 Frank Miedema1* Mette D. Hazenberg2 Kiki Tesselaar1 Debbie van Baarle1 Rob J. de Boer3 José A. M. Borghans1
Frank Miedema1* Mette D. Hazenberg2 Kiki Tesselaar1 Debbie van Baarle1 Rob J. de Boer3 José A. M. Borghans1
- 1Department of Immunology, University Medical Center Utrecht, Utrecht, Netherlands
- 2Department of Internal Medicine and Hematology, Academic Medical Center, Amsterdam, Netherlands
- 3Theoretical Biology and Bioinformatics, Utrecht University, Utrecht, Netherlands
In the past decade, evidence has accumulated that human immunodeficiency virus (HIV)-induced chronic immune activation drives progression to AIDS. Studies among different monkey species have shown that the difference between pathological and non-pathological infection is determined by the response of the immune system to the virus, rather than its cytopathicity. Here we review the current understanding of the various mechanisms driving chronic immune activation in HIV infection, the cell types involved, its effects on HIV-specific immunity, and how persistent inflammation may cause AIDS and the wide spectrum of non-AIDS related pathology. We argue that therapeutic relief of inflammation may be beneficial to delay HIV-disease progression and to reduce non-AIDS related pathological side effects of HIV-induced chronic immune stimulation.
Chronic Immune Activation is the Primary Driver in HIV Pathogenesis
Upon discovery of the virus that causes AIDS, the name human immunodeficiency virus (HIV) was coined because the virus eventually causes severe immune deficiency. This was based on the clinical symptoms with which end-stage HIV-infected patients presented and on the gradual decline of CD4+ T-cell numbers in the blood, which is still considered a hallmark of HIV-disease progression. The finding that HIV is confined to CD4+ leukocytes and is cytopathic for CD4+ T cells established the hypothesis that HIV causes immune deficiency by directly killing CD4+ T cells and impeding CD4+ T-cell renewal (1). The molecular mechanisms involved in CD4+ T-cell killing by HIV infection have been studied in great detail, leading to novel insights into the down-stream effects of abortive infection and viral integration on cell death (2–4). However, increased apoptosis rates in HIV-infected individuals are not confined to infected CD4+ T cells, but are also observed in non-infected CD4+ T cells and in cell types that are not even targets for HIV infection, suggesting that the cytopathic effects of HIV are not the full story (5, 6).
Paradoxically, HIV induces strong cellular immune responses, both with respect to magnitude and breadth (7–11), and even in progressive HIV infection, high avidity HIV-specific CD8+ T cells are being induced (12). Both CD4+ and CD8+ T cells are more activated in acute and chronic HIV infection, and hence proliferate rapidly and have a short half life. This explains why both T-cell production and death rates are increased throughout HIV infection (13, 14). At first, the high division rate of CD4+ T cells in untreated HIV-infected patients was interpreted to reflect a homeostatic response to the loss of CD4+ T cells (15–18). Studies in patients on combination anti-retroviral therapy (cART) pointed out, however, that T-cell proliferation rates drop concomitant with the loss of virus, even when CD4+ T-cell numbers are still far below healthy control levels, suggesting that the increased T-cell division rates are caused by the virus itself. It became clear that chronic immune activation is a hallmark of pathogenic HIV infection, exemplified by the increased expression of soluble and cellular immune activation markers, including IFNα, TNFα, and sTNFR and the increased fraction of activated CD8+ T cells; markers that have long been used as surrogate markers for HIV-disease progression (19–27). In fact, the level of immune activation is the best predictor of progression to AIDS (28, 29) and death (22, 30–32), independent of HIV viral load. HIV-2 infection is characterized by an overall slower progression rate, lower viral loads, and higher CD4+ T-cell numbers than HIV-1 infection (33). Yet, the cytopathicity of HIV-2 for human CD4+ lymphoid cells is not lower compared to HIV-1 (34). A striking difference between the two viral subtypes is that the level of immune activation is lower in HIV-2 compared to HIV-1 infection, although expression patterns and prognostic values for immune activation markers were found to be similar when patients with HIV-1 or HIV-2 infection were matched for CD4+ T-cell depletion levels (35, 36) These observations were paralleled by insights from simian immunodeficiency virus (SIV) infection in sooty mangabeys (SMs) and African green monkeys (AGMs). SIV infection in these animals is characterized by high viral loads without high levels of immune activation, and does not lead to AIDS, which will be discussed in detail in the Box 1 below (37, 38). Together, these observations have gradually shifted the paradigm from the classical hypothesis that viral cytopathicity is the primary driver of CD4+ T-cell depletion and immune deficiency, to the hypothesis that chronic immune activation is the cause of T-cell depletion and immune deficiency (35, 39).
Box 1. Damage control in non-pathogenic SIV infection.
In pathogenic SIV infection in rhesus macaques (RMs), high levels of immune activation are associated with progression to AIDS. SIV infection in sooty mangabeys (SMs) and African green monkeys (AGMs), in contrast, do not lead to AIDS despite high viral loads (37, 87–91). Interestingly, SM do not mount stronger cytotoxic T-cell or neutralizing antibody responses to SIV compared to RM, and productively infected CD4+ T cells in SIV-infected SM and RM have similar life spans (92–94). Several lines of evidence show that systemically LPS induces features of pathogenic SIV infection (95), that pre-existing microbial translocation and loss of GI integrity in pigtail macaques was associated with faster SIV disease progression (96). In non-pathogenic, like in pathogenic SIV infection, however, a severe depletion of memory T cells in the gut occurs, apparently without causing generalized immune activation in non-pathogenic SIV infection (54, 55).
As the dynamics of virus and virus-infected CD4+ T cells in these animal models of SIV infection are comparable, excessive indirect activation-induced killing of T cells in rhesus macaques has been proposed to be the major pathological difference (37, 38, 87, 97–100). Indeed, despite the fact that RM develop strong immune responses upon SIV infection, these responses fail to clear the virus, resulting in persistently high levels of immune activation throughout infection (38, 101).
Compelling evidence has been obtained for a SM-specific polymorphism in TLR signaling, leading to attenuated production of type I IFN by pDCs induced via TLR7/9 activation in SIV-infected SM (102, 103). The gene involved, IRF-7, is a signaling protein downstream of TLR7 and 9. Interestingly, TLR7- and 9-induced production of TNFα appeared to be unaffected in SM, which agrees with the fact that TNFα release is mediated by the NF-κB and not by the IRF-7 pathway. This observation suggests that release of type I IFNs, but not TNFα, may be critical for SIV pathogenesis, which makes IFNα and IRF-7 potential drug targets. Despite the inability of SM to produce high levels of type I IFNs upon TLR7/9 activation by SIV, peak viremia during acute SIV infection in these animals is accompanied by clear signs of an innate and adaptive immune response, including the induction of IFN-stimulated genes (ISGs) (104, 105). Gene expression profiling showed the induction of ISGs, acute inflammatory genes, and genes associated with chemotaxis and neutrophil recruitment, DC activation and maturation, apoptosis, and cytotoxic T-cell responses during the acute phase of both pathogenic and non-pathogenic SIV infection (83, 104–106). In SM and AGM, expression of ISGs returns to normal levels after 30 days of infection. Since this decline in inflammation is paralleled by a gene expression program of immune regulatory genes, including genes that down-regulate T-cell responses [e.g., indolamine 2,3 dioxygenase (IDO), IL-10, LAG3, and PD-L1] and genes that down-regulate IFN responses (e.g., adenosine deaminase), it has been proposed that active downregulation may be involved (83, 104). Further detailed mechanistic studies are required to reveal whether – and if so which – specific down-regulatory pathways are involved. Of note, also host genes implied in intracellular viral restriction are rapidly up-regulated in non-pathogenic infection (83, 104).
If type I IFN is one of the main causes of immune activation in HIV and SIV infection, it remains puzzling how the clear difference in IFNα production by pDC from SIV-infected SM and RM can be reconciled with the apparent similarity of immune responses, and specifically the expression of IFN-inducible genes, observed during acute SIV infection of both species. There is, however, evidence that upregulation of ISGs in acutely SIV-infected SM is induced even though IFNα production by their pDCs is severely diminished (66, 83, 104). Interestingly, Favre and colleagues (66) found upregulation of IFNα, but not IL-12 and IL-6, in acute SIV infection in AGM, although IFNα release was very limited in duration compared to the sustained release of all three cytokines in pathogenic SIV infection. Also the detailed characteristics of immune activation in acutely SIV-infected RM and SM are quite different. Acute SIV infection in SM (and AGM) is not accompanied by increased CD4+ T-cell turnover, but strong increases in CD8+ T-cell activation, division (Ki67 expression) and apoptosis have been observed (99, 102, 107, 108). Thus, both timing and quality of gene expression of pro-inflammatory cytokines seem to be critically different between pathogenic and non-pathogenic SIV infection (109). Taken together, current data are compatible with the idea that SM and AGM respond to SIV with a limited and transient innate response and with an adaptive response that is mainly restricted to CD8+ T cells. In pathogenic SIV infection, an excessive innate response is generated with sustained IFNα and ISG induction which induces proliferation of NK cells and a broad SIV-specific and bystander CD4+ and CD8+ T-cell response (83, 102, 108). It could be that in SM and AGM, low and transient type I IFN responses during acute SIV infection induce a different gene expression program, allowing for resolution and/or downregulation of the immune response during subsequent chronic SIV infection.
It has been proposed that damage control in SIV-infected SM may in part be due to the preservation of central memory CD4+ T cells (Tcm) which are thought to provide protection against the harmful side effects of bacterial translocation (110). Depletion of memory T cells from the gut and bacterial translocation occur only transiently during acute SIV infection in SMs (54). In contrast to rhesus macaques, SMs are able to avoid epithelial barrier breakdown and thereby limit the undesired side effects of bacterial translocation during chronic SIV infection (111). SM are able to spare Tcm from viral infection because of low CCR5 expression (112), while in AGM Tcm may be protected against SIV infection by CD4 downregulation (113). In pathogenic SIV and HIV infection, in contrast, Tcm are thought to be selectively lost through viral infection (112) However, the observation that the number of activated and naive T cells, and not the number of Tcm, is predictive for HIV-disease progression does not support the idea that Tcm numbers are most critical (114). High levels of immune activation in pathogenic SIV infection may promote SIV infection of Tcm, resulting in Tcm depletion which may contribute to the vicious cycle of loss of immune control. Further investigations are needed to better quantify the contribution of the various mechanisms that cause CD4+ Tcm activation and death.
In conclusion, non-pathogenic SIV infection of SM and AGM are examples of a pathogen-host symbiosis with an established state of tolerance. This is not immunological tolerance in the strict sense, but a state of tolerance in which the host resists the pathological effects of the virus by avoiding excessive inflammation (115, 116). Further investigation into the various and potentially different mechanisms by which SM and AGM avoid chronic immune activation is warranted and of great importance for our understanding and the treatment of HIV disease.
Causes of Immune Activation in HIV Infection
It has long been known that innate and adaptive immunity get activated upon acute HIV infection, as extensively described and reviewed elsewhere (35, 39–46). Chronic HIV infection is now known to be characterized by increased expression of pro-inflammatory cytokines, including type I IFNs, IL-6, TGFβ, IL-8, IL-1α, and IL-1β, serum markers of inflammation including sCD14, CRP, cystatin C, D-dimers, and activation of the coagulation system (47). In the last couple of years much attention has focused at the causes of immune activation in HIV infection, with a redirection of research focus from T-cell immunity to innate immunity.
Breach of Gastro-Intestinal Immunity
In the late 1990s, acute SIV infection in rhesus macaques (RMs) was shown to induce a severe and rapid depletion of memory CD4+ T cells from the gut (48). Later, in both humans and monkeys, it was found that this breach of the gut immune system resulted in a significant increase in bacterial components, including lipopolysaccharide (LPS), in the blood (49–51). LPS is a known activator of innate immune cells via Toll-like receptor (TLR) 4, and LPS concentrations in the circulation of HIV-infected individuals correlated strongly with T-cell activation levels (51, 52). It was concluded that translocation of immune stimulatory bacterial products contributes to systemic immune activation, via TLR activation of various leukocyte populations. LPS was used as an indicator for bacterial translocation, but other bacterial products, such as flagellin, peptidoglycan, and bacterial CpG-rich DNA domains that are recognized by TLR2, 5, and 9 respectively, may also contribute to immune activation. It was proposed that the early attack on the memory CD4+ T-cell population in the gut may be a critical determinant of disease progression (53). However, also in non-pathogenic SIV infection a severe depletion of memory T cells in the gut occurs, apparently without causing generalized immune activation (54, 55). Moreover, an attenuated variant of pathogenic SIVmac239 was shown to spare mucosal CD4+ T cells and yet to cause T-cell activation, CD4+ T-cell loss, and progression to AIDS without any signs of microbial translocation (56), showing that immune activation due to gut damage is not required to develop AIDS. On the other hand, in patients on cART, with very low HIV viral load, residual levels of bacterial translocation were positively correlated with immune activation levels suggesting that bacterial translocation may be a dominant driver of immune activation in patients treated with anti-viral drugs (57–63).
The breach of gut integrity in pathogenic SIV and HIV infection has been shown to be associated with depletion of CD4+ Th17 cells, a cell type that is normally abundant in the mucosa and is known to be involved in immunity to commensal bacteria (64). It is assumed that the immune system normally keeps a delicate balance between T regulatory (Treg) cells and Th17 cells, to protect against pathogens but avoid collateral damage from excessive immune responses (65). The selective loss of Th17 CD4+ T cells from the gut – possibly due to selective infection – has therefore been held responsible for the long-term loss of the intestinal integrity and thereby for chronic immune activation in pathogenic HIV infection (64, 66, 67). More recently, depletion of IL-21-producing CD4+ T cells has been observed in both the blood and rectal mucosa of SIV-infected RMs (68). Treatment of these animals with IL-21 resulted in the maintenance of intestinal Th17 cells, and a reduction of microbial translocation and systemic inflammation (69). The dynamics of the Th17/Treg balance and the role of Th17 cells and Th17-derived cytokines in HIV infection is currently subject of intensive study.
Single-Stranded RNA, Toll-Like Receptors, and Type I IFN Production
In 2004 it was reported that TLR7 and 8 recognize RNA from various viruses (70, 71), and it has been demonstrated that single-stranded (ss) HIV RNA directly activates the innate immune system via these TLRs (72, 73). After endosomal binding of ssHIV RNA to TLR7, HIV induces the release of type I interferons by plasmacytoid dendritic cells (pDCs) through the upregulation of TRAIL (72–75). Single stranded HIV RNA has also been shown to activate NK cells in a TLR7 and 8 dependent way, and this process is dependent on cell–cell contact between pDCs and monocytes (76). Finally, pro-inflammatory responses can be induced through intracellular recognition of HIV DNA intermediates. These intermediates can be the result of abortive HIV infection of CD4+ T cells, and induce the production of IFN-β and IL-1β (4). In agreement with these in vitro observations, gene expression analyses of lymphocytes from HIV-infected persons were shown to have a dominant signature of IFN-stimulated genes (ISGs) (77, 78). Immediately after start of cART – when virus production and viral load rapidly decline – markers of T-cell activation, expression of pro-inflammatory cytokines such as IFNα, IL-6, IL-1-β, and macrophage inflammatory protein-1α, adhesion molecules VCAM-1 and ICAM-1, and the levels of soluble markers for endothelial cell and coagulation activation are all rapidly and strongly reduced, although not to normal levels (15, 18, 73, 79–81). These data suggest that HIV itself, most likely through its ssRNA or DNA intermediates, is an important driver of immune activation in untreated HIV infection.
Type I IFNs provide an important link between chronic innate and adaptive immune activation in HIV infection, because they induce activation and maturation of pDCs, NK cells, T cells, and B cells (82). Gene expression profile data from pathogenic and non-pathogenic SIV-infected primates suggest that persistent release of type I IFNs is a particular feature of pathogenic infection (83). It is well established that pDCs are mass producers of type I IFNs (82). At a certain point, pDCs typically become refractory to restimulation by TLR ligands, thereby avoiding excessive immune activation and collateral damage in the course of viral infection (84, 85). Bhardwaj and colleagues (86) nicely showed that HIV, in contrast to other TLR7 agonists such as influenza virus and herpes simplex virus, induces a partially matured phenotype in pDCs. Because of this phenotype, pDCs are not rendered refractory and continue to produce type I IFNs during ongoing HIV exposure.
Interestingly, and similar to what is observed in SIV-infected SMs (102, 104) and AGMs (83), chronically HIV-infected individuals who do not progress to AIDS despite their high viral loads turned out to have very low levels of proliferating and activated T cells (117) correlating with relatively low levels of ISGs and immune activation gene expression in CD8+ T cells (118). A recent study confirmed the central role of IFNα in HIV-1 infection by showing that IFNα is the dominant type I IFN detectable in the plasma of HIV-infected individuals and that its levels correlate with immune activation and depletion of CD4+ T cells (119). In addition, it was shown that pDCs derived from women produce more IFNα in response to HIV-1 than pDCs from men, resulting in higher levels of T-cell activation (120, 121). This may at least in part explain the observation that HIV-infected women with a given viral load have a 1.6-fold higher risk to develop AIDS than men, and despite having lower viral loads on average, typically progress faster to AIDS than men (122).
It has been reported that pDCs from SMs have a species-specific inability to produce high levels of type I IFN (102, 103) related to sequence polymorphisms in IRF-7, a signaling protein downstream of TLR7 and 9 (see Box 1). Also in humans, polymorphisms of IRF-7 have been reported that are associated with the level of HIV-induced IFNα production by pDCs in vitro and with CD8+ T-cell activation in vivo (123). These data stress the importance of the IRF-7 pathway in HIV pathogenesis, although there is no definite proof yet that IRF-7 itself is responsible for the induction of different responses in different individuals. Together, these observations suggest that the continuous release of type I IFNs plays a critical role in SIV and HIV pathogenesis. Future studies should point out what the direct and indirect role of IRF-7 polymorphisms is in determining the set point level of chronic immune activation in HIV-infected subjects, and should clarify the potential of IFNα and IRF-7 as drug targets (Figure 1).
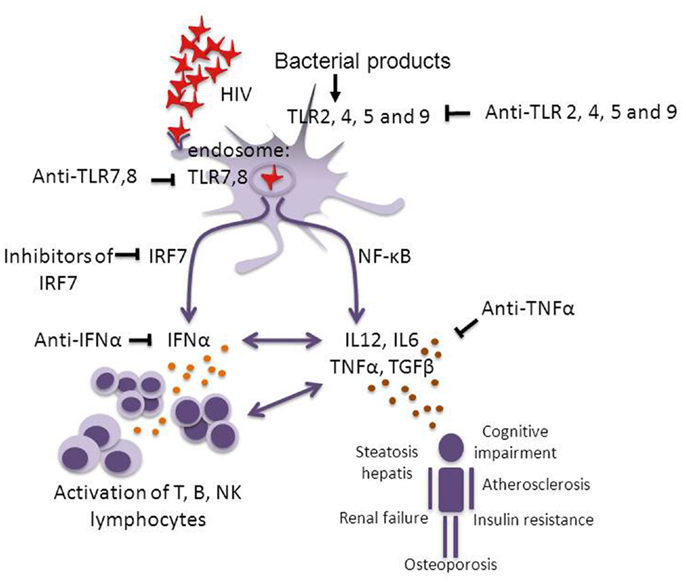
Figure 1. Pathways of chronic immune activation and its down-stream effects in HIV infection. HIV infection induces chronic immune activation through activation of the innate and the adaptive immune system, via single-stranded (ss) RNA and possibly through intracellular viral DNA which activate pDCs via endosomal TLR7 and 8. This activation leads to the induction of IFNα via the IRF-7 pathway and the induction of IL-6, IL-12, TNFα, and TGFβ through the NF-κB pathway. Continuous activation of the lymphocyte compartment leads to attrition of the T-cell pool (14, 15) and “immune paralysis” (e.g., impaired CTL responses). Bacterial translocation may be another source of TLR activation via TLR2, 4, 5, and 9 (49–51). Over time also non-AIDS related complications develop. Potential targets for therapeutic interventions with inflammation to diminish pathology are indicated. It has been shown that blocking the effect of TLR7 and 9 significantly reduces HIV-induced immune activation (124). Studies in pathogenic and non-pathogenic SIV infection suggest that blocking IRF-7 or IFNα should be investigated. In rheumatoid arthritis patients who were treated with TNFα inhibiting agents (infliximab, etanercept) it was shown that blocking the effect of TNFα reversed the increased incidence of cardiovascular complications and insulin resistance. In analogy, the potential for a therapy interfering with TNFα in HIV infection should be tested (125–127).
Pathogenic Effects of Immune Activation and Inflammation
The key role of chronic immune activation in HIV and SIV pathogenesis is now commonly accepted, as it is so clearly associated with CD4+ T-cell decline and progression to AIDS. The clinical outcome of HIV infection, however, does not only depend on CD4+ T-cell loss, but also on non-immunological side effects of chronic immune activation.
Inflammation Drives CD4+ T-Cell Depletion and Loss of HIV-Specific Immunity
A large body of work has suggested that chronic immune activation in HIV infection has deleterious effects on immune function in general, as well as on HIV-specific immunity by inducing persistent activation and maturation of all sorts of innate and adaptive immune cells (82). Through continuous activation and differentiation of T cells, chronic HIV infection gradually depletes the naive CD4+ and naive CD8+ T-cell pools (31, 35, 43, 128, 129). Intrinsically different responses of the distinct T-cell lineages to activation may determine clonal expansion and contraction (130), and thereby the sensitivity of the different T-cell populations to chronic activation-induced cell loss, although the molecular basis for these differences remains unclear. Thymic and T-cell progenitor dysfunction, most likely caused by aberrantly high levels of pro-inflammatory cytokines expressed during untreated HIV infection, have been reported (43, 131) and the loss of such progenitor cells could aggravate the depleting effects of chronic immune activation on the adaptive immune system. Moreover, continuous inflammation in lymph nodes has been suggested to result in TGFβ-induced collagen deposition, fibrosis, and pathological changes in lymph node architecture, possibly adding to impaired T-cell proliferation and survival (132–134). Continuous activation has recently been shown to induce upregulation of inhibitory receptors such as programed death-1 (PD-1), CTLA-4, and Tim-3, which may interfere with ongoing HIV-specific T-cell responses, and ultimately lead to T-cell anergy and loss of HIV-specific T cells (135–137). Similarly, B-cell dysfunction, which is observed immediately after acute HIV infection (138), is closely related to chronic activation of the B-cell compartment. Increased B-cell turnover and differentiation is associated with the phenotypic and functional B-cell abnormalities characteristic for untreated HIV infection (139–142). A recent study showed the downregulation of the regulatory receptor B- and T-lymphocyte attenuator (BTLA) and the upregulation of PD-1 on B cells in HIV infection (143). Interestingly, a direct down-regulating effect of type I IFN on BTLA expression on CD4+ and CD8+ T cells has been reported, which may directly contribute to T-cell hyperactivation (144). Recently evidence was reported for a link between PD-1L on follicular Th cells and impairment of B-cells function (145).
Persistent immune activation has also been shown to have deleterious effects on HIV-specific CD4+ (7, 146–153) and CD8+ T-cell immunity (154–160), amongst others by preventing the establishment of IL-2-producing memory CD4+ and CD8+ T cells (146, 151–153). HIV-specific cytotoxic T-cell responses are generally considered to play an important role in anti-HIV immunity. Certain HLA alleles clearly correlate with viral load set point and disease progression. In line with this, the major genetic factors related to HIV-1 control coming out of a genome wide association study (GWAS) were shown to affect HLA–viral peptide interaction (161). There is accumulating evidence that Gag-specific CTL responses which preferentially target conserved epitopes have a protective effect (162–171). However, in two large prospective cohort studies, CD4+ and CD8+ HIV Gag-specific T-cell immunity within the first year after HIV seroconversion were not found to be predictive for disease progression (172, 173). This observation was confirmed in a longitudinal study in an African cohort (174). Also in these studies, immune activation turned out to be the strongest risk factor for disease progression, stronger than, and independent of, viral load (172, 173). It is important to consider the possibility that the typical association between strong CTL responses and a lack of HIV-disease progression that is observed in cross-sectional studies, may merely reflect the preservation of CTL responses in the absence of chronic immune activation rather than a protective effect of CTL themselves (175).
HIV-Induced Inflammation and HIV-Associated Non-AIDS Disease
Increasing insight in the source and the role of inflammation in HIV pathogenesis has been paralleled by recent progress in our understanding of the role of inflammation in a much wider spectrum of clinical conditions than infectious diseases. After the introduction of anti-retroviral therapy for HIV infection, several case studies suggested that patients treated with cART had an increased risk to develop sub-clinical atherosclerosis and acute myocardial infarction (176–179). Initial studies reported that the increased risk of cardiovascular disease was associated with specific classes of anti-viral drugs (180). Later studies revealed that cardiovascular risk was in fact larger in untreated compared to treated HIV infection (181, 182), but also in patients on cART, the risk for cardiovascular disease is higher than expected based on traditional cardiovascular risk factors alone. In addition to cardiovascular disease, HIV infection poses patients at increased risk to develop a number of other non-AIDS related complications, such as non-alcoholic steatohepatitis, renal dysfunction, osteoporosis, insulin resistance, metabolic syndrome, and cognitive impairment (47). It has been shown that soluble mediators released by activated immune cells, such as IL-6, IL-1, and TNFα, also act on non-immune tissue cells with various tissue-dependent pathological effects. In a broad variety of clinical conditions, including obesity, atherosclerosis, neurodegenerative disease, and autoimmune diseases, chronic inflammatory processes are now recognized to play a major role (183), and it has been postulated that most non-AIDS defining complications of HIV infection are related to the chronic inflammatory state induced by HIV (Figure 1) (184, 185). This hypothesis is strengthened by recent observations in patients with rheumatoid arthritis (RA). Both HIV infection and RA are characterized by a chronic inflammatory state and increased levels of pro-inflammatory cytokines like TNFα, IL-1β, and IL-6, and also in RA patients the incidence of non-primary disease related complications such as cardiovascular disease, osteoporosis, non-alcoholic fatty liver disease (NAFLD), and cognitive impairment are more prevalent than among the general population (125–127). Thus, clinical symptoms that initially seemed unrelated are now being recognized as part of the total complex of HIV-associated disease and appear to have a common underlying pathogenesis of chronic inflammation and excessive immune activation (186, 187). Preliminary data suggest a central role for TNFα in HIV-associated non-AIDS disease but it remains to be determined to what extent other pro-inflammatory cytokines, perhaps acting via TNFα, are involved.
HIV in Comparison to Other Persistent Viral Infections
These novel insights into HIV pathogenesis prompt the question as to how HIV differs from most other viruses. We believe that HIV pathogenesis is caused by a combination of specific characteristics. Most importantly HIV infects CD4+ T helper cells. In addition a variety of cells that express CD4 and one of the HIV coreceptors can be infected albeit at very low levels. Thereby, the virus is not confined to a single organ and may induce a variety of systemic immune responses. HIV induces much higher levels of cytokines during acute infection compared to hepatitis B or hepatitis C (41). HIV is virtually insensitive to control by neutralizing antibodies and cellular immunity because of various mechanisms, including the glycan shield surrounding the HIV virion (188) and the high mutation rate of the virus, which allows for rapid immune escape. After acute HIV infection, virus- and host-specific set points are established that determine the subsequent clinical course based on the level, and probably the type, of immune activation that is induced. Like other viruses, HIV induces type I IFN release by pDCs. The fact that HIV is targeted to pDCs by virtue of their expression of CD4, and the recent finding that HIV does not induce full maturation of pDC, which prevents these cells to become refractory to restimulation, as outlined above (86), may turn out to be critical factors driving persistent IFN release and thereby chronic activation of the innate and the adaptive immune system in HIV patients, resulting in exhaustion of immunity and broad spectrum end-organ immune pathology. Even though immune responses in acute hepatitis B and hepatitis C virus infection may differ from those in acute HIV infection, in individuals who do not clear hepatitis viral infection and who convert to chronic hepatitis, persistently increased immune activation levels have been reported. In analogy to what is observed in HIV patients, also non-hepatitis related conditions, such as metabolic syndrome and cardiovascular disease, occur more frequently in chronic hepatitis patients than in the general population, even when corrected for traditional risk factors for, e.g., cardiovascular disease (189). Strikingly, peripheral blood naive T-cell numbers in chronic hepatitis C virus-infected patients were found to be significantly lower than in healthy individuals, and associated with increased levels of inflammation (190). Thus, while immune responses during acute infection may differ between HIV, hepatitis B, and hepatitis C virus infection, leading to clearance of the virus in the majority of hepatitis B infected patients and a subset of hepatitis C infected individuals, once chronic inflammation has been established, its effects tend to be similar for the three patient groups. Other viral infections, like Epstein–Barr virus (EBV) and cytomegalovirus (CMV) are incomparable to HIV or chronic hepatitis infection, because after an acute phase these infections convert into a truly latent stage, during which no virus is detectable in the peripheral blood of immunocompetent individuals.
The Immune Activation Hypothesis Reduced to Practice
Boosting Immunity
Great effort has been put over the years into approaches to therapeutically strengthen anti-viral immune responses. Thus far, however, there is little proof for beneficial effects, and in fact the possibility of induction of adverse effects is an important concern. Therapeutic vaccination, with DNA and live viral vector based vaccines and combinations thereof, has had only transient and small effects on viral load (191, 192). In one trial in which therapeutic vaccination was followed by interruption of cART, viral rebound was larger and time to restart therapy shorter, than in the non-vaccinated group (193). With respect to prophylactic vaccines, CTL-based vaccines may have some potential if they manage to consistently lower the viral set point. However, upon infection such vaccines will at best reduce and not completely prevent chronic immune activation driven pathogenesis and should therefore not be considered as curative. In fact, the strongest protective effect is to be expected from HIV vaccines that stimulate HLA-B57, B58, or B27 restricted T-cell responses, as they are associated with significantly lower viral loads. Such vaccines would however only help the carriers of protective HLA molecules, most of which already experience much slower disease progression upon HIV infection. In order to develop CTL vaccines that are applicable to a wider patient population it is of vital importance to gain better insight into the mechanisms responsible for the relative protection conferred by these protective HLA molecules.
For boosting of immunity and enhancement of CD4+ T-cell production, IL-2 has been administered in large scale multi-center international trials in patients with and without cART with substantial increases in CD4+ T-cell counts but no beneficial clinical effects (194). Administration of IL-7 (195, 196) or human growth hormone (197) has been tried out in small cohorts, with successful effects on naive and central memory T-cell numbers, but again without significant clinical effects. As these biological compounds are known to have strong activating effects on the peripheral T-cell compartment (198) their administration is not without risk, and one should be aware of possible adverse effects in the long run. Immune stimulating therapy should in any case be restricted to patients on cART, although even on cART (residual) immune activation is correlated with poor immune reconstitution (199). To enhance anti-HIV responses, blockade of inhibitory ligand-receptor interactions, such as PD-1, CTLA-4, and Tim-3, has been proposed (200). Some positive results have been obtained with PD-1 blockade in SIV-infected macaques, which has been shown to lead to improved virus-specific CD8+ T-cell responses, reduction in plasma viral load and prolonged survival (201), and to reduced hyperactivation and bacterial translocation (202). However, experiments with CTLA-4 blockade have demonstrated that the effects of inhibitory receptor blockade may even be deleterious, leading to increased T-cell activation and viral replication (203). Great care therefore needs to be taken with approaches that may increase the level of CD4+ T-cell proliferation, and in our opinion should never be applied without cART.
Taken together, therapeutic interventions aiming at enhancing anti-HIV T-cell immunity may not have the desired beneficial effect in the majority of people, and may even have adverse long-term effects through the immune stimulation they induce. It has been argued that since our understanding of virus-specific cellular immunity – and in particular its repertoire, its functional and kinetic requirements, and its regulation and tissue distribution – are still far from complete, the real correlate of immune protection against AIDS is still to be discovered (204). Indeed, not all immune activation needs to be equally pathogenic, and we cannot exclude the possibility that induction of HIV-specific T-cell responses without excessive and chronic release of type I IFNs and other cytokines might be favorable to the host for control of HIV. However, as pDC activation is believed to be required for the induction of an adequate adaptive T-cell response, induction of strong HIV-specific immune responses without chronic release of type I IFNs may be an impossible combination; in fact, pDC activation may collaterally cause the very same pathology that the adaptive immune response should prevent. Irrespective of the hypothesis of what is causing AIDS pathogenesis, of all vaccination strategies, prophylactic vaccines that are able to induce a strong broadly neutralizing antibody response at this time seem to be most promising to induce protective immunity to HIV infection in a large number of individuals (205).
Therapeutic Damage Control
Another correlate of the immune activation hypothesis is that immune suppressive therapy might have beneficial clinical effects because it reduces the deleterious effects of immune activation. Immune suppressive drugs like cyclosporin (206, 207) and mycophenolic acid (208), that are used to prevent T-cell activation in organ transplant rejection, have been experimentally tried in HIV infection. In combination with cART, variable effects on T-cell turnover, activation, and CD4+ T-cell numbers were shown (206–208).
Given the recent insight that not activation of CD4+ and CD8+ T cells via TCR, but instead TLR activation, release of type I IFNs and expression of IFNα/β inducible genes may contribute more to systemic immune activation in HIV infection, the latter proteins and genes may be more relevant targets for therapeutic interventions (Figure 1). TLR antagonists and inhibitors are currently an area of intense investigation and it is to be expected that many will become available for phase I/II or experimental proof of concept clinical trials in the very near future (209, 210). Indeed, in a preliminary study in which chloroquine, an inhibitor of endosomal TLR3, 7, 8, and 9 was administered to HAART-naive HIV-infected patients, significantly lower immune activation levels were observed, as reflected by decreased levels of T-cell division and expression of activation markers (124). Although these findings need to be reconfirmed and more clinical studies are needed, this study suggests that interference with HIV-induced TLR7/9 activation is feasible. Because of the clear association between immune activation and clinical outcome such interventions may be promising. Also IRF-7, which selectively induces IFNα but not TNFα or IL-12 production, is a potential drug target and treatment with IFNα neutralizing antibodies or blocking TNFα or the TNFα-R are feasible options to be explored in order to decrease inflammation and tissue-related pathology. Indeed, targeting TNFα in pathogenic SIV infection in RMs by administration of adalimumab (Humira) has been shown to reduce systemic inflammation and many of its down-stream effects (211).
Humanized anti-IFNα monoclonal antibodies have been developed and have been tested in phase I trials in patients suffering from systemic lupus erythematosus (SLE) and psoriasis, autoimmune diseases in which IFNα is believed to play a critical role. In SLE but not psoriasis one dose of anti-IFNα monoclonal antibody resulted in downregulation of IFN-inducible gene expression with beneficial clinical effects (212, 213). No evidence for adverse effects, such as an increase in viral infections or viral reactivation was observed which opens up the possibility to consider application of anti-IFNα treatment to HIV-infected patients to neutralize over-expression of IFNα. Induction of anti-IFNα antibodies by immunization with inactivated IFNα to inhibit progression to AIDS has been investigated in a large multicentre study, and beneficial effects on CD4+ T-cell decline and markers of clinical progression were reported in patients that developed anti-IFNα antibodies (214). Although these studies have never been repeated, the recently obtained insights into the role of IFNα in HIV-disease progression warrant future research in this direction.
Paradoxically, IFNα administration has been investigated in the pre-cART era as a treatment option for HIV infection with or without Kaposi sarcoma (215, 216). Although IFNα treatment showed the expected anti-viral effect, leading to lower viral loads, this type of treatment became of less interest when cART became available. In addition, IFNα treatment induced flu-like syndrome, immune activation, and T-cell depletion when given to HIV patients co-infected with HCV (217–219).
In RA patients who were treated with TNFα inhibiting agents (such as infliximab or etanercept) it was shown that blocking the effect of TNFα reversed the increased incidence of cardiovascular complications and insulin resistance (125–127). Anecdotal reports have shown the safety of anti-TNFα treatment in RA patients who were also HIV infected and on HAART (220). A non-specific intervention aimed at lowering immune activation and its side effects, such as cardiovascular disease, might be the addition of statins to standard anti-retroviral regimens, as has been suggested for treatment of RA (221).
In addition to potentially improving HIV-treatment options, the interventions suggested above will provide us with a wealth of data allowing dissection of the relative contribution of different cytokines such as IFNα and TNFα to immune activation and end-organ immune pathology in HIV infection. It should be noted however that, given the complex interrelationship between potentially protective immune responses and the damage induced by chronic immune activation, any of these interventions could in principle also aggravate HIV-induced pathology. Therefore, a combination with HAART seems at this time the best approach.
Conclusion
We review compelling evidence for CD4+ T-cell loss in HIV infection caused by various down-stream effects of persistent and strong innate immune activation. Immune activation is induced by HIV ssRNA and possibly its DNA intermediates and to some extent by translocation of bacterial products from the gut. This CD4+ T-cell death is occurring in addition to CD4+ T-cell loss due to direct HIV-induced cell killing. We conclude that immune activation is most likely the main cause of CD4+ T-cell depletion, loss of HIV-specific immunity and HIV-associated non-AIDS disease, also in patients on cART. Although much knowledge is still lacking, we are beginning to understand which receptors and active molecules are most likely dominant in the cellular and molecular pathways involved in HIV pathology. This new perspective has major implications for HIV vaccinology, but also opens up novel therapeutic options that may be explored in the near future.
Search strategy and selection criteria: references for this article were identified through searches of PubMed for articles published from 1985, by use of the terms HIV, SIV, AIDS, immune activation, immunity, pathogenesis. Articles resulting from these searches and relevant references cited in those articles were reviewed. Articles published in English were included.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our work is supported by the Dutch Aids Fonds (to Kiki Tesselaar), the Netherlands Organization for Scientific Research (now, grant 917.96.350 to Mette D. Hazenberg, 836.07.002 to José A. M. Borghans, and 016.048.603 to Rob J. de Boer), and by a Utrecht University High Potential Grant (to Debbie van Baarle).
References
1. Fauci AS, Pantaleo G, Stanley S, Weissman D. Immunopathogenic mechanisms of HIV infection. Ann Intern Med (1996) 124:654–63. doi: 10.7326/0003-4819-124-7-199604010-00006
2. Cooper A, Garcia M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature (2013) 498(7454):376–9. doi:10.1038/nature12274
3. Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature (2002) 417(6884):95–8. doi:10.1038/417095a
4. Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, et al. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell (2010) 143(5):789–801. doi:10.1016/j.cell.2010.11.001
5. Finkel TH, Tudor-Williams G, Banda NK, Cotton MF, Curiel T, Monks C, et al. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat Med (1995) 1:129–34. doi:10.1038/nm0295-129
6. Meyaard L, Otto SA, Jonker RR, Mijnster MJ, Keet RPM, Miedema F. Programmed death of T cells in HIV-1 infection. Science (1992) 257:217–9. doi:10.1126/science.1352911
7. Pitcher CJ, Quittner C, Peterson DM, Connors M, Koup RA, Maino V, et al. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nat Med (1999) 5(5):518–25. doi:10.1038/8400
8. Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science (1996) 274:94–6. doi:10.1126/science.274.5284.94
9. Ogg GS, Jin X, Bonhoeffer S, Moss P, Nowak M, Monard S, et al. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J Virol (1999) 73:797–800.
10. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med (2004) 10(3):282–9. doi:10.1038/nm992
11. De Boer RJ, Mohri H, Ho DD, Perelson AS. Turnover rates of B cells, T cells, and NK cells in simian immunodeficiency virus-infected and uninfected rhesus macaques. J Immunol (2003) 170(5):2479–87.
12. Draenert R, Verrill CL, Tang Y, Allen TM, Wurcel AG, Boczanowski M, et al. Persistent recognition of autologous virus by high-avidity CD8 T cells in chronic, progressive human immunodeficiency virus type 1 infection. J Virol (2004) 78(2):630–41. doi:10.1128/JVI.78.2.630-641.2004
13. Hellerstein MK, Hoh RA, Hanley MB, Cesar D, Lee D, Neese RA, et al. Subpopulations of long-lived and short-lived T cells in advanced HIV-1 infection. J Clin Invest (2003) 112(6):956–66. doi:10.1172/JCI17533
14. Grossman Z, Paul WE. The impact of HIV on naïve T-cell homeostasis. Nat Med (2000) 6:976–7. doi:10.1038/79667
15. Hazenberg MD, Stuart JW, Otto SA, Borleffs JC, Boucher CA, de Boer RJ, et al. T cell division in human immunodeficiency virus (HIV-1)-infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active anti-retroviral therapy. Blood (2000) 95(1):249–55.
16. Mohri H, Perelson AS, Tung K, Ribeiro RM, Ramratnam B, Markowitz M, et al. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by anti-retroviral therapy. J Exp Med (2001) 194:1277–87. doi:10.1084/jem.194.9.1277
17. Kovacs JA, Lempicki RA, Sidorov IA, Adelsberger JW, Herpin B, Metcalf JA, et al. Identification of dynamically distinct subpopulations of T lymphocytes that are differentially affected by HIV. J Exp Med (2001) 194:1731–41. doi:10.1084/jem.194.12.1731
18. Lempicki RA, Kovacs JA, Baseler MW, Adelsberger JW, Dewar RL, Natarajan V, et al. Impact of HIV-1 infection and highly active antiretroviral therapy on the kinetics of CD4+ and CD8+ T cell turnover in HIV-infected patients. Proc Natl Acad Sci U S A (2000) 97:13778–83. doi:10.1073/pnas.250472097
19. Fahey JL, Taylor JMG, Detels R, Hofmann B, Melmed R, Nishanian P, et al. The prognostic value of cellular and serologic markers in infection with human immunodeficiency virus type 1. N Engl J Med (1990) 322:166–72. doi:10.1056/NEJM199001183220305
20. Hofmann B, Wang YX, Cumberland WG, Detels R, Bozorgmehri M, Fahey JL. Serum beta 2-microglobulin level increases in HIV infection: relation to seroconversion, CD4 T-cell fall and prognosis. AIDS (1990) 4(3):207–14. doi:10.1097/00002030-199003000-00005
21. Bofill M, Mocroft A, Lipman M, Medina E, Borthwick NJ, Sabin CA, et al. Increased numbers of primed activated CD8+CD38+CD45RO+ T cells predict the decline of CD4+ T cells in HIV-1-infected patients. AIDS (1996) 10:827–34. doi:10.1097/00002030-199607000-00005
22. Giorgi JV, Liu Z, Hultin LE, Cumberland WG, Hennessey K, Detels R. Elevated levels of CD38+ CD8+ T cells in HIV infection add to the prognostic value of low CD4+ T cell levels: results of 6 years of follow-up. The Los Angeles Center, Multicenter AIDS Cohort Study. J Acquir Immune Defic Syndr (1993) 6:904–12.
23. Lien E, Aukrust P, Sundan A, Muller F, Froland SS, Espevik T. Elevated levels of serum-soluble CD14 in human immunodeficiency virus type 1 (HIV-1) infection: correlation to disease progression and clinical events. Blood (1998) 92(6):2084–92.
24. Godfried MH, van der Poll T, Jansen J, Romijin JA, Schattenkerk JK, Endert E, et al. Soluble receptors for tumour necrosis factor: a putative marker of disease progression in HIV infection. AIDS (1993) 7(1):33–6. doi:10.1097/00002030-199301000-00005
25. Zangerle R, Fuchs D, Sarcletti M, Gallati H, Reibnegger G, Wachter H, et al. Increased concentrations of soluble tumor necrosis factor receptor 75 but not of soluble intercellular adhesion molecule-1 are associated with the decline of CD4+ lymphocytes in HIV infection. Clin Immunol Immunopathol (1994) 72(3):328–34. doi:10.1006/clin.1994.1149
26. Zangerle R, Fuchs D, Reibnegger G, Fritsch P, Wachter H. Markers for disease progression in intravenous drug users infected with HIV-1. AIDS (1991) 5(8):985–91. doi:10.1097/00002030-199108000-00010
27. von Sydow M, Sonnerborg A, Gaines H, Strannegard O. Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res Hum Retroviruses (1991) 7(4):375–80. doi:10.1089/aid.1991.7.375
28. Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, et al. Shorter survival in advanced Human Immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis (1999) 179:859–70. doi:10.1086/314660
29. Zangerle R, Steinhuber S, Sarcletti M, Dierich MP, Wachter H, Fuchs D, et al. Serum HIV-1 RNA levels compared to soluble markers of immune activation to predict disease progression in HIV-1-infected individuals. Int Arch Allergy Immunol (1998) 116(3):228–39. doi:10.1159/000023949
30. Liu Z, Cumberland WG, Hultin LE, Kaplan AH, Detels R, Giorgi JV. CD8+ T lymphocyte activation in HIV-1 disease reflects an aspect of pathogenesis distinct from viral burden and immunodeficiency. J Acquir Immune Defic Syndr (1998) 18:332–40. doi:10.1097/00042560-199808010-00004
31. Hazenberg MD, Otto SA, van Benthem BH, Roos MT, Coutinho RA, Lange JM, et al. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS (2003) 17(13):1881–8. doi:10.1097/00002030-200309050-00006
32. Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood (2004) 104(4):942–7. doi:10.1182/blood-2003-09-3333
33. Marlink R, Kanki P, Thior I, Travers K, Eisen G, Siby T, et al. Reduced rate of disease development after HIV-2 infection as compared to HIV-1. Science (1994) 265:1587–90. doi:10.1126/science.7915856
34. Schramm B, Penn ML, Palacios EH, Grant RM, Kirchhoff F, Goldsmith MA. Cytopathicity of human immunodeficiency virus type 2 (HIV-2) in human lymphoid tissue is coreceptor dependent and comparable to that of HIV-1. J Virol (2000) 74:9594–600. doi:10.1128/JVI.74.20.9594-9600.2000
35. Grossman Z, Meier-Schellersheim M, Sousa AE, Victorino RMM, Paul WE. CD4 T-cell depletion in HIV infection: are we closer to understanding the cause? Nat Med (2002) 8:319–23. doi:10.1038/nm0402-319
36. Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RM. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J Immunol (2002) 169(6):3400–6.
37. Chakrabarti LA, Lewin SR, Zhang L, Gettie A, Luckay A, Martin LN, et al. Normal T cell turnover in sooty mangabeys harboring active simian immunodeficiency virus infection. J Virol (2000) 74(3):1209–23. doi:10.1128/JVI.74.3.1209-1223.2000
38. Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, McClure HM, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity (2003) 18(3):441–52. doi:10.1016/S1074-7613(03)00060-8
39. Hazenberg MD, Hamann D, Schuitemaker H, Miedema F. T cell depletion in HIV-1 infection: how CD4+ T cells go out of stock. Nat Immunol (2000) 1(4):285–9. doi:10.1038/79724
40. Pedersen C, Lindhardt BO, Jensen BL, Lauritzen E, Gerstoft J, Dickmeiss E, et al. Clinical course of primary HIV infection: consequences for subsequent course of infection. BMJ (1989) 299(6692):154–7. doi:10.1136/bmj.299.6692.154
41. Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol (2009) 83(8):3719–33. doi:10.1128/JVI.01844-08
42. Gaines H, von Sydow MA, von Stedingk LV, Biberfeld G, Bottiger B, Hansson LO, et al. Immunological changes in primary HIV-1 infection. AIDS (1990) 4(10):995–9. doi:10.1097/00002030-199010000-00008
43. Douek DC, Picker LJ, Koup RA. T cell dynamics in HIV-1 infection 14. Annu Rev Immunol (2003) 21:265–304. doi:10.1146/annurev.immunol.21.120601.141053
44. Clark DR, De Boer RJ, Wolthers KC, Miedema F. T cell dynamics in HIV-1 infection. Adv Immunol (1999) 73:301–27. doi:10.1016/S0065-2776(08)60789-0
45. Elbim C, Pillet S, Prevost MH, Preira A, Girard PM, Rogine N, et al. Redox and activation status of monocytes from human immunodeficiency virus-infected patients: relationship with viral load. J Virol (1999) 73(6):4561–6.
46. Elbim C, Prevot MH, Bouscarat F, Franzini E, Chollet-Martin S, Hakim J, et al. Polymorphonuclear neutrophils from human immunodeficiency virus-infected patients show enhanced activation, diminished fMLP-induced L-selectin shedding, and an impaired oxidative burst after cytokine priming. Blood (1994) 84(8):2759–66.
47. Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med (2011) 62:141–55. doi:10.1146/annurev-med-042909-093756
48. Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science (1998) 280(5362):427–31. doi:10.1126/science.280.5362.427
49. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med (2004) 200(6):749–59. doi:10.1084/jem.20040874
50. Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, et al. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature (2005) 434(7037):1148–52.
51. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med (2006) 12(12):1365–71. doi:10.1038/nm1511
52. Gordon SN, Cervasi B, Odorizzi P, Silverman R, Aberra F, Ginsberg G, et al. Disruption of intestinal CD4+ T cell homeostasis is a key marker of systemic CD4+ T cell activation in HIV-infected individuals. J Immunol (2010) 185(9):5169–79. doi:10.4049/jimmunol.1001801
53. Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol (2006) 7(3):235–9. doi:10.1038/ni1316
54. Gordon SN, Klatt NR, Bosinger SE, Brenchley JM, Milush JM, Engram JC, et al. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J Immunol (2007) 179(5):3026–34.
55. Pandrea IV, Gautam R, Ribeiro RM, Brenchley JM, Butler IF, Pattison M, et al. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J Immunol (2007) 179(5):3035–46.
56. Breed MW, Jordan AP, Aye PP, Lichtveld CF, Midkiff CC, Schiro FR, et al. Loss of a tyrosine-dependent trafficking motif in the simian immunodeficiency virus envelope cytoplasmic tail spares mucosal CD4 cells but does not prevent disease progression. J Virol (2013) 87(3):1528–43. doi:10.1128/JVI.01928-12
57. Marchetti G, Bellistri GM, Borghi E, Tincati C, Ferramosca S, La FM, et al. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. AIDS (2008) 22(15):2035–8. doi:10.1097/QAD.0b013e3283112d29
58. Deeks SG, Phillips AN. HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ (2009) 338:a3172. doi:10.1136/bmj.a3172
59. Kalayjian RC, Machekano RN, Rizk N, Robbins GK, Gandhi RT, Rodriguez BA, et al. Pretreatment levels of soluble cellular receptors and interleukin-6 are associated with HIV disease progression in subjects treated with highly active antiretroviral therapy. J Infect Dis (2010) 201(12):1796–805. doi:10.1086/652750
60. Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis (2009) 199(8):1177–85. doi:10.1086/597476
61. Cassol E, Malfeld S, Mahasha P, van der Merwe MS, Cassol S, Seebregts C, et al. Persistent microbial translocation and immune activation in HIV-1-infected South Africans receiving combination antiretroviral therapy. J Infect Dis (2010) 202(5):723–33. doi:10.1086/655229
62. Wallet MA, Rodriguez CA, Yin L, Saporta S, Chinratanapisit S, Hou W, et al. Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T-cell activation following therapy. AIDS (2010) 24(9):1281–90. doi:10.1097/QAD.0b013e328339e228
63. Baroncelli S, Galluzzo CM, Pirillo MF, Mancini MG, Weimer LE, Andreotti M, et al. Microbial translocation is associated with residual viral replication in HAART-treated HIV+ subjects with <50copies/ml HIV-1 RNA. J Clin Virol (2009) 46(4):367–70. doi:10.1016/j.jcv.2009.09.011
64. Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood (2008) 112(7):2826–35. doi:10.1182/blood-2008-05-159301
65. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell (2010) 140(6):845–58. doi:10.1016/j.cell.2010.02.021
66. Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog (2009) 5(2):e1000295. doi:10.1371/journal.ppat.1000295
67. Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med (2008) 14(4):421–8. doi:10.1038/nm1743
68. Micci L, Cervasi B, Ende ZS, Iriele RI, Reyes-Aviles E, Vinton C, et al. Paucity of IL-21-producing CD4(+) T cells is associated with Th17 cell depletion in SIV infection of rhesus macaques. Blood (2012) 120(19):3925–35. doi:10.1182/blood-2012-04-420240
69. Pallikkuth S, Micci L, Ende ZS, Iriele RI, Cervasi B, Lawson B, et al. Maintenance of intestinal Th17 cells and reduced microbial translocation in SIV-infected rhesus macaques treated with interleukin (IL)-21. PLoS Pathog (2013) 9(7):e1003471. doi:10.1371/journal.ppat.1003471
70. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (2004) 303(5663):1526–9. doi:10.1126/science.1093620
71. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303(5663):1529–31. doi:10.1126/science.1093616
72. Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via toll-like receptor-viral RNA interactions. J Clin Invest (2005) 115(11):3265–75. doi:10.1172/JCI26032
73. Meier A, Alter G, Frahm N, Sidhu H, Li B, Bagchi A, et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded toll-like receptor ligands. J Virol (2007) 81(15):8180–91. doi:10.1128/JVI.00421-07
74. Fonteneau JF, Larsson M, Beignon AS, McKenna K, DaSilva I, Amara A, et al. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J Virol (2004) 78(10):5223–32. doi:10.1128/JVI.78.10.5223-5232.2004
75. Hardy AW, Graham DR, Shearer GM, Herbeuval JP. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by toll-like receptor 7-induced IFN-alpha. Proc Natl Acad Sci U S A (2007) 104(44):17453–8. doi:10.1073/pnas.0707244104
76. Alter G, Suscovich TJ, Teigen N, Meier A, Streeck H, Brander C, et al. Single-stranded RNA derived from HIV-1 serves as a potent activator of NK cells. J Immunol (2007) 178(12):7658–66.
77. Sedaghat AR, German J, Teslovich TM, Cofrancesco J Jr, Jie CC, Talbot CC Jr, et al. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. J Virol (2008) 82(4):1870–83. doi:10.1128/JVI.02228-07
78. Hyrcza MD, Kovacs C, Loutfy M, Halpenny R, Heisler L, Yang S, et al. Distinct transcriptional profiles in ex vivo CD4+ and CD8+ T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J Virol (2007) 81(7):3477–86. doi:10.1128/JVI.01552-06
79. Cohen Stuart JW, Hazenberg MD, Hamann D, Otto SA, Borleffs JC, Miedema F, et al. The dominant source of CD4+ and CD8+ T-cell activation in HIV infection is antigenic stimulation. J Acquir Immune Defic Syndr (2000) 25(3):203–11. doi:10.1097/00126334-200011010-00001
80. Bucy RP, Hockett RD, Derdeyn CA, Saag MS, Squires K, Sillers M, et al. Initial increase in blood CD4+ lymphocytes after HIV antiretroviral therapy reflects redistribution from lymphoid tissues. J Clin Invest (1999) 103(10):1391–8. doi:10.1172/JCI5863
81. Wolf K, Tsakiris DA, Weber R, Erb P, Battegay M. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J Infect Dis (2002) 185(4):456–62. doi:10.1086/338572
82. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol (2005) 23:307–36. doi:10.1146/annurev.immunol.23.021704.115843
83. Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest (2009) 119(12):3544–55. doi:10.1172/JCI40093
84. Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood (2006) 107(6):2423–31. doi:10.1182/blood-2005-07-2709
85. Bjorck P. Dendritic cells exposed to herpes simplex virus in vivo do not produce IFN-alpha after rechallenge with virus in vitro and exhibit decreased T cell alloreactivity. J Immunol (2004) 172(9):5396–404.
86. O’Brien M, Manches O, Sabado RL, Baranda SJ, Wang Y, Marie I, et al. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-alpha-producing and partially matured phenotype. J Clin Invest (2011) 121(3):1088–101. doi:10.1172/JCI44960
87. Rey-Cuille MA, Berthier JL, Bomsel-Demontoy MC, Chaduc Y, Montagnier L, Hovanessian AG, et al. Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease. J Virol (1998) 72(5):3872–86.
88. Hartung S, Boller K, Cichutek K, Norley SG, Kurth R. Quantitation of a lentivirus in its natural host: simian immunodeficiency virus in African green monkeys. J Virol (1992) 66(4):2143–9.
89. Goldstein S, Ourmanov I, Brown CR, Beer BE, Elkins WR, Plishka R, et al. Wide range of viral load in healthy African green monkeys naturally infected with simian immunodeficiency virus. J Virol (2000) 74(24):11744–53. doi:10.1128/JVI.74.24.11744-11753.2000
90. Diop OM, Gueye A, Dias-Tavares M, Kornfeld C, Faye A, Ave P, et al. High levels of viral replication during primary simian immunodeficiency virus SIVagm infection are rapidly and strongly controlled in African green monkeys. J Virol (2000) 74(16):7538–47. doi:10.1128/JVI.74.16.7538-7547.2000
91. Broussard SR, Staprans SI, White R, Whitehead EM, Feinberg MB, Allan JS. Simian immunodeficiency virus replicates to high levels in naturally infected African green monkeys without inducing immunologic or neurologic disease. J Virol (2001) 75(5):2262–75. doi:10.1128/JVI.75.5.2262-2275.2001
92. Dunham R, Pagliardini P, Gordon S, Sumpter B, Engram J, Moanna A, et al. The AIDS resistance of naturally SIV-infected sooty mangabeys is independent of cellular immunity to the virus. Blood (2006) 108(1):209–17. doi:10.1182/blood-2005-12-4897
93. Li B, Stefano-Cole K, Kuhrt DM, Gordon SN, Else JG, Mulenga J, et al. Nonpathogenic simian immunodeficiency virus infection of sooty mangabeys is not associated with high levels of autologous neutralizing antibodies. J Virol (2010) 84(12):6248–53. doi:10.1128/JVI.00295-10
94. Sodora DL, Allan JS, Apetrei C, Brenchley JM, Douek DC, Else JG, et al. Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts. Nat Med (2009) 15(8):861–5. doi:10.1038/nm.2013
95. Pandrea I, Gaufin T, Brenchley JM, Gautam R, Monjure C, Gautam A, et al. Cutting edge: experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J Immunol (2008) 181(10):6687–91.
96. Canary LA, Vinton CL, Morcock DR, Pierce JB, Estes JD, Brenchley JM, et al. Rate of AIDS progression is associated with gastrointestinal dysfunction in simian immunodeficiency virus-infected pigtail macaques. J Immunol (2013) 190(6):2959–65. doi:10.4049/jimmunol.1202319
97. Milush JM, Reeves JD, Gordon SN, Zhou D, Muthukumar A, Kosub DA, et al. Virally induced CD4+ T cell depletion is not sufficient to induce AIDS in a natural host. J Immunol (2007) 179(5):3047–56.
98. Barry AP, Silvestri G, Safrit JT, Sumpter B, Kozyr N, McClure HM, et al. Depletion of CD8+ cells in sooty mangabey monkeys naturally infected with simian immunodeficiency virus reveals limited role for immune control of virus replication in a natural host species. J Immunol (2007) 178(12):8002–12.
99. Gordon SN, Dunham RM, Engram JC, Estes J, Wang Z, Klatt NR, et al. Short-lived infected cells support virus replication in sooty mangabeys naturally infected with simian immunodeficiency virus: implications for AIDS pathogenesis. J Virol (2008) 82(7):3725–35. doi:10.1128/JVI.02408-07
100. Kornfeld C, Ploquin MJ, Pandrea I, Faye A, Onanga R, Apetrei C, et al. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J Clin Invest (2005) 115(4):1082–91. doi:10.1172/JCI200523006
101. Silvestri G, Feinberg MB. Turnover of lymphocytes and conceptual paradigms in HIV infection. J Clin Invest (2003) 112(6):821–4. doi:10.1172/JCI200319799
102. Mandl JN, Barry AP, Vanderford TH, Kozyr N, Chavan R, Klucking S, et al. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med (2008) 14(10):1077–87. doi:10.1038/nm.1871
103. Mandl JN, Akondy R, Lawson B, Kozyr N, Staprans SI, Ahmed R, et al. Distinctive TLR7 signaling, type I IFN production, and attenuated innate and adaptive immune responses to yellow fever virus in a primate reservoir host. J Immunol (2011) 186(11):6406–16. doi:10.4049/jimmunol.1001191
104. Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest (2009) 119(12):3556–72. doi:10.1172/JCI40115
105. Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, et al. Down-regulation of robust acute type I IFN responses distinguishes non-pathogenic SIV infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol (2010) 84(15):7886–91. doi:10.1128/JVI.02612-09
106. Lederer S, Favre D, Walters KA, Proll S, Kanwar B, Kasakow Z, et al. Transcriptional profiling in pathogenic and non-pathogenic SIV infections reveals significant distinctions in kinetics and tissue compartmentalization. PLoS Pathog (2009) 5(2):e1000296. doi:10.1371/journal.ppat.1000296
107. Estes JD, Gordon SN, Zeng M, Chahroudi AM, Dunham RM, Staprans SI, et al. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J Immunol (2008) 180(10):6798–807.
108. Meythaler M, Martinot A, Wang Z, Pryputniewicz S, Kasheta M, Ling B, et al. Differential CD4+ T-lymphocyte apoptosis and bystander T-cell activation in rhesus macaques and sooty mangabeys during acute simian immunodeficiency virus infection. J Virol (2009) 83(2):572–83. doi:10.1128/JVI.01715-08
109. Manches O, Bhardwaj N. Resolution of immune activation defines nonpathogenic SIV infection. J Clin Invest (2009) 119(12):3512–5. doi:10.1172/JCI41509
110. Brenchley JM, Silvestri G, Douek DC. Nonprogressive and progressive primate immunodeficiency lentivirus infections. Immunity (2010) 32(6):737–42. doi:10.1016/j.immuni.2010.06.004
111. Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog (2010) 6(8):e1001052. doi:10.1371/journal.ppat.1001052
112. Paiardini M, Cervasi B, Reyes-Aviles E, Micci L, Ortiz AM, Chahroudi A, et al. Low levels of SIV infection in sooty mangabey central memory CD4(+) T cells are associated with limited CCR5 expression. Nat Med (2011) 17(7):830–6. doi:10.1038/nm.2395
113. Beaumier CM, Harris LD, Goldstein S, Klatt NR, Whitted S, McGinty J, et al. CD4 downregulation by memory CD4+ T cells in vivo renders African green monkeys resistant to progressive SIVagm infection. Nat Med (2009) 15(8):879–85. doi:10.1038/nm.1970
114. Ganesan A, Chattopadhyay PK, Brodie TM, Qin J, Gu W, Mascola JR, et al. Immunologic and virologic events in early HIV infection predict subsequent rate of progression. J Infect Dis (2010) 201(2):272–84. doi:10.1086/649430
115. Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol (2008) 8(11):889–95. doi:10.1038/nri2432
116. Medzhitov R. Damage control in host-pathogen interactions. Proc Natl Acad Sci U S A (2009) 106(37):15525–6. doi:10.1073/pnas.0908451106
117. Choudhary SK, Vrisekoop N, Jansen CA, Otto SA, Schuitemaker H, Miedema F, et al. Low immune activation despite high levels of pathogenic human immunodeficiency virus type 1 results in long-term asymptomatic disease. J Virol (2007) 81(16):8838–42. doi:10.1128/JVI.02663-06
118. Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, Martinez R, et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Invest (2011) 121(6):2391–400. doi:10.1172/JCI45235
119. Hardy GA, Sieg S, Rodriguez B, Anthony D, Asaad R, Jiang W, et al. Interferon-alpha is the primary plasma type-I IFN in HIV-1 infection and correlates with immune activation and disease markers. PLoS One (2013) 8(2):e56527. doi:10.1371/journal.pone.0056527
120. Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, et al. Sex differences in the toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med (2009) 15(8):955–9. doi:10.1038/nm.2004
121. Chang JJ, Woods M, Lindsay RJ, Doyle EH, Griesbeck M, Chan ES, et al. Higher expression of several interferon-stimulated genes in HIV-1-infected females after adjusting for the level of viral replication. J Infect Dis (2013) 208(5):830–8. doi:10.1093/infdis/jit262
122. Sterling TR, Vlahov D, Astemborski J, Hoover DR, Margolick JB, Quinn TC. Initial plasma HIV-1 RNA levels and progression to AIDS in women and men. N Engl J Med (2001) 344(10):720–5. doi:10.1056/NEJM200103083441003
123. Chang J, Lindsay RJ, Kulkarni S, Lifson JD, Carrington M, Altfeld M. Polymorphisms in interferon regulatory factor 7 reduce interferon-alpha responses of plasmacytoid dendritic cells to HIV-1. AIDS (2011) 25(5):715–7. doi:10.1097/QAD.0b013e328343c186
124. Murray SM, Down CM, Boulware DR, Stauffer WM, Cavert WP, Schacker TW, et al. Reduction of immune activation with chloroquine therapy during chronic HIV infection. J Virol (2010) 84(22):12082–6. doi:10.1128/JVI.01466-10
125. Gonzalez-Gay MA, Gonzalez-Juanatey C, Vazquez-Rodriguez TR, Miranda-Filloy JA, Llorca J. Insulin resistance in rheumatoid arthritis: the impact of the anti-TNF-alpha therapy. Ann N Y Acad Sci (2010) 1193:153–9. doi:10.1111/j.1749-6632.2009.05287.x
126. Moreland LW, Curtis JR. Systemic nonarticular manifestations of rheumatoid arthritis: focus on inflammatory mechanisms. Semin Arthritis Rheum (2009) 39(2):132–43. doi:10.1016/j.semarthrit.2008.08.003
127. McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol (2009) 6(6):410–7. doi:10.1038/nrcardio.2009.57
128. Roederer M, Gregson Dubs J, Anderson MT, Raju PA, Herzenberg LA, Herzenberg L. CD8 naive T cell counts decrease progressively in HIV-infected adults. J Clin Invest (1995) 95:2061–6. doi:10.1172/JCI117892
129. Picker LJ, Hagen SI, Lum R, Reed-Inderbitzin EF, Daly LM, Sylwester AW, et al. Insufficient production and tissue delivery of CD4+ memory T cells in rapidly progressive simian immunodeficiency virus infection. J Exp Med (2004) 200(10):1299–314. doi:10.1084/jem.20041049
130. Ribeiro RM, Mohri H, Ho DD, Perelson AS. In vivo dynamics of T cell activation, proliferation, and death in HIV-1 infection: why are CD4+ but not CD8+ T cells depleted? Proc Natl Acad Sci U S A (2002) 99(24):15572–7. doi:10.1073/pnas.242358099
131. Clark DR, Repping S, Pakker NG, Prins JM, Notermans DW, Wit FW, et al. T-cell progenitor function during progressive human immunodeficiency virus-1 infection and after antiretroviral therapy. Blood (2000) 96(1):242–9.
132. Schacker TW, Nguyen PL, Beilman GJ, Wolinsky S, Larson M, Reilly C, et al. Collagen deposition in HIV-1 infected lymphatic tissues and T cell homeostasis. J Clin Invest (2002) 110(8):1133–9. doi:10.1172/JCI200216413
133. Estes JD, Wietgrefe S, Schacker T, Southern P, Beilman G, Reilly C, et al. Simian immunodeficiency virus-induced lymphatic tissue fibrosis is mediated by transforming growth factor beta 1-positive regulatory T cells and begins in early infection. J Infect Dis (2007) 195(4):551–61. doi:10.1086/510852
134. Zeng M, Smith AJ, Wietgrefe SW, Southern PJ, Schacker TW, Reilly CS, et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest (2011) 121(3):998–1008. doi:10.1172/JCI45157
135. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443(7109):350–4. doi:10.1038/nature05115
136. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med (2006) 12(10):1198–202. doi:10.1038/nm1106-1329b
137. Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol (2007) 8(11):1246–54. doi:10.1038/ni1515
138. Terpstra FG, Al BJ, Roos MT, De Wolf F, Goudsmit J, Schellekens PT, et al. Longitudinal study of leukocyte functions in homosexual men seroconverted for HIV-1: rapid and persistent loss of B-cell function after HIV-1 infection. Eur J Immunol (1989) 19:667–73. doi:10.1002/eji.1830190415
139. Lane HC, Depper JL, Greene WC, Whalen G, Waldmann TA, Fauci AS. Qualitative analysis of immune function in patients with the acquired immunodeficiency syndrome. N Engl J Med (1985) 313:79–84. doi:10.1056/NEJM198507113130204
140. Martinez-Maza O, Crabb E, Mitsuyasu RT, Fahey JL, Giorgi JV. Infection with the human immunodeficiency virus (HIV) is associated with in vivo increase in B lymphocyte activation and immaturity. J Immunol (1987) 138:3720–4.
141. Cagigi A, Nilsson A, De Milito A, Chiodi F. B cell immunopathology during HIV-1 infection: lessons to learn for HIV-1 vaccine design. Vaccine (2008) 26(24):3016–25. doi:10.1016/j.vaccine.2007.11.063
142. Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol (2009) 9(4):235–45. doi:10.1038/nri2524
143. Boliar S, Murphy MK, Tran TC, Carnathan DG, Armstrong WS, Silvestri G, et al. B-lymphocyte dysfunction in chronic HIV-1 infection does not prevent cross-clade neutralization breadth. J Virol (2012) 86(15):8031–40. doi:10.1128/JVI.00771-12
144. Zhang Z, Xu X, Lu J, Zhang S, Gu L, Fu J, et al. B and T lymphocyte attenuator down-regulation by HIV-1 depends on type I interferon and contributes to T-cell hyperactivation. J Infect Dis (2011) 203(11):1668–78. doi:10.1093/infdis/jir165
145. Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med (2013) 19(4):494–9. doi:10.1038/nm.3109
146. Younes SA, Yassine-Diab B, Dumont AR, Boulassel MR, Grossman Z, Routy JP, et al. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J Exp Med (2003) 198(12):1909–22. doi:10.1084/jem.20031598
147. McNeil AC, Shupert WL, Iyasere CA, Hallahan CW, Mican JA, Davey RT Jr, et al. High-level HIV-1 viremia suppresses viral antigen-specific CD4(+) T cell proliferation. Proc Natl Acad Sci U S A (2001) 98(24):13878–83. doi:10.1073/pnas.251539598
148. Kaufman D, Lichterfeld M, Altfeld M, Allen TM, Johnston M, Lee P, et al. Limited durability of immune control following treated acute HIV infection. PLoS Med (2004) 1(2):e36. doi:10.1371/journal.pmed.0010036
149. Jansen CA, De Cuyper IM, Steingrover R, Jurriaans S, Sankatsing SUC, Prins JM, et al. Analysis of the effect of highly active anti-retroviral therapy during acute HIV-1 infection on HIV-specific CD4+ T-cell functions. AIDS (2005) 19:1145–54. doi:10.1097/01.aids.0000176214.17990.94
150. Jansen CA, Piriou E, De Cuyper IM, van Dort K, Lange JM, Miedema F, et al. Long-term highly active antiretroviral therapy in chronic HIV-1 infection: evidence for reconstitution of antiviral immunity. Antivir Ther (2006) 11(1):105–16.
151. Harari A, Petitpierre S, Vallelian F, Pantaleo G. Skewed representation of functionally distinct populations of virus-specific CD4 T cells in HIV-1-infected subjects with progressive disease: changes after antiretroviral therapy. Blood (2004) 103(3):966–72. doi:10.1182/blood-2003-04-1203
152. Harari A, Vallelian F, Pantaleo G. Phenotypic heterogeneity of antigen-specific CD4 T cells under different conditions of antigen persistence and antigen load. Eur J Immunol (2004) 34(12):3525–33. doi:10.1002/eji.200425324
153. Harari A, Vallelian F, Meylan PR, Pantaleo G. Functional heterogeneity of memory CD4 T cell responses in different conditions of antigen exposure and persistence. J Immunol (2005) 174(2):1037–45.
154. Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol (2002) 3(11):1061–8. doi:10.1038/ni845
155. Champagne P, Ogg GS, King AS, Knabenhans C, Ellefsen K, Nobile M, et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature (2001) 410:106–11. doi:10.1038/35065118
156. Betts MR, Krowka JF, Kepler TB, Davidian M, Christopherson C, Kwok S, et al. Human immunodeficiency virus type 1-specific cytotoxic T lymphocyte activity is inversely correlated with HIV type 1 viral load in HIV type 1-infected long-term survivors. AIDS Res Hum Retroviruses (1999) 15(13):1219–28. doi:10.1089/088922299310313
157. Betts MR, Ambrozak DR, Douek DC, Bonhoeffer S, Brenchley JM, Casazza JP, et al. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: relationship to viral load in untreated HIV infection. J Virol (2001) 75(24):11983–91. doi:10.1128/JVI.75.24.11983-11991.2001
158. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med (2002) 8(4):379–85. doi:10.1038/nm0402-379
159. van Baarle D, Tsegaye A, Miedema F, Akbar A. Significance of senescence for virus-specific memory T cell responses: rapid ageing during chronic stimulation of the immune system. Immunol Lett (2005) 97(1):19–29. doi:10.1016/j.imlet.2004.10.003
160. Klein MR, van Baalen CA, Holwerda AM, Kerkhof-Garde SR, Bende RJ, Keet IPM, et al. Kinetics of Gag-specific CTL responses during the clinical course of HIV-1 infection: a longitudinal analysis of rapid progressors and long-term asymptomatics. J Exp Med (1995) 181:1365–72. doi:10.1084/jem.181.4.1365
161. Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science (2010) 330(6010):1551–7. doi:10.1126/science.1195271
162. Borghans JA, Molgaard A, de Boer RJ, Kesmir C. HLA alleles associated with slow progression to AIDS truly prefer to present HIV-1 p24. PLoS One (2007) 2(9):e920. doi:10.1371/journal.pone.0000920
163. Dahirel V, Shekhar K, Pereyra F, Miura T, Artyomov M, Talsania S, et al. From the cover: coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc Natl Acad Sci U S A (2011) 108(28):11530–5. doi:10.1073/pnas.1105315108
164. Buseyne F, Le CJ, Corre B, Porrot F, Burgard M, Rouzioux C, et al. Inverse correlation between memory Gag-specific cytotoxic T lymphocytes and viral replication in human immunodeficiency virus-infected children. J Infect Dis (2002) 186(11):1589–96. doi:10.1086/345482
165. Edwards BH, Bansal A, Sabbaj S, Bakari J, Mulligan MJ, Goepfert PA. Magnitude of functional CD8+ T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J Virol (2002) 76(5):2298–305. doi:10.1128/jvi.76.5.2298-2305.2002
166. Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med (2007) 13(1):46–53. doi:10.1038/nm1520
167. Masemola A, Mashishi T, Khoury G, Mohube P, Mokgotho P, Vardas E, et al. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J Virol (2004) 78(7):3233–43. doi:10.1128/JVI.78.7.3233-3243.2004
168. Novitsky V, Gilbert P, Peter T, McLane MF, Gaolekwe S, Rybak N, et al. Association between virus-specific T-cell responses and plasma viral load in human immunodeficiency virus type 1 subtype C infection. J Virol (2003) 77(2):882–90. doi:10.1128/JVI.77.2.882-890.2003
169. Zuniga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, et al. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J Virol (2006) 80(6):3122–5. doi:10.1128/JVI.80.6.3122-3125.2006
170. Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, et al. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog (2012) 8(11):e1003041. doi:10.1371/journal.ppat.1003041
171. Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, et al. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J Exp Med (2008) 205(5):1009–17. doi:10.1084/jem.20072457
172. Schellens IM, Borghans JA, Jansen CA, De Cuyper IM, Geskus RB, van Baarle D, et al. Abundance of early functional HIV-specific CD8+ T cells does not predict AIDS-free survival time. PLoS One (2008) 3(7):e2745. doi:10.1371/journal.pone.0002745
173. Jansen CA, De Cuyper IM, Hooibrink B, van der Bij AK, van Baarle D, Miedema F. Prognostic value of HIV-1 Gag-specific CD4+ T-cell responses for progression to AIDS analysed in a prospective cohort study. Blood (2005) 107(4):1427–33. doi:10.1182/blood-2005-07-2907
174. Brumme Z, Wang B, Nair K, Brumme C, de Pierres C, Reddy S, et al. Impact of select immunologic and virologic biomarkers on CD4 cell count decrease in patients with chronic HIV-1 subtype C infection: results from Sinikithemba Cohort, Durban, South Africa. Clin Infect Dis (2009) 49(6):956–64. doi:10.1086/605503
175. Jansen CA, van Baarle D, Miedema F. HIV-specific CD4+ T cells and viremia: who’s in control? Trends Immunol (2006) 27(3):119–24. doi:10.1016/j.it.2006.01.004
176. Henry K, Melroe H, Huebsch J, Hermundson J, Levine C, Swensen L, et al. Severe premature coronary artery disease with protease inhibitors. Lancet (1998) 351(9112):1328. doi:10.1016/S0140-6736(05)79053-X
177. Holmberg SD, Moorman AC, Williamson JM, Tong TC, Ward DJ, Wood KC, et al. Protease inhibitors and cardiovascular outcomes in patients with HIV-1. Lancet (2002) 360(9347):1747–8. doi:10.1016/S0140-6736(02)11672-2
178. Mary-Krause M, Cotte L, Simon A, Partisani M, Costagliola D. Increased risk of myocardial infarction with duration of protease inhibitor therapy in HIV-infected men. AIDS (2003) 17(17):2479–86. doi:10.1097/00002030-200311210-00010
179. Friis-Moller N, Sabin CA, Weber R, d’Arminio MA, El-Sadr WM, Reiss P, et al. Combination antiretroviral therapy and the risk of myocardial infarction. N Engl J Med (2003) 349(21):1993–2003. doi:10.1056/NEJMoa030218
180. Friis-Moller N, Reiss P, Sabin CA, Weber R, Monforte A, El-Sadr W, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med (2007) 356(17):1723–35. doi:10.1056/NEJMoa062744
181. Bozzette SA, Ake CF, Tam HK, Chang SW, Louis TA. Cardiovascular and cerebrovascular events in patients treated for human immunodeficiency virus infection. N Engl J Med (2003) 348(8):702–10. doi:10.1056/NEJMoa022048
182. El-Sadr WM, Lundgren JD, Neaton JD, Gordin F, Abrams D, Arduino RC, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med (2006) 355(22):2283–96. doi:10.1056/NEJMoa062360
183. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell (2010) 140(6):771–6. doi:10.1016/j.cell.2010.03.006
184. Hsue PY, Hunt PW, Sinclair E, Bredt B, Franklin A, Killian M, et al. Increased carotid intima-media thickness in HIV patients is associated with increased cytomegalovirus-specific T-cell responses. AIDS (2006) 20(18):2275–83. doi:10.1097/QAD.0b013e3280108704
185. Hsue PY, Hunt PW, Schnell A, Kalapus SC, Hoh R, Ganz P, et al. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS (2009) 23(9):1059–67. doi:10.1097/QAD.0b013e32832b514b
186. Tebas P, Henry WK, Matining R, Weng-Cherng D, Schmitz J, Valdez H, et al. Metabolic and immune activation effects of treatment interruption in chronic HIV-1 infection: implications for cardiovascular risk. PLoS One (2008) 3(4):e2021. doi:10.1371/journal.pone.0002021
187. Phillips AN, Neaton J, Lundgren JD. The role of HIV in serious diseases other than AIDS. AIDS (2008) 22(18):2409–18. doi:10.1097/QAD.0b013e3283174636
188. Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, et al. Antibody neutralization and escape by HIV-1. Nature (2003) 422(6929):307–12. doi:10.1038/nature01470
189. Targher G, Bertolini L, Padovani R, Rodella S, Arcaro G, Day C. Differences and similarities in early atherosclerosis between patients with non-alcoholic steatohepatitis and chronic hepatitis B and C. J Hepatol (2007) 46(6):1126–32. doi:10.1016/j.jhep.2007.01.021
190. Yonkers NL, Sieg S, Rodriguez B, Anthony DD. Reduced naive CD4 T cell numbers and impaired induction of CD27 in response to T cell receptor stimulation reflect a state of immune activation in chronic hepatitis C virus infection. J Infect Dis (2011) 203(5):635–45. doi:10.1093/infdis/jiq101
191. Levy Y, Gahery-Segard H, Durier C, Lascaux AS, Goujard C, Meiffredy V, et al. Immunological and virological efficacy of a therapeutic immunization combined with interleukin-2 in chronically HIV-1 infected patients. AIDS (2005) 19(3):279–86.
192. Levy Y, Durier C, Lascaux AS, Meiffredy V, Gahery-Segard H, Goujard C, et al. Sustained control of viremia following therapeutic immunization in chronically HIV-1-infected individuals. AIDS (2006) 20(3):405–13. doi:10.1097/01.aids.0000206504.09159.d3
193. Autran B, Murphy RL, Costagliola D, Tubiana R, Clotet B, Gatell J, et al. Greater viral rebound and reduced time to resume antiretroviral therapy after therapeutic immunization with the ALVAC-HIV vaccine (vCP1452). AIDS (2008) 22(11):1313–22. doi:10.1097/QAD.0b013e3282fdce94
194. Abrams D, Levy Y, Losso MH, Babiker A, Collins G, Cooper DA, et al. Interleukin-2 therapy in patients with HIV infection. N Engl J Med (2009) 361(16):1548–59. doi:10.1056/NEJMoa0903175
195. Levy Y, Lacabaratz C, Weiss L, Viard JP, Goujard C, Lelievre JD, et al. Enhanced T cell recovery in HIV-1-infected adults through IL-7 treatment. J Clin Invest (2009) 119(4):997–1007. doi:10.1172/JCI38052
196. Sportes C, Hakim FT, Memon SA, Zhang H, Chua KS, Brown MR, et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J Exp Med (2008) 205(7):1701–14. doi:10.1084/jem.20071681
197. Napolitano LA, Schmidt D, Gotway MB, Ameli N, Filbert EL, Ng MM, et al. Growth hormone enhances thymic function in HIV-1-infected adults. J Clin Invest (2008) 118(3):1085–98.
198. Sereti I, Dunham RM, Spritzler J, Aga E, Proschan MA, Medvik K, et al. IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood (2009) 113(25):6304–14. doi:10.1182/blood-2008-10-186601
199. Deeks SG, Barbour JD, Grant RM, Martin JN. Duration and predictors of CD4 T-cell gains in patients who continue combination therapy despite detectable plasma viremia. AIDS (2002) 16(2):201–7. doi:10.1097/00002030-200201250-00009
200. Kaufmann DE, Walker BD. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J Immunol (2009) 182(10):5891–7. doi:10.4049/jimmunol.0803771
201. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature (2009) 458(7235):206–10. doi:10.1038/nature07662
202. Dyavar SR, Velu V, Titanji K, Bosinger SE, Freeman GJ, Silvestri G, et al. PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J Clin Invest (2012) 122(5):1712–6. doi:10.1172/JCI60612
203. Cecchinato V, Tryniszewska E, Ma ZM, Vaccari M, Boasso A, Tsai WP, et al. Immune activation driven by CTLA-4 blockade augments viral replication at mucosal sites in simian immunodeficiency virus infection. J Immunol (2008) 180(8):5439–47.
204. Virgin HW, Walker BD. Immunology and the elusive AIDS vaccine. Nature (2010) 464(7286):224–31. doi:10.1038/nature08898
205. Burton DR, Weiss RA. AIDS/HIV. A boost for HIV vaccine design. Science (2010) 329(5993):770–3. doi:10.1126/science.1194693
206. Rizzardi GP, Harari A, Capiluppi B, Tambussi G, Ellefsen K, Ciuffreda D, et al. Treatment of primary HIV-1 infection with cyclosporin A coupled with highly active antiretroviral therapy. J Clin Invest (2002) 109(5):681–8. doi:10.1172/JCI0214522
207. Markowitz M, Vaida F, Hare CB, Boden D, Mohri H, Hecht FM, et al. The virologic and immunologic effects of cyclosporine as an adjunct to antiretroviral therapy in patients treated during acute and early HIV-1 infection. J Infect Dis (2010) 201(9):1298–302. doi:10.1086/651664
208. Vrisekoop N, Sankatsing SU, Jansen CA, Roos MT, Otto SA, Schuitemaker H, et al. Short communication: no detrimental immunological effects of mycophenolate mofetil and HAART in treatment-naive acute and chronic HIV-1-infected patients. AIDS Res Hum Retroviruses (2005) 21(12):991–6. doi:10.1089/aid.2005.21.991
209. Hennessy EJ, Parker AE, O’Neill LA. Targeting toll-like receptors: emerging therapeutics? Nat Rev Drug Discov (2010) 9(4):293–307. doi:10.1038/nrd3203
210. Hedayat M, Netea MG, Rezaei N. Targeting of toll-like receptors: a decade of progress in combating infectious diseases. Lancet Infect Dis (2011) 11(9):702–12. doi:10.1016/S1473-3099(11)70099-8
211. Tabb B, Morcock DR, Trubey CM, Quinones OA, Hao XP, Smedley J, et al. Reduced inflammation and lymphoid tissue immunopathology in rhesus macaques receiving anti-tumor necrosis factor treatment during primary simian immunodeficiency virus infection. J Infect Dis (2013) 207(6):880–92. doi:10.1093/infdis/jis643
212. Bissonnette R, Papp K, Maari C, Yao Y, Robbie G, White WI, et al. A randomized, double-blind, placebo-controlled, phase I study of MEDI-545, an anti-interferon-alfa monoclonal antibody, in subjects with chronic psoriasis. J Am Acad Dermatol (2010) 62(3):427–36. doi:10.1016/j.jaad.2009.05.042
213. Merrill JT, Wallace DJ, Petri M, Kirou KA, Yao Y, White WI, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon {alpha} monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann Rheum Dis (2011) 70(11):1905–13. doi:10.1136/ard.2010.144485
214. Gringeri A, Musicco M, Hermans P, Bentwich Z, Cusini M, Bergamasco A, et al. Active anti-interferon-alpha immunization: a European-Israeli, randomized, double-blind, placebo-controlled clinical trial in 242 HIV-1 – infected patients (the EURIS study). J Acquir Immune Defic Syndr Hum Retrovirol (1999) 20(4):358–70. doi:10.1097/00042560-199904010-00006
215. Tavel JA, Huang CY, Shen J, Metcalf JA, Dewar R, Shah A, et al. Interferon-alpha produces significant decreases in HIV load. J Interferon Cytokine Res (2010) 30(7):461–4. doi:10.1089/jir.2009.0090
216. Kovacs JA, Deyton L, Davey R, Falloon J, Zunich K, Lee D, et al. Combined zidovudine and interferon-alpha therapy in patients with Kaposi sarcoma and the acquired immunodeficiency syndrome (AIDS). Ann Intern Med (1989) 111(4):280–7. doi:10.7326/0003-4819-111-4-280
217. Pesce A, Taillan B, Rosenthal E, Garnier G, Vinti H, Dujardin P, et al. Opportunistic infections and CD4 lymphocytopenia with interferon treatment in HIV-1 infected patients. Lancet (1993) 341(8860):1597. doi:10.1016/0140-6736(93)90736-Z
218. Landau A, Batisse D, Duong Van Huyen JP, Piketty C, Bloch F, Pialoux G, et al. Efficacy and safety of combination therapy with interferon-alpha2b and ribavirin for chronic hepatitis C in HIV-infected patients. AIDS (2000) 14(7):839–44. doi:10.1097/00002030-200005050-00010
219. Arizcorreta A, Marquez M, Fernandez-Gutierrez C, Guzman EP, Brun F, Rodriguez-Iglesias M, et al. T cell receptor excision circles (TRECs), CD4+, CD8+, and their CD45RO+, and CD45RA+, subpopulations in hepatitis C virus (HCV)-HIV-co-infected patients during treatment with interferon alpha plus ribavirin: analysis in a population on effective antiretroviral therapy. Clin Exp Immunol (2006) 146(2):270–7.
220. Cepeda EJ, Williams FM, Ishimori ML, Weisman MH, Reveille JD. The use of anti-tumour necrosis factor therapy in HIV-positive individuals with rheumatic disease. Ann Rheum Dis (2008) 67(5):710–2. doi:10.1136/ard.2007.081513
Keywords: AIDS, pathogenesis, immune activation, TLR, Immunity, therapy
Citation: Miedema F, Hazenberg MD, Tesselaar K, van Baarle D, de Boer RJ and Borghans JAM (2013) Immune activation and collateral damage in AIDS pathogenesis. Front. Immunol. 4:298. doi: 10.3389/fimmu.2013.00298
Received: 18 July 2013; Accepted: 09 September 2013;
Published online: 26 September 2013.
Edited by:
Teunis Geijtenbeek, University of Amsterdam, NetherlandsReviewed by:
Cynthia Ann Derdeyn, Emory University, USAWilliam Anderson Paxton, Academic Medical Center, Netherlands
Copyright: © 2013 Miedema, Hazenberg, Tesselaar, van Baarle, de Boer and Borghans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank Miedema, Department of Immunology, University Medical Center Utrecht, Heidelberglaan 100, 3584CX, Utrecht, Netherlands e-mail: f.miedema@umcutrecht.nl