 Paola Italiani
Paola Italiani Giacomo Della Camera
Giacomo Della Camera Diana Boraschi
Diana Boraschi- Institute of Biochemistry and Cell Biology (IBBC), National Research Council (CNR), Naples, Italy
The capacity of engineered nanoparticles to activate cells of the innate immune system, in particular monocytes and macrophages, is considered at the basis of their toxic/inflammatory effects. It is, however, evident that even nanoparticles that do not directly induce inflammatory activation, and are therefore considered as safe, can nevertheless induce epigenetic modifications and affect metabolic pathways in monocytes and macrophages. Since epigenetic and metabolic changes are the main mechanisms of innate memory, we had previously proposed that nanoparticles can induce/modulate innate memory, that is, have the ability of shaping the secondary response to inflammatory challenges. In light of new data, it is now possible to support the original hypothesis and show that different types of nanoparticles can both directly induce innate memory, priming macrophages for a more potent response to subsequent stimuli, and modulate bacteria-induced memory by attenuating the priming-induced enhancement. This evidence raises two important issues. First, in addition to overt toxic/inflammatory effects, we should consider evaluating the capacity to induce innate memory and the related epigenetic and metabolic changes in the immunosafety assessment of nanomaterials, since modulation of innate memory may be at the basis of long-term unwanted immunological effects. The other important consideration is that this capacity of nanomaterials could open a new avenue in immunomodulation and the possibility of using engineered nanomaterials for improving immune responses to vaccines and resistance to infections, and modulate anomalous immune/inflammatory reactions in chronic inflammatory diseases, autoimmunity, and a range of other immune-related pathologies.
Introduction
In the last decades two important research fields have crossed paths: nanotechnology and immunology. On the one hand, nanotechnologies have experienced a huge development in many different application areas, including nanomedicine. Many nanoparticles (NP) have been engineered for appropriate modulation of their physical-chemical properties (e.g., nano-size, chemical composition, crystallinity, inorganic or organic functionalization, solubility, shape, aggregation behavior, electronic surface charge, and flexibility), usefulness for diagnostics (e.g., molecular imaging), and therapeutic (e.g., drug delivery system) purposes (“theranostic nanoparticles”) (1). On the other hand, the necessity to use NP that are safe for the organism has required to improve our knowledge on the interactions between NP and the immune system, in particular the impact on innate immunity (2, 3). Indeed, the innate immune system is the first line of defense of the organism, and the cells of the innate immune system are the first that interact with NP introduced into the body (3). As a result of this interaction, NP could either raise no response or trigger an innate/inflammatory reaction, based on whether or not the innate cells “sense” the particles as potentially dangerous. In nanomedicine, this is a crucial issue for two reasons: 1. to avoid an immune reaction against NP that could provoke damage in the body; 2. to avoid immune surveillance and clearance by the immune cells at the expense of the NP efficacy as diagnostic or therapeutic tools. Over the last years, the interaction between NP and the immune system has been widely investigated, and it is currently of great interest for the possibility of using immunomodulatory NP formulations in the therapy of a wide range of diseases. In fact, if on the one hand NP can directly activate undesirable immune effects (4), on the other hand they can be manipulated to attain reduction of their immunotoxicity (5) and tailored in order to get therapeutically desirable immunomodulation (i.e., immunostimulation or immunosuppression) (3, 6, 7).
It is well-known that invertebrates can adapt in virtually any ecosystem and survive infections by only relying on innate immunity (8). This implies the capacity to mount a faster and more effective response upon re-exposure to a stimulus (“immune training/priming”) although they do not have an adaptive immune system. This kind of improved secondary defensive response in invertebrates is considered an adaptive aspect of innate immunity and the demonstration of a bona fide immune memory. The same phenomenon has been observed in the innate immune system of mammals, in parallel to the much better-known immune memory displayed by the adaptive immune system. Indeed, both the innate and the adaptive immune system (concomitantly present in higher vertebrates) aim to improve their efficacy following a primary exposure to pathogens or other challenges (e.g., endogenous molecules, such as oxLDL, high level of glucose, uric acid, etc.) (9–11) by altering the type and magnitude of response to secondary threats toward stronger and more effective responses (“potentiation”) or weaker and less self-damaging reactions (“tolerance”) (12–14). The concept that innate immune cells can react differently to secondary challenges can be defined as innate immune memory. At variance with adaptive immune memory, the innate memory is non-specific, that is, the cells are more or less reactive regardless of the origin/nature of the secondary stimulus. The higher or lower reactivity is assessed in terms of production of inflammatory and anti-inflammatory factors, and a metabolic and epigenetic reprogramming of the cells seems to be the molecular mechanism underlying this functional plasticity/flexibility (15).
The establishment of innate immune memory has a profound impact on the effectiveness of human immune responses. Indeed, innate memory seems to provide an explanation for the heterologous effects of vaccines, as suggested in the case of human volunteers immunized with the BCG vaccine against Mycobacterium tuberculosis (attenuated Mycobacterium bovis strain Bacillus Calmette-Guérin bacteria), who became less susceptible to unrelated subsequent infections, possibly because of their potentiated and thus more robust innate immune response (16, 17). However, while innate memory is expected to generate improved defense to new challenges, it is possible that it could also cause detrimental effects in inducing and/or maintaining autoimmune and autoinflammatory diseases, as recently discussed elsewhere (18).
Thus, there will be a tremendous benefit in understanding 1) which metabolic and epigenetic changes could possibly be targeted to either promote or prevent innate memory and 2) which agents may initiate or modulate innate memory toward potentiation or tolerance, to be used as therapeutic tools. In this perspective, NP appear to be good candidates for the design of new therapeutic strategies for modulation of innate memory (19, 20). Indeed, NP can be tailored/functionalized to modulate innate memory responses and act as immunosuppressors in diseases with exacerbated immune response/inflammation (autoimmune disorders and auto-inflammatory diseases, atherosclerosis, multiple sclerosis, diabetes, etc.) or as immunostimulants in conditions with reduced/compromised immune responsiveness (e.g., certain types of cancer, sepsis, or infections). Thus, the ability of NP to modulate the molecular mechanisms involved in the development of innate memory would provide an avenue for improving the therapy of these diseases.
On this basis, we should revise our approach in examining the effects of NP on innate immune responses by including the effects on the establishment and modulation of innate memory. In nanosafety studies, this becomes particularly important for avoiding detrimental NP effect on human health, in particular in immune-related diseases. For instance, if NP induce a potentiated memory response in autoimmune patients or a tolerant response in immunosuppressed patients, this will exacerbate the patients' conditions. On the other hand, in the case of nanomedicine it would be particularly promising having the possibility to modulate precisely the immune memory so as to potentiate secondary responses in immunosuppressed patients and, vice versa, reduce reactivity in autoimmune conditions.
To date, nanoimmunosafety studies have paid little/no attention to the possible effects of NP on innate immune memory. While a large body of work has focused on the NP effects on innate immune cells (3), understanding the effects of NP beyond this primary response is still overlooked, although it would lead to a truly complete view of the consequences of NP exposure in humans (21). Three years ago, we hypothesized that NP could have effects on the induction or modulation of the innate memory of human mononuclear phagocytes (21). After 3 years, we can support our original hypothesis in light of new knowledge regarding the molecular mechanisms underlying the development of innate memory (i.e., the metabolic and epigenetic reprogramming of the cell) and regarding the NP effects on the metabolism and the epigenetic modifications in innate immune cells (in particular, monocytes/macrophages). If NP can have an effect on the metabolism and can induce epigenetics changes, and if the molecular mechanisms involved in the development of innate memory are indeed metabolic and epigenetic changes, for transitive property we suppose that NP could have an effect on innate memory, and therefore this effect should be taken into account in the evaluation of the immunosafety/toxicity of NP. Moreover, we assume that the development of innate memory is not only restricted to microbial agents or endogenous molecules as mentioned before but also to non-biological stressors such as NP. The objective/focus of our review is to summarize the data supporting our deductive reasoning and to support the hypothesis with the very first experimental evidence of a direct effect of NP on the induction of innate memory in human monocytes/macrophages.
Innate Memory and Epigenetic Reprogramming
Epigenetics is referred to as “a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence” (22). Epigenetic modifications involved in development of innate memory are essentially three: DNA methylation, post-translational modifications of histones, and regulation of non-coding RNAs (ncRNAs), that is, short (miRNA) and long (lncRNA) RNA. All of them are involved in the regulation of gene expression.
Innate immune memory shows as an enhanced or reduced reactivity of innate immune cells upon a second challenge, which mostly includes an increased or decreased production of inflammation-related factors. This production depends on the coordinated regulation of expression of an array of genes, pointing at the importance of epigenetic mechanisms in the establishment of innate memory. The epigenetic reprogramming of innate cells in innate immune memory has been extensively reviewed elsewhere (15, 23). Here, we briefly report the main epigenetic marks identified so far in blood monocytes and bone marrow-derived macrophages, the innate cells that are the best-known effectors in the context of innate immune memory.
A high level of trimethylation of histone 3 lysine 4 (H3K4me3) on gene promoters is generally associated with a robust transcription uniformly across various cell types (24, 25). In the case of human monocytes primed in vitro with β-glucan from Candida albicans, it has been observed that cells present an accumulation of H3K4me3, acetylation of histone 3 lysine 27 (H3K27ac), and monomethylation of histone 3 lysine 4 (H3K4me1) on enhancers of immune genes. Upon removal of the priming stimulus, H3K27ac was lost over time, but H3K4me3 remained on the chromatin ensuring a potentiated immunological response to a second stimulation. H3K4m1 was also maintained over time, although not all genome regions retained this mark (26–28). The same marks have been observed in murine hematopoietic progenitors (29) stimulated with β-glucan in vitro, in myeloid progenitors and bone marrow-derived macrophages from BCG vaccinated mice (30), and in monocytes isolated from healthy volunteers after vaccination with BCG (31), supporting a role for these specific modifications in the epigenetically regulated establishment of innate memory. These epigenetic changes allow the transcriptional machinery to access DNA thereby promoting an enhanced transcription of inflammatory genes such as TNF-α and IL-6 (31). Thus, these epigenetic marks appear to be associated to a particular type of memory, that is, the potentiation of the secondary response. Conversely, it has been proposed that the lack (or delayed establishment) of the same epigenetic changes after cell priming with lipopolysaccharides (LPS) from Escherichia coli could be responsible of the other type of innate memory, that is, tolerance (27). This is further suggested by the finding that β-glucan could increase H3K27ac on enhancers of LPS-tolerized genes along with the re-establishment of the macrophage capacity to produce inflammatory cytokines after a re-exposure to LPS. These findings also highlight the possible reversibility of innate memory phenotypes (27). Moreover, H3K4me3 is selectively present only on promoters of genes that maintained an active expression (non-tolerized genes) during LPS tolerance induced in murine macrophage (32).
To date, the role of DNA methylation in innate immune memory is poorly understood. A distinct methylation pattern was observed in in vitro LPS-primed human monocytes after 6 days from stimulation, while β-glucan-treated cells present a methylome similar to that of naïve macrophages (27).
Likewise, the contribution of miRNAs to the development of innate memory remains largely unexplored. A recent study reported how specific miRNAs can regulate tolerance in murine macrophages in vitro. The authors identified miR-221 and miR-222 as regulators of the functional reprogramming of mouse bone marrow-derived macrophages during LPS-induced tolerance in vitro. Prolonged stimulation with LPS leads to increased expression of miR miR-222, which downregulates the Brg1 gene and provokes the silencing of a subset of inflammatory genes through SWItch/Sucrose Non-Fermentable (SWI/SNF) and STAT-mediated chromatin remodeling. The involvement of miR-221 and miR-222 is confirmed also in patients with sepsis, with an increase in their expression in peripheral blood mononuclear cells correlated with immunosuppression and organ damage (33).
lncRNAs have only recently started to be investigated in the context of innate memory (34, 35). lncRNA seems to facilitate the H3K4me3 epigenetic priming in innate immune cells, such as monocytes (34). 3D chromatin topology can place lncRNAs proximal to innate immune gene promoters, and, through formation of lncRNA/protein complexes, lncRNAs can regulate gene transcription by influencing the access of transcription-regulating proteins to their target genes. Indeed, recent data indicate that genes encoding inflammatory cytokines (e.g., IL-1β, IL-6) and chemokines (e.g., IL-8, CXCL1, 2, 3) engage in chromosomal contacts with a special type of lncRNAs, the immune-gene priming lncRNAs (IPLs). For example, one of these IPLs, UMLILO, acts in cis (i.e., on the same molecule of DNA as the genes to be transcribed) and points the adapter protein WDR5 and the histone methyltransferase MLL1 complex to the inflammatory gene promoters thereby enabling their H3K4me3 epigenetic priming, prior to their transcriptional activation, an event that leads to an enhanced transcription of the immune genes in response to a secondary challenge (36).
Recently, it has been also speculated that another type of lncRNA, the enhanced RNAs (eRNAs), may play a role in memory immune responses (15). eRNAs derive from the transcription of enhancers, are cell-specific, and are involved in the regulation of chromosomal looping and in the transcription of tissue-specific genes/contacts between target genes and enhancers. For example, eRNAs can interact with mediator complex or Yin Yang 1 to regulate chromosomal contacts between target genes and enhancers (37, 38). Also, eRNAs can interact with proteins such as p300 and CBP (cyclic adenosine monophosphate response element-binding protein), able to stimulate catalytic histone acetyltransferase activity at specific genomic loci (39). As a consequence, the gene transcription is activated. Therefore, eRNAs may play a role in innate memory responses by regulating looping at key enhancers and histone acetylation. Moreover, latent enhancers (a sub-class of enhancers), upon LPS stimulation, acquire the typical histone modifications (H3K4me1 and H3K27Ac) associated with an active enhancer region and that persist after the removal of the stimulus in mouse bone marrow-derived macrophages (40).
Innate Memory and Metabolic Reprogramming
Along with epigenetic changes, another hallmark characteristic associated with the induction of innate memory is the metabolic reprogramming of innate cells.
Changes in cellular metabolism are associated with different immune functions (41) and macrophage functional phenotypes (42). In the case of mononuclear phagocytes, activated monocytes and classically activated macrophages (M1) are characterized by an enhanced glycolytic metabolic state for ATP production, an activated pentose phosphate pathway (PPP), an impaired oxidative phosphorylation (OXPHOS), and an anabolic repurposing of metabolites of the tricarboxylic acid cycle (TCA), with pyruvate fermented in lactate. On the other hand, naïve quiescent monocytes and alternatively activated (IL-4-induced) macrophages (M2) preferentially make use of OXPHOS for ATP production, while the concomitant presence of glycolytic pathways generate the pyruvate for fueling the TCA cycle.
Human monocytes stimulated in vitro with β-glucan or BCG show a metabolic shift similar to that observed in classically activated macrophages (43, 44), that is, elevated consumption of glucose by glycolysis, conversion of pyruvate to lactate, and decreased OXPHOS with reduced oxygen consumption (Warburg effect). In both cases (BCG- and β-glucan-primed monocytes), the activation of the Akt-mTOR-HIF1α pathway drives the shift from OXPHOS to aerobic glycolysis (43, 44). Moreover, in β-glucan- and BCG-primed monocytes the metabolites of TCA cycle (citrate, succinate, malate, fumarate, and 2-hydroxyglutarate) are increased probably thanks to increased glutamine metabolism (glutaminolysis) (43, 45). Glutamine is metabolized first into glutamate and then in α-ketoglutarate and can become a source of succinate, fumarate, and citrate, which in turn replenish the TCA cycle (46). Furthermore, β-glucan-primed monocytes show an up-regulation of genes involved in the cholesterol biosynthesis. The activation of the cholesterol biosynthesis pathway, but not its actual synthesis, is also involved in the metabolic reprogramming of primed cells, as it has been shown that blocking mevalonate (a metabolite of the cholesterol synthesis pathway) with statins prevents the induction of innate memory (47). Although the fatty acid synthesis also increases in macrophages upon LPS stimulation, the blockade of this synthesis does not affect the memory phenotype in monocytes primed with β-glucan and restimulated with LPS (44).
Recently, it has also been shown that the liver X receptor (LXR), a key regulator of cholesterol and fatty acid homeostasis, may play a role in the establishment of innate memory (48). Human monocytes primed in vitro with LXR agonists and challenged after 5 days with a TLR2 agonist, Pam3Cys, showed an inflammatory activation accompanied by epigenetic reprogramming (increased H3K27ac and H3K4me3 on IL-6 and TNF-α promoters) and metabolic reprogramming (increased lactate production and decreased oxygen consumption) with respect to unprimed cells. Moreover, the authors observed an increase, after priming with LXR agonists, in the expression of several genes involved in the synthesis and metabolism of acetyl-CoA and that acetyl-CoA is able per se to prime monocytes and induce a memory phenotype upon challenge with TLR2 agonist. All these effects depend on both mevalonate pathways and IL-1β signaling. In fact, the inflammatory memory phenotype induced by priming with LXR agonists is attenuated by inhibition of mevalonate pathways with fluvastatin, an agent that competitively inhibits hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase, which in turn catalyzes the conversion of HMG-CoA to mevalonic acid, the rate-limiting step in cholesterol biosynthesis. Likewise, blocking of IL-1β with its receptor antagonist, IL-1Ra, has the same inhibitory effect.
Metabolic changes in blood monocytes or macrophages during sepsis, in mice and humans, mirror the same shift toward aerobic glycolysis during the hyperinflammatory phase and a decreased OXPHOX and TCA cycle during the hypoinflammatory phase (49). These aspects are extensively reviewed elsewhere (50).
Furthermore, sepsis is able to induce an inflammatory memory phenotype in bone marrow monocytes. Indeed, post-septic naïve bone marrow monocytes and blood monocytes isolated from mice showed an increased cytokine production when stimulated with LPS in vitro (51).
Interplay Between Epigenetic and Metabolic Reprogramming
As mentioned above, the epigenetic reprogramming involved in the development of innate memory encompasses mainly methylation and acetylation of histones and the activity of lncRNA, while the metabolic reprogramming results in changes to glycolysis, TCA cycle, glutaminolysis, and cholesterol metabolism. Different genes involved in these pathways are quickly upregulated upon stimulation, causing changes in the levels of intracellular metabolites. The metabolic reprogramming and epigenetic reprogramming are strictly linked because the former supplies the metabolites and co-factors critical for the induction and preservation of the latter in the establishment of a memory phenotype. This immunometabolic cross-talk seems to be a common denominator across species, from nematodes to vertebrates, and the two reprogramming may represent ancestral and conserved mechanisms underlying the development of innate memory (52). The cross-talk between metabolism and epigenetics occurs because the stimulation of innate immune cells with microbial or endogenous ligands modifies the epigenetic landscape of metabolic regulators. Indeed, the genes involved in the metabolism of glucose, glutamine, and cholesterol and in the TCA cycle pathway are upregulated after stimulation, causing changes in the levels of intracellular intermediates (36). These various metabolites/intermediates in turn act as co-factors or can influence the functionality of the enzymes responsible for epigenetic modifications (53, 54), as illustrated in the following examples. Metabolic intermediates, such as acetyl-CoA produced from glycolysis and glutaminolysis, act as donors of acetyl groups for histone acetylation (55). An intermediate of the TCA cycle, α-ketoglutarate, is important for the functions of ten-eleven translocation (TET) proteins involved in DNA methylation (56) and consequently involved in β-glucan- and BCG-induced long-term reprogramming of myeloid progenitors in the mouse bone marrow (29, 30) and is also a cofactor of the JmjC domain-containing histone demethylases (KDMs) (57). S-adenosyl methionine is a co-substrate of histone and DNA methyl transferases, being the donor of methyl groups (58). Succinate and fumarate can inhibit demethylation reactions (56), and in particular fumarate (derived from glutaminolysis for the replenishment of intermediates of the TCA cycle) inhibits the H3K4 demethylase KDM5, favoring the enrichment of H3K4me3 at the promoters of inflammatory genes (44). Furthermore, succinate, fumarate, and mevalonate stabilize HIF1α, thereby sustaining Akt-mTOR-HIF1α signaling (43, 46) that, as mentioned before, is responsible of the switch from oxidative phosphorylation to aerobic glycolysis in human macrophages.
These few examples underline the complex and intimate relationship between the cellular metabolic machinery and the epigenetic modulators. Exhaustive details on such relationship are reported elsewhere (15, 42, 53).
Nanoparticles and Epigenetics
The effects of engineered NP and nanomaterials on epigenetic modifications has been extensively and excellently reviewed (59–63). It is quite clear that NP can be considered potential epimutagens (although a lot still remains to be unknown). Thus, in the evaluation of NP safety, epigenetic effects should be listed among potential health hazards, in addition to genotoxic and cytotoxic effects (64, 65). Most studies on the epigenetic effects of NP have been carried out in vitro on human tumors or transformed cell lines (e.g., A549, HaCaT, MCF-7, BEAS-2B, HepG2, 293T, and GLC-82) and in vivo in mice or on exposed subjects/workers. Epigenetic changes induced by NP can vary depending on NP characteristics such as size, shape, chemical structure, surface functionalization, exposure dose, short-term or long-term exposure, and type of cells.
We refer to aforementioned literature for a broader vision and more detailed reading on the NP effects on epigenetic changes, while here we briefly report work that directly or indirectly pertains to the innate immune system. Although many studies describe cytotoxicity induced by NP for several cell types, no data are available that could link NP-induced toxicity to epigenetic alterations.
There is evidence that some NP can enter the nucleus and interact with positively charged core histones, as it has been proven for anionic Cd-Te quantum dots (QD). The QD enter the nucleus of THP-1 cells, where they preferentially bind to core histones as opposed to other nuclear macromolecules, such as DNA and RNA, and change their physical-chemical properties leading to an increased formation of QD/protein aggregates (66).
Among epigenetic modifications, NP can induce both global DNA methylation and methylation at the level of specific genes. Notably, NP-induced DNA methylation can differ depending on NP and cell type of NP and cells exposed. For example, a study has investigated the effects of the exposure to NP of copper oxide (CuO) or titanium dioxide nanoparticles (TiO2) on the cellular epigenome of human and murine macrophage-like tumor cells (THP-1 and RAW264.7, respectively) (67). In particular, the authors have addressed the methylation status of the two most abundant transposable elements (TE) in the human genome (LINE1 and Alu) and in the mouse genome (SINEB1 and SINEB2). They observed that CuO NP induced hypermethylation in LINE1 and Alu elements in THP-1, while CuO and TiO2 NP enhanced the methylation level of SINEB1 in RAW264.7.
In another study, despite no significant difference in global DNA methylation, it was possible to observe DNA methylation changes in the promoter CpG regions of genes of enzymes involved in the epigenetic regulation, DNA repair (i.e., DNMT1, HDAC4, ATM), and gene of TGF-β repressor (SKI) in blood cells of workers exposed to multi-walled carbon nanotubes (MWCNT) with respect to unexposed control individuals (68). Furthermore, workers exposed to particulate matter and polycyclic aromatic hydrocarbons exhibited, in their peripheral blood leukocytes, significantly higher average levels of methylation at the promoter of tumor-suppressor genes, such as CDKN2A, APC, and MLH1, and significant hypomethylation at the repetitive DNA sequence LINE-1 (a sequence involved in DNA folding/packaging) in comparison to unexposed workers (69).
Another study showed DNA methylation changes in human monocyte-like cells (THP-1) after incubation with either single-walled or multi-walled CNT. By assessing methylation of single CpG sites, it was observed that CNT induced gene-specific differential methylation, with promoter hypomethylation evident for a thousand different genes, compared with the control samples. Some of these genes were associated with alterations in some signaling pathways, such as the PI3K-AKT-mTOR, JAK-STAT, MAP, and VEGF pathways, and platelet activation. The authors discussed the possible contribution of these epigenetic alterations on genes involved in macrophage polarization and hypothesized a mixed M1/M2 macrophage functional phenotype upon CNT exposure (70).
Using the LUminometric Methylation Assay (LUMA) and the 5-methylcytosine (5-mC) quantification assay, Brown et al. (71) elucidated the effect of oro-pharyngeal instillation of MWCNT on global DNA methylation and specifically on genes associated with inflammation (TNF-α and IFN-γ) and with fibrosis (Thy-1) in C57BL/6 mice. They found that MWCNT leads to DNA hypomethylation at the inflammatory gene promoters and hypermethylation at the fibrotic gene promoter in the lung and to a reduction of global DNA methylation in lung and circulating white blood cells, 7 days post-exposure, which coincided with development of MWCNT-induced fibrosis.
More research is needed to establish a specific role for dysregulated ncRNA upon in vivo and in vitro exposure to NP. Among the few data available, a recent study has shown that miR-350 may promote apoptosis in RAW264.7 cells through the negative regulation of the PIK3R3 gene and that exposure to TiO2 NP caused an increase in miR-350 and a decrease in PIK3R3 (72).
Another study investigated significant changes in mRNA and ncRNA expression profiles in the blood of workers exposed to MWCNT compared to non-exposed group. Gene set enrichment analysis and pathway analysis showed that differentially expressed sets of miRNAs and their target genes were mainly involved in cell cycle regulation/progression/control, apoptosis, and proliferation and revealed the potential of MWCNT to trigger pulmonary effects and carcinogenic outcomes in humans (73).
Interestingly, upon exposure to silver, titanium dioxide, and zinc oxide NP, differentiated macrophage-like THP-1 cells presented a profile of miRNA and miRNA variants, called isomiRs, that was unique to each NP type, and an identified co-regulated miR-mRNA cluster (encompassing hsa-miR-142-5p,−342-3p,−5100,−6087,−6894-3p, and−7704) seems to be a potential biomarker of metal-based nanoparticle exposure (74).
Nanoparticles and Cell Metabolism
While the effects of NP on monocytes/macrophages activation have been widely investigated, those on cellular metabolism is still poorly explored (75). Here we will focus on some evidence of the NP effects on metabolic pathways of human or murine monocytes and macrophages extrapolated from activation/cytotoxicity studies. Chen et al. (76) reported metabolic dysfunctions in macrophages exposed to TiO2 NP. In particular, the authors observed that both the levels of ATP and the metabolic flux at the level of most TCA metabolites were attenuated in a dose-dependent manner in RAW264.7 and bone marrow-derived macrophages (BMDM) after 24 h exposure to NP. These results suggest that TiO2 NP could cause a significant mitochondrial dysfunction in macrophages, through the down-regulation of the TCA cycle and ATP production.
In another study, cells of mouse macrophage-like line J774A.1 were exposed to a high dose of silver NP for 24 h, and the NP effects have been evaluated immediately after treatment (24 h) and after a recovery period of 72 h after NP removal (77). The authors observed that, although specialized macrophage functions (e.g., phagocytosis) are restored during the recovery period, lipopolysaccharide-induced cytokine (e.g., IL-6 and TNF-α) and nitric oxide production, some enzymatic activities involved in the TCA cycle (e.g., NADPH-dependent isocitrate dehydrogenase and malate dehydrogenase) and glucose consumption, did not return to basal level, showing that some effects of silver NP persist after cessation of exposure.
Another study on the murine macrophage RAW264.7 cell line showed that exposure to silk, poly(lactic-co-glycolic acid) (PLGA), and silica NP (considered good candidates for nanomedicine applications) induced some metabolic changes that were independent of the NP type; that is, higher glucose consumption and lactate production (indicative of increased glycolytic activity), high level of itaconate and succinate (two intermediate metabolites of TCA cycle), decreased amino acids levels (e.g., aspartate and glutamate), and ATP decrease over time. Other metabolic changes were specific for individual NP type, such as glutaminolysis (reduced or enhanced or unchanged based on NP types) and a variation of the creatine kinase/phosphocreatine pathway (enhanced or unchanged based on NP types) (78).
In another study on murine macrophage-like RAW264.7 cells, the cell metabolic profiles have been evaluated upon exposure to unmodified or PEGylated silk fibroin NP (another promising nanomedicine) (79). Exposure to unmodified NP caused an increase in glycolysis and reprogramming of the TCA cycle (with a significant increase in lactate, itaconate, and succinate), an increase in the creatine kinase/phosphocreatine pathway, a decrease in ATP level and in the levels of several amino acids (aspartate, glutamine, glutamate, and alanine) and an increase in others (glycine, lysine, branched chain, and aromatic amino acids), and an increase in cholesterol and phosphatidylcholine together with decrease in unsaturated fatty acids. By contrast, milder effects were observed for all these metabolic pathways in macrophages stimulated with PEGylated silk fibroin NP. Thus, NP appear to have a direct effect on the metabolism of innate immune cells. In addition, there is evidence that NP can also modulate cell metabolism by affecting/interfering with biological processes such as autophagy and differentiation/polarization.
For example, Wu et al. (80) demonstrated that dextran-coated superparamagnetic iron oxide NP (Dex-SPION) induced autophagosome accumulation in human monocytes. By inhibiting the autophagy, they observed increased production of inflammatory cytokines (TNFα, IL-1β, and IL-6) upon exposure to Dex-SPION, suggesting that the induction of autophagy by these NP has an important role in the modulation of human monocytes' function. Autophagy as a highly conserved metabolic process aiming to sequester (in autophagosomes) and digest (by lysosomal pathways) injured organelles and proteins in all eukaryotic cells (81) and is strictly linked to metabolic changes during monocyte differentiation and macrophage polarization (82). Although underlying mechanisms are still debated, it seems that autophagy is strictly linked to the metabolic changes during immune cell differentiation, and autophagy and metabolism are both regulated through AMPK and mTOR signaling cascades (82). This would imply that NP effects on autophagy will impact cell metabolism.
Many studies reveal an immunomodulatory role for NP (e.g., Au NPs, SiO2 NP, functionalized polystyrene, and molybdenum disulfide quantum dots) that can drive macrophages polarization toward M1- or M2-like phenotypes (83–85). M1-like cells are classically activated inflammatory macrophages mainly involved in host defense to infections and tumors, while M2 defines alternatively activated macrophages mainly involved in type 2 inflammation, anti-inflammatory responses, wound-healing, tissue remodeling, and tumor growth (86–89). Considering the significant metabolic differences between M1 and M2 macrophages, the effects of NP on macrophage polarization should be considered in terms of their possible effects in inducing metabolic reprogramming and, accordingly, in the development of a memory phenotype in these cells.
A proven relationship between metabolism and cellular redox state also exists (90, 91). Innate immune cells, especially macrophages and neutrophils, produce toxic reactive oxygen and nitrogen species (ROS and RNS). As an example, when circulating monocytes migrate into inflamed or infected tissues, they differentiate in M1 macrophages that release superoxide anion radicals, nitric oxide, and hydrogen peroxide (H2O2) to support pathogen killing (oxidative burst). The production of ROS requires a rapid burst of energy (glycolysis and rapid availability of NADPH), and, in turn, ROS production is required for the switch from oxidative phosphorylation to glycolysis (for details refer to 90). However, an excessive accumulation of ROS and NOS (oxidative stress) can be dangerous also for macrophages and other surrounding host cells, and, if maintained over time, it can lead to oxidation of proteins, lipids, carbohydrates, and DNA, causing metabolic dysfunction and DNA damage. The oxidative modifications of these molecules may provoke a re-programming of monocytes/macrophages. For example, an oxidative modification, S-glutathionylation (reaction between oxidized thiol group of protein and glutathione for the formation of mixed disulfides), occurs in proteins of both human monocytic cell line (THP-1) stimulated with H2O2 and after metabolic stress due to low-density lipoprotein (LDL) and high glucose exposure and in peritoneal macrophages of mice exposed to long-term metabolic stress. This modification alters the expression and function of proteins involved in the metabolism and inflammatory functions of monocytes/macrophages (92). ROS can provoke DNA damage directly or influence the methylome causing oxidative DNA lesions and can indirectly affect epigenetic mechanisms. These alterations are frequently associated to aging and immunosenescence (93–95).
These scenarios offer other levels at which NP can affect innate memory. NP can induce oxidative stress, and, as a consequence, ROS can oxidize enzymes involved in metabolism and lead to DNA damage. There is abundant evidence in the literature on the capacity of different NP to modulate ROS generation in innate immune cells. For example, SiO2, Au, FexOy, and carbon-based NP increase ROS production in macrophages (96–100). Moreover, it is worth noting that DNA damage due to NP-mediated oxidative stress in blood cells could be particularly important in age-related hematologic abnormalities. For example, it could affect TET2, one of the genes involved in clonal hematopoiesis, whose mutations are associated with increased risk of hematologic cancer and cardiovascular diseases (101, 102). The effects of NP on oxidative stress have been well-investigated and reviewed elsewhere (103–105).
The role of NP-induced ROS in the functional re-programming of immune cells toward a memory phenotype has not been investigated yet, despite its expected relevance.
These few studies, limited in number and mostly targeting transformed murine cells, nevertheless provide evidence that NP can have an effect in the metabolic reprogramming of monocytes/macrophages. The metabolic effects of NP should be thoroughly addressed, in future studies, especially because even NP that are considered non-toxic might have long-term effects on cellular metabolism and energy homeostasis (75).
Table 1 summarizes the main effects of NP on epigenetic and metabolic reprogramming of innate immune cells, macrophage polarization, and ROS/RNS production.
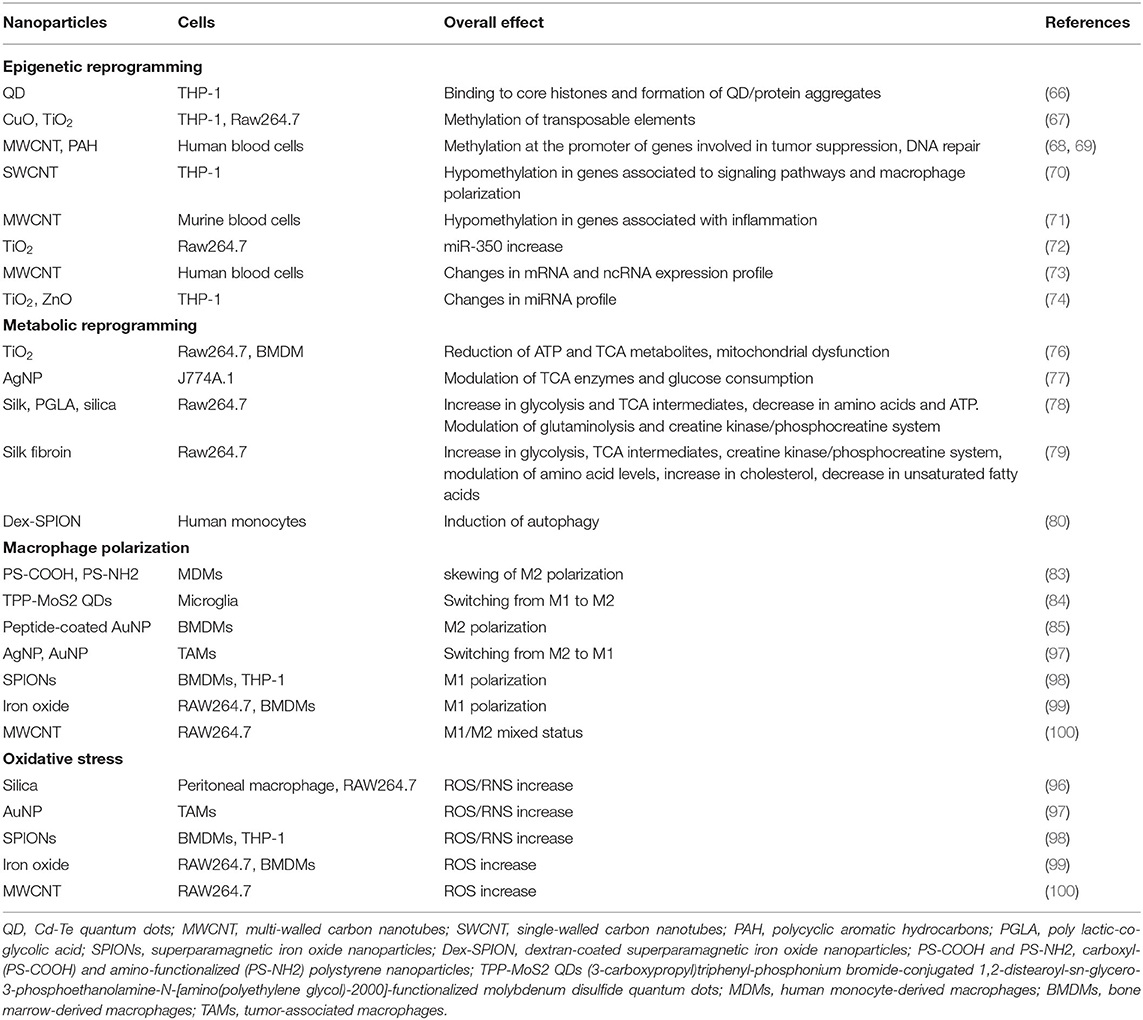
Table 1. Main effects of NP on epigenetic and metabolic reprogramming of innate immune cells.
Most studies investigating the immune effects of NP on epigenetics and metabolism are performed using in vitro models based on human or murine cancer or immortalized cell lines. Cell lines have the advantages of easy handling and high reproducibility, but they have the disadvantage to be poorly predictive of human primary cell response (due to physiological and metabolic differences with normal primary immune cells) (106, 107), and, obviously, they often do not fully represent what occurs in vivo. In the use of primary murine cells there are important differences between primary murine and human immune cells, for example, differences in LPS-induced immunometabolism (108) or in inducibility of expression of genes involved in the inflammatory response (109). Thus, the use of blood isolated from subjects exposed to NP, and the use of primary immune cells isolated from healthy donors and stimulated in vitro with NP, remains the preferred choice, despite the limits due to the lack of in vivo system complexity and donor variability. However, in case of innate memory, the donor variability due to the individual history of previous infections/exposure is a critical issue for understanding the need for a personalized nano-immunotoxicological assessment.
Nanoparticles Can Induce and Modulate Innate Immune Memory
In light of what is discussed and reviewed so far, it is reasonable to speculate that NP may be able to induce and modulate innate immune memory by regulating epigenetic and metabolic reprogramming of innate immune cells, thereby influencing their ability to react to a second exposure (Figure 1, upper part). Indeed, recent evidence shows that this is the case. This evidence comes from NP of interest for biomedical applications, that is, gold (Au) NP and pristine graphene (pGr). As reported in our previous publication (21), preliminary studies show that Au NP could modulate the memory induced by LPS priming in human primary monocytes in vitro and that this response could differ in different individuals, likely due to individual immunological history of previous infections. More recently, we have shown that Au NP have a significant effect in reducing the BCG-induced memory in human primary monocytes in vitro. At variance with the tolerance induced by LPS priming, monocytes primed with BCG showed an enhanced production of inflammatory (IL-6 and TNF-α) and anti-inflammatory cytokines (IL-1Ra and IL-10) when challenged with LPS. Although Au NP did not have a direct effect in inducing a memory response per se, when monocytes were primed with BCG and Au NP together, a reduction in cytokine production in response to LPS challenge could be observed, compared to that in cells primed with BCG alone, suggesting that Au NP interfered with the induction of memory by BCG and shifted the memory response of monocytes toward a tolerant phenotype (110) (Figure 1, bottom part). This study provides the first evidence that Au NP can influence the development of innate immune memory triggered by microbial molecules or vaccines and affect the monocyte response to subsequent challenges.
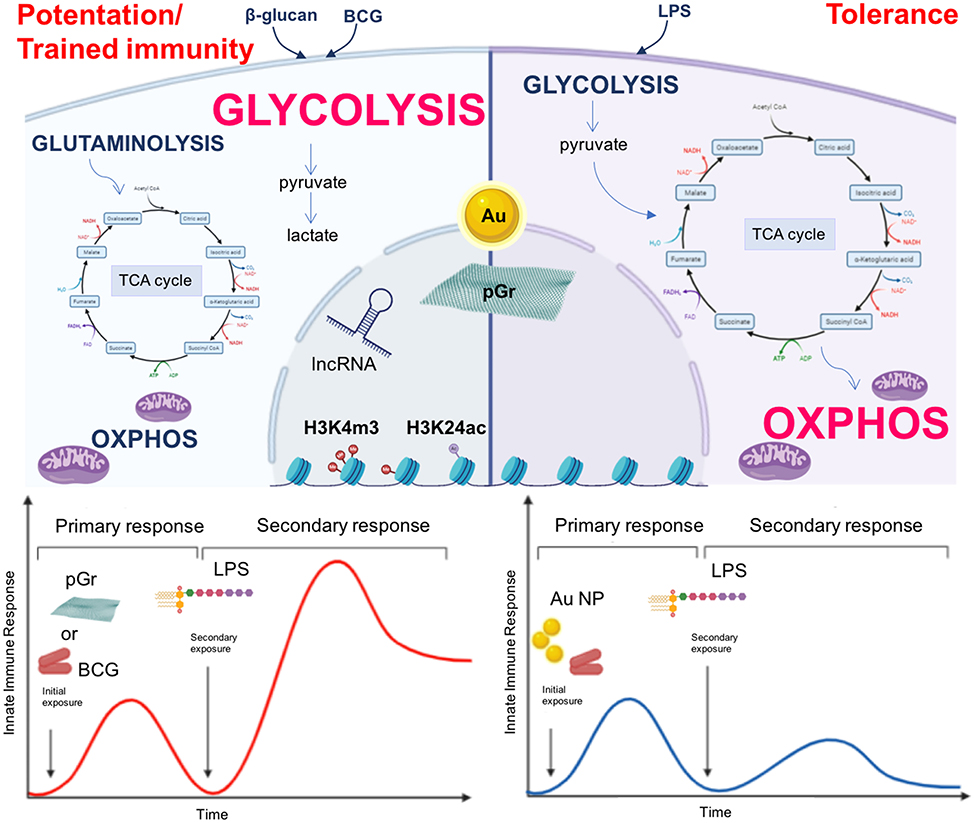
Figure 1. Nanoparticles as inducers of innate immune memory. Upper part: β-glucan or BCG-primed monocytes (left) have an enhanced glycolytic metabolic state and an impaired oxidative phosphorylation (OXPHOS). The tricarboxylic acid cycle (TCA) is fueled by metabolites derived from glutaminolysis, while pyruvate is mainly fermented into lactate. On the other hand, LPS-primed cells (right) preferentially use OXPHOS, and the glycolytic pathway generates pyruvate for fueling the TCA cycle. The epigenetic reprogramming occurring in β-glucan- and BCG-primed monocytes mainly encompasses histone methylation and acetylation and involves lncRNA, modifications that are lacking (or established late) in LPS-primed monocytes. NP (Au, pGr) may play a role in the induction and modulation of innate immune memory by regulating epigenetic and metabolic reprogramming of innate immune cells (dashed arrows). Lower part: BCG-primed monocytes (left) show an increased reactivity after a subsequent exposure to LPS. When monocytes are primed with BCG in the presence of Au NP, they show a reduced reactivity (right). Thus, Au NP are able to reduce the BCG-induced memory response in human primary monocytes by shifting the memory effect from potentiation/trained immunity to tolerance. By contrast, pGr (left) is able to prime directly murine BMDM, which responds to a subsequent LPS challenge with a potentiated reaction.
In another recent study, Lebre et al. (111) proved that pGr can directly prime mouse BMDM in vitro. BMDM exposed to pGr for 24 h and, after 5 days, stimulated with LPS produced significantly higher amounts of the inflammatory cytokines IL-6 and TNF-α and lower amounts of the regulatory cytokine IL-10 compared to unprimed cells (Figure 1, bottom part). Challenge with other Toll-like receptor (TLR) agonists, such as CpG (TLR9 agonist) or the R848 (TLR7/8 agonist), yielded the same memory response in pGR-primed macrophages as that obtained with LPS, that is, enhancement of IL-6 secretion and decrease in IL-10 secretion. The induction of memory by pGr occurred through epigenetic reprograming, since preincubation of BMDM with inhibitors of histone methyltransferase or histone demethylase abolished the priming effect of pGr in terms of enhancement of LPS-induced IL-6 production. Interestingly, the same inhibitors did not have effect on LPS-induced IL-10 and TNF-α, suggesting a different regulatory mechanism for each inflammatory and anti-inflammatory factor.
Collectively, these data provide evidence that NP are able to reprogram macrophages to respond differentially to a second stimulation. However, while pGr can apparently induce innate memory per se, Au NP do not have a direct effect but can modulate the memory induced by another stimulus.
The molecular mechanisms by which Au NP affect innate memory induction have not been fully investigated yet, but it is reasonable to hypothesize that they can involve metabolic reprogramming more than epigenetic changes.
Conclusions
In the last decades, engineered NP have found wide application in medicine, as diagnostic and therapeutic tools. As a consequence, the evaluation of nanoimmunosafety has become crucial in order to avoid unwanted immune system activation, which could contribute to chronic inflammation, autoimmunity, or allergy. Conversely, the ability of NP to interact with innate immunity could be exploited and, through rational engineering, directed toward an intentional enhancement or suppression of immune reactions to treat a range of disease conditions and in preventive vaccination strategies. The recent evidence that innate memory exists also in vertebrates and that it is based on the epigenetic and metabolic reprogramming of innate cells is opening a new area of investigation in the assessment of NP immune interactions. Some studies have shown that NP can induce epigenetic modifications (DNA methylation, histone modification, and non-coding RNA modulation) and metabolic changes in monocytes and macrophages, the major innate immune cells. Other studies have revealed that certain types of NP can induce innate memory or modulate the memory induced by bacterial priming, in in vitro systems using mouse macrophages or human monocytes.
In this context, a thorough investigation is needed to examine the epigenetic and metabolic profiles induced in monocytes/macrophages by NP exposure and correlate them to functional memory profiles. Thus, both the immunosafety of NP and their medical exploitation have to be re-considered in terms of induction of epigenetic and metabolic changes and of induction/regulation of innate memory. This information could pave the way to new nanotechnological applications in medicine.
Author Contributions
PI wrote the manuscript. GDC contributed to writing. DB revised the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the EU Commission H2020 projects ENDONANO (GA 812661) and PANDORA (GA671881), the Italian MIUR InterOmics Flagship projects MEMORAT and MAME, and the Italian MIUR/PRIN-20173ZECCM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Eun-Kyung L, Taekhoon K, Soonmyung P, Seungjoo Haam°Yong-Min H, Kwangyeol L. Nanomaterials for theranostics: recent advances and future challenges. Chem Rev. (2015) 115:327–94. doi: 10.1021/cr300213b
2. Fadeel B. Hide and seek: nanomaterial interactions with the immune system. Front Immunol. (2019) 10:133. doi: 10.3389/fimmu.2019.00133
3. Boraschi D, Italiani P, Palomba R, Decuzzi P, Duschl A, Fadeel B, et al. Nanoparticles and innate immunity: new perspectives on host defence. Semin Immunol. (2017) 34:33–51. doi: 10.1016/j.smim.2017.08.013
4. Dobrovolskaia MA, Shurin M, Shvedova AA. Current understanding of interactions between nanoparticles and the immune system. Toxicol Appl Pharmacol. (2016) 299:78–89. doi: 10.1016/j.taap.2015.12.022
5. Muhammad Q, Jang Y, Kang SH, Moon J, Kim WJ, Park H. Modulation of immune responses with nanoparticles and reduction of their immunotoxicity. Biomater Sci. (2020) 8:1490–501. doi: 10.1039/c9bm01643k
6. Fang RH, Zhang L. Nanoparticle-based modulation of the immune system. Annu Rev Chem Biomol Eng. (2016) 7:305–26. doi: 10.1146/annurev-chembioeng-080615-034446
7. Smith DM, Simon JK, Baker JR. Applications of nanotechnology for immunology. Nat Rev Immunol. (2013) 13:592–605. doi: 10.1038/nri3488
8. Gourbal B, Pinaud S, Beckers GJM, Van Der Meer JWM, Conrath U, Netea MG. Innate immune memory: an evolutionary perspective. Immunol Rev. (2018) 283:21–40. doi: 10.1111/imr.12647
9. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–75.e14. doi: 10.1016/j.cell.2017.12.013
10. Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. (2018) 61:942–53. doi: 10.1007/s00125-017-4526-6
11. Crişan TO, Cleophas MCP, Novakovic B, Erler K, van de Veerdonk FL, Stunnenberg HG, et al. Uric acid priming in human monocytes is driven by the AKT–PRAS40 autophagy pathway. Proc Natl Acad Sci USA. (2017) 114:5485–90. doi: 10.1073/pnas.1620910114
12. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. (2011) 9:355–61. doi: 10.1016/j.chom.2011.04.006
13. Boraschi D, Italiani P. Innate immune memory: time for adopting a correct terminology. Front Immunol. (2018) 9:799. doi: 10.3389/fimmu.2018.00799
14. Seeley JJ, Ghosh S. Molecular mechanisms of innate memory and tolerance to LPS. J Leukoc Biol. (2017) 101:107–19. doi: 10.1189/jlb.3MR0316-118RR
15. Domínguez-Andrés J, Fanucchi S, Joosten LAB, Mhlanga MM, Netea MG. Advances in understanding molecular regulation of innate immune memory. Curr Opin Cell Biol. (2020) 63:68–75. doi: 10.1016/j.ceb.2019.12.006
16. Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang S-Y, Oosting M, et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe. (2018) 23:89–100. e5. doi: 10.1016/j.chom.2017.12.010
17. Walk J, de Bree LCJ, Graumans W, Stoter R, van Gemert G-J, van de Vegte-Bolmer M, et al. Outcomes of controlled human malaria infection after BCG vaccination. Nat Commun. (2019) 10:874. doi: 10.1038/s41467-019-08659-3
18. Arts RJW, Joosten LAB, Netea MG. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front Immunol. (2018) 9:298. doi: 10.3389/fimmu.2018.00298
19. Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Pérez-Medina C, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity. (2018) 49:819–28.e6. doi: 10.1016/j.immuni.2018.09.008
20. Mulder WJM, Ochando J, Joosten LAB, Fayad ZA, Netea MG. Therapeutic targeting of trained immunity. Nat Rev Drug Discov. (2019) 18:553–66. doi: 10.1038/s41573-019-0025-4
21. Italiani P, Boraschi D. Induction of innate immune memory by engineered nanoparticles: a hypothesis that may become true. Front Immunol. (2017) 8:734. doi: 10.3389/fimmu.2017.00734
22. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. (2009) 23:781–83. doi: 10.1101/gad.1787609
23. van der Heijden CDCC, Noz MP, Joosten LAB, Netea MG, Riksen NP, Keating ST. Epigenetics and trained immunity. Antioxid Redox Signal. (2018) 29:1023–40. doi: 10.1089/ars.2017.7310
24. Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. (2013) 152:1021–36. doi: 10.1016/j.cell.2013.01.052
25. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. (2007) 25:15–30. doi: 10.1016/j.molcel.2006.12.014
26. Quintin J, Saeed S, Martens JH, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
27. Novakovic B, Habibi E, Wang SY, Arts RJ, Davar R, Megchelenbrink W, et al. Beta-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. (2016) 167: 1354–68.e1314. doi: 10.1016/j.cell.2016.09.034
28. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.112/science.1251086
29. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–61. doi: 10.1016/j.cell.2017.11.034
30. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. (2018) 172:176–90. doi: 10.1016/j.cell.2017.12.031
31. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
32. Foster SL, Hargreaves DC, Medzhitov R. Gene specific control of inflammation by TLR-induced chromatin modifications. Nature. (2007) 447:972–78. doi: 10.1038/nature05836
33. Seeley JJ, Baker RG, Mohamed G, Bruns T, Hayden MS, Deshmukh SD, et al. Induction of innate immune memory via microRNA targeting of chromatin remodelling factors. Nature. (2018) 559:114–19. doi: 10.1038/s41586-018-0253-5
34. Fanucchi S, Mhlanga MM. Lnc-ing trained immunity to chromatin architecture. Front Cell Dev Biol. (2019) 7:2. doi: 10.3389/fcell.2019.00002
35. Fok ET, Davignon L, Fanucchi S, Mhlanga MM. The lncRNA connection between cellular metabolism and epigenetics in trained immunity. Front Immunol. (2018) 9:3184. doi: 10.3389/fimmu.2018.03184
36. Fanucchi S, Fok ET, Dalla E, Shibayama Y, Börner K, Chang EY, et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat Genet. (2018) 51:138–50. doi: 10.1038/s41588-018-0298-2
37. Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, et al. Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature. (2013) 494:497–501. doi: 10.1038/nature11884
38. Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, et al. Transcription factor trapping by RNA in gene regulatory elements. Science. (2015) 350:978–81. doi: 10.1126/science.aad3346
39. Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, Berger SL. RNA binding to CBP stimulates histone acetylation and transcription. Cell. (2017) 168:135–49. e22. doi: 10.1016/j.cell.2016.12.020
40. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. (2013) 152:157–71. doi: 10.1016/j.cell.2012.12.018
41. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
42. Van den Bossche J, O'Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. (2017) 38: 395–406. doi: 10.1016/j.it.2017.03.001
43. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR-and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
44. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. (2016) 17:2562–71. doi: 10.1016/j.celrep.2016.11.011
45. Arts RJW, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metabol. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
46. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. (2012) 15:110–21. doi: 10.1016/j.cmet.2011.12.009
47. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Yang Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–46.e9. doi: 10.1016/j.cell.2017.11.025
48. Sohrabi Y, Sonntag GVH, Braun LC, Lagache SMM, Liebmann M, Klotz L, et al. LXR activation induces a proinflammatory trained innate immunity-phenotype in human monocytes. Front Immunol. (2020) 11:353. doi: 10.3389/fimmu.2020.00353
49. Vidula Vachharajani V, McCall CE. Epigenetic and metabolic programming of innate immunity in sepsis. Innate Immun. (2019) 25:267–79. doi: 10.1177/1753425919842320
50. Arts RJW, Gresnigt MS, Joosten LAB, Netea MG. Cellular metabolism of myeloid cells in sepsis. J Leukoc Biol. (2017) 101:151–64. doi: 10.1189/jlb.4MR0216-066R
51. Bomans K, Schenz J, Sztwiertnia I, Schaack D, Weigand MA, Uhle F. Sepsis induces a long-lasting state of trained immunity in bone marrow monocytes. Front Immunol. (2018) 9:2685. doi: 10.3389/fimmu.2018.02685
52. Penkov S, Mitroulis I, Hajishengallis G, Chavakis T. Immunometabolic crosstalk: an ancestral principle of trained immunity? Trends Immunol. (2019) 40:1–11. doi: 10.1016/j.it.2018.11.002
53. Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol. (2012) 227:3169–77. doi: 10.1002/jcp.24054
54. Sohrabi Y, Godfrey R, Findeisen HM. Altered cellular metabolism drives trained immunity. Trends Endocrinol Metab. (2018) 29:602–5. doi: 10.1016/j.tem.2018.03.012
55. Simithy J, Sidoli S, Yuan Z-F, Coradin M, Bhanu NV, Marchione DM, et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat Commun. (2017) 8:1141. doi: 10.1038/s41467-017-01384-9
56. Lio C-WJ, Rao A. TET enzymes and 5hmC in adaptive and innate immune systems. Front Immunol. (2019) 10:210. doi: 10.3389/fimmu.2019.00210
57. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of a-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. (2012) 26:1326–38. doi: 10.1101/gad.191056.112
58. Trievel RC, Scheiner S. Crystallographic and computational characterization of methyl tetrel bonding in S-Adenosylmethionine-dependent methyltransferases. Molecules. (2018) 23:2965. doi: 10.3390/molecules23112965
59. Arts RJ, Joosten LA, Netea MG. Immunometabolic circuits in trained immunity. Semin Immunol. (2016) 28:425–30. doi: 10.1016/j.smim.2016.09.002
60. Gedda MR, Babele PK, Zahra K, Madhukar P. Epigenetic aspects of engineered nanomaterials: is the collateral damage inevitable? Front Bioeng Biotechnol. (2019) 7:228. doi: 10.3389/fbioe.2019.00228
61. Sierra MI, Valdés A, Fernández AF, Torrecillas R, Fraga MF. The effect of exposure to nanoparticles and nanomaterials on the mammalian epigenome. Int J Nanomedicine. (2016) 11:6297–306. doi: 10.2147/IJN.S120104
62. Yu J, Loh XL, Luo Y, Ge S, Fan X, Ruan J. Insights into the epigenetic effects of nanomaterials on cells. Biomater Sci. (2020) 8:763–75. doi: 10.1039/c9bm01526d
63. Wong BSE, Qidong Hu Q, Baeg GH. Epigenetic modulations in nanoparticle-mediated toxicity. Food Chem Toxicol. (2017) 109:746–52. doi: 10.1016/j.fct.2017.07.006
64. Smolkova B, Dusinska M, Gabelova A. Nanomedicine and epigenome. Possible health risks. Food Chem Toxicol. (2017) 109:780e796. doi: 10.1016/j.fct.2017.07.020
65. Smolkova B, Dusinska M, Gabelova A. Epigenetic effects of nanomaterials. In: Nriagu J, editor Encyclopedia of Environmental Health. 2nd ed. Amsterdam: Elsevier (2018). p. 1–8.
66. Conroy J, Byrne SJ, Gun'ko YK, Rakovich YP, Donegan JF, Davies A, et al. CdTe nanoparticles display tropism to core histones and histone-rich cell organelles. Small. (2008) 4:2006–15. doi: 10.1002/smll.200800088
67. Lu X, Miousse IR, Pirela SV, Melnyk S, Koturbash I, Demokritou P. Short-term exposure to engineered nanomaterials affects cellular epigenome. Nanotoxicology. (2016) 10:140–50. doi: 10.3109/17435390.2015
68. Ghosh M, Öner D, Poels K, Tabish AM, Vlaanderen J, Pronk A, et al. Changes in DNA methylation induced by multi-walled carbon nanotube exposure in the workplace. Nanotoxicology. (2017) 11:1195–210. doi: 10.1080/17435390.2017.1406169
69. Silva IR, Ramos MCAS, Arantes LMRB, Lengert AVH, Oliveira MA, Cury FP, et al. Evaluation of DNA methylation changes and micronuclei in workers exposed to a construction environment. Int J Environ Res Public Health. (2019) 16:902. doi: 10.3390/ijerph16060902
70. Öner D, Moisse M, Ghosh M, Duca RC, Poels K, Luyts K, et al. Epigenetic effects of carbon nanotubes in human monocytic cells. Mutagenesis. (2017) 32:181–91. doi: 10.1093/mutage/gew053
71. Brown TA, Lee JW, Holian A, Porter V, Fredriksen H, Kim M, et al. Alterations in DNA methylation corresponding with lung inflammation and as a biomarker for disease development after MWCNT exposure. Nanotoxicology. (2016) 10:453–61. doi: 10.3109/17435390.2015.1078852
72. Sui Y, Fu Y, Zhang Y, Ma S, Yin L, Pu Y, Liang G. Molecular mechanism for miR-350 in regulating of titanium dioxide nanoparticles in macrophage RAW264.7 cells. Chem Biol Interact. (2018) 280:77–85. doi: 10.1016/j.cbi.2017.12.020
73. Shvedova AA, Yanamala N, Kisin ER, Khailullin TO, Birch ME, Fatkhutdinova LM. Integrated analysis of dysregulated ncRNA and mRNA expression profiles in humans exposed to carbon nanotubes. PLoS ONE. (2016) 11:e0150628. doi: 10.1371/journal.pone.0150628
74. Ndika J, Seemab U, Poon WL, Fortino V, El-Nezami H, Karisola P, et al. Silver, titanium dioxide, and zinc oxide nanoparticles trigger miRNA/isomiR expression changes in THP-1 cells that are proportional to their health hazard potential. Nanotoxicology. (2019) 13:1380–95. doi: 10.1080/17435390.2019.1661040
75. Chen N, Wang H, Huang Q, Li J, Yan J, He D, et al. Long-term effects of nanoparticles on nutrition and metabolism. Small. (2014) 10: 3603–11q. doi: 10.1002/smll.201303635
76. Chen Q, Wang N, Zhu M, Lu J, Zhong H, Xue X, et al. TiO2 nanoparticles cause mitochondrial dysfunction, activate inflammatory responses, and attenuate phagocytosis in macrophages: a proteomic and metabolomic insight. Redox Biol. (2018) 15:266–76. doi: 10.1016/j.redox.2017.12.011
77. Dalzon B, Torres A, Diemer H, Ravanel S, Collin-Faure V, Pernet-Gallay K, et al. How reversible are the effects of silver nanoparticles on macrophages? A proteomic-instructed view. Environ Sci Nano. (2019) 6:3133. doi: 10.1039/c9en00408d
78. Saborano R, Wongpinyochit T, Totten JD, Johnston BF, Seib FP, Duarte IF. Metabolic reprogramming of macrophages exposed to silk, poly(lactic-co-glycolic acid), and silica nanoparticles. Adv Healthc Mater. (2017) 6:1601240. doi: 10.1002/adhm.201601240
79. Totten JD, Wongpinyochit T, Carrola J, Duarte IF, Seib FP. PEGylation-dependent metabolic rewiring of macrophages with silk fibroin nanoparticles. ACS Appl Mater Interfaces. (2019) 11:14515–25. doi: 10.1021/acsami.8b18716
80. Wu Q, Jin R, Feng T, Liu L, Yang L, Tao Y, et al. Iron oxide nanoparticles and induced autophagy in human monocytes. Int J Nanomedicine. (2017) 12:3993–4005. doi: 10.2147/IJN.S135189
81. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
82. Riffelmacher T, Richter FC, Simon AK. Autophagy dictates metabolism and differentiation of inflammatory immune cells. Autophagy. (2018) 14:199–206. doi: 10.1080/15548627.2017.1362525
83. Fuchs AK, Syrovets T, Haas KA, Loos C, Musyanovych A, Mailänder V, et al. Carboxyl-and amino-functionalized polystyrene nanoparticles differentially affect the polarization profile of M1 and M2 macrophage subsets. Biomaterials. (2016) 85:78–87. doi: 10.1016/j.biomaterials.2016.01.064
84. Ren C, Li D1, Zhou Q1, Hu X. Mitochondria-targeted TPP-MoS2 with dual enzyme activity provides efficient neuroprotection through M1/M2 microglial polarization in an Alzheimer's disease model. Biomaterials. (2020) 232:119752. doi: 10.1016/j.biomaterials.2019.119752
85. Wang L, Zhang H, Sun L, Gao W, Xiong Y, Ma A, et al. Manipulation of macrophage polarization by peptide-coated gold nanoparticles and its protective effects on acute lung injury. J Nanobiotechnol. (2020) 18:38. doi: 10.1186/s12951-020-00593-7
86. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
87. Gordon S, Mantovani A. Diversity and plasticity of mononuclear phagocytes. Eur J Immunol. (2011) 41:2470–2. doi: 10.1002/eji.201141988
88. Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun. (2014) 6:716–26. doi: 10.1159/000364945
89. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/fimmu.2014.00514
90. Griffiths HR, Gao D, Pararasa C. Redox regulation in metabolic programming and inflammation. Redox Biol. (2017) 12:50–7. doi: 10.1016/j.redox.2017.01.023
91. Virág L, Jaén RI, Regdon Z, Boscá L, Prieto P. Self-defense of macrophages against oxidative injury: fighting for their own survival. Redox Biol. (2019) 26:101261. doi: 10.1016/j.redox.2019.101261
92. Ullevig SL, Kim HS, Short JD, Tavakoli S, Weintraub ST, Downs K, et al. Protein S-glutathionylation mediates macrophage responses to metabolic cues from the extracellular environment. Antioxid Redox Signal. (2016) 25:836–51. doi: 10.1089/ars.2015.6531
93. Cencioni C, Spallotta F, Martelli F, Valente S, Mai A, Zeiher AM, et al. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int J Mol Sci. (2013) 14:17643–63. doi: 10.3390/ijms140917643
94. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. (2016) 166:822–39. doi: 10.1016/j.cell.2016.07.050
95. Ben-Avraham D, Muzumdar RH, Atzmon G. Epigenetic genome-wide association methylation in aging and longevity. Epigenomics. (2012) 4:503–9. doi: 10.2217/epi.12.41
96. Park EJ, Park K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol Lett. (2009) 184:18–25. doi: 10.1016/j.toxlet.2008.10.012
97. Pal R, Chakraborty B, Nath A, Singh LM, Ali M, Rahman DS, et al. Noble metal nanoparticle-induced oxidative stress modulates tumor associated macrophages (TAMs) from an M2 to M1 phenotype: an in vitro approach. Int Immunopharmacol. (2016) 38:332–41. doi: 10.1016/j.intimp.2016.06.006
98. Rojas JM, Sanz-Ortega L, Mulens-Arias V, Gutiérrez L, Pérez-Yagüe S, Barber DF. Superparamagnetic iron oxide nanoparticle uptake alters M2 macrophage phenotype, iron metabolism, migration and invasion. Nanomedicine. (2016) 12:1127–38. doi: 10.1016/j.nano.2015.11.020
99. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. (2016) 11:986–94. doi: 10.1038/nnano.2016.168
100. Meng J, Li X, Wang C, Guo H, Liu J, Xu H. Carbon nanotubes activate macrophages into a M1/M2 mixed status: recruiting naïve macrophages and supporting angiogenesis. ACS Appl Mater Interfaces. (2015) 7:3180–8. doi: 10.1021/am507649n
101. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
102. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. (2017) 355:842–47. doi: 10.1126/science.aag1381
103. Mendoza RP, Brown JM. Engineered nanomaterials and oxidative stress: current understanding and future challenges. Curr Opin Toxicol. (2019) 13:74–80. doi: 10.1016/j.cotox.2018.09.001
104. Manke A, Wang L, Rojanasakul Y. Mechanisms of nanoparticle-induced oxidative stress and toxicity. Biomed Res Int. (2013) 2013:942916. doi: 10.1155/2013/942916
105. Khanna P, Ong C, Bay BH, Baeg GH. Nanotoxicity: an interplay of oxidative stress, inflammation and cell death. Nanomaterials. (2015) 5:1163–80. doi: 10.3390/nano5031163
106. Boraschi D, Italiani P. Model validity in nanoimmunosafety: advantages and disadvantages of in vivo vs. in vitro models, and human vs. animal models. Current Bionanotechnol. (2016) 2:71–6. doi: 10.2174/2213529402666160601121721
107. Wilding JL, Bodmer WF. Cancer cell lines for drug discovery and development. Cancer Res. (2014) 74:2377–84. doi: 10.1158/0008-5472.CAN-13-2971
108. Vijayana V, Pradhan P, Braud L, Fuchs HR, Gueler F, Motterlini R, et al. Human and murine macrophages exhibit differential metabolic responses to lipopolysaccharide—a divergent role for glycolysis. Redox Biol. (2019) 22:101147. doi: 10.1016/j.redox.2019.101147
109. Rico D, Vaquerizas JM, Dopazo H, Bosca L. Identification of conserved domains in the promoter regions of nitric oxide synthase2: implications for the species-specific transcription and evolutionary differences. BMC Genomics. (2008) 8:271. doi: 10.1186/1471-2164-8-271
110. Swartzwelter BJ, Barbero F, Verde A, Mangini M, Pirozzi M, De Luca AC, et al. Gold nanoparticles modulate BCG-induced innate immune memory in human monocytes by shifting the memory response towards tolerance. Cells. (2020) 9:284. doi: 10.3390/cells9020284
Keywords: innate memory, monocytes, macrophages, nanoparticles, epigenetics, metabolism
Citation: Italiani P, Della Camera G and Boraschi D (2020) Induction of Innate Immune Memory by Engineered Nanoparticles in Monocytes/Macrophages: From Hypothesis to Reality. Front. Immunol. 11:566309. doi: 10.3389/fimmu.2020.566309
Received: 27 May 2020; Accepted: 24 August 2020;
Published: 06 October 2020.
Edited by:
Martin Rottenberg, Karolinska Institutet (KI), SwedenReviewed by:
Maria Lerm, Linköping University, SwedenReto Asmis, Wake Forest School of Medicine, United States
Copyright © 2020 Italiani, Della Camera and Boraschi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Italiani, italianipaola@gmail.com; paola.italiani@ibbc.cnr.it