 Kokouvi Kassegne1*
Kokouvi Kassegne1* Komi Komi Koukoura2
Komi Komi Koukoura2 Hai-Mo Shen3,4,5Shen-Bo Chen3,4,5Hai-Tian Fu1
Hai-Mo Shen3,4,5Shen-Bo Chen3,4,5Hai-Tian Fu1 Yong-Quan Chen1,6
Yong-Quan Chen1,6 Xiao-Nong Zhou3,4,5
Xiao-Nong Zhou3,4,5 Jun-Hu Chen3,4,5*
Jun-Hu Chen3,4,5* Yang Cheng1*
Yang Cheng1*- 1Laboratory of Pathogen Infection and Immunity, Department of Public Health and Preventive Medicine, Wuxi School of Medicine, Jiangnan University, Wuxi, China
- 2Laboratoire des Sciences Biomédicales, Alimentaires et Santé Environnementale, Département des Analyses Biomédicales, Ecole Supérieure des Techniques Biologiques et Alimentaires, Université de Lomé, Lomé, Togo
- 3National Institute of Parasitic Diseases, Chinese Centre for Disease Control and Prevention, Chinese Centre for Tropical Diseases Research, WHO Collaborating Centre for Tropical Diseases, National Centre for International Research on Tropical Diseases, Ministry of Science and Technology, Key Laboratory of Parasite and Vector Biology, Ministry of Health, Shanghai, China
- 4National Institute of Parasitic Diseases, Chinese Centre for Disease Control and Prevention—Shenzhen Centre for Disease Control and Prevention Joint Laboratory for Imported Tropical Disease Control, Shanghai, China
- 5The School of Global Health, Chinese Centre for Tropical Diseases Research, Shanghai JiaoTong University School of Medicine, Shanghai, China
- 6School of Food Science and Technology, State Key Laboratory of Food Science and Technology, Jiangnan University, Wuxi, China
Malaria is a public health concern worldwide, and Togo has proven to be no exception. Effective approaches to provide information on biological insights for disease elimination are therefore a research priority. Local selection on malaria pathogens is due to multiple factors including host immunity. We undertook genome-wide analysis of sequence variation on a sample of 10 Plasmodium falciparum (Pf) clinical isolates from Togo to identify local-specific signals of selection. Paired-end short-read sequences were mapped and aligned onto > 95% of the 3D7 Pf reference genome sequence in high fold coverage. Data on 266 963 single nucleotide polymorphisms were obtained, with average nucleotide diversity π = 1.79 × 10−3. Both principal component and neighbor-joining tree analyses showed that the Togo parasites clustered according to their geographic (Africa) origin. In addition, the average genome-wide diversity of Pf from Togo was much higher than that from other African samples. Tajima’s D value of the Togo isolates was −0.56, suggesting evidence of directional selection and/or recent population expansion. Against this background, within-population analyses identifying loci of balancing and recent positive selections evidenced that host immunity has been the major selective agent. Importantly, 87 and 296 parasite antigen genes with Tajima’s D values > 1 and in the top 1% haplotype scores, respectively, include a significant representation of membrane proteins at the merozoite stage that invaded red blood cells (RBCs) and parasitized RBCs surface proteins that play roles in immunoevasion, adhesion, or rosetting. This is consistent with expectations that elevated signals of selection due to allele-specific acquired immunity are likely to operate on antigenic targets. Collectively, our data suggest a recent expansion of Pf population in Togo and evidence strong host immune selection on membrane/surface antigens reflected in signals of balancing/positive selection of important gene loci. Findings from this study provide a fundamental basis to engage studies for effective malaria control in Togo.
Introduction
Malaria clinical presentation ensues when Plasmodium parasites invade and destroy red blood cells (RBCs). Fever and chills occur at the time of rupture of infected RBCs (iRBCs) containing merozoites that are freed to invade uninfected RBCs (1, 2). Failure to receive prompt treatment may lead to dyserythropoietic anaemia or severe malaria. P. falciparum (Pf) is the most dangerous malaria parasite because of the high level of mortality with which it is associated, its widespread resistance to antimalarial medicines, and its dominance in the world’s most malarious continent, Africa (3–5).
In Togo, malaria transmission occurs most of each year. Although decades of control efforts have reduced the disease burden, the entire country’s population is still at risk of falciparum malaria infection (6). In addition, challenges in parasite control would have made the infection a public health concern and may aggravate the difficulty of treatment. Clinical spectrum of malaria in Togo usually ranges from asymptomatic carriage of malaria parasites to a febrile disease that may evolve into a severe, life-threatening illness, making the infection a major cause of morbidity and mortality, especially in children (7, 8). Antimalarial drug resistance (e.g., parasite resistance to chloroquine or pyrimethamine) has been experienced across Africa. In early investigations in Togo, clinical and parasitological therapeutic failure tests of artemether-lumefarine (AL) and artesunate-amodiaquine (ASAQ) for 3% and 3.8%, respectively, have been observed (6), and they drew the entire country’s attention to an eventual resistance to artemisinins. However, in a recent study, therapeutic efficacy of AL and ASAQ was shown without delay in the clearance of mutant parasites (9). Pf surface-exposed proteins are targets of host immune responses, and repeated exposures to the parasite in endemic areas induce a slow and gradual development of acquired immunity to clinical malaria, which is usually evidenced as a decline in the prevalence of clinical episodes (10, 11). Hence, acquisition of information on both immunity-related antigens and drug resistance genes for effective interventions to sustain and drive forward the struggle against malaria parasite in Togo is therefore a research priority.
Complete sequencing of the Pf genome has boosted post-genomic studies of malaria (12). It provides fundamental knowledge for better understanding of the cellular and molecular mechanisms of infection and immunity to develop new control methods, including new drugs and vaccines, improved diagnostics, and effective vector control techniques. With rapid development of sequencing technologies (13), hundreds of falciparum isolate genomic data worldwide had been investigated and shared by large collaborative initiatives such as the MalariaGEN Pf Community Project and the Pf3k Consortium. Application of the genomic approaches in the analysis of whole genome variations–generated high-density single nucleotide polymorphisms (SNPs) of the parasite has mostly focused on vaccine antigen genes and drug-resistant genes. However, to date, nothing is known on genomes of malaria isolates in Togo, and this could limit the joint research with those in other endemic areas in the sub-Saharan Africa region.
In this study, we performed the first whole-genome sequencing (WGS) of Pf clinical isolates from Togo. With the aim to contribute to accelerating the pursuit of effective malaria control, we applied genomic approaches in the analysis of whole genome variations–generated high-density SNPs to provide biological insights on target genes, especially those under host immune selection.
Materials and Methods
Sampling Sites and Ethics Statement
Malaria transmission in Togo occurs for most of each year with seasonal outbreaks (9), and populations are served by health facilities experienced in the management of malaria cases. For this study, clinical samples were collected at health centres in urban areas of Agou-Gadzépé (7°28’01’’ N; 1°55’01’’ E) and Atakpamé (7°52’87’’ N; 1° 13’05’’ E) in Agou and Ogou prefectures, respectively, in the Plateaux Region (Figure 1) in 2017 and 2018. Samples collection was made under a study protocol approved by the Togolese Ministry of Health’s Bioethics Committee following institutional ethical guidelines by the ethics committee at National Institute of Parasitic Diseases, Chinese Centre for Disease Control and Prevention. Informed consent was obtained from all subjects prior to sample collection.
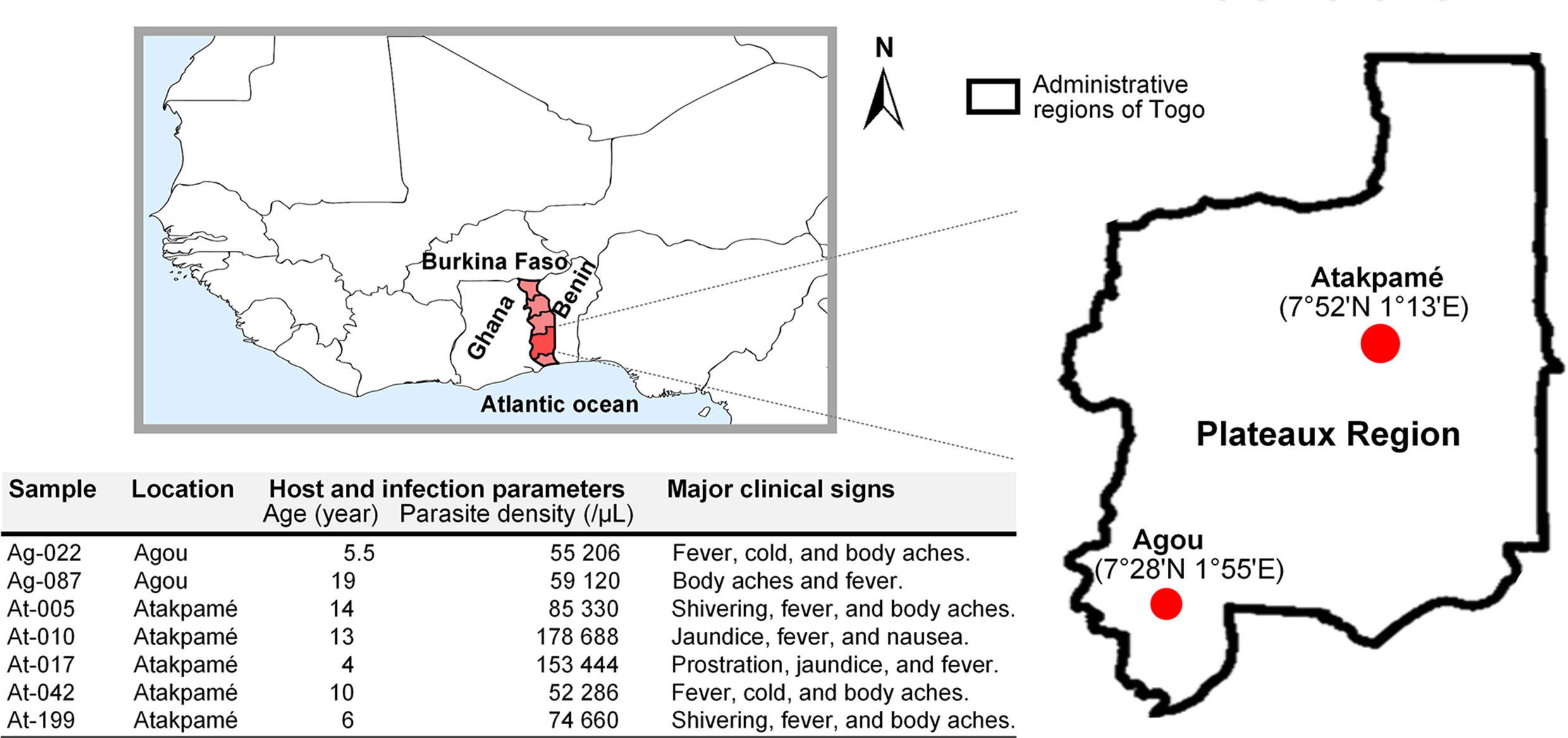
Figure 1 Sampling location in Togo and information about clinical samples used in this study.
Genomic Data
For our analyses, genome and annotation data of Pf 3D7 strain (the most complete whole genome standard reference) from PlasmoDB database (http://plasmodb.org/plasmo/) (14) were downloaded. In addition, raw sequences from 62 genome data of falciparum clinical isolates from Africa [n = 32 (Congo DR, Gambia, Ghana, Guinea, Malawi, Mali, Nigeria, and Senegal)] and Asia [n = 30 (Bangladesh, Cambodia, China-Myanmar Border (CMB), Laos, Myanmar, Thailand, and Viet Nam)] were also referenced (15–17).
Sampling of Malaria Parasites and Extraction of Genomic DNA
Malaria-naturally exposed subjects who received parasitological diagnosis using Giemsa-stained thick blood smear microscopy under 1000x magnification were referred to our study. Whole blood specimens from subjects who were diagnosed with the presence of Pf asexual parasitaemia (parasites counted per 200 leukocytes and parasite density calculated as the number of parasites per microliter by assuming a fixed leukocyte count of 8000 cells/μL of blood) were sampled as dried blood spots (DBSs) on Whatman FTA cards (GE Healthcare) as recommended by the manufacturer. Genomic DNA was extracted [using the QIAGEN DNeasy Blood & Tissue Kit (Qiagen), according to the manufacturer’s instructions] from DBSs and monospecies infection was confirmed by polymerase chain reaction (PCR). Ten clinical samples with high parasitaemia (parasite density > 50000/μL), and qualitatively and quantitatively good enough, were selected to ensure the integrity of sequencing.
Whole-Genome Sequencing
WGS of Pf clinical isolates from Togo was performed by OE Biotech (Shanghai). Extracted genomic DNA was sheared into 150 bp fragments using a Covaris instrument. The fragmented DNA molecules were used to construct Illumina-sequencing libraries with TruSeq DNA LT Sample Prep Kit (Illumina). All libraries were sequenced on the Illumina HiSeq X10 platform according to the manufacturer’s protocol (18), using the direct sequencing approach, as described previously (17). All reads were filtered by removing the adapter sequences and low quality sequences were removed with Trimmomatic-3.0. (19). The sequencing reads have been submitted to the Short Read Archive of the National Centre for Biotechnology Information.
Identification of SNPs and Population Structure
All sequenced reads from the 10 samples were mapped to the Pf 3D7 genome using Burrows-Wheeler Aligner and Sequence Alignment/Map (SAMtools-1.3) (20). Samples with average coverage < 95% sequences mapping over 3D7 reference genome were removed. For high-quality SNP calling, sequencing reads were genotyped using an in-house pipeline based on GATK best practices and SnpEff workflows (21), with Pf3K known-sites (15).
Principal component analysis (PCA) and neighbor-joining were performed to investigate major geographical division of population structure. PCA and a neighbor-joining tree of all samples were undertaken via SPSS-Ver25 and Mega-Ver6.0 programs, respectively, to compare Pf SNPs from Togo isolates with those from the 62 isolates collected worldwide (15–17).
Tests for Signatures of Selection
For SNPs in all populations, nucleotide diversity (π) was estimated for the whole genome mutation rate in 4 kb sliding window and 2 kb step across each chromosome in Arlequin-Ver3.5 (22). To distinguish between genes evolving neutrally and under selective pressures, or genetic hitchhiking, Tajima’s D value (TD) for each sliding window and the corresponding gene was also calculated.
In addition, long-range haplotype diversity approach integrated haplotype score (iHS) was employed to identify genes under recent positive selection. iHS compares integrated extended-haplotype homozygosity (EHH) values between alleles at a given SNP (23). iHS computation was based on the Togo clinical isolates by tracking the decay of haplotype homozygosity for both the ancestral and derived haplotypes extending from every SNP site (24). For this test, we restricted the analyses to SNPs with inferred ancestral states with minor allele frequencies equal to or higher than 5% (25). iHS scores were estimated using Selscan-Ver1.10a (26).
To assess whether genes associated with putative functions were enriched among the group of genes with high Tajima’s D values (> 1.0) or high |iHS| (top 1% score), gene ontology (GO) term analysis was conducted. Genes with a TD > 1.0 were classed as genes of potential interest for GO analysis. Analysis was performed using GO Enrichment tool of PlasmoDB (http://plasmodb.org/plasmo/, PlasmoDB Ver-46). The adjusted P values were also generated from Fisher’s exact test, and the statistical significance was set for P < 0.05.
Results
Genetic Diversity of Falciparum Isolates From Togo
We used a direct sequencing approach that requires only high parasitaemia for malaria parasites without leukocytes filtration (17) to sequence clinical isolates of Pf genomes from Togo. Among the 10 clinical samples that were sequenced, results of seven were good enough and provided enough coverage (> 95% sequences mapping over the 3D7 reference genome) (Table 1). The remaining three samples mapped onto only 58.89%, 56.43%, and 46.83% (unshown data) and failed for further analysis. In this study, the Togo isolates generated between 55 and 176 M paired-end reads of 150 bp from each of the samples, globally. All sequencing reads have been deposited to the National Centre for Biotechnology Information (NCBI) Short Read Archive (Bio-Project Accession Number: PRJNA616298). A variable proportion of reads (3.8–14.2%) from all the isolates were mapped to the reference and aligned onto at least 95% of the reference 3D7 strain genome in high fold coverage (7.2–33.9x).
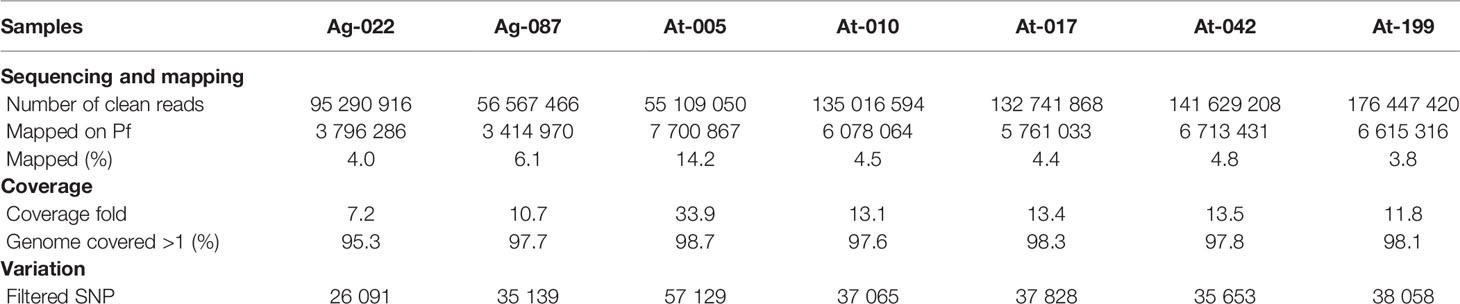
Table 1 Sequencing and mapping summary of Pf genome of seven clinical isolates from Togo.
For analysis of polymorphism, a total of 266963 SNPs common loci were available for analysis after quality filtering (Table 1). The list of the SNPs for all the isolates is provided in Supplementary Table 1. Of the 266963 SNPs, excluding the low-frequency SNPs (103497 SNPs with minor allele frequency < 5%), a total of 163466 SNPs across the seven isolates were identified and could be mapped to coding sequences. In addition, SNPs were identified across 4614 genes on 14 chromosomes in the samples and 931 genes had more than five SNPs (Figure 2A). These genes were considered informative for comparisons of polymorphic nucleotide sites.
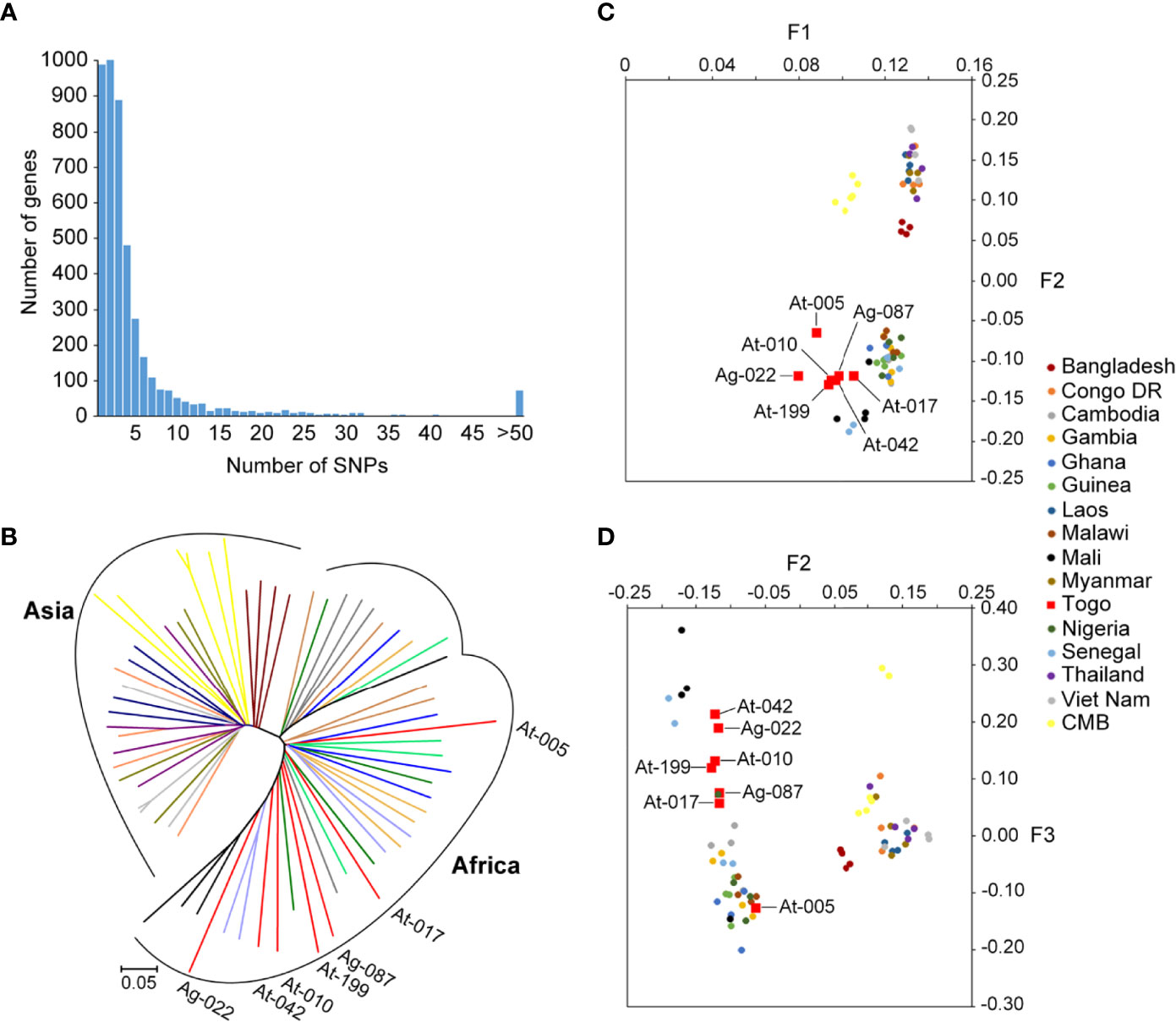
Figure 2 SNPs frequency distribution in samples and genomic relationships among Pf reference strains and Togo isolates. (A) Distribution numbers of genes with each given number of SNPs in the population sample of seven Pf clinical isolates from Togo. From the 4614 genes analyzed in total from 14 chromosomes, 20.2% (931/4614) had more than five SNPs. (B) Neighbor-joining tree of Pf constructed from the SNPs occurring in at least half of the samples. Lineages are colored according to geographic origin. Branch lengths indicate considerable diversity in Pf strains. Annotated branches represent the Togo isolates. (C, D) Principal component analysis based on common SNP loci in Togo clinical isolates and reference strains. Colors correspond to the geographic origin of the samples, of which the Togo isolates are highlighted in red. (C) The major fact (F1) of differentiation of the PCA identified clearly the two groups of isolates that clustered according to their geographic origin. (D) The second and third facts (F2 and F3) defined a distinct South-Asia cluster and distinguished the African samples better according to their locations.
Comparison of Genetic Diversity of the Isolates Among Different Endemic Regions
Overall genome-wide π of Pf clinical isolates from Togo were estimated at 1.79 × 10−3. However, genetic diversity was lower in intronic regions but higher in exonic and intergenic regions (Supplementary Table 1). Supplementary Figure 1 shows the π map of the isolates across 14 chromosomes. Interestingly, we observed that Togo samples have genes with higher SNPs, suggesting a greater genetic diversity than that reported from other African samples (π = 1.03 × 10−3) (27), but lower than that of isolates from CMB (π = 2.87 × 10−2) (17).
We then performed PCA and neighbor-joining analyses of all strains to assess major geographical difference. As part of Africa isolates, the Togo isolates illustrated a higher discrepancy than the 3D7 strain genome. Neighbor-joining displayed a tree with two distinct branches separating two major clades that correspond to the Asia and Africa geographical groups of samples (Figure 2B). There was evidence of clear distinction of the isolates from the two regions, and African isolates displayed sub-clusters to form two (or three) monophyletic clades. Furthermore, we found that the outcome from PCA was similar to that of the neighbor-joining analysis. The major axis of differentiation (F1) of the PCA distinguished clearly two major Asia and Africa groups of isolates, which is in accordance with their geographical origins (Figure 2C). Similar observation was noted in recent studies on Pf isolates from CMB (17, 28). In addition, among the Africa samples, Togo samples exhibited greater genetic diversity than has been reported from other African regions. The second and third principal components (F2 and F3) defined a distinct South-Asian cluster and distinguished the African samples better according to their locations, where Togo samples were well differentiated from other African samples (Figure 2D). Furthermore, Togo isolates were widely separated in our PCA result, suggesting high diversity of Pf from Togo.
Signatures of Selection in the Isolates From Togo
We investigated signatures of selection of the parasite in this sub-Saharan Africa region. TD of the Togo isolates was −0.56 across the entire genome (Figure 3A), indicating a population history of purifying selection and/or recent population expansion. To study allele frequency distributions for individual genes, with the gene transfer format file containing information about gene structure, we annotated genes and then calculated TD for the individual genes. Of the 5601 genes analyzed, the TD obtained were mostly negative (3759 genes, average TD = -0.95). Such predominantly negative values are consistent with previous analyses indicating a historical population expansion of Pf in Africa (29), and it suggests that these genes were under selective sweep (directional selection) (17, 28, 30). We found 931 genes that each had at least five SNPs. A list of the top 250 lowest values for genes with at least one SNP (n = 4614) is provided in Supplementary Table 2. Against this background, 746/4614 genes (16.2%) had positive TD values (Supplementary Table 3), of which 140 genes had values > 1 (87 genes coding proteins with known functions), suggesting signals of balancing selection for these genes (28, 31).
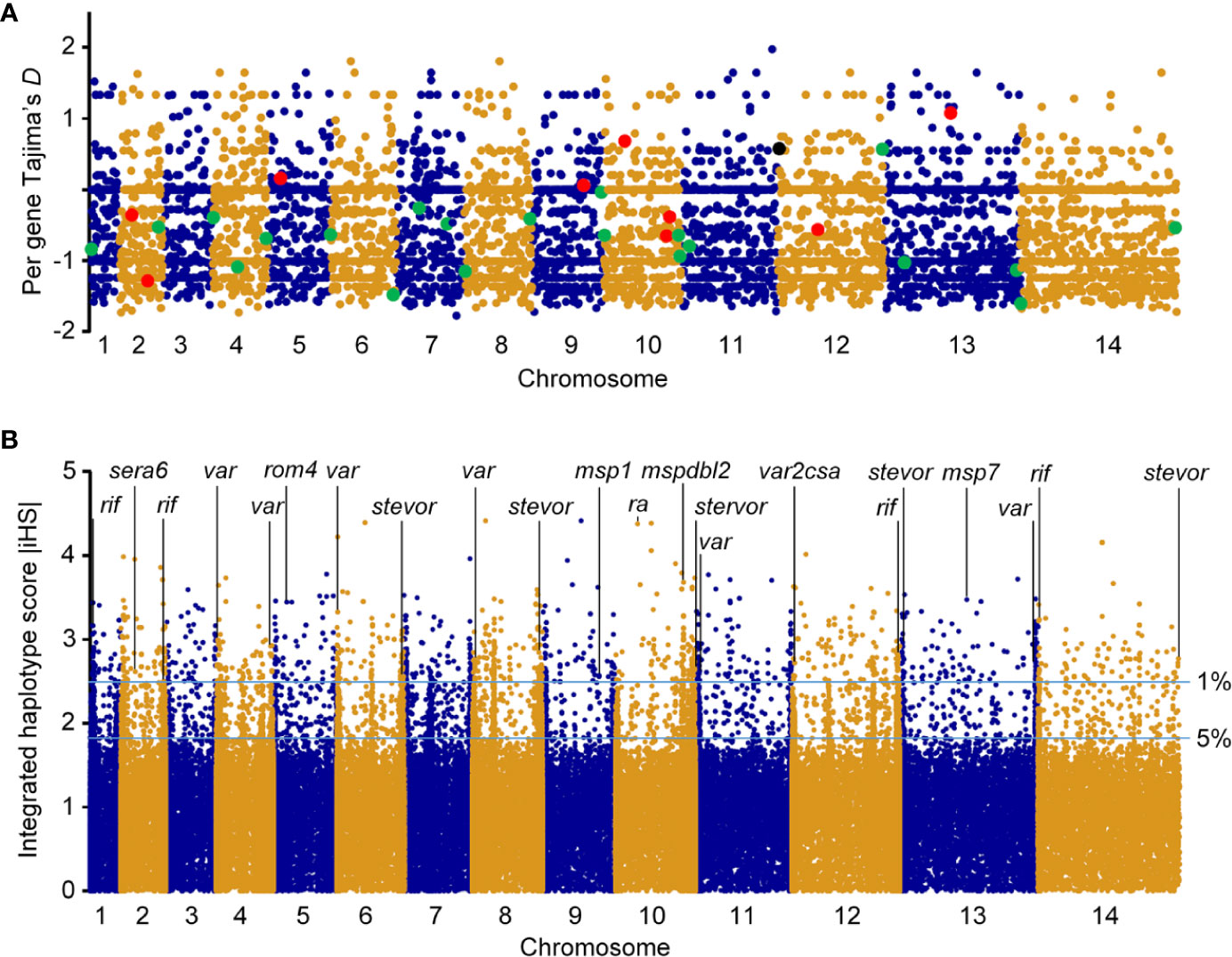
Figure 3 Genomic map of Pf isolates from Togo. Paired-end short-read sequencing produced high-quality data for a population sample of seven falciparum clinical isolates from Togo, with genome-wide average mapping depth to the 3D7 reference strain genome. (A) Tajima’s D values map of falciparum clinical isolates from Togo. Values for all gene SNPs were plotted and arranged according to their chromosomal positions (blue and gold colors indicate consecutive chromosomes numbered from the smallest upwards). TD for RBC invasion-related antigen genes are shown in enlarged red dyes and those for genes implicated in parasite-mediate immunoevasion, cytoadhesion, resetting/sequestration, or deformability of RBC/rigidity, are indicated in enlarged green dyes. Pregnancy malaria-related var2csa is also highlighted (black dye). (B) Top |iHS| hits in Pf isolates from Togo with SNPs minor allele frequency ⩾ 5%. x axis indicates individual chromosomes in alternating colors of their SNPs; y axis is the value of |iHS|. Plot of genome-wide |iHS| scores shows regions of the genome that have windows of elevated values with high scoring (top 1% of |iHS| values) for important gene loci highlighted, consistent with the operation of recent positive directional selection. The horizontal lines represent values of 2.49223 and 1.8157 used to define windows containing SNPs with overlapping regions of EHH.
Mean pairwise divergence was higher in a significant representation of genes that encode membrane proteins expressed at the merozoite stage that invades RBCs (for example, merozoite surface proteins, MSPs; serine repeat antigens, SERAs; rhomboid proteases, ROMs; duffy binding-like merozoite surface proteins, MSPDBLs; rhoptry associated adhesins, RA) (32, 33), and parasitized RBC surface proteins that play roles in disease severity—immune evasion, rosetting, or cytoadherence to microvasculature (repetitive interspersed family of polypeptides, RIFINs; erythrocyte membrane protein 1, PfEMP1; and subtelomeric variant open reading frames, STEVORs) (34–37). Importantly, there was evidence of balancing selection on particular genes including antigen genes related to RBC invasion, including those with solid balancing selection (reflected in high TD) [sera5 (TD = 1.42); apical asparagine-rich protein, aarp (TD = 1.34); ferlin-like protein, flp (TD=1.29); msp3 (TD = 1.28); and msp7 (TD = 1.08)] (Table 2) and those with TD < 1 [phospholipase, pl; erythrocyte binding antigen-175, eba175; reticulocyte binding protein 2 homologue a, rh2a; ra; 6-cysteine protein, pf41; apical membrane antigen 1, ama1; sera4; glutamate-rich protein, glurp; merozoite TRAP-like protein, mtrap; rom4; membrane associated erythrocyte binding-like protein, maebel; rhoptry neck protein 2, ron2; subtilisin-like protease 1, sub1; and msp1] (Supplementary Table 3). Most of these antigen genes were reported previously for the balancing selection (28, 31, 38, 39) and were significantly enriched by GO analysis (P < 0.0001). Although antigen genes associated with parasite-mediated immune evasion, adhesion, or rosetting were found in the positive TD list [13 rifs (PF3D7_1000600, PF3D7_1254800, PF3D7_0713000, PF3D7_0632100, PF3D7_1040900, PF3D7_1300400, PF3D7_1254700, PF3D7_1150300, PF3D7_0808800, PF3D7_0114700, PF3D7_1101300, PF3D7_1100300, and PF3D7_0401300), three vars (PF3D7_0302300, PF3D7_1200600, and PF3D7_0601400) and one stevor (PF3D7_1479900)] (Supplementary Table 3) and were all highly significantly enriched by GO analysis (P < 0.0001), only the var PF3D7_0302300 is likely under strong balancing selection (TD = 1.33) (Table 2). Interestingly, PF3D7_1200600 (TD = 0.57) is the Pf var gene (var2csa) implicated in pregnancy malaria (34). GO analysis showed significant drug resistance enrichment for amino acid transporter aat1 and bifunctional farnesyl/geranylgeranyl diphosphate synthase fpps/ggpps, which got TD > 1.

Table 2 Ten Pf genes with Tajima’s D scores > 1 enriched by GO analysis in Togo isolates.
Antigenic variation within Pf surface-exposed putative proteins is a target of host immune selection. Therefore, we applied iHS for all SNPs from Pf isolate genomes to investigate genome-wide evidence for positive selection (Figure 3B). We identified all 14 chromosomal regions with loci above the top 5% value (|iHS| > 1.8157) of the randomly expected distribution including 646 genes (Supplementary Table 4). Using |iHS| = 2.49223 (top 1% expected distribution) as a strong hits threshold, we identified 306 genes under significant positive selection (38, 40), of which 296 had at least five SNPs (Supplementary Table 5).
This analysis identified the selection signals for important genes with loci above the top 1% iHS score (|iHS| > 2.49223), including 10 RBC invasion-related antigen genes [msp1, msp4, msp7, msp9, ron2, mspdbl1, mspdbl2, ra, sera6, and rom4] (Supplementary Table 6). From these, msp1, msp7, mspdbl1, mspdbl2, ra, and sera6 were highly significantly enriched by GO analysis (P < 0.0001) (Table 3) and have been reported previously as promising subunit candidates for a malaria multicomponent vaccine (32, 39, 41). Similarly, 134 genes implicated in roles for immune evasion, RBCs aggregation, or cytoadherence to microvasculature (73 rifs, 50 vars, and 12 stevors) were identified (Supplementary Table 7), of which six rifs, seven vars, and eight stevors (Table 3) were reported previously as targets of acquired immunity and may serve to prevent severe malaria (42–46). Furthermore, the var2csa that is implicated in pregnancy placental malaria (34, 47) was also observed in the top highest haplotype scores. Overall, iHS values aligned onto those obtained from Tajima’s D analysis. We also found signals of positive selection in genes that may be related to drug resistance (n = 8) within the top 1% iHS (Supplementary Table 8). From these, two genes (abc transporter I family member 1, abcI3 and AP2 domain transcription factor, apiap2) (Table 3) were significantly enriched by GO analysis (P < 0.001) and were previously reported (48, 49). But, no selection signals were observed around the five known Pf drug resistance genes that include the chloroquine resistance transporter (crt), multidrug resistance-1 (mdr1), dihydrofolate reductase (dhfr), dihydropteroate synthase (dhps), and kelch 13 (k13).
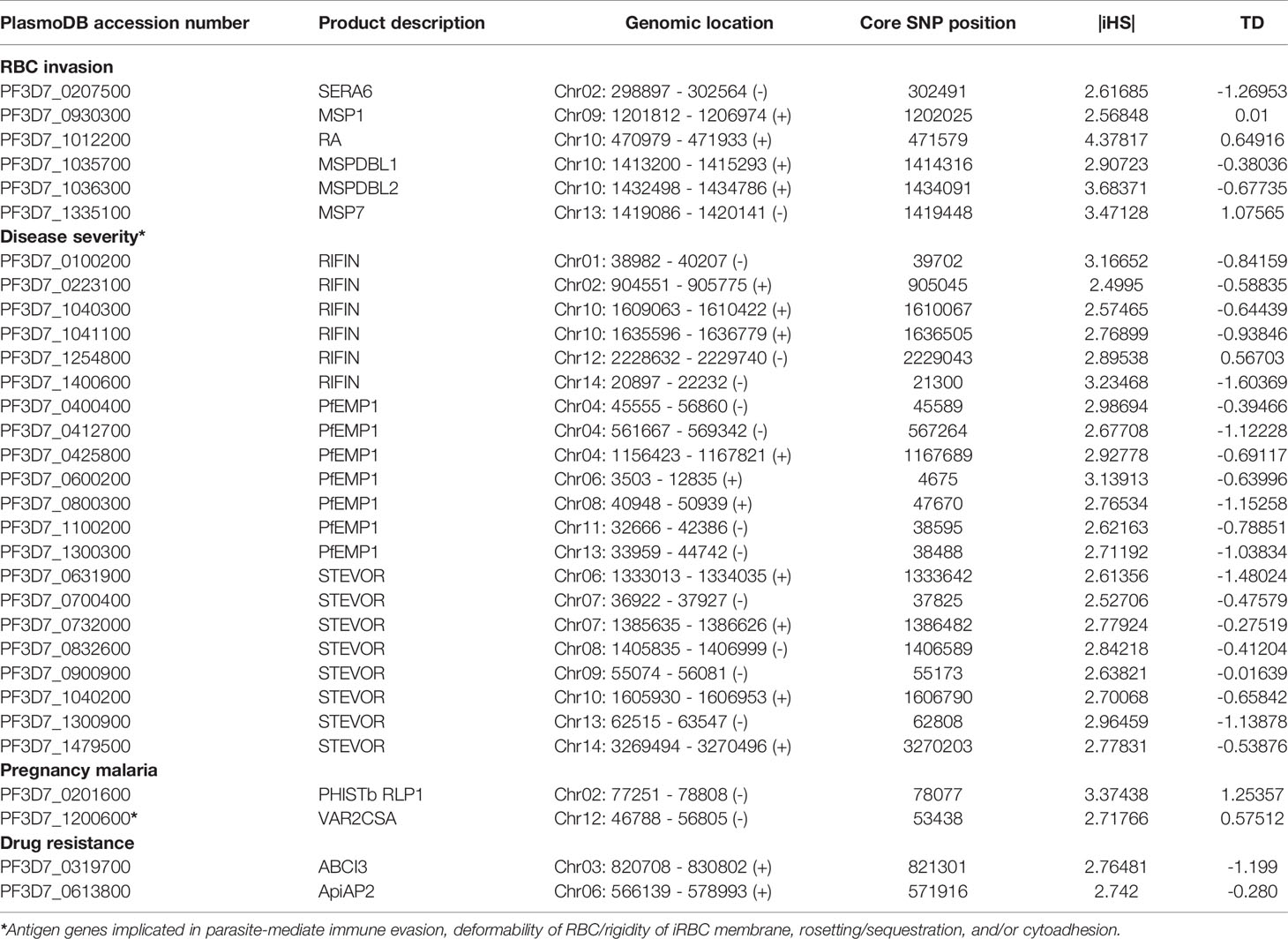
Table 3 List of important Pf top 1% |iHS|-related genes enriched by GO analysis in Togo isolates.
Discussion
P. falciparum originated in Africa and spread to other continents as human migration gradually formed new populations (29). In this study, both the PCA and neighbor-joining tree analyses showed that the parasites derived from Togo clustered according to their geographic origin and distinguished two major clades that correspond to the Asia and Africa geographical groups of samples (17, 38). In addition, our data revealed the average nucleotide diversity of Pf from Togo is much higher than that from other African samples, but it is lower than the parasite from the CMB, probably due to the historically different antimalarial drugs used in that area (17). However, locally varying selection on pathogens due to differences in host immunity may be the major factor for the high nucleotide diversity observed in Togo isolates in comparison to other Africa isolates.
The purpose of the Tajima test is to detect deviation from neutrality, in other words, to indicate processes such as balancing selection, selective sweeps, and population expansion. This study revealed that some particular antigen genes that are related to RBC invasion and disease severity, and known to be polymorphic and under balancing selection by host immune system (31, 39), got TD < 0; suggesting selective sweep (directional selection) and/or recent population expansion. Interestingly, previous scans for evidence of positive selection on Pf have clearly identified loci that have undergone selective sweeps (38, 49, 50) as well as loci that are apparently under balancing selection, including those encoding targets of acquired immunity (31). In addition, some other investigations have observed multiple genes under recent positive selection by computation of iHS in other parasite populations (39, 40, 51, 52). Therefore, here, we applied iHS as a complementary analysis to assess signals of host immune selection.
In Pf isolates from Togo, within genes that are likely under signals of recent positive selection, host immunity-related antigen genes have been the major selective agents. In terms of the top outlier genes (top 1% |iHS| as a strong hits threshold and GO enrichment analysis), 31 of the 306 genes with known functions included six RBC invasion-linked antigen genes (msp1, msp7, mspdbl1, mspdbl2, ra, and sera6) (32, 41) and 22 antigen genes (six rifs, seven vars, and eight stevors) that are associated with roles in evasion to host immunity, rosetting or cytoadhesion (35–37), among which is var2csa, a pregnancy placental malaria-related gene (34, 47) (Table 3). Potential interest for GO analysis for genes under balancing selection by host immune system revealed six genes related to RBC invasion (aarp, flp, msp3, msp7, pl, and sera5) (32), one var (PF3D7_0302300) associated with pathogenesis (GO: 0009405), and phistb rpl1 that is implicated in placental cytoadherence to microvasculature (47).
Interestingly, we found that most of the gene family members with elevated |iHS| are located close to each other on the chromosome. For example, from three sera genes that are contiguously arranged on chromosome two, sera6 was involved in the top 1% SNP locus (|iHS| = 2.61685) and the remaining other two were also included in the 5% iHS list. This was also observed on chromosome two between mps4 involved in the top 1% |iHS| (|iHS| = 2.83572) and msp2 included in the 5% |iHS| (|iHS| = 2.08998). Following similar observation with eight serine-repeat antigen genes in P. vivax isolates (25), this could be explained by the process of positive natural selection increasing the prevalence of both selected variant as well as of nearby variants, generating local regions of extended haplotypes.
We identified genes that are likely to have been under exceptionally strong recent positive selection. Given these genes encode membrane/surface proteins, they would have been under high selection from the host immune system as potential selective targets of host immunity, and this may explain the high iHS scores that we observed (39, 41, 42). For example, highly elevated |iHS| associated with the gene encoding the MSP1 antigen was consistent with that from a previous report on Pf isolates from Gambia and Guinea, as this gene has a complex pattern of polymorphism that is likely to result from different selective processes (38). The MSP1, a core member of band 3 co-ligand complex during RBC invasion (32), has been validated as one of the leading blood-stage malaria vaccine antigens with sequences incorporated in experimental vaccine trials (41). In addition, highly supported windows of elevated iHS scores were also observed on chromosomes two and 10, incorporating the sera6 and a cluster of different antigen genes (including ra, mspdbl1, and mspdbl2), respectively. Similarly, genes under high operation of positive selection in the Togo isolates include those encoding known surface antigens such as vars (PF3D7_1100200, PF3D7_0425800, PF3D7_1300300, and PF3D7_0400400) and promising targets of immunity that require further studies [members of rif (PF3D7_0223100, PF3D7_1400600, and PF3D7_0100200) and stevor (PF3D7_1040200, PF3D7_0631900, PF3D7_1300900, and PF3D7_0832600) families]. They are known to bind to cerebral endothelial/RBC surface receptors and have been identified or reported previously as immune targets that may serve to prevent severe malaria (43–45, 53, 54).
This analysis failed to detect selection signals for some important antigen genes such as lsa3, ama1, msp2, msp3, eba175, or circumsporozoite protein, csp [which have been entered vaccine-stage development (39, 41)], and rif (PF3D7_0100400, PF3D7_0401600, or PF3D7_1254800), vars (PF3D7_1150400, PF3D7_0533100, or PF3D7_0412700), or stevors (PF3D7_1254100 or PF3D7_0300400), to mention a few (43, 44, 53, 55), which have been identified or validated as targets of acquired immunity for vaccine development (39, 42). The reason could be that iHS may not be suitable for detecting positive selection for those SNPs that have reached fixation in a local population (28). Another possible explanation could be that they may be less targeted by host immunity in Togo subjects, given malaria transmission intensity and parasite genetic diversity are known to vary greatly among different parts of Africa due to variation in rainfall abundance and seasonality (39). However, immunological investigations using higher numbers of samples are needed in the future.
Positively skewed allele frequency distributions indicating the operation of balancing selection of Pf genes in other parasite populations have been reported (31, 38, 39, 56). In this study, the phistb rlp1 encoding PHISTb domain-containing RESA-like protein 1 at the surface of iRBCs, which was reported previously as most likely under balancing selection (31, 38), was also identified. It interacts with VAR2CSA and modulates knob-associated heat-shock protein 40 expression on the iRBC surface, and thus may regulate VAR2CSA expression to confer stable chondroitin sulfate A binding capacity and the parasite’s cytoadherence (47). The var2csa was also detected among genes under strong positive selection in the Togo isolates. It encodes a particular parasite adhesion molecule (PfEMP1) expressed on the surface of iRBCs for roles in sequestration of Pf-iRBCs in the placenta, which occurs as a result of its binding to host receptors such as chondroitin sulphate A. Signals of strong balancing selection were evident in a similar subset of genes in Togo and other West Africa isolates. This is consistent with expectations that balancing selection due to allele frequency-dependent acquired immune responses is likely to operate on antigenic targets in Togo subjects (38). Such evidence could lead to studies for a vaccine to induce antibodies to prevent placental adhesion/sequestration by reducing the maternal anaemia and infant deaths that are associated with malaria in pregnancy (34, 39).
Furthermore, we found high |iHS| for two particularly important antigen genes (msp7 and phistb rlp1), although they appear to being under balancing selection. The msp7 in association with msp1, is important in invasion of mature RBCs and has been reported as a potential target of acquired immunity (32). Following similar observation with csp gene in P. knowlesi isolates (30), these genes could be targets of both balancing and directional selection due to their location within an elevated window of haplotype homozygosity on chromosomes, or might have hitchhiked to intermediate allele frequencies by a linked locus under selection within population-specific isolates.
Of the eight Pf drug-resistant genes identified within elevated iHS regions in Togo samples, none of the five known drug resistance genes (crt, mdr1, dhfr, dhps, and k13) were included, suggesting that Togo population is not under important antimalarial drug selection. This is consistent with a recent study in Togo that has shown therapeutic efficacy of AL and ASAQ without delay in the clearance of mutant parasites (9). However, GO analysis for the drug-resistant genes that we identified by iHS computation within the top 1% |iHS| (abcI3 and apiap2) or with TD > 1 (aat1 and fpps/ggpps) were highly significantly (P < 0.001) enriched. In addition, our study suggested additional drug resistance genes under strong positive selection (Supplementary Table 8), which have been reported previously (48, 49).
Conclusion
This study assessed the first whole-genome sequences of Pf isolates from Togo. Our results showed that the parasites derived from Togo clustered according to their geographic origin and suggest greater genetic diversity of Pf isolates in Togo than seen in other African countries. In addition, Tajima’s D values were predominantly negative, consistent with directional selection and/or a history of recent expansion of Pf population in Togo. Against this background, there was evidence of balancing and positive selections on particular genes. Loci showing evidence of recent positive selection and balancing selection attest that host immunity has been the major selective agent. This is reflected in a significant representation of genes that encode membrane proteins expressed at the merozoite stage that invades RBCs and parasitized RBC surface proteins implicated in roles for immunoevasion, rosetting, or cytoadhesion. Our study would contribute with insightful information on the current epidemiological scenario of malaria in Togo and provides a fundamental basis to engage studies for effective malaria control in Togo.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA616298.
Ethics Statement
Permission was obtained from all malaria subjects before collecting specimens. Blood collection was made with informed consent from all individuals or their parents, under a study protocol reviewed and approved by the Togo Ministry of Health’s Bioethics Committee (Authorisation N°019/2019/MSHP/CBRS), following institutional ethical guidelines by the ethics committee at National Institute of Parasitic Diseases, Chinese Centre for Disease Control and Prevention.
Author Contributions
KK, J-HC, and YC conceptualized the study. KK and KKK collected and analyzed the specimens. H-MC performed bioinformatics analysis. S-BC and H-TF participated in the experiments. KK interpreted the data and wrote the manuscript. KK, Y-QC, and Z-NZ revised the manuscript critically for intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported in part by the National Research and Development Plan of China (Grant No. 2018YFE0121600 and 2016YFC1202000), the National Natural Science Foundation of China (Grant No. 81601787, 81871681), the Natural Science Foundation of Jiangsu Province (Grant No. BK20160192), the Fundamental Research Funds for the Central Universities funded by the Ministry of Education of China (Grant No. JUSRP51710A), the Bill & Melinda Gates Foundation (Grant No. OPP1161962), the National First-Class Discipline Program of Food Science and Technology (Grant No. JUFSTR20180101), the Scientific Research Project of Public Health Research Centre of Jiangnan University (Grant No. 1285210162190530), and the Project of Shanghai Science and Technology Commission (Grant No. 18490741100). The sponsor played no roles in the study design or in the collection, analysis, or interpretation of the data, in writing the report, or in the decision to submit the article for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Kokou Sepenou Noussougnon and Koffigan Ananou for their help in collecting clinical samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.552698/full#supplementary-material
References
1. Miller LH, Good MF, Milon G. Malaria pathogenesis. Science (1994) 264(5167):1878–83. doi: 10.1126/science.8009217
2. Plewes K, Turner GDH, Dondorp AM. Pathophysiology, clinical presentation, and treatment of coma and acute kidney injury complicating falciparum malaria. Curr Opin Infect Dis (2018) 31(1):69–77. doi: 10.1097/QCO.0000000000000419
3. WHO. World malaria report (2018). Available at: http://www.who.int/malaria/publications/world-malaria-report-2018/report/en/ (Accessed March 30, 2020).
4. Kassegne K, Zhang T, Chen SB, Xu B, Dang ZS, Deng WP, et al. Study roadmap for high-throughput development of easy to use and affordable biomarkers as diagnostics for tropical diseases: a focus on malaria and schistosomiasis. Infect Dis Poverty (2017) 6(1):130. doi: 10.1186/s40249-017-0344-9
5. Uwase J, Chu R, Kassegne K, Lei Y, Shen F, Fu H, et al. Immunogenicity analysis of conserved fragments in Plasmodium ovale species merozoite surface protein 4. Malar J (2020) 19(1):126. doi: 10.1186/s12936-020-03207-7
6. WHO. Malaria profile in Togo (2018). Available at: https://www.who.int/malaria/publications/country-profiles/profile_tgo_en.pdf?ua=1 (Accessed March 30, 2020).
7. Gbadoé AD, Kini-Caussi M, Koffi S, Traoré H, Atakouma DY, Tatagan-Agbi K, et al. Evolution of severe pediatric malaria in Togo between 2000 and 2002. Med Mal Infect (2006) 36(1):52–4. (in French). doi: 10.1016/j.medmal.2005.10.006
8. Eliades MJ, Wolkon A, Morgah K, Crawford SB, Dorkenoo AM, Sodahlon Y, et al. Burden of malaria at community level in children less than 5 years of age in Togo. Am J Trop Med Hyg (2006) 75(4):622–9. doi: 10.4269/ajtmh.2006.75.622
9. Dorkenoo AM, Yehadji D, Agbo YM, Layibo Y, Agbeko F, Adjeloh P, et al. Therapeutic efficacy trial of artemisinin-based combination therapy for the treatment of uncomplicated malaria and investigation of mutations in k13 propeller domain in Togo, 2012-2013. Malar J (2016) 15:331. doi: 10.1186/s12936-016-1381-8
10. Trape JF, Rogier C, Konate L, Diagne N, Bouganali H, Canque B, et al. The Dielmo project: a longitudinal study of natural malaria infection and the mechanisms of protective immunity in a community living in a holoendemic area of Senegal. Am J Trop Med Hyg (1994) 51(2):123–37. doi: 10.4269/ajtmh.1994.51.123
11. Bloland PB, Boriga DA, Ruebush TK, McCormick JB, Roberts JM, Oloo AJ, et al. Longitudinal cohort study of the epidemiology of malaria infections in an area of intense malaria transmission II. Descriptive epidemiology of malaria infection and disease among children. Am J Trop Med Hyg (1999) 60(4):641–8. doi: 10.4269/ajtmh.1999.60.641
12. Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature (2002) 419(6906):498–511. doi: 10.1038/nature01097
13. Chen SB, Wang Y, Kassegne K, Xu B, Shen HM, Chen JH, et al. Whole-genome sequencing of a Plasmodium vivax clinical isolate exhibits geographical characteristics and high genetic variation in China-Myanmar border area. BMC Genomics (2017) 18(1):131. doi: 10.1186/s12864-017-3523-y
14. Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res (2009) 37(Database issue):D539–43. doi: 10.1093/nar/gkn814
15. Manske M, Miotto O, Campino S, Auburn S, Almagro-Garcia J, Maslen G, et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature (2012) 487(7407):375–9. doi: 10.1038/nature11174
16. Lu F, Culleton R, Zhang M, Ramaprasad A, von Seidlein L, Zhou H, et al. Emergence of Indigenous Artemisinin-Resistant Plasmodium falciparum in Africa. N Engl J Med (2017) 376(10):991–3. doi: 10.1056/NEJMc1612765
17. Shen HM, Chen SB, Cui YB, Xu B, Kassegne K, Abe EM, et al. Whole-genome sequencing and analysis of Plasmodium falciparum isolates from China-Myanmar border area. Infect Dis Poverty (2018) 7(1):118. doi: 10.1186/s40249-018-0493-5
18. Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature (2008) 456(7218):53–9. doi: 10.1038/nature07517
19. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (2014) 30(15):2114–20. doi: 10.1093/bioinformatics/btu170
20. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (2009) 25(16):2078–9. doi: 10.1093/bioinformatics/btp352
21. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res (2010) 20(9):1297–303. doi: 10.1101/gr.107524.110
22. Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour (2010) 10(3):564–7. doi: 10.1111/j.1755-0998.2010.02847.x
23. Sabeti PC, Reich DE, Higgins JM, Lohmueller J, Hostetter E, Cotsapas C, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature (2002) 419(6909):832–7. doi: 10.1038/nature01140
24. Voight BF, Kudaravalli S, Wen X. Pritchard JK. A map of recent positive selection in the human genome. PloS Biol (2006) 4(3):e72. doi: 10.1371/journal.pbio.0040072
25. Shen HM, Chen SB, Wang Y, Xu B, Abe EM, Chen JH, et al. Genome-wide scans for the identification of Plasmodium vivax genes under positive selection. Malar J (2017) 16(1):238. doi: 10.1186/s12936-017-1882-0
26. Szpiech ZA, Hernandez RD. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol (2014) 31(10):2824–7. doi: 10.1093/molbev/msu211
27. Nygaard S, Braunstein A, Malsen G, Van Dongen S, Gardner PP, Krogh A, et al. Long- and short-term selective forces on malaria parasite genomes. PloS Genet (2010) 6(9):e1001099. doi: 10.1371/journal.pgen.1001099
28. Ye R, Tian Y, Huang Y, Zhang Y, Wang J, Sun X, et al. Genome-Wide Analysis of Genetic Diversity in Plasmodium falciparum Isolates From China-Myanmar Border. Front Genet (2019) 10:1065. doi: 10.3389/fgene.2019.01065
29. Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Krettli AU, et al. Early origin and recent expansion of Plasmodium falciparum. Science (2003) 300(5617):318–21. doi: 10.1126/science.1081449
30. Assefa S, Lim C, Preston MD, Duffy CW, Nair MB, Adroub SA, et al. Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi. Proc Natl Acad Sci U S A (2015) 112(42):13027–32. doi: 10.1073/pnas.1509534112
31. Amambua-Ngwa A, Tetteh KK, Manske M, Gomez-Escobar N, Stewart LB, Deerhake ME, et al. Population genomic scan for candidate signatures of balancing selection to guide antigen characterization in malaria parasites. PloS Genet (2012) 8(11):e1002992. doi: 10.1371/journal.pgen.1002992
32. Cowman AF, Berry D, Baum J. The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J Cell Biol (2012) 198(6):961–71. doi: 10.1083/jcb.201206112
33. Kassegne K, Abe EM, Cui YB, Chen SB, Xu B, Deng WP, et al. Contribution of Plasmodium immunomics: potential impact for serological testing and surveillance of malaria. Expert Rev Proteomics (2019) 16(2):117–29. doi: 10.1080/14789450.2019.1554441
34. Rowe JA, Kyes SA. The role of Plasmodium falciparum var genes in malaria in pregnancy. Mol Microbiol (2004) 53(4):1011–9. doi: 10.1111/j.1365-2958.2004.04256.x
35. Wahlgren M, Goel S, Akhouri RR. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat Rev Microbiol (2017) 15(8):479–91. doi: 10.1038/nrmicro.2017.47
36. Niang M, Bei AK, Madnani KG, Pelly S, Dankwa S, Kanjee U, et al. STEVOR is a Plasmodium falciparum erythrocyte binding protein that mediates merozoite invasion and rosetting. Cell Host Microbe (2014) 16(1):81–93. doi: 10.1016/j.chom.2014.06.004
37. Goel S, Palmkvist M, Moll K, Joannin N, Lara P, Akhouri RR, et al. RIFINs are adhesins implicated in severe Plasmodium falciparum malaria. Nat Med (2015) 21(4):314–7. doi: 10.1038/nm.3812
38. Mobegi VA, Duffy CW, Amambua-Ngwa A, Loua KM, Laman E, Nwakanma DC, et al. Genome-wide analysis of selection on the malaria parasite Plasmodium falciparum in West African populations of differing infection endemicity. Mol Biol Evol (2014) 31(6):1490–9. doi: 10.1093/molbev/msu106
39. Conway DJ. Paths to a malaria vaccine illuminated by parasite genomics. Trends Genet (2015) 31(2):97–107. doi: 10.1016/j.tig.2014.12.005
40. Mu J, Awadalla P, Duan J, McGee KM, Keebler J, Seydel K, et al. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat Genet (2007) 39(1):126–30. doi: 10.1038/ng1924
41. Kassegne K, Abe EM, Chen JH, Zhou XN. Immunomic approaches for antigen discovery of human parasites. Expert Rev Proteomics (2016) 13(12):1091–101. doi: 10.1080/14789450.2016.1252675
42. Chan JA, Fowkes FJ, Beeson JG. Surface antigens of Plasmodium falciparum-infected erythrocytes as immune targets and malaria vaccine candidates. Cell Mol Life Sci (2014) 71(19):3633–57. doi: 10.1007/s00018-014-1614-3
43. Zhou AE, Berry AA, Bailey JA, Pike A, Dara A, Agrawal S, et al. Antibodies to Peptides in Semiconserved Domains of RIFINs and STEVORs Correlate with Malaria Exposure. mSphere (2019) 4(2):e00097-19. doi: 10.1128/mSphere.00097-19
44. Travassos MA, Niangaly A, Bailey JA, Ouattara A, Coulibaly D, Lyke KE, et al. Children with cerebral malaria or severe malarial anaemia lack immunity to distinct variant surface antigen subsets. Sci Rep (2018) 8(1):6281. doi: 10.1038/s41598-018-24462-4
45. Quintana MDP, Ch’ng JH, Moll K, Zandian A, Nilsson P, Idris ZM, et al. Antibodies in children with malaria to PfEMP1, RIFIN and SURFIN expressed at the Plasmodium falciparum parasitized red blood cell surface. Sci Rep (2018) 8(1):3262. doi: 10.1038/s41598-018-21026-4
46. Nilsson Bark SK, Ahmad R, Dantzler K, Lukens AK, De Niz M, Szucs MJ, et al. Quantitative Proteomic Profiling Reveals Novel Plasmodium falciparum Surface Antigens and Possible Vaccine Candidates. Mol Cell Proteomics (2018) 17(1):43–60. doi: 10.1074/mcp.RA117.000076
47. Goel S, Muthusamy A, Miao J, Cui L, Salanti A, Winzeler EA, et al. Targeted disruption of a ring-infected erythrocyte surface antigen (RESA)-like export protein gene in Plasmodium falciparum confers stable chondroitin 4-sulfate cytoadherence capacity. J Biol Chem (2014) 289(49):34408–21. doi: 10.1074/jbc.M114.615393
48. Cowell AN, Istvan ES, Lukens AK, Gomez-Lorenzo MG, Vanaerschot M, Sakata-Kato T, et al. Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science (2018) 359(6372):191–9. doi: 10.1126/science.aan4472
49. Park DJ, Lukens AK, Neafsey DE, Schaffner SF, Chang HH, Valim C, et al. Sequence-based association and selection scans identify drug resistance loci in the Plasmodium falciparum malaria parasite. Proc Natl Acad Sci U S A (2012) 109(32):13052–7. doi: 10.1073/pnas.1210585109
50. Chang HH, Park DJ, Galinsky KJ, Schaffner SF, Ndiaye D, Ndir O, et al. Genomic sequencing of Plasmodium falciparum malaria parasites from Senegal reveals the demographic history of the population. Mol Biol Evol (2012) 29(11):3427–39. doi: 10.1093/molbev/mss161
51. Ocholla H, Preston MD, Mipando M, Jensen AT, Campino S, MacInnis B, et al. Whole-genome scans provide evidence of adaptive evolution in Malawian Plasmodium falciparum isolates. J Infect Dis (2014) 210(12):1991–2000. doi: 10.1093/infdis/jiu349
52. Staff PG. Correction: Imputation-Based Population Genetics Analysis of Plasmodium falciparum Malaria Parasites. PloS Genet (2016) 12(8):e1006300. doi: 10.1371/journal.pgen.1006300
53. Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T, et al. Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature (2017) 552(7683):101–5. doi: 10.1038/nature24994
54. Tan J, Pieper K, Piccoli L, Abdi A, Perez MF, Geiger R, et al. A LAIR1 insertion generates broadly reactive antibodies against malaria variant antigens. Nature (2016) 529(7584):105–9. doi: 10.1038/nature16450
55. Turner L, Wang CW, Lavstsen T, Mwakalinga SB, Sauerwein RW, Hermsen CC, et al. Antibodies against PfEMP1, RIFIN, MSP3 and GLURP are acquired during controlled Plasmodium falciparum malaria infections in naive volunteers. PloS One (2011) 6(12):e29025. doi: 10.1371/journal.pone.0029025
Keywords: Plasmodium falciparum, genomes, balancing selection, directional selection, immunity, Togo
Citation: Kassegne K, Komi Koukoura K, Shen H-M, Chen S-B, Fu H-T, Chen Y-Q, Zhou X-N, Chen J-H and Cheng Y (2020) Genome-Wide Analysis of the Malaria Parasite Plasmodium falciparum Isolates From Togo Reveals Selective Signals in Immune Selection-Related Antigen Genes. Front. Immunol. 11:552698. doi: 10.3389/fimmu.2020.552698
Received: 16 April 2020; Accepted: 02 September 2020;
Published: 23 October 2020.
Edited by:
Antonio Barragan, Stockholm University, SwedenReviewed by:
Akira Kaneko, Karolinska Institutet (KI), SwedenYaming Cao, China Medical University, China
Moses Okpeku, University of KwaZulu-Natal, South Africa
Copyright © 2020 Kassegne, Komi Koukoura, Shen, Chen, Fu, Chen, Zhou, Chen and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kokouvi Kassegne, ephremk@hotmail.fr; Jun-Hu Chen, chenjh@nipd.chinacdc.cn; Yang Cheng, woerseng@126.com