 Sam Blanchett
Sam Blanchett Yves Dondelinger
Yves Dondelinger Alessandro Barbarulo1
Alessandro Barbarulo1 Mathieu J. M. Bertrand
Mathieu J. M. Bertrand Benedict Seddon
Benedict Seddon- 1Institute of Immunity and Transplantation, The Pears Building, University College London, London, United Kingdom
- 2Vlaams Instituut voor Biotechnologie (VIB) Center for Inflammation Research, Ghent, Belgium
- 3Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium
The Inhibitor of Kappa B Kinase (IKK) complex is a critical regulator of NF-κB activation. More recently, IKK has also been shown to repress RIPK1 dependent extrinsic cell death pathways by directly phosphorylating RIPK1 at serine 25. In T cells, IKK expression is essential for normal development in the thymus, by promoting survival of thymocytes independently of NF-κB activation. RIPK1 undergoes extensive phosphorylation following TNF stimulation in T cells, though which targets are required to repress RIPK1 has not been defined. Here, we show that TNF induced phosphorylation of RIPK1 at S25 is IKK dependent. We test the relevance of this phosphorylation event in T cells using mice with a RIPK1S25D phosphomimetic point mutation to endogenous RIPK1. We find that this mutation protects T cells from TNF induced cell death when IKK activity is inhibited in vitro, and can rescues development of IKK deficient thymocytes in vivo to a degree comparable with kinase dead RIPK1D138N. Together, these data show that phosphorylation of RIPK1S25 by IKK represents a key regulatory event promoting survival of T cells by IKK.
Introduction
The inhibitor of kappa-B kinase (IKK) complex is a trimeric complex of two kinases, IKK1 (IKKα) and IKK2 (IKKβ), and a third regulatory component, NEMO (IKKγ). It is classically described as the prototypic activator of NF-κB transcriptional pathway. IKK phosphorylates inhibitory IκB proteins, targeting them for degradation by the proteasome and thereby releasing NF-κB dimers to enter the nucleus. As such, IKK function has long been presumed to be mediated by its activation of NF-κB family of transcription factors, that play critical roles in controlling development and function of many cell types (1).
In mouse T cells, studies of tissue specific knockouts reveal complex overlapping and distinct roles for IKK and NF-κB in T cell biology. Ablation of the IKK complex, either by deletion of NEMO (2), or combined loss of IKK1 and IKK2 subunits (3) results in a developmental arrest in single positive (SP) thymocytes at the immature HSAhi stage. Similar developmental blocks are observed in mice lacking the upstream activator of IKK, TAK1 (4–6). In contrast, expression of NF-κB REL subunits is redundant for thymic development and generation of mature peripheral T cells (7, 8). An explanation for this apparent contradiction comes from two key observations. First, the trigger for NF-κB activation via IKK in developing thymocytes is not TCR but TNF. Blockade of TNF signalling rescues development in IKK1/2 deficient thymocytes (3). Second, recent studies reveal that the IKK complex has two functions during TNF signalling in T cells - activating NF-κB and directly repressing cell death by inhibiting the serine threonine kinase, RIPK1 (8, 9).
Ligation of TNFR1 causes recruitment of TRADD, TRAF2, and the serine/threonine kinase RIPK1. The ubiquitin ligases TRAF2, cellular inhibitor of apoptosis proteins (cIAPs) and the linear ubiquitin chain assembly complex (LUBAC), add ubiquitin chain modifications to themselves and RIPK1, creating a scaffold that allows recruitment and activation of the TAB/TAK and IKK complexes that in turn activate NF-κB. This is termed complex I [reviewed in (10, 11)]. A failure to maintain the stability of this complex results in the formation of cell death inducing complexes. In the presence of IAP inhibitors, IKK inhibitors or TAK1 inhibitors (12–14), a cytosolic protein complex (called complex II) composed of FADD, CASPASE 8 and RIPK1 forms that induces apoptosis, a function dependent upon RIPK1 kinase activity (9, 11, 15). Phosphorylation of RIPK1 by IKK blocks RIPK1 kinase activity and therefore its capacity to induce apoptosis (14, 16). In thymocytes, it is this function of IKK, and not NF-κB activation, that is critical for their survival and accounts for the phenotype observed in IKK deficiency (8).
Since the recognition that RIPK1 activity can be directly controlled through phosphorylation, there has been considerable interest in better understanding those kinases that target RIPK1 and the impact on RIPK1 function. RIPK1 appears to undergo extensive phosphorylation on multiple residues during TNF signalling (17) that have been implicated in repressing the kinase activity of RIPK1. In addition to autophosphorylation (18), TAK1, TBK1, MK2 and IKK have all been shown in various cell types and contexts to mediate repressive phosphorylation of RIPK1 (14, 19–24). IKK has been shown to phosphorylate RIPK1 both at Ser6 and Ser25 (14), and it is this latter site that has been shown to be important to prevent cell death in MEFs and during Yersinia infection (16), in which effective immune control of infection depends on RIPK1 triggered cell death processes in myeloid cells (25). In T cells, the minimal requirements for repression of RIPK1 have not been defined. In the present study, we probed the mechanism by which IKK represses RIPK1 specifically in T cells by asking whether phosphorylation of Ser25 of RIPK1 was required and sufficient for the pro-survival functions of IKK function in T cells, both during TNF stimulation in vitro, and in mice with IKK deficient T cells in vivo.
Results
IKK function and expression is essential for phosphorylation of RIPK1Ser25
Our previous work shows that following TNF stimulation in T cells, phosphorylation of RIPK1 within complex I is dependent upon IKK (8). We therefore asked whether Ser 25 of RIPK1 was phosphorylated in mouse T cells and if it was dependent on IKK activity. Mouse thymocytes were stimulated with TNF and then cytosolic extracts analysed by immunoblotting. Shortly after TNF stimulation and mouse thymocytes, RIPK1 is recruited to TNFR1 complex I and becomes modified with extensive ubiquitin chain additions and phosphorylation residues. To detect pS25 RIPK1, a phospho-specific anti-RIPK1 pSer25 anti-sera was used to immunoprecipitate pS25 RIPK1 from cell extracts (16). Immunoprecipitates were subsequently treated to remove ubiquitin and phospho groups and then total RIPK1 detected by immunoblotting. Following TNF stimulation of WT thymocytes, pS25 RIPK1 was readily detectable (Figure 1A). Pretreatment of cells with pan-IKK inhibitor, IKK16, prior to TNF stimulation, prevented detection of pS25 RIPK1, demonstrating that pS25 was IKK dependent. To confirm the requirement for IKK genetically, we wished to assess TNF stimulation of thymocytes lacking IKK expression. However, since IKK deletion results in a complete loss of mature thymocytes, we analysed Chukfx/fx Ikbkbfx/fx huCD2iCre (IKK1/2ΔCD2) mice that express a kinase dead RIPK1D138N mutant. Inactivation of RIPK1 in vivo in this way in IKK1/2ΔCD2 RIPK1D138N mice rescues thymocytes from cell death, and permits analysis of RIPK1 recruitment to TNF induced complex I (8). Following TNF stimulation, pS25 RIPK1 was readily detectable in thymocytes from Cre -ve litter mate controls. In contrast, no pS25 RIPK1 was detected following TNF stimulation of IKK1/2 deficient thymocytes (Figure 1B). Together, these data demonstrate that Ser25 of RIPK1 is a target of phosphorylation following TNF stimulation of thymocytes, and that phosphorylation is strictly IKK dependent.
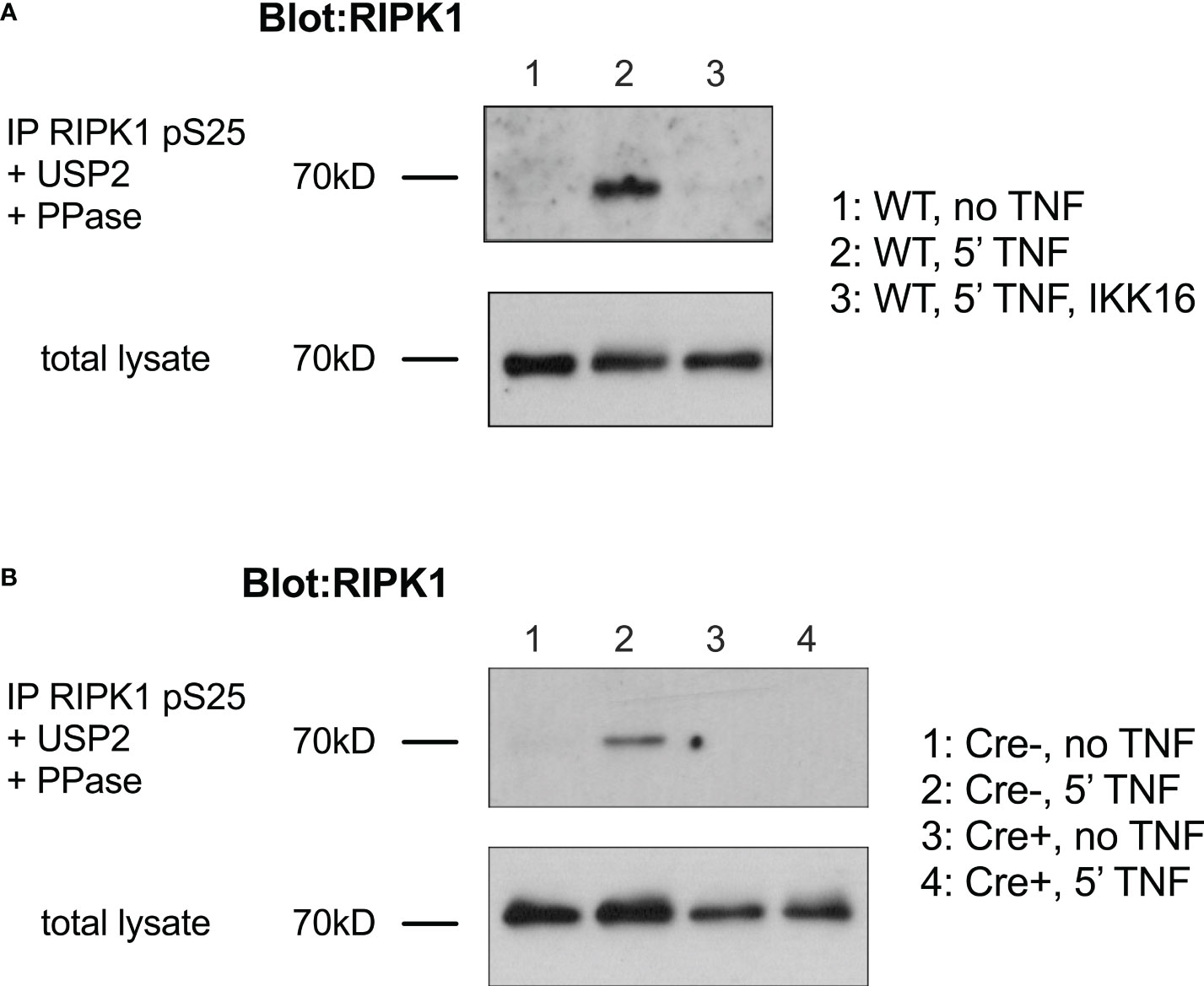
Figure 1 Phosphorylation of RIPK1 Ser25 following TNF stimulation is IKK dependent in T cells. (A) Thymocytes from WT mice were stimulated for 5’ with PBS (lane 1) or TNF (lane 2-3) (5ng/ml) following pre-incubation for 30’ with IKK16 (2µM, lane 3). Cell were lysed and immunoprecipitated with anti-phospho-Ser25 RIPK1 (RIPK1 pS25), followed by treatment with USP2 and PPase. i.p. were separated by gel electrophoresis and RIPK1 protein analysed by western blot. Corresponding total cell lysates were run prior to immunoprecipitation as loading control. (B) Thymocytes from Cre+ and Cre- littermate IKK1/2ΔCD2 RIPK1D138N mice were stimulated with TNF and immunopreciptates of anti-RIPK1 pS25 analysed by immunoblotting as described in (A). Data are representative of one other experiment.
TNF induced cell death of thymocytes is completely blocked in RIPK1S25D expressing mice
We next wanted to ask whether IKK dependent phospshorylation of RIPK1Ser25 was sufficient for repression of TNF induced cell death by IKK. To do this, we took advantage of a recently described knockin mutant mouse strain that expresses an RIPK1S25D mutant (16). In this strain, Ser25 has been mutated to aspartic acid. Aspartic acid and phosphorylated serine closely resemble one another chemically, and this mutation has been shown to have the predicted phosphomimetic properties (16). We therefore asked whether this phosphomimetic of RIPK1 Ser25 was sufficient to prevent RIPK1 induced cell death in the absence of IKK activity in T cells. Since IKK1/2ΔCD2 mice lack mature thymocytes and have no peripheral T cells, we took a combined genetic and pharmacological approach to test the efficacy of RIPK1S25D in blocking cell death. We generated IKK1ΔCD2 RIPK1S25D that specifically lack IKK1 but express IKK2. Redundancy between IKK1 and IKK2 permits near normal development and generation of T cells in the absence of IKK1 expression (3, 8, 26). Application of the highly specific IKK2 inhibitor, Bl605906, (27) to IKK1 deficient T cells results in a robust blockade of IKK activity.
We first tested the capacity of RIPK1S25D to inhibit TNF induced apoptosis in thymocytes in vitro. Our previous work shows that thymocytes become progressively more dependent upon IKK signalling to promote survival as they transition through development, correlating with induction of RIPK1 protein expression (8). CD4+CD8+ double positive (DP) thymocytes and immature HSAhi CD4+ single positive (CD4iSP) thymocytes do not require IKK for survival in vivo. In contrast, immature HSAhi CD8+ SP (CD8iSP) thymocytes and both mature HSAlo CD4+ (CD4mSP) and HSAlo CD8+ SP (CD8mSP) thymocytes are all highly dependent on IKK expression for their survival in vivo (3). To assess TNF induced death, thymocytes from WT, IKK1ΔCD2 and IKK1ΔCD2RIPK1S25D mice were cultured overnight with different doses of TNF in the presence and absence of IKK2 inhibitor and cell death assessed by flow cytometry. IKK2 inhibition alone did not render any subset of thymocytes susceptible to TNF induced cell death (Figure 2A). In contrast, in the presence of IKK2 inhibitor, CD8iSP, CD8mSP and CD4mSP underwent increasing extents of cell death with increasing doses of TNF. Cell death was RIPK1 kinase dependent, since further application of RIPK1 inhibitor, Nec1, completely blocked cell death in cultures. In contrast, no TNF inducible cell death was observed in cultures of thymocytes from IKK1ΔCD2 RIPK1S25D mice with IKK2 inhibitor. We presume that cell death was mediated by apoptosis rather than necroptosis, since thymocytes do not express MLKL (8), and previous studies show that T cells become susceptible to necroptosis only following activation (28).
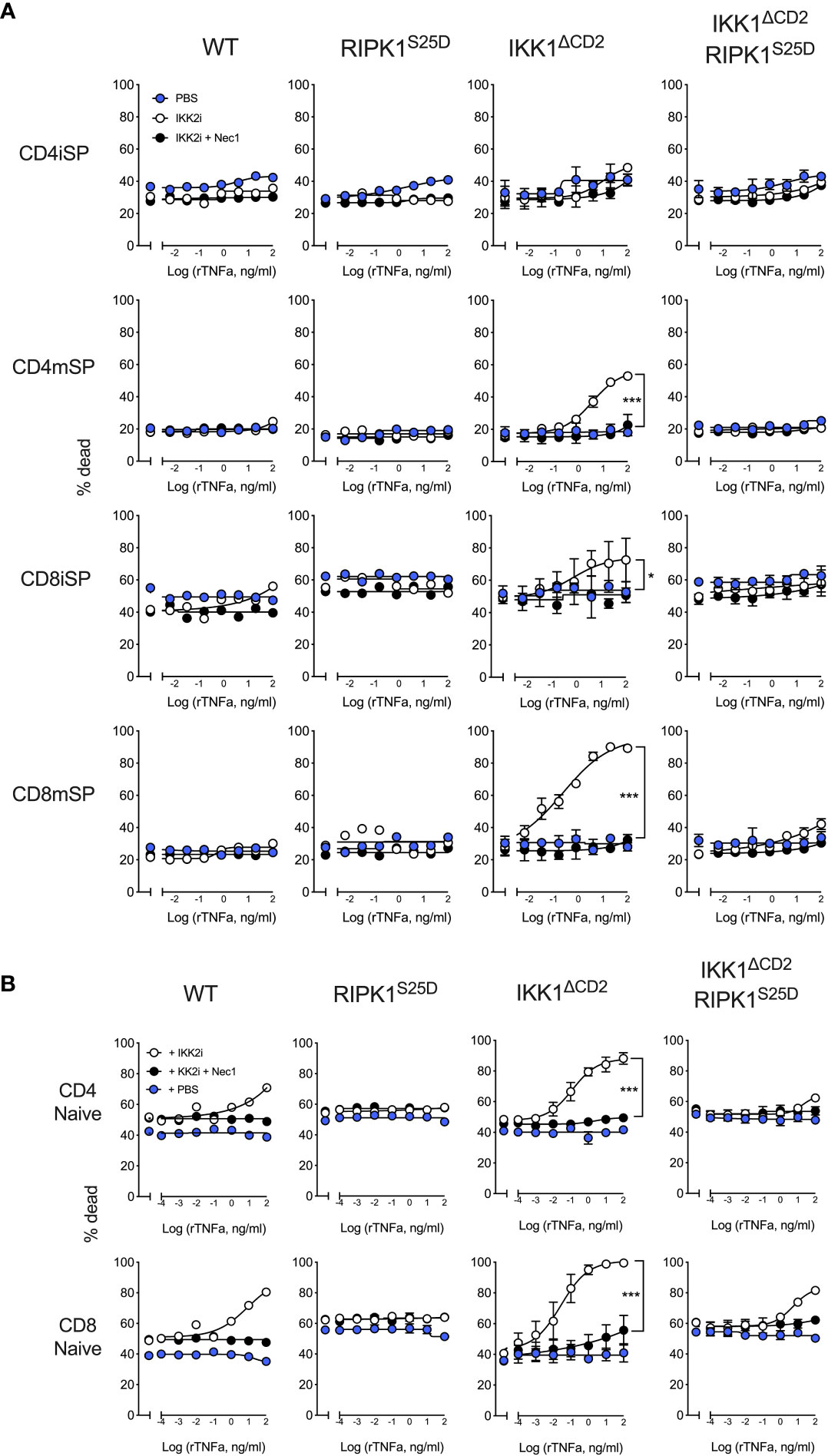
Figure 2 TNF induced cell death in the absence of IKK kinase activity is blocked by RIPK1S25D in thymocytes and peripheral naive T cells. Thymus and LN cells from IKK1ΔCD2 and IKK1ΔCD2RIPK1S25D mice, and iCre -ve litter mates (WT or RIPK1S25D columns), were cultured for 24h at different doses of TNF with addition of either IKK2i (10µM), IKK2i + Nec1 (10nM), or PBS as control. Cultured cells were analysed by flow cytometry and % dead cells calculated and graphed for the indicated populations of thymocytes (A) and peripheral naive T cells (B). Data are representative of three or more independent replicate experiments. Error bars show s.d. Significant differences between PBS and IKK2i conditions were determined by 2-way ANOVA. * < 0.05, *** < 0.001.
TNF induced cell death of peripheral T cells is largely blocked by RIPK1S25D
We next tested whether RIPK1S25D was sufficient to also protect mature peripheral T cells from TNF induced cell death. In contrast to thymocytes, IKK2 inhibitor alone was able to render WT T cells susceptible to RIPK1 dependent TNF induced cell death at higher concentrations of TNF. In contrast, T cells from RIPK1S25D mice expressing normal IKK, were resistant to cell death in the face of IKK2 inhibition (Figure 2B). Similarly, IKK2 inhibition of IKK1 deficient peripheral T cells from IKK1ΔCD2 mice resulted in high levels of cell death, even at lower concentrations of TNF. In this IKK1 deficient setting, RIPK1S25D rescued both CD4+ and CD8+ T cells from TNF induced cell death at all but the highest concentrations of TNF, where reproducible low levels of cell death were observed. To test whether the low level of cell death observed in cultures of T cells from IKK1ΔCD2RIPK1S25D mice with IKK2 inhibitor were RIPK1 dependent, we also cultured cells in the presence of Nec1. This showed that the low level of cell death of TNF stimulated IKK1ΔCD2RIPK1S25D was in fact RIPK1 dependent, suggesting some residual kinase activity of RIPK1S25D.
RIPK1S25D rescues development of IKK deficient thymocytes
To test the capacity of RIPK1S25D to block cell death in vivo, we asked whether the block in T cell development in IKK1/2ΔCD2 mice could be overcome by the phosphomimetic RIPK1 mutant. We therefore generated IKK1/2ΔCD2RIPK1S25D and compared their phenotype with IKK1/2ΔCD2 mice expressing a kinase dead RIPK1D138N mutant. As previously reported, numbers of DP and CD4iSP thymocytes are normal in the absence of IKK expression, while CD8iSP, CD8mSP and CD4mSP subsets are profoundly reduced in numbers (Figure 3). Kinase dead RIPK1 rescues numbers of CD4mSP and CD8iSP to near normal levels, while CD8mSP numbers are restored to levels approximately half way between those in IKK1/2ΔCD2 and WT (8). Analysing thymus of IKK1/2ΔCD2RIPK1S25D mice revealed an identical pattern of rescue to the kinase dead RIPK1 expressing mice. CD4mSP and CD8iSP numbers were indistinguishable from IKK expressing controls, while CD8mSPs underwent a clear but incomplete rescue of cellularity (Figure 3). These results suggest that RIPK1S25D mutant is sufficient to repress RIPK1 dependent cell death in vivo in the absence of IKK expression.
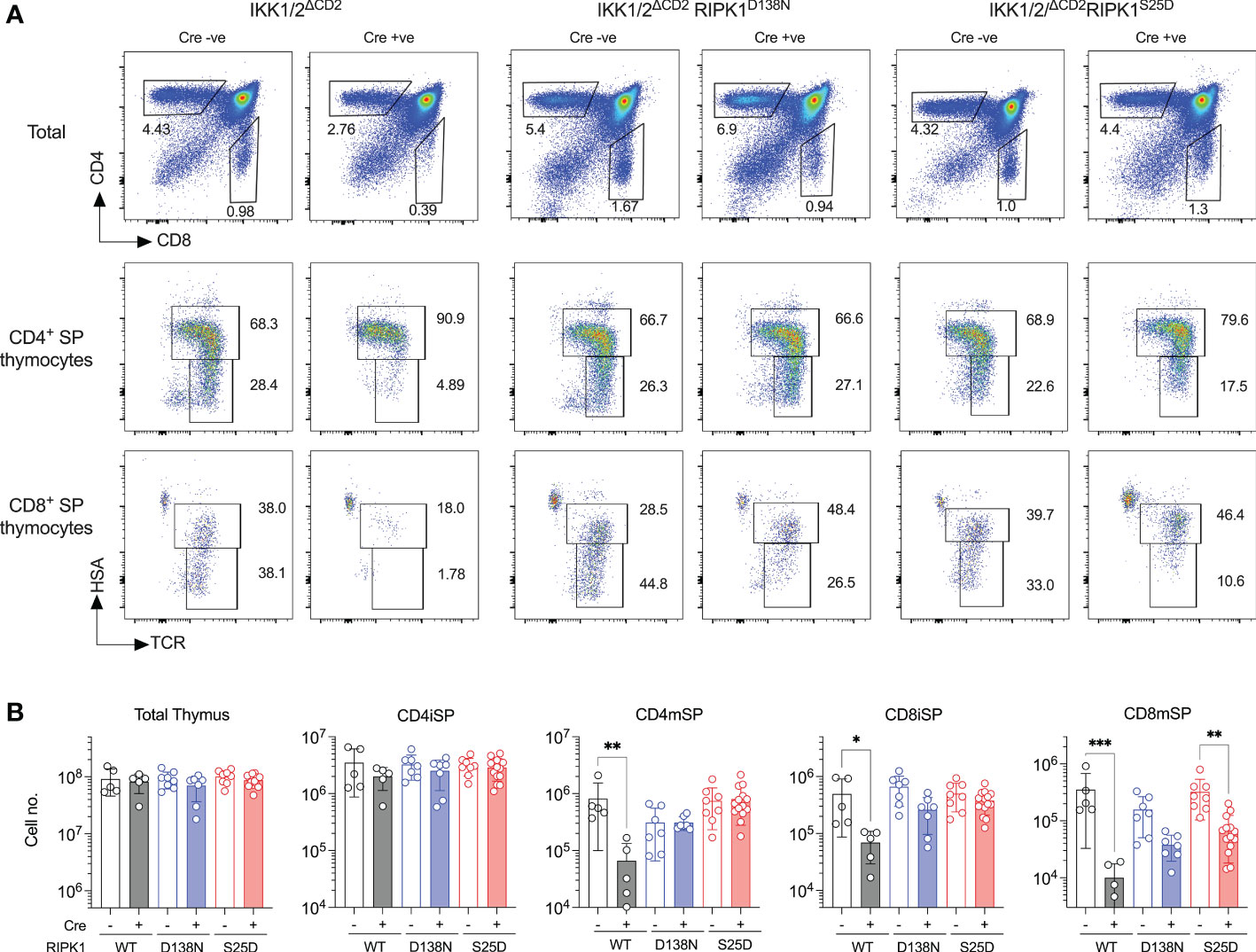
Figure 3 RIPK1S25D rescues IKK deficient thymocytes from cell death in vivo. Single cell suspensions were prepared from the thymus of Cre -ve (n=5) and Cre +ve (n=5) IKK1/2ΔCD2, Cre -ve (n=7) and Cre +ve (n=7) IKK1/2ΔCD2 RIPK1D138N and Cre -ve (n=8) and Cre +ve (n=14) IKK1/2ΔCD2 RIPK1S25D mice, enumerated and analyse by flow cytometry. (A) Flow plots show phenotype of the indicated populations (rows) from the indicated mice (columns) and gates used to define immature HSAhi CD4+ TCRhi SP (CD4iSP), mature HSAlo CD4+ TCRhi SP (CD4mSP) immature HSAhi CD8+ TCRhi SP (CD8iSP) and HSAlo CD8+ TCRhi SP (CD8mSP) thymic subpopulations. (B) Bar charts show the total cell numbers for the indicated populations recovered from different strains. Data are pooled from five independent experiments. Mann-Witney student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
RIPK1S25D rescues CD4+ but not CD8+ peripheral T cells in IKK1/2ΔCD2 mice
Earlier work shows that kinase dead RIPK1 permits a substantial rescue of CD4+ naive T cell compartment and a small but significant rescue of CD8+ naive T cells (8). We therefore compared the size and composition of the peripheral T cell compartments of IKK1/2ΔCD2RIPK1D138N and IKK1/2ΔCD2RIPK1S25D mice. Substantial naive CD4+ compartments were evident in both IKK1/2ΔCD2RIPK1D138N and IKK1/2ΔCD2RIPK1S25D mice (Figure 4A), with similar numbers of recovered in both strains (Figure 4B). Significant rescue of CD8+ naive T cells was evident, albeit at a lower level than observed in the CD4+ compartment, in IKK1/2ΔCD2RIPK1D138N mice. There was some evidence of rescue in CD8+ naive T cell numbers in IKK1/2ΔCD2RIPK1S25D mice, but not to significant levels. The various IKK1/2ΔCD2 strains also carried a Rosa26RYFP Cre reporter construct. This is useful in the context of gene deletions resulting in strong developmental blocks, because rare cells in which iCre fails to completely delete conditional genes, so called ‘escapants’, can have a competitive advantage over gene deleted counterparts, and may accumulate in peripheral lymphoid tissues. Indeed, there are substantial fractions of YFP-ve cells amongst the naive T cells present in periphery of IKK1/2ΔCD2 mice, indicating the presence of such escapants. Analysing YFP expression by naive T cells in IKK1/2ΔCD2RIPK1D138N and IKK1/2ΔCD2RIPK1S25D mice revealed a strong restoration of YFP expressing cells, further indicating a degree of rescue of gene deleted populations in these mice. In contrast, amongst effector phenotype and CD25+ Treg populations, YFP expression remained less than 5% in all IKK deficient strains (data not shown) indicating no rescue of these subsets by either RIPK1S25D or RIPK1D138N mutants. Nevertheless, rescue of CD4+ naive T cells in both RIPK1 mutant strains was far more extensive than observed in CD8+ subsets.
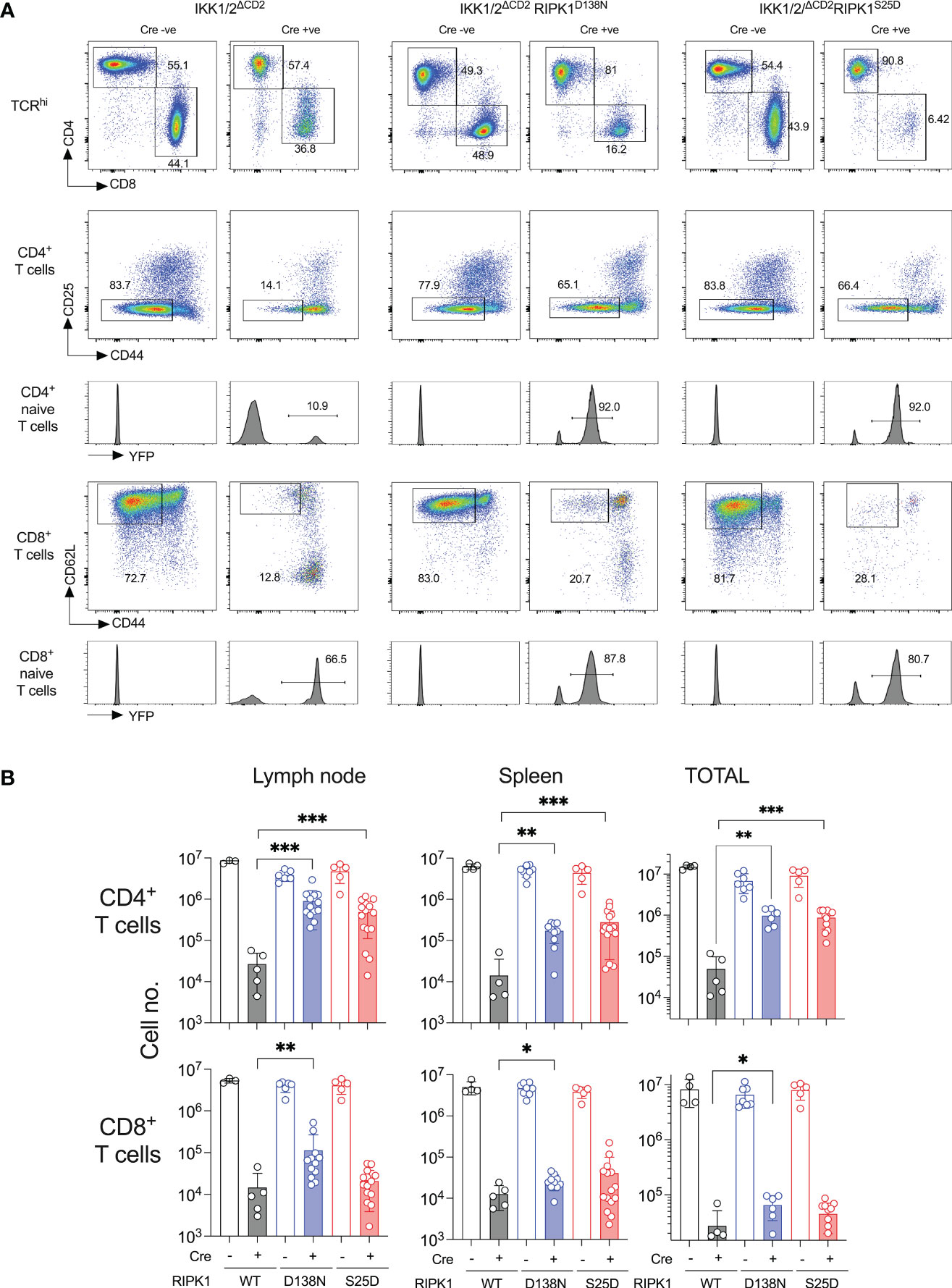
Figure 4 RIPK1S25D rescues IKK deficient CD4+ but not CD8+ naive T cells from cell death in vivo. LN and spleen were recovered from the same groups of mice described in Figure 3 and single cell suspensions analysed by flow cytometry. (A) Flow plots show the phenotype of lymph node cells and gates used to identify CD4+ naive T cells as CD4+ TCRhi CD44lo CD25- and CD8+ naive T cells as CD8+ TCRhi CD44lo CD25- cells. Histograms are of YFP expression by naive gated T cells from the corresponding strains. YFP is only activated in Cre expressing strains. (B) Bar charts show the numbers of CD4+ and CD8+ naive T cells recovered from lymph nodes, spleen and both combined, from the indicated strains. Cell recoveries from Cre+ and corresponding Cre- strains were all highly significant differences (p<0.0001). Significant differences between Cre+ strains are indicated on plots. Data are pooled from five independent experiments. * p<0.05, ** p<0.01, *** p<0.001.
Discussion
Here, we investigated the mechanisms by which IKK signalling regulates RIPK1 dependent cell death in T cells, specifically whether phosphorylation of Ser25 of RIPK1 by IKK was sufficient to mediate repression of RIPK1. We make the following novel findings. We show that RIPK1 Ser25 is a target of phosphorylation in T cells following TNF stimulation, and that phosphorylation is dependent upon both IKK expression and kinase activity. Analysing mice expressing a phosphomimetic S25D mutant of RIPK1, suggests that phosphorylation of Ser25 is sufficient to almost completely block TNF induced cell in the absence of IKK activity in vitro and show that the same mutant mediates a near identical rescue of thymocyte development in IKK deficient T cells as kinase dead RIPK1D138N mutant. Together, these results highlight Ser25 of RIPK1 as a critical regulatory target of IKK kinase activity in T cells.
In thymocytes, RIPK1S25D was sufficient to completely block TNF induced cell death in vitro. In contrast, peripheral T cells from RIPK1S25D mice, under conditions of IKK inhibition, underwent a low level of TNF induced cell death that was apparently RIPK1 kinase dependent, since death was blocked by Nec1. This highlights some features of IKK regulated cell death in T cells noted elsewhere (29), specifically that repression of cell death by the IKK complex in peripheral T cells has more stringent requirements for IKK activity than in thymocytes. There is redundancy between IKK1 and IKK2 to control RIPK1 in thymocytes. The activity of IKK1 alone appears sufficient to control RIPK1 mediated cell death in thymocytes, since IKK2 inhibitor did not sensitise any population to cell death, and similarly, IKK1 deficient thymocytes were resistant to TNF induced cell death. In contrast, IKK2 inhibition did sensitise WT peripheral T cells, and to a greater extent in CD8+ T cells than in CD4+ T cells. Therefore, the incomplete inhibition of cell death by RIPK1S25D amongst CD8+ T cells may reflect more active engagement of extrinsic cell death pathways specifically in CD8+ T cells. Ripk1 expression is induced during thymic development and reaches a greater maximal level in CD8 lineage cells than CD4. Higher intracellular concentrations of RIPK1 may increase the likelihood of triggering cell death pathways given appropriate stimuli, and may require a higher threshold IKK kinase activity to maintain effective repression. This would explain why active repression of TNF induced cell death is more easily perturbed by IKK2 inhibitor in CD8 peripheral T cells.
The impact of RIPK1S25D mutant upon TNF induced cell death we observed in vitro, strongly correlated with the capacity of this mutant to rescue thymocytes and T cells from the consequences of IKK ablation in vivo. CD4mSP thymocytes and peripheral naive CD4+ T cells were equally well rescued by RIPK1S25D and RIPK1D138N. In contrast, the failure of RIPK1S25D to completely block TNF induced cell death following IKK inhibition in CD8+ T cells in vitro was reflected in the in vivo phenotype of IKK1/2ΔCD2RIPK1S25D mice. While RIPK1S25D and RIPK1D138N mediated comparable rescue of CD8+ thymic subsets, we did not observed any significant rescue of CD8+ naive T cells by RIPK1S25D, although enrichment of YFP expressing cells in this compartment was suggestive of some low level of rescue. These observations suggest that the modest level of TNF induced cell death observed in vitro in RIPK1S25D mice was physiologically relevant, under conditions of complete IKK ablation in vivo, and highlights that reactivity of T cells to TNF stimulation in vitro can be accurately predictive in vivo behaviour.
Blockade of TNF induced cell death in RIPK1S25D cells by Nec1, revealed that kinase activity of the RIPK1S25D mutant is not completely repressed by this mutation. Ser25 of RIPK1 is within the kinase domain of RIPK1, next to highly conserved Glycine residues thought to facilitate the γ- phosphate of ATP for catalysis. The phosphorylation of Ser25 is thought to sterically interfere with ATP substrate access to the enzymatic domain of RIPK1. It remains unclear whether the S25D RIPK1 mutation represents a perfect phosphomimetic, and that RIPK1 with phospho-Ser25 alone retains some level of kinase activity or whether RIPK1 induced cell death simply represents imperfect repression of kinase activity by S25D mutation. Other studies show that Ser6, that is also phosphorylated by IKK, does not contribute to repression of RIPK1, either alone or in combination with Ser25 (16). However, while RIPK1S25D alone mediates very substantial protection of T cells from the impact of IKK blockade, and suggests that survival of T cells requires tonic phosphophorylation of RIPK1 on this residue, it is possible that optimal repression also requires inhibitory modifications by other kinases to other residues. It will be important in future studies to determine the minimal modifications required for repression of RIPK1, as well as the minimal deregulation required to unleash RIPK1 induced cell death. For instance, TAK1 is required to activate IKK complex but has also been implicated in other cell types of directly phosphorylating RIPK1 and mediating repression. TAK1 deletion in T cells results in a related, yet distinct, arguably more profound thymic block than observed following IKK deletion (6). While a failure to activate IKK undoubtedly contributes to the phenotype of TAK1 deletion, the capacity of TAK1 to activate other pathways, such as p38-MK2 that results in phosphorylation of RIPK1 (19, 21), may create a compound phenotype, that may include targeting RIPK1, both indirectly through IKK and MK2 activation, and also by direct kinase activity. It is unclear whether optimal RIPK1 repression in T cells requires cooperative activity of both IKK and TAK1, or whether these kinases activities mediate repression independently of one another. Disentangling such regulatory networks controlling RIPK1 activity remain important future challenges.
Materials and methods
Mice
Mice with conditional alleles of Ikbkb (30) and/or Chuk (31) were intercrossed with mice expressing Cre under the control of the human CD2 (huCD2iCre) (32), and with mice with a D138N mutation in Ripk1 (RIPK1D138N) (33) or with a Ripk1S25D mutation (16). Chukfx/fx huCD2iCre (IKK1ΔCD2), Chukfx/fx Ikbkbfx/fx huCD2iCre (IKK1/2ΔCD2), Chukfx/fx Ikbkbfx/fx huCD2iCre RIPK1D138N (IKK1/2ΔCD2 RIPK1D138N), Chukfx/fx Ikbkbfx/fx huCD2iCre RIPK1S25D (IKK1/2ΔCD2 RIPK1D138N), Chukfx/fx huCD2iCre RIPK1S25D(IKK1ΔCD2 RIPK1S25D) strains were bred in the Comparative Biology Unit of the Royal Free UCL campus and at Charles River laboratories, Manston, UK. Animal experiments were performed according to institutional guidelines and Home Office regulations.
Flow cytometry and electronic gating strategies
Flow cytometric analysis was performed with 2-5 x 106 thymocytes, 1-5 x 106 lymph node or spleen cells. Cell concentrations of thymocytes, lymph nodes (superficial cervical, mandibular, axillary, superficial inguinal, and mesenteric chain) and spleen cells were determined with a Scharf Instruments Casy Counter. Cells were incubated with saturating concentrations of antibodies in 100 μl of Dulbecco’s phosphate-buffered saline (PBS) containing bovine serum albumin (BSA, 0.1%) for 1hour at 4°C followed by two washes in PBS-BSA. Panels used the following mAb: EF450-conjugated antibody against CD25(ThermoFisher Scientific), PE-conjugated antibody against CD127 (ThermoFisher Scientific), BV785-conjugated CD44 antibody (Biolegend), BV650-conjugated antibody against CD4 (Biolegend), BUV395-conjugated antibody against CD8 (BD Biosciences), BUV737-conjugated antibody against CD24 (BD Biosciences), PerCP-cy5.5-conjugated antibody against TCR (Tonbo Biosciences). Cell viability was determined using LIVE/DEAD cell stain kit (Invitrogen Molecular Probes), following the manufacturer’s protocol. multi-color flow cytometric staining was analyzed on a LSRFortessa (Becton Dickinson) instrument, and data analysis and color compensations were performed with FlowJo V10 software (TreeStar). Naive peripheral T cells were identified by gating CD4+ or CD8+ subsets with TCRhi CD44lo CD25lo. Mature CD4+ and CD8+ SP thymocytes were identified as TCRhiCD4+CD8-HSAlo and TCRhiCD4-CD8+HSAlo respectively.
In vitro culture
Thymocytes and LN T cells were cultured at 37°C with 5% CO2 in RPMI-1640 (Gibco, Invitrogen Corporation, CA) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco Invitrogen), 0.1% (v/v) 2-mercaptoethanol βME (Sigma Aldrich) and 1% (v/v) penicillin-streptomycin (Gibco Invitrogen). Recombinant TNF (Peprotech) was supplemented to cultures at 20ng/ml, unless otherwise stated, with PBS used as vehicle. Inhibitors were used at the following concentrations, unless otherwise stated: IKK2 inhibitor BI605906 (Tocris Bio-techne) (IKK2i) (10µM in 0.1% DMSO vehicle), Nec1 (10µM in 0.1% DMSO) (Insight biotechnology).
Immunoblotting
Thymocytes (2 x 107/condition) were stimulated with TNF (50ng/ml) for 5 minutes, washed two times in ice-cold PBS and lysed in NP-40 lysis buffer. pSer25 RIPK1 complexes were immunoprecipitated, followed by deubiquitylated (by USP2 treatment) and dephosphorylated (by lambda protein phosphatase treatment) as described elsewhere (16). Enzymatic reactions were allowed to proceed for 30 min at 37°C and subsequently quenched by the addition of 12.5 µL 5x laemmli buffer. IPs and total cell lysates were analyzed by standard immunoblotting. Generation of anti-pSer25 RIPK1 sera and its utility for detecting pSer25 RIPK1 by immunoprecipitation is characterised and described in detail elsewhere (16).
Statistics
Statistical analysis, line fitting, regression analysis, and figure preparation were performed using Graphpad Prism 8. Column data compared by unpaired Mann-Witney student’s t test. * p<0.05, ** p<0.01, *** p<0.001, **** p < 0.0001.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by United Kingdom Home Office under project license PPL PP2330953.
Author contributions
Conceptualization: MB and BS; Methodology: SB, YD, AB, MJMB, and BS; Investigation: SB, YD, and AB; Visualization: SB, YD, and BS; Funding acquisition: BS; Project administration: SB and BS; Supervision: BS; Writing – original draft: BS; Writing – review and editing: BS, SB, YD, and MB. All authors contributed to the article and approved the submitted version.
Funding
The work in the Seddon lab is supported by the Medical Research Council UK under programme codes MR/P011225/1 and MR/N013867/1. Research in the lab of MJMB is supported by the VIB, by Ghent University (iBOF ATLANTIS), by grants from the FWO (G035320N, G044518N, EOS G0G6618N, EOS G0I5722N) and from the Flemish Government (Methusalem BOF16/MET_V/007 - attributed to P. Vandenabeele).
Acknowledgments
We thank UCL Comparative Biology Unit staff for assistance with mouse breeding and maintenance. We thank the following for generously sharing of their mouse strains: Prof Manolis Pasparakis for Chuk conditional strain, Prof Michael Karin for Ikbkb conditional strain, Prof Vishva Dixit for the RIPK1D138N strain.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol (2004) 25:280–8. doi: 10.1016/j.it.2004.03.008
2. Schmidt-Supprian M, Courtois G, Tian J, Coyle AJ, Israel A, Rajewsky K, et al. Mature T cells depend on signaling through the IKK complex. Immunity (2003) 19:377–89. doi: 10.1016/S1074-7613(03)00237-1
3. Webb LV, Ley SC, Seddon B. TNF activation of NF-kappaB is essential for development of single-positive thymocytes. J Exp Med (2016) 213:1399–407. doi: 10.1084/jem.20151604
4. Liu HH, Xie M, Schneider MD, Chen ZJ. Essential role of TAK1 in thymocyte development and activation. Proc Natl Acad Sci United States America (2006) 103:11677–82. doi: 10.1073/pnas.0603089103
5. Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol (2006) 7:851–8. doi: 10.1038/ni1355
6. Xing Y, Wang X, Jameson SC, Hogquist KA. Late stages of T cell maturation in the thymus involve NF-kappaB and tonic type I interferon signaling. Nat Immunol (2016) 17:565–73. doi: 10.1038/ni.3419
7. Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-kappaB transcription-Factor-Dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity (2017) 47:450–465.e455.
8. Webb LV, Barbarulo A, Huysentruyt J, Vanden Berghe T, Takahashi N, Ley S, et al. Survival of single positive thymocytes depends upon developmental control of RIPK1 kinase signaling by the IKK complex independent of NF-κB. Immunity (2019) 50:348–361.e344. doi: 10.1016/j.immuni.2019.01.004
9. Ting AT, Bertrand MJM. More to life than NF-kappaB in TNFR1 signaling. Trends Immunol (2016) 37:535–45. doi: 10.1016/j.it.2016.06.002
10. Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal (2010) 3:re4. doi: 10.1126/scisignal.3115re4
11. Annibaldi A, Meier P. Checkpoints in TNF-induced cell death: Implications in inflammation and cancer. Trends Mol Med (2018) 24:49–65. doi: 10.1016/j.molmed.2017.11.002
12. Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell (2008) 133:693–703. doi: 10.1016/j.cell.2008.03.036
13. Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ (2013) 20:1381–92. doi: 10.1038/cdd.2013.94
14. Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, et al. NF-kappaB-Independent role of IKKalpha/IKKbeta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell (2015) 60:63–76. doi: 10.1016/j.molcel.2015.07.032
15. Dondelinger Y, Vandenabeele P, Bertrand MJ. Regulation of RIPK1's cell death function by phosphorylation. Cell Cycle (2016) 15:5–6. doi: 10.1080/15384101.2015.1112688
16. Dondelinger Y, Delanghe T, Priem D, Wynosky-Dolfi MA, Sorobetea D, Rojas-Rivera D, et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun (2019) 10:1729. doi: 10.1038/s41467-019-09690-0
17. Delanghe T, Dondelinger Y, Bertrand MJM. RIPK1 Kinase-Dependent Death: A Symphony of Phosphorylation Events. Trends Cell Biol (2020) 30:189–200.
18. Laurien L, Nagata M, Schunke H, Delanghe T, Wiederstein JL, Kumari S, et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat Commun (2020) 11:1747. doi: 10.1038/s41467-020-15466-8
19. Dondelinger Y, Delanghe T, Rojas-Rivera D, Priem D, Delvaeye T, Bruggeman I, et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol (2017) 19:1237–47. doi: 10.1038/ncb3608
20. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun (2017) 8:359. doi: 10.1038/s41467-017-00406-w
21. Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol Cell (2017) 66:698–710.e695. doi: 10.1016/j.molcel.2017.05.003
22. Mohideen F, Paulo JA, Ordureau A, Gygi SP, Harper JW. Quantitative phospho-proteomic analysis of TNFalpha/NFkappaB signaling reveals a role for RIPK1 phosphorylation in suppressing necrotic cell death. Mol Cell Proteomics (2017) 16:1200–16. doi: 10.1074/mcp.M117.068189
23. Annibaldi A, Wicky John S, Vanden Berghe T, Swatek KN, Ruan J, Liccardi G, et al. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol Cell (2018) 69:566–580.e565. doi: 10.1016/j.molcel.2018.01.027
24. Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell (2018) 174:1477–1491.e1419. doi: 10.1016/j.cell.2018.07.041
25. Peterson LW, Philip NH, Delaney A, Wynosky-Dolfi MA, Asklof K, Gray F, et al. RIPK1-dependent apoptosis bypasses pathogen blockade of innate signaling to promote immune defense. J Exp Med (2017) 214:3171–82. doi: 10.1084/jem.20170347
26. Chen X, Willette-Brown J, Wu X, Hu Y, Howard OM, Hu Y, et al. IKKalpha is required for the homeostasis of regulatory T cells and for the expansion of both regulatory and effector CD4 T cells. FASEB J (2015) 29:443–54. doi: 10.1096/fj.14-259564
27. Zhang J, Clark K, Lawrence T, Peggie MW, Cohen P. An unexpected twist to the activation of IKKbeta: TAK1 primes IKKbeta for activation by autophosphorylation. Biochem J (2014) 461:531–7. doi: 10.1042/BJ20140444
28. Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med (2011) 208:633–41. doi: 10.1084/jem.20110251
29. Carty F, Layzell S, Barbarulo A, Webb L, Seddon B. (2022). Tonic IKK signalling regulates naive T cell survival in vivo by both repressing RIPK1 dependent extrinsic cell death pathways and independent activation of NFκB. bioRxiv
30. Li ZW, Omori SA, Labuda T, Karin M, Rickert RC. IKK beta is required for peripheral b cell survival and proliferation. J Immunol (2003) 170:4630–7. doi: 10.4049/jimmunol.170.9.4630
31. Gareus R, Huth M, Breiden B, Nenci A, Rosch N, Haase I, et al. Normal epidermal differentiation but impaired skin-barrier formation upon keratinocyte-restricted IKK1 ablation. Nat Cell Biol (2007) 9:461–9. doi: 10.1038/ncb1560
32. De Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, et al. Transgenic mice with hematopoietic and lymphoid specific expression of cre. Eur J Immunol (2003) 33:314–25. doi: 10.1002/immu.200310005
Keywords: T cells, extrinsic cell death, RIPK1, IKK complex, TNF
Citation: Blanchett S, Dondelinger Y, Barbarulo A, Bertrand MJM and Seddon B (2022) Phosphorylation of RIPK1 serine 25 mediates IKK dependent control of extrinsic cell death in T cells. Front. Immunol. 13:1067164. doi: 10.3389/fimmu.2022.1067164
Received: 11 October 2022; Accepted: 08 November 2022;
Published: 01 December 2022.
Edited by:
Jongdae Lee, Guangzhou Medical University, ChinaReviewed by:
Yenkel Grinberg-Bleyer, U1052 Centre de Recherche en Cancerologie de Lyon (INSERM), FranceZhishan Ding, Zhejiang Chinese Medical University, China
Copyright © 2022 Blanchett, Dondelinger, Barbarulo, Bertrand and Seddon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benedict Seddon, benedict.seddon@ucl.ac.uk