 Tom Macleod1,2
Tom Macleod1,2 Anna Berekmeri2
Anna Berekmeri2 Charlie Bridgewood2
Charlie Bridgewood2 Martin Stacey1
Martin Stacey1 Dennis McGonagle2,3
Dennis McGonagle2,3 Miriam Wittmann2,3*
Miriam Wittmann2,3*- 1School of Molecular and Cellular Biology, University of Leeds, Leeds, United Kingdom
- 2Leeds Institute of Rheumatic and Musculoskeletal Medicine (LIRMM), University of Leeds, Leeds, United Kingdom
- 3National Institute for Health Research (NIHR) Leeds Biomedical Research Centre (BRC), The Leeds Teaching Hospitals, Leeds, United Kingdom
The skin barrier would not function without IL-1 family members, but their physiological role in the immunological aspects of skin barrier function are often overlooked. This review summarises the role of IL-1 family cytokines (IL-1α, IL-1β, IL-1Ra, IL-18, IL-33, IL-36α, IL-36β, IL-36γ, IL-36Ra, IL-37 and IL-38) in the skin. We focus on novel aspects of their interaction with commensals and pathogens, the important impact of proteases on cytokine activity, on healing responses and inflammation limiting mechanisms. We discuss IL-1 family cytokines in the context of IL-4/IL-13 and IL-23/IL-17 axis-driven diseases and highlight consequences of human loss/gain of function mutations in activating or inhibitory pathway molecules. This review highlights recent findings that emphasize the importance of IL-1 family cytokines in both physiological and pathological cutaneous inflammation and emergent translational therapeutics that are helping further elucidate these cytokines.
Introduction
The IL-1 family is a large group of cytokines, all of which are expressed in human epidermis, and have well established roles in skin immunopathology. Collectively these cardinal immunological functions encompass anti-microbial host protection and barrier function maintenance or restoration via inflammation and inflammation resolution mechanisms. In addition to established pathological roles this family of cytokines are vital for maintaining the skin’s immunological barrier and wound repair, yet this physiological role often gets overlooked. This review aims to give an overview of the role IL-1 family proteins play in skin, focusing on novel findings in their physiological role, and provides context for a deeper understanding of pathway derangement with resultant cutaneous disease.
The IL-1 Family Cytokines
The IL-1 superfamily (Figure 1) can be split into three smaller subfamilies according to structural similarities amongst the cytokine IL-1 consensus sequence and their primary ligand binding receptors: the IL-1, IL-18 and IL-36 subfamilies (1). The IL-1 subfamily has three agonists (IL-1β, IL-1α and IL-33) and a receptor antagonist (IL-1Ra). The IL-18 subfamily contains IL-18 as well as the anti-inflammatory cytokine IL-37 and IL-18 binding protein (IL-18BP). The IL-36 subfamily consists of three agonists – IL-36α, IL-36β and IL-36γ – and the antagonist IL-36Ra. It also contains the cytokine IL-38 whose function is not wholly understood yet is considered predominantly anti-inflammatory.
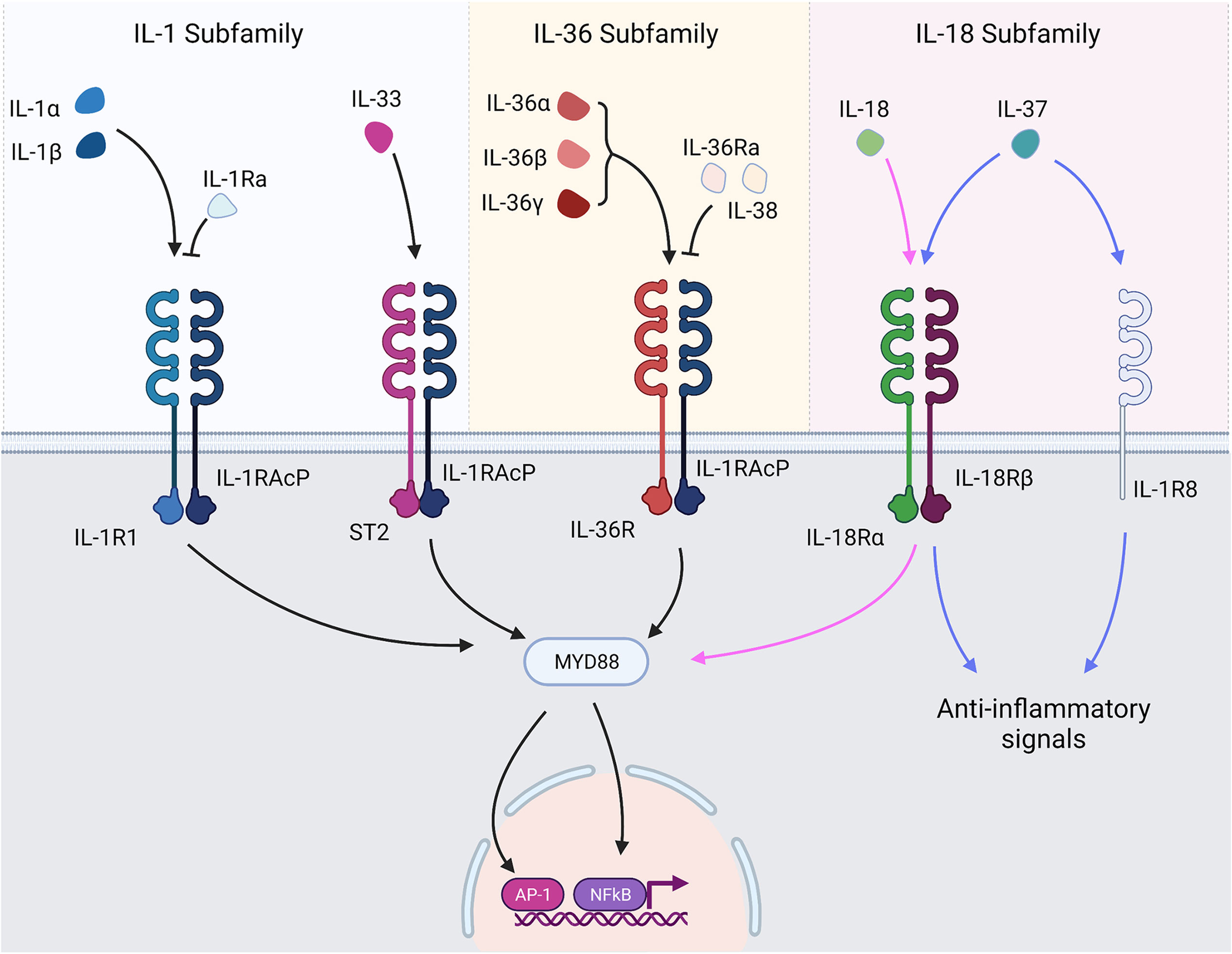
Figure 1 IL-1 superfamily cytokines and receptors. IL-1 family cytokines bind their respective receptors and transduce signals through TIR/MyD88 signalling pathways. The major signalling pathways result in activation of NF-κB and AP-1 and subsequent transcription of genes under their control. Receptor antagonists bind their respective receptors but prevent the recruitment of the accessory protein, thereby preventing signal transduction. IL-37 exerts anti-inflammatory signals following interaction with IL-1R8 and IL-18Rα.
With the exception of IL-1Ra, all IL-1 family cytokines lack an N-terminal signal peptide and are therefore not secreted in a classical manner (2). Each cytokine is produced as an immature precursor that undergoes maturation by proteolytic truncation, the specificity of which differs from cytokine to cytokine. The activity of IL-1 family cytokines is hugely affected by this proteolytic maturation. Some cytokines such as IL-1α and IL-33 have biological activity in their immature form, yet others such as IL-1β and the IL-36 cytokines must undergo maturation to become active (1–3). Within the proteolytic maturation itself there exists more levels of regulation as variation in the specific truncations generated can affect the biological activity of the resulting mature cytokine. Therefore, changes to the proteolytic environment to which the IL-1 family cytokines are exposed can significantly affect the resulting inflammatory outcome and their subsequent impact on skin.
Signalling within the IL-1 family is a highly conserved process, with agonists binding their cognate receptors causing signal transduction via Toll/Interleukin-1 Receptor (TIR) and MyD88-dependent signalling pathways culminating in the activation of NF-κB and MAPK and subsequent NF-κB and AP-1 dependent gene transcription (4). As a result, many IL-1 family members generate similar pro-inflammatory responses. Nuances in cellular and tissue compartment expression, activation of IL-1 family cytokines and the responsive cell types, however, means IL-1 family cytokines can generate a huge variation of cellular responses and impart a broad impact on the immune system. The IL-1 family cytokines signal through IL-1 family receptors, some of which are shared amongst cytokines (Figure 1). IL-1 receptor biology is a complex topic in itself that is beyond the remit of this review and has been extensively reviewed elsewhere (5). However, to provide a contextual overview, all IL-1 family cytokines transduce signals in a similar manner. Agonists bind their cognate receptor, which subsequently recruits a membrane-bound accessory protein. Interaction between the ligand-bound receptor and the recruited accessory protein allows a signal to be transduced to the cell (6). As outlined in greater detail by Figure 1, some of the receptors are shared amongst IL-1 family cytokines. IL-1α, and IL-1β bind IL-1R1, IL-33 binds ST2, IL-18 and IL-37 bind IL-18Ra, IL-37 also binds a decoy receptor IL-1R8 through which it mediates some anti-inflammatory activity, and the IL-36 cytokines all bind IL-36R.
All IL-1 family cytokines are expressed, with some variation, within the skin. IL-1α is constitutively expressed by keratinocytes, yet is retained as intracellular stores (7, 8). IL-1β has a more specialised expression profile across the body, however in the skin most cell types present can express the cytokine, notably keratinocytes, fibroblasts and both resident and infiltrating myeloid cells (9, 10). Unlike IL-1α, IL-1β is only expressed following cell activation. IL-33 is only weakly expressed by keratinocytes in healthy human skin, but is strongly induced in the epidermis of inflamed skin (11). IL-18 is also expressed throughout the epidermis. Similarly to IL-1α, IL-18 is constitutively expressed by keratinocytes and retained within cells as intracellular stores, yet is only cleaved into its mature form following cell activation in the same manner as IL-1β (12). IL-37 exists in 5 isoforms termed IL-37a-e as a result of alternative splicing. The largest and most common isoform, IL-37b, is expressed by keratinocytes, monocytes and other immune cells within skin (13). Of the IL-36 cytokines, IL-36γ is the most strongly expressed in skin. Tape strip analysis of human skin show basal levels of IL-36γ are present in the outer layers of the epidermis, even in healthy skin (14). IL-36γ is constitutively expressed at low levels by keratinocytes, and strongly upregulated following activation (15, 16). The IL-36 cytokines are not constitutively expressed by other cell types present in skin but are inducible in myeloid cells following activation (17). In addition to functioning as cytokines, IL-1α, IL-33 and some isoforms of IL-37 are known to translocate to the nucleus where they can regulate gene expression through their DNA-binding capabilities (8, 13, 18).
Sensing Environmental Threats
A number of IL-1 family proteins have a significant role in detecting environmental threats encountered in skin. Many have been added to the group of damage-associated molecular patterns (DAMPs) or “alarmins”.
Protease Sensors
Several factors in IL-1 family member biology and expression characteristics make these cytokines excellent sensors of protease activity, alerting the immune system to potential threats. Although IL-1α, IL-33 and IL-37 exhibit a reduced level of activity in their immature form, IL-1β, IL-18, and the IL-36 cytokines require N-terminal truncation in order to engage their respective receptors (19). Proteolytic activation of IL-1 family cytokines is a tightly controlled process in order to license inflammation only when necessary. IL-1β and IL-18 activity is controlled by regulation of caspase-1, which itself remains inactive until inflammasome assembly occurs (20). The IL-36 agonists require precise cleavage to achieve maturity; truncation a single amino acid upstream or downstream of the correct site will generate an inactive cytokine (2). Indeed, as will be discussed later, a breakdown in the regulation of IL-1 proteolysis can lead to significant inflammatory disease.
Nevertheless, despite strict endogenous controls on the activity of IL-1 family cytokines, several have been shown to be activated by exogenous pathogen-derived and environmental proteases. IL-1β and IL-36γ are both processed to their active forms by the protease SpeB, secreted by Streptococcus pyogenes during infection (15). IL-36γ is also processed to its active form by Alp1, secreted by Aspergillus fumigatus, and both IL-36γ and IL-36α have been shown to be activated by Staphylococcus aureus and Trichophyton rubrum culture filtrates (15). Active IL-1β and IL-36 cytokines promote the development of Type I and Type III immune responses, which will facilitate clearance of bacterial and fungal pathogens. As such, the requirement of IL-1 family cytokines to undergo proteolytic cleavage to reach biological maturity also enables the cytokines to function as sensors of exogenous proteases and signal the presence of a threat.
Alarmin Function
IL-1α has long been associated with an alarmin role owing to its constitutive expression maintaining an intracellular store of the cytokine, and its ability to bind its receptor and transduce signals in its immature form. In the event of cellular damage or programmed cell death IL-1α is released into the extracellular space and able to warn surrounding cells of damage or infection (21). Furthermore, the skin barrier is subjected to mechanical stressing as a physiological phenomenon and this too regulates the release of IL-1α (22). Whilst IL-1α has been long established in this role, recent work supports the case that other IL-1 family members may also function in an alarmin capacity despite not wholly fitting the classical definition.
IL-36γ, in particular, is well suited as an epidermal alarmin as it is constitutively expressed in healthy human skin and is found even in the upper layers of the stratum corneum, where it is most likely to come into contact with invading pathogens following injury (14, 16). Its sensitivity to exogenous proteolytic activation and presence in the epidermal layers as a store of inactive inflammatory cytokine means that a small amount of pathogen-derived protease capable of activating IL-36γ can rapidly generate a strong inflammatory signal without relying on protein synthesis. Figure 2 outlines the mechanism by which IL-1 family cytokines may sense environmental threats and warn the surrounding tissue to potential damage or infection.
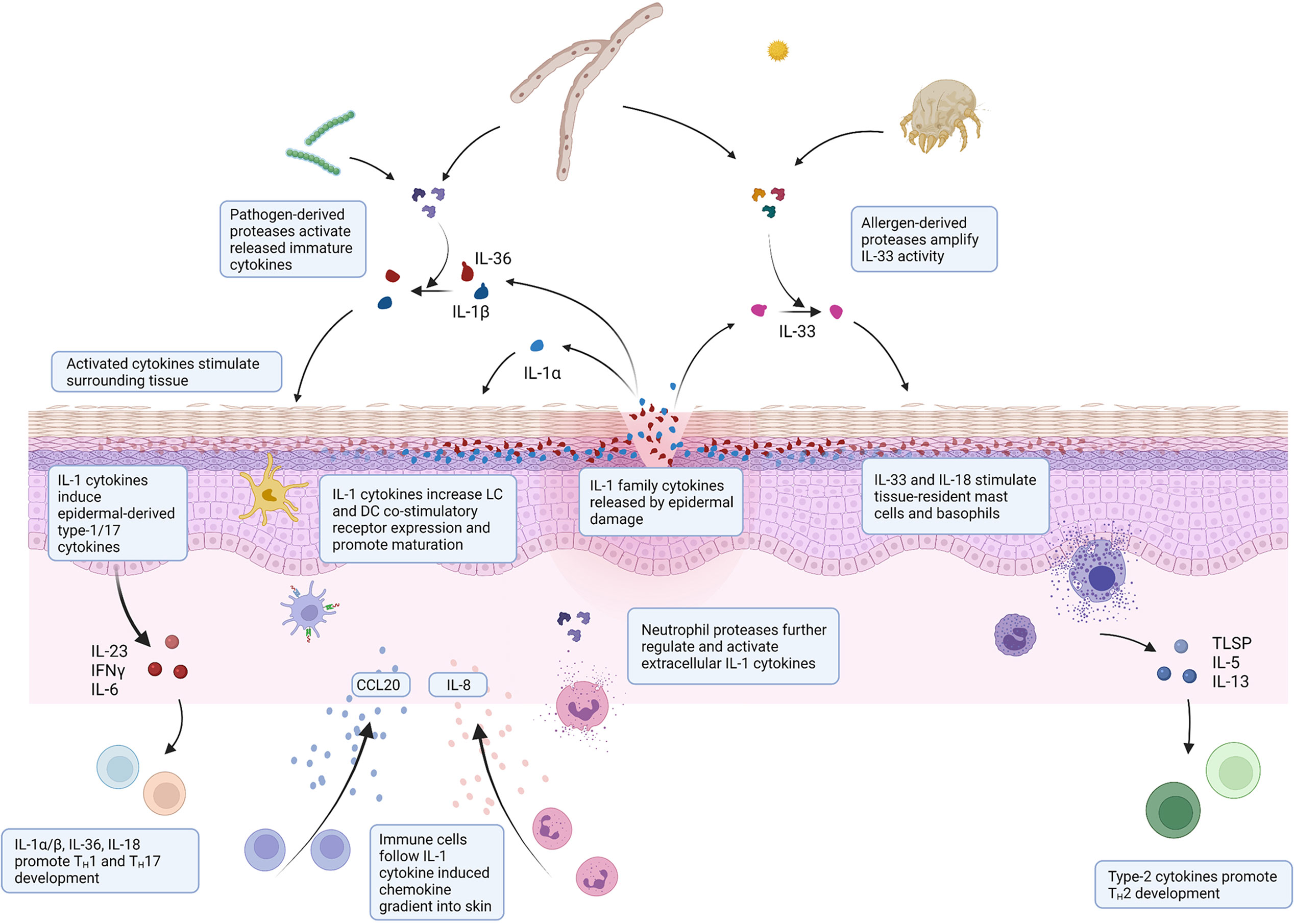
Figure 2 Environmental interaction of IL-1 family cytokines in skin injury. Schematic illustrating the mechanisms by which IL-1 family cytokines can sense environmental threats and orchestrate a subsequent immunological response. Epidermal damage through infection or injury releases IL-1 cytokines. Immature cytokines are activated by environmental proteases expressed by pathogens during infection or allergens present in the environment. Active released cytokines stimulate surrounding epidermis and resident immune cells to facilitate immune cell recruitment to mount an appropriate immune response.
Although not all IL-1 family cytokines act with an alarmin function in skin, several still have a crucial role in communicating the presence of environmental threats through their regulation by inflammasomes. Upon recognition of PAMPs and DAMPs by inflammasome sensors such as NLRP3, NLRC4, NAIP and AIM2, inflammasome complexes form and act as platforms for the activation of IL-1β, IL-18 and IL-37 (20). Inflammasome-mediated activation of the pore-forming protein gasdermin D then facilitates release of these cytokines into the surrounding tissue, thereby signalling the presence of an environmental threat (23). Whilst IL-1β must be induced and expressed prior to activation and secretion, IL-18 is constitutively expressed in the epidermis and even secreted in its immature form and is therefore available for activation immediately upon inflammasome formation (24). Furthermore, the inflammasome complexes themselves can be released into the surrounding extracellular tissue following activation which allows for maturation of extracellular IL-18 and IL-1β, amplifying the inflammatory response (25).
Microbial Interaction
In addition to alerting the skin to potential threats, many aspects of IL-1 family biology also help the immune system discriminate harmful pathogenic microbes from commensals. IL-1β has been shown to have a role facilitating the discrimination of pathogens from commensals in skin through its influence on peripheral tolerance. Colonisation of skin by commensals during early life gives rise to commensal-specific regulatory T cell populations, which provide the host with commensal-specific tolerance and prevent unwanted inflammation (26). Recent evidence in mouse models shows IL-1β can influence which colonising microbes the immune system tolerates by negatively regulating Treg cells (27). During early life colonisation of skin by S. aureus, virulence factors and cytotoxic proteins expressed by S. aureus that commensals lack induce expression of IL-1β. The induced IL-1β prevents the expansion of S. aureus-specific Treg cells, meaning subsequent exposure to S. aureus does not result in the expansion of a strong tolerogenic population of Treg cells, thereby allowing an inflammatory response to the skin pathogen (27).
Unlike IL-1β and IL-18, IL-36 cytokines are not activated by caspase-1 following inflammasome activation (3, 28). Whilst the mechanisms of IL-36 cytokine release have not been definitively outlined, cell death induced by pathogen-mediated damage has been demonstrated as one method. Indeed, the literature highlights a trend of IL-1 family cytokines to be released and activated by proteins associated with pathogen virulence. Phenol-soluble modulin (PSM)α virulence peptides secreted by S. aureus release IL-1α and IL-36 cytokines from keratinocytes through cellular damage (29, 30). As previously highlighted, the virulence factors SpeB and Alp 1 secreted during infection by S. pyogenes and A. fumigatus respectively activate IL-36γ, and SpeB has been shown to activate extracellular pro-IL-1β (15, 31). Recently the Yersinia pestis virulence factor YopJ, which inhibits NF-kB signalling, has been shown to trigger release of IL-1β and IL-18 via caspase-8-dependent gasdermin D activation (32, 33). By requiring either active or pathogen-dependent release from a damaged or activated cell, or activation by a pathogen-derived protease, or a combination of the two, these cytokines help the skin’s immune system discriminate the harmful from the harmless and allow appropriate anti-microbial or wound-healing responses.
IL-33 is an immunological outlier of the IL-1 family of cytokines given it is associated with the paradigmatic Type II diseases rather than with the innate immunopathology driven diseases associated with the other members. It has been shown to contribute to the downregulation of both filaggrin and human beta defensin 2 expression (34, 35). However, IL-33 shares many characteristics with its fellow IL-1 family cytokines in its alarmin function and as a sensor of environmental threats. IL-33 also appears to have a role in detecting cellular stress. Keratinocytes increase expression of IL-33 when experiencing hypo-osmotic stress. The induced IL-33 was observed to localise at the nucleus rather than being secreted, leading the authors to hypothesise that the increased expression acts as a failsafe in circumstances where stress progresses to damage, thus releasing IL-33 and inducing inflammation (36). In addition to the above mentioned IL-33 functions, this may be of relevance in atopic dermatitis (AD) and eczema where a loss of barrier integrity leaves the skin more exposed to external osmotic stress. Indeed, exposure to fresh water is known to exacerbate AD and eczema (37). A similar concept of increasing alarmin expression as a failsafe has also been suggested to occur with IL-1α, whereby the presence of the commensal Staphylococcus epidermidis allows the epidermis to maintain a level of intracellular IL-1α, keeping it primed for an efficient response in the event of damage (38). Similar to IL-1 and IL-36 cytokines, IL-33 has been shown to be truncated to potently active forms by numerous exogenous proteases, including pollen- and house dust mite-derived proteases (39). IL-33 is retained in the cell nucleus and cytosol as an intracellular store, is active in its immature form, and is released upon cell damage or programmed cell death (40). Yet, proteolytic truncation can dramatically increase its biological activity and will therefore generate a more potent alarmin once exposed to the external environment. In contrast to the other IL-1 family cytokines, IL-33 drives Type II immune responses, so susceptibility to proteolytic activation by an alternative set of exogenous proteases may function as a method of fine-tuning the inflammatory alarm depending upon the proteolytic environment experienced by the alarmin once released from the cell.
Pathological Consequences
Whilst IL-1 family cytokines are well suited as sensors of potential threats, their sensitivity to proteolytic activation and interaction with environmental microbes may also lead to the development of pathological inflammation. IL-36γ and Cutibacterium acnes have also been implicated in the pathomechanism of drug-induced acneiform skin toxicity (41). Satoh et al. report that EGFR/MEK inhibition in patients colonised with the commensal causes synergistic expression of IL-36γ. C. acnes mediated activation of NF-kB combined with increased expression of Krüppel-like factor 4 as a result of EGFRi/MEKi treatment strongly upregulated IL-36γ expression, which in turn lead to inflammatory skin-based toxicity (41). S. pyogenes tonsillar infection has a well-established strong aetiological association with guttate psoriasis – an acute eruptive form of psoriasis seen largely in children – and is the strongest environmental factor in triggering and exacerbating psoriatic skin lesions (42, 43). It has therefore been hypothesised that S. pyogenes-mediated proteolysis of IL-1β and IL-36γ during infection may trigger inflammation and lead to the development of inflammatory disease. The importance of balanced proteolytic regulation between host and commensal in epidermal homeostasis is underlined in patients with Netherton Syndrome (NS), in which the endogenous protease inhibitor LEKTI-1 is non-functional. Patients with NS suffer predominantly IL-17- and IL-36-driven cutaneous inflammation in conjunction with a S. aureus and S. epidermidis dominated microbiome (44, 45). A recent study conducted in mice has demonstrated the lack of adequate endogenous protease inhibition imparted by LEKTI-1 promotes dysbiosis, which in turn alters the proteolytic balance of the epidermis through the secretion of proteolytic virulence factors, inducing skin damage and promoting inflammation (45).
The pathological consequence of protease sensitivity is not limited to exogenous proteases as many endogenous proteases are also capable of truncating IL-1 family precursors to biologically active forms. IL-1α can be truncated to a more potent form by numerous proteases including calpain, granzyme B and neutrophil proteases to a more biologically potent form (46). Yet ‘unplanned’ truncation by endogenous proteases often generates less biologically active forms than their intended activators. IL-1β, for example, can be truncated by neutrophil elastase, proteinase-3 and cathepsin G, however the resultant forms are less active than caspase-1-truncated IL-1β (47–49). Nevertheless these mechanisms of activation are implicated in a number of inflammatory conditions. Proteases secreted by neutrophils, which are prominent in psoriasis plaques, have been shown to truncate immature IL-1 family cytokines including IL-1α, IL-1β, and the IL-36 cytokines, thereby potentially amplifying the inflammatory environment once recruited to the skin (50). Cathepsin G, neutrophil elastase and proteinase-3 have been demonstrated to activate IL-36α and IL-36γ (16, 51). There is some debate over the inflammatory impact of neutrophil-derived proteases as they are also involved in activating immature IL-36Ra and may also be involved in proteolytically de-activating IL-1 family cytokines following prolonged exposure (3, 52). Given the abundance of neutrophils and IL-36γ in psoriasis plaques, it seems likely neutrophils will exacerbate IL-36-mediated inflammation in psoriasis, however the contradictions present in the literature demonstrate there is still a lack of understanding on the overall neutrophil-mediated impact on IL-1 and IL-36 mediated inflammation in vivo.
In addition to promoting inflammatory disease, proteolysis and microbe-induced expression of IL-1 family members can promote atopic disease. Whilst IL-18 is primarily associated with promoting Type I immune responses, IL-18 secretion is known to be induced by S. aureus colonisation in cases of AD (53). This is particularly interesting when considered in conjunction with mast cell digestion allowing activation of extracellular IL-18 in the absence of caspase-1 activity (54). In a mouse model of atopic dermatitis, epicutaneous S. aureus has been shown to induce IL-36-dependent signalling and a subsequent increase in IgE and IL-4 production (55). However, as yet there are no data implicating this mechanism is present in humans and there is no comprehensive link between IL-36 signalling and factors driving immunoglobulin class switching.
IL-1α has been demonstrated to be instrumental in the development of chronic inflammation in filaggrin-deficient mice. In these mice, skin injury and dysbiosis drive IL-1α expression and secretion from keratinocytes, which in turn causes chronic inflammation (56). IL-33 has also been implicated in the environmental allergen-mediated triggering of allergic inflammation. Recent evidence shows environmental allergens including house dust mites, Alternaria alternata, Aspergillus fumigatus and cat dander can initiate intracellular cleavage and subsequent secretion of active IL-33 as a result of RIPK-caspase-8 ripoptosome activation in epithelial cells (57). The released active IL-33 is then free to induce pro-atopic responses from surrounding tissue. Considering the evidence that IL-33 is also susceptible to proteolytic activation by many proteases produced by these environmental allergens when in the extracellular space, it seems likely pathological IL-33-mediated signalling would result from exposure to these allergens.
Orchestrating Immune Responses
Once released and activated, IL-1 family cytokines help to orchestrate an appropriate immune response through interaction with their local cellular environment (Figure 2). IL-1 family cytokines have an impact to some extent on all cells within the skin and elicit a response from neighbouring keratinocytes and infiltrating immune cells. In doing so they influence both the innate and adaptive response.
Effects on the Keratinocyte Barrier
Within the epidermis, keratinocytes are strong responders to IL-1 family cytokines. Given their abundance in comparison to infiltrating immune cells, they are arguably the most important initial responders to IL-1 family cytokines and act to both fight infection through release of anti-microbial peptides and amplify inflammatory signals through release of a range of chemokines and cytokines. As many IL-1 family cytokines transduce signal through MyD88, keratinocytes respond to pro-inflammatory IL-1 family cytokines in a very similar fashion. Indeed, a recent study examining transcriptional differences in keratinocytes following IL-1β and IL-36 cytokine stimulation found nearly all IL-1β responses were replicated by IL-36 stimulation (58). In response to IL-1α, IL-1β, and IL-36 stimulation, keratinocytes express and release a host of antimicrobial peptides including human beta defensins, LL-37, and S100 proteins which directly combat infection (59, 60). They increase protein levels of many key pro-inflammatory cytokines including IL-23, IL-17C, TNFα, as well as other IL-1 family cytokines that will further amplify an inflammatory response (1, 61). Keratinocytes also secrete chemokines following stimulation with IL-1 family cytokines. IL-1α, IL-1β, and IL-36 cytokines all induce secretion of IL-8 (CXCL8) and CCL2 (aka MCP-1) – strong inducers of neutrophil and myeloid cell chemotaxis respectively – and CCL20 – responsible for the influx of cells expressing CCR6, which include TH17, gamma delta (γδ) T cells and Type III innate lymphoid cells (ILC3s) (58, 62). Keratinocytes also secrete CXCL10 in response to IL-18 stimulation; a chemoattractant of CXCR3-expressing cells including CD8 and CD4 T cells (63).
In addition to inducing expression and secretion of cytokines, chemokines and peptides from keratinocytes, IL-1 family cytokines can also induce cellular changes in keratinocytes that influence their barrier function. Recently IL-1β has been shown to promote cell-intrinsic immunity in epithelial cells by inducing activation of IRF3 and therefore driving an innate anti-viral response. IL-1β was found to induce the release of mitochondrial DNA into the cytosol of stimulated cells, subsequently activating the cGAS-STING pathway and activating IRF3 (64). There is evidence to suggest IL-1α stimulation of keratinocytes reduces the adherence of S. pyogenes, thereby reducing the likelihood of invading bacteria establishing an infection. This was hypothesised to be a result of changes in their expression of receptors utilised by S. pyogenes in adhering to keratinocytes (65). IL-18 stimulation of keratinocytes is known to increase expression of molecules including MHC-II, increasing their capacity to present antigens to infiltrating T cells (63).
Effects on the Skin’s Immune Compartment
In addition to their effects on the epidermis, the IL-1 family cytokines have a significant effect on the immune compartment. Langherans cells (LCs) and dendritic cells (DCs) reside in the epidermis and dermis, respectively, sampling their environment for threats to respond to. LCs express high levels of IL-36R and IL-1R in comparison to blood-derived myeloid cells and respond to stimulation by secreting pro-inflammatory mediators and expressing co-stimulatory receptors. Whilst traditionally IL-1β has been thought of as a crucial cytokine along with TNFα for the maturation and migration of LCs (66), IL-36β has been shown to be equally as potent as IL-1β in stimulating LCs (67). Dendritic cells, on the other hand, are slightly more varied in their expression and response to IL-1β and IL-36 cytokines. CD3a+ DCs express high levels of IL-36R yet have limited expression of IL-1RAcP, and conversely CD14+ DCs express high levels of IL-1RAcP but do not express IL-36R. As a result, dermal DCs are not very responsive to IL-36 stimulation but respond well to IL-1β by similarly expressing pro-inflammatory cytokines and co-stimulatory receptors (67).
Neutrophils and macrophages recruited to the skin during inflammation are also influenced by IL-1 family cytokines. IL-1α, IL-1β and IL-18 have been shown to activate neutrophils, increasing oxidative burst and inducing calcium-dependent degranulation (68, 69). Macrophages react variably to IL-1 family cytokines depending on their differentiation. M0 macrophages are responsive to both IL-1β and IL-36 cytokines, but more responsive to IL-1β than IL-36. M1 macrophages respond to IL-1β but not to IL-36, whilst M2 macrophages are equally responsive to both IL-36 cytokines and IL-1β (67). IL-18, originally termed IFN-gamma (IFN-γ) inducing factor, plays a significant role in the activation of mononuclear cells facilitating improved clearance of both intracellular and extracellular bacterial infections as a result of its induction of IFN-y from multiple cellular sources (70).
Innate lymphoid cells (ILCs) both reside within the dermis and epidermis as tissue-resident cells and are actively recruited to the skin during inflammation. IL-1β has recently been shown to facilitate plasticity in ILCs. ILC2s stimulated with IL-1β begin to express low levels of T-bet and IL-12Rβ2, allowing a phenotypic switch to ILC1s following IL-12 stimulation (71). Similarly, ILC2s treated with IL-1β, TGF-b and IL-23 can adopt an ILC3 phenotype by expressing RORγt and producing IL-17 (72). IL-18 in combination with IL-15 has been shown to be important in the proliferation of ILC3s and can contribute to their role in epithelial barrier function by inducing expression of IL-22 (73).
In addition to their effects on cells of the innate immune system, IL-1 family cytokines significantly influence adaptive immunity, both directly and indirectly. IL-1β is well established to potentiate production of IFN-γ and act as a driver of TH17 differentiation through direct stimulation (74). Recently, IL-1β has also been shown to promote TH17 differentiation indirectly via activation of DCs. IL-1β-primed DCs increased expression of CD14 and induced RORγt expression and IL-17 production in CD4 memory T cells in a CD14 dependent manner (75). Whilst murine T cells are well established as expressing IL-36R and are directly responsive to IL-36 stimulation, the sensitivity of human T cells to IL-36 cytokines is less clear. Early research could not demonstrate a direct effect of IL-36 stimulation on human T cells, however a later report by Penha et al. show expression of IL-36R in blood- and intestinal-derived T cells and dose-dependent CD4 T cell expansion following stimulation with IL-36β (62, 67, 76). IL-36 cytokines have also similarly been shown to promote CD4 T cell proliferation through their activation of DCs, other antigen presenting cells, and the induction of polarising cytokines (62, 77, 78). Interestingly, in mice IL-36γ has been shown to prevent the development of Treg cells, instead promoting development of TH9 effector cells (79).
Unlike the other IL-1 family members, IL-33’s influence on the immune compartment drives Type II immunity and is strongly linked with promoting TH2-driven diseases such as AD. IL-33 has been shown to induce the expression of CCL17 from AD-derived keratinocytes; a chemokine responsible for attracting CCR4 positive cells associated with Type II immune responses (80). Mast cells undergo activation and maturation following IL-33 stimulation and become more responsive to IgE and IgG. These cells also respond to both IL-18 and IL-33 by producing Type II polarising cytokines such as TSLP, IL-4, and IL-13 (81–83) and therefore promote TH2 development. TH2 cells themselves are also known to express ST2 and respond directly to IL-33 stimulation by producing IL-5 and IL-13 (84, 85).
In addition to driving TH2 development, IL-33 has been well described as a driver of ILC2 proliferation (86). ILC2s are thought to play a prominent role in the pathogenesis of AD. Their numbers are found at increased levels in the skin of AD patients when compared to that of psoriasis patients (87). Indeed mouse models of AD induced by transgenic IL-33 expression can be effectively treated by the specific depletion of ILC2s (88). Despite their role in promoting pathological Type II inflammation, IL-33 and ILC2s are also associated with protection. Numerous publications demonstrate their importance for protection against gut tissue damage and gastro-intestinal infections (89–91), and similarly has been shown as promoting ILC2-mediated wound healing in skin (92).
Resolving and Regulating IL-1 Family Inflammation
All IL-1 family members are tightly controlled (93) on multiple levels including RNA stability, translation, secretion and activation by proteases as well as soluble receptors, receptor antagonists and binding proteins (Table 1). This significant level of control may also illustrate the powerful pro-inflammatory properties of IL-1 family members and disturbance in any of these mechanisms can lead to auto-inflammatory (sometimes clinically dramatic) disease manifestations.

Table 1 Table showing the regulatory mechanisms controlling IL-1 family mediated signalling.
There is limited knowledge about how counter-regulatory IL-1 family mechanisms are activated and regulated. While counter-regulatory activity happens on the level of receptor expression, soluble receptor release, signalling molecule stability/ubiquitination, translocation, activation and mRNA stability, here we highlight protein antagonists for each IL-1 family member. The cutaneous dynamics of all counter-regulatory mechanisms have not been well investigated and as such no complete picture is available for the IL-1 family relevant resolution phase of inflammatory epithelial responses in vivo. For blood derived monocytes, persistent versus resolving inflammation experimental setups have been investigated (96) and showed that IL-1β, IL-1Ra and soluble receptors are all released between 4 and 14h during the course of both resolving and persistent inflammation in vitro.
IL-1Ra is the first described endogenous receptor antagonist. Different variants, including an intracellular form lacking a leader sequence (97), have been described which add to its complex biology. In the skin, IL-1Ra is produced abundantly by keratinocytes, infiltrating neutrophils and infiltrating or resident antigen presenting cells. IL-1Ra binds to the IL-1R1 but fails to induce recruitment of the co-receptor IL-1R3 (aka IL-1RAcP), thus preventing signal transduction via the IL-1R. This is how it works as a competitive antagonist for both IL-1α and IL-1β. IL-1R type 2 (CD121b) is a decoy receptor which binds to IL-1α and IL-1β with high affinity and IL-1RA with lower affinity. While IL-1R2 interacts with IL-1R3 this interaction does not result in signal transduction. In addition to its membrane bound form IL-1R2 has also been found as a soluble protein either as a result of shedding or alternative splicing and has been shown to be active in the cytosol binding to IL-1α and preventing its cleavage and activation. Similarly, soluble receptors have been described for IL-33, sequestering extracellular IL-33 and limiting subsequent signalling (98). All IL-1α/β relevant molecules mentioned, including the IL-1RA and type 2 IL-1R are expressed by keratinocytes (99). The IL-36 cytokines are also regulated through a receptor antagonist, IL-36Ra. IL-36Ra functions in an analogous manner to IL-1Ra, binding its cognate receptor IL-36R without recruiting the accessory protein IL-1RAcP, thus preventing signal transduction.
IL-38 is another anti-inflammatory cytokine belonging to the IL-36 sub-family. Whilst this cytokine is thought to be an important regulator of IL-36 dependent inflammation and has been shown to be a general regulator of IL-1-family mediated inflammation, its precise mechanism of action is not wholly understood. There is evidence to suggest it may directly antagonise IL-36R like IL-36Ra, however this has not yet been conclusively demonstrated. Nevertheless, it has been shown to inhibit IL-36γ-induced NF-kB activity in keratinocytes and seems to be important in regulating inflammation in psoriasis. Its expression is lower in psoriatic skin than in healthy skin and increases following anti-IL-17A treatment in a manner that positively correlates with therapeutic efficacy (100).
While those cells producing IL-1α/β also harbour the full repertoire of antagonistic molecules the same is not necessarily true for the IL-18 system. Here keratinocytes are recognised producers of IL-18 as of relevance in psoriasis, cutaneous lupus and chronic eczema, however a main source of IL-18BP in the skin organ are fibroblasts (101) which are not known to secrete IL-18. Yet it has recently been shown that the cleaved pro-peptide from IL-18 can interact with mature IL-18, potentially abrogating its ability to engage with its receptor, highlighting an innate method of limiting IL-18 signalling without requiring expression of IL-18BP (102).
It is of interest that another anti-inflammatory molecule impacting on IL-1 family members, IL-37 (103), uses the IL-18R in addition to SIGIRR (IL-1R8) for its downstream effects (104). IL-1R8 interacts with IRAK and TRAF6 thus competing with IL-1 or TLR activated signalling. As previously mentioned, Gudjonsson et al. have clearly shown that the signalling events induced by IL-36 are mostly identical to IL-1 induced signalling (58). The difference in their pathophysiology relies on the compartment where they are expressed and events leading to cleavage of pro-forms into bioactive molecules. Regarding IL-1R8, it can attenuate inflammatory responses induced by IL-1, IL-18, IL-33, IL-36, and TLR. A recent study in SIGIRR-/- mice demonstrates its importance in regulating IL-36-mediated inflammation (105). The anti-inflammatory molecule IL-37 can also bind to IL-18BP and seems to enhance its effects. IL-37 along with all IL-36 members and the IL-36 receptor antagonist IL-36Ra are all expressed by human keratinocytes.
Studies examining the regulation of IL-33 have revealed some interesting mechanisms of limiting IL-1 cytokine activity. Whilst proteolytic regulation of the IL-1 family cytokines is often associated with their activation, it has been demonstrated that IL-33, already active in its native form, is inactivated following truncation by caspase-3 and caspase-7 during apoptosis (106, 107). Interestingly, this inhibition is mediated by truncation of a specific amino acid sequence that is absent in all other IL-1 family cytokines, suggesting specific regulation of IL-33 during apoptosis (106). Intriguingly IL-33 has recently been demonstrated to exhibit an intrinsic method of limiting prolonged activity. IL-33 quickly loses its biological function following release into the extracellular environment as oxidation of critical cysteine residues in IL-33 causes a conformational switch to an inactive form (108). As other IL-1 family cytokines also contain free cysteine residues it is possible other IL-1 family members are regulated in a similar manner. Indeed Cohen et al. demonstrate IL-1α, IL-18, IL-36α and IL-36γ all exhibit similar oxidative changes when exposed to an oxidative environment (108).
Within the skin compartment much inflammatory activity related to the IL-1 family is based in the epidermis. As mentioned, dermal resident cells such as fibroblasts are less able to produce IL-1 family agonists but contribute to antagonist secretion including IL-18BP and IL-1Ra. Thus, infiltrating macrophages aside, the overall property of the dermal versus epidermal compartment is more inflammation limiting than pro-inflammatory with regard to IL-1 family molecules.
The importance of both adequate regulation and resolution of inflammation by the IL-1 family cytokines is most clearly demonstrated when examining monogenic mutations that directly affect the regulation of IL-1 family cytokines (summarized in Figure 3). This is most striking in conditions that result from the loss of the IL-1 family antagonists. Deficiency of the IL-1 Receptor Antagonist (DIRA) is an autosomal recessive disease caused by mutations of IL1RN that results in non-expression of the IL-1 receptor antagonist and subsequent unopposed IL-1 receptor activation by IL-1α and IL-1β (109). DIRA is most commonly associated with a pustular rash, joint swelling and periostitis. DIRA has been successfully treated with the anti-IL-1 biologics Anakinra (recombinant IL-1Ra) (110, 111), Canakinumab (112) (IL-1β blocking antibody) and Rilonacept (fusion protein of IL-1 receptor and IgG Fc) (Figure 4) (113).
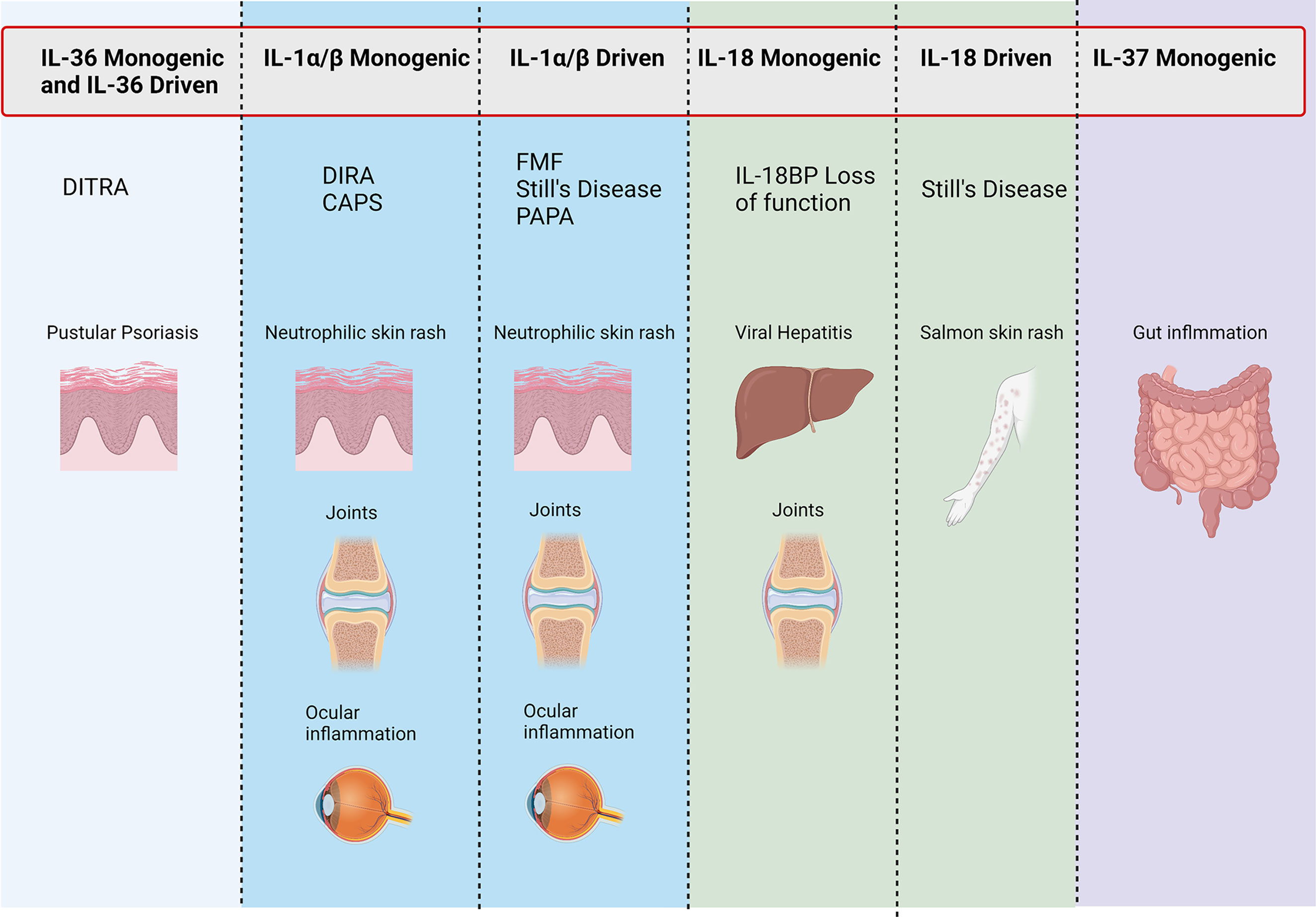
Figure 3 Conditions associated with IL-1 family genetic mutations. Figure outlining symptoms associated with disease-causing mutations in monogenic and IL-1 family driven conditions.
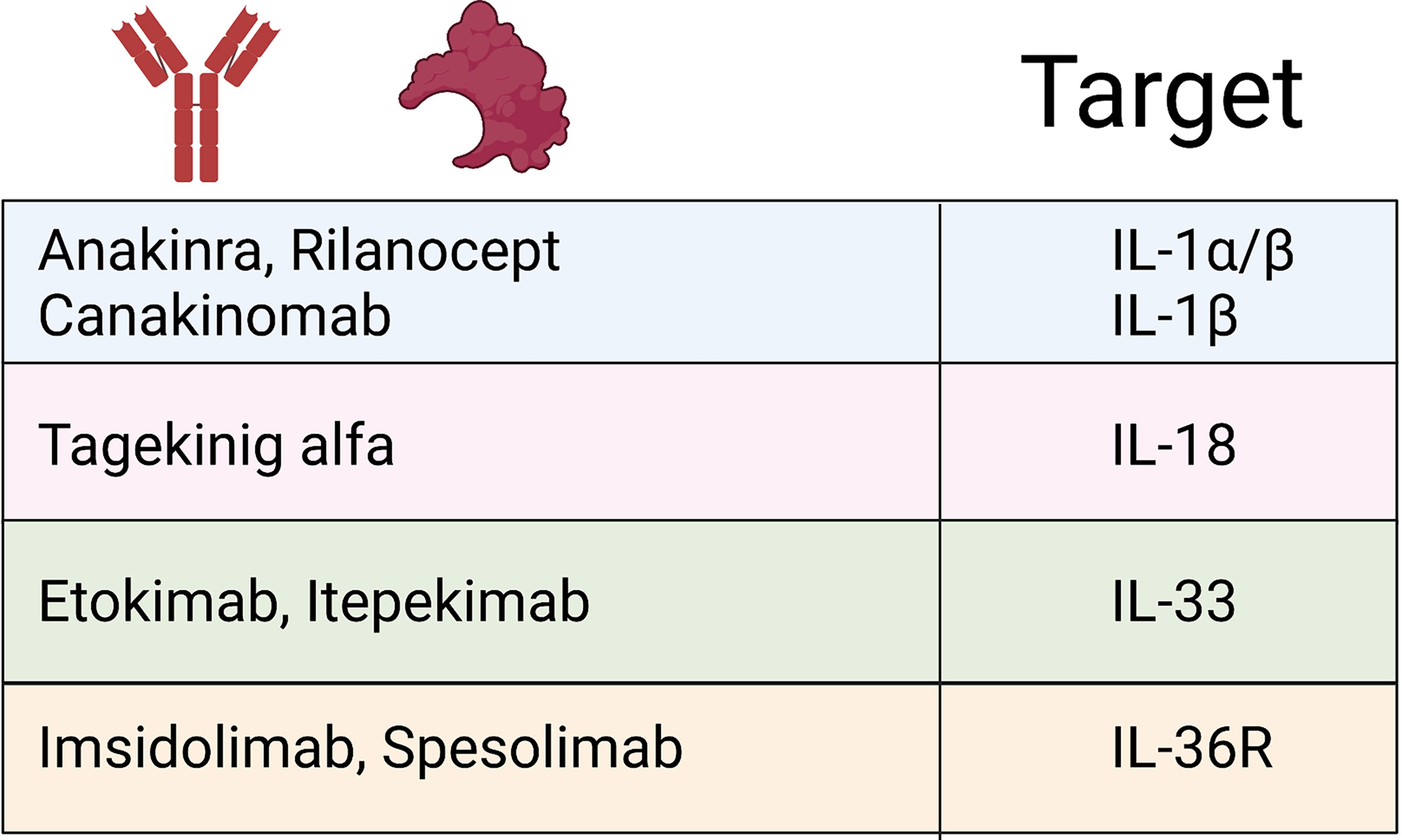
Figure 4 IL-1 family biologics. Schematic outlining current FDA-approved biologics targeting IL-1 family cytokines and signalling pathways that have demonstrated efficacy treating IL-1 cytokine-driven conditions.
Similarly, loss of function mutations in IL36RN, which encodes IL-36Ra, leads to the development of DITRA (Deficiency of interleukin-36 receptor antagonist) in which unchecked IL-36 signalling causes significant systemic inflammation (114). In addition to the immediate inflammatory consequence of enhanced IL-36 signalling, the lack of IL-36-specific inhibition has been shown to promote extensive antigen-dependent expansion of TH17 cells and subsequent production of IL-17A, promoting T cell-mediated inflammation (115). Affected patients develop GPP psoriasis with a systemic inflammation and fever. GPP has successfully been treated with anti-IL-36R therapy (Figure 4) (116). If left untreated, both DIRA and DITRA can prove fatal, demonstrating the importance of proper regulation of IL-1 and IL-36 signalling (117, 118).
Numerous genetic defects that occur in the inflammasome-activation pathway demonstrate the importance of properly regulated inflammasome activation. Examples of both gain of function mutations in inflammasome activation and loss of function mutations in inflammasome regulation have been well reported. Cryopyrin-Associated Periodic Syndrome (CAPS) represents autosomal dominant gain of function mutations that results in over activation of the inflammasome and increased conversion of pro-caspase-1 to activate caspase-1 and this results in increased IL-1β and IL-18 protein levels (119, 120). These conditions include Familial Cold Autoinflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS) caused by mutations in the CIAS1 gene (NLRP3) which encodes cryopyrin (121–123). CINCA (chronic infantile neurologic, cutaneous and arthritis syndrome) was later revealed to be caused by a spontaneous mutation in the same gene and called, Neonatal Onset Multisystem Inflammatory Disease (NOMID) (124, 125). Recent publications have also identified gain of function mutations in NLRC4 resulting in similar aberrant activation of the NLRC4 inflammasome (126). Amongst severe systemic symptoms such as macrophage activation syndrome and periodic fever, patients also exhibit severe urticarial rashes that are marked by significant neutrophil infiltrates. Interestingly, patients with NLRC4 mutations exhibit higher levels of IL-1β and IL-18 than those with NLRP3 mutations, and have been shown to respond well to IL-18BP treatment (127–129). CAPS patients respond well to IL-1 blockade, with Anakinra, Rilonacept and Canakinumab all showing efficacy (130–135).
FMF (Familial Mediterranean Fever) results from a breakdown in regulation of inflammasome activity caused by mutations in the MEFV gene which encodes pyrin (136, 137). The precise role of pyrin in the pathogenesis of FMF is heavily debated. Pyrin interacts with ASC, a component of the inflammasome and sequesters ASC, making unable to form an inflammasome (138, 139). Wildtype pyrin has also been demonstrated to bind to caspase-1 and inhibit IL-1β secretion (138, 140). FMF is commonly treated with colchicine, however, there are reports of IL-1 blockade being successful in the form of Anakinra and Canakinumab (141, 142).
Whilst not common, there are also examples of IL-37 and IL-18BP loss of function giving rise to inflammatory disease. An infant with non-functional IL-37 has been identified to suffer inflammatory bowel disease, however, as yet shows no sign of skin manifestations. The loss of functional IL-18BP resulted in a case of fulminant viral hepatitis and the death of the patient (143, 144).
The IL-1 family cytokines are also well known to have central roles in complex multifactorial inflammatory conditions such as psoriasis, ichthyosis conditions, hidradenitis suppurativa (HS) and AD. As these topics have been extensively reviewed elsewhere (93, 145, 146) this review shall highlight recent examples of inflammatory conditions involving IL-1 family cytokines.
Systemic-onset juvenile idiopathic arthritis (sJIA) and adult-onset Still’s disease (AoSD) are rare systemic auto-inflammatory polygenic disorders, which are accompanied by a broad spectrum of manifestations. The diseases, considered part of a spectrum continuum, are associated with fever, arthritis and skin rashes that are salmon pink in appearance. Skin biopsies show neutrophilic inflammation (147). Both diseases considered can significantly overlap with macrophage activation syndrome (MAS) and feature a cytokine storm. The causes of these entities and immunogenetics are complex and are discussed elsewhere (148). However, a central mechanism appears to be the dysregulated production of IFN-y, IL-1, IL-6 and IL-18, the latter driving IFN-y (148). Targeting of IL-1α/β (Anakinra (149) or Canakinumab (150), as well as recombinant IL-18BP (151) (tadekinig alfa) has shown some success. A hair follicle disease hidradenitis suppurativa (HS), which is dominated by pathological neutrophils, has recently been shown to significantly involve the IL-1 and IL-36 pathways in driving its proinflammatory pathways (152, 153) IL-1β-driven transcription present in HS lesions was found to induce immune cell infiltration to lesions and extracellular matrix degradation (152). IL-36 cytokines were also found to be significantly upregulated, and their regulatory counterparts downregulated, in lesional HS (153).
IL-1 Family Cytokines in the Promotion of Wound Healing
Wound healing is essential in maintaining the skin integrity and barrier function. It is a complex process, defined by four consecutive but overlapping stages, starting with haemostasis, followed by inflammation, proliferation, and finally tissue remodelling (154, 155). Imbalance in any of these stages can lead to delayed or impaired wound healing. IL-1 family members are known to play a key role in the wound healing process. IL-1α and IL-1β levels are immediately increased following tissue injury, reaching their peak in the first 12 – 24h, underscoring their alarmin function, and return to normal once the proliferation stage of wound healing is complete (156). Interestingly, reinforcing the importance of proteolytic regulation of IL-1 family cytokines, thrombin has recently been demonstrated to truncate IL-1α to a more potent form following its activation in the coagulation cascade (157). Indeed, a mouse model expressing IL-1α that cannot be truncated by thrombin exhibits delayed wound closure (157).
As already alluded to, IL-1 cytokines promote the initial neutrophil recruitment to the site of injury, facilitating the removal of debris and the prevention of a potential bacterial infection (158). Similarly to TNFα, both IL-1α and IL-1β facilitate wound healing by inducing fibroblast and keratinocyte proliferation, the production and degradation of extracellular matrix (ECM) proteins (collagens, elastins, fibronectins and laminins), fibroblast chemotaxis, and by exerting immune modulatory functions (156). The majority of these effects are obtained via the production of various growth factors by IL-1 stimulated cells. In vitro experiments showed that IL-1 produced by human keratinocytes stimulate the production of transforming growth factor alpha (TGF-α) in an autocrine fashion (159). TGF-α increases keratinocyte migration (160) and proliferation (161–163) as well as their expression of K6 and K16, markers of a hyperproliferative state (164). Furthermore, keratinocyte derived IL-1 stimulates fibroblasts to produce other growth factors with important roles in keratinocyte proliferation, including keratinocyte growth factor (KGF) (165–167) and granulocyte–macrophage colony-stimulating factor (GM-CSF), creating a double paracrine loop (168, 169). In addition, IL-1 indirectly induces dermal fibroblast and arterial smooth muscle cell proliferation by stimulating their production of platelet-derived growth factor (PDGF) AA (170). The newly forming tissue during wound repair has a high oxygen demand, therefore, neovascularisation is an essential part of normal wound healing. IL-1β has been shown to induce vascular endothelial growth factor (VEGF) production by keratinocytes, a key factor in neovascularisation, although to a lesser extent compared to TNFα (171, 172). Interestingly, the bacterial microbiome has been shown to be important in promoting wound healing by inducing IL-1α and IL-1β expression during wounding. A recent study has shown a low bacterial quantity and diversity in mice skin correlates with poor wound-induced hair follicle neogenesis (WIHN), and that increasing bacterial counts, even pathological S. aureus, improves WIHN (173). This microbiome-induced promotion of skin regeneration was shown to be dependent on keratinocyte-specific IL-1β signalling. Furthermore, in a small human trial the use of topical antibiotics prior to wounding was demonstrated to prolong the healing process (173).
The various growth factors resulting from the keratinocyte-fibroblast crosstalk are essential in the proliferation of the epithelial stem cell pool (174). Epithelial stem cell mobilisation is crucial in the proliferative phase of wound healing. Progenitor cells derived from interfollicular epidermal and hair follicle stem cells are mobilised to rapidly restore skin integrity (175, 176). However, the signals that trigger the activation of these stem cells proximal to the wound are not fully understood. Recent work conducted in mice indicates the synergistic involvement of IL-1α and IL-7, secreted by keratinocytes, in the expansion of epidermal γδ T cells (177). γδ T cells are resident epidermal cells, which upon activation secrete KGF and induce the proliferation of epidermal stem cells (178, 179). Moreover, it has been shown that in mice IL-1β is indispensable in the recall of inflammatory memory of epidermal stem cells, facilitating wound healing following repeated insults (180).
Interestingly, IL-1β and IL-18 have recently been implicated in the activation of cutaneous mucosal associated invariant T (MAIT) cells following their interaction with local microbiota, which results in the promotion of wound healing. Following interaction with S. epidermidis in human epidermis, cutaneous MAIT cells have been shown to expand, produce IL-17A and exhibit a transcriptional profile consistent with leukocyte activation and tissue repair in a manner dependent on IL-1 and IL-18 signalling (181). Local IL-1α signalling was shown to be crucial for MAIT cell IL-17A production, whilst IL-18 was required for their expansion. As previously mentioned, the presence of S. epidermidis allows the epidermis to maintain a level of IL-1α expression, thus is primed for allowing IL-1 signalling following tissue damage, promoting MAIT cell activation and subsequent tissue repair. IL-1 family cytokines have also been implicated in promoting wound healing through their impact on commensal-specific tissue resident T cells (TRM). A recent study identified S. epidermidis-specific CD8+ TC17 cells to co-express GATA3 and constitutively express a Type II transcriptome, including IL5 and IL13. Despite their expression of the Type II transcriptome, protein synthesis was only licenced following stimulation by tissue damage-associated alarmins (182). Stimulation of S. epidermidis-specific TC17 cells with IL-1α, IL-33 and IL-18 in conjunction with T cell receptor activation induced production of IL-13, which has been shown to promote wound healing (183). Indeed, the authors demonstrated these Tc17 cells were able to recover S. epidermidis-accelerated wound healing in IL-13 deficient mice (182).
Less is known on the role of other IL-1 family members in wound healing apart for an important role for IL-33 that is upregulated following cutaneous wounding in mice, peaking 5 days post-injury (92). Unlike IL-1β, IL-33 promotes reepithelization by facilitating the shift from a pro-inflammatory to an anti-inflammatory macrophage profile and stimulates ECM production in both normal (184) and diabetic mice (185). Furthermore, it enhances neoangiogenesis via the production of VEGF and von Willebrand factor (185). IL-33-sensitive ILC2 cells have also been shown to play a role in the wound healing process. Rak et al. identified an increase in ST2+ ILC2 numbers at sites of healing wounds in both human and mouse cutaneous tissue. The authors demonstrated that IL-33 knockout mice exhibited significantly smaller numbers of activated ILC2s at wound sites and were significantly slower in closing wounds (92). Oshio et al. also demonstrate IL-33 knockout mice exhibit slower wound closure, and identify the importance of nuclear IL-33 in limiting excessive NF-kB-mediated inflammation (186). IL-36 cytokines have also been implicated in wound healing as a result of injury. Jiang et al. demonstrate that release of TLR3 agonists following tissue damage induces expression of IL-36γ, which in turn induces expression of REG3A in keratinocytes to promote re-epithelialization and wound healing (187).
A wound healing response does not always have a beneficial outcome. Tissue trauma and the subsequent wound healing response can contribute to resurgence of dormant tumours under certain circumstances. The production of growth factors, increase in inflammatory mediators and promotion of re-epithelialisation and proliferation have all been implicated in promoting metastasis and tumour dormancy escape (188). Furthermore, inflammation induced in wound healing does not always resolve completely, and can in some cases lead to the development of chronic wounds (189). Indeed, whilst IL-1 family cytokines are important in the wound healing process, their dysregulation can contribute to pathological outcomes. IL-1 family members have been found to play roles in such inflammatory override, with an essential role being attributed to IL-1β (190). It has been shown that non-microbial danger signals resulting from hyperglycaemia and obesity can lead to inflammasome activation and release of IL-1β and IL-18 from a number of different cell types, including monocytes/macrophages, known to be involved in all stages of wound healing (191–196). IL-1β then sustains the inflammasome activity of macrophages, creating a positive feedback loop (197). Moreover, a recent study in diabetic mice showed that beside maintaining an inflammatory macrophage profile, increased levels of IL-1β also result in the persistence of inflammatory cells at the wound site, senescence of fibroblasts and high levels of matrix metalloproteinases (190), which in turn degrade growth factors and extracellular matrix proteins with an important role in wound healing.
Murine models showed that topical inhibition of caspase 1, the inflammasome, IL-1β or its signalling through IL-1R1, results in the switch from a sustained pro-inflammatory macrophage stage to an anti-inflammatory macrophage profile, reduced levels of proinflammatory cytokines (IL-1β, TNFα, IL-6, CXCL1), chemokines (the murine homologue of IL-8, MIP-1α), and matrix metalloproteases, but increased the levels of anti-inflammatory cytokines (TGF-β1, IL-4, IL-10), tissue inhibitor of metallopeptidase-1 (TIMP-1) and growth factors (FGF-2, PDGF-BB, VEGF-A), facilitating effective wound healing (190, 197–200). Treatment with IL-1RA has been also shown to reduce postoperative wound pain due to the reduced inflammatory response (200). These studies highlight IL-1β as the biggest culprit in the maintenance of chronic, non-healing wounds in diabetic patients, suggesting the inflammasome as a potential therapeutic target in the treatment of non-healing diabetic wounds.
Whilst IL-36 signalling has been shown to have some beneficial roles in the wound healing process, excessive signalling also seems detrimental. Studies performed in mice lacking expression of IL-36Ra demonstrated these mice experienced a 3-day delay in wound closure as a result. Similar to IL-1β, the delay in wound closure was associated with an increased influx of inflammatory cells such as neutrophils and macrophages and increased expression of inflammatory cytokines. Interestingly, normal wound healing was restored in this mouse model by inhibition of TLR4, which the authors attributed to the inhibition of hyaluronic acid-mediated activation of TLR4 and subsequent induction of IL-36 signalling (201).
Concluding Remarks
This review has provided an overview of the role IL-1 family cytokines play in maintaining the skin’s immunological barrier emphasising recent findings that contribute to our understanding. As has been outlined above, IL-1 family cytokines significantly impact on all stages of an immune response in skin, from sensing threats and alerting surrounding tissue to danger, to facilitating the closure of a wound and restoring the skin’s barrier function. However, IL-1 family cytokines are potent cytokines that require tight regulation and balanced signalling. As has been highlighted, a breakdown in adequate regulation often contributes to, if not causes, disease. The dynamic nature of their regulation, particularly at the post-translational level via proteolysis, makes understanding their inflammatory outcome in a given biological system difficult. Yet, given the significant impact they have on almost every aspect of immune function in skin, it is important to consider.
Author Contributions
TM prepared and wrote the manuscript. AB, CB, and MW contributed to the content of the review. MS, DM and MW directed focus. All authors listed contributed intellectually and approved the manuscript for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All figures were created with BioRender.com
References
1. Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity (2013) 39(6):1003–18. doi: 10.1016/j.immuni.2013.11.010
2. Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, et al. Interleukin-36 (IL-36) Ligands Require Processing for Full Agonist (IL-36alpha, IL-36beta, and IL-36gamma) or Antagonist (IL-36Ra) Activity. J Biol Chem (2011) 286(49):42594–602. doi: 10.1074/jbc.M111.267922
3. Ainscough JS, Macleod T, McGonagle D, Brakefield R, Baron JM, Alase A, et al. Cathepsin S is the Major Activator of the Psoriasis-Associated Proinflammatory Cytokine IL-36gamma. Proc Natl Acad Sci USA (2017) 114(13):E2748–57. doi: 10.1073/pnas.1620954114
4. Narayanan KB, Park HH. Toll/interleukin-1 Receptor (TIR) Domain-Mediated Cellular Signaling Pathways. Apoptosis (2015) 20(2):196–209. doi: 10.1007/s10495-014-1073-1
5. Sims JE, Smith DE. The IL-1 Family: Regulators of Immunity. Nat Rev Immunol (2010) 10(2):89–102. doi: 10.1038/nri2691
6. Fields JK, Gunther S, Sundberg EJ. Structural Basis of IL-1 Family Cytokine Signaling. Front Immunol (2019) 10:1412. doi: 10.3389/fimmu.2019.01412
7. Di Paolo NC, Shayakhmetov DM. Interleukin 1alpha and the Inflammatory Process. Nat Immunol (2016) 17(8):906–13. doi: 10.1038/ni.3503
8. Werman A, Werman-Venkert R, White R, Lee JK, Werman B, Krelin Y, et al. The Precursor Form of IL-1alpha is an Intracrine Proinflammatory Activator of Transcription. Proc Natl Acad Sci USA (2004) 101(8):2434–9. doi: 10.1073/pnas.0308705101
9. Watanabe H, Gaide O, Petrilli V, Martinon F, Contassot E, Roques S, et al. Activation of the IL-1beta-Processing Inflammasome is Involved in Contact Hypersensitivity. J Invest Dermatol (2007) 127(8):1956–63. doi: 10.1038/sj.jid.5700819
10. Tardif F, Ross G, Rouabhia M. Gingival and Dermal Fibroblasts Produce Interleukin-1 Beta Converting Enzyme and Interleukin-1 Beta But Not Interleukin-18 Even After Stimulation With Lipopolysaccharide. J Cell Physiol (2004) 198(1):125–32. doi: 10.1002/jcp.10400
11. Sundnes O, Pietka W, Loos T, Sponheim J, Rankin AL, Pflanz S, et al. Epidermal Expression and Regulation of Interleukin-33 During Homeostasis and Inflammation: Strong Species Differences. J Invest Dermatol (2015) 135(7):1771–80. doi: 10.1038/jid.2015.85
12. Companjen AR, van der Velden VH, Vooys A, Debets R, Benner R, Prens EP. Human Keratinocytes are Major Producers of IL-18: Predominant Expression of the Unprocessed Form. Eur Cytokine Netw (2000) 11(3):383–90.
13. Boraschi D, Lucchesi D, Hainzl S, Leitner M, Maier E, Mangelberger D, et al. IL-37: A New Anti-Inflammatory Cytokine of the IL-1 Family. Eur Cytokine Netw (2011) 22(3):127–47. doi: 10.1684/ecn.2011.0288
14. Berekmeri A, Latzko A, Alase A, Macleod T, Ainscough JS, Laws P, et al. Detection of IL-36gamma Through Noninvasive Tape Stripping Reliably Discriminates Psoriasis From Atopic Eczema. J Allergy Clin Immunol (2018) 142(3):988–91.e4. doi: 10.1016/j.jaci.2018.04.031
15. Macleod T, Ainscough JS, Hesse C, Konzok S, Braun A, Buhl AL, et al. The Proinflammatory Cytokine IL-36gamma Is a Global Discriminator of Harmless Microbes and Invasive Pathogens Within Epithelial Tissues. Cell Rep (2020) 33(11):108515. doi: 10.1016/j.celrep.2020.108515
16. Johnston A, Xing X, Wolterink L, Barnes DH, Yin Z, Reingold L, et al. IL-1 and IL-36 are Dominant Cytokines in Generalized Pustular Psoriasis. J Allergy Clin Immunol (2017) 140(1):109–20. doi: 10.1016/j.jaci.2016.08.056
17. Gabay C, Towne JE. Regulation and Function of Interleukin-36 Cytokines in Homeostasis and Pathological Conditions. J Leukoc Biol (2015) 97(4):645–52. doi: 10.1189/jlb.3RI1014-495R
18. Bessa J, Meyer CA, de Vera Mudry MC, Schlicht S, Smith SH, Iglesias A, et al. Altered Subcellular Localization of IL-33 Leads to non-Resolving Lethal Inflammation. J Autoimmun (2014) 55:33–41. doi: 10.1016/j.jaut.2014.02.012
19. Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity (2015) 42(6):991–1004. doi: 10.1016/j.immuni.2015.06.003
20. Zheng D, Liwinski T, Elinav E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov (2020) 6:36. doi: 10.1038/s41421-020-0167-x
21. Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, et al. The Interleukin-1alpha Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front Immunol (2013) 4:391. doi: 10.3389/fimmu.2013.00391
22. Bronneberg D, Spiekstra SW, Cornelissen LH, Oomens CW, Gibbs S, Baaijens FP, et al. Cytokine and Chemokine Release Upon Prolonged Mechanical Loading of the Epidermis. Exp Dermatol (2007) 16(7):567–73. doi: 10.1111/j.1600-0625.2007.00566.x
23. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an Executor of Pyroptosis and Required for Interleukin-1beta Secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
24. Puren AJ, Fantuzzi G, Dinarello CA. Gene Expression, Synthesis, and Secretion of Interleukin 18 and Interleukin 1beta are Differentially Regulated in Human Blood Mononuclear Cells and Mouse Spleen Cells. Proc Natl Acad Sci USA (1999) 96(5):2256–61. doi: 10.1073/pnas.96.5.2256
25. Baroja-Mazo A, Martin-Sanchez AF, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 Inflammasome is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat Immunol (2014) 15(8):738–48. doi: 10.1038/ni.2919
26. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A Wave of Regulatory T Cells Into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity (2015) 43(5):1011–21. doi: 10.1016/j.immuni.2015.10.016
27. Leech JM, Dhariwala MO, Lowe MM, Chu K, Merana GR, Cornuot C, et al. Toxin-Triggered Interleukin-1 Receptor Signaling Enables Early-Life Discrimination of Pathogenic Versus Commensal Skin Bacteria. Cell Host Microbe (2019) 26(6):795–809 e5. doi: 10.1016/j.chom.2019.10.007
28. Martin U, Scholler J, Gurgel J, Renshaw B, Sims JE, Gabel CA. Externalization of the Leaderless Cytokine IL-1F6 Occurs in Response to Lipopolysaccharide/ATP Activation of Transduced Bone Marrow Macrophages. J Immunol (2009) 183(6):4021–30. doi: 10.4049/jimmunol.0803301
29. Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, et al. Staphylococcus Aureus Virulent PSMalpha Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe (2017) 22(5):667–77.e5. doi: 10.1016/j.chom.2017.10.008
30. Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, et al. Staphylococcus Aureus Epicutaneous Exposure Drives Skin Inflammation via IL-36-Mediated T Cell Responses. Cell Host Microbe (2017) 22(5):653–66.e5. doi: 10.1016/j.chom.2017.10.006
31. LaRock CN, Todd J, LaRock DL, Olsen J, O’Donoghue AJ, Robertson AAB, et al. IL-1β is an Innate Immune Sensor of Microbial Proteolysis. Sci Immunol (2016) 1(2):eaah3539. doi: 10.1126/sciimmunol.aah3539
32. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen Blockade of TAK1 Triggers Caspase-8-Dependent Cleavage of Gasdermin D and Cell Death. Science (2018) 362(6418):1064–9. doi: 10.1126/science.aau2818
33. Chen KW, Demarco B, Ramos S, Heilig R, Goris M, Grayczyk JP, et al. RIPK1 Activates Distinct Gasdermins in Macrophages and Neutrophils Upon Pathogen Blockade of Innate Immune Signaling. Proc Natl Acad Sci USA (2021) 118(28):e2101189118. doi: 10.1073/pnas.2101189118
34. Seltmann J, Roesner LM, von Hesler FW, Wittmann M, Werfel T. IL-33 Impacts on the Skin Barrier by Downregulating the Expression of Filaggrin. J Allergy Clin Immunol (2015) 135(6):1659–61.e4. doi: 10.1016/j.jaci.2015.01.048
35. Alase A, Seltmann J, Werfel T, Wittmann M. Interleukin-33 Modulates the Expression of Human Beta-Defensin 2 in Human Primary Keratinocytes and may Influence the Susceptibility to Bacterial Superinfection in Acute Atopic Dermatitis. Br J Dermatol (2012) 167(6):1386–9. doi: 10.1111/j.1365-2133.2012.11140.x
36. Pietka W, Khnykin D, Bertelsen V, Lossius AH, Stav-Noraas TE, Hol Fosse J, et al. Hypo-Osmotic Stress Drives IL-33 Production in Human Keratinocytes-An Epidermal Homeostatic Response. J Invest Dermatol (2019) 139(1):81–90. doi: 10.1016/j.jid.2018.07.023
37. Kippenberger S, Loitsch S, Guschel M, Muller J, Kaufmann R, Bernd A. Hypotonic Stress Induces E-Cadherin Expression in Cultured Human Keratinocytes. FEBS Lett (2005) 579(1):207–14. doi: 10.1016/j.febslet.2004.11.077
38. Rademacher F, Simanski M, Hesse B, Dombrowsky G, Vent N, Glaser R, et al. Staphylococcus Epidermidis Activates Aryl Hydrocarbon Receptor Signaling in Human Keratinocytes: Implications for Cutaneous Defense. J Innate Immun (2019) 11(2):125–35. doi: 10.1159/000492162
39. Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental Allergens Induce Allergic Inflammation Through Proteolytic Maturation of IL-33. Nat Immunol (2018) 19(4):375–85. doi: 10.1038/s41590-018-0067-5
40. Haraldsen G, Balogh J, Pollheimer J, Sponheim J, Kuchler AM. Interleukin-33 - Cytokine of Dual Function or Novel Alarmin? Trends Immunol (2009) 30(5):227–33. doi: 10.1016/j.it.2009.03.003
41. Satoh TK, Mellett M, Meier-Schiesser B, Fenini G, Otsuka A, Beer HD, et al. IL-36gamma Drives Skin Toxicity Induced by EGFR/MEK Inhibition and Commensal Cutibacterium Acnes. J Clin Invest (2020) 130(3):1417–30. doi: 10.1172/JCI128678
42. Telfer NR, Chalmers RJ, Whale K, Colman G. The Role of Streptococcal Infection in the Initiation of Guttate Psoriasis. Arch Dermatol (1992) 128(1):39–42. doi: 10.1001/archderm.128.1.39
43. Thorleifsdottir RH, Sigurdardottir SL, Sigurgeirsson B, Olafsson JH, Sigurdsson MI, Petersen H, et al. Patient-Reported Outcomes and Clinical Response in Patients With Moderate-To-Severe Plaque Psoriasis Treated With Tonsillectomy: A Randomized Controlled Trial. Acta Derm Venereol (2017) 97(3):340–5. doi: 10.2340/00015555-2562
44. Barbieux C, Bonnet des Claustres M, Fahrner M, Petrova E, Tsoi LC, Gouin O, et al. Netherton Syndrome Subtypes Share IL-17/IL-36 Signature With Distinct IFN-Alpha and Allergic Responses. J Allergy Clin Immunol (2021). doi: 10.1016/j.jaci.2021.08.024
45. Williams MR, Cau L, Wang Y, Kaul D, Sanford JA, Zaramela LS, et al. Interplay of Staphylococcal and Host Proteases Promotes Skin Barrier Disruption in Netherton Syndrome. Cell Rep (2020) 30(9):2923–33.e7. doi: 10.1016/j.celrep.2020.02.021
46. Chiu JW, Binte Hanafi Z, Chew LCY, Mei Y, Liu H. IL-1alpha Processing, Signaling and Its Role in Cancer Progression. Cells (2021) 10(1):92. doi: 10.3390/cells10010092
47. Black RA, Kronheim SR, Cantrell M, Deeley MC, March CJ, Prickett KS, et al. Generation of Biologically Active Interleukin-1 Beta by Proteolytic Cleavage of the Inactive Precursor. J Biol Chem (1988) 263(19):9437–42. doi: 10.1016/S0021-9258(19)76559-4
48. Hazuda DJ, Strickler J, Kueppers F, Simon PL, Young PR. Processing of Precursor Interleukin 1 Beta and Inflammatory Disease. J Biol Chem (1990) 265(11):6318–22. doi: 10.1016/S0021-9258(19)39328-7
49. Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, et al. Converting Enzyme-Independent Release of Tumor Necrosis Factor Alpha and IL-1beta From a Stimulated Human Monocytic Cell Line in the Presence of Activated Neutrophils or Purified Proteinase 3. Proc Natl Acad Sci USA (1999) 96(11):6261–6. doi: 10.1073/pnas.96.11.6261
50. Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, et al. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity But Poor Effectors of Microbial Killing. Cell Rep (2018) 22(11):2937–50. doi: 10.1016/j.celrep.2018.02.062
51. Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-Derived Proteases Escalate Inflammation Through Activation of IL-36 Family Cytokines. Cell Rep (2016) 14(4):708–22. doi: 10.1016/j.celrep.2015.12.072
52. Macleod T, Doble R, McGonagle D, Wasson CW, Alase A, Stacey M, et al. Neutrophil Elastase-Mediated Proteolysis Activates the Anti-Inflammatory Cytokine IL-36 Receptor Antagonist. Sci Rep (2016) 6:24880. doi: 10.1038/srep24880
53. Terada M, Tsutsui H, Imai Y, Yasuda K, Mizutani H, Yamanishi K, et al. Contribution of IL-18 to Atopic-Dermatitis-Like Skin Inflammation Induced by Staphylococcus Aureus Product in Mice. Proc Natl Acad Sci USA (2006) 103(23):8816–21. doi: 10.1073/pnas.0602900103
54. Omoto Y, Tokime K, Yamanaka K, Habe K, Morioka T, Kurokawa I, et al. Human Mast Cell Chymase Cleaves Pro-IL-18 and Generates a Novel and Biologically Active IL-18 Fragment. J Immunol (2006) 177(12):8315–9. doi: 10.4049/jimmunol.177.12.8315
55. Patrick GJ, Liu H, Alphonse MP, Dikeman DA, Youn C, Otterson JC, et al. Epicutaneous Staphylococcus Aureus Induces IL-36 to Enhance IgE Production and Ensuing Allergic Disease. J Clin Invest (2021) 131(5):e143334. doi: 10.1172/JCI143334
56. Archer NK, Jo JH, Lee SK, Kim D, Smith B, Ortines RV, et al. Injury, Dysbiosis, and Filaggrin Deficiency Drive Skin Inflammation Through Keratinocyte IL-1alpha Release. J Allergy Clin Immunol (2019) 143(4):1426–43.e6. doi: 10.1016/j.jaci.2018.08.042
57. Dai X, Tohyama M, Murakami M, Shiraishi K, Liu S, Mori H, et al. House Dust Mite Allergens Induce Interleukin 33 (IL-33) Synthesis and Release From Keratinocytes via ATP-Mediated Extracellular Signaling. Biochim Biophys Acta Mol Basis Dis (2020) 1866(5):165719. doi: 10.1016/j.bbadis.2020.165719
58. Swindell WR, Beamer MA, Sarkar MK, Loftus S, Fullmer J, Xing X, et al. RNA-Seq Analysis of IL-1B and IL-36 Responses in Epidermal Keratinocytes Identifies a Shared MyD88-Dependent Gene Signature. Front Immunol (2018) 9:80. doi: 10.3389/fimmu.2018.00080
59. Albanesi C, Madonna S, Gisondi P, Girolomoni G. The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front Immunol (2018) 9:1549. doi: 10.3389/fimmu.2018.01549
60. Li N, Yamasaki K, Saito R, Fukushi-Takahashi S, Shimada-Omori R, Asano M, et al. Alarmin Function of Cathelicidin Antimicrobial Peptide LL37 Through IL-36gamma Induction in Human Epidermal Keratinocytes. J Immunol (2014) 193(10):5140–8. doi: 10.4049/jimmunol.1302574
61. Goldstein JD, Bassoy EY, Caruso A, Palomo J, Rodriguez E, Lemeille S, et al. IL-36 Signaling in Keratinocytes Controls Early IL-23 Production in Psoriasis-Like Dermatitis. Life Sci Alliance (2020) 3(6):e202000688. doi: 10.26508/lsa.202000688
62. Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, et al. IL-36 Promotes Myeloid Cell Infiltration, Activation, and Inflammatory Activity in Skin. J Immunol (2014) 192(12):6053–61. doi: 10.4049/jimmunol.1301481
63. Wittmann M, Purwar R, Hartmann C, Gutzmer R, Werfel T. Human Keratinocytes Respond to Interleukin-18: Implication for the Course of Chronic Inflammatory Skin Diseases. J Invest Dermatol (2005) 124(6):1225–33. doi: 10.1111/j.0022-202X.2005.23715.x
64. Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M. Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell (2019) 74(4):801–15.e6. doi: 10.1016/j.molcel.2019.02.038
65. Darmstadt GL, Fleckman P, Rubens CE. Tumor Necrosis Factor-Alpha and Interleukin-1alpha Decrease the Adherence of Streptococcus Pyogenes to Cultured Keratinocytes. J Infect Dis (1999) 180(5):1718–21. doi: 10.1086/315066
66. Cumberbatch M, Dearman RJ, Kimber I. Langerhans Cells Require Signals From Both Tumour Necrosis Factor-Alpha and Interleukin-1 Beta for Migration. Immunology (1997) 92(3):388–95. doi: 10.1046/j.1365-2567.1997.00360.x
67. Dietrich D, Martin P, Flacher V, Sun Y, Jarrossay D, Brembilla N, et al. Interleukin-36 Potently Stimulates Human M2 Macrophages, Langerhans Cells and Keratinocytes to Produce Pro-Inflammatory Cytokines. Cytokine (2016) 84:88–98. doi: 10.1016/j.cyto.2016.05.012
68. Leung BP, Culshaw S, Gracie JA, Hunter D, Canetti CA, Campbell C, et al. A Role for IL-18 in Neutrophil Activation. J Immunol (2001) 167(5):2879–86. doi: 10.4049/jimmunol.167.5.2879
69. Smith RJ, Bowman BJ, Speziale SC. Interleukin-1 Stimulates Granule Exocytosis From Human Neutrophils. Int J Immunopharmacol (1986) 8(1):33–40. doi: 10.1016/0192-0561(86)90070-6
70. Nakanishi K. Unique Action of Interleukin-18 on T Cells and Other Immune Cells. Front Immunol (2018) 9:763. doi: 10.3389/fimmu.2018.00763
71. Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 Is a Critical Regulator of Group 2 Innate Lymphoid Cell Function and Plasticity. Nat Immunol (2016) 17(6):646–55. doi: 10.1038/ni.3447
72. Golebski K, Ros XR, Nagasawa M, van Tol S, Heesters BA, Aglmous H, et al. IL-1beta, IL-23, and TGF-Beta Drive Plasticity of Human ILC2s Towards IL-17-Producing ILCs in Nasal Inflammation. Nat Commun (2019) 10(1):2162. doi: 10.1038/s41467-019-09883-7
73. Victor AR, Nalin AP, Dong W, McClory S, Wei M, Mao C, et al. IL-18 Drives ILC3 Proliferation and Promotes IL-22 Production via NF-kappaB. J Immunol (2017) 199(7):2333–42. doi: 10.4049/jimmunol.1601554
74. Renne J, Schafer V, Werfel T, Wittmann M. Interleukin-1 From Epithelial Cells Fosters T Cell-Dependent Skin Inflammation. Br J Dermatol (2010) 162(6):1198–205. doi: 10.1111/j.1365-2133.2010.09662.x
75. Ilarregui JM, van Beelen AJ, Fehres CM, Bruijns SC, Garcia-Vallejo JJ, van Kooyk Y. New Roles for CD14 and IL-Beta Linking Inflammatory Dendritic Cells to IL-17 Production in Memory CD4(+) T Cells. Immunol Cell Biol (2016) 94(10):907–16. doi: 10.1038/icb.2016.66
76. Penha R, Higgins J, Mutamba S, Barrow P, Mahida Y, Foster N. IL-36 Receptor is Expressed by Human Blood and Intestinal T Lymphocytes and is Dose-Dependently Activated via IL-36beta and Induces CD4+ Lymphocyte Proliferation. Cytokine (2016) 85:18–25. doi: 10.1016/j.cyto.2016.05.023
77. Vigne S, Palmer G, Martin P, Lamacchia C, Strebel D, Rodriguez E, et al. IL-36 Signaling Amplifies Th1 Responses by Enhancing Proliferation and Th1 Polarization of Naive CD4+ T Cells. Blood (2012) 120(17):3478–87. doi: 10.1182/blood-2012-06-439026
78. Vigne S, Palmer G, Lamacchia C, Martin P, Talabot-Ayer D, Rodriguez E, et al. IL-36R Ligands are Potent Regulators of Dendritic and T Cells. Blood (2011) 118(22):5813–23. doi: 10.1182/blood-2011-05-356873
79. Harusato A, Abo H, Ngo VL, Yi SW, Mitsutake K, Osuka S, et al. IL-36gamma Signaling Controls the Induced Regulatory T Cell-Th9 Cell Balance via NFkappaB Activation and STAT Transcription Factors. Mucosal Immunol (2017) 10(6):1455–67. doi: 10.1038/mi.2017.21
80. Du HY, Fu HY, Li DN, Qiao Y, Wang QW, Liu W. The Expression and Regulation of Interleukin-33 in Human Epidermal Keratinocytes: A New Mediator of Atopic Dermatitis and Its Possible Signaling Pathway. J Interferon Cytokine Res (2016) 36(9):552–62. doi: 10.1089/jir.2015.0159
81. Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can Promote Survival, Adhesion and Cytokine Production in Human Mast Cells. Lab Invest (2007) 87(10):971–8. doi: 10.1038/labinvest.3700663
82. Ho LH, Ohno T, Oboki K, Kajiwara N, Suto H, Iikura M, et al. IL-33 Induces IL-13 Production by Mouse Mast Cells Independently of IgE-FcepsilonRI Signals. J Leukoc Biol (2007) 82(6):1481–90. doi: 10.1189/jlb.0407200
83. Kaieda S, Wang JX, Shnayder R, Fishgal N, Hei H, Lee RT, et al. Interleukin-33 Primes Mast Cells for Activation by IgG Immune Complexes. PloS One (2012) 7(10):e47252. doi: 10.1371/journal.pone.0047252
84. Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 Induces Antigen-Specific IL-5+ T Cells and Promotes Allergic-Induced Airway Inflammation Independent of IL-4. J Immunol (2008) 181(7):4780–90. doi: 10.4049/jimmunol.181.7.4780
85. Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a Chemoattractant for Human Th2 Cells. Eur J Immunol (2007) 37(10):2779–86. doi: 10.1002/eji.200737547
86. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate Production of T(H)2 Cytokines by Adipose Tissue-Associated C-Kit(+)Sca-1(+) Lymphoid Cells. Nature (2010) 463(7280):540–4. doi: 10.1038/nature08636
87. Mashiko S, Mehta H, Bissonnette R, Sarfati M. Increased Frequencies of Basophils, Type 2 Innate Lymphoid Cells and Th2 Cells in Skin of Patients With Atopic Dermatitis But Not Psoriasis. J Dermatol Sci (2017) 88(2):167–74. doi: 10.1016/j.jdermsci.2017.07.003
88. Imai Y, Yasuda K, Nagai M, Kusakabe M, Kubo M, Nakanishi K, et al. IL-33-Induced Atopic Dermatitis-Like Inflammation in Mice Is Mediated by Group 2 Innate Lymphoid Cells in Concert With Basophils. J Invest Dermatol (2019) 139(10):2185–94.e3. doi: 10.1016/j.jid.2019.04.016
89. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 Promotes an Innate Immune Pathway of Intestinal Tissue Protection Dependent on Amphiregulin-EGFR Interactions. Proc Natl Acad Sci USA (2015) 112(34):10762–7. doi: 10.1073/pnas.1509070112
90. Clark JT, Christian DA, Gullicksrud JA, Perry JA, Park J, Jacquet M, et al. IL-33 Promotes Innate Lymphoid Cell-Dependent IFN-Gamma Production Required for Innate Immunity to Toxoplasma Gondii. Elife (2021) 10:e65614. doi: 10.7554/eLife.65614
91. Frisbee AL, Saleh MM, Young MK, Leslie JL, Simpson ME, Abhyankar MM, et al. IL-33 Drives Group 2 Innate Lymphoid Cell-Mediated Protection During Clostridium Difficile Infection. Nat Commun (2019) 10(1):2712. doi: 10.1038/s41467-019-10733-9
92. Rak GD, Osborne LC, Siracusa MC, Kim BS, Wang K, Bayat A, et al. IL-33-Dependent Group 2 Innate Lymphoid Cells Promote Cutaneous Wound Healing. J Invest Dermatol (2016) 136(2):487–96. doi: 10.1038/JID.2015.406
93. Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The Interleukin (IL)-1 Cytokine Family–Balance Between Agonists and Antagonists in Inflammatory Diseases. Cytokine (2015) 76(1):25–37. doi: 10.1016/j.cyto.2015.06.017
94. Bayraktar R, Bertilaccio MTS, Calin GA. The Interaction Between Two Worlds: MicroRNAs and Toll-Like Receptors. Front Immunol (2019) 10:1053. doi: 10.3389/fimmu.2019.01053
95. Boraschi D, et al. The Family of the Interleukin-1 Receptors. Immunol Rev (2018) 281(1):197–232. doi: 10.1111/imr.12606
96. Italiani P, Mosca E, Della Camera G, Melillo D, Migliorini P, Milanesi L, et al. Profiling the Course of Resolving vs. Persistent Inflammation in Human Monocytes: The Role of IL-1 Family Molecules. Front Immunol (2020) 11:1426. doi: 10.3389/fimmu.2020.01426
97. Martin P, Palmer G, Rodriguez E, Palomo J, Lemeille S, Goldstein J, et al. Intracellular IL-1 Receptor Antagonist Isoform 1 Released From Keratinocytes Upon Cell Death Acts as an Inhibitor for the Alarmin IL-1alpha. J Immunol (2020) 204(4):967–79. doi: 10.4049/jimmunol.1901074
98. Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 Blocks Interleukin-33 Signaling in Allergic Airway Inflammation. J Biol Chem (2007) 282(36):26369–80. doi: 10.1074/jbc.M704916200
99. Rauschmayr T, Groves RW, Kupper TS. Keratinocyte Expression of the Type 2 Interleukin 1 Receptor Mediates Local and Specific Inhibition of Interleukin 1-Mediated Inflammation. Proc Natl Acad Sci USA (1997) 94(11):5814–9. doi: 10.1073/pnas.94.11.5814
100. Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, et al. IL-38 has an Anti-Inflammatory Action in Psoriasis and its Expression Correlates With Disease Severity and Therapeutic Response to Anti-IL-17A Treatment. Cell Death Dis (2018) 9(11):1104. doi: 10.1038/s41419-018-1143-3
101. Wittmann M, Doble R, Bachmann M, Pfeilschifter J, Werfel T, Muh H. IL-27 Regulates IL-18 Binding Protein in Skin Resident Cells. PloS One (2012) 7(6):e38751. doi: 10.1371/journal.pone.0038751
102. Tsutsumi N, Yokota A, Kimura T, Kato Z, Fukao T, Shirakawa M, et al. An Innate Interaction Between IL-18 and the Propeptide That Inactivates its Precursor Form. Sci Rep (2019) 9(1):6160. doi: 10.1038/s41598-019-42661-5
103. Nold MF, Nold-Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA. IL-37 is a Fundamental Inhibitor of Innate Immunity. Nat Immunol (2010) 11(11):1014–22. doi: 10.1038/ni.1944
104. Nold-Petry CA, Lo CY, Rudloff I, Elgass KD, Li S, Gantier MP, et al. IL-37 Requires the Receptors IL-18Ralpha and IL-1r8 (SIGIRR) to Carry Out its Multifaceted Anti-Inflammatory Program Upon Innate Signal Transduction. Nat Immunol (2015) 16(4):354–65. doi: 10.1038/ni.3103
105. Giannoudaki E, Stefanska AM, Lawler H, Leon G, Hernandez Santana YE, Hassan N, et al. SIGIRR Negatively Regulates IL-36-Driven Psoriasiform Inflammation and Neutrophil Infiltration in the Skin. J Immunol (2021) 207(2):651–60. doi: 10.4049/jimmunol.2100237
106. Cayrol C, Girard JP. The IL-1-Like Cytokine IL-33 is Inactivated After Maturation by Caspase-1. Proc Natl Acad Sci USA (2009) 106(22):9021–6. doi: 10.1073/pnas.0812690106
107. Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of Interleukin-33 Bioactivity Through Proteolysis by Apoptotic Caspases. Immunity (2009) 31(1):84–98. doi: 10.1016/j.immuni.2009.05.007
108. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the Alarmin IL-33 Regulates ST2-Dependent Inflammation. Nat Commun (2015) 6:8327. doi: 10.1183/13993003.congress-2015.OA292
109. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An Autoinflammatory Disease With Deficiency of the Interleukin-1–Receptor Antagonist. N Engl J Med (2009) 360(23):2426–37. doi: 10.1056/NEJMoa0807865
110. Brau-Javier CN, Gonzales-Chavez J, Toro JR. Chronic Cutaneous Pustulosis Due to a 175-Kb Deletion on Chromosome 2q13: Excellent Response to Anakinra. Arch Dermatol (2012) 148(3):301–4. doi: 10.1001/archdermatol.2011.2857
111. Schnellbacher C, Ciocca G, Menendez R, Aksentijevich I, Goldbach-Mansky R, Duarte AM, et al. Deficiency of Interleukin-1 Receptor Antagonist Responsive to Anakinra. Pediatr Dermatol (2013) 30(6):758–60. doi: 10.1111/j.1525-1470.2012.01725.x
112. Ulusoy E, Karaca NE, El-Shanti H, Kilicoglu E, Aksu G, Kutukculer N. Interleukin-1 Receptor Antagonist Deficiency With a Novel Mutation; Late Onset and Successful Treatment With Canakinumab: A Case Report. J Med Case Rep (2015) 9(1):1–4. doi: 10.1186/s13256-015-0618-4
113. Garg M, de Jesus AA, Chapelle D, Dancey P, Herzog R, Rivas-Chacon R, et al. Rilonacept Maintains Long-Term Inflammatory Remission in Patients With Deficiency of the IL-1 Receptor Antagonist. JCI Insight (2017) 2(16):e94838. doi: 10.1172/jci.insight.94838
114. Setta-Kaffetzi N, Navarini AA, Patel VM, Pullabhatla V, Pink AE, Choon SE, et al. Rare Pathogenic Variants in IL36RN Underlie a Spectrum of Psoriasis-Associated Pustular Phenotypes. J Invest Dermatol (2013) 133(5):1366–9. doi: 10.1038/jid.2012.490
115. Arakawa A, Vollmer S, Besgen P, Galinski A, Summer B, Kawakami Y, et al. Unopposed IL-36 Activity Promotes Clonal CD4(+) T-Cell Responses With IL-17a Production in Generalized Pustular Psoriasis. J Invest Dermatol (2018) 138(6):1338–47. doi: 10.1016/j.jid.2017.12.024
116. Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N Engl J Med (2019) 380(10):981–3. doi: 10.1056/NEJMc1811317
117. Mendonca LO, Malle L, Donovan FX, Chandrasekharappa SC, Montealegre Sanchez GA, Garg M, et al. Deficiency of Interleukin-1 Receptor Antagonist (DIRA): Report of the First Indian Patient and a Novel Deletion Affecting IL1RN. J Clin Immunol (2017) 37(5):445–51. doi: 10.1007/s10875-017-0399-1
118. Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, et al. Interleukin-36-Receptor Antagonist Deficiency and Generalized Pustular Psoriasis. N Engl J Med (2011) 365(7):620–8. doi: 10.1056/NEJMoa1013068
119. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature (2006) 440(7081):228–32. doi: 10.1038/nature04515
120. Janssen R, Verhard E, Lankester A, Ten Cate R, van Dissel JT, et al. Enhanced Interleukin-1beta and Interleukin-18 Release in a Patient With Chronic Infantile Neurologic, Cutaneous, Articular Syndrome. Arthritis Rheum (2004) 50(10):3329–33. doi: 10.1002/art.20494
121. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a New Gene Encoding a Putative Pyrin-Like Protein Causes Familial Cold Autoinflammatory Syndrome and Muckle–Wells Syndrome. Nat Genet (2001) 29(3):301–5. doi: 10.1038/ng756
122. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 Forms an IL-1β-Processing Inflammasome With Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity (2004) 20(3):319–25. doi: 10.1016/S1074-7613(04)00046-9
123. Muckle TJ. The ‘Muckle-Wells’ Syndrome. Br J Dermatol (1979) 100(1):87–92. doi: 10.1111/j.1365-2133.1979.tb03572.x
124. Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De Novo CIAS1 Mutations, Cytokine Activation, and Evidence for Genetic Heterogeneity in Patients With Neonatal-Onset Multisystem Inflammatory Disease (NOMID): A New Member of the Expanding Family of Pyrin-Associated Autoinflammatory Diseases. Arthritis Rheum (2002) 46(12):3340–8. doi: 10.1002/art.10688
125. Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic Infantile Neurological Cutaneous and Articular Syndrome is Caused by Mutations in CIAS1, a Gene Highly Expressed in Polymorphonuclear Cells and Chondrocytes. Am J Hum Genet (2002) 71(1):198–203. doi: 10.1086/341357
126. Bardet J, Laverdure N, Fusaro M, Picard C, Garnier L, Viel S, et al. NLRC4 GOF Mutations, a Challenging Diagnosis From Neonatal Age to Adulthood. J Clin Med (2021) 10(19):4369. doi: 10.3390/jcm10194369
127. Romberg N, Vogel TP, Canna SW. NLRC4 Inflammasomopathies. Curr Opin Allergy Clin Immunol (2017) 17(6):398–404. doi: 10.1097/ACI.0000000000000396
128. Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-Threatening NLRC4-Associated Hyperinflammation Successfully Treated With IL-18 Inhibition. J Allergy Clin Immunol (2017) 139(5):1698–701. doi: 10.1016/j.jaci.2016.10.022
129. Novick D, Dinarello CA. IL-18 Binding Protein Reverses the Life-Threatening Hyperinflammation of a Baby With the NLRC4 Mutation. J Allergy Clin Immunol (2017) 140(1):316. doi: 10.1016/j.jaci.2017.02.037
130. Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-Onset Multisystem Inflammatory Disease Responsive to Interleukin-1beta Inhibition. N Engl J Med (2006) 355(6):581–92. doi: 10.1056/NEJMoa055137
131. Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of Clinical Features in Muckle-Wells Syndrome and Response to Anakinra. Arthritis Rheum (2004) 50(2):607–12. doi: 10.1002/art.20033
132. Lovell DJ, Bowyer SL, Solinger AM. Interleukin-1 Blockade by Anakinra Improves Clinical Symptoms in Patients With Neonatal-Onset Multisystem Inflammatory Disease. Arthritis Rheum (2005) 52(4):1283–6. doi: 10.1002/art.20953
133. Hoffman HM, Throne ML, Amar N, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and Safety of Rilonacept (Interleukin-1 Trap) in Patients With Cryopyrin-Associated Periodic Syndromes: Results From Two Sequential Placebo-Controlled Studies. Arthritis Rheum (2008) 58(8):2443–52. doi: 10.1002/art.23687
134. Maksimovic L, Stirnemann J, Caux F, Ravet N, Rouaghe S, Cuisset L, et al. New CIAS1 Mutation and Anakinra Efficacy in Overlapping of Muckle–Wells and Familial Cold Autoinflammatory Syndromes. Rheumatology (Oxford) (2008) 47(3):309–10. doi: 10.1093/rheumatology/kem318
135. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of Canakinumab in the Cryopyrin-Associated Periodic Syndrome. N Engl J Med (2009) 360(23):2416–25. doi: 10.1056/NEJMoa0810787
136. Ancient Missense Mutations in a New Member of the RoRet Gene Family are Likely to Cause Familial Mediterranean Fever. The International FMF Consortium. Cell (1997) 90(4):797–807. doi: 10.1016/S0092-8674(00)80539-5
137. French FMF Consortium. A Candidate Gene for Familial Mediterranean Fever. Nat Genet (1997) 17(1):25–31. doi: 10.1038/ng0997-25
138. Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, et al. The B30.2 Domain of Pyrin, the Familial Mediterranean Fever Protein, Interacts Directly With Caspase-1 to Modulate IL-1beta Production. Proc Natl Acad Sci USA (2006) 103(26):9982–7. doi: 10.1073/pnas.0602081103
139. Lachmann HJ, Sengül B, Yavuzşen TU, Booth DR, Booth SE, Bybee A, et al. Clinical and Subclinical Inflammation in Patients With Familial Mediterranean Fever and in Heterozygous Carriers of MEFV Mutations. Rheumatol (Oxford) (2006) 45(6):746–50. doi: 10.1093/rheumatology/kei279
140. Papin S, Cuenin S, Agostini L, Martinon F, Werner S, Beer HD, et al. The SPRY Domain of Pyrin, Mutated in Familial Mediterranean Fever Patients, Interacts With Inflammasome Components and Inhibits proIL-1beta Processing. Cell Death Differ (2007) 14(8):1457–66. doi: 10.1038/sj.cdd.4402142
141. Ozdogan H, Ugurlu S. Canakinumab for the Treatment of Familial Mediterranean Fever. Expert Rev Clin Immunol (2017) 13(5):393–404. doi: 10.1080/1744666X.2017.1313116
142. Belkhir R, Moulonguet-Doleris L, Hachulla E, Prinseau J, Baglin A, Hanslik T. Treatment of Familial Mediterranean Fever With Anakinra. Ann Intern Med (2007) 146(11):825–6. doi: 10.7326/0003-4819-146-11-200706050-00023
143. Belkaya S, Michailidis E, Korol CB, Kabbani M, Cobat A, Bastard P, et al. Inherited IL-18BP Deficiency in Human Fulminant Viral Hepatitis. J Exp Med (2019) 216(8):1777–90. doi: 10.1084/jem.20190669
144. Zhang ZZ, Zhang Y, He T, Sweeney CL, Baris S, Karakoc-Aydiner E, et al. Homozygous IL37 Mutation Associated With Infantile Inflammatory Bowel Disease. Proc Natl Acad Sci USA (2021) 118(10):e2009217118. doi: 10.1073/pnas.2009217118
145. Sims J, Towne J, Blumberg H. 11 IL-1 Family Members in Inflammatory Skin Disease. Ernst Schering Res Found Workshop (2006) 56):187–91. doi: 10.1007/3-540-37673-9_11
146. Hahn M, Frey S, Hueber AJ. The Novel Interleukin-1 Cytokine Family Members in Inflammatory Diseases. Curr Opin Rheumatol (2017) 29(2):208–13. doi: 10.1097/BOR.0000000000000361
147. Cozzi A, Papagrigoraki A, Biasi D, Colato C, Girolomoni G. Cutaneous Manifestations of Adult-Onset Still’s Disease: A Case Report and Review of Literature. Clin Rheumatol (2016) 35(5):1377–82. doi: 10.1007/s10067-014-2614-2
148. McGonagle D, Ramanan AV, Bridgewood C. Immune Cartography of Macrophage Activation Syndrome in the COVID-19 Era. Nat Rev Rheumatol (2021) 17(3):145–57. doi: 10.1038/s41584-020-00571-1
149. Castañeda S, Atienza-Mateo B, Martín-Varillas JL, Serra López-Matencio JM, González-Gay MA. Anakinra for the Treatment of Adult-Onset Still’s Disease. Expert Rev Clin Immunol (2018) 14(12):979–92. doi: 10.1080/1744666X.2018.1536548
150. Sfriso P, Bindoli S, Doria A, Feist E, Galozzi P. Canakinumab for the Treatment of Adult-Onset Still’s Disease. Expert Rev Clin Immunol (2020) 16(2):129–38. doi: 10.1080/1744666X.2019.1707664
151. Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, et al. Open-Label, Multicentre, Dose-Escalating Phase II Clinical Trial on the Safety and Efficacy of Tadekinig Alfa (IL-18BP) in Adult-Onset Still’s Disease. Ann Rheum Dis (2018) 77(6):840–7. doi: 10.1136/annrheumdis-2017-212608
152. Witte-Handel E, Wolk EK, Tsaousi A, Irmer ML, Mossner R, Shomroni O, et al. The IL-1 Pathway Is Hyperactive in Hidradenitis Suppurativa and Contributes to Skin Infiltration and Destruction. J Invest Dermatol (2019) 139(6):1294–305. doi: 10.1016/j.jid.2018.11.018
153. Hessam S, Sand M, Gambichler T, Skrygan M, Ruddel I, Bechara FG. Interleukin-36 in Hidradenitis Suppurativa: Evidence for a Distinctive Proinflammatory Role and a Key Factor in the Development of an Inflammatory Loop. Br J Dermatol (2018) 178(3):761–7. doi: 10.1111/bjd.16019
154. Behm B, Babilas P, Landthaler M, Schreml S. Cytokines, Chemokines and Growth Factors in Wound Healing. J Eur Acad Dermatol Venereol (2012) 26(7):812–20. doi: 10.1111/j.1468-3083.2011.04415.x
155. Hantash BM, Zhao L, Knowles JA, Lorenz HP. Adult and Fetal Wound Healing. Front Biosci (2008) 13(1):51–61. doi: 10.2741/2559
156. Hübner G, Brauchle M, Smola H, Madlener M, Fässler R, Werner S. Diffirenttial Regulation of Pro-Inflammatory Cytokines During Wound Healing in Normal and Glucocorticoid-Treated mice. Cytokine (1996) 8(7):548–56. doi: 10.1006/cyto.1996.0074
157. Burzynski LC, Humphry M, Pyrillou K, Wiggins KA, Chan JNE, Figg N, et al. The Coagulation and Immune Systems Are Directly Linked Through the Activation of Interleukin-1alpha by Thrombin. Immunity (2019) 50(4):1033–42 e6. doi: 10.1016/j.immuni.2019.03.003
158. Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Perspective Article: Growth Factors and Cytokines in Wound Healing. Wound Repair Regen (2008) 16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x
159. Lee SW, Morhenn VB, Ilnicka M, Eugui EM, Allison AC. Autocrine Stimulation of Interleukin-1 Alpha and Transforming Growth Factor Alpha Production in Human Keratinocytes and its Antagonism by Glucocorticoids. J Invest Dermatol (1991) 97(1):106–10. doi: 10.1111/1523-1747.ep12478503
160. Li Y, Fan J, Chen M, Li W, Woodley DT. Transforming Growth Factor-Alpha: A Major Human Serum Factor That Promotes Human Keratinocyte Migration. J Invest Dermatol (2006) 126(9):2096–105. doi: 10.1038/sj.jid.5700350
161. Barrandon Y, Green H. Cell Migration is Essential for Sustained Growth of Keratinocyte Colonies: The Roles of Transforming Growth Factor-Alpha and Epidermal Growth Factor. Cell (1987) 50(7):1131–7. doi: 10.1016/0092-8674(87)90179-6
162. Gottlieb AB, Chang CK, Posnett DN, Fanelli B, Tam JP. Detection of Transforming Growth Factor Alpha in Normal, Malignant, and Hyperproliferative Human Keratinocytes. J Exp Med (1988) 167(2):670–5. doi: 10.1084/jem.167.2.670
163. Cha D, O'Brien P, O'Toole EA, Woodley DT, Hudson LG. Enhanced Modulation of Keratinocyte Motility by Transforming Growth Factor-Alpha (TGF-Alpha) Relative to Epidermal Growth Factor (EGF). J Invest Dermatol (1996) 106(4):590–7. doi: 10.1111/1523-1747.ep12345083
164. Jiang CK, Magnaldo T, Ohtsuki M, Freedberg IM, Bernerd F, Blumenberg M. Epidermal Growth Factor and Transforming Growth Factor Alpha Specifically Induce the Activation- and Hyperproliferation-Associated Keratins 6 and 16. Proc Natl Acad Sci USA (1993) 90(14):6786–90. doi: 10.1073/pnas.90.14.6786
165. Brauchle M, Angermeyer K, Hübner G, Werner S. Large Induction of Keratinocyte Growth Factor Expression by Serum Growth Factors and Pro-Inflammatory Cytokines in Cultured Fibroblasts. Oncogene (1994) 9(11):3199–204.
166. Chedid M, Rubin JS, Csaky KG, Aaronson SA. Regulation of Keratinocyte Growth Factor Gene Expression by Interleukin 1. J Biol Chem (1994) 269(14):10753–7. doi: 10.1016/S0021-9258(17)34123-6
167. Maas-Szabowski N, Stark H-J, Fusenig NE. Keratinocyte Growth Regulation in Defined Organotypic Cultures Through IL-1-Induced Keratinocyte Growth Factor Expression in Resting Fibroblasts. J Invest Dermatol (2000) 114(6):1075–84. doi: 10.1046/j.1523-1747.2000.00987.x
168. Smola H, Thiekötter G, Fusenig NE. Mutual Induction of Growth Factor Gene Expression by Epidermal-Dermal Cell Interaction. J Cell Biol (1993) 122(2):417–29. doi: 10.1083/jcb.122.2.417
169. Werner S, Smola H. Paracrine Regulation of Keratinocyte Proliferation and Differentiation. Trends Cell Biol (2001) 11(4):143–6. doi: 10.1016/S0962-8924(01)01955-9
170. Raines EW, Dower SK, Ross R. Interleukin-1 Mitogenic Activity for Fibroblasts and Smooth Muscle Cells is Due to PDGF-AA. Science (1989) 243(4889):393–6. doi: 10.1126/science.2783498
171. Frank S, Hübner G, Breier G, Longaker MT, Greenhalgh DG, Werner S. Regulation of Vascular Endothelial Growth Factor Expression in Cultured Keratinocytes. Implications for Normal and Impaired Wound Healing. J Biol Chem (1995) 270(21):12607–13. doi: 10.1074/jbc.270.21.12607
172. Fahey E, Doyle SL. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front Immunol (2019) 10:1426–6. doi: 10.3389/fimmu.2019.01426
173. Wang G, Sweren E, Liu H, Wier E, Alphonse MP, Chen R, et al. Bacteria Induce Skin Regeneration via IL-1beta Signaling. Cell Host Microbe (2021) 29(5):777–91.e6. doi: 10.1016/j.chom.2021.03.003
174. Lee P, Lee D-J, Chan C, Chen S-W, Ch’en I, Jamora C. Dynamic Expression of Epidermal Caspase 8 Simulates a Wound Healing Response. Nature (2009) 458(7237):519–23. doi: 10.1038/nature07687
175. Blanpain C, Fuchs E. Epidermal Homeostasis: A Balancing Act of Stem Cells in the Skin. Nat Rev Mol Cell Biol (2009) 10(3):207–17. doi: 10.1038/nrm2636
176. Lau K, Paus R, Tiede S, Day P, Bayat A. Exploring the Role of Stem Cells in Cutaneous Wound Healing. Exp Dermatol (2009) 18(11):921–33. doi: 10.1111/j.1600-0625.2009.00942.x
177. Lee P, Gund R, Dutta A, Pincha N, Rana I, Ghosh S, et al. Stimulation of Hair Follicle Stem Cell Proliferation Through an IL-1 Dependent Activation of γδt-Cells. Elife (2017) 6:e28875. doi: 10.7554/eLife.28875
178. Jameson J, Havran WL. Skin Gammadelta T-Cell Functions in Homeostasis and Wound Healing. Immunol Rev (2007) 215:114–22. doi: 10.1111/j.1600-065X.2006.00483.x
179. Munoz LD, Sweeney MJ, Jameson JM. Skin Resident γδ T Cell Function and Regulation in Wound Repair. Int J Mol Sci (2020) 21(23):9286. doi: 10.3390/ijms21239286
180. Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, et al. Inflammatory Memory Sensitizes Skin Epithelial Stem Cells to Tissue Damage. Nature (2017) 550(7677):475–80. doi: 10.1038/nature24271
181. Constantinides MG, Link VM, Tamoutounour S, Wong AC, Perez-Chaparro PJ, Han SJ, et al. MAIT Cells are Imprinted by the Microbiota in Early Life and Promote Tissue Repair. Science (2019) 366(6464):eaax6624. doi: 10.1126/science.aax6624
182. Harrison OJ, Linehan JL, Shih HY, Bouladoux N, Han SJ, Smelkinson M, et al. Commensal-Specific T Cell Plasticity Promotes Rapid Tissue Adaptation to Injury. Science (2019) 363(6422):eaat6280. doi: 10.1126/science.aat6280
183. Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, et al. IL-13 Activates a Mechanism of Tissue Fibrosis That Is Completely TGF-Beta Independent. J Immunol (2004) 173(6):4020–9. doi: 10.4049/jimmunol.173.6.4020
184. Yin H, Li X, Hu S, Liu T, Yuan B, Gu H, et al. IL-33 Accelerates Cutaneous Wound Healing Involved in Upregulation of Alternatively Activated Macrophages. Mol Immunol (2013) 56(4):347–53. doi: 10.1016/j.molimm.2013.05.225
185. He R, Yin H, Yuan B, Liu T, Luo L, Huang P, et al. IL-33 Improves Wound Healing Through Enhanced M2 Macrophage Polarization in Diabetic Mice. Mol Immunol (2017) 90:42–9. doi: 10.1016/j.molimm.2017.06.249
186. Oshio T, Komine M, Tsuda H, Tominaga SI, Saito H, Nakae S, et al. Nuclear Expression of IL-33 in Epidermal Keratinocytes Promotes Wound Healing in Mice. J Dermatol Sci (2017) 85(2):106–14. doi: 10.1016/j.jdermsci.2016.10.008
187. Jiang Z, Liu Y, Li C, Chang L, Wang W, Wang Z, et al. IL-36gamma Induced by the TLR3-SLUG-VDR Axis Promotes Wound Healing via REG3A. J Invest Dermatol (2017) 137(12):2620–9. doi: 10.1016/j.jid.2017.07.820
188. Dillekas H, Straume O. The Link Between Wound Healing and Escape From Tumor Dormancy. Surg Oncol (2019) 28:50–6. doi: 10.1016/j.suronc.2018.11.009
189. Wilkinson HN, Hardman MJ. Wound Healing: Cellular Mechanisms and Pathological Outcomes. Open Biol (2020) 10(9):200223. doi: 10.1098/rsob.200223
190. Tan JL, Lash B, Karami R, Nayer B, Lu Y-Z, Piotto C, et al. Restoration of the Healing Microenvironment in Diabetic Wounds With Matrix-Binding IL-1 Receptor Antagonist. Commun Biol (2021) 4(1):422. doi: 10.1038/s42003-021-01913-9
191. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat Med (2011) 17(2):179–88. doi: 10.1038/nm.2279
192. Koenen TB, Stienstra R, van Tits LJ, de Graaf J, Stalenhoef AF, Joosten LA, et al. Hyperglycemia Activates Caspase-1 and TXNIP-Mediated IL-1beta Transcription in Human Adipose Tissue. Diabetes (2011) 60(2):517–24. doi: 10.2337/db10-0266
193. Dasu MR, Devaraj S, Jialal I. High Glucose Induces IL-1beta Expression in Human Monocytes: Mechanistic Insights. Am J Physiol Endocrinol Metab (2007) 293(1):E337–46. doi: 10.1152/ajpendo.00718.2006
194. Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F, et al. Monocytes From Patients With Rheumatoid Arthritis and Type 2 Diabetes Mellitus Display an Increased Production of Interleukin (IL)-1β via the Nucleotide-Binding Domain and Leucine-Rich Repeat Containing Family Pyrin 3(NLRP3)-Inflammasome Activation: A Possible Implication for Therapeutic Decision in These Patients. Clin Exp Immunol (2015) 182(1):35–44. doi: 10.1111/cei.12667
195. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 Inflammasome Activation in Patients With Type 2 Diabetes. Diabetes (2013) 62(1):194–204. doi: 10.2337/db12-0420
196. Koh TJ, DiPietro LA. Inflammation and Wound Healing: The Role of the Macrophage. Expert Rev Mol Med (2011) 13:e23–3. doi: 10.1017/S1462399411001943
197. Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ. Sustained Inflammasome Activity in Macrophages Impairs Wound Healing in Type 2 Diabetic Humans and Mice. Diabetes (2014) 63(3):1103–14. doi: 10.2337/db13-0927
198. Yan C, Gao N, Sun H, Yin J, Lee P, Zhou L, et al. Targeting Imbalance Between IL-1β and IL-1 Receptor Antagonist Ameliorates Delayed Epithelium Wound Healing in Diabetic Mouse Corneas. Am J Pathol (2016) 186(6):1466–80. doi: 10.1016/j.ajpath.2016.01.019
199. Mirza RE, Fang MM, Ennis WJ, Koh TJ. Blocking Interleukin-1β Induces a Healing-Associated Wound Macrophage Phenotype and Improves Healing in Type 2 Diabetes. Diabetes (2013) 62(7):2579–87. doi: 10.2337/db12-1450
200. Hu Y, Liang D, Li X, Liu HH, Zhang X, Zheng M, et al. The Role of Interleukin-1 in Wound Biology. Part II: In Vivo and Human Translational Studies. Anesth Analg (2010) 111(6):1534–42. doi: 10.1213/ANE.0b013e3181f691eb
Keywords: IL-36 cytokine family, IL-1 family cytokines, IL-33, proteolytic regulation, IL-1alpha, IL-1beta, pathogen/commensal discrimination, IL-18
Citation: Macleod T, Berekmeri A, Bridgewood C, Stacey M, McGonagle D and Wittmann M (2021) The Immunological Impact of IL-1 Family Cytokines on the Epidermal Barrier. Front. Immunol. 12:808012. doi: 10.3389/fimmu.2021.808012
Received: 02 November 2021; Accepted: 06 December 2021;
Published: 23 December 2021.
Edited by:
Ryan O’Shaughnessy, Queen Mary University of London, United KingdomReviewed by:
Georg Varga, University Hospital Muenster, GermanyNathan K. Archer, Johns Hopkins Medicine, United States
Copyright © 2021 Macleod, Berekmeri, Bridgewood, Stacey, McGonagle and Wittmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam Wittmann, M.wittmann@leeds.ac.uk