 Sebastian Scheer
Sebastian Scheer Colby Zaph
Colby Zaph- Infection and Immunity Program, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, VIC, Australia
G9a (KMT1C, EHMT2) is a lysine methyltransferase (KMT) whose primary function is to di-methylate lysine 9 of histone H3 (H3K9me2). G9a-dependent H3K9me2 is associated with gene silencing and acts primarily through the recruitment of H3K9me2-binding proteins that prevent transcriptional activation. Gene repression via G9a-dependent H3K9me2 is critically required in embryonic stem (ES) cells for the development of cellular lineages by repressing expression of pluripotency factors. In the immune system, lymphoid cells such as T cells and innate lymphoid cells (ILCs) can differentiate from a naïve state into one of several effector lineages that require both activating and repressive mechanisms to maintain the correct gene expression program. Furthermore, the long-term immunity to re-infection is mediated by memory T cells, which also require specific gene expression and repression to maintain a quiescent state. In this review, we examine the molecular machinery of G9a-dependent functions, address the role of G9a in lymphoid cell differentiation and function, and identify potential functions of T cells and ILCs that may be controlled by G9a. Together, this review will highlight the dynamic nature of G9a-dependent H3K9me2 in the immune system and shed light on the nature of repressive epigenetic modifications in cellular lineage choice.
The mammalian immune system is made up of a large number of cell types that have the ability to respond to external environmental cues and adopt a wide variety of cell fates. These lineage decisions are critical for the development of proper immune responses to pathogens as well as important for the resolution of inflammatory responses. Despite the importance of these cell fate decisions, the molecular mechanisms that control them are still not completely described nor understood. In addition to expression of cell lineage-specific master regulatory transcription factors (TFs) (1), the epigenetic landscape of the chromatin is emerging as a central control point in cellular lineage differentiation.
In the immune system, lymphoid cells such as B cells, T cells, and innate lymphoid cells (ILCs) have the capacity to respond to the external environment by modulating the expression of lineage-specific factors that are critical for protective immunity to a wide variety of pathogens. For example, the description of CD4 T helper (Th) cell subsets by Mosmann and Coffman in 1986 has provided a fundamental framework for the division of labor mediated by lymphoid cells (2). Naive Th cells respond to signals from innate immune cells—primarily secreted cytokines—to differentiate into one of several ‘lineages’ that differ in the expression of TFs, cytokines, and cell surface molecules (3). For example, Th1 cells differentiate from naive Th cells in the presence of IL-12, express the TFs STAT4 and T-bet, leading to the production of IFN-γ. In contrast, IL-4 signaling promotes STAT6 and GATA3 expression in Th cells, resulting in IL-4- and IL-13-producing Th2 cells. In recent years, additional Th cell subsets have been identified that derive from a naïve precursor cell, which include Th17 cells (that express the TF RORγt and secrete IL-17A) and Treg cells (that express the TF FOXP3 and secrete TGF-β). More recently, ILCs that have similar patterns of differentiation and gene expression have been identified. Although the differentiation of ILCs appears to occur earlier than Th cells with ‘committed’ progenitors exiting the bone marrow (4), distinct ILC subsets that are closely related to Th1, Th2, and Th17 cells (ILC1s, ILC2s, and ILC3s, respectively) have been described. Strikingly, the TF expression patterns are highly conserved between Th cells and ILCs, suggesting that generalized molecular mechanisms control lymphoid cell differentiation and function. Understanding the molecular mechanisms of immune cell differentiation will provide the basis for development of new therapies to promote immunity to infection as well as prevent inflammatory diseases caused by dysregulated immune responses.
Epigenetic Regulation of Gene Expression
Epigenetic regulation encapsulates a wide range of mechanisms that can result in heritable changes in gene expression. From physical localization of genes within the nucleus to post-translational modifications of DNA and histones, epigenetic mechanisms can profoundly influence gene expression and alter cellular lineage development and function. The site-specific methylation and demethylation of CpG motifs in DNA by DNA methyltransferases (DNMTs) and the TET family of proteins is perhaps the best studied epigenetic mechanism that directly regulates gene expression (5). Although DNA methylation has been implicated in lymphoid cell responses (6–8), this review will focus on the posttranslational methylation of histones.
Regulation of gene expression by histone modifying enzymes is an important mechanism that regulates cellular development and differentiation. Histones can be modified posttranslationally by phosphorylation, acetylation, ubiquitination, sumoylation, and methylation (9). In particular, methylation of histone lysine residues is an important regulator of gene expression. Mixed lineage leukemia-1 (MLL1)-dependent H3K4me3 (10) and enhancer of zeste homolog-2 (EZH2)-dependent H3K27me3 (11) are the best known modifications and are associated with gene expression and repression, respectively (12–14). Other histone-methylation sites have been shown to be critical as well, including H3K9, with G9a-dependent H3K9me2 and Suv39h1/2-mediated H3K9me3 playing important roles in cell differentiation and function (15–18). Of these, H3K9me2 has been shown to modify euchromatin and dynamically regulate gene expression in differentiating cells.
In embryonic stem (ES) cells, it has been proposed that H3K9me2 marks increase across the genome as cells differentiate and acquire lineage specificity (19) although this is contentious (20). Specifically, H3K9me2 is found enriched at lineage non-specific genes, suggesting that acquisition of H3K9me2 is critical for gene silencing during differentiation (21). However, there is very little known about the role of G9a in cells of the immune system. In lymphoid cells such as T cells and ILCs, it is clear that G9a-dependent H3K9me2 is critical for cellular differentiation and function, although the mechanisms differ from ES cells. In this review, we focus on the role of the histone lysine methyltransferase G9a in lymphoid cell responses in health and disease.
G9a is Required for Dimethylation of H3K9
G9a (Ehmt2) was first identified as a gene located in the major histocompatibility complex (MHC) locus in mice and human leukocyte antigen (HLA) locus in humans and was also called HLA-B-associated transcript 8 (BAT8) (22–25). The Ehmt2 gene is located in the ~700 kb (mouse)/~1.1 Mb (human) MHC/HLA Class III region that contains over 60 genes (26) including cytokines (TNF-α and TNF-β), complement proteins C2 and C4, heat shock proteins (HSP70), and enzymes (steroid 21-hydroxylase Cyp21). The Ehmt2 gene is made up of 28 exons that code for a 1,263 amino acid protein. A splice form lacking exon 10 that codes for 34 amino acids has also been identified, although the functional significance is still unknown (24).
The related protein G9a-like protein (GLP, EHMT1) whose gene is not located in the MHC/HLA locus forms a heterodimer with G9a in vivo and is critically required for the H3K9me2 methylation activity (27). Genetic deletion of either protein results in a significant reduction in H3K9me2, suggesting that both subunits are essential to the enzymatic activity (28). Mutation of the active sites has shown that the methyltransferase activity of G9a plays a larger role in H3K9me2 methylation in vivo (29). However, global gene expression analysis of neurons of mice with targeted deletions of either G9a or GLP identified differences that may be due to differential requirement of each subunit in gene-specific expression (30). Further, loss of GLP is associated with Kleefstra Syndrome, a rare genetic disease that is characterized by intellectual disability and other social and physical impairments (31). There has been no analysis of the function of immune cells in Kleefstra Syndrome patients. Thus, although GLP may play a specific role in regulation of gene expression, it remains to be directly tested.
G9a is a 1,263 amino acid protein with several distinct domains (Figure 1). G9a does not contain a DNA-binding domain and must rely on cofactors for its localization to specific genes. Functionally, the C-terminal SET domain contains the lysine methyltransferase activity that defines the major function of this family of proteins. The SET domain of G9a is able to mono- and dimethylate H3K9 but is less efficient in mediating trimethylation (32). Consistent with this, deletion of G9a leads to a global reduction in H3K9me2 while H3K9me3 is largely unaffected (18, 28). Unlike other members of the SET domain family, G9a also has unique domains that provide additional functions. First, G9a has a series of eight 33-amino acid repeats that have homology to the ankyrin repeat domain of Drosophila Notch (25). This region was further shown to act as a domain that could specifically bind to dimethylated lysine residues, providing a protein that can not only generate a specific posttranslational modification but also bind to that modification (33). Interestingly, although the ankyrin repeats of G9a have an affinity for H3K9me2, GLP binds to H3K9me1 with higher affinity (33) and mice that carry a knock in of G9a with non-functional ankyrin repeats develop normally while mice with a mutated GLP have severe developmental defects resulting in perinatal lethality (34). These results further demonstrate that G9a and GLP have some non-overlapping roles in vivo. G9a also has a stretch of 25 glutamic acid residues as well as a Cysteine-rich region, whose functions remain unknown.
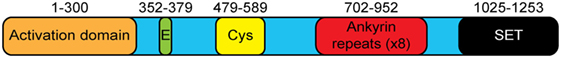
Figure 1. The structure of G9a. G9a is a 1,253 amino acid protein that has several distinct domains including an N-terminal activation domain, glutamate-rich (23 consecutive Glu residues) and cysteine-rich regions of unknown function, eight ankyrin repeat units (binding of dimethylated lysine residues), and a C-terminal enzymatic SET domain.
Although G9a has predominantly been studied in the context of gene repression via its methyltransferase activity on histones, it is clear that G9a also has a role in gene activation under certain conditions (35–37), which is methyltransferase-independent (discussed below). This function has been mapped to the N-terminus of the protein as the first 280 amino acids are sufficient to promote gene expression by acting as a scaffold to recruit transcriptional coactivators such as CARM1 and p300 (36, 38). Thus, G9a is a complex protein that is involved in gene repression and activation through distinct mechanisms.
G9a is the Major H3K9 Dimethyltransferase
G9a is the enzyme that is responsible for the dimethylation of H3K9, a hallmark of silenced euchromatin (18, 28, 39–41). H3K9me2 acts as a binding site for heterochromatin protein 1 (HP1) that recruits transcriptional repressors to prevent gene activation (42). Although H3K9me2 is the main product of G9a-dependent methylation, the G9a/GLP complex has also been described to methylate H1 (43, 44) and contributes to the methylation of H3K27 (39, 45). In addition, G9a has been shown to have activity against several non-histone proteins including itself (46) though the most well-studied aspect of G9a biology is the H3K9me2-dependent repression of gene expression. From genetic and biochemical studies, it is clear that G9a-dependent H3K9me2 is associated with genomic regions that are expressed at low levels (21) but the mechanisms that regulate the dynamic methylation patterns mediated by G9a still remain unclear. Indeed, as G9a lacks a domain that would promote direct interaction with DNA or chromatin, G9a has to rely on the DNA-binding capacity of its interaction partners.
In ES cells, G9a-dependent H3K9me2 is linked to de novo DNA methylation (47, 48). DNA methylation of endogenous retroelements, and a subset of non-repetitive sequences including CpG-rich promoters, is reduced in G9a-deficient cells, and Dnmt3a recruitment to retrotransposons is decreased in these cells. However, the interaction between G9a, H3K9me2, and DNMTs was absent in differentiated cells (48), suggesting that functional G9a–DNMT interactions are not maintained past development. Related to this function of G9a, the sustained silencing of pluripotency-associated genes in G9a-deficient ES cells is impaired and results in the reversal from the differentiated into a pluripotent state in a significant fraction of cells (49, 50). This effect has been exploited in the generation of induced pluripotent stem cells as inclusion of a G9a-specific chemical inhibitor BIX-01298 can replace viral transduction of Sox2 in fibroblasts (51).
Taken together, a general theme of G9a playing a role in the epigenetic silencing of cell-type inappropriate genes has emerged from studies in ES cells (52, 53). However, the role of G9a in immune cell function is less well understood.
G9a Can Methylate Non-Histone Proteins
In addition to its role as a histone lysine methyltransferase, several studies have shown that G9a is also able to methylate a wide range of non-histone targets, including G9a itself (46), CDYL1, WIZ and ACINUS, C/EBPβ, CSB, histone deacetylase (HDAC)1, mAM, KLF12, SIRT1, Reptin, MyoD, p21, and p53 (54). Although the precise role of posttranslational methylation on protein function remains unclear, methylation of non-histone proteins may affect protein stability, protein–protein interactions, subcellular localization, or function (55). Nevertheless, the precise physiological role of G9a-dependent methylation of non-histone proteins remains unclear and will not be discussed in depth in this review.
G9a has Methyltransferase-Independent Activities
Apart from its ability to methylate substrates, G9a has also been shown to have methyltransferase-independent activities through the N-terminal domain of the G9a protein (35, 36, 38). Work from the Stallcup group was the first to show that in contrast to expectations, G9a was a strong coactivator of nuclear hormone receptor activity including androgen, estrogen, and glucocorticoid receptors by associating with the transcriptional coactivators GRIP1, CARM1, and p300 (35), and in the case of estrogen, with the receptor itself (36). G9a has also been shown to positively regulate gene expression at the β-globin gene locus (37). Regulation of the β-globin gene locus is a well-characterized system to examine the role of locus control regions (LCRs) in tissue- and stage-specific expression of genes (56). In red blood cells, the β-globin locus is regulated partially through the addition and removal of histone modifications including H3K4me3 and H3K9me2 (57). Knockdown of G9a in adult erythroid progenitor cells led to the heightened mis-expression of fetal β-globin as well as a significant reduction in adult β-globin gene expression (37), further demonstrating inhibitory and activating effects of G9a. Similar to studies with nuclear hormone receptors, the activation of gene expression by G9a was independent of methyltransferase activity. Importantly, the β-globin gene locus is regulated in a similar manner to the type 2 response Il4-Il5-Il13 locus (58) and may provide a common mechanistic link to regulate gene expression. Thus, in addition to its repressive functions, it is clear that G9a can positively influence gene expression at select genetic loci.
G9a in the Immune System
The vast majority of studies on the function of G9a have been carried out in ES cells and very little is known about the role of G9a in innate and adaptive immune cells. However, the ability of immune cells to respond to the external environment and differentiate into functionally distinct cell lineages is reminiscent of the cellular plasticity of ES cells and suggests that epigenetic mechanisms may be a conserved regulatory mechanism in these cell types.
In innate immune cells such as macrophages, G9a-dependent H3K9me2 has been associated with gene repression during endotoxin tolerance (59–61). Macrophages that are chronically stimulated with lipopolysaccharide (LPS) become unresponsive to further LPS stimulation through the acquisition of H3K9me2 at repressed genetic loci (61). In tolerized macrophages, G9a has been shown to interact with the TF ATF7 as well as several members of the NF-κB family including RelB, RelA, c-Rel, and NF-κB1 (59–61). It is proposed that G9a is recruited to specific loci by these factors to deposit H3K9me2, leading to gene repression. However, a direct role for G9a in macrophages during endotoxin tolerance has not been tested. Nevertheless, these studies identify a role for G9a in gene silencing during cellular responses to inflammatory signals. Consistent with this role in promoting tolerance, G9a has been also shown to limit JAK/STAT signaling in Drosophila following viral infections (62). In the absence of G9a, viral infection leads to increased lethality in flies but is not due to increased pathogen burden but due to heightened expression of JAK/STAT-dependent target genes. Thus, G9a is an important regulator of innate inflammatory gene expression.
G9a has also been implicated in several aspects of T cell biology. Although genome-wide studies mapping the binding of G9a or the deposition of H3K9me2 in immune cells has not been carried out due to technical reasons, a descriptive genome-wide analysis of H3K9me2 marks in resting human lymphocytes using ChIP-on-chip methods demonstrated that this epigenetic mark is enriched on genes that are associated with several specific pathways including T cell receptor signaling, IL-4 signaling, and GATA3 transcription (63). Furthermore, lymphocytes isolated from patients with type I diabetes displayed a distinct H3K9me2 profile, with genetic regions that had increased (CXCL3, CTLA-4, SLC17A4) and reduced (RARA, CAMK4, TNF) levels of H3K9me2 (64). Thus, G9a-mediated H3K9me2-dependent regulation of T cell responses may be associated with T cell function as well as development of inflammatory diseases such as diabetes.
More recently, cell lineage-specific deletion of G9a has been used to delineate the role of G9a in immune cells. Three independent strains of mice with a “floxed” G9a allele (G9afl/fl mice) have been generated (16, 65, 66). Crossing G9afl/fl mice with Cd4-Cre mice or Lck-Cre mice results in a T cell-specific deletion of G9a (G9aΔT mice). G9aΔT mice are born and develop normally and have no discernable defects in the generation of T cells in the thymus, spleen, or lymph nodes (16, 67), suggesting that unlike ES cells G9a is dispensable for cellular development of peripheral naive T cells. However, upon activation of T cells in vitro or in vivo, G9a was shown to play a critical role in regulating the function of Th cells. Consistent with the ability of G9a to promote and repress gene expression, G9a-deficient Th cells had a failure to produce certain cytokines while overproducing others (15, 16). Under distinct differentiating conditions, G9a was differentially required to activate or repress specific gene programs.
Th1 cells that produce IFN-γ are critical for immunity against intracellular pathogens including bacteria, viruses, and protozoan parasites (68). Strikingly, the absence of G9a in T cells has no effect on the development or magnitude of Th1 cell responses in vitro or in vivo (16). There was no difference in the frequency of Th1 cells that developed following activation of G9a-deficient T cells or wild type T cells in the presence of a G9a-specific inhibitor under Th1 cell-promoting conditions (16). Thus, from our understanding so far, G9a is dispensable for Th1 cell responses.
Immunity to infection with parasitic helminths such as the whipworm Trichuris muris is associated with a polarized type 2 cytokine response, with the production of IL-4, IL-5, and IL-13 by Th cells leading to mucus production, intestinal epithelial cell turnover, and worm expulsion (69). Infection of G9aΔT mice with Trichuris resulted in significantly reduced frequencies of protective Th2 cells, heightened frequencies of non-protective Th1 cells, and susceptibility to infection (16). This was consistent with a reduction in the production of type II cytokines by Th cells after in vitro activation. A role for G9a in repressing promiscuous type I cytokine gene expression in Th2 cells was only observed temporarily, as H3K9me2 is replaced by H3K27me3 at the Ifng locus during differentiation (70). Thus, G9a is required for activation of the type II cytokine gene program. G9a-deficient Th2 cells express comparable levels of the major Th2 cell transcriptional factors including GATA3 and STAT6 but fail to produce type II cytokines, suggesting that G9a is a central component of the transcriptional machinery for type II cytokines. As the Il4-Il5-Il13 locus is similar in genetic structure to the β-globin locus, it is perhaps not surprising that the ability of G9a to transactivate the type II cytokine gene locus was also independent of its methyltransferase activity (16). Taken together, these results identify a role for G9a as a critical component of the Th2 cell regulatory machinery.
In contrast to Th2 cells, G9a plays an important role in limiting Th17 and Treg cell differentiation that is dependent upon its methyltransferase activity (15). Activation of G9a-deficient Th cells under Th17- or Treg cell-promoting conditions resulted in a significant increase in the frequencies of IL-17A-producing and FOXP3-expressing cells, respectively (15). Inhibition of G9a methyltransferase activity with the small-molecule inhibitors BIX-01294 or UNC0638 also resulted in enhanced Th17 and Treg cell differentiation. Unlike the proposed role for G9a in ES cells, H3K9me2 is not deposited at lineage-promiscuous genes to control lineage differentiation in Th cells. Instead, G9a-dependent H3K9me2 is found at high levels in naive undifferentiated Th cells and is rapidly lost at lineage-specific and -non-specific loci after T cell activation (15). The loss of H3K9me2 alone is insufficient to promote gene expression, providing an explanation for the lack of a phenotype in the steady state. Thus, in naive T cells, H3K9me2 and G9a act as an additional layer of negative regulation to maintain cells in a naive state. Mechanistically, loss of G9a-dependent H3K9me2 results in an increase in the accessibility of the chromatin to transactivating factors, which leads to heightened responsiveness to external signals such as cytokines. In the case of Th17 and Treg cells, G9a-deficient Th cells are ~40 times more sensitive to TGF-β (15). Taken together, these results suggest that G9a, through the deposition of H3K9me2 at a wide variety of immune genes, is specifically important in naive T cells to repress gene expression, possibly by limiting accessibility of TFs and coactivators to specific genetic loci.
Unlike naive T cells, G9a has been proposed to repress expression of CD25 and CD27, cell surface receptors that are associated with cell activation and proliferation, in memory T cells during viral infection (71). Through a specific interaction with the transcriptional repressor Blimp-1 (PRDM1), G9a (but not EZH2) is recruited to specific genetic loci resulting in gene repression. In the absence of Blimp-1, reduced levels of H3K9me2, H3K9me3, and H3K27me3 is observed, suggesting that G9a is critical for the silencing of genes during memory T cell differentiation and development. Whether G9a is required for memory T cell generation has not been examined directly; however, this is an active area of research, which will shed further light on the role of G9a in T cell memory development. Thus, in the absence of G9a, other repressive modifications such as Suv39h1/2-dependent H3K9me3 and EZH2-dependent H3K27me3 may compensate. Although, the details remain unclear, the identification of the epigenetic mechanisms in memory T cell development and function will be critical for the optimal design of vaccines as well as the development of therapeutic strategies to target dysregulated memory T cell responses in inflammatory diseases.
In addition to T cells, G9a also plays a critical role in the development and differentiation of ILCs (72). Mice with a hematopoietic cell-specific deletion of G9a (generated by crossing G9afl/fl mice with Vav-Cre mice) have reduced numbers of group 2 ILCs (ILC2s) and increased frequencies of ILC3s in all tissues examined. Although the small number of ILC2s present in lung tissue are phenotypically normal, they are dysfunctional, failing to produce effector cytokines IL-5 and IL-13 following stimulation with the activating cytokine IL-33 or following intranasal administration of the protease allergens papain or house dust mite antigen (72). Genome-wide expression analysis of bone marrow-derived ILC2 precursors identified a global shift in expression from ILC2-specific transcripts to ILC3-specific genes, placing G9a and H3K9me2 as a central regulator of the ILC2/ILC3 lineage choice.
In contrast to T cells and ILCs, G9a appears to play a minor role in B cell development and function (52, 67). An early study using a V–D–J minilocus suggested that G9a inhibited germline transcription and recombination (73). However, mice with a B cell-specific deletion of G9a (Mb1-Cre) fail to show any overt phenotypes, including displaying normal V–D–J recombination (67). The absence of G9a did result in a skewed usage of the κ light chain over the λ light chain, as well as a slight reduction in IL-4- and LPS-induced proliferation and differentiation into plasma cells. This reduction in plasma cell differentiation is consistent with a study showing that G9a directly interacts with Blimp-1, a critical plasma cell differentiation factor (74). Thus, G9a is dispensable for normal B cell development and has little effect on most functions of B cells. However, it is possible that G9a has a subtle role in specific aspects of B cell biology that remain to be determined.
Together, these results demonstrate that G9a is an important regulator of immune cell function. However, the precise mechanisms are yet to be defined. In the following sections, immune functions regulated by proteins known to interact with G9a will be discussed and potential mechanisms for G9a-dependent regulation will be proposed.
Interactions and Potential Roles of G9a
From the studies outlined above, it is clear that G9a is a central regulatory node in the establishment of the epigenetic landscape of cells and is critical for shaping cellular identity. However, how G9a mediates its function is poorly understood. As G9a lacks a DNA-binding domain, it is dependent upon additional cofactors for its localization at specific genetic loci. Several classes of G9a-interacting proteins have been identified (Figure 2). Strikingly, many of the G9a cofactors have important roles in immune cell development and function, potentially offering a mechanistic explanation for the phenotypes associated with loss of G9a in lymphoid cells.
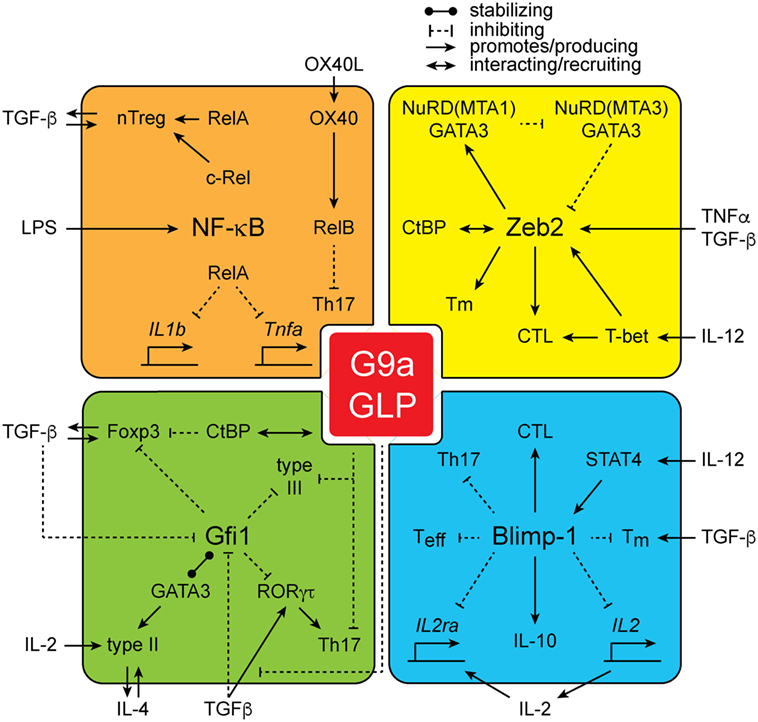
Figure 2. The interactions of G9a. G9a interacts with a large number of proteins including NF-kB family members RelA, c-Rel, and RelB, as well as zinc finger E-box-binding (Zeb2), growth factor independent 1 (Gfi1), and Blimp-1.
Growth Factor Independent 1
The transcriptional repressor growth factor independent 1 (Gfi1) is an important regulator of immune cell development and function (75). Gfi1 is able to bind to a large number of promoters and enhancers (76) and plays a central role in gene silencing through the recruitment of repressive modulators including histone methyltransferases, HDACs, and histone demethylases (77–79). Gfi1 has been shown to directly interact with G9a (78, 80) and Gfi1-deficient cells display a significant decrease in H3K9 methylation (78). Further, the phenotypes associated with Gfi1 deficiency in the immune system are strikingly similar to those observed in G9a-deficient mice, suggesting that Gfi1 is central to the function of G9a.
Gfi1 was first identified as a factor that, when induced or overexpressed in an IL-2-dependent T cell line, led to IL-2-independent growth (81). Furthermore, Gfi1 was shown to reduce the requirement for IL-2 by modulating the cell cycle regulation of T cells (82, 83). More recently, Gfi1 has been described to have multiple effects that echo the phenotypes seen in mice lacking G9a in T cells. First, Gfi1 is critical for promoting the differentiation of Th2 cells through a variety of mechanisms including increasing GATA3 stability, enhancing Th2 cell proliferation, inhibiting Th1 cell differentiation, and promoting expression of type 2 cytokines (84–87). Under Th2 cell-polarizing conditions, Gfi1 was found to be highly upregulated by IL-4 in a STAT6-dependent manner (84) and retroviral overexpression of Gfi1 in Th2 cells resulted in increased proliferation and survival (84). Gfi1-deficient T cells failed to optimally produce IL-4 after in vitro stimulation or following infection with the helminth parasite Schistosoma mansoni (87). Mechanistically, Gfi1 inhibits the proteasomal degradation of GATA3 through its N-terminal Snail/Gfi1 (SNAG) domain (85). As Th2 cell differentiation is impaired in G9a-deficient T cells (16), it is possible that G9a–Gfi1–GATA3 interactions are critical for the establishment of a transcriptional module that results in activation of the type 2 cytokine locus. Based on the role of the G9a N-terminus in activating nuclear hormone receptor-dependent gene expression by acting as scaffold (38), these results suggest that in Th2 cells, the N-terminus of G9a may aid in recruitment of Gfi1, GATA3, and potentially other factors required for optimal Th2 cell development.
Growth factor independent 1 has also been implicated in the differentiation of Th17 and Treg cells (88). Downregulation of Gfi1 expression by TGF-β is critical to allow expression of IL-17A/F in Th17 cells and CD103 in Treg cells (88) as well as surface expression of the ectonucleotidases CD39 and CD73 (89). Gfi1 potentially recruits the lysine demethylase LSD1 to these genetic loci to reduce the activating methylation marks. Upon stimulation with TGF-β, Gfi1 expression is reduced allowing optimal Th17 and Treg cell differentiation. Gfi1-deficient T cells display increased production of IL-17A and increased FOXP3 expression in response to TGF-β, which is identical to G9a-deficient T cells (15). Further, similar to the dysregulated expression of IL-17A observed in G9a-deficient Th2 cells (16), Gfi1 is required to silence IL-17A expression in Th2 cells (88). Thus, it is intriguing to hypothesize that Gfi1–G9a interactions are critical to restrain Th17 and Treg cell responses, linking transcriptional repression to epigenetic gene silencing.
Growth factor independent 1 is also an important regulator of ILC2 development and function (90). Expression of Gfi1 is correlated to the expression of the IL-33 receptor (Il1rl1, ST2) and GATA3. Loss of Gfi1 in ILC2s leads to impaired expression of GATA3 and an upregulation of IL-17A expression. This is reminiscent of the role of G9a in ILC biology, where G9a is required to repress ILC3-specific genes during ILC2 development (72). However, the effects of G9a and potentially Gfi1 appear to be dependent upon the methyltransferase-dependent gene repressive effects unlike in T cells.
Taken together, these results suggest that G9a–Gfi1 interactions are critical for their functions in T cells and ILCs. Future studies defining the molecular basis for these interactions may provide novel therapeutics to inhibit dysregulated Th2 cell responses that are associated with diseases such as asthma and allergy.
Zinc Finger E-Box-Binding Protein 2
Zinc finger E-box-binding proteins (Zeb1 and Zeb2) are TFs that have primarily been associated with TGF-β-dependent epithelial–mesenchymal transition in tumor cells (91, 92). Zeb proteins repress expression of several epithelial genes such as E-cadherin through the recruitment of repressive molecules including C-terminal binding protein (CtBP) and components of the nucleosome remodeling deacetylase complex (NuRD) including HDAC1. Recently, in a proteomic screen of G9a-interacting proteins in activated and endotoxin-tolerant macrophages, G9a was found to be strongly associated with components of several complexes that regulate chromatin structure including the Swi/SNF complex, NuRD complex, and CtBP/CoREST complexes (61). In breast cancer cells, G9a was shown to be a component of a complex containing Zeb2 and the NuRD component MTA1 (93). Interestingly, this was a highly dynamic complex that switched between a GATA3/G9a/MTA3 complex with a Zeb2/G9a/MTA1 complex that cross-regulated each other. A potential complex of G9a, Zeb2, and GATA3 that interacts with the NuRD complex may provide a molecular mechanism for how G9a, independent of its methyltransferase activity, regulates type 2 gene expression in Th2 cells. It is clear that nucleosome remodeling is an important aspect of type 2 cytokine expression (94, 95); however, the precise molecular mechanisms remain elusive. In addition, a direct role for Zeb2 in Th2 cell differentiation has not been evaluated.
Zinc finger E-box-binding protein has been shown to regulate the development of protective CD8 T cells during viral infection (96, 97). Following infection with lymphocytic choriomeningitis virus (LCMV), Zeb2 expression is induced in effector CD8 T cells that express the surface marker KLRG1 and produce IFN-γ. The upregulation of Zeb2 is dependent upon the TF T-bet (Tbx21) and expression of Zeb2 is critical for the terminal differentiation of effector CD8 T cells that are required for immunity to viral infection. As GATA3 has been implicated in CD8 T cell function (98, 99), it is possible that a Zeb2/G9a/GATA3 complex is critical for the function of CD8 T cell function during viral infection. Further, as TGF-β has been shown to play an important role in shaping an effective CD8 T cell response and memory formation (100–103), and G9a is critical for regulating TGF-β responsiveness (15), the intersection of these pathways may prove important for defining the molecular mechanisms of T cell biology during infection.
NF-κB
The NF-κB family of TFs plays a central and critical role in all aspects of immune cell biology (104). Both, the canonical (c-Rel, p65/RelA, and p50/NF-κB1) and non-canonical (RelB and NF-κB2) family members have been shown to be important regulators of immune cell function (105, 106). In a proteomic screen for G9a-binding partners, it was found that several members of the NF-κB family (RelB, c-Rel, RelA, and NF-κB1) are highly enriched for binding to G9a in macrophages (61), and RelB had been previously shown to associate with G9a during endotoxin tolerance, which was associated with gene silencing (59). In addition, the ankyrin repeats of GLP have been shown to directly recognize and bind to a monomethylated lysine residue in RelA, which directly links the G9a/GLP complex to attenuate cell proliferation and inflammatory responses in immunologically important genes such as Il1b and Tnfa (107). Together, these studies suggest that induction of NF-κB by LPS results in the recruitment of G9a and gene silencing, leading to endotoxin tolerance. Thus, under these circumstances, G9a-dependent dimethylation of H3K9 is important for gene repression during cellular activation.
In T cells, NF-κB family members have been shown to be important for Treg cell development and function (108–113) as well as implicated in Th17 cell responses (114, 115). In the absence of c-Rel or RelA, thymic-derived natural Treg (nTreg) cells have a severe developmental defect and show reduced ability to suppress inflammatory responses. In contrast, the absence of G9a had no effect on nTreg cell development or function and the development of peripherally-activated Treg (pTreg) cells was enhanced in the absence of G9a (15). Thus, whether G9a/NF-κB interactions are required for Treg cell function is unclear.
However, the interaction between G9a and the non-canonical family member RelB may prove to be more important in T cell biology. Recently, RelB was shown to have an important function in limiting the development of Th17 cells (114). Costimulation of Th17 cells through OX40–OX40L interactions resulted in a significant reduction in IL-17A expression. This effect was mediated by RelB-dependent recruitment of G9a to the Il17a locus, resulting in repression of Il17a expression. Further, in the absence of RelB, Th17 cells show reduced pathogenicity and inflammation in experimental autoimmune encephalomyelitis (EAE) (114). Thus, it is likely that RelB/G9a interactions, possibly downstream of OX40/OX40L signaling, are required for optimal development of Th17 cells.
It is clear that there is a significant interaction between NF-κB and G9a, although the precise molecular mechanisms in distinct immune cells are yet to be elucidated. Nevertheless, placing G9a in the context of NF-kB signaling suggests that there is a close relationship between inflammatory signal transduction pathways and the epigenetic machinery to control gene expression during an immune response.
Blimp-1
Blimp-1 (Prdm1) is a zinc finger protein which directly interacts with G9a and has been shown to be central to the development of immune responses. Blimp-1 is a transcriptional repressor that contains a non-catalytic PR domain that is related to the SET methyltransferase domain, and a zinc-finger DNA-binding domain (74). In the absence of Blimp-1, mice develop a lethal multiorgan inflammatory disease caused by an accumulation of effector and memory T cells (116), thus making Blimp-1 an important regulator of the adaptive immune response. Blimp-1 is also required for repression of Il2 transcription in T cells, resulting in an autoregulatory loop that controls immune responses (117, 118). In addition, Blimp-1 is crucial for the IL-27-dependent induction of IL-10 by Th1 cells (so-called Tr1 cells) following infection with Toxoplasma gondii or influenza A virus (119, 120). Moreover, Blimp-1 is critical for the development of terminally differentiated effector CD8 T cells (121) and controls the development of “exhausted” CD8 T cells during chronic viral infection (122). Thus, Blimp-1 plays an important role in T cell-mediated immunity to infection.
In CD8 T cells, Blimp-1 has been directly linked to G9a-dependent H3K9me2-mediated repression (71). Following viral infection, Blimp-1 recruits G9a to repress expression of CD25 and CD27 and limit the expansion and proliferation of CD8 T cells. However, in addition to H3K9me2, G9a was also associated with increased levels of H3K9me3 and H3K27me3, modifications that are not mediated by G9a, suggesting that additional methyltransferases were associated with Blimp-1 and involved in gene silencing during effector differentiation. Thus, in addition to G9a, other mechanisms are likely to be required for the repression of lineage non-specific genes in differentiating cells.
In Th cells, similar to results in G9aΔT mice (15), mice with a T cell-specific deletion of Blimp-1 have increased frequencies of Th17 cells in the intestinal mucosa (123). Furthermore, IL-23-induced Blimp-1 has been shown to be critical for the development of inflammatory Th17 cells that are required for the pathogenesis of EAE (124). Thus, mice lacking Blimp-1 in T cells fail to develop EAE. Although it remains unknown whether Blimp-1 and G9a directly interact in CD4 T cells, it is not outrageous to suggest that G9a/Blimp-1 interactions may be critical for the development of pathogenic Th17 cells, and blockade of this interaction may provide a new therapeutic strategy to prevent inflammatory diseases associated with dysregulated Th17 cell responses.
Inhibition of G9a Activity by Chemical Probes
Currently, there are several chemical probes that specifically target the methyltransferase activity of G9a. BIX-01294 was first identified in a high-throughput screen (125) and was shown to bind to the SET domain of G9a and GLP in the peptide-binding site, preventing methylation (126). BIX-01294 was subsequently optimized through structure–activity relationships to generate UNC0224, UNC0321, and E72 that showed increased activity and specificity (127, 128). The further development of UNC0638 and UNC0642 resulted in a potent, specific, stable, and cell-permeable inhibitor of G9a (129). However, UNC0642 has poor pharmacokinetics for in vivo use. More recently, an additional inhibitor of G9a that is unrelated to UNC0642, called A-366, was discovered through an independent high-throughput screen (130, 131). Treatment of mice with A-366 showed no overt toxicity and was able to reduce the growth of tumor xenografts (131). Thus, although A-366 has not been tested in the context of inflammatory disease, these results suggest that inhibition of G9a could prove to be a significant therapeutic strategy to modulate immune responses.
Concluding Remarks
It is clear that G9a is a central control point in lymphoid cell development, differentiation, and function. Acting through its diverse binding partners, G9a can repress and activate gene programs associated with a wide variety of immune responses. As G9a has been shown to be amenable to drug inhibition, blocking the function of G9a may provide a new therapeutic modality to modulate a wide variety of inflammatory diseases. For example, both Blimp-1 and G9a are required to limit Th17 cell development in a cell-intrinsic manner (15, 123). However, the increased frequency of Th17 cells does not result in enhanced pathogenicity of inflammatory diseases such as EAE or intestinal inflammation (15, 124), demonstrating that reducing the activity of Blimp-1 or G9a, or by inhibiting their interaction, may be a viable method to reduce the development of pathogenic Th17 cells. Future studies will define the precise role of G9a in immune cell development, differentiation, and function and determine the relevance of G9a as a drug target to treat inflammatory disease.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. F. Antignano for providing comments on the manuscript. This work is supported by the Australian National Health and Medical Research Council (project grants 1104433 and 1104466). CZ is a veski innovation fellow.
References
1. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations*. Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
2. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136:2348–57.
3. Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol (2002) 2:933–44. doi:10.1038/nri954
4. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17:765–74. doi:10.1038/ni.3489
5. Schübeler D. Function and information content of DNA methylation. Nature (2015) 517:321–6. doi:10.1038/nature14192
6. Young HA, Ghosh P, Ye J, Lederer J, Lichtman A, Gerard JR, et al. Differentiation of the T helper phenotypes by analysis of the methylation state of the IFN-gamma gene. J Immunol (1994) 153:3603–10.
7. Makar KW, Wilson CB. DNA methylation is a nonredundant repressor of the Th2 effector program. J Immunol (2004) 173:4402–6. doi:10.4049/jimmunol.173.7.4402
8. Makar KW, Perez-Melgosa M, Shnyreva M, Weaver WM, Fitzpatrick DR, Wilson CB. Active recruitment of DNA methyltransferases regulates interleukin 4 in thymocytes and T cells. Nat Immunol (2003) 4:1183–90. doi:10.1038/ni1004
9. Berger SL. The complex language of chromatin regulation during transcription. Nature (2007) 447:407–12. doi:10.1038/nature05915
10. Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet (2015) 47:1149–57. doi:10.1038/ng.3385
11. Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell (2007) 129:823–37. doi:10.1016/j.cell.2007.05.009
12. Huang Y, Min S, Lui Y, Sun J, Su X, Liu Y, et al. Global mapping of H3K4me3 and H3K27me3 reveals chromatin state-based regulation of human monocyte-derived dendritic cells in different environments. Genes Immun (2012) 13:311–20. doi:10.1038/gene.2011.87
13. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity (2009) 30:155–67. doi:10.1016/j.immuni.2008.12.009
14. Russ BE, Olshanksy M, Smallwood HS, Li J, Denton AE, Prier JE, et al. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity (2014) 41:853–65. doi:10.1016/j.immuni.2014.11.001
15. Antignano F, Burrows K, Hughes MR, Han JM, Kron KJ, Penrod NM, et al. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J Clin Invest (2014) 124:1945–55. doi:10.1172/JCI69592
16. Lehnertz B, Northrop JP, Antignano F, Burrows K, Hadidi S, Mullaly SC, et al. Activating and inhibitory functions for the histone lysine methyltransferase G9a in T helper cell differentiation and function. J Exp Med (2010) 207:915–22. doi:10.1084/jem.20100363
17. Yokochi T, Poduch K, Ryba T, Lu J, Hiratani I, Tachibana M, et al. G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proc Natl Acad Sci U S A (2009) 106:19363–8. doi:10.1073/pnas.0906142106
18. Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev (2002) 16:1779–91. doi:10.1101/gad.989402
19. Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet (2009) 41:246–50. doi:10.1038/ng.297
20. Filion GJ, van Steensel B. Reassessing the abundance of H3K9me2 chromatin domains in embryonic stem cells. Nat Genet (2010) 42:4; author rely 5–6. doi:10.1038/ng0110-4
21. Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev (2011) 25:781–8. doi:10.1101/gad.2027411
22. Spies T, Bresnahan M, Strominger JL. Human major histocompatibility complex contains a minimum of 19 genes between the complement cluster and HLA-B. Proc Natl Acad Sci U S A (1989) 86:8955–8. doi:10.1073/pnas.86.22.8955
23. Kendall E, Sargent CA, Campbell RD. Human major histocompatibility complex contains a new cluster of genes between the HLA-D and complement C4 loci. Nucleic Acids Res (1990) 18:7251–7. doi:10.1093/nar/18.24.7251
24. Brown SE, Campbell RD, Sanderson CM. Novel NG36/G9a gene products encoded within the human and mouse MHC class III regions. Mamm Genome (2001) 12:916–24. doi:10.1007/s00335-001-3029-3
25. Milner CM, Campbell RD. The G9a gene in the human major histocompatibility complex encodes a novel protein containing ankyrin-like repeats. Biochem J (1993) 290(Pt 3):811–8. doi:10.1042/bj2900811
26. Xie T, Rowen L, Aguado B, Ahearn ME, Madan A, Qin S, et al. Analysis of the gene-dense major histocompatibility complex class III region and its comparison to mouse. Genome Res (2003) 13:2621–36. doi:10.1101/gr.1736803
27. Ogawa H, Ishiguro K-I, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science (2002) 296:1132–6. doi:10.1126/science.1069861
28. Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev (2005) 19:815–26. doi:10.1101/gad.1284005
29. Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J (2008) 27:2681–90. doi:10.1038/emboj.2008.192
30. Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ, et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron (2009) 64:678–91. doi:10.1016/j.neuron.2009.11.019
31. Willemsen MH, Vulto-van Silfhout AT, Nillesen WM, Wissink-Lindhout WM, van Bokhoven H, Philip N, et al. Update on Kleefstra syndrome. Mol Syndromol (2012) 2:202–12. doi:10.1159/000335648
32. Collins RE, Tachibana M, Tamaru H, Smith KM, Jia D, Zhang X, et al. In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J Biol Chem (2005) 280:5563–70. doi:10.1074/jbc.M410483200
33. Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, et al. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol (2008) 15:245–50. doi:10.1038/nsmb.1384
34. Liu S, Ye D, Guo W, Yu W, He Y, Hu J, et al. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget (2015) 6:6887–901. doi:10.18632/oncotarget.3159
35. Lee DY, Northrop JP, Kuo M-H, Stallcup MR. Histone H3 lysine 9 methyltransferase G9a is a transcriptional coactivator for nuclear receptors. J Biol Chem (2006) 281:8476–85. doi:10.1074/jbc.M511093200
36. Purcell DJ, Jeong KW, Bittencourt D, Gerke DS, Stallcup MR. A distinct mechanism for coactivator versus corepressor function by histone methyltransferase G9a in transcriptional regulation. J Biol Chem (2011) 286:41963–71. doi:10.1074/jbc.M111.298463
37. Chaturvedi CP, Hosey AM, Palii C, Perez-Iratxeta C, Nakatani Y, Ranish JA, et al. Dual role for the methyltransferase G9a in the maintenance of beta-globin gene transcription in adult erythroid cells. Proc Natl Acad Sci U S A (2009) 106:18303–8. doi:10.1073/pnas.0906769106
38. Bittencourt D, Wu DY, Jeong KW, Gerke DS, Herviou L, Ianculescu I, et al. G9a functions as a molecular scaffold for assembly of transcriptional coactivators on a subset of glucocorticoid receptor target genes. Proc Natl Acad Sci U S A (2012) 109:19673–8. doi:10.1073/pnas.1211803109
39. Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem (2001) 276:25309–17. doi:10.1074/jbc.M101914200
40. Peters AH, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AA, Perez-Burgos L, et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell (2003) 12:1577–89. doi:10.1016/S1097-2765(03)00477-5
41. Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell (2003) 12:1591–8. doi:10.1016/S1097-2765(03)00479-9
42. Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol (2004) 5:296–304. doi:10.1038/nrm1355
43. Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, et al. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the Jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem (2009) 284:8395–405. doi:10.1074/jbc.M807818200
44. Weiss T, Hergeth S, Zeissler U, Izzo A, Tropberger P, Zee BM, et al. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics Chromatin (2010) 3:7. doi:10.1186/1756-8935-3-7
45. Wu H, Chen X, Xiong J, Li Y, Li H, Ding X, et al. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res (2011) 21:365–7. doi:10.1038/cr.2010.157
46. Chin HG, Esteve PO, Pradhan M, Benner J, Patnaik D, Carey MF, et al. Automethylation of G9a and its implication in wider substrate specificity and HP1 binding. Nucleic Acids Res (2007) 35:7313–23. doi:10.1093/nar/gkm726
47. Leung DC, Dong KB, Maksakova IA, Goyal P, Appanah R, Lee S, et al. Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc Natl Acad Sci U S A (2011) 108:5718–23. doi:10.1073/pnas.1014660108
48. Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J (2008) 27:2691–701. doi:10.1038/emboj.2008.193
49. Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol (2006) 8:188–94. doi:10.1038/ncb1353
50. Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol (2008) 15:1176–83. doi:10.1038/nsmb.1476
51. Shi Y, Do JT, Desponts C, Hahm HS, Schöler HR, Ding S. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell (2008) 2:525–8. doi:10.1016/j.stem.2008.05.011
52. Gyory I, Wu J, Fejér G, Seto E, Wright KL. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat Immunol (2004) 5:299–308. doi:10.1038/ni1046
53. Roopra A, Qazi R, Schoenike B, Daley TJ, Morrison JF. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol Cell (2004) 14:727–38. doi:10.1016/j.molcel.2004.05.026
54. Chen WL, Sun HP, Li DD, Wang ZH, You QD, Guo XK. G9a – an appealing antineoplastic target. Curr Cancer Drug Targets (2016) 16:1–15. doi:10.2174/1568009616666160512145303
55. Carr SM, Poppy Roworth A, Chan C, La Thangue NB. Post-translational control of transcription factors: methylation ranks highly. FEBS J (2015) 282:4450–65. doi:10.1111/febs.13524
56. Levings PP, Bungert J. The human beta-globin locus control region. Eur J Biochem (2002) 269:1589–99. doi:10.1046/j.1432-1327.2002.02797.x
57. Bulger M, Schübeler D, Bender MA, Hamilton J, Farrell CM, Hardison RC, et al. A complex chromatin landscape revealed by patterns of nuclease sensitivity and histone modification within the mouse beta-globin locus. Mol Cell Biol (2003) 23:5234–44. doi:10.1128/MCB.23.15.5234-5244.2003
58. Lee GR, Fields PE, Flavell RA. Regulation of IL-4 gene expression by distal regulatory elements and GATA-3 at the chromatin level. Immunity (2001) 14:447–59. doi:10.1016/S1074-7613(01)00125-X
59. Chen X, El Gazzar M, Yoza BK, McCall CE. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem (2009) 284:27857–65. doi:10.1074/jbc.M109.000950
60. Yoshida K, Maekawa T, Zhu Y, Renard-Guillet C, Chatton B, Inoue K, et al. The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat Immunol (2015) 16:1034–43. doi:10.1038/ni.3257
61. Liu C, Yu Y, Liu F, Wei X, Wrobel JA, Gunawardena HP, et al. A chromatin activity-based chemoproteomic approach reveals a transcriptional repressome for gene-specific silencing. Nat Commun (2014) 5:5733. doi:10.1038/ncomms6733
62. Merkling SH, Bronkhorst AW, Kramer JM, Overheul GJ, Schenck A, Van Rij RP. The epigenetic regulator G9a mediates tolerance to RNA virus infection in Drosophila. PLoS Pathog (2015) 11:e1004692. doi:10.1371/journal.ppat.1004692
63. Miao F, Wu X, Zhang L, Riggs AD, Natarajan R. Histone methylation patterns are cell-type specific in human monocytes and lymphocytes and well maintained at core genes. J Immunol (2008) 180:2264–9. doi:10.4049/jimmunol.180.4.2264
64. Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes (2008) 57:3189–98. doi:10.2337/db08-0645
65. Sampath SC, Marazzi I, Yap KL, Sampath SC, Krutchinsky AN, Mecklenbräuker I, et al. Methylation of a histone mimic within the histone methyltransferase G9a regulates protein complex assembly. Mol Cell (2007) 27:596–608. doi:10.1016/j.molcel.2007.06.026
66. Tachibana M, Nozaki M, Takeda N, Shinkai Y. Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J (2007) 26:3346–59. doi:10.1038/sj.emboj.7601767
67. Thomas LR, Miyashita H, Cobb RM, Pierce S, Tachibana M, Hobeika E, et al. Functional analysis of histone methyltransferase g9a in B and T lymphocytes. J Immunol (2008) 181:485–93. doi:10.4049/jimmunol.181.1.485
68. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood (2008) 112:1557–69. doi:10.1182/blood-2008-05-078154
69. Cliffe LJ, Grencis RK. The Trichuris muris system: a paradigm of resistance and susceptibility to intestinal nematode infection. Adv Parasitol (2004) 57:255–307. doi:10.1016/S0065-308X(04)57004-5
70. Chang S, Aune TM. Dynamic changes in histone-methylation “marks” across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nat Immunol (2007) 8:723–31. doi:10.1038/ni1473
71. Shin HM, Kapoor VN, Guan T, Kaech SM, Welsh RM, Berg LJ. Epigenetic modifications induced by Blimp-1 regulate CD8(+) T cell memory progression during acute virus infection. Immunity (2013) 39:661–75. doi:10.1016/j.immuni.2013.08.032
72. Antignano F, Braam M, Hughes MR, Chenery AL, Burrows K, Gold MJ, et al. G9a regulates group 2 innate lymphoid cell development by repressing the group 3 innate lymphoid cell program. J Exp Med (2016) 213:1153–62. doi:10.1084/jem.20151646
73. Osipovich O, Milley R, Meade A, Tachibana M, Shinkai Y, Krangel MS, et al. Targeted inhibition of V(D)J recombination by a histone methyltransferase. Nat Immunol (2004) 5:309–16. doi:10.1038/ni1042
74. Turner CA Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell (1994) 77:297–306. doi:10.1016/0092-8674(94)90321-2
75. van der Meer LT, Jansen JH, van der Reijden BA. Gfi1 and Gfi1b: key regulators of hematopoiesis. Leukemia (2010) 24:1834–43. doi:10.1038/leu.2010.195
76. Zweidler-Mckay PA, Grimes HL, Flubacher MM, Tsichlis PN. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol (1996) 16:4024–34. doi:10.1128/MCB.16.8.4024
77. Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell (2007) 27:562–72. doi:10.1016/j.molcel.2007.06.039
78. Vassen L, Fiolka K, Möröy T. Gfi1b alters histone methylation at target gene promoters and sites of γ-satellite containing heterochromatin. EMBO J (2006) 25:2409–19. doi:10.1038/sj.emboj.7601124
79. Lee MC, Kuo YY, Chou WC, Hou HA, Hsiao M, Tien HF. Gfi-1 is the transcriptional repressor of SOCS1 in acute myeloid leukemia cells. J Leukoc Biol (2014) 95:105–15. doi:10.1189/jlb.0912475
80. Duan Z, Zarebski A, Montoya-Durango D, Grimes HL, Horwitz M. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol Cell Biol (2005) 25:10338–51. doi:10.1128/MCB.25.23.10338-10351.2005
81. Gilks CB, Bear SE, Grimes HL, Tsichlis PN. Progression of interleukin-2 (IL-2)-dependent rat T cell lymphoma lines to IL-2-independent growth following activation of a gene (Gfi-1) encoding a novel zinc finger protein. Mol Cell Biol (1993) 13(3):1759–68. doi:10.1128/MCB.13.3.1759
82. Grimes HL, Chan TO, Zweidler-McKay PA, Tong B, Tsichlis PN. The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol Cell Biol (1996) 16:6263–72. doi:10.1128/MCB.16.11.6263
83. Zörnig M, Schmidt T, Karsunky H, Grzeschiczek A, Möröy T. Zinc finger protein GFI-1 cooperates with myc and pim-1 in T-cell lymphomagenesis by reducing the requirements for IL-2. Oncogene (1996) 12:1789–801.
84. Zhu J, Guo L, Min B, Watson CJ, Hu-Li J, Young HA, et al. Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity (2002) 16:733–44. doi:10.1016/S1074-7613(02)00317-5
85. Shinnakasu R, Yamashita M, Kuwahara M, Hosokawa H, Hasegawa A, Motohashi S, et al. Gfi1-mediated stabilization of GATA3 protein is required for Th2 cell differentiation. J Biol Chem (2008) 283:28216–25. doi:10.1074/jbc.M804174200
86. Suzuki J, Maruyama S, Tamauchi H, Kuwahara M, Horiuchi M, Mizuki M, et al. Gfi1, a transcriptional repressor, inhibits the induction of the T helper type 1 programme in activated CD4 T cells. Immunology (2016) 147:476–87. doi:10.1111/imm.12580
87. Zhu J, Jankovic D, Grinberg A, Guo L, Paul WE. Gfi-1 plays an important role in IL-2-mediated Th2 cell expansion. Proc Natl Acad Sci U S A (2006) 103:18214–9. doi:10.1073/pnas.0608981103
88. Zhu J, Davidson TS, Wei G, Jankovic D, Cui K, Schones DE, et al. Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103 +inducible regulatory T cells. J Exp Med (2009) 206:329–41. doi:10.1084/jem.20081666
89. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Végran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity (2012) 36:362–73. doi:10.1016/j.immuni.2011.12.019
90. Spooner CJ, Lesch J, Yan D, Khan AA, Abbas A, Ramirez-Carrozzi V, et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol (2013) 14:1229–36. doi:10.1038/ni.2743
91. Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop – a motor of cellular plasticity in development and cancer? EMBO Rep (2010) 11:670–7. doi:10.1038/embor.2010.117
92. Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by EF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol Biol Cell (2007) 18:3533–44. doi:10.1091/mbc.E07-03-0249
93. Si W, Huang W, Zheng Y, Yang Y, Liu X, Shan L, et al. Dysfunction of the reciprocal feedback loop between GATA3- and ZEB2-nucleated repression programs contributes to breast cancer metastasis. Cancer Cell (2015) 27:822–36. doi:10.1016/j.ccell.2015.04.011
94. Monticelli S, Lee DU, Nardone J, Bolton DL, Rao A. Chromatin-based regulation of cytokine transcription in Th2 cells and mast cells. Int Immunol (2005) 17:1513–24. doi:10.1093/intimm/dxh329
95. Ansel KM, Lee DU, Rao A. An epigenetic view of helper T cell differentiation. Nat Immunol (2003) 4:616–23. doi:10.1038/ni0703-616
96. Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, Nguyen JV, et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J Exp Med (2015) 212:2027–39. doi:10.1084/jem.20150194
97. Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, et al. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Cell Biol (2015) 211:2113OIA258. doi:10.1084/jem.20150186
98. Wang Y, Misumi I, Gu A-D, Curtis TA, Su L, Whitmire JK, et al. GATA-3 controls the maintenance and proliferation of T cells downstream of TCR and cytokine signaling. Nat Immunol (2013) 14:714–22. doi:10.1038/ni.2623
99. Nguyen ML, Hatton L, Li J, Olshansky M, Kelso A, Russ BE, et al. Dynamic regulation of permissive histone modifications and GATA3 binding underpin acquisition of granzyme A expression by virus-specific CD8(+) T cells. Eur J Immunol (2016) 46:307–18. doi:10.1002/eji.201545875
100. Ma C, Zhang N. Transforming growth factor-beta signaling is constantly shaping memory T-cell population. Proc Natl Acad Sci U S A (2015) 112:11013–7. doi:10.1073/pnas.1510119112
101. Zhang N, Bevan MJ. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. doi:10.1016/j.immuni.2013.08.019
102. Zhang N, Bevan MJ. TGF-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat Immunol (2012) 13:667–73. doi:10.1038/ni.2319
103. Takai S, Schlom J, Tucker J, Tsang KY, Greiner JW. Inhibition of TGF-β1 signaling promotes central memory T cell differentiation. J Immunol (2013) 191:2299–307. doi:10.4049/jimmunol.1300472
104. Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol (2009) 27:693–733. doi:10.1146/annurev.immunol.021908.132641
105. Gerondakis S, Fulford TS, Messina NL, Grumont RJ. NF-κB control of T cell development. Nat Immunol (2014) 15:15–25. doi:10.1038/ni.2785
106. Gerondakis S, Siebenlist U. Roles of the NF-κB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol (2010) 2:a000182. doi:10.1101/cshperspect.a000182
107. Levy D, Kuo AJ, Chang Y, Schaefer U, Kitson C, Cheung P, et al. Lysine methylation of the NF-κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat Immunol (2011) 12:29–36. doi:10.1038/ni.1968
108. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med (2009) 206:3001–14. doi:10.1084/jem.20091411
109. Vang KB, Yang J, Pagán AJ, Li LX, Wang J, Green JM, et al. Cutting edge: CD28 and c-Rel-dependent pathways initiate regulatory T cell development. J Immunol (2010) 184:4074–7. doi:10.4049/jimmunol.0903933
110. Deenick EK, Elford AR, Pellegrini M, Hall H, Mak TW, Ohashi PS. c-Rel but not NF-kappaB1 is important for T regulatory cell development. Eur J Immunol (2010) 40:677–81. doi:10.1002/eji.201040298
111. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity (2009) 31:932–40. doi:10.1016/j.immuni.2009.10.006
112. Visekruna A, Huber M, Hellhund A, Bothur E, Reinhard K, Bollig N, et al. c-Rel is crucial for the induction of Foxp3(+) regulatory CD4(+) T cells but not T(H)17 cells. Eur J Immunol (2010) 40:671–6. doi:10.1002/eji.200940260
113. Messina N, Fulford T, O’Reilly L, Loh WX, Motyer JM, Ellis D, et al. The NF-κB transcription factor RelA is required for the tolerogenic function of Foxp3(+) regulatory T cells. J Autoimmun (2016) 70:52–62. doi:10.1016/j.jaut.2016.03.017
114. Xiao X, Shi X, Fan Y, Wu C, Zhang X, Minze L, et al. The costimulatory receptor OX40 inhibits interleukin-17 expression through activation of repressive chromatin remodeling pathways. Immunity (2016) 44:1271–83. doi:10.1016/j.immuni.2016.05.013
115. Chen G, Hardy K, Pagler E, Ma L, Lee S, Gerondakis S, et al. The NF-κB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J Immunol (2011) 187:4483–91. doi:10.4049/jimmunol.1101757
116. Kallies A, Hawkins ED, Belz GT, Metcalf D, Hommel M, Corcoran LM, et al. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat Immunol (2006) 7:466–74. doi:10.1038/ni1321
117. Martins GA, Cimmino L, Liao J, Magnusdottir E, Calame K. Blimp-1 directly represses Il2 and the Il2 activator Fos, attenuating T cell proliferation and survival. J Exp Med (2008) 205:1959–65. doi:10.1084/jem.20080526
118. Wang L, van Panhuys N, Hu-Li J, Kim S, Le Gros G, Min B. Blimp-1 induced by IL-4 plays a critical role in suppressing IL-2 production in activated CD4 T cells. J Immunol (2008) 181:5249–56. doi:10.4049/jimmunol.181.8.5249
119. Neumann C, Heinrich F, Neumann K, Junghans V, Mashreghi M-F, Ahlers J, et al. Role of Blimp-1 in programing Th effector cells into IL-10 producers. J Exp Med (2014) 211:1807–19. doi:10.1084/jem.20131548
120. Perona-Wright G, Kohlmeier JE, Bassity E, Freitas TC, Mohrs K, Cookenham T, et al. Persistent loss of IL-27 responsiveness in CD8+ memory T cells abrogates IL-10 expression in a recall response. Proc Natl Acad Sci U S A (2012) 109:18535–40. doi:10.1073/pnas.1119133109
121. Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity (2009) 31:296–308. doi:10.1016/j.immuni.2009.05.014
122. Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, et al. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity (2009) 31:309–20. doi:10.1016/j.immuni.2009.06.019
123. Salehi S, Bankoti R, Benevides L, Willen J, Couse M, Silva JS, et al. B lymphocyte-induced maturation protein-1 contributes to intestinal mucosa homeostasis by limiting the number of IL-17-producing CD4+ T cells. J Immunol (2012) 189:5682–93. doi:10.4049/jimmunol.1201966
124. Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, et al. Interleukin-23-induced transcription factor Blimp-1 promotes pathogenicity of T helper 17 cells. Immunity (2016) 44:131–42. doi:10.1016/j.immuni.2015.11.009
125. Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell (2007) 25:473–81. doi:10.1016/j.molcel.2007.01.017
126. Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol (2009) 16:312–7. doi:10.1038/nsmb.1560
127. Liu F, Chen X, Allali-Hassani A, Quinn AM, Wasney GA, Dong A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem (2009) 52:7950–3. doi:10.1021/jm901543m
128. Chang Y, Ganesh T, Horton JR, Spannhoff A, Liu J, Sun A, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J Mol Biol (2010) 400:1–7. doi:10.1016/j.jmb.2010.04.048
129. Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang X-P, et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem (2013) 56:8931–42. doi:10.1021/jm401480r
130. Sweis RF, Pliushchev M, Brown PJ, Guo J, Li F, Maag D, et al. Discovery and development of potent and selective inhibitors of histone methyltransferase g9a. ACS Med Chem Lett (2014) 5:205–9. doi:10.1021/ml400496h
Keywords: G9a, T cells, innate lymphoid cells, epigenetics, immunological memory, mouse models, infection, inflammation
Citation: Scheer S and Zaph C (2017) The Lysine Methyltransferase G9a in Immune Cell Differentiation and Function. Front. Immunol. 8:429. doi: 10.3389/fimmu.2017.00429
Received: 19 December 2016; Accepted: 27 March 2017;
Published: 11 April 2017
Edited by:
Benjamin Youngblood, St Jude Children’s Research Hospital, USAReviewed by:
Rhys Allan, Walter and Eliza Hall Institute of Medical Research, AustraliaScott Hale, University of Utah, USA
Copyright: © 2017 Scheer and Zaph. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Colby Zaph, colby.zaph@monash.edu