitoff, Y. A., Fedyakov, M. A., et al. (2019). The spectrum of pathogenic variants of the ATP7B gene in Wilson disease in the Russian Federation. J. Trace Elem. Med. Biol. 59, 126420. doi: 10.1016/j.jtemb.2019.126420
Baralle, D., and Buratti, E. (2017). RNA splicing in human disease and in the clinic. Clin. Sci. 131, 355–368. doi: 10.1042/cs20160211
Bryksin, A. V., and Matsumura, I. (2010). Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques 48, 463–465. doi: 10.2144/000113418
Byrne, J. A., Strautnieks, S. S., Ihrke, G., Pagani, F., Knisely, A. S., Linton, K. J., et al. (2009). Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology 49, 553–567. doi: 10.1002/hep.22683
Cartegni, L., and Krainer, A. R. (2002). Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 30, 377–384. doi: 10.1038/ng854
Cartegni, L., Wang, J., Zhu, Z., Zhang, M. Q., and Krainer, A. R. (2003). ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 31, 3568–3571. doi: 10.1093/nar/gkg616
Claverie-Martin, F., Gonzalez-Paredes, F. J., and Ramos-Trujillo, E. (2015). Splicing defects caused by exonic mutations in PKD1 as a new mechanism of pathogenesis in autosomal dominant polycystic kidney disease. RNA Biol. 12, 369–374. doi: 10.1080/15476286.2015.1014291
Dal Mas, A., Fortugno, P., Donadon, I., Levati, L., Castiglia, D., and Pagani, F. (2015). Exon-specific U1s correct SPINK5 Exon 11 skipping caused by a synonymous substitution that affects a bifunctional splicing regulatory. Elem. Hum. Mutat. 36, 504–512. doi: 10.1002/humu.22762
Diao, S. Q., Hong, M. F., Huang, Y. Q., Wei, Z. S., Su, Q. X., Peng, Z. X., et al. (2014). Identification and characterization of a novel splice-site mutation in the Wilson disease gene. J. Neurol. Sci. 345, 154–158. doi: 10.1016/j.jns.2014.07.031
Dong, Y., Ni, W., Chen, W. J., Wan, B., Zhao, G. X., Shi, Z. Q., et al. (2016). Spectrum and classification of ATP7B variants in a large cohort of chinese patients with Wilson’s disease guides genetic diagnosis. Theranostics 6, 638–649. doi: 10.7150/thno.14596
El-Mougy, F. A., Sharaf, S. A., Elsharkawy, M. M., Mandour, I. A., El-Essawy, R. A., Eldin, A. M., et al. (2014). Gene mutations in Wilson disease in Egyptian children: report on two novel mutations. Arab. J. Gastroenterol. 15, 114–118. doi: 10.1016/j.ajg.2014.10.005
Garanto, A., Duijkers, L., Tomkiewicz, T. Z., and Collin, R. W. J. (2019). ABCA4 antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic variant c.4539+2001G>A in Stargardt disease. Genes 10, 452. doi: 10.3390/genes10060452
Gong, Q., and Zhou, Z. (2018). Nonsense-Mediated mRNA Decay of hERG Mutations in Long QT Syndrome. Methods Mol. Biol. 1684, 37–49. doi: 10.1007/978-1-4939-7362-0_4
Hedera, P. (2019). Wilson’s disease: a master of disguise. Park. Relat. Disord. 59, 140–145. doi: 10.1016/j.parkreldis.2019.02.016
Li, H., Tao, R., Liu, L., and Shang, S. (2019). Population screening and diagnostic strategies in screening family members of Wilson’s disease patients. Ann. Transl. Med. 7, S59. doi: 10.21037/atm.2019.03.54
Lutsenko, S., Barnes, N. L., Bartee, M. Y., and Dmitriev, O. Y. (2007). Function and regulation of human copper-transporting ATPases. Physiol. Rev. 87, 1011–1046. doi: 10.1152/physrev.00004.2006
Qian, Z., Cui, X., Huang, Y., Liu, Y., Li, N., Zheng, S., et al. (2019). Novel mutations found in the ATP7B gene in Chinese patients with Wilson’s disease. Mol. Genet. Genomic Med. 7, e649. doi: 10.1002/mgg3.649
Sandahl, T. D., Laursen, T. L., Munk, D. E., Vilstrup, H., Weiss, K. H., and Ott, P. (2019). The prevalence of Wilson disease. An update. Hepatology 71, 722–732. doi: 10.1002/hep.30911
Singh RnandSingh, N. N. (2018). Mechanism of splicing regulation of spinal muscular atrophy genes. Adv. Neurobiol. 20, 31–61. doi: 10.1007/978-3-319-89689-2_2
Takeuchi, Y., Mishima, E., Shima, H., Akiyama, Y., Suzuki, C., Suzuki, T., et al. (2015). Exonic mutations in the SLC12A3 gene cause exon skipping and premature termination in Gitelman syndrome. J. Am. Soc. Nephrol. 26, 271–279. doi: 10.1681/asn.2013091013
Telianidis, J., Hung, Y. H., Materia, S., and Fontaine, S. L. (2013). Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front. Aging Neurosci. 51:44. doi: 10.3389/fnagi.2013.00044
Tiulpakov, A., Zubkova, N., Makretskaya, N., Krasnova, T. S., Melnikova, A. I., Fedyaeva, A. S., et al. (2020). Minigene splicing assessment of 20 novel synonymous and intronic glucokinase gene variants identified in patients with maturity-onset diabetes of the young. Hum. Mutat. 41, 129–132. doi: 10.1002/humu.23919
Walshe, J. M. (2017). History of Wilson disease: a personal account. Handb. Clin. Neurol. 142, 1–5. doi: 10.1016/b978-0-444-63625-6.00001-x
Wan, L., Tsai, C. H., Hsu, C. M., Huang, C. C., Yang, C. C., Liao, C. C., et al. (2010). Mutation analysis and characterization of alternative splice variants of the Wilson disease gene ATP7B. Hepatology. 52, 1662–1670. doi: 10.1002/hep.23865
Wang, C., Zhou, W., Huang, Y., Yin, H., Jin, Y., Jia, Z., et al. (2018). Presumed missense and synonymous mutations in ATP7B gene cause exon skipping in Wilson disease. Liver Int. 38, 1504–1513.
Wheway, G., Lord, J., and Baralle, D. (2019). Splicing in the pathogenesis, diagnosis and treatment of ciliopathies. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 194433. doi: 10.1016/j.bbagrm.2019.194433
Xie, J. J., and Wu, Z. Y. (2017). Wilson’s disease in China. Neurosci. Bull. 33, 323–330. doi: 10.1007/s12264-017-0107-4
Keywords: Wilson disease, ATP7B gene, minigene assays, splicing aberrations, synonymous mutations
Citation: Zhou X, Zhou W, Wang C, Wang L, Jin Y, Jia Z, Liu Z and Zheng B (2021) A Comprehensive Analysis and Splicing Characterization of Naturally Occurring Synonymous Variants in the ATP7B Gene. Front. Genet. 11:592611. doi: 10.3389/fgene.2020.592611
Received: 14 August 2020; Accepted: 16 December 2020;
Published: 25 February 2021.
Edited by:
Y-h. Taguchi, Chuo University, JapanReviewed by:
Eugenia Fraile-Bethencourt, Oregon Health and Science University, United StatesElena Ramos-Trujillo, University of La Laguna, Spain
Rusdy Ghazali Malueka, Gadjah Mada University, Indonesia
Copyright © 2021 Zhou, Zhou, Wang, Wang, Jin, Jia, Liu and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bixia Zheng, zbx_88117@163.com; Zhifeng Liu, zfliu@njmu.edu.cn
†These authors have contributed equally to this work