 Kevin P. Kotredes
Kevin P. Kotredes Brianna Thomas
Brianna Thomas Ana M. Gamero
Ana M. Gamero- Department of Medical Genetics and Molecular Biochemistry, Temple University School of Medicine, Philadelphia, PA, USA
The immune system of the gastrointestinal (GI) tract manages the significant task of recognizing and eliminating pathogens while maintaining tolerance of commensal bacteria. Dysregulation of this delicate balance can be detrimental, resulting in severe inflammation, intestinal injury, and cancer. Therefore, mechanisms to relay important signals regulating cell growth and immune reactivity must be in place to support GI homeostasis. Type I interferons (IFN-I) are a family of pleiotropic cytokines, which exert a wide range of biological effects including promotion of both pro- and anti-inflammatory activities. Using animal models of colitis, investigations into the regulation of intestinal epithelium inflammation highlight the role of IFN-I signaling during fine modulation of the immune system. The intestinal epithelium of the gut guides the immune system to differentiate between commensal and pathogenic microbiota, which relies on intimate links with the IFN-I signal-transduction pathway. The current paradigm depicts an IFN-I-induced antiproliferative state in the intestinal epithelium enabling cell differentiation, cell maturation, and proper intestinal barrier function, strongly supporting its role in maintaining baseline immune activity and clearance of damaged epithelia or pathogens. In this review, we will highlight the importance of IFN-I in intestinal homeostasis by discussing its function in inflammation, immunity, and cancer.
Introduction
The gastrointestinal (GI) tract has the greatest mucosal surface area of any organ system shared with the environment, interacting with a wide array of microbes and chemical irritants. These interactions with colonizing bacteria, especially early in life, are fundamental in developing proper gut health (1). The intestines of newborns are initially sterile (2), but become colonized immediately after birth, upon exposure to their new environment. The establishment of healthy intestinal microbiota can be hindered due to lack of exposure to commensal bacteria or upon treatment with antibiotic medications (3). This appears to be very important as there is mounting evidence that resident microbiota play an important role in shaping the function of the GI tract. The initiation and progression of human inflammatory bowel diseases (IBDs) are reliant on the dysregulation of complex interactions among genetic, environmental, and immune factors, as well as physical barriers within the intestinal mucosa. The physical barrier between the external environment and internal tissue is the first line of defense against microbial pathogens, toxins, and other environmental factors (4). This protective barrier is provided by the inner lining of the intestine, a single-cell layer of intestinal epithelial cells (IECs), and their specialized subtypes (e.g., Paneth, goblet, or enteroendocrine cells) (5). IECs serve an essential role as regulators of mucosal immune responses (6) and as cohabitants within the intestinal environs, which can be colonized by commensal or pathogenic bacteria, fungi, and viruses (7). Paneth cells, in particular, play a vital role in gut homeostasis (8–10) at least via expression of IFN-I and interferon-stimulated genes (ISGs) (11, 12). Ingested antigens and constituents of commensal bacteria are constantly testing the immune system of the gut. Responses to antigens can be either positive or negative, inducing an antigen-specific state of immunity (13). Cytokines like IFN-I act as initial signaling mechanisms within this innate immune system determining the durability and specificity of the response. Together, a series of direct responses and feedback loops are in place for maintaining gut homeostasis—preventing tissue damage, hyperplasia, malignancy, and ultimately cancer.
Type I Interferons (IFN-I)
The innate immune system is a remarkable network that has evolved to protect the host against disease. It has the ability to detect a wide range of microbial markers and, in response, rapidly activate a number of inflammatory and antimicrobial pathways. Part of this sophisticated system involves the family of IFN-I (IFN-α or IFN-β). These immunomodulatory cytokines are broadly expressed as α-helical cytokines transcribed from 13 homologous IFN-α genes (IFN-α1 and -α13 are the same) and a single IFN-β gene (14). They play a critical role as first line of defense by promoting and shaping antiviral and antibacterial immunity. Constitutive, baseline expression of IFN-I is very low in the intestines, typical of most tissues (12, 15–18). IFN induction is a rapid event that can be triggered in response to viral attack (via recognition of cytosolic viral double-stranded RNA, 5′triphosphate single-stranded RNA, or viral DNA) and bacterial infections (via recognition of lipopolysaccharide, lipoprotein, or flagellin, for example) (19). Each response is activated by specific pattern-recognition receptors (PRRs), like RIG-like helicases and toll-like receptors (TLRs), expressed by different cell types (20). Secreted IFN-I then activates autocrine and paracrine signaling cascades via the heterodimeric IFN-I receptor complex (14). IFN-I bind to and activate the cognate cell surface receptor consisting of the IFNAR1 and IFNAR2 chains, which induce downstream signaling via tyrosine phosphorylation of JAK kinases (JAK1 and TYK2). Activated JAKs then phosphorylate the transcription factors STAT1 and STAT2 in the cytoplasm that in association with IRF9 from the heterotrimeric complex ISGF3. ISGF3 translocates to the nucleus and binds to the promoters of IFN target genes and activates the transcription of many ISGs (21). These ISGs drive immunomodulatory antiviral (22), antiproliferative (23), antibacterial (24), and antitumor actions (15) throughout the body, including the GI tract (18).
IFN-I as Anti-Inflammatory Immunomodulators
Type I interferons not only function as signaling molecules of innate immunity but also promote the activation of adaptive immunity. It is well-established that systemic IFN-I can influence CD4+ T cell differentiation and function via their effects on dendritic cells (DCs). IFN-I drive DC activation and maturation (25), MHC II expression, and production of IL-12 (26, 27), to augment T helper (Th)1 cell responses. In addition, IFN-I can act directly on T cells to inhibit their expansion from lymph nodes, thus promoting DC–T cell interactions (28). Several studies also show that IFN-I enhance natural killer (NK), B, and CD8+ T cell activity (29, 30). By contrast, other studies present a different side of IFN-I—as key factors in the attenuation of an active immune response. Primarily, IFN-I increase the susceptibility of lymphocytes and macrophages to apoptosis (24, 31–34). IFN-I also inhibit the expression of IL-8, a chemotactic cytokine responsible for recruiting neutrophils and leukocytes to areas of inflammation (35, 36), and of IL-17, via inhibition of Th17 differentiation (37, 38). IFN-I antagonize the effects of local IL-17 by downregulating the expression of IL-1β, IL-23, and osteopontin, and by inducing the production of the anti-inflammatory cytokine IL-27 in DCs (38, 39). Induction of IFN-I in macrophages by bacterial infection reduces IL-17A/F variant expression, followed by a decrease in IL-17A(+) γδ T cells, further highlighting the role of IFN-I on T cell populations during infection (40). Further, IFN-I can inhibit the secretion of IL-1β, both by inhibiting production of pro-IL-1β and blocking pro-IL-1β cleavage to mature IL-1β via impeding inflammasome activation (41). To suppress inflammation, IFN-I also induce the secretion of anti-inflammatory cytokines (e.g., IL-10, IL-27, and IL-1RA) from phagocytes via expression of inhibitory feedback SOCS and PIAS proteins in T cells and phagocytes (42–44). Additionally, IFN-I suppress IFN-γ-induced MHC II expression by downregulating IFNGR1 levels as a negative feedback mechanism (45, 46), and high levels of IFN-I can inhibit IL-12 production during certain viral infections (47). IFN-I also inhibit inflammatory responses by inducing tristetraprolin, a strong suppressor of TNF-α and IFN-γ (48, 49).
Alterations to the IFNAR1 gene have been linked to susceptibility for IBD and changes to microbiome populations (50, 51), thus providing supporting evidence that IFN-I contribute to immune defenses against conditionally pathogenic microbiota and intestinal inflammation (52). In a T cell adoptive transfer model of colitis, signaling through host hematopoietic cell Ifnar1 was necessary to deter development of colitis. Ifnar1−/−-recipient mice developed severe colitis, compared with Ifnar1+/+ mice, when inoculated with CD4+ T cells from a WT mouse (18). Phagocytes collected from the colonic lamina propria (LP) of Ifnar1−/− mice produced less IL-10, IL-1RA, and IL-27 than did cells from WT mice (18) demonstrating an important role for IFN-I signaling driving the expression of anti-inflammatory cytokines by gut phagocytes and maintenance of intestinal T cell homeostasis. Oral administration of the colonic irritant, dextran sulfate sodium (DSS) is another well-established model of acute colitis as it produces submucosal inflammation and ulceration in the gut thereby providing a “leaky” epithelial cell-lining ideal for translocation of luminal microbiota into the LP (53). Ifnar1−/− mice are found more susceptible to DSS-induced colitis pointing to conventional DCs as critical players in attenuating inflammation (16, 18, 54, 55). However, a later study found that deletion of Ifnar1 in LysM+ myeloid cells, but not in conventional DCs exacerbated DSS-induced colitis (56). These differing results could be attributed to the mouse model employed. In the first study, Abe et al. used transgenic DTR mice with intact Ifnar1 to deplete CD11c+ DCs via administration of diphtheria toxin. By contrast, Rauch et al. used mice with conditional deletion of Ifnar1 in DCs or in myeloid cells. Nevertheless, both studies agree on the protective effect of IFN-I by suppressing IL-1 production during inflammation of the gut. Altogether, IFN-I activate and orchestrate different programs to keep inflammation under control.
IFN-I are Instrumental in Maintaining Homeostasis in the Gut
Balance of the microbiome within the small and large intestine is important for not only maintenance of the intestinal epithelium, proper digestion, and nutrient uptake but is also strongly tied to immunity, inflammation, and cancer risk (57, 58). Both pro- and anti-inflammatory cytokines are chief among these immunomodulatory agents, including IFN-I, in regulating the growth and renewal of IECs (59–61). IFN-I are constitutively expressed in the intestines by LP CD11c+ DCs (16, 18, 62). The LP is the layer of connective tissue underneath the intestinal epithelium, enriched in immune cells. In the colon, CD11c+ DCs cells express mRNA for IFN-α5-, IFN-α9-, and IFN-I/ISGS3-induced genes thus indicative of active IFN-I production and signaling. Proper regulation of epithelial cell turnover in the intestinal lining is important for balance between replacement of damaged/sloughed cells and hyperplasia, which leads to pre-cancerous polyp formation (61, 63). Secretion of IFN-α has been shown as an important regulator of epithelial apoptosis. IFN-α administration prevented epithelial cell apoptosis in an Escherichia coli-induced mouse model of disease (64). Basolateral IFN-α also polarized monolayers of IECs, protected these cells against apoptosis, and promoted disruption of epithelial tight junctions (54). Moreover, IFN-α can induce the expression of GBP-1 (64), shown to prevent apoptosis, and promote intestinal epithelial barrier integrity (65). Prevention of apoptosis by IFN-α-induced GBP-1 subsequently inhibited endothelial cell angiogenesis (66, 67). In a study conducted in mice with deleted Ifnar1 in the intestines, loss of IFN-I signaling increased the number of Paneth cells and hyperproliferation of epithelial cells with no signs of spontaneous inflammation or enhanced susceptibility to DSS, when compared to littermate controls (50, 51). Most recently, Fuchs et al. reported that increased protein levels of IFNAR1 in vivo [via deletion in the intestine of casein kinase 1α (CK1α), which controls the ubiquitination and degradation of both β-catenin and the IFNAR1] led to an increased ISG transcriptional signature (52) highlighting baseline IFN-I signaling in the intestinal epithelium. Deletion of CK1α in the intestines of Ifnar1−/− mice resulted in decreased levels of p21, inhibited p53 activation, and unrestricted IEC proliferation resulting in loss of gut barrier function and prompt animal death. Hence, IFN-I enable enhanced maturation, differentiation, and establishment of the cohesive epithelial barrier in the gut highlighting the contribution of IFN-I signaling to the control of IEC proliferation and function. As such, IFN-I are vital in maintenance of the host-microbiota equilibrium and constraining IEC proliferation and viability.
The microbiome in the gut plays an important role in the pathogenesis of IBD. This is evidenced by a variety of animal models in which development of intestinal inflammation is completely abolished under germ-free conditions (68). In healthy individuals, the gut microenvironment exists in a continuous state of controlled inflammation, despite the presence of potent antigen-presenting cells, like DCs. DCs are important for controlling T cell-mediated antigen response (69) and are the major source of TLR-driven IFN-I production (70). Conventional DCs have been attributed with inhibition of DSS-induced colitis, in part, to IFN-I production (14, 55). IFN-I regulated colonic recruitment of neutrophils and monocytes, as well as activation of pro-inflammatory macrophages (55). Additionally, Ifnar1 loss in myeloid cells promoted colitis via increased IL-1 production (56), a pro-inflammatory cytokine produced by activated macrophages (71). Interestingly, in celiac disease (an IBD driven by strong T cell activation toward gluten), the role of IFN-I appears reversed. In humans, mucosal DC populations are increased in celiac disease patients (72). Activated mature DCs from these patients maintained higher IFN-α transcripts, as well as for IL-18 and IL-23, two cytokines responsible for Th1 polarization and subsequent IFN-γ production. Furthermore, IFN-α blockade inhibited IFN-γ transcripts in ex vivo-organ culture of celiac biopsy specimens challenged with gluten (72). Yet in mouse models of colitis pretreated with synthetic bacterial DNA, increased anti-inflammatory IL-10 and decreased IFN-γ production were reported (73). Along these same lines, a human ulcerative colitis (UC) study showed a correlation between IFN-I response and Th17 differentiation and suppression of IL-17 production (74). Th17 cells are central effectors that produce pro-inflammatory cytokines, particularly IL-17 in the gut (75, 76). IL-17 then induces the secretion of chemokines and antimicrobial peptides to create a mucosal barrier to eliminate pathogens; however, excessive IL-17 production exacerbates inflammation thereby promoting pathogen colonization (77).
T regulatory (Treg) cells play a central role in suppressing the development of intestinal inflammation and IBD (78–80). Tregs maintain intestinal homeostasis under conditions of continuous challenge with inflammatory microbes. Induction of Treg populations by recombinant bacterial DNA analogs was TGF-β- and IFN-I-dependent in a mouse model of IBD (81). Maintenance of the Treg population in the gut is mediated by IFN-I signaling driving the expression of Foxp3 in colonic Tregs (82). Continuous Foxp3 expression is necessary for the development and regulatory function of Tregs (83, 84). IFN-I limit inflammation by eliciting production of the regulatory cytokine IL-10 or by enhancing the activity of Treg cells (79, 85). Additionally, apoptotic resident intestinal DCs help regulate the populations of Tregs in the intestine via production of IFN-β (86). In IBD patients, Th1 and Th17 constitute a major driving force in the disease process in the inflamed mucosa characterized by high surface expression of activated CD69 (87, 88). Expression of CD69 is strongly induced by IFN-I (28). Several studies in mice indicate a role of CD69 in the regulation of arthritis (89), asthma (90), myocarditis (91), pathogen clearance (92), and tumor immunity (93). Commensal bacteria in the intestinal tract are shown to induce CD69 expression in CD4+ T cells. Secretion of the regulatory cytokine TGF-β1 by CD4+ T cells decreased, whereas production of the pro-inflammatory cytokines (IFN-γ, TNF-α, and IL-21) increased, upon deletion of CD69. CD69−/− cells showed impaired IFN-β1 induction by TLR3 ligand polyI:C. CD4+ T cells lacking CD69 expression were hindered in their ability to mature into Tregs (Foxp3+) leading to accelerated colitis (94).
IFN-I Confer Protection Against Colitis
Toll-like receptors play an important role in innate immunity by recognizing structurally conserved bacterial and viral components. TLRs are important transmembrane-signaling PRRs involved in inducing inflammation and are pivotal in the establishment of adaptive immunity. In addition to innate immune cells such as macrophages and DCs, IECs express a spectrum of TLRs (95). TLR signaling can induce strong production of several inflammatory cytokines, including IFN-I (96). TLR2 and TLR4 recognize bacterial cell wall components at the cell surface, while TLR3, TLR7, and TLR9 recognize bacterial or viral nucleic acids in endosomes after phagocytosis of bacteria or viruses (97). Activation of DCs via TLRs contributes to both rapid anti-pathogen responses and maintenance of homeostatic protective immunity (98). This is partly mediated by the direct production of cytokines necessary for the development of downstream humoral and cell-mediated immunity. Imiquimod, a TLR7 agonist, has been shown to ameliorate DSS-induced acute colitis by inducing the expression of IFN-I in the colonic mucosa (99). When administered as a preventive measure, ligands for TLR9 (CpG) or TLR3 (polyI:C) also induced IFN-I and lessened disease severity of DSS-induced colitis (54, 100). Administration of neutralizing antibodies against IFN-I also impeded these downstream anti-inflammatory effects via TLR9, thus highlighting the importance of IFN-I signaling in maintaining intestinal homeostasis and providing avenues for future therapeutics (54, 101). The activation of TLR9 by CpG dinucleotides initiates a cascade of innate and adaptive immune responses, at least partially mediated by secretion of IFN-I and IFN-γ, that results in cell-mediated Th1 and humoral immune reactions (102). The TLR9 signaling pathway can induce the production of inflammatory cytokines through nuclear factor κB and interferon regulatory factor (IRF)-5, and IFN-I through IRF7 (96). In other studies, comparison of transcriptome profiles from gnotobiotic mice, which lack commensal bacteria that constitute the microbiome, to three bacterial colonization models—specific pathogen-free mice, ex-germ-free mice with bacterial reconstitution at the time of delivery, and ex-germ-free mice with bacterial reconstitution at 5 weeks of age—showed that TLR-driven expression of Irf3, a crucial rate-limiting transcription factor in the induction of IFN-I, was essential for normal development of the host immune system (103). Commensal bacteria triggered the production of IFN-β via recognition of dsRNA by TLR3, which in turn protected mice from experimental colitis (104).
Inflammatory bowel disease is a group of intestinal chronic inflammatory conditions mainly UC and Crohn’s disease (CD) that affects part or the entire GI tract. The precise cause is unknown, but evidence overwhelmingly suggests symptoms arise from either pathogenic or commensal intestinal bacteria triggering an abnormal immune response. IFN-α-secreting DCs in gut-associated lymphoid tissues (GALTs) regulate differentiation of Tregs (105). GALTs are primary locations of host encounter with exogenous antigens and pathogens. Interaction of GALT with microbiota regulates both the size and duration of systemic immune responses (106, 107). The commensal microflora constituting the microbiome of the intestinal tract is strictly entwined in the well-being of the host. In particular, the balance of bacterial populations is directly related to IBD, though additional host-driven genetic predispositions are also suspected. Genome-wide association studies have implicated the locus containing IFNAR1 as a genetic risk factor for developing human IBD (50). In patients with IBD, chronic inflammation is a major risk factor for the development of GI malignancies (108). Patients suffering from IBD typically use non-specific medications to manage the symptoms and include steroids, 5-aminosalacylic acid derivative, immunosuppressants, or antibodies against TNF-α (109). Systemic administration of IFN-I to treat IBD patients has been evaluated and the results vary in suppressing disease burden (110–113). UC is associated with increased expression of IL-13 in NK T cells from the mucosa of the GI tract (114–116). IFN-I have been shown to deter IL-4/IL-13 transcription and secretion (117) by, as well as blocking of signaling in, human CD4+ T cells (118). In one small study, the majority of UC patients treated systemically with interferon-β-a1 showed reduced disease burden using rectal bleeding as a clinical measure. In the responder group, the clinical effect of IFN-I therapy correlated with decreased IL-13 production by LP mononuclear cells. By contrast, the non-responders had significantly higher production of IL-17 and IL-6 compared to responders (119). In cases where IFN-I therapy exacerbated the disease, parallel diseases in the patient may have complicated the correlated observations (111). Initial studies in an experimental model of colitis depicted the benefits of IFN-I in regulating intestinal growth, via apoptotic turnover of old cells or constitution of the hematopoietic cell population in the gut (54), but subsequent studies could not produce a therapeutic effect from IFN-I in IBD patients (120, 121). In an animal study, the therapeutic potential of IFN-β-secreting Lactobacillus (La-IFN-β) by delivering IFN-β in the gut prior to the induction of colitis was evaluated (122). Unexpectedly, this preventive measure heightened sensitivity to DSS when compared to mice pretreated with control Lactobacillus. Colitic mice that received La-IFN-β had increased intestinal secretion of TNF-α, IFN-γ, IL-17A, and IL-13 and decreased Treg populations in their small intestine. Intestinal DCs from La-IFN-β-treated mice and bone marrow-derived DCs exposed La-IFN-β showed decreased IFNAR1 expression. The underlying causes for the differing results of these various studies have yet to be identified. Further muddying the waters, conventional DCs can either enhance or inhibit DSS-induced colitis, independently of T cells, contingent on their manner of activation (55), emphasizing again the importance of IFN-I-driven immunoregulation in the gut.
IFN-I in Colorectal Cancer
Like many other cancer types, colorectal cancer development has an inflammatory component. In fact, the risk of patients with IBD to develop CRC is strongly linked to the duration of the disease, anatomical extent, and severity of colonic inflammation (123). It is estimated that as much as 15% of all IBD patients will die of colitis-associated cancer (CAC), although early diagnosis and proper treatment of IBD symptoms can reduce the risk of CAC (124). IFN-I promote the recruitment and activation of tumor-parallel immune cells, the presence of which is believed to improve the prognostic pathological assessments of CRC (125). Aside from the immune-compartment-driven inflammation referenced earlier, genetic alterations within IFN-I signaling cascades have been implicated in CRC. To model CAC in rodents, the axozymethane (AOM)/DSS protocol was developed and is widely used to study colorectal cancer (126). Mice are given a single intraperitoneal injection of the carcinogen AOM, which is known to cause activating mutations in β-catenin, Kras and upregulation of Cox2, and iNOS (127). Addition of DSS given in multiple cycles generates a chronic inflammatory environment that reliably accelerates the carcinogenic effect of a single dose of AOM by dramatically shortening the duration of time for tumors to arise. Using this model, loss of Ifnar1 in IECs was reported to increase inflammation and severity of colitis. This poses cancer risk as evidenced by Ifnar1-expressing mice displaying decreased GI tumor burden corresponding with decreased mucosal inflammation (51). However, findings from our lab employing the same CAC model revealed a distinct and unexpected phenotype, in that loss of Stat2 (an essential component in IFN-I signaling) reduced tumor burden and inflammation in the colon (128). To further establish the role of IFN-I in CRC, additional animal studies are warranted using the sporadic model of CRC, which also has a strong link to inflammation (129).
Another link to consider in CRC is the study of single-nucleotide polymorphisms (SNPs) in IFN-I-related genes that include STAT1, JAKs, IRFs, IFN-γ, and IFN-γR, which have been associated with increased CRC risk and disease progression (130, 131). In stark contrast, SNPs in IFNA7 and IFNA14 genes have been found associated with overall survival, more specifically in CRC patients without distant metastasis at time of diagnosis (132). These genes are located nearby several transcription factor-binding sites, but remains unknown how IFNA7 and IFNA14 directly influence overall survival, though they may still be regarded as potential CRC patient biomarkers. SNPs in IFNAR1 were also found associated with CRC risk (132), but how they affect IFN-I signaling and inflammation as a whole in the gut remains to be evaluated.
In humans, CRC tumor specimens show elevated mRNA expression of TLR9, IFNAR1, and IL-6, indicating that IFN-I-signaling components and effectors may be good predictors for overall survival (133). Other contrasting studies, however, find that TLR9 expression is decreased in hyperplastic and villous polyps from patients who develop CRC, further supporting a possible protective role for TLR9 expression against malignant transformation in colorectal mucosa (134). To add to the complexity of the role of signaling components of IFN-I and gene products of IFN-I, a recent study reported that in vitro formation of colorectal tumor spheroids, in the absence of IFN-I treatment, induced transcription of ISGs via IRF9/STAT2 (135). In vitro-tumor spheroids are characterized by non-proliferating, metabolically stressed cells in the hypoxic inner core, surrounded by actively proliferating cells in the outer layers. Knockdown of STAT2, but especially IRF9 inhibited accumulation of three ISGs: IFI27, IFITM1, and OAS1, whereas STAT1 knockdown had no effect. In addition, expression of IRF9 in this 3D model resulted in a significant decrease in the sensitivity of CRC cells to multiple chemotherapeutic drugs (135). Another ISG, ISG15, functions as a ubiquitin-like modifier, able to form covalent conjugates called “protein ISGylation” on many cellular proteins leading to a cellular stress response and increased inflammation. Elevated ISGylation has recently been proposed to promote intestinal inflammation and CAC in mice (136). In other studies, ATG16LI, which is not an ISG, has been found to regulate autophagy as well as innate immunity. A non-synonymous ATG16LI polymorphism carrying a T300A amino acid substitution is implicated in CD. Paradoxically, this SNP in CRC patients was found associated with increased overall survival and reduced metastasis. Data show an elevated IFN-I transcriptional signature and mitochondrial antiviral signaling suggesting that ATG16L1 T300A could be regulating IFN-I production (137). Further investigation is warranted to fully understand the contributions of IFN-I-related signaling pathways in CRC.
Concluding Remarks
Type I interferons are broadly expressed cytokines that drive innate immunity, responding to pathogenic attack or injury with both pro- and anti-inflammatory responses (summarized in Figure 1). This remarkable and well-orchestrated task is facilitated by the production of other cytokines and chemokines to eradicate the invading microorganism and begin the process of wound healing. IFN-I are the bridge between innate and adaptive immunity via promotion of DC maturation leading to disease-specific education and expansion of T cells. This process removes pathogenic microbes while promoting beneficial commensals. Similarly, IFN-I regulate epithelial cell apoptosis to promote intestinal barrier function. Deregulated immune responses to commensal bacteria that penetrate the intestinal epithelium barrier is believed to be the main cause of IBD, primarily UC and CD, which leave patients more vulnerable to CAC. Intensive research has been performed in experimental mouse models of colitis, however, to better understand the complex IFN-I-driven immunological effects, more studies are needed to better explain the diverse clinical results of IFN-I when evaluated in the setting of IBD. In fact, some patients appeared to respond better than others to IFN-I treatment, implying that additional factors must be identified to determine their regulatory role in IFN-I signaling. Most IECs appear capable of producing sufficient IFN-I, as well as other cytokines, and considering the important observations already made with Ifnar1−/− mice, the influences of STAT proteins and IFN-I-related proteins and parallel signaling pathways, will need to be taken into account and studied in more depth in future studies of intestinal immunity and homeostasis.
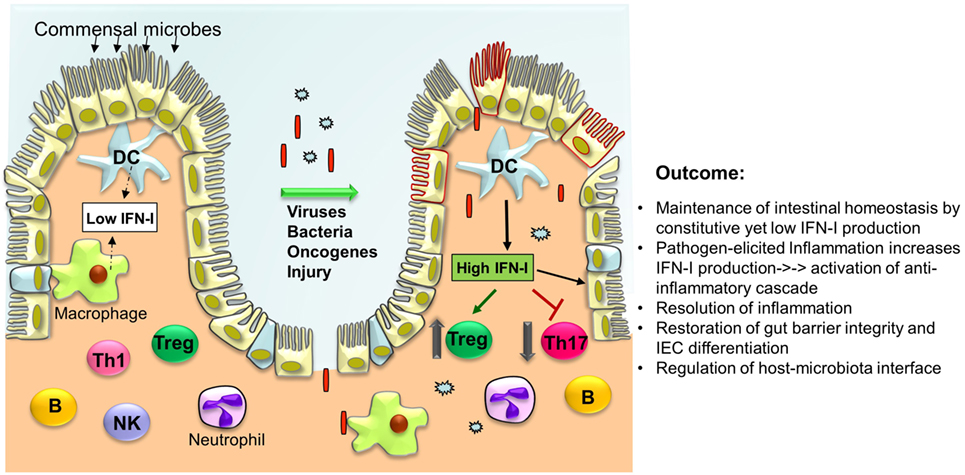
Figure 1. Type I interferons (IFN-I) orchestrate a series of intracellular events in immune cells and intestinal epithelial cells (IECs) to cease inflammation resulting in the regeneration of the intestinal epithelium and restoration of the gut barrier. Under normal conditions, low levels of IFN-I are secreted by lamina propria dendritic cells (DCs) and other phagocytes. In response to microbial attack and/or tissue injury, production of IFN-I by these cells is increased that in turn act on T cells to suppress Th17 cell differentiation while promoting Treg expansion thereby limiting inflammation. IFN-I also inhibit the production of pro-inflammatory cytokines (i.e., IL-1β, IL-8, IL-23) and induce the production of anti-inflammatory mediators (IL-1RA, IL-10, IL-27). Furthermore, IFN-I act on IEC and Paneth cells to restrict proliferation and favor their differentiation to establish gut barrier integrity.
Author Contributions
KK and AG wrote and edited the review. BT assisted with literature searches and organization of review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the W.W. Smith Charitable Trust and Temple University Bridge Funding Program to AG.
References
1. Mshvildadze M, Neu J, Mai V. Intestinal microbiota development in the premature neonate: establishment of a lasting commensal relationship? Nutr Rev (2008) 66:658–63. doi:10.1111/j.1753-4887.2008.00119.x
2. Gewolb IH, Schwalbe RS, Taciak VL, Harrison TS, Panigrahi P. Stool microflora in extremely low birthweight infants. Arch Dis Child Fetal Neonatal Ed (1999) 80:F167–73. doi:10.1136/fn.80.3.F167
3. Fanaro S, Chierici R, Guerrini P, Vigi V. Intestinal microflora in early infancy: composition and development. Acta Paediatr (2003) 91:48–55. doi:10.1111/j.1651-2227.2003.tb00646.x
4. Catalioto RM, Maggi CA, Giuliani S. Intestinal epithelial barrier dysfunction in disease and possible therapeutical interventions. Curr Med Chem (2011) 18:398–426. doi:10.2174/092986711794839179
5. Van Der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol (2009) 71:241–60. doi:10.1146/annurev.physiol.010908.163145
6. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28:573–621. doi:10.1146/annurev-immunol-030409-101225
7. Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe (2008) 3:417–27. doi:10.1016/j.chom.2008.05.001
8. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi:10.1038/nature07416
9. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134:743–56. doi:10.1016/j.cell.2008.07.021
10. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature (2013) 503:272–6. doi:10.1038/nature12599
11. Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett (1998) 441:106–10. doi:10.1016/S0014-5793(98)01514-2
12. Munakata K, Yamamoto M, Anjiki N, Nishiyama M, Imamura S, Iizuka S, et al. Importance of the interferon-alpha system in murine large intestine indicated by microarray analysis of commensal bacteria-induced immunological changes. BMC Genomics (2008) 9:192. doi:10.1186/1471-2164-9-192
13. Scurlock AM, Vickery BP, Hourihane JO, Burks AW. Pediatric food allergy and mucosal tolerance. Mucosal Immunol (2010) 3:345–54. doi:10.1038/mi.2010.21
14. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12:125–35. doi:10.1038/nri3133
15. Gresser I, Belardelli F. Endogenous type I interferons as a defense against tumors. Cytokine Growth Factor Rev (2002) 13:111–8. doi:10.1016/S1359-6101(01)00035-1
16. Chirdo FG, Millington OR, Beacock-Sharp H, Mowat AM. Immunomodulatory dendritic cells in intestinal lamina propria. Eur J Immunol (2005) 35:1831–40. doi:10.1002/eji.200425882
17. Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol (2009) 183:3229–36. doi:10.4049/jimmunol.0804277
18. Kole A, He J, Rivollier A, Silveira DD, Kitamura K, Maloy KJ, et al. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. J Immunol (2013) 191:2771–9. doi:10.4049/jimmunol.1301093
19. Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol (2013) 34:67–73. doi:10.1016/j.it.2012.10.004
20. Cho H, Kelsall BL. The role of type I interferons in intestinal infection, homeostasis, and inflammation. Immunol Rev (2014) 260:145–67. doi:10.1111/imr.12195
21. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi:10.1038/nri1604
22. Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A (2012) 109:4239–44. doi:10.1073/pnas.1114981109
23. Bekisz J, Baron S, Balinsky C, Morrow A, Zoon KC. Antiproliferative properties of type I and type II interferon. Pharmaceuticals (2010) 3:994–1015. doi:10.3390/ph3040994
24. Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol (2005) 5:675–87. doi:10.1038/nri1684
25. Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood (2002) 99:3263–71. doi:10.1182/blood.V99.9.3263
26. Le Bon A, Schiavoni G, D’agostino G, Gresser I, Belardelli F, Tough DF. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity (2001) 14:461–70. doi:10.1016/S1074-7613(01)00126-1
27. Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, et al. A type I interferon autocrine-paracrine loop is involved in toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med (2005) 201:1435–46. doi:10.1084/jem.20041964
28. Desbien AL. Analysis of the innate response to adjuvants: characterization of the draining lymph node by fluorescence-activated cell sorting. Methods Mol Biol (2017) 1494:305–12. doi:10.1007/978-1-4939-6445-1_23
29. Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res (2011) 17:2619–27. doi:10.1158/1078-0432.CCR-10-1114
30. Welsh RM, Bahl K, Marshall HD, Urban SL. Type 1 interferons and antiviral CD8 T-cell responses. PLoS Pathog (2012) 8:e1002352. doi:10.1371/journal.ppat.1002352
31. Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med (2004) 200:535–40. doi:10.1084/jem.20040769
32. Gold JA, Hoshino Y, Hoshino S, Jones MB, Nolan A, Weiden MD. Exogenous gamma and alpha/beta interferon rescues human macrophages from cell death induced by Bacillus anthracis. Infect Immun (2004) 72:1291–7. doi:10.1128/IAI.72.3.1291-1297.2004
33. O’connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med (2004) 200:437–45. doi:10.1084/jem.20040712
34. Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol (2008) 181:2092–102. doi:10.4049/jimmunol.181.3.2092
35. Oliveira IC, Sciavolino PJ, Lee TH, Vilcek J. Downregulation of interleukin 8 gene expression in human fibroblasts: unique mechanism of transcriptional inhibition by interferon. Proc Natl Acad Sci U S A (1992) 89:9049–53. doi:10.1073/pnas.89.19.9049
36. Nozell S, Laver T, Patel K, Benveniste EN. Mechanism of IFN-beta-mediated inhibition of IL-8 gene expression in astroglioma cells. J Immunol (2006) 177:822–30. doi:10.4049/jimmunol.177.2.822
37. Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol (2009) 65:499–509. doi:10.1002/ana.21652
38. Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-beta inhibits human Th17 cell differentiation. J Immunol (2009) 183:5418–27. doi:10.4049/jimmunol.0803227
39. Shinohara ML, Kim JH, Garcia VA, Cantor H. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity (2008) 29:68–78. doi:10.1016/j.immuni.2008.05.008
40. Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol (2010) 184:3755–67. doi:10.4049/jimmunol.0902065
41. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34:213–23. doi:10.1016/j.immuni.2011.02.006
42. Wang Q, Miyakawa Y, Fox N, Kaushansky K. Interferon-alpha directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood (2000) 96:2093–9.
43. Molnarfi N, Hyka-Nouspikel N, Gruaz L, Dayer JM, Burger D. The production of IL-1 receptor antagonist in IFN-beta-stimulated human monocytes depends on the activation of phosphatidylinositol 3-kinase but not of STAT1. J Immunol (2005) 174:2974–80. doi:10.4049/jimmunol.174.5.2974
44. Chang EY, Guo B, Doyle SE, Cheng G. Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J Immunol (2007) 178:6705–9. doi:10.4049/jimmunol.178.11.6705
45. Ling PD, Warren MK, Vogel SN. Antagonistic effect of interferon-beta on the interferon-gamma-induced expression of Ia antigen in murine macrophages. J Immunol (1985) 135:1857–63.
46. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med (2010) 207:327–37. doi:10.1084/jem.20091746
47. Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, et al. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med (2002) 195:517–28. doi:10.1084/jem.20011672
48. Sauer I, Schaljo B, Vogl C, Gattermeier I, Kolbe T, Muller M, et al. Interferons limit inflammatory responses by induction of tristetraprolin. Blood (2006) 107:4790–7. doi:10.1182/blood-2005-07-3058
49. Ogilvie RL, Sternjohn JR, Rattenbacher B, Vlasova IA, Williams DA, Hau HH, et al. Tristetraprolin mediates interferon-gamma mRNA decay. J Biol Chem (2009) 284:11216–23. doi:10.1074/jbc.M901229200
50. Jostins L, Ripke S, Weersma RK, Duerr RH, Mcgovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi:10.1038/nature11582
51. Tschurtschenthaler M, Wang J, Fricke C, Fritz TM, Niederreiter L, Adolph TE, et al. Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut (2014) 63:1921–31. doi:10.1136/gutjnl-2013-305863
52. Katlinskaya YV, Katlinski KV, Lasri A, Li N, Beiting DP, Durham AC, et al. Type I interferons control proliferation and function of the intestinal epithelium. Mol Cell Biol (2016) 36:1124–35. doi:10.1128/MCB.00988-15
53. Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol (2002) 20:495–549. doi:10.1146/annurev.immunol.20.100301.064816
54. Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest (2005) 115:695–702. doi:10.1172/JCI200522996C1
55. Abe K, Nguyen KP, Fine SD, Mo JH, Shen C, Shenouda S, et al. Conventional dendritic cells regulate the outcome of colonic inflammation independently of T cells. Proc Natl Acad Sci U S A (2007) 104:17022–7. doi:10.1073/pnas.0708469104
56. Rauch I, Hainzl E, Rosebrock F, Heider S, Schwab C, Berry D, et al. Type I interferons have opposing effects during the emergence and recovery phases of colitis. Eur J Immunol (2014) 44:2749–60. doi:10.1002/eji.201344401
57. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi:10.1016/j.cell.2010.01.025
58. Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology (2010) 138(2101–2114):e2105. doi:10.1053/j.gastro.2010.01.058
59. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity (2012) 37:158–70. doi:10.1016/j.immuni.2012.04.011
60. McAleer JP, Kolls JK. Maintaining poise: commensal microbiota calibrate interferon responses. Immunity (2012) 37:10–2. doi:10.1016/j.immuni.2012.07.001
61. Santaolalla R, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol (2012) 28:124–9. doi:10.1097/MOG.0b013e3283506559
62. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26:503–17. doi:10.1016/j.immuni.2007.03.006
63. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell (2006) 127:469–80. doi:10.1016/j.cell.2006.10.018
64. Mirpuri J, Brazil JC, Berardinelli AJ, Nasr TR, Cooper K, Schnoor M, et al. Commensal Escherichia coli reduces epithelial apoptosis through IFN-alphaA-mediated induction of guanylate binding protein-1 in human and murine models of developing intestine. J Immunol (2010) 184:7186–95. doi:10.4049/jimmunol.0903116
65. Schnoor M, Betanzos A, Weber DA, Parkos CA. Guanylate-binding protein-1 is expressed at tight junctions of intestinal epithelial cells in response to interferon-gamma and regulates barrier function through effects on apoptosis. Mucosal Immunol (2009) 2:33–42. doi:10.1038/mi.2008.62
66. Briken V, Ruffner H, Schultz U, Schwarz A, Reis LF, Strehlow I, et al. Interferon regulatory factor 1 is required for mouse Gbp gene activation by gamma interferon. Mol Cell Biol (1995) 15:975–82. doi:10.1128/MCB.15.2.975
67. Pammer J, Reinisch C, Birner P, Pogoda K, Sturzl M, Tschachler E. Interferon-alpha prevents apoptosis of endothelial cells after short-term exposure but induces replicative senescence after continuous stimulation. Lab Invest (2006) 86:997–1007. doi:10.1038/labinvest.3700461
68. Bleich A, Mahler M. Environment as a critical factor for the pathogenesis and outcome of gastrointestinal disease: experimental and human inflammatory bowel disease and Helicobacter-induced gastritis. Pathobiology (2005) 72:293–307. doi:10.1159/000091327
69. Cong Y, Brandwein SL, Mccabe RP, Lazenby A, Birkenmeier EH, Sundberg JP, et al. CD4+ T cells reactive to enteric bacterial antigens in spontaneously colitic C3H/HeJBir mice: increased T helper cell type 1 response and ability to transfer disease. J Exp Med (1998) 187:855–64. doi:10.1084/jem.187.6.855
70. Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-beta in toll-like receptor-stimulated dendritic cell activation. Int Immunol (2002) 14:1225–31. doi:10.1093/intimm/dxf089
71. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
72. Di Sabatino A, Pickard KM, Gordon JN, Salvati V, Mazzarella G, Beattie RM, et al. Evidence for the role of interferon-alfa production by dendritic cells in the Th1 response in celiac disease. Gastroenterology (2007) 133:1175–87. doi:10.1053/j.gastro.2007.08.018
73. Obermeier F, Dunger N, Strauch UG, Grunwald N, Herfarth H, Scholmerich J, et al. Contrasting activity of cytosin-guanosin dinucleotide oligonucleotides in mice with experimental colitis. Clin Exp Immunol (2003) 134:217–24. doi:10.1046/j.1365-2249.2003.02288.x
74. Moschen AR, Geiger S, Krehan I, Kaser A, Tilg H. Interferon-alpha controls IL-17 expression in vitro and in vivo. Immunobiology (2008) 213:779–87. doi:10.1016/j.imbio.2008.07.022
75. Yen D, Cheung J, Scheerens H, Poulet F, Mcclanahan T, Mckenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest (2006) 116:1310–6. doi:10.1172/JCI21404
76. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139:485–98. doi:10.1016/j.cell.2009.09.033
77. Blaschitz C, Raffatellu M. Th17 cytokines and the gut mucosal barrier. J Clin Immunol (2010) 30:196–203. doi:10.1007/s10875-010-9368-7
78. Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med (2000) 192:295–302. doi:10.1084/jem.192.2.295
79. Levings MK, Sangregorio R, Galbiati F, Squadrone S, De Waal Malefyt R, Roncarolo MG. IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J Immunol (2001) 166:5530–9. doi:10.4049/jimmunol.166.9.5530
80. Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol (2001) 166:7282–9. doi:10.4049/jimmunol.166.12.7282
81. Bleich A, Janus LM, Smoczek A, Westendorf AM, Strauch U, Mahler M, et al. CpG motifs of bacterial DNA exert protective effects in mouse models of IBD by antigen-independent tolerance induction. Gastroenterology (2009) 136:278–87. doi:10.1053/j.gastro.2008.09.022
82. Lee SE, Li X, Kim JC, Lee J, Gonzalez-Navajas JM, Hong SH, et al. Type I interferons maintain Foxp3 expression and T-regulatory cell functions under inflammatory conditions in mice. Gastroenterology (2012) 143:145–54. doi:10.1053/j.gastro.2012.03.042
83. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
84. Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity (2009) 30:616–25. doi:10.1016/j.immuni.2009.04.009
85. Aman MJ, Tretter T, Eisenbeis I, Bug G, Decker T, Aulitzky WE, et al. Interferon-alpha stimulates production of interleukin-10 in activated CD4+ T cells and monocytes. Blood (1996) 87:4731–6.
86. Nakahashi-Oda C, Udayanga KG, Nakamura Y, Nakazawa Y, Totsuka N, Miki H, et al. Apoptotic epithelial cells control the abundance of Treg cells at barrier surfaces. Nat Immunol (2016) 17:441–50. doi:10.1038/ni.3345
87. Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta-converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A (2001) 98:13249–54. doi:10.1073/pnas.231473998
88. Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut (2009) 58:1152–67. doi:10.1136/gut.2008.163667
89. Sancho D, Gomez M, Viedma F, Esplugues E, Gordon-Alonso M, Garcia-Lopez MA, et al. CD69 downregulates autoimmune reactivity through active transforming growth factor-beta production in collagen-induced arthritis. J Clin Invest (2003) 112:872–82. doi:10.1172/JCI200319112
90. Martin P, Gomez M, Lamana A, Matesanz Marin A, Cortes JR, Ramirez-Huesca M, et al. The leukocyte activation antigen CD69 limits allergic asthma and skin contact hypersensitivity. J Allergy Clin Immunol (2010) 126(2):.e1–3. doi:10.1016/j.jaci.2010.05.010
91. Cruz-Adalia A, Jimenez-Borreguero LJ, Ramirez-Huesca M, Chico-Calero I, Barreiro O, Lopez-Conesa E, et al. CD69 limits the severity of cardiomyopathy after autoimmune myocarditis. Circulation (2010) 122:1396–404. doi:10.1161/CIRCULATIONAHA.110.952820
92. Vega-Ramos J, Alari-Pahissa E, Valle JD, Carrasco-Marin E, Esplugues E, Borras M, et al. CD69 limits early inflammatory diseases associated with immune response to Listeria monocytogenes infection. Immunol Cell Biol (2010) 88:707–15. doi:10.1038/icb.2010.62
93. Esplugues E, Vega-Ramos J, Cartoixa D, Vazquez BN, Salaet I, Engel P, et al. Induction of tumor NK-cell immunity by anti-CD69 antibody therapy. Blood (2005) 105:4399–406. doi:10.1182/blood-2004-10-3854
94. Radulovic K, Manta C, Rossini V, Holzmann K, Kestler HA, Wegenka UM, et al. CD69 regulates type I IFN-induced tolerogenic signals to mucosal CD4 T cells that attenuate their colitogenic potential. J Immunol (2012) 188:2001–13. doi:10.4049/jimmunol.1100765
95. Lee J, Mo JH, Katakura K, Alkalay I, Rucker AN, Liu YT, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol (2006) 8:1327–36. doi:10.1038/ncb1500
96. Kaisho T, Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol (2006) 117:979–87; quiz 988. doi:10.1016/j.jaci.2006.02.023
97. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi:10.1016/j.cell.2006.02.015
98. Beutler B, Eidenschenk C, Crozat K, Imler JL, Takeuchi O, Hoffmann JA, et al. Genetic analysis of resistance to viral infection. Nat Rev Immunol (2007) 7:753–66. doi:10.1038/nri2174
99. Sainathan SK, Bishnupuri KS, Aden K, Luo Q, Houchen CW, Anant S, et al. Toll-like receptor-7 ligand imiquimod induces type I interferon and antimicrobial peptides to ameliorate dextran sodium sulfate-induced acute colitis. Inflamm Bowel Dis (2012) 18:955–67. doi:10.1002/ibd.21867
100. Vijay-Kumar M, Wu H, Aitken J, Kolachala VL, Neish AS, Sitaraman SV, et al. Activation of toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm Bowel Dis (2007) 13:856–64. doi:10.1002/ibd.20142
101. Lee J, Rachmilewitz D, Raz E. Homeostatic effects of TLR9 signaling in experimental colitis. Ann N Y Acad Sci (2006) 1072:351–5. doi:10.1196/annals.1326.022
102. Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene (2008) 27:161–7. doi:10.1038/sj.onc.1210911
103. Yamamoto M, Yamaguchi R, Munakata K, Takashima K, Nishiyama M, Hioki K, et al. A microarray analysis of gnotobiotic mice indicating that microbial exposure during the neonatal period plays an essential role in immune system development. BMC Genomics (2012) 13:335. doi:10.1186/1471-2164-13-335
104. Kawashima T, Kosaka A, Yan H, Guo Z, Uchiyama R, Fukui R, et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity (2013) 38:1187–97. doi:10.1016/j.immuni.2013.02.024
105. Bilsborough J, George TC, Norment A, Viney JL. Mucosal CD8alpha+ DC, with a plasmacytoid phenotype, induce differentiation and support function of T cells with regulatory properties. Immunology (2003) 108:481–92. doi:10.1046/j.1365-2567.2003.01606.x
106. Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity (2010) 32:815–27. doi:10.1016/j.immuni.2010.06.001
107. Schwarzer M, Repa A, Daniel C, Schabussova I, Hrncir T, Pot B, et al. Neonatal colonization of mice with Lactobacillus plantarum producing the aeroallergen Bet v 1 biases towards Th1 and T-regulatory responses upon systemic sensitization. Allergy (2011) 66:368–75. doi:10.1111/j.1398-9995.2010.02488.x
108. Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: the role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol (2016) 22:4794–801. doi:10.3748/wjg.v22.i20.4794
109. Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis (2006) 12(Suppl 1):S3–9. doi:10.1097/01.MIB.0000195385.19268.68
110. Musch E, Andus T, Malek M. Induction and maintenance of clinical remission by interferon-beta in patients with steroid-refractory active ulcerative colitis-an open long-term pilot trial. Aliment Pharmacol Ther (2002) 16:1233–9. doi:10.1046/j.1365-2036.2002.01264.x
111. Watanabe T, Inoue M, Harada K, Homma N, Uchida M, Ogata N, et al. A case of exacerbation of ulcerative colitis induced by combination therapy with PEG-interferon alpha-2b and ribavirin. Gut (2006) 55:1682–3. doi:10.1136/gut.2006.105197
112. Musch E, Andus T, Malek M, Chrissafidou A, Schulz M. Successful treatment of steroid refractory active ulcerative colitis with natural interferon-beta – an open long-term trial. Z Gastroenterol (2007) 45:1235–40. doi:10.1055/s-2007-963378
113. Rodrigues S, Magro F, Soares J, Nunes AC, Lopes S, Marques M, et al. Case series: ulcerative colitis, multiple sclerosis, and interferon-beta 1a. Inflamm Bowel Dis (2010) 16:2001–3. doi:10.1002/ibd.21242
114. Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity (2002) 17:629–38. doi:10.1016/S1074-7613(02)00453-3
115. Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest (2004) 113:1490–7. doi:10.1172/JCI19836
116. Fuss IJ, Strober W. The role of IL-13 and NK T cells in experimental and human ulcerative colitis. Mucosal Immunol (2008) 1(Suppl 1):S31–3. doi:10.1038/mi.2008.40
117. Kaser A, Molnar C, Tilg H. Differential regulation of interleukin 4 and interleukin 13 production by interferon alpha. Cytokine (1998) 10:75–81. doi:10.1006/cyto.1997.0270
118. Dickensheets HL, Venkataraman C, Schindler U, Donnelly RP. Interferons inhibit activation of STAT6 by interleukin 4 in human monocytes by inducing SOCS-1 gene expression. Proc Natl Acad Sci U S A (1999) 96:10800–5. doi:10.1073/pnas.96.19.10800
119. Mannon PJ, Hornung RL, Yang Z, Yi C, Groden C, Friend J, et al. Suppression of inflammation in ulcerative colitis by interferon-beta-1a is accompanied by inhibition of IL-13 production. Gut (2011) 60:449–55. doi:10.1136/gut.2010.226860
120. Pena-Rossi C, Schreiber S, Golubovic G, Mertz-Nielsen A, Panes J, Rachmilewitz D, et al. Clinical trial: a multicentre, randomized, double-blind, placebo-controlled, dose-finding, phase II study of subcutaneous interferon-beta-la in moderately active ulcerative colitis. Aliment Pharmacol Ther (2008) 28:758–67. doi:10.1111/j.1365-2036.2008.03778.x
121. Pena Rossi C, Hanauer SB, Tomasevic R, Hunter JO, Shafran I, Graffner H. Interferon beta-1a for the maintenance of remission in patients with Crohn’s disease: results of a phase II dose-finding study. BMC Gastroenterol (2009) 9:22. doi:10.1186/1471-230X-9-22
122. McFarland AP, Savan R, Wagage S, Addison A, Ramakrishnan K, Karwan M, et al. Localized delivery of interferon-beta by Lactobacillus exacerbates experimental colitis. PLoS One (2011) 6:e16967. doi:10.1371/journal.pone.0016967
123. Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med (2015) 372:1441–52. doi:10.1056/NEJMra1403718
124. Van Der Kraak L, Gros P, Beauchemin N. Colitis-associated colon cancer: is it in your genes? World J Gastroenterol (2015) 21:11688–99. doi:10.3748/wjg.v21.i41.11688
125. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol (2014) 232:199–209. doi:10.1002/path.4287
126. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci (2003) 94:965–73. doi:10.1111/j.1349-7006.2003.tb01386.x
127. Takahashi M, Wakabayashi K. Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci (2004) 95:475–80. doi:10.1111/j.1349-7006.2004.tb03235.x
128. Gamero AM, Young MR, Mentor-Marcel R, Bobe G, Scarzello AJ, Wise J, et al. STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev Res (2010) 3:495–504. doi:10.1158/1940-6207.CAPR-09-0105
129. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature (2012) 491:254–8. doi:10.1038/nature11465
130. Slattery ML, Lundgreen A, Bondurant KL, Wolff RK. Interferon-signaling pathway: associations with colon and rectal cancer risk and subsequent survival. Carcinogenesis (2011) 32:1660–7. doi:10.1093/carcin/bgr189
131. Slattery ML, Lundgreen A, Kadlubar SA, Bondurant KL, Wolff RK. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol Carcinog (2013) 52:155–66. doi:10.1002/mc.21841
132. Lu S, Pardini B, Cheng B, Naccarati A, Huhn S, Vymetalkova V, et al. Single nucleotide polymorphisms within interferon signaling pathway genes are associated with colorectal cancer susceptibility and survival. PLoS One (2014) 9:e111061. doi:10.1371/journal.pone.0111061
133. Chang LC, Fan CW, Tseng WK, Chein HP, Hsieh TY, Chen JR, et al. IFNAR1 is a predictor for overall survival in colorectal cancer and its mRNA expression correlated with IRF7 but not TLR9. Medicine (2014) 93:e349. doi:10.1097/MD.0000000000000349
134. Eiro N, Gonzalez L, Gonzalez LO, Andicoechea A, Fernandez-Diaz M, Altadill A, et al. Study of the expression of toll-like receptors in different histological types of colorectal polyps and their relationship with colorectal cancer. J Clin Immunol (2012) 32:848–54. doi:10.1007/s10875-012-9666-3
135. Kolosenko I, Fryknas M, Forsberg S, Johnsson P, Cheon H, Holvey-Bates EG, et al. Cell crowding induces interferon regulatory factor 9, which confers resistance to chemotherapeutic drugs. Int J Cancer (2015) 136:E51–61. doi:10.1002/ijc.29161
136. Fan JB, Miyauchi-Ishida S, Arimoto K, Liu D, Yan M, Liu CW, et al. Type I IFN induces protein ISGylation to enhance cytokine expression and augments colonic inflammation. Proc Natl Acad Sci U S A (2015) 112:14313–8. doi:10.1073/pnas.1505690112
Keywords: interferon, intestine, inflammation, microbiome, epithelium
Citation: Kotredes KP, Thomas B and Gamero AM (2017) The Protective Role of Type I Interferons in the Gastrointestinal Tract. Front. Immunol. 8:410. doi: 10.3389/fimmu.2017.00410
Received: 17 November 2016; Accepted: 22 March 2017;
Published: 06 April 2017
Edited by:
Charles Dudley Mills, BioMedical Consultants, USAReviewed by:
Laurel L. Lenz, University of Colorado Denver School of Medicine, USAAlessandra Mancino, Istituto Europeo di Oncologia, Italy
Copyright: © 2017 Kotredes, Thomas and Gamero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana M. Gamero, gameroa@temple.edu