 Zenglei Zhang
Zenglei Zhang Lin Zhao
Lin Zhao Xingyu Zhou
Xingyu Zhou Xu Meng
Xu Meng Xianliang Zhou
Xianliang Zhou- Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Hypertension is regarded as the most prominent risk factor for cardiovascular diseases, which have become a primary cause of death, and recent research has demonstrated that chronic inflammation is involved in the pathogenesis of hypertension. Both innate and adaptive immunity are now known to promote the elevation of blood pressure by triggering vascular inflammation and microvascular remodeling. For example, as an important part of innate immune system, classically activated macrophages (M1), neutrophils, and dendritic cells contribute to hypertension by secreting inflammatory cy3tokines. In particular, interferon-gamma (IFN-γ) and interleukin-17 (IL-17) produced by activated T lymphocytes contribute to hypertension by inducing oxidative stress injury and endothelial dysfunction. However, the regulatory T cells and alternatively activated macrophages (M2) may have a protective role in hypertension. Although inflammation is related to hypertension, the exact mechanisms are complex and unclear. The present review aims to reveal the roles of inflammation, immunity, and oxidative stress in the initiation and evolution of hypertension. We envisage that the review will strengthen public understanding of the pathophysiological mechanisms of hypertension and may provide new insights and potential therapeutic strategies for hypertension.
1 Introduction
Hypertension has become the leading global risk factor for cardiovascular diseases (CVDs) (1), and 1.15 billion adult individuals have been confirmed with hypertension according to related data (2). In addition, the American Heart Association and the American College of Cardiology formulated strict new diagnostic criteria that are expected to extend the number of patients with hypertension by two–threefold (3, 4). Many common and effective drugs, including angiotensin-converting enzyme inhibitors (ACEIs)/angiotensin II (Ang II) type 1 receptor blockers (ARBs), beta-blockers, calcium channel blockers (CCBs), and diuretics, have been widely confirmed to control raised blood pressure (5), causing a significant reduction of mortality associated with hypertension; however, hypertension still remains a major public health problem and health care burden in both developed and developing countries. Blood pressure is still poorly controlled in approximately half of patients who are receiving treatment (6), so potential therapeutic targets are urgently needed.
The pathogenesis of hypertension is complex and there are many risk factors. Accumulating evidence confirms that an activated inflammatory response and immune system are an indispensable part in the genesis and evolution of hypertension and are associated with hypertensive complications, such as myocardial infraction, hemorrhagic stroke, and renal injury (7). The elevated level of inflammatory biomarkers, including C-reactive protein (CRP) and cytokines, have been detected in patients with hypertension, revealing that the immune system is involved in hypertension as a low-grade inflammatory condition which is a chronic and continuous process and is more characterized by the increase of inflammatory cells and inflammatory mediators compared with infectious diseases, and usually does not show significant symptoms (5, 8, 9). Elevated BP by many risk factors including genetic susceptibility and several environmental factors can induce organ injury promoting the formation of damage-associated molecular patterns (DAMPs) and new antigens which are regarded as the main impetus for the low-grade inflammation (10). Inappropriate immune activation may act in the kidney, microvascular, nervous system, and even gut microbiome to promote the elevation of blood pressure (11–17), especially salt-sensitive hypertension (18, 19). Inflammation is generally considered an immune response and thus, in the early stage, mainly engages the innate immune system (IIS) and is subsequently followed by the adaptive immune response. Although interactions occur between these responses, they have different effects on disease progression and clinical significance; therefore, we will discuss these separately in this review.
In 1967, Okuda and Grollman first reported there was a relationship between the immune and vascular systems based on the induction of artery hypertension in rats after they had received lymphocytes derived from rats with hypertension (20). According to the surface markers and functions, macrophages are classified into proinflammatory M1 macrophages and anti-inflammatory M2 macrophages. The recent study has confirmed the damage effect of M1 macrophages and the inconsiderably protective role of M2 macrophages in hypertension (10). Moreover, increasing evidence showed that hypertension has a chronic inflammatory status characterized as the transmigration, accumulation, and activation of inflammatory cells, and the proinflammatory cytokines and free radicals produced by activated innate immune cells and endothelial cells (21). In the present review, we will discuss how the immune system and associated inflammation and oxidative stress affect the procession of hypertension and will summarize the potential therapeutic targets in hypertension.
2 The relationship of inflammation, immunity, and oxidative stress
Although numerous risk factors promote the genesis and progress of hypertension, the role of inflammation, immunity, and oxidative stress have been overwhelming confirmed by evidence from many laboratories worldwide (22–26). The causal state of circulating immune cells, such as monocytes, neutrophils, and lymphocytes, has been demonstrated (27), and the imbalanced activated immune system is known to produce an inflammatory condition with an increasing amount of proinflammatory cytokines. First, leukocytes gather via cytokine and chemokine signaling and roll on the vascular endothelium regulated by E and P selectin in the early phase of vascular inflammation. The interaction of leucocyte integrins and the intracellular adhesion molecules (ICAMs) 1–5 and vascular cell adhesion molecule 1 (VCAM-1) then play a crucial part in the subsequent processes (28). Cytokines and chemokines promote oxidative stress, a typical characteristic of essential hypertension. Reactive oxygen species (ROS) are the major effector of the oxidative stress injury and consequently may have a crucial role in connecting inflammation, immune system, and hypertension (10). A low inflammatory condition, resulting from massive production of the superoxide anion (·O2) and hydrogen peroxide (H2O2) by endothelial cells, monocytes, and macrophages, promotes oxidative stress and causes vascular dysfunction and target-organ damage in the process of hypertension (29). The activated renin-angiotensin-aldosterone system (RAAS) is a crucial regulator of oxidative stress, and results in the inflammatory damage of vessels (30). As the major component of the RAAS, Ang II promotes vascular inflammation by activating nicotinamide adenine dinucleotide phosphate oxidase oxidases (NOXs) and increasing the expression of endothelin-1 (ET-1), causing the production of a large number of proinflammatory mediators that contribute to the endothelial dysfunction. The main function of NOXs are producing ROS. For example, the low level of ROS produced by NOX2 in physiological state is closely related to the process of cell proliferation and differentiation. However, excessive ROS by activated NOXs is responsible for the CVDs, especially hypertension. Although 7 isoforms of NOXs have been reported, NOX2 and 4 are considered as having tight association with CVDs. NOX 2 and 4 are mainly expressed in endothelial cells and cardiomyocytes (31). NOX2 causes vascular oxidative stress via producing superoxide directly, while NOX4 mainly depend on the production of H2O2 through the rapid dismutation of superoxide into H2O2 (32). The role that activated NOX2 contributes to hypertension via mediating oxidative stress injury and further promoting endothelial dysfunction has been observed (33, 34). According to the results from Toral M et al., the NOX2 inhibitor decreased vascular ROS production and restored endothelial dysfunction in the hypertensive mice (34). NOX4 facilitates vascular hyperproliferation and microvascular remodeling through the induced hyperoxidation and ER stress (35). Furthermore, Ang II, ET-1, and aldosterone may be responsible for impaired remodeling of vessels in a large extent via upregulating expression of chemokines in vascular smooth muscle cells and endothelial cells (36, 37). In addition, activated neutrophils can release ROS as well as proinflammatory cytokines, causing significant oxidative stress. The positive association between the concentration of CRP and the level of oxidative stress has been confirmed (38), and Savoia et al. reported that CRP may upregulate expression of angiotensin type 1 receptors, which could then modulate the formation of ROS (39).
The kidneys have an indispensable role in modulating blood pressure, and renal hypertension resulting from chronic kidney injury and renal artery stenosis is one of most common secondary hypertension diseases globally (40). ROS has been detected in the early stage of chronic kidney disease and exasperates renal function contributing to oxidative stress and chronic inflammation (41, 42), which further accelerates the process of hypertension.
In summary, oxidative stress may be a trigger as well as the result of both inflammation and imbalance in the immune system, contributing to the vascular injury and remodeling, and results in the progression of hypertension.
3 Pathophysiological mechanisms of essential hypertension
Essential hypertension is a disease of unknown etiology that includes the complex interaction of many factors, especially those of genetic and environmental origin. However, it is unclear how these factors increase blood pressure. Increasing evidence indicates that essential hypertension is not a homogeneous disease and that the etiology and pathogenesis vary among individuals. Furthermore, hypertension has a long course and generally slow progression. The mechanisms of initiation, maintenance, and acceleration are different in different stages, and there are interactions among various pathogenic mechanisms (e.g., oxidative stress and inflammation and the immune system) (43). Therefore, hypertension is a multi-factor, -link, and -stage disease that is also affected by differences between individuals. In this section, we intend to briefly summarize the potential pathophysiological process of essential hypertension.
The main mechanisms of hypertension include microvascular remodeling, imbalance of the autonomic nervous system (ANS), and activation of the RAAS (43). The increase of peripheral resistance in small arteries, ranging from 100 to 300 µm in diameter, is the central mechanism and the most significant characteristic in essential hypertension (44). As mentioned above, ROS, activated immune cells, and inflammation, can stimulate endothelial cells to produce and release large amounts of vasoactive substances, such as nitric oxide (NO), prostaglandin-I-2 (PGI2), ET-1, and endothelium-dependent vasoconstrictor factor (EDGF) (44). Age and various cardiovascular risk factors, including dyslipidemia, elevated blood sugar, and smoking, lead to abnormal blood endothelial cell function, increase the production of oxygen free radicals, enhance NO inactivation, vascular inflammation, and oxidative stress response, and influence the elastic function and structure of arteries. With the decreased elasticity of the aorta, the pulse wave conduction velocity increases, and the phase of the reflected wave arriving at the central aorta advances from diastole to systole. The occurrence of a delayed systolic pressure peak can elevate systolic blood pressure and reduce diastolic blood pressure. Changes in the structure (sparse number of tubes or increased wall/lumen ratio) and function (decreased elasticity and increased resistance) and function of resistance arterioles affect peripheral pressure responses.
The position of the origin or the intensity of the reflected wave also has an important part in increasing the pulse pressure. In the nervous system, various reasons can cause changes in the function of the cerebral subcortical nerve center leading to abnormal concentrations and activities of various neuropeptides and neurotransmitters, including norepinephrine, epinephrine, dopamine, serotonin, vasopressin, enkephalin, and brain natriuretic peptide as well as changes in the central renin-angiotensin system. These changes eventually hyperactivate the sympathetic nerve system, increasing the concentrations of plasma catecholamines and constriction of arteries and causing resistance and increased blood pressure (45, 46).
AT-II is the main effector of the RAAS and is involved in a range of processes that can increase blood pressure. These include acting on angiotensin receptor 1, making arteriole smooth muscle contract, stimulating the glomerular zona of adrenal cortex to secrete aldosterone, and promoting the release of norepinephrine through the positive feedback in sympathetic nerve terminal presynaptic membrane. Recently, many tissues, including the blood vessel wall, heart, central nervous system, kidney, and adrenal gland, have been found to contain various components of the RAAS (47). The role of tissue RAAS in the function and structure of the heart and blood vessels may have a greater impact on the occurrence and maintenance of hypertension (44). In addition, water and sodium retention and insulin resistance can contribute to elevating blood pressure.
4 Inflammation in hypertension
Inflammation is a rapid, nonspecific defense response of the body that acts to maintain hemostasis via monitoring and clearing of foreign bodies. This involves coordinating vascular endothelial cells and circulating inflammatory cells and cytokines (48). Although inflammation has beneficial effects, including eradicating pathogens and protecting organs from damage, imbalanced regulation may cause serious and sustained inflammatory response that can in turn cause progressive tissue injury, organ dysfunction, and fibrosis, and even systemic inflammatory response syndrome. Overwhelming evidence shows that low-grade chronic inflammation contributes to the initiation and maintenance of essential hypertension (49, 50). A recent study reported that the chronic inflammatory condition induced by excessive and prolonged stimulation of IIS could lead to vascular endothelial cell damage (51). In this section, we mainly discuss the role of inflammasomes and inflammatory cytokines and of neuroinflammation in the progress of essential hypertension.
4.1 Inflammasomes
The inflammasomes are cytosolic protein complexes, such as NLR-family pyrin domain-containing protein (NLRP)1 and NLRP3 mainly expressed in innate immune cells (like monocytes and macrophages) and endothelial cells, that identify pathogens and activate inflammatory responses, including the intracellular IIS receptors (52, 53). As inflammasomes are major components of the IIS, they mediate important inflammatory responses and pyroptosis, which are tightly associated with endothelial dysfunction. Improperly activated inflammasomes are involved in the potential pathogenesis of many inflammations related diseases (54). NLRP3 is the most characteristic inflammasome among the pattern recognition receptors (PRRs) that recognize damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) to initiate and promote an inflammation response (55). Briefly, a wide range of DAMPs or PAMPs stimulate NLRP3 activation by inducing ion transporting (e.g., K+ efflux and Ca2+ influx) and lysosomal leakage. The activated NLRP3 inflammasome then triggers caspases to induce cleavage of inactive proinflammatory cytokine precursors, such as pro-IL-1β, pro-IL-18, and pro-IL-37 (Figure 1) (48). Numerous studies show that activated NLRP3 inflammasomes are tightly associated with many chronic inflammatory and metabolic diseases, such as ischemic heart diseases and diabetes mellitus, stroke, atherosclerosis, and hypertension (56–59). Studies have suggested that inflammasomes and their related cytokines are tightly associated with raised blood pressure. According to Dalekos et al., an elevated serum concentration of IL-1β was detected in patients with hypertension (60). Endothelial cells are not only the action target of IL-1β but also produce IL-1β (61). In vitro experiments show that monocytes derived from hypertensive patients can release a high level of IL-1β in response to the stimulation of Ang II or lipopolysaccharide (LPS) (62). In addition, the production of mitochondrial ROS is a vital component of cellular oxidative stress, and NLRP3 activation is indispensable in this process. More and more agonists have been reported to produce vascular inflammation and injury by triggering NLRP3 inflammasome activation. According to Xie et al., the NLRP3 inflammasome is involved in visfatin-mediated vascular injury, which may cause atherosclerosis (63). IL-1β and IL-18 would then be released into the blood flow to activate more inflammatory cells and thereby expand the inflammatory response when inflammasomes are activated.
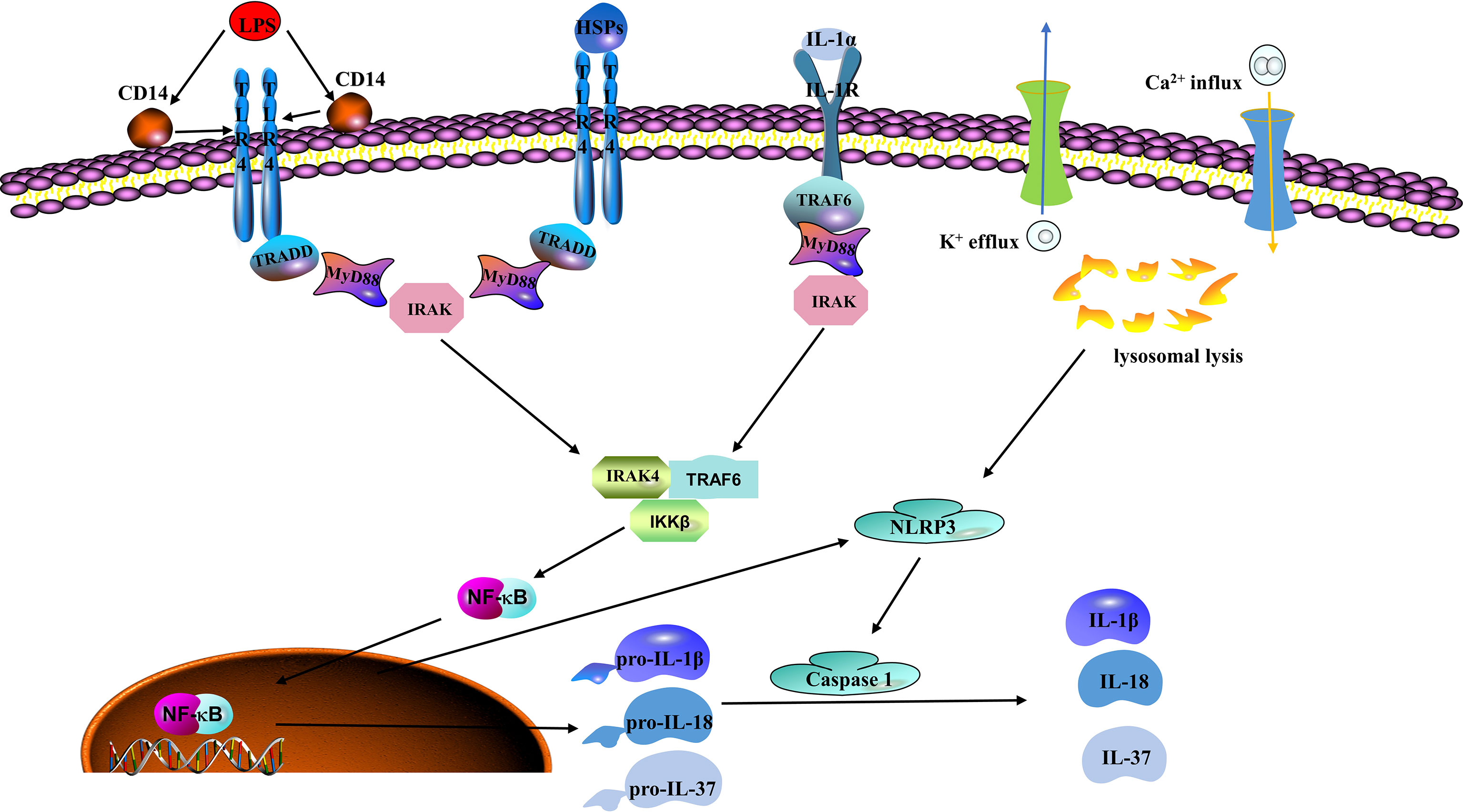
Figure 1 Classic mechanisms of inflammasome activation. Lipopolysaccharide (LPS), which is regarded as the prototype of pathogen associated molecular patterns (PAMPs) and heat shock proteins (HSPs) as a typical representative of damage associated molecular patterns (DAMPs), can be recognized by toll-like receptor 4 (TLR4). Nuclear factor kappa B (NFκB) mediates these signals to produce NOD-like receptor family pyrin domain containing 3 (NLRP3), pro-IL-1β, pro-IL-18, and pro-IL-37. Next, NLRP3 is activated by potassium (K+) efflux, calcium (Ca2+) influx, and lysosomal leakage to recruit cysteine-requiring aspartate protease-1 (caspase-1). Caspase-1 then induces the maturation and release of pro-IL-1β, pro-IL-18, and pro-IL-37 and other related inflammatory agents that cause the inflammatory reaction and participate in the occurrence and development of the disease.
Recent research suggests that endothelial dysfunction is associated with activated NLRP3 inflammasomes through the release of HMGB1. Activated NLRP3 inflammasome and subsequent release of HMGB1 have been considered as the underlying cause of cell-to-cell junction breaks in mouse vascular endothelial cells treated with high-glucose levels (64). Interestingly, NLRP3 deficiency has a significantly protective role via preventing tight junction disruption in diabetic mice and ameliorating endothelial permeability in endothelial cells in vitro (64), and inhibiting the NLRP3 inflammasome by pharmacological inhibition also produced potent therapeutic effects (65). More recently, knockdown of NLRP3 also exhibited remarkable effect on lowering blood pressure, improving vascular remodeling and insulin resistance, and ameliorating or delaying the atherosclerosis via regulating metabolism, relieving oxidative stress and reducing release of inflammatory cytokines (58, 66, 67). WEHD, a caspase-1 inhibitor, could significantly reduce inflammasome activity and prohibited ATP-induced hypertension in mice (68).
4.2 Role of cytokines in raising blood pressure
Traditionally, essential hypertension was thought to be caused by hemodynamic changes, but in recent years, numerous studies have revealed that inflammatory cytokines play a significant part in promoting the progression of hypertension via affecting vascular and renal function. Cytokines are a major component of the immune system and the main performer of inflammatory response, which connects them with vascular remodeling and hypertension. Until recently, the connection between how inflammation and immunity affect blood pressure levels was not well understood. According to previous observations, numerous T cells and monocytes/macrophages accumulated in vessels and kidneys in the mice model with renal ischemia-reperfusion injury (69). Based on other’s and our own research, these cells have been shown to secret potent cytokines that impair organs, especially the heart and kidneys. Modern experimental technologies have demonstrated that a variety of cytokines are involved in the initiation and progression of hypertension (Table 1).

Table 1 Examples of inflammatory cytokines involved in hypertension.
4.2.1 Interleukin 17
IL-17 is derived from TH17 cells, an important subset of CD4+ T cells, and includes several isoforms, including IL-17A and IL-17F; several B cells and CD8+ cells also produce IL-17 (84). IL-17A is generally considered to act synergistically with other cytokines (e.g., TNF) during inflammatory response. IL-17A and TNF-α have been demonstrated to synergistically enhance lung inflammation and expression of C-C motif chemokine ligand 2 (CCL2) in an NADPH oxidase-dependent manner (70), which affects vascular functions by influencing gene expression (71). Moreover, IL-17A is reported to be involved in multiple inflammatory diseases, such as asthma, psoriasis, and systemic lupus erythematosus (84). Considering the important position of IL-17 in inflammation-related diseases, research on anti-IL-17 monoclonal antibodies (Ixekizumab and Secukinumab) and anti-IL-17 receptor antibodies (Brodalumab) has become a scientific hotspot in recent years, and these antibodies have shown satisfactory treatment effects in clinical trials (85, 86).
Growing evidence has linked IL-17A to increased blood pressure, although the exact mechanisms are unclear. Plasma IL-17A concentrations were significantly increased in Ang II-induced animal models with hypertension. In contrast, Ang II infusion could not impair endothelial function or result in an elevation of blood pressure in IL-17A-/- mice. A similar result was observed when the level of oxidative stress was lowered and infiltration of T cells decreased in IL-17A-/- mice compared with that in the control group. Treatment with anti-IL-17 antibody was found to significantly lower blood pressure and reduce collagen deposition in mouse heart and kidneys. Consistent with these findings, when co-cultured human vascular smooth muscle cells (VSMCs) with TNF-α in vitro, IL-17A facilitate the expression of various cytokines and chemokines, such as CCL7 and 8, CXCL2 and CSF3, which could recruit more inflammatory cells. Subsequently, another exact role of IL-17A in hypertension has been demonstrated by Nguyen et al. that IL-17A could reduce the production of NO by phosphorylating threonine 495 of endothelial nitric oxide synthase (eNOS) in porcine aortic endothelial cells (87). And the study also showed intravenous administration of IN-17A could elevate the blood pressure modestly in the normal mice. Taking together, inhibition of IL-23/IL-17 axis has been regarded as promising therapeutic target in the future precise therapy of hypertension (72).
4.2.2 Interleukin 6
IL-6 is a 21 kDa glycoprotein secreted by monocytes, macrophages, and dendritic cells, which can activate related genes and upregulate expression of receptors involved in cell proliferation, differentiation, and apoptosis. Once the receptor recognizes the IL-6 signals, the related cellular events, such as activation of Janus kinases and Ras-mediated signal pathways, are initiated immediately (88). IL-6 is responsible for regulating the expression of the acute-phase plasma proteins in liver cells. In addition, IL-6 could facilitate the production of IL-17 via polarizing CD4+ T cells. Mounting evidence indicates that IL-6 has a crucial role in aspects of the chronic inflammatory response, such as hypertension, rheumatoid arthritis, and ischemic heart disease. Blocking IL-6-related signaling pathways by using the human monoclonal antibody Tocilizumab has shown considerable clinically therapeutic effect on rheumatoid and juvenile arthritis, and related clinical trials are being performed in other inflammatory diseases (89). Therefore, IL-6 may well have a potential therapeutic role in CVD treatment, especially with hypertension.
Accumulating evidence strongly indicates that IL-6 signaling pathways are a vital link in Ang II-induced hypertensive animal models, as the level of IL-6 had a positive relationship with blood pressure and was significantly lower when the Ang-II receptor was blocked (73). In vivo animal studies show that injection of Ang II can increase plasma IL-6 concentration and blood pressure, whereas the blood pressure-raising effect of Ang-II was clearly weakened in IL-6-/- mice (74, 75), and was also blocked by spironolactone, suggesting the activation of mineralocorticoid receptors. In addition, evidence from in vitro experiments indicated that IL-6 promotes the expression and activity of sodium channels in mouse cortical collecting duct cells (90), implicating that IL-6 may have the ability to facilitate water and sodium retention in vivo. Thus, these research suggested that IL-6 may a potential and promising therapeutic targets in lowering blood pressure leading to the inhibition of the activity of mineralocorticoid receptors and sodium channels in duct cells.
4.2.3 Tumor necrosis factor-α
TNF-α is a well-established inflammatory cytokine in the acute phase response, whose expression has been shown to increase in human and rodent hypertension studies (91). The role of TNF-α signaling in modulating many secondary inflammatory processes, such as cytokine secretion, cell differentiation, and apoptosis, is complex and cell-type and dose-dependent (92). Clinical trial data show that elevated plasma IL-6 concentrations are tightly associated with the severity of refractory hypertension and the six-year risk of death (93). In contrast, TNF-α inhibitors could lower the level of blood pressure in patients and animal models (76, 77). Although a variety of cells can secrete TNF-α, which can activate other immune cells through TNF-α receptors (TNFR1 and TNFR2), it is mainly secreted by monocytes and macrophages. The activation of TNF-α receptors is responsible for cell apoptosis, NADPH oxidase activation, and nuclear factor kappa B (NFκB) activation (78). An accumulating body of studies suggest that NFκB and NADPH oxidase activation promote hypertension by upregulating the expression of chemokines and adhesion molecules in blood vessels, facilitating microvascular remodeling and sodium retention (79). Superoxide production from NADPH oxidase could react with endothelial NO to form the strong oxidant peroxynitrite. This can severely affect vasodilation and cause a significant elevation of blood pressure because of the decrease in NO levels. Moreover, TNF-α can affect the promoter of eNOS and help destabilize the eNOS mRNA structure, which eventually leads to the massive degradation of eNOS and reduction of NO synthesis (80, 81). In kidneys, TNF-α can inhibit eNOS activity in the renal medulla in a Rho-kinase dependent fashion, thereby reducing production of NO which inhibits sodium reabsorption at several sites along the renal tubule (94, 95), and can result in renal injury directly which shift the pressure natriuresis curve to promote the elevation of blood pressure. Thus, TNF-α plays an important role in elevating blood pressure. Unfortunately, TNF-α inhibitors are currently mainly used to treat autoimmune diseases, and there is no evidence that they can be used to lower blood pressure and improve cardiovascular prognosis, but that is worthy to explore and evaluate in the future.
4.2.4 Interferon gamma
IFN-γ is the only known member of the type II family of interferons and is mainly produced by T helper (Th) 1 cells. IFN-γ is not only an important part of adaptive immune responses but also indispensable in protecting hosts from infection. William et al. reported that IFN-γ-producing CD4+ and CD8+ T cells are consistently increased in hypertensive mice (21), but, according to the results from Ishimitsu et al., subcutaneous injections of IFN-γ could lower blood pressure and alleviate proteinuria and glomerular injury in Dahl salt sensitive rats (96). However, the effect on lowering blood pressure did not observe in the mice with essential hypertension (96). The study neglected the role of endogenous IFN-γ in hypertension, which might be responsible for the conflicting results. The previous study suggested that IFN-γR deficiency can significantly attenuate ventricular hypertrophy and ventricular electrical remodeling (82). Although high doses of angiotensin were injected, blood pressure levels did not increase significantly, but the degree of renal fibrosis was reduced while the glomerular filtration rate was maintained in mice lacking the IFN-γ receptor 1 compared with that in wild-type mice (82). One of the most important mechanisms whereby IFN-γ triggers hypertension is that IFN-γ can increase the expression of angiotensinogen of rat renal proximal tubule cells in a JAK2/STAT3-dependent manner (97), which consequently increases blood volume. Therefore, it is reasonable to infer that interferon-secreting T cells may establish a link with RAAS by regulating the production of angiotensinogen, resulting in an increase in blood pressure.
4.2.5 Interleukin 1 beta
IL-1β is known as an important component of the IL-1 family of interleukins and mainly derived from monocytes, T cells and neutrophils. A growing amount of evidence has confirmed the association between IL-1β and hypertension (98). Specifically, it has been demonstrated that IL-1β could upregulate many proinflammatory genes, including IL-6, IL-17 and IFN-γ, resulting in further tissue injury and inflammation related events, like hypertension and myocardial infarction (99–101). High levels of IL-1β have been detected in the serum of patients with essential hypertension in recent studies (102), indicating the role of IL-1β in elevating blood pressure. Considering the tight association with hypertension and the essential role in inflammation, more and more studies on how the IL-1β involves in pathophysiological mechanisms of hypertension have been performed and explored. According to a previous study, IL-1β not only triggers the inflammatory response directly but also mediates the phenotype and functions of VSMCs and eventually leads to vascular remodeling on inflammatory-dependent or independent mechanisms (103). A recent study which aimed to evaluate the effect of an IL-1R1 receptor Inhibitor (Anakinra) on lowering the blood pressure in patients with obesity suggested Anakinra could significantly lower the systolic blood pressure and peripheral vascular resistance (104), which supported that the IL-1β could affect the progression of hypertension. Another study suggests that IL-1β derived from renal tubular epithelial cells of diabetic db/db mice could polarize the naïve macrophages into M1 subtype which releases a large amount of IL-6 resulting in salt-sensitive hypertension, but could be blunted when inhibited the synthesis of IL-1β or knocked out the IL-1 R1 in immune cells (83). Thus collectively, IL-1β plays a crucial role in the progression of essential hypertension and may be a novel promising therapeutic targets of hypertension.
4.3 Gut microbiota and neuroinflammation in hypertension
Recently, we have gained greater understanding of the role of gut microbiota and neuroinflammation in the pathogenesis of hypertension. Interaction between the gut microbiota and epithelial cells of the gut-brain axis is involved in regulating ANS activity to control blood pressure. The endocrine pathway of hypertension mainly involves the activation of the hypothalamus-pituitary-adrenal axis. The complex interactions of immune-regulatory organs such as bone marrow, gut, and spleen, which can enhance vascular tone and contraction, can contribute to increases in peripheral resistance and blood pressure.
In addition, increased neural activity may result in neuroinflammatory events, activating microglia, producing more proinflammatory molecules, and an inflammatory environment in autonomic brain regions. Disorder of the gut-brain axis, including gut microbiota dysbiosis, gut epithelial injury, and deranged brain input, is responsible for hypertension through inflammatory mediators, metabolites, circulating bacteria, and altered afferent information, causing neuroinflammation and disorder of the ANS. This in turn can negatively impact gut functions and associated microflora to create a vicious spiral. In this section, we address the role of an impaired gut-brain axis in the pathophysiology of hypertension.
The ANS can regulate the arterial blood pressure by controlling the vasomotor activity of sympathetic and parasympathetic nerves. Numerous inflammatory cytokines, including IL-1β (105), TNF-α, IL-6 (88), and IFN-γ (91), were detected in animal brains in hypertension models, and these cytokines have been demonstrated to raise blood pressure by increasing sympathetic nerve activity. Furthermore, the impaired blood-brain barrier is another important cause of neuroinflammation. For example, increasing evidence suggests that Ang-II as well as gut microbes and their metabolites affect the blood-brain barrier permeability, which allows more cytokines to enter the brain. Once the blood-brain barrier is disturbed, circulating inflammatory factors and toxins can enter autonomic brain regions, interfering with normal neural activity and causing hypertension (106). Microglial cells are the important part of the IIS in the brain, and responsible for homeostasis of the central nervous system via clearing senescent and apoptotic cells (107). Inhibition of microglia activation helps to decrease hypertension and alleviate sympathetic activation and peripheral inflammation (108). Interestingly, specific deletion of microglia could significantly relieve the Ang II-induced elevation of blood pressure and inflammation (109).
In recent years, the gut microbiota has become a hot research topic in CVDs and metabolic diseases and is considered as the most promising interventive target of chronic diseases in the coming decades. Rapid developments and major breakthroughs in sequencing technologies have helped recognize the important role of the gut microbiota on body homeostasis, especially regarding obesity, insulin resistance, and CVDs. Animal and human experiments consistently showed that patients with hypertension had significantly lower gut flora richness compared with that of normotensive subjects (110). Two concurrent studies have reported the relationship between gut microbiome composition and hypertension (111, 112). A recent study by Mell et al. highlighted differences in the cecal microflora between salt-sensitive and -tolerant strains in Dahl rats (111). Moreover, changes in the variety and abundance of several short chain fatty acids in the plasma were observed after cecal transplantation. Thus, there is reason to believe microbial composition may affect the levels of short chain fatty acids in the plasma via roles in metabolism. Significant dysbiosis due to decreased microbial richness, diversity, homogeneity, and increased Firmicutes/Bacteroides ratios in hypertensive animals have been observed by comparing changes in the fecal microflora in spontaneously hypertensive and chronic Ang II-infused hypertensive rats (112). Another difference between the microbiota of normotensive and hypertensive patients is the production of LPS. Veillonellae are enriched in gut microbes in human hypertensive patients and produce LPS. Biofilms encapsulate bacterial communities in an extracellular matrix produced by bacteria, increasing their adhesion, and creating an environment that is more suitable for their grow. LPS-producing bacteria in biofilms were more inflammatory than Gram-negative bacteria were without biofilm protection (113, 114). This shows that the hypertensive microbiota are more inflammatory throughout the body and, because of the arrival of LPS in the brain, are more responsible for the possibility of ANS activation and hypertension.
5 Immunity in hypertension
The human immune system includes the IIS and the adaptive immune system (AIS), and the components of these function in an extremely complex and complementary manner to exterminate invaders and rescue injured tissue, thereby maintaining homeostasis. Most immune cells become involved in the initiation, progress, and maintenance of hypertension (Figure 2). Briefly, in the presence of genetic susceptibility, several environmental factors, especially salt and environmental stress, can cause a small rise in arterial blood pressure responsible for activating sympathetic nerves or inhibiting parasympathetic nerves. Next, elevated blood pressure causes or aggravates tissue damage and together with oxidative stress damage caused by Ang II and ET-1 is conducive to the formation of DAMPs and new antigens, and thereby initiates subsequent inflammatory activation. DAMPs trigger the activation of the IIS by recognizing and activating the Toll-like receptors (TLRs) on antigen-presenting cells while the new antigens enhance the immunogenicity of dendritic cells and facilitate their production and release of proinflammatory cytokines, which not only enable the proliferation and activation of T cells but also further produce more cytokines. DAMPS and new antigens can activate γδ T cells, which can activate T lymphocytes directly. Several infectious diseases can also enhance the activation of the IIS in a PAMP-dependent manner. Innate immune cells and γδ T cells release proinflammatory cytokines as well as autoantibodies, directly or through the activated AIS, producing vascular and renal damage, which is a feedforward process contributing to a progressive raise in blood pressure. In addition, additional effects, such high salt intake, may reactivate the T effector memory cells in the lymphoid organs, which will induce a more intense inflammatory response and cause more severe tissue damage. M2 macrophages and Treg cells have a role in anti-inflammatory response during this process, although their effect is very weak and not enough to reduce the organ damage.
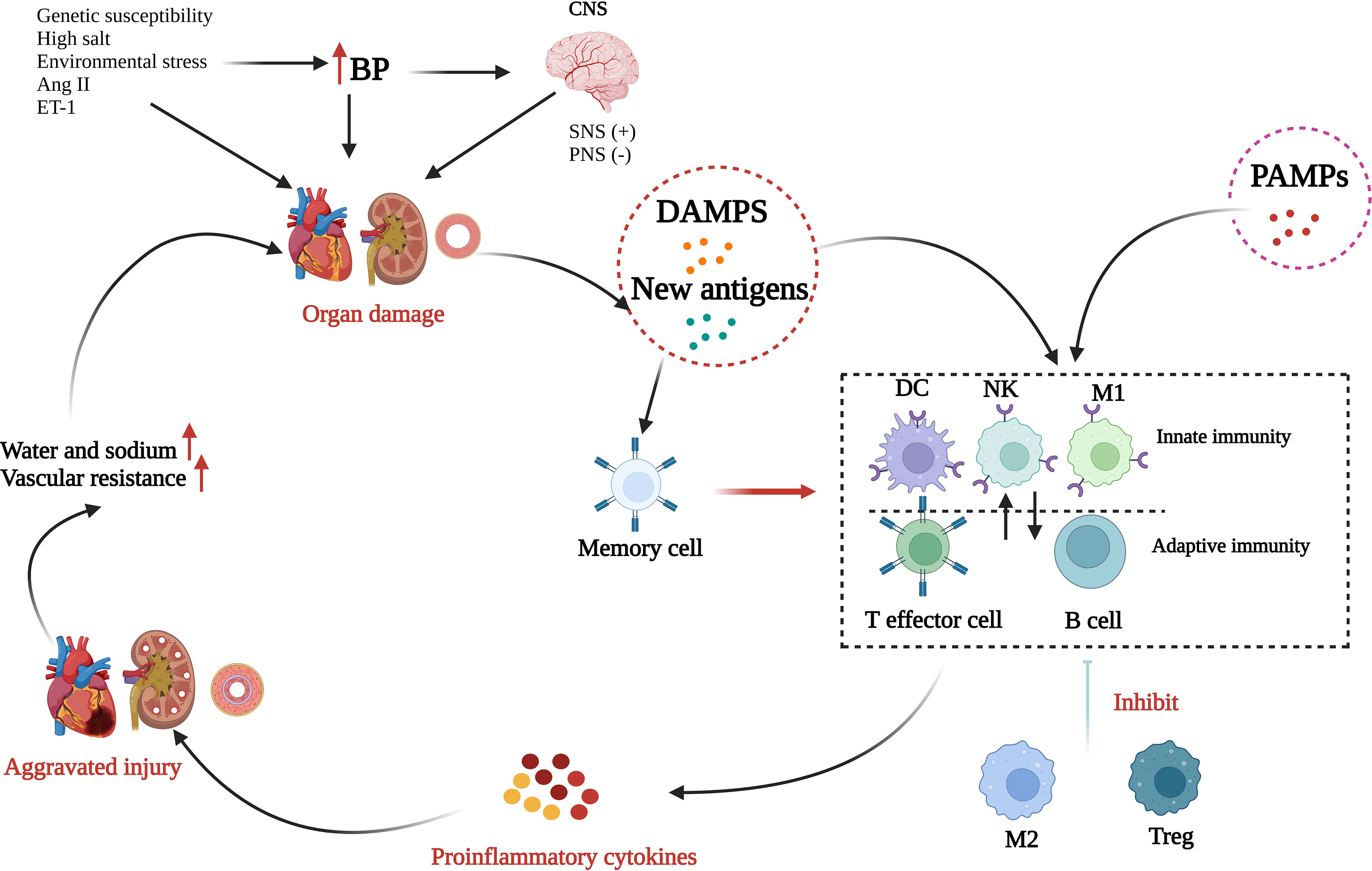
Figure 2 Role of inflammation and immunity in hypertension. Many risk factors, such as genetic susceptibility, high salt, and environmental stress, can elevate blood pressure (BP) and consequently activate the sympathetic nerve system (SNS) or inhibit the parasympathetic nerve system (PNS). Over time, elevated BP can induce organ injury promoting the formation of damage-associated molecular patterns (DAMPs) and new antigens. In addition, several infectious diseases can enhance the activation of the innate immune system through the binding of pathogen-associated molecular patterns (PAMPs) to toll-like receptors. DAMPs, new antigens, and PAMPs activate innate immunity which interacts with adaptive immunity. Next, numerous proinflammatory cytokines are released and result in further organ injury. Through alternatively activated macrophages (M2) and Tregs have roles in anti-inflammatory response, the effect is too weak to reduce the organ damage. Finally, peripheral vascular resistance and blood volume increases to result in hypertension (Created with BioRender.com).
5.1 Innate immune system
Innate immunity is the rapid, nonspecific defense response to a variety of exogenous stimuli that is often considered as the early stage of inflammation. The key effectors are mainly monocytes, M1 macrophages, natural killer cells, and neutrophils. Monocytes/macrophages have been revealed to be tightly associated with the microvascular remodeling. In a rodent model with a deficiency in macrophage colony stimulating factor, Ang II failed to induce endothelial injury and vascular oxidative stress or elevate blood pressure (115). Consistently, removing lysozyme M-positive myelomonocytic cells prevented mice from suffering severe hypertension (116). As mentioned above, macrophages are crucial to the progress of hypertension and, although the exact mechanisms remain elusive, increasing evidence indicates that a large amount of ROS derived from activated macrophages may cause irreversible vascular endothelial cell damage and microvascular remodeling, causing increased peripheral arteriolar resistance (116, 117). Experiments have shown that activated monocytes can produce endothelial dysfunction. Chemokines become involved in the pathological process of hypertension by recruiting immune cells to the site of endothelial injury, causing further impairment of endothelial function. An increase in the number of monocytes and neutrophils with the CXCL1 receptor CCR2 can be clearly detected in patients with hypertension, suggesting that blocking CCR2 may alleviate inflammation and control blood pressure. Neutrophils are an important component of the IIS and produce large quantities of cytokines and superoxide anion when stimulated (118). Ang-II could connect thromboinflammation with essential hypertension by inducing neutrophil to release extracellular traps. The use of plasma from untreated patients with hypertension to stimulate neutrophils in normal patients can facilitate the production of endothelial collagen, thereby causing vascular remodeling and injury (119). More recently, the gene SH2B3 (LNK) that encodes SH2B adaptor protein 3 have been confirmed that regulate the immune cell (notably T cells and macrophages) development, differentiation, and signaling in the hypertension according to the genome-wide association studies (120). Therefore, elucidating molecular mechanisms where SH2B3 regulating immune cells in hypertension may found novel therapeutic targets for hypertension.
5.2 Adaptive immune system
Increasing evidence suggests that the activated AIS promotes the pathogenesis of hypertension and aggravates targeted organ damage. The AIS executes a highly specific response and is regulated by activated T and B lymphocytes. According to specific markers on the cells, T lymphocytes are divided into two subpopulations: CD4+ and CD8+ T lymphocytes. Although it has been confirmed that both types of T cells are involved in the initiation and progression of hypertension, results based on in vivo models showed that CD4+ T cells are likely to the key factor in promoting hypertension (121). However, Trott et al. reported that CD8+ T cells can be a crucial motivating force in experimental hypertension. Once the endogenous or exogenous antigens presented by antigen-presenting cells are recognized, lymphocytes are activated and differentiated into T effector cells or Treg cells (122). Under the action of inflammatory cytokines or chemokines, these activated T lymphocytes target the site of inflammation. The balance among T cell subsets can influence inflammatory responses. Kassan et al. (123) described the relationship between Tregs and vascular dysfunction in patients with hypertension and found that endothelial damage, plaque rupture, and arterial occlusion mainly resulted from the imbalance of Treg cells.
The hypothesis that T lymphocytes are the important participant in the pathogenesis of hypertension was proposed many years ago but failed to receive enough attention initially. It was not until the past 20 years that experimental evidence confirmed the exact role of T lymphocytes in elevating blood pressure and promoting vascular injury (76). Elevated blood pressure may further activate the immune response via modifying self-antigens or generating new antigens. Studies have shown that several T lymphocyte subsets may promote the occurrence of hypertension and vascular remodeling (124). T- and B-lymphocyte-deficient mice, produced by silencing recombination-activated gene 1 (RAG1−/−), exhibited a blunted hypertensive response compared with that in control mice in the Ang II-induced hypertension animal model. Adoptive transfer of effector T cells, but not B cells, to the RAG1−/− mice restored the effects of Ang II (76). Moreover, in mice lacking T cells, there was insufficient infiltration of innate immune cells in vascular pathological sections, which was possibly related to the absence of cytokines derived from Th cells (76).
Studies have demonstrated that T lymphocyte-mediated immune responses can be induced by oxidative stress. The role of Th17, an important part of effector T cells, in hypertension has been identified by Madhur et al. (125). Th17 not only promotes but can also inhibit inflammation. Using the Ang II-induced hypertension animal model, improved vascular function, lowered oxidative stress levels, and decreased T lymphocyte infiltration have been observed in IL-17−/− mice compared with these parameters in control mice (125). Madhur et al. revealed that the blood pressure level is positively correlated with the amount of circulating Th17 cells and that the inhibition of IL-17 contributes to the improvement of hypertension, which is consistent with previous research. In addition, enhanced acquired immunity due to genetic susceptibility, and vascular inflammation due to decreased Treg immunosuppressive function may contribute to hypertension (126). Viel et al. proposed a famous hypothesis that the genetic predisposition based on loci on chromosome 2 where many proinflammatory genes locate enhances adaptive immunity (127).
6 Oxidative stress in hypertension
Oxygen molecules can easily form free radicals because of their special electronic arrangement structure. These oxygen molecules are called oxygen free radicals (OFR) and include the superoxide anion, hydroxyl radical, and NO free radical. The hydroxyl radical is the most active OFR found so far. Furthermore, ROS, which includes H2O2 and O3 as well as OFR, is a critical signaling molecule that mediates the activation of transcription factors, induction of immune response genes, and the phosphorylation of kinases (128). Cumulative evidence from humans and animals suggests that ROS plays an important part in regulating endothelial cell function and vascular remodeling (129, 130). In the development of hypertension, the interaction of ROS and humoral factors such as ET-1 and Ang-II increases the production of ROS, especially the superoxide anion produced by the uncoupling of NOXs family and eNOS (130). Recently, studies have suggested that hypertension is associated with the decrease in NO and the increase in oxidative stress (131). ROS directly inhibits the activity of NO (132). In addition, ROS can stimulate the PI3K/Akt-MAPK pathway related to redox transcription factors causing overexpression of redox genes and thereby inhibiting the expression of eNOS mRNA and eNOS activity, which reduces the availability of NO. NADPH oxidase is regarded as the most important provider of ROS in vascular walls and endothelial cells and has an indispensable role in the pathogenesis of endothelial dysfunction and vascular remodeling. There is another crucial source of eNOS, xanthine oxidase and mitochondrial uncoupling, that helps to explain the increase in ROS production in different vascular diseases (133, 134). Previous evidence demonstrated that ROS regulates the arrangement of various proteins and the role of signal pathways in cells and that this redox biology is precisely and spatially regulated to influence the individual healthy conditions. Moreover, disordered physiological ROS production can cause a variety of diseases, including hypertension. During hypertension, neurohumoral factors can stimulate receptors located on the cell membrane to activate NADPH oxidase and mitochondria to produce ROS, such as highly active superoxide anions, which can then initiate cellular phosphorylation pathways (135). This subsequently initiates gene expression of factors such as p53, activating protein-1 (AP-1), nuclear E2-related factor 2 (Nrf2), and NFκB. Eventually, these changes result in endothelial dysfunction and hypertension (136, 137).
7 Novel potentially therapeutic strategies in hypertension
Multiple animal experiments have proved that an insufficiency of vascular endothelial cells is closely related to the increase in arterial blood pressure. Considering the pathogenesis, prevalence, and severity of hypertension, it is critical to find novel and potential therapeutic strategies to lower blood pressure. In this section, we will summarize and explore the role of anti-inflammation and antioxidant therapies for hypertension (Figure 3).
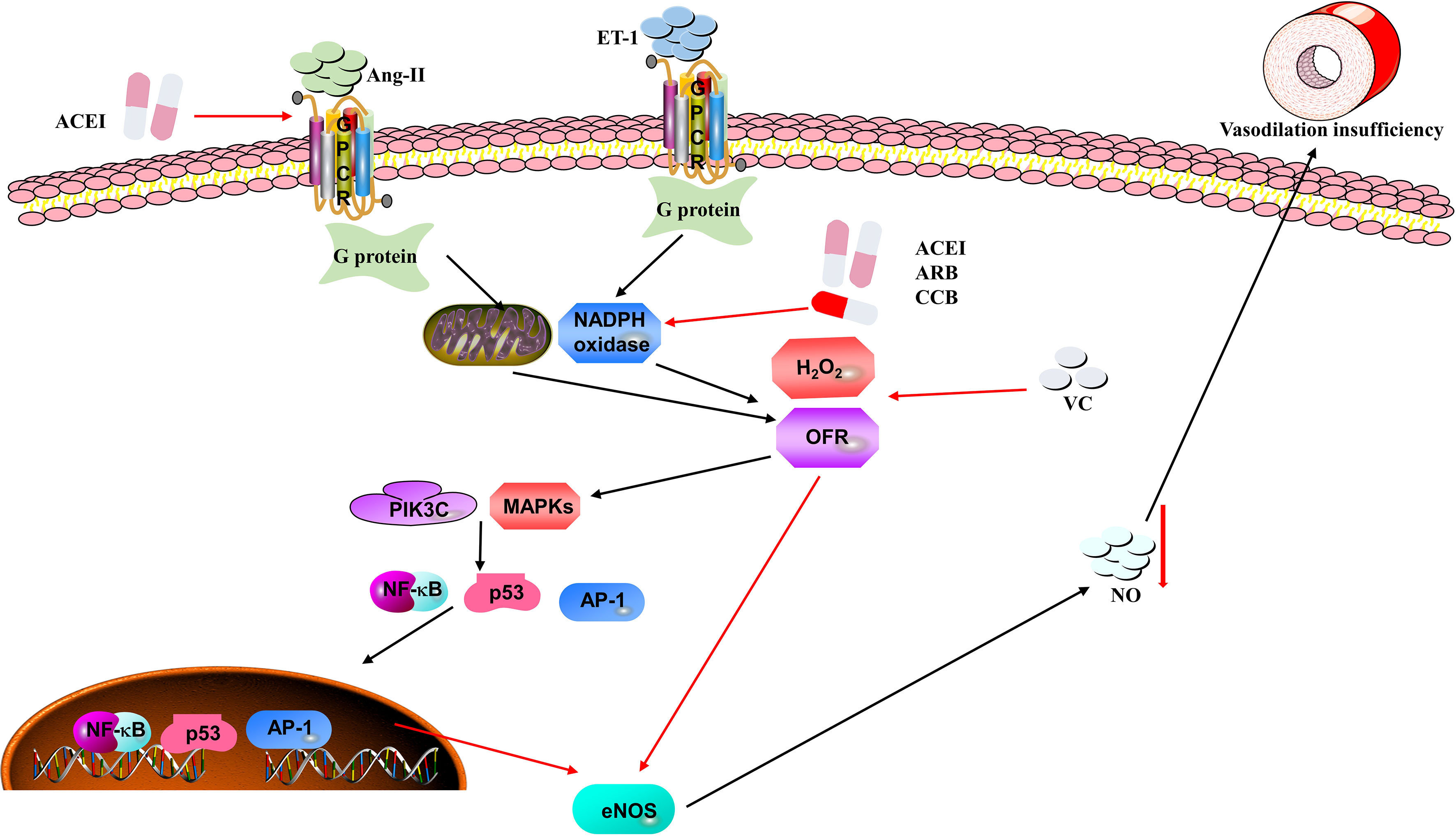
Figure 3 Mechanisms of oxidative stress causing hypertension in vascular endothelial cells. Ang II and ET-1 can stimulate NADPH oxidase and mitochondria to produce ROS (e.g., H2O2 and OFR), which are recognized by their receptors on the endothelial cells. ROS stimulate the PI3K/Akt-MAPK pathway to inhibit the expression of eNOS mRNA and eNOS activity, thus reducing the availability of NO, which results in vasodilation insufficiency and elevation of blood pressure. Moreover, angiotensin converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), calcium channel blockers (CCBs) and vitamin C (VC) may be potential therapeutic strategies in the process of oxidative stress.
7.1 Anti-inflammation therapeutic role in hypertension
As mentioned above, chronic inflammation has a close association with elevated blood pressure. Clinical studies indicate that elevated CRP levels at baseline are more likely due to an increased risk of hypertension (138). Although inflammatory activation is closely associated with elevated blood pressure, so far there is no obvious data supporting that routine use of anti-inflammatory drugs can treat hypertension. Table 2 describes several drugs that may lower blood pressure via an anti-inflammatory effect. A preliminary analysis of one clinical study shows that minocycline, a broad-spectrum tetracycline antibiotic has a continuous antihypertensive effect in patients with hypertension (139). Mycophenolate mofetil is a well-known immunosuppressant, whose active metabolite inhibits the activation and synthesis of T and B lymphocytes by inhibiting dihydrolactate dehydrogenase and thereby decreases the synthesis of pyrimidine nucleotides, which can significantly lower blood pressure in salt-sensitive hypertension (140). In addition, a significant effect of mycophenolate mofetil on lowering blood pressure has been observed in patients suffering from psoriasis and rheumatoid arthritis (141). Another observational study showed that long-term use of immunosuppressive drugs can alleviate arterial stiffness and lower blood pressure in patients with chronic kidney disease (142). A monoclonal antibody against IL-17A could lower baseline blood pressure in patients with psoriasis (143). In addition, an IL-1 receptor antagonist (anakinra) could significantly reduce the blood pressure after a 14-day treatment via specifically inhibiting the actions of IL-1α and IL-1β (145, 146), but no significant antihypertensive effect was observed with canakinumab (an IL-1β antagonist) in large-scale randomized controlled trials in patients with previous myocardial infarction and elevated hsCRP (147). The study suggests that the mechanisms of reducing major adverse cardiovascular events are not associated with the change of blood pressure. The main reason that canakinumab does not lower the blood pressure may be related to the enrolled populations of whom 80% are patients with hypertension and were taking antihypertensive drugs, which may mask the real antihypertensive effect of canakinumab. Statins are widely used lipid-controlling drugs in clinics and can significantly reduce cholesterol levels, thereby reducing the risk of CVDs and improving prognosis. Statins also show multiple effects and have particular benefits on the cardiovascular system. Mounting evidence has demonstrated that statins have mild antihypertensive effects, especially in patients with intractable blood pressure and with hypertensive target organ damage (102, 144). The underlying mechanism whereby statins reduce blood pressure may be associated with the protective effect on the vascular endothelium by inhibiting the production of ROS, reducing the circulating level of proinflammatory cytokines, and inhibiting the expression of adhesion molecules on vascular endothelial cells and smooth muscle cells.

Table 2 Anti-inflammation therapeutic strategies in hypertension.
7.2 The value of antioxidant therapy in hypertension
Antioxidants are substances that effectively trap ROS, so they can reduce oxidative damage and may lower blood pressure (Table 3). The use of antioxidants, such as vitamin C and E, have been confirmed as effective therapeutic strategies in lowering the level of blood pressure. Vitamin C as an enzyme manager, increases eNOS activity and reduces ROS, and has been proved to improve vasodilation in hypertensive patients (158). Studies from humans and animals have shown that vitamin C can enhance endothelial function in a variety of situations (159). The effect of antioxidants on lowering blood pressure and enhancing vascular function has been confirmed in the hypertensive animals (160). One possible reason is that the free contractile response to norepinephrine is increased in patients with hypertension, which can be reduced by vitamin C (148). However, in several large-scale clinical trials, the effect of supplementation of vitamin C on blood pressure was unpredictable and more experiments are needed to confirm the exact effect (161–163), which may result from the reduced bioavailability of NO. B-cell lymphoma 6 (BCL6) is known as a key intervention target for autoimmune diseases by inhibiting production of ROS and apoptosis (149); however, it is unclear whether this can reduce hypertension. Chen et al. recently reported that BCL6 can lower blood pressure by suppressing vascular smooth muscle cell proliferation, attenuating oxidative stress injury, and microvascular remodeling in the Ang-II induced hypertensive rats (150).
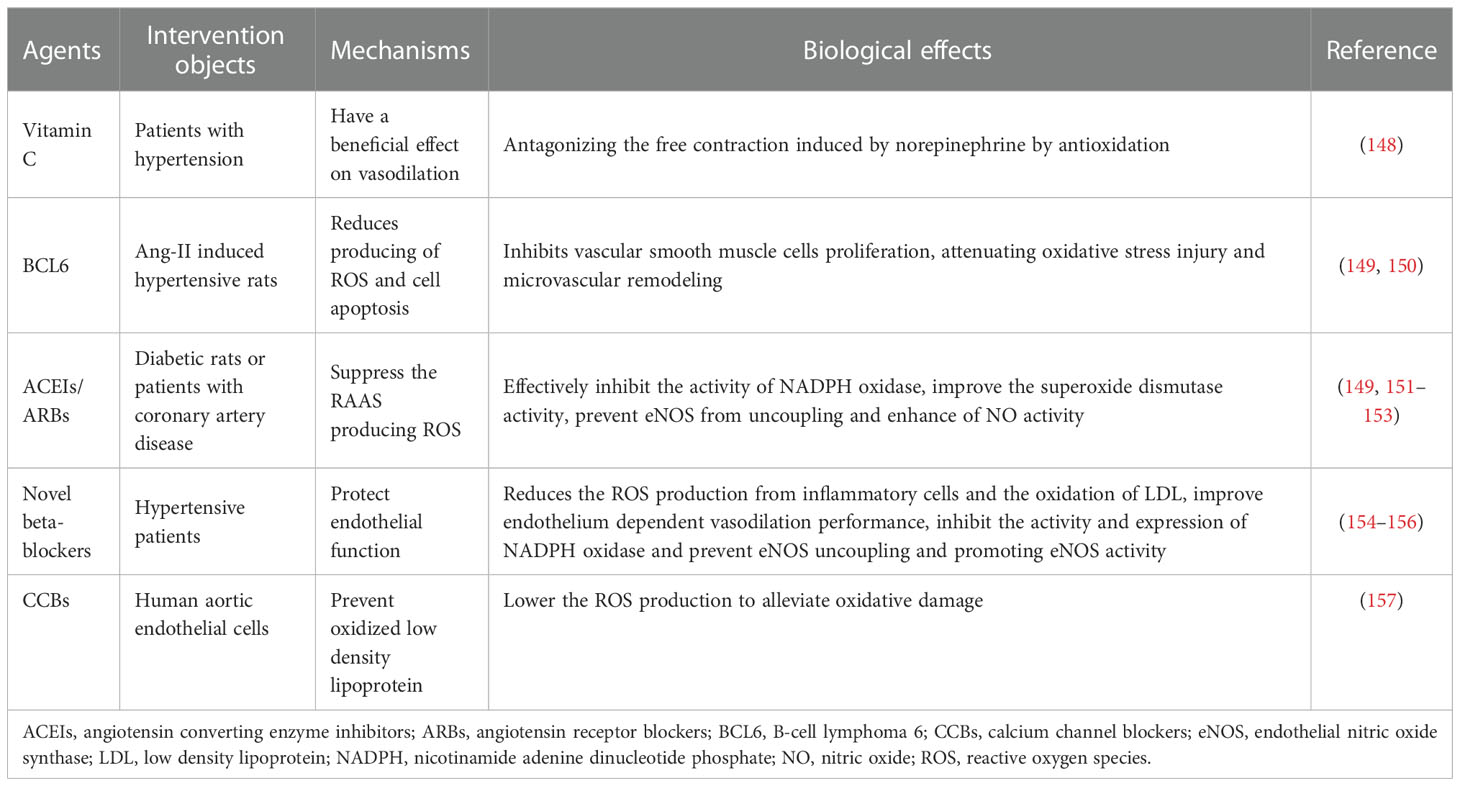
Table 3 Antioxidant therapy in hypertension.
In addition, several antihypertensive drugs currently used in the clinic have a significant and specific effect on decreasing the incidence of hypertension independent of the expected mechanism. Next, we will mainly discuss the ACEI, ARB, beta-blockers, and CCBs. RAAS has a crucial part in the production of ROS during the process of hypertension. NOXs are recognized as one of most important sources of ROS in the endothelium and are mainly induced by Ang-II (149). Both ACEI and ARB can not only effectively inhibit the activity of NOXsbut improve the superoxide dismutase activity (151). In addition, they can prevent eNOS from uncoupling and enhance NO activity (152, 153). The novel beta-blockers, nebivolol and carvedilol, have been reported to have antioxidant properties. According to the results from clinical and experimental studies, these drugs have favorable protective effects on endothelial function and do not depend on the activity of beta-blockers. The antioxidant effect of carvedilol is mainly based on reducing the ROS production from inflammatory cells and the oxidation of low-density lipoprotein (LDL). Carvedilol and nebivolol could improve endothelium dependent vasodilation performance in patients with hypertension (154). Moreover, nebivolol exhibits a protective effect in the endothelial function and lowers blood pressure through suppressing the activity and expression of NADPH oxidase, thereby preventing eNOS uncoupling and promoting eNOS activity to produce more NO (155, 156). CCBs, especially dihydropyridines (like nifedipine) could directly reduce production of ROS to protect the endothelial function. CCBs have been demonstrated to prevent oxidized LDL, which can promote ROS production to trigger oxidative damage from causing endothelial dysfunction (157). Therefore, in addition to the direct effect of lowering blood pressure, dihydropyridine, CCBs play an additional antihypertensive effect by improving the function of vascular endothelial cells through antioxidation.
8 Conclusion
Accumulating evidence shows that hypertension is a chronic inflammatory condition that involves the migration, accumulation, and activation of immune cells and the production of ROS. Although many studies have been performed, the specific molecular mechanisms of inflammation and immunity that affect elevated blood pressure remain unclear. Inflammatory or oxidative stress can damage vascular endothelial cells and cause microcirculation remodeling; however, whether this can increase blood pressure through other mechanisms requires more research. Although there are many drugs that can treat hypertension, a deeper understanding of the mechanism of elevated blood pressure and the discovery of more action targets will be more conducive to earlier detection and intervention of hypertension and thereby significantly reduce the occurrence of cardiovascular adverse events. As emphasized in this review, the occurrence of hypertension is complex, so it is necessary to understand the pathological mechanism of the different stages. In addition, there is also an urgent requirement for the research and development of new antihypertensive drugs for different targets.
Author contributions
(I) Conception and design: All authors. (II) Administrative support: None. (III) Provision of study materials or patients: None. (IV) Collection and assembly of data: None. (V) Data analysis and interpretation: None. (VI) All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Key Research and Development Program of China (2016YFC1300100), Nonprofit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019XK320057), CAMS Innovation Fund for Medical Science (CIFMS, 2022-I2M-C&T-A-010).
Acknowledgments
We thank Mike Herbert, PhD from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: A comparative risk assessment. Lancet Diabetes Endocrinol (2014) 2:634–47. doi: 10.1016/S2213-8587(14)70102-0
2. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in blood pressure from 1975 to 2015: A pooled analysis of 1479 population-based measurement studies with 19·1 million participants. Lancet (2017) 389(10064):37–55. doi: 10.1016/S0140-6736(16)31919-5
3. Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: Executive summary: A report of the American college of Cardiology/American heart association task force on clinical practice guidelines. Hypertension (2018) 71(6):1269–324. doi: 10.1161/HYP.0000000000000066
4. Elijovich F, Laffer CL, Sahinoz M, Pitzer A, Ferguson JF, Kirabo A. The gut microbiome, inflammation, and salt-sensitive hypertension. Curr Hypertens Rep (2020) 22(10):79. doi: 10.1007/s11906-020-01091-9
5. Lu X, Crowley SD. Inflammation in salt-sensitive hypertension and renal damage. Curr Hypertens Rep (2018) 20(12):103. doi: 10.1007/s11906-018-0903-x
6. Egan BM, Zhao Y, Axon R. US Trends in prevalence, awareness, treatment, and control of hypertension, 1988-2008. JAMA (2010) 303(20):2043–50. doi: 10.1001/jama.2010.650
7. Barrows IR, Ramezani A, Raj DS. Inflammation, immunity, and oxidative stress in hypertension-partners in crime? Adv Chronic Kidney Dis (2019) 26(2):122–30. doi: 10.1053/j.ackd.2019.03.001
8. Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: Recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep (2016) 18(3):21. doi: 10.1007/s11906-016-0628-7
9. Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med (2018) 215(1):21–33. doi: 10.1084/jem.20171773
10. Rizzoni D, De Ciuceis C, Szczepaniak P, Paradis P, Schiffrin EL, Guzik TJ. Immune system and microvascular remodeling in humans. Hypertension (2022) 79(4):691–705. doi: 10.1161/HYPERTENSIONAHA.121.17955
11. Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, et al. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol (2002) 161(5):1679–93. doi: 10.1016/S0002-9440(10)64445-8
12. Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, et al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int (2001) 59(6):2222–32. doi: 10.1046/j.1523-1755.2001.00737.x
13. Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM, et al. Resistance artery remodeling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: Evidence from m-CSF-deficient mice. Am J Physiol Heart Circ Physiol (2007) 292(4):H1789–95. doi: 10.1152/ajpheart.01118.2006
14. Kossmann S, Hu H, Steven S, Schonfelder T, Fraccarollo D, Mikhed Y, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem (2014) 289(40):27540–50. doi: 10.1074/jbc.M114.604231
15. Wiig H, Schroder A, Neuhofer W, Jantsch J, Kopp C, Karlsen TV, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest (2013) 123(7):2803–15. doi: 10.1172/JCI60113
16. Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, et al. Renal denervation prevents immune cell activation and renal inflammation in angiotensin II-induced hypertension. Circ Res (2015) 117(6):547–57. doi: 10.1161/CIRCRESAHA.115.306010
17. Zaldivia MTK, Rivera J, Hering D, Marusic P, Sata Y, Lim B, et al. Renal denervation reduces monocyte activation and monocyte-platelet aggregate formation: An anti-inflammatory effect relevant for cardiovascular risk. Hypertension (2017) 69(2):323–31. doi: 10.1161/HYPERTENSIONAHA.116.08373
18. O’Donnell M, Mente A, Yusuf S. Sodium intake and cardiovascular health. Circ Res (2015) 116(6):1046–57. doi: 10.1161/CIRCRESAHA.116.303771
19. Wright JT Jr, Rahman M, Scarpa A, Fatholahi M, Griffin V, Jean-Baptiste R, et al. Determinants of salt sensitivity in black and white normotensive and hypertensive women. Hypertension (2003) 42(6):1087–92. doi: 10.1161/01.HYP.0000101687.89160.19
20. Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Texas Rep Biol Med (1967) 25:257–64.
21. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res (2015) 116(6):1022–33. doi: 10.1161/CIRCRESAHA.116.303697
22. Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension (2015) 65(3):569–76. doi: 10.1161/HYPERTENSIONAHA.114.04975
23. Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation (2010) 122(24):2529–37. doi: 10.1161/CIRCULATIONAHA.109.930446
24. Wu KL, Chan SH, Chan JY. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflamm (2012) 9:212. doi: 10.1186/1742-2094-9-212
25. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature (2003) 421(6921):384–8. doi: 10.1038/nature01339
26. Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme m-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation (2011) 124(12):1370–81. doi: 10.1161/CIRCULATIONAHA.111.034470
27. Siedlinski M, Jozefczuk E, Xu X, Teumer A, Evangelou E, Schnabel RB, et al. White blood cells and blood pressure: A mendelian randomization study. Circulation (2020) 141:1307–17. doi: 10.1161/CIRCULATIONAHA.119.045102
28. Xiao L, Harrison DG. Inflammation in hypertension. Can J Cardiol (2020) 36:635–47. doi: 10.1016/j.cjca.2020.01.013
29. Muñoz M, López-Oliva ME, Rodríguez C, Martínez MP, Sáenz-Medina J, Sánchez A, et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol (2020) 28:101330. doi: 10.1016/j.redox.2019.101330
30. Schiffrin EL. Vascular remodeling in hypertension: mechanisms and treatment. Hypertension (2012) 59:367–74. doi: 10.1161/HYPERTENSIONAHA.111.187021
31. Zhang Y, Murugesan P, Huang K, Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat Rev Cardiol (2020) 17(3):170–94. doi: 10.1038/s41569-019-0260-8
32. Takac I, Schröder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, et al. The e-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem (2011) 286(15):13304–13. doi: 10.1074/jbc.M110.192138
33. Ma MM, Gao M, Guo KM, Wang M, Li XY, Zeng XL, et al. TMEM16A contributes to endothelial dysfunction by facilitating Nox2 NADPH oxidase-derived reactive oxygen species generation in hypertension. Hypertension (2017) 69(5):892–901. doi: 10.1161/HYPERTENSIONAHA.116.08874
34. Toral M, Romero M, Rodríguez-Nogales A, Jiménez R, Robles-Vera I, Algieri F, et al. Lactobacillus fermentum improves tacrolimus-induced hypertension by restoring vascular redox state and improving eNOS coupling. Mol Nutr Food Res (2018):e1800033. doi: 10.1002/mnfr.201800033
35. Camargo LL, Harvey AP, Rios FJ, Tsiropoulou S, Da Silva RNO, Cao Z, et al. Vascular nox (NADPH oxidase) compartmentalization, protein hyperoxidation, and endoplasmic reticulum stress response in hypertension. Hypertension (2018) 72(1):235–46. doi: 10.1161/HYPERTENSIONAHA.118.10824
36. Mikolajczyk TP, Szczepaniak P, Vidler F, Maffia P, Graham GJ, Guzik TJ. Role of inflammatory chemokines in hypertension. Pharmacol Ther (2021) 223:107799. doi: 10.1016/j.pharmthera.2020.107799
37. Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D, et al. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J (2016) 30:1987–99. doi: 10.1096/fj.201500088R
38. Yasunari K, Maeda K, Nakamura M, Yoshikawa J. Oxidative stress in leukocytes is a possible link between blood pressure, blood glucose, and c-reacting protein. Hypertension (2002) 39(3):777–80. doi: 10.1161/hy0302.104670
39. Savoia C, Schiffrin EL. Inflammation in hypertension. Curr Opin Nephrol Hypertens (2006) 15:152–8. doi: 10.1097/01.mnh.0000203189.57513.76
40. Foley RN, Collins AJ. End-stage renal disease in the united states: An update from the united states renal data system. J Am Soc Nephrol (2007) 18(10):2644–8. doi: 10.1681/ASN.2007020220
41. Dounousi E, Papavasiliou E, Makedou A, Ioannou K, Katopodis KP, Tselepis A, et al. Oxidative stress is progressively enhanced with advancing stages of CKD. Am J Kidney Dis (2006) 48(5):752–60. doi: 10.1053/j.ajkd.2006.08.015
42. Stuveling EM, Hillege HL, Bakker SJ, Gans RO, De Jong PE, De Zeeuw D. C-reactive protein is associated with renal function abnormalities in a non-diabetic population. Kidney Int (2003) 63(2):654–61. doi: 10.1046/j.1523-1755.2003.00762.x
43. Li Y, Wei B, Liu X, Shen XZ, Shi P. Microglia, autonomic nervous system, immunity and hypertension: Is there a link? Pharmacol Res (2020) 155:104451. doi: 10.1016/j.phrs.2019.104451
44. Usui T, Okada M, Hara Y, Yamawaki H. Death-associated protein kinase 3 mediates vascular inflammation and development of hypertension in spontaneously hypertensive rats. Hypertension (2012) 60(4):1031–9. doi: 10.1161/HYPERTENSIONAHA.112.200337
45. Valensi P. Autonomic nervous system activity changes in patients with hypertension and overweight: Role and therapeutic implications. Cardiovasc Diabetol (2021) 20(1):170. doi: 10.1186/s12933-021-01356-w
46. Noll G, Wenzel RR, Binggeli C, Corti C, Lüscher TF. Role of sympathetic nervous system in hypertension and effects of cardiovascular drugs. Eur Heart J (1998) 19 Suppl F:F32–8.
47. Ferrario CM, Mullick AE. Renin angiotensin aldosterone inhibition in the treatment of cardiovascular disease. Pharmacol Res (2017) 125(pt A):57–71. doi: 10.1016/j.phrs.2017.05.020
48. Olivares-Silva F, De Gregorio N, Espitia-Corredor J, Espinoza C, Vivar R, Silva D, et al. Resolvin-D1 attenuation of angiotensin II-induced cardiac inflammation in mice is associated with prevention of cardiac remodeling and hypertension. Biochim Biophys Acta Mol Basis Dis (2021) 1867(12):166241. doi: 10.1016/j.bbadis.2021.166241
49. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol (2020) 21(1):30–41. doi: 10.1038/s41590-019-0548-1
50. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension (2017) 70:660–7. doi: 10.1161/HYPERTENSIONAHA.117.07802
51. Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol (2019) 19(8):517–32. doi: 10.1038/s41577-019-0160-5
52. Próchnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab (2017) 26(1):71–93. doi: 10.1016/j.cmet.2017.06.018
53. Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis (2020) 11(9):776. doi: 10.1038/s41419-020-02985-x
54. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol (2009) 10(3):241–7. doi: 10.1038/ni.1703
55. Zhang Z, Tang J, Cui X, Qin B, Zhang J, Zhang L, et al. New insights and novel therapeutic potentials for macrophages in myocardial infarction. Inflammation (2021) 44(5):1696–712. doi: 10.1007/s10753-021-01467-2
56. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481(7381):278–86. doi: 10.1038/nature10759
57. Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism (2017) 74:1–9. doi: 10.1016/j.metabol.2017.06.002
58. Sun HJ, Ren XS, Xiong XQ, Chen YZ, Zhao MX, Wang JJ, et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis (2017) 10):e3074. doi: 10.1038/cddis.2017.470
59. Zhao Z, Wang Y, Zhou R, Li Y, Gao Y, Tu D, et al. A novel role of NLRP3-generated IL-1β in the acute-chronic transition of peripheral lipopolysaccharide-elicited neuroinflammation: Implications for sepsis-associated neurodegeneration. J Neuroinflamm (2020) 17(1):64. doi: 10.1186/s12974-020-1728-5
60. Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin-1beta in the systemic circulation of patients with essential hypertension: Additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med (1997) 129:300–8. doi: 10.1016/S0022-2143(97)90178-5
61. Wang JG, Williams JC, Davis BK, Jacobson K, Doerschuk CM, Ting JP, et al. Monocytic microparticles activate endothelial cells in an IL-1β-dependent manner. Blood (2011) 118(8):2366–74. doi: 10.1182/blood-2011-01-330878
62. Dörffel Y, Franz S, Pruss A, Neumann G, Rohde W, Burmester GR, et al. Preactivated monocytes from hypertensive patients as a factor for atherosclerosis? Atherosclerosis (2001) 157(1):151–60. doi: 10.1016/S0021-9150(00)00674-2
63. Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol (2014) 184(5):1617–28. doi: 10.1016/j.ajpath.2014.01.032
64. Chen Y, Wang L, Pitzer AL, Li X, Li PL, Zhang Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J M ol Med (Berl) (2016) 94(12):1335–47. doi: 10.1007/s00109-016-1481-5
65. Toldo S, Mezzaroma E, Buckley LF, Potere N, Di Nisio M, Biondi-Zoccai G, et al. Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol Ther (2022) 236:108053. doi: 10.1016/j.pharmthera.2021.108053
66. Orecchioni M, Kobiyama K, Winkels H, Ghosheh Y, McArdle S, Mikulski Z, et al. Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science (2022) 375(6577):214–21. doi: 10.1126/science.abg3067
67. Sharma BR, Kanneganti TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol (2021) 22(5):550–9. doi: 10.1038/s41590-021-00886-5
68. Xia M, Abais JM, Koka S, Meng N, Gehr TW, Boini KM, et al. Characterization and activation of NLRP3 inflammasomes in the renal medulla in mice. Kidney Blood Press Res (2016) 41(2):208–21. doi: 10.1159/000443424
69. Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: Versatile players in renal inflammation and fibrosis. Nat Rev Nephrol (2019) 15(3):144–58. doi: 10.1038/s41581-019-0110-2
70. Sharma AK, Mulloy DP, Le LT, Laubach VE. NADPH oxidase mediates synergistic effects of IL-17 and TNF-alpha on CXCL1 expression by epithelial cells after lung ischemia-reperfusion. Am J Physiol Lung Cell Mol Physiol (2014) 306:L69–79. doi: 10.1152/ajplung.00205.2013
71. Oh S, Yang JY, Park CH, Son KH, Byun K. Dieckol reduces muscle atrophy by modulating angiotensin type II type 1 receptor and NADPH oxidase in spontaneously hypertensive rats. Antioxid (Basel) (2021) 10(10):1561. doi: 10.3390/antiox10101561
72. Higaki A, Mahmoud AUM, Paradis P, Schiffrin EL. Role of interleukin-23/interleukin-17 axis in T-cell-mediated actions in hypertension. Cardiovasc Res (2021) 117:1274–83. doi: 10.1093/cvr/cvaa257
73. Vázquez-Oliva G, Fernández-Real JM, Zamora A, Vilaseca M, Badimón L. Lowering of blood pressure leads to decreased circulating interleukin-6 in hypertensive subjects. J Hum Hypertens (2005) 19(6):457–62. doi: 10.1038/sj.jhh.1001845
74. Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS, et al. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol (2006) 290(3):H935–40. doi: 10.1152/ajpheart.00708.2005
75. Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: Role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension (2010) 56(5):879–84. doi: 10.1161/HYPERTENSIONAHA.110.158071
76. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med (2007) 204(10):2449–60. doi: 10.1084/jem.20070657
77. Yoshida S, Takeuchi T, Kotani T, Yamamoto N, Hata K, Nagai K, et al. Infliximab, a TNF-α inhibitor, reduces 24-h ambulatory blood pressure in rheumatoid arthritis patients. J Hum Hypertens (2014) 28(3):165–9. doi: 10.1038/jhh.2013.80
78. Kleinbongard P, Heusch G, Schulz R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther (2010) 127(3):295–314. doi: 10.1016/j.pharmthera.2010.05.002
79. Landry DB, Couper LL, Bryant SR, Lindner V. Activation of the NF-kappa b and I kappa b system in smooth muscle cells after rat arterial injury. induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am J Pathol (1997) 151(4):1085–95.
80. Neumann P, Gertzberg N, Johnson A. TNF-alpha induces a decrease in eNOS promoter activity. Am J Physiol Lung Cell Mol Physiol (2004) 286:L452–9. doi: 10.1152/ajplung.00378.2002
81. Alonso J, Sanchez de Miguel L, Monton M, Casado S, Lopez-Farre A. Endothelial cytosolic proteins bind to the 3’ untranslated region of endothelial nitric oxide synthase mRNA: Regulation by tumor necrosis factor alpha. Mol Cell Biol (1997) 17:5719–26. doi: 10.1128/MCB.17.10.5719
82. Markó L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, et al. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension (2012) 60(6):1430–6. doi: 10.1161/HYPERTENSIONAHA.112.199265
83. Veiras LC, Bernstein EA, Cao D, Okwan-Duodu D, Khan Z, Gibb DR, et al. Tubular IL-1β induces salt sensitivity in diabetes by activating renal macrophages. Circ Res (2022) 131(1):59–73. doi: 10.1161/CIRCRESAHA.121.320239
84. Gu C, Wu L, Li X. IL-17 family: Cytokines, receptors and signaling. Cytokine (2013) 64:477–85. doi: 10.1016/j.cyto.2013.07.022
85. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med (2014) 371(4):326–38. doi: 10.1056/NEJMoa1314258
86. Mease PJ, Genovese MC, Greenwald MW, Ritchlin CT, Beaulieu AD, Deodhar A, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med (2014) 370(24):2295–306. doi: 10.1056/NEJMoa1315231
87. Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res (2001) 88(11):E68–75. doi: 10.1161/hh1101.092677
88. Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J (1998) 334(Pt 2):297–314. doi: 10.1042/bj3340297
89. Ogata A, Tanaka T. Tocilizumab for the treatment of rheumatoid arthritis and other systemic autoimmune diseases: Current perspectives and future directions. Int J Rheumatol (2012) 2012:946048. doi: 10.1155/2012/946048
90. Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, et al. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol (2010) 299(2):R590–5. doi: 10.1152/ajpregu.00207.2009
91. Segiet A, Smykiewicz P, Kwiatkowski P, Żera T. Tumour necrosis factor and interleukin 10 in blood pressure regulation in spontaneously hypertensive and normotensive rats. Cytokine (2019) 113:185–94. doi: 10.1016/j.cyto.2018.07.003
92. Aggarwal BB. Signalling pathways of the TNF superfamily: A double-edged sword. Nat Rev Immunol (2003) 3(9):745–56. doi: 10.1038/nri1184
93. Barbaro NR, Harrison DG. Markers or makers: Inflammatory cytokines in treatment-resistant hypertension. Hypertension (2019) 73(4):767–9. doi: 10.1161/HYPERTENSIONAHA.119.12604
94. Ramseyer VD, Hong NJ, Garvin JL. Tumor necrosis factor alpha decreases nitric oxide synthase type 3 expression primarily via Rho/Rho kinase in the thick ascending limb. Hypertension (2012) 59:1145–50. doi: 10.1161/HYPERTENSIONAHA.111.189761
95. Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: Clinical implications. Annu Rev Physiol (2011) 73:359–76. doi: 10.1146/annurev-physiol-012110-142247
96. Ishimitsu T, Uehara Y, Numabe A, Tsukada H, Ogawa Y, Iwai J, et al. Interferon gamma attenuates hypertensive renal injury in salt-sensitive Dahl rats. Hypertension (1992) 19(6 Pt 2):804–8. doi: 10.1161/01.HYP.19.6.804
97. Satou R, Miyata K, Gonzalez-Villalobos RA, Ingelfinger JR, Navar LG, Kobori H. Interferon-γ biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J (2012) 26(5):1821–30. doi: 10.1096/fj.11-195198
98. Krishnan SM, Ling YH, Huuskes BM, Ferens DM, Saini N, Chan CT, et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc Res (2019) 115(4):776–87. doi: 10.1093/cvr/cvy252
99. Sims JE, Smith DE. The IL-1 family: Regulators of immunity. Nat Rev Immunol (2010) 10(2):89–102. doi: 10.1038/nri2691
100. Xu D, Mu R, Wei X. The roles of IL-1 family cytokines in the pathogenesis of systemic sclerosis. Front Immunol (2019) 10:2025. doi: 10.3389/fimmu.2019.02025
101. Dinarello C, Arend W, Sims J, Smith D, Blumberg H, O’Neill L, et al. IL-1 family nomenclature. Nat Immunol (2010) 11(11):973. doi: 10.1038/ni1110-973
102. Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR. IL-1β and IL-18: Inflammatory markers or mediators of hypertension? Br J Pharmacol (2014) 171(24):5589–602. doi: 10.1111/bph.12876
103. Postlethwaite AE, Raghow R, Stricklin GP, Poppleton H, Seyer JM, Kang AH. Modulation of fibroblast functions by interleukin 1: Increased steady-state accumulation of type I procollagen messenger RNAs and stimulation of other functions but not chemotaxis by human recombinant interleukin 1 alpha and beta. J Cell Biol (1988) 106(2):311–8. doi: 10.1083/jcb.106.2.311
104. Urwyler SA, Ebrahimi F, Burkard T, Schuetz P, Poglitsch M, Mueller B, et al. IL (Interleukin)-1 receptor antagonist increases ang (Angiotensin [1-7]) and decreases blood pressure in obese individuals. Hypertension (2020) 75(6):1455–63. doi: 10.1161/HYPERTENSIONAHA.119.13982
105. Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension (2010) 56:297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409
106. Santisteban MM, Kim S, Pepine CJ, Raizada MK. Brain-Gut-Bone marrow axis: Implications for hypertension and related therapeutics. Circ Res (2016) 118(8):1327–36. doi: 10.1161/CIRCRESAHA.116.307709
107. Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: New roles for the synaptic stripper. Neuron (2013) 77(1):10–8. doi: 10.1016/j.neuron.2012.12.023
108. Cui C, Xu P, Li G, Qiao Y, Han W, Geng C, et al. Vitamin d receptor activation regulates microglia polarization and oxidative stress in spontaneously hypertensive rats and angiotensin II-exposed microglial cells: Role of renin-angiotensin system. Redox Biol (2019) 26:101295. doi: 10.1016/j.redox.2019.101295
109. Shen XZ, Li Y, Li L, Shah KH, Bernstein KE, Lyden P, et al. Microglia participate in neurogenic regulation of hypertension. Hypertension (2015) 66:309–16. doi: 10.1161/HYPERTENSIONAHA.115.05333
110. Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G, et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome (2017) 5(1):14. doi: 10.1186/s40168-016-0222-x
111. Mell B, Jala VR, Mathew AV, Byun J, Waghulde H, Zhang Y, et al. Evidence for a link between gut microbiota and hypertension in the dahl rat. Physiol Genomics (2015) 47:187–97. doi: 10.1152/physiolgenomics.00136.2014
112. Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, et al. Gut dysbiosis is linked to hypertension. Hypertension (2015) 65:1331–40. doi: 10.1161/HYPERTENSIONAHA.115.05315
113. Hetemäki I, Jian C, Laakso S, Mäkitie O, Pajari AM, de Vos WM, et al. Fecal bacteria implicated in biofilm production are enriched and associate to gastrointestinal symptoms in patients with APECED - a pilot study. Front Immunol (2021) 12:668219. doi: 10.3389/fimmu.2021.668219
114. Srivastava A, Gupta J, Kumar S, Kumar A. Gut biofilm forming bacteria in inflammatory bowel disease. Microb Pathog (2017) 112:5–14. doi: 10.1016/j.micpath.2017.09.041
115. De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: Evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol (2005) 25(10):2106–13. doi: 10.1161/01.ATV.0000181743.28028.57
116. Wenzel P, Rossmann H, Müller C, Kossmann S, Oelze M, Schulz A, et al. Heme oxygenase-1 suppresses a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur Heart J (2015) 36(48):3437–46. doi: 10.1093/eurheartj/ehv544
117. Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol (2012) 3:128. doi: 10.3389/fphys.2012.00128
118. Higaki A, Caillon A, Paradis P, Schiffrin EL. Innate and innate-like immune system in hypertension and vascular injury. Curr Hypertens Rep (2019) 21:4. doi: 10.1007/s11906-019-0907-1
119. Chrysanthopoulou A, Gkaliagkousi E, Lazaridis A, Arelaki S, Pateinakis P, Ntinopoulou M, et al. Angiotensin II triggers release of neutrophil extracellular traps, linking thromboinflammation with essential hypertension. JCI Insight (2021) 6:e148668. doi: 10.1172/jci.insight.148668
120. Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, PhysGen Knockout Program. CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension (2014) 63:559–64. doi: 10.1161/HYPERTENSIONAHA.113.02191
121. Itani HA, McMaster WG Jr, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, et al. Activation of human T cells in hypertension: Studies of humanized mice and hypertensive humans. Hypertension (2016) 68:123–32. doi: 10.1161/HYPERTENSIONAHA.116.07237
122. Leibowitz A, Schiffrin EL. Immune mechanisms in hypertension. Curr Hypertens Rep (2011) 13:465–72. doi: 10.1007/s11906-011-0224-9
123. Kassan M, Wecker A, Kadowitz P, Trebak M, Matrougui K. CD4+CD25+Foxp3 regulatory T cells and vascular dysfunction in hypertension. J Hypertens (2013) 31:1939–43. doi: 10.1097/HJH.0b013e328362feb7
124. Schiffrin EL. Mechanisms of remodelling of small arteries, antihypertensive therapy and the immune system in hypertension. Clin Invest Med (2015) 38:E394–402. doi: 10.25011/cim.v38i6.26202
125. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension (2010) 55:500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094
126. Schiffrin EL. T Lymphocytes: A role in hypertension? Curr Opin Nephrol Hypertens (2010) 19:181–6. doi: 10.1097/MNH.0b013e3283360a2e
127. Viel EC, Lemarié CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol (2010) 298:H938–44. doi: 10.1152/ajpheart.00707.2009
128. Dröge W. Free radicals in the physiological control of cell function. Physiol Rev (2002) 82(1):47–95. doi: 10.1152/physrev.00018.2001
129. Montezano AC, Touyz RM. Oxidative stress, noxs, and hypertension: Experimental evidence and clinical controversies. Ann Med (2012) 44:S2–16. doi: 10.3109/07853890.2011.653393
130. Rodrigo R, González J, Paoletto F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens Res (2011) 34(4):431–40. doi: 10.1038/hr.2010.264
131. Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension (2004) 44:248–52. doi: 10.1161/01.HYP.0000138070.47616.9d
132. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ Res (2000) 87:840–4. doi: 10.1161/01.RES.87.10.840
133. Rochette L, Lorin J, Zeller M, Guilland JC, Lorgis L, Cottin Y, et al. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: possible therapeutic targets? Pharmacol Ther (2013) 140(3):239–57. doi: 10.1016/j.pharmthera.2013.07.004
134. Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol (2006) 26(11):2439–44. doi: 10.1161/01.ATV.0000243924.00970.cb
135. Majzunova M, Dovinova I, Barancik M, Chan JY. Redox signaling in pathophysiology of hypertension. J BioMed Sci (2013) 20(1):69. doi: 10.1186/1423-0127-20-69
136. Balakumar P, Jagadeesh G. Multifarious molecular signaling cascades of cardiac hypertrophy: Can the muddy waters be cleared? Pharmacol Res (2010) 62(5):365–83. doi: 10.1016/j.phrs.2010.07.003
137. Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol (2000) 20(10):2175–83. doi: 10.1161/01.ATV.20.10.2175
138. Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA (2003) 290(22):2945–51. doi: 10.1001/jama.290.22.2945
139. Qi Y, Aranda JM, Rodriguez V, Raizada MK, Pepine CJ. Impact of antibiotics on arterial blood pressure in a patient with resistant hypertension-a case report. Int J Cardiol (2015) 201:157–8. doi: 10.1016/j.ijcard.2015.07.078
140. Fanouriakis A, Tziolos N, Bertsias G, Boumpas DT. Update on the diagnosis and management of systemic lupus erythematosus. Ann Rheum Dis (2021) 80(1):14–25. doi: 10.1136/annrheumdis-2020-218272
141. Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol (2006) 17(12 Suppl 3):S218–25. doi: 10.1681/ASN.2006080918
142. Ferro CJ, Edwards NC, Hutchison C, Cockwell P, Steeds RP, Savage CO, et al. Does immunosuppressant medication lower blood pressure and arterial stiffness in patients with chronic kidney disease? An observational study. Hypertens Res (2011) 34(1):113–9. doi: 10.1038/hr.2010.193
143. Makavos G, Ikonomidis I, Andreadou I, Varoudi M, Kapniari I, Loukeri E, et al. Effects of interleukin 17A inhibition on myocardial deformation and vascular function in psoriasis. Can J Cardiol (2020) 36(1):100–11. doi: 10.1016/j.cjca.2019.06.021
144. Boutouyrie P, Chowienczyk P, Humphrey JD, Mitchell GF. Arterial stiffness and cardiovascular risk in hypertension. Circ Res (2021) 128(7):864–86. doi: 10.1161/CIRCRESAHA.121.318061
145. Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA heart study. Eur Heart J (2015) 36(6):377–84. doi: 10.1093/eurheartj/ehu272
146. Alfaidi MA, Chamberlain J, Rothman A, Crossman D, Villa-Uriol MC, Hadoke P, et al. Dietary docosahexaenoic acid reduces oscillatory wall shear stress, atherosclerosis, and hypertension, most likely mediated via an IL-1-Mediated mechanism. J Am Heart Assoc (2018) 7(13):e008757. doi: 10.1161/JAHA.118.008757
147. Rothman AM, MacFadyen J, Thuren T, Webb A, Harrison DG, Guzik TJ, et al. Effects of interleukin-1β inhibition on blood pressure, incident hypertension, and residual inflammatory risk: A secondary analysis of CANTOS. Hypertension (2020) 75(2):477–82. doi: 10.1161/HYPERTENSIONAHA.119.13642
148. Li Y, Zafar S, Salih Ibrahim RM, Chi HL, Xiao T, Xia WJ, et al. Exercise and food supplement of vitamin c ameliorate hypertension through improvement of gut microflora in the spontaneously hypertensive rats. Life Sci (2021) 269:119097. doi: 10.1016/j.lfs.2021.119097
149. Cardenas MG, Oswald E, Yu W, Xue F, MacKerell AD Jr, Melnick AM. The expanding role of the BCL6 oncoprotein as a cancer therapeutic target. Clin Cancer Res (2017) 23(4):885–93. doi: 10.1158/1078-0432.CCR-16-2071
150. Chen D, Zang YH, Qiu Y, Zhang F, Chen AD, Wang JJ, et al. BCL6 attenuates proliferation and oxidative stress of vascular smooth muscle cells in hypertension. Oxid Med Cell Longev (2019) 2019:5018410. doi: 10.1155/2019/5018410
151. Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ Res (2000) 86(5):494–501. doi: 10.1161/01.RES.86.5.494
152. Wenzel P, Schulz E, Oelze M, Müller J, Schuhmacher S, Alhamdani MS, et al. AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radic Biol Med (2008) 45(5):619–26. doi: 10.1016/j.freeradbiomed.2008.05.009
153. Hornig B, Landmesser U, Kohler C, Ahlersmann D, Spiekermann S, Christoph A, et al. Comparative effect of ace inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: Role of superoxide dismutase. Circulation (2001) 103(6):799–805. doi: 10.1161/01.CIR.103.6.799
154. Zepeda RJ, Castillo R, Rodrigo R, Prieto JC, Aramburu I, Brugere S, et al. Effect of carvedilol and nebivolol on oxidative stress-related parameters and endothelial function in patients with essential hypertension. Basic Clin Pharmacol Toxicol (2012) 111(5):309–16. doi: 10.1111/j.1742-7843.2012.00911.x
155. Mollnau H, Schulz E, Daiber A, Baldus S, Oelze M, August M, et al. Nebivolol prevents vascular NOS III uncoupling in experimental hyperlipidemia and inhibits NADPH oxidase activity in inflammatory cells. Arterioscler Thromb Vasc Biol (2003) 23(4):615–21. doi: 10.1161/01.ATV.0000065234.70518.26
156. Münzel T, Gori T. Nebivolol: The somewhat-different beta-adrenergic receptor blocker. J Am Coll Cardiol (2009) 54(16):1491–9. doi: 10.1016/j.jacc.2009.05.066
157. Matsubara M, Hasegawa K. Benidipine, a dihydropyridine-calcium channel blocker, prevents lysophosphatidylcholine-induced injury and reactive oxygen species production in human aortic endothelial cells. Atherosclerosis (2005) 178(1):57–66. doi: 10.1016/j.atherosclerosis.2004.08.020
158. Ulker S, McKeown PP, Bayraktutan U. Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. Hypertension (2003) 41(3):534–9. doi: 10.1161/01.HYP.0000057421.28533.37
159. Duffy SJ, Gokce N, Holbrook M, Hunter LM, Biegelsen ES, Huang A, et al. Effect of ascorbic acid treatment on conduit vessel endothelial dysfunction in patients with hypertension. Am J Physiol Heart Circ Physiol (2001) 280(2):H528–34. doi: 10.1152/ajpheart.2001.280.2.H528
160. Atarashi K, Ishiyama A, Takagi M, Minami M, Kimura K, Goto A, et al. Vitamin e ameliorates the renal injury of Dahl salt-sensitive rats. Am J Hypertens (1997) 10(5 Pt 2):116S–9S. doi: 10.1016/S0895-7061(97)00088-5
161. Duffy SJ, Gokce N, Holbrook M, Huang A, Frei B, Keaney JF Jr, et al. Treatment of hypertension with ascorbic acid. Lancet (1999) 354(9195):2048–9. doi: 10.1016/S0140-6736(99)04410-4
162. Block G, Mangels AR, Norkus EP, Patterson BH, Levander OA, Taylor PR. Ascorbic acid status and subsequent diastolic and systolic blood pressure. Hypertension (2001) 37(2):261–7. doi: 10.1161/01.HYP.37.2.261
Keywords: hypertension, inflammation, immunity, oxidative stress, cytokines, cardiovascular diseases
Citation: Zhang Z, Zhao L, Zhou X, Meng X and Zhou X (2023) Role of inflammation, immunity, and oxidative stress in hypertension: New insights and potential therapeutic targets. Front. Immunol. 13:1098725. doi: 10.3389/fimmu.2022.1098725
Received: 15 November 2022; Accepted: 21 December 2022;
Published: 10 January 2023.
Edited by:
Jianmin Chen, Queen Mary University of London, United KingdomReviewed by:
Alberto N. Peón, Sociedad Española de Beneficencia, MexicoAisah Aniisah Aubdool, Queen Mary University of London, United Kingdom
Copyright © 2023 Zhang, Zhao, Zhou, Meng and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianliang Zhou, zhouxianliang0326@hotmail.com; Xu Meng, mengxu1219@hotmail.com
†These authors have contributed equally to this work