 Chenglin Duan1,2
Chenglin Duan1,2 Jingjing Shi1
Jingjing Shi1 Guozhen Yuan1
Guozhen Yuan1 Xintian Shou1,2
Xintian Shou1,2 Ting Chen1,2
Ting Chen1,2 Xueping Zhu1Yihan Yang1,2
Xueping Zhu1Yihan Yang1,2 Yuanhui Hu1*
Yuanhui Hu1*- 1Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 2Beijing University of Chinese Medicine, Beijing, China
Background: Traditional observational studies have demonstrated an association between heart failure and Alzheimer’s disease. The strengths of observational studies lie in their speed of implementation, cost, and applicability to rare diseases. However, observational studies have several limitations, such as uncontrollable confounders. Therefore, we employed Mendelian randomization of genetic variants to evaluate the causal relationships existing between AD and HF, which can avoid these limitations.
Materials and Methods: A two-sample bidirectional MR analysis was employed. All datasets were results from the UK’s Medical Research Council Integrative Epidemiology Unit genome-wide association study database, and we conducted a series of control steps to select the most suitable single-nucleotide polymorphisms for MR analysis, for which five primary methods are offered. We reversed the functions of exposure and outcomes to explore the causal direction of HF and AD. Sensitivity analysis was used to conduct several tests to avoid heterogeneity and pleiotropic bias in the MR results.
Results: Our MR studies did not support a meaningful causal relationship between AD on HF (MR-Egger, p = 0.634 > 0.05; weighted median (WM), p = 0.337 > 0.05; inverse variance weighted (IVW), p = 0.471 > 0.05; simple mode, p = 0.454 > 0.05; weighted mode, p = 0.401 > 0.05). At the same time, we did not find a significant causal relationship between HF and AD with four of the methods (MR-Egger, p = 0.195 > 0.05; IVW, p = 0.0879 > 0.05; simple mode, p = 0.170 > 0.05; weighted mode, p = 0.110 > 0.05), but the WM method indicated a significant effect of HF on AD (p = 0.025 < 0.05). Because the statistical powers of IVW and MR-Egger are more than that of WM, we think that there is no causal effect of HF on AD. Sensitivity analysis and horizontal pleiotropy were not detected in the MR analysis.
Conclusion: Our results did not provide significant evidence indicating any causal relationships between HF and AD in the European population. Therefore, more large-scale datasets or datasets related to similar factors are expected for further MR analysis.
Introduction
The occurrence of heart failure (HF) and Alzheimer’s disease (AD) may result in a series of severe medical and financial problems for ordinary families and societies in general. The combination of HF and AD raises the cost of health care, and the present study provides sufficient evidence to show that this increase in cost is approximately fourfold if someone suffers from the combination of HF and AD (Chhatre et al., 2009). In addition, we have found increases in rehospitalization rates and mortality in patients who have been affected simultaneously by HF and memory dysfunction (Vogels et al., 2007; Pressler et al., 2010). In 1997, “cardiogenic dementia” was first proposed by researchers (Lancet, 1977). Since then, a growing body of studies have shown that there exists a close relationship between HF and cognitive impairment (Cohen and Mather, 2007). Along with advances in research techniques and the unremitting efforts of clinicians, there is much persuasive evidence for a relationship between HF and AD due to similar risk and genetic factors, including gender, age, and obesity (Kivipelto et al., 2005; Vina and Lloret, 2010; Roger, 2013). Moreover, genetic evidence also suggests that the Apo E4 (apolipoprotein E4) allele and variants in the presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes have close contact in the development of HF and AD (Pang and Baum, 2000; Van Uden et al., 2000; Li et al., 2006; Gianni et al., 2010; Iadecola, 2013). In recent years, there has been a growing belief that HF is one of the risk factors for AD (Qiu et al., 2006). Although researchers have found similar mechanistic pathways for HF and AD, there is a lack of sufficient evidence to estimate the causal interrelationships between HF and AD.
Moreover, the causal relationship between HF and AD based on summary-level and genome-wide association study (GWAS) datasets has not been explored. Therefore, exploring this relationship between HF and AD and the causal direction of its effects is essential for clinicians to pursue. By determining these causal relationships, clinicians can provide a relatively accurate prognosis for patients.
The strength of observational studies is that they are rapid and inexpensive to conduct. Moreover, they play an essential role in exploring new hypotheses for the causal relationships existing between exposure and outcomes, which provides important hypotheses for further studies. Observational studies allow for studies of rare diseases in small sample sizes (Lewallen and Courtright, 1998). However, it is well known that observational studies still have several limitations as regards evaluating causal effects that demand prompt solutions and bias in controlling for confounders (Greenland and Morgenstern, 2001). Moreover, because of the inherent weaknesses of traditional observational studies, the estimated effects of HF and AD are subject to certain deficiencies. Underlying these limitations, compared to observational studies, the use of Mendelian randomization (MR), which employs single-nucleotide polymorphisms (SNPs) as instrumental variables (IVs), is essential in order to overcome the limitations of confounders, reverse causality, and bias in observational studies (Davey Smith and Hemani, 2014; Davies et al., 2018).
MR is a persuasive genetic assessment tool that gives a robust estimate of exposures and outcomes (Davey Smith and Hemani, 2014; Davies et al., 2018) when there is a lack of high-quality randomized controlled trials. In MR analysis, instrumental variables evaluate the causal relationships between exposures and outcomes, which need to meet three critical assumptions: 1) genetic variants are strongly associated with exposure, 2) genetic variants are independent of the confounders of exposure and outcome, and 3) genetic variants are related to outcome only via exposure (Davey Smith and Hemani, 2014; Davies et al., 2018). The MR method uses summary GWAS datasets to estimate the causal relationships between exposure and outcome rather than genetic variants from individuals (Hemani et al., 2018). A bidirectional MR determines the causal direction of the relationship between two sets of independent genetic variants related to two phenotypes (Richmond and Davey Smith, 2019). Therefore, we applied a two-sample bidirectional MR method to analyze the causal relationships between and the causal direction of HF and AD.
Materials and Methods
A two-sample bidirectional MR analysis is shown in Supplementary Figure S1, where panel (A) shows the causal effect of AD traits on HF and panel (B) shows the causal effect of HF traits on AD.
Data Source
Two GWAS datasets were used for the MR analysis. When allele frequencies and exposure or outcome distributions vary substantially between different ethnic groups, population stratification will arise. Population stratification generates an association between exposure and outcome at the summary level of the population (Larsson et al., 2019). Therefore, in order to avoid the bias of population stratification, we can select ethnically homogeneous groups to further the MR analysis. In order to avoid population stratification, only genetic variants derived from European ancestries were employed in the analysis. The IEU GWAS database was developed by the UK Medical Research Council (MRC) Integrative Epidemiology Unit (IEU) at the University of Bristol (Hemani et al., 2018). As of September 2017, the IEU GWAS database contained 1,673 publicly available GWAS datasets, which are constantly being updated and expanded. The datasets have been applied to MR analysis by numerous researchers (Hemani et al., 2018). All database datasets are publicly available for download and use, and our approach relied on summary-level GWAS data to obtain MR estimates. We obtained datasets for HF and AD derived from the IEU GWAS database. The GWAS dataset for AD came from the Alzheimer Disease Genetics Consortium (ADGC), the European Alzheimer’s Disease Initiative (EADI), the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE), and the Genetic and Environmental Risk in AD/Defining Genetic, Polygenic, and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES) and included 63,926 individuals (21,982 cases vs. 41,944 controls) of European ancestry. We also obtained information for 10,528,610 SNPs (Kunkle et al., 2019) (https://gwas.mrcieu.ac.uk). The GWAS dataset for HF was retrieved from the IEU GWAS database, which contained 977,323 individuals of European descent; we also obtained detailed information for 7,773,021 SNPs (Shah et al., 2020) (https://gwas.mrcieu.ac.uk). In order to avoid deviations arising from population stratification, we used only datasets for people with European ancestry.
SNP Selection
Based on the above two datasets, we formulated a series of quality control standards to screen SNPs that were used as IVs. First, to meet assumption 1 (IVs are strongly associated with exposure), we selected IVs that achieved a genome-wide significance level (p < 5 × 10–8) from exposures (AD or HF). Second, linkage disequilibrium (LD) reflects the association between alleles at linked loci, which destroys the randomness of genetic variants (Collins, 2009). In the process of data processing, to meet IV assumptions 2 or 3, LD should be removed. Therefore, some parameters were set to eliminate the genetic variants with possible LD, that is, LD was pruned using the default settings of the clump data function of the TwoSampleMR package (physical distance threshold, 10,000 kb; r2 < 0.001). Third, the PhenoScanner database (Kamat et al., 2019) contains a large amount of data to link SNPs with diseases and phenotypes, and we used the PhenoScanner tool to exclude any of the selected SNPs that were associated with possible mechanistic pathways of outcomes (HF or AD). To test whether the selected SNPs were associated with other risk factors for HF, we input the selected SNPs into the tool one by one (p < 5 × 10–8 indicated that this SNP was strongly associated with phenotypes) and set the rest of the parameters to default values. Last, the above-selected SNPs with a minor allele frequency (MAF) of <0.01 were also not considered in order to avoid potential bias from the original datasets owing to low confidence. If a proxy variant could not be found for a target variant from the study of the outcome, the variant should be excluded. At the same time, one SNP should be eliminated for being palindromic with intermediate allele frequencies when harmonizing exposure and outcome using the harmonise_data function of the TwoSampleMR package. F statistics were also calculated in order to evaluate the strength of association between SNPs and exposure, and we set the threshold for the F statistic to be > 10. One of the SNPs with a value of <10 was deemed to be a “weak instrument” and was ruled out of our MR analysis (Burgess et al., 2017). The F statistic was calculated using the formula F = β2/SE2, where β represents the effect on the risk of exposure and SE is the standard error (Bowden et al., 2016b).
MR Analysis
In the MR analysis, the statistical analysis was performed using R software version 4.1.0 along with the R packages Two samples used are MR and MR-PRESSO (Hemani et al., 2018; Verbanck et al., 2018), and the codes used in our study are publicly available.
Statistical Analysis for MR
The IVW method was implemented as the primary MR method for combining the Wald ratios of the causal effects between exposure and outcome, and it can provide the most accurate estimate for all selected SNPs (Burgess et al., 2013). The IVW method was found to be the most predictable if directional pleiotropy is non-existent. The MR-Egger method provides a causal estimate using the slope of the weighted linear regression against horizontal pleiotropy (Bowden et al., 2016b). The weighted median estimator gives a credible estimate of effect when up to half of the weight is derived from valid IVs (Bowden et al., 2016a). The simple mode and weighted mode can also be applied to MR analysis. The five methods mentioned earlier were used in our MR analysis. In bidirectional MR studies, we reverse the functions of exposure and outcome because exposure and outcome are a two-way street. Therefore, we sought to estimate AD based on the risk of HF. At the same time, we performed a bidirectional MR analysis to determine the causal effect of HF on AD.
Sensitivity Analysis
The existence of heterogeneity among IVs is an indicator of violations of the MR assumptions (Bowden et al., 2017). The IVW and MR-Egger methods were used to estimate the level of heterogeneity among the IVs. Cochran’s Q test and the I2 statistic calculated the level of heterogeneity among the causal estimates for the selected SNPs using the IVW and MR-Egger methods: p < 0.05 and I2 > 50% reflect the existence of significant heterogeneity (Zhang et al., 2015). Furthermore, to perform sensitivity analysis, we created a leave-one-out plot (Burgess and Thompson, 2017) to ensure that the effect was not disproportionately reliant on any particular SNP. If the estimated effect changes very significantly when one of the genetic variants is removed, that genetic variant is an outlier or sensitive, and the MR analysis is repeated. Last, the potentially unbalanced horizontal pleiotropy tests among the genetic variants were performed by means of MR-Egger, MR-PRESSO, and funnel plots. For the MR-Egger intercept, p < 0.05 is an indicator of genetic variants with directional pleiotropy (Burgess and Thompson, 2017). MR-PRESSO detects the potential pleiotropy, which is adequately powered among even a tiny subset of loci (Verbanck et al., 2018). In addition, MR-Egger regression generally had lower precision than the MR-PRESSO outliner test (Verbanck et al., 2018). The tests of sensitivity mentioned above are two-sided in our bidirectional MR analysis.
Results
Effect of AD Traits on HF
In selecting the IVs from the GWAS dataset for AD, we acquired 21 IVs without LD (r2 < 0.001) that were within the physical distance threshold (10,000 kb) and achieved genome-wide significance (p < 5 × 10–8). We used the PhenoScanner tool to rule out five SNPs that were associated with potential pleiotropic effects (rs34665982 and rs12151021 are associated with hemoglobin concentration, rs7412 and rs147711004 with coronary artery disease, and rs9381563 with reticulocytes). Of the SNPs mentioned earlier, we detected two SNPs (rs72654445 and rs139136389) not found in HF within the MAF threshold (>0.01). At the same time, two SNPs (rs11257242 and rs114812713) were removed for being palindromic with intermediate allele frequencies by the TwosampleMR function of the R package. After rigorous calculations, the F statistic for individual SNPs ranged from 31.9 to 466.8, and a weak instrument did not exist. By a series of SNP selection quality control steps, 12 SNPs were selected for further MR analysis (Table 1; Supplementary Figure S2A).

TABLE 1. Detailed information of selected SNPs for MR analysis of the causal effect of AD on HF.
Five methods (MR-Egger, WM, IVW, simple mode, and weighted mode) and 12 significant IVs were employed to analyze the causal effects of AD on HF, which were found to be consistent among the five MR methods, and this result did not suggest a significant association (MR-Egger, p = 0.634 > 0.05; WM, p = 0.337 > 0.05; IVW, p = 0.471 > 0.05; simple mode, p = 0.454 > 0.05; weighted mode, p = 0.401 > 0.05) (Table 2; Supplementary Figure S3).
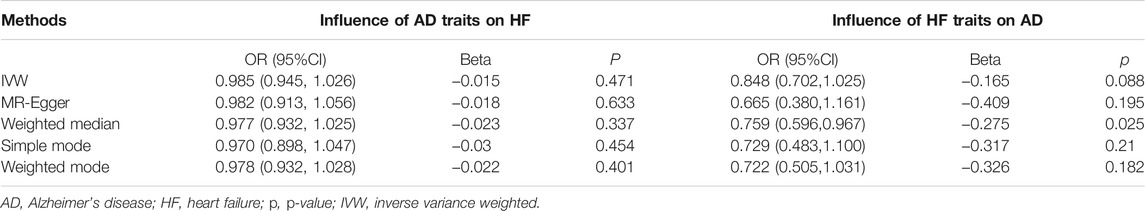
TABLE 2. Bidirectional MR results between HF and AD.
The heterogeneity test did not show significant heterogeneity among the 12 IVs with the MR-Egger and IVW methods (MR-Egger: Q = 16.857, I2 ≈ 35% < 50%; p = 0.776 > 0.05, and IVW: Q = 16.874, I2 ≈ 35% < 50%; p = 0.112 > 0.05) (Table 3). We complemented the leave-one-out cross-validation to calculate the level of sensitivity; the effects were not disproportionately driven by a particular SNP (Supplementary Figure S4). The test for MR-Egger regression was not significant (MR-Egger intercept = 0.0006, p = 0.921 > 0.05), which indicated that the results were not biased by horizontal pleiotropy. A funnel plot was also created to test for the absence of horizontal pleiotropy (Table 3; Supplementary Figure S5). The MR-PRESSO global test and the MR-PRESSO outliner test did not find any heterogeneity or outliners.

TABLE 3. Heterogeneity test and horizontal pleiotropy test.
Effect of HF Traits on AD
In the bidirectional MR analysis, we reversed the location of exposure and outcome and repeated the steps in Effect of AD Traits on HF above. With the LD threshold (r2 < 0.001, kb = 10,000), we reached the level of genome-wide significance (p < 5 × 10–8). A total of 12 HF IVs were employed in this study, which were not associated with potential pleiotropy by the PhenoScanner tool. All the 12 selected IVs were found in HF within the MAF threshold (>0.01). However, three SNPs (rs1556516, rs4746140, and rs4766578) were removed from the study for being palindromic with intermediate allele frequencies. The F statistic for individual SNPs ranged from 30.9 to 83.1 (F statistic >10). Thus, nine SNPs were selected for further analysis (Table 4; Supplementary Figure S2B).

TABLE 4. Detailed information of selected SNPs for MR analysis of the causal effect of HF on AD.
There was no evidence suggesting a significant association between HF traits on AD using the four methods (MR-Egger, p = 0.195 > 0.05; IVW, p = 0.088 > 0.05; simple mode, p = 0.170 > 0.05; weighted mode, p = 0.110 > 0.05). However, the WM method indicated a significant effect of HF on AD (p = 0.025 < 0.05). The IVW and MR-Egger methods were more persuasive than the WM method (Bowden et al., 2016b). Therefore, we believe that HF does not have a causal effect on AD (Table 2; Supplementary Figure S6).
The MR-Egger and IVW methods revealed no heterogeneity among the nine IVs (MR-Egger: Q = 4.192, I2 ≈ −0.90 < 50%, p = 0.757 > 0.05 and IVW: Q = 5.019, I2 ≈ −0.60 < 50%, p = 0.755 > 0.05) (Table 3). We did not find that the effects were driven by any SNPs with the leave-one-out cross-validation (Supplementary Figure S7). As for horizontal pleiotropy, we conducted MR-Egger regression and created a funnel plot (MR-Egger intercept = 0.017, p = 0.393 > 0.05), which did not reveal any significant results (Table 3; Supplementary Figure S8). Moreover, we did not find the presence of heterogeneity and outliners when we used the MR-PRESSO global test and the MR-PRESSO outliner test.
Discussion
To the best of our knowledge, our study is the first to use a two-sample and bidirectional method to determine whether there is a causal relationship between heart failure (HF) and Alzheimer’s disease (AD) in European populations. Although we applied a series of quality control steps in our analysis, the results of our study did not support the hypothesis that there is a meaningful causal relationship between genetically predicted HF and AD because the statistical analysis did not provide sufficiently conclusive results to support the hypothesis. On the one hand, the conclusions of previous observational epidemiological studies suggesting a significant association between HF and AD may have resulted from the presence of uncontrolled biases or cofounders; on the other hand, because of the lack of sufficient GWAS studies of HF and AD, our results need to be carefully explained. In addition, our MR analysis indicates only an average lifetime risk, and it cannot explain whether the existence of HF (or AD) in a particular period has any effect on the level of risk of AD (or HF). Moreover, with regard to the selected IVs for AD or HF used in this study, it is the biological correlation with HF or AD that is important, and the biological actors in this case are still imprecisely known. Moreover, the selected SNPs had limitations and we explored only a small part of the variance in HF and AD, although we used several methods to exclude horizontal pleiotropy and weak instrument bias.
To ascertain the causal relationships between AD and HF, obtaining large-scale GWAS datasets derived from AD or HF may be effective. Moreover, this study included only European populations and not other ethnic groups, so causal relationships between HF and AD may well be found when other ethnicities (African and Asian) are examined. Moreover, in observational studies, there are many similar risks and genetic factors that provide relevant evidence to prove an association between HF and AD, for example, obesity, age, Apo E4, etc. Therefore, in future MR studies, valid results may be obtained if we conduct further MR analysis on large-scale summary-level GWAS datasets of risk factors (age and obesity) or genetic factors derived from Apo E4.
Overall, in this bidirectional MR study, we failed to find persuasive evidence suggesting a causal relationship between genetic liability to AD and HF. In future studies, in order to explore the relationship between HF and AD, high-quality clinical trials and excellent laboratory studies will be indispensable. From the researcher’s point of view, MR is a relatively superior method for understanding the relationships between various diseases (Wootton et al., 2018). We hope that this method will improve the quality of the control steps.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
CD, JS, and GY collected the data, analyzed the data with the R package, and wrote the manuscript. XS, XZ, TC, and YY supervised the process of MR analysis and verified the data. YH provided constructive suggestions during the editing of the manuscript. All authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 82074409).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.772343/full#supplementary-material
References
Bowden, J., Davey Smith, G., Haycock, P. C., and Burgess, S. (2016a). Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 40 (4), 304–314. doi:10.1002/gepi.21965
Bowden, J., Del Greco, M. F., Minelli, C., Davey Smith, G., Sheehan, N. A., and Thompson, J. R. (2016b). Assessing the Suitability of Summary Data for Two-Sample Mendelian Randomization Analyses Using MR-Egger Regression: the Role of the I2 Statistic. Int. J. Epidemiol. 45 (6), 1961–1974. doi:10.1093/ije/dyw220
Bowden, J., Del Greco, M. F., Minelli, C., Davey Smith, G., Sheehan, N., and Thompson, J. (2017). A Framework for the Investigation of Pleiotropy in Two-Sample Summary Data Mendelian Randomization. Stat. Med. 36 (11), 1783–1802. doi:10.1002/sim.7221
Burgess, S., Butterworth, A., and Thompson, S. G. (2013). Mendelian Randomization Analysis with Multiple Genetic Variants Using Summarized Data. Genet. Epidemiol. 37 (7), 658–665. doi:10.1002/gepi.21758
Burgess, S., Small, D. S., and Thompson, S. G. (2017). A Review of Instrumental Variable Estimators for Mendelian Randomization. Stat. Methods Med. Res. 26 (5), 2333–2355. doi:10.1177/0962280215597579
Burgess, S., and Thompson, S. G. (2017). Interpreting Findings from Mendelian Randomization Using the MR-Egger Method. Eur. J. Epidemiol. 32 (5), 377–389. doi:10.1007/s10654-017-0255-x
Chhatre, S., Weiner, M. G., Jayadevappa, R., and Johnson, J. C. (2009). Incremental burden of Congestive Heart Failure Among Elderly with Alzheimer’s. Aging Ment. Health 13 (4), 628–634. doi:10.1080/13607860902774469
Cohen, M. B., and Mather, P. J. (2007). A Review of the Association between Congestive Heart Failure and Cognitive Impairment. Am. J. Geriatr. Cardiol. 16 (3), 171–174. doi:10.1111/j.1076-7460.2007.06563.x
Collins, A. (2009). Allelic Association: Linkage Disequilibrium Structure and Gene Mapping. Mol. Biotechnol. 41 (1), 83–89. doi:10.1007/s12033-008-9110-3
Davey Smith, G., and Hemani, G. (2014). Mendelian Randomization: Genetic Anchors for Causal Inference in Epidemiological Studies. Hum. Mol. Genet. 23 (R1), R89–R98. doi:10.1093/hmg/ddu328
Davies, N. M., Holmes, M. V., and Davey Smith, G. (2018). Reading Mendelian Randomisation Studies: a Guide, Glossary, and Checklist for Clinicians. Bmj 362, k601. doi:10.1136/bmj.k601
Gianni, D., Li, A., Tesco, G., McKay, K. M., Moore, J., Raygor, K., et al. (2010). Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation 121 (10), 1216–1226. doi:10.1161/circulationaha.109.879510
Greenland, S., and Morgenstern, H. (2001). Confounding in Health Research. Annu. Rev. Public Health 22, 189–212. doi:10.1146/annurev.publhealth.22.1.189
Hemani, G., Zheng, J., Elsworth, B., Wade, K. H., Haberland, V., Baird, D., et al. (2018). The MR-Base Platform Supports Systematic Causal Inference across the Human Phenome. Elife 7. doi:10.7554/eLife.34408
Iadecola, C. (2013). The Pathobiology of Vascular Dementia. Neuron 80 (4), 844–866. doi:10.1016/j.neuron.2013.10.008
Kamat, M. A., Blackshaw, J. A., Young, R., Surendran, P., Burgess, S., Danesh, J., et al. (2019). PhenoScanner V2: an Expanded Tool for Searching Human Genotype-Phenotype Associations. Bioinformatics 35 (22), 4851–4853. doi:10.1093/bioinformatics/btz469
Kivipelto, M., Ngandu, T., Fratiglioni, L., Viitanen, M., Kareholt, I., Winblad, B., et al. (2005). Obesity and Vascular Risk Factors at Midlife and the Risk of Dementia and Alzheimer Disease. Arch. Neurol. 62 (10), 1556–1560. doi:10.1001/archneur.62.10.1556
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 51 (3), 414–430. doi:10.1038/s41588-019-0358-2
Larsson, S. C., Michaëlsson, K., and Burgess, S. (2019). Mendelian Randomization in the Bone Field. Bone 126, 51–58. doi:10.1016/j.bone.2018.10.011
Lewallen, S., and Courtright, P. (1998). Epidemiology in Practice: Case-Control Studies. Community Eye Health 11 (28), 57–58.
Li, D., Parks, S. B., Kushner, J. D., Nauman, D., Burgess, D., Ludwigsen, S., et al. (2006). Mutations of Presenilin Genes in Dilated Cardiomyopathy and Heart Failure. Am. J. Hum. Genet. 79 (6), 1030–1039. doi:10.1086/509900
Pang, C. P., and Baum, L. (2000). Lipoproteins and Related Molecules in Alzheimer’s Disease. Microsc. Res. Tech. 50 (4), 259–260. doi:10.1002/1097-0029(20000815)50:4<259:Aid-jemt1>3.0.Co;2-9
Pressler, S. J., Kim, J., Riley, P., Ronis, D. L., and Gradus-Pizlo, I. (2010). Memory Dysfunction, Psychomotor Slowing, and Decreased Executive Function Predict Mortality in Patients with Heart Failure and Low Ejection Fraction. J. Card. Fail. 16 (9), 750–760. doi:10.1016/j.cardfail.2010.04.007
Qiu, C., Winblad, B., Marengoni, A., Klarin, I., Fastbom, J., and Fratiglioni, L. (2006). Heart Failure and Risk of Dementia and Alzheimer Disease: a Population-Based Cohort Study. Arch. Intern. Med. 166 (9), 1003–1008. doi:10.1001/archinte.166.9.1003
Richmond, R. C., and Davey Smith, G. (2019). Commentary: Orienting Causal Relationships between Two Phenotypes Using Bidirectional Mendelian Randomization. Int. J. Epidemiol. 48 (3), 907–911. doi:10.1093/ije/dyz149
Roger, V. L. (2013). Epidemiology of Heart Failure. Circ. Res. 113 (6), 646–659. doi:10.1161/circresaha.113.300268
Shah, S., Henry, A., Roselli, C., Lin, H., Sveinbjörnsson, G., Fatemifar, G., et al. (2020). Genome-wide Association and Mendelian Randomisation Analysis Provide Insights into the Pathogenesis of Heart Failure. Nat. Commun. 11 (1), 163. doi:10.1038/s41467-019-13690-5
Van Uden, E., Kang, D. E., Koo, E. H., and Masliah, E. (2000). LDL Receptor-Related Protein (LRP) in Alzheimer’s Disease: towards a Unified Theory of Pathogenesis. Microsc. Res. Tech. 50 (4), 268–272. doi:10.1002/1097-0029(20000815)50:4<268:Aid-jemt3>3.0.Co;2-1
Verbanck, M., Chen, C. Y., Neale, B., and Do, R. (2018). Detection of Widespread Horizontal Pleiotropy in Causal Relationships Inferred from Mendelian Randomization between Complex Traits and Diseases. Nat. Genet. 50 (5), 693–698. doi:10.1038/s41588-018-0099-7
Vina, J., and Lloret, A. (2010). Why Women Have More Alzheimer’s Disease Than Men: Gender and Mitochondrial Toxicity of Amyloid-Beta Peptide. J. Alzheimers Dis. 20, S527–S533. doi:10.3233/jad-2010-100501
Vogels, R. L. C., Scheltens, P., Schroeder-Tanka, J. M., and Weinstein, H. C. (2007). Cognitive Impairment in Heart Failure: A Systematic Review of the Literature. Eur. J. Heart Fail. 9 (5), 440–449. doi:10.1016/j.ejheart.2006.11.001
Wootton, R. E., Lawn, R. B., Millard, L. A. C., Davies, N. M., Taylor, A. E., Munafò, M. R., et al. (2018). Evaluation of the Causal Effects between Subjective Wellbeing and Cardiometabolic Health: Mendelian Randomisation Study. Bmj 362, k3788. doi:10.1136/bmj.k3788
Keywords: Mendelian randomization, heart failure, Alzheimer’s disease, genome-wide association study, single-nucleotide polymorphisms, causal effects
Citation: Duan C, Shi J, Yuan G, Shou X, Chen T, Zhu X, Yang Y and Hu Y (2022) Causal Association Between Heart Failure and Alzheimer’s Disease: A Two-Sample Bidirectional Mendelian Randomization Study. Front. Genet. 12:772343. doi: 10.3389/fgene.2021.772343
Received: 09 September 2021; Accepted: 18 November 2021;
Published: 11 January 2022.
Edited by:
Pinyi Lu, Frederick National Laboratory for Cancer Research (NIH), United StatesReviewed by:
Hui Lu, Capital Medical University, ChinaHaoxiang Cheng, Icahn School of Medicine at Mount Sinai, United States
Zhaotong Lin, University of Minnesota Twin Cities, United States
Copyright © 2022 Duan, Shi, Yuan, Shou, Chen, Zhu, Yang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanhui Hu, huiyuhui55@sohu.com