 Jana Beicht1†
Jana Beicht1† Mareike Kubinski1†
Mareike Kubinski1† Isabel Zdora2,3
Isabel Zdora2,3 Christina Puff2
Christina Puff2 Jeannine Biermann1Thomas Gerlach1
Jeannine Biermann1Thomas Gerlach1 Wolfgang Baumgärtner2,3
Wolfgang Baumgärtner2,3 Gerd Sutter4,5
Gerd Sutter4,5 Albert D. M. E. Osterhaus1
Albert D. M. E. Osterhaus1 Chittappen Kandiyil Prajeeth1
Chittappen Kandiyil Prajeeth1 Guus F. Rimmelzwaan1*
Guus F. Rimmelzwaan1*- 1Research Center for Emerging Infections and Zoonoses, University of Veterinary Medicine Hannover, Foundation, Hannover, Germany
- 2Department of Pathology, University of Veterinary Medicine Hannover, Foundation, Hannover, Germany
- 3Center for Systems Neuroscience, Hannover Graduate School for Neurosciences, Infection Medicine, and Veterinary Sciences (HGNI), Hannover, Germany
- 4Division of Virology, Institute for Infectious Diseases and Zoonoses, Ludwig Maximilian University (LMU) Munich, Munich, Germany
- 5German Center for Infection Research (DZIF), Partner Site Munich, Munich, Germany
Introduction: Tick-borne encephalitis virus (TBEV) is one of the most relevant tick-transmitted neurotropic arboviruses in Europe and Asia and the causative agent of tick-borne encephalitis (TBE). Annually more than 10,000 TBE cases are reported despite having vaccines available. In Europe, the vaccines FSME-IMMUN® and Encepur® based on formaldehyde-inactivated whole viruses are licensed. However, demanding vaccination schedules contribute to sub-optimal vaccination uptake and breakthrough infections have been reported repeatedly. Due to its immunogenic properties as well as its role in viral replication and disease pathogenesis, the non-structural protein 1 (NS1) of flaviviruses has become of interest for non-virion based flavivirus vaccine candidates in recent years.
Methods: Therefore, immunogenicity and protective efficacy of TBEV NS1 expressed by neuraminidase (NA)-deficient Influenza A virus (IAV) or Modified Vaccinia virus Ankara (MVA) vectors were investigated in this study.
Results: With these recombinant viral vectors TBEV NS1-specific antibody and T cell responses were induced. Upon heterologous prime/boost regimens partial protection against lethal TBEV challenge infection was afforded in mice.
Discussion: This supports the inclusion of NS1 as a vaccine component in next generation TBEV vaccines.
1 Introduction
The neurotropic tick-borne encephalitis virus (TBEV) belongs to the genus Flavivirus of the Flaviviridae family and is one of the most important tick-transmitted viruses in Europe and Asia. Since 2000, more than 50,000 confirmed tick-borne encephalitis (TBE) cases have been reported in Europe and the number of countries reporting cases is increasing [reviewed in (1)]. Its positive-sensed single-stranded RNA genome encodes for a polyprotein that is cleaved co- and post-translationally by viral and cellular proteases into three structural proteins (capsid C, envelope E, precursor membrane/membrane prM/M) and seven non-structural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). Three main TBEV subtypes are distinguished, the Far-Eastern, the European and the Siberian subtype, which differ in geographical spread and virulence. More recently, novel Baikalian and the Himalayan subtypes have been identified (2–4). TBEV is mainly transmitted by Ixodes spp. ticks (5) and causes asymptomatic infections in humans in 70-98% of cases depending on viral (e.g. dose, virulence of TBEV strain) and host factors (e.g. age) [reviewed in (6)]. However, patients can also present with a wide range of clinical signs, from mild febrile illness to severe neurological disease including meningitis, encephalitis and myelitis, summarized as TBE. Up to 50% of patients with TBE can suffer from long-term sequelae [reviewed in (7)]. Specific antiviral treatment of TBE is not available in Europe [reviewed in (6)] and therefore, vaccination is the most important protective measure. Worldwide, six inactivated TBEV vaccines have been licensed. In Europe, FSME-IMMUN® (Pfizer) and Encepur® (Bavarian Nordic), both based on European TBEV strains, are being used. For primary vaccination, three vaccine doses are required with the need of booster vaccinations every 3-5 years depending on the age of the vaccinee. Although these vaccines are considered safe and effective with high seroconversion rates (8), vaccine breakthrough infections have been reported to occur frequently (9–14). Of interest, TBE in patients with vaccination breakthrough has been described to be more severe than after infection in unvaccinated patients (9, 12), reviewed in (6)]. TBEV vaccination aims primarily at the induction of virus-neutralizing antibodies to the E protein. In addition, it has been shown that the currently used vaccines also induce virus-specific CD4+ T cell responses [reviewed in (15)]. Some studies indicate that the use of European licensed vaccines also induce NS1-specific antibodies, however, this is still matter of debate (16–18). NS1 plays an important role in the TBEV replication cycle and may contribute to the pathogenesis of TBEV infections as was described for other flaviviruses [reviewed in (19)]. NS1 is involved in viral replication and virus assembly intracellularly, but it is also found on the cell surface and can be secreted into extracellular space in its oligomeric form (20). Although the mechanisms of protection are not fully understood, it was shown previously that immunization with TBEV NS1 is able to partly protect mice against challenge infection (21–27). For the induction of TBEV NS1-specific immunity, synthetic peptides (25, 26) and recombinant viral vectors based on adenoviruses (21, 22, 27) and vaccinia viruses (VACV) (23, 24) have been used. The use of viral vectors offers the advantage that they can induce both humoral and cell-mediated immunity, although the latter has not been studied in great detail.
The use of NS1 as vaccine antigen may offer some advantages over the use of the E protein. NS1 vaccination will not result in the induction of virion-reactive antibodies and consequently, the risk of antibody-dependent enhancement (ADE) of infection is reduced (28). Although clear evidence for ADE of TBEV infection in vivo is lacking, ADE has been shown in vitro (29–32).
In the present study, we constructed and characterized Influenza A virus (IAV)- and Modified Vaccinia virus Ankara (MVA)-based vectors expressing the TBEV NS1 protein (IAV-NS1 and MVA-NS1) and tested their immunogenicity and protective efficacy in a mouse model. Recombinant IAVs, member of the Orthomyxoviridae family, have been used for the development of vaccine candidates against a variety of different viruses, and immunogenicity was demonstrated in pre-clinical studies [reviewed in (33)]. MVA, a member of the Poxviridae family, has a longstanding record as a safe and effective viral vaccine vector with extensive use in clinical trials [reviewed in (34)]. To investigate whether TBEV NS1-specific antibody and T cell responses can be improved, we also evaluated heterologous prime/boost vaccination regimens with IAV and MVA as viral vectors. Since the order of prime/boost administration can be important [reviewed in (35)], prime immunization with MVA-NS1 followed by boost immunization with IAV-NS1 and vice versa was tested. Heterologous prime/boost regimens with MVA-NS1 and IAV-NS1 proved to be highly immunogenic, induced NS1-specific antibodies, CD4+ and CD8+ T cell responses and afforded partial protection against a lethal TBEV challenge in mice.
2 Materials and methods
2.1 Cell culture
All cell lines were cultivated at 37°C in 5% CO2. Primary chicken embryo fibroblast (CEF) cells were generated from 10-11 days old chicken embryos (specific pathogen-free eggs purchased from VALO BioMedia GmbH) and cultured in Minimum Essential Medium Eagle (Sigma-Aldrich) with 10% fetal bovine serum (FBS), 100 IU/ml penicillin and 100 µg/ml streptomycin (Pen/Strep, Sigma-Aldrich), and 1% MEM non-essential amino acid solution (NEAA, Sigma-Aldrich). HEK293T and MDCK cells were propagated in Dulbecco’s modified Eagle medium (DMEM, Gibco™) supplemented with 10% FBS, Pen/Strep and 2 mM GlutaMAX™ (Gibco™). HeLa cells were cultivated in DMEM with 10% FBS, Pen/Strep, 2 mM GlutaMAX™ and 1% MEM NEAA solution. A549 cells were maintained in F-12 Nut Mix (1X) + GlutaMAX-I (Gibco™) supplemented with 10% FBS, Pen/Strep, 2 mM GlutaMAX™ and 20 mM HEPES. For IAV infection, 10% FBS was replaced by 0.1% bovine serum albumin and 1 µg/ml Trypsin-TPCK (Sigma-Aldrich) was added freshly to the media. For MVA and TBEV infection media, 10% FBS was replaced by 2% FBS. Cell lines were tested negative for mycoplasma before use (MycoStrip™–Mycoplasma detection Kit, InvivoGen).
2.2 Viruses
TBEV strain Neudoerfl (European subtype) was provided by the Department of Microbiology of the German Armed Forces, Munich, Germany. The reverse genetics pHW2000 plasmids containing the individual gene segments of A/Puerto Rico/8/1934 (H1N1) (PR8) were provided by Richard Webby and Robert Webster, St. Jude Children’s Research Hospital, Memphis, TN, USA. Plasmids encoding for TBEV Neudoerfl NS1 (based on UniProtKB: P14336) and the SARS-CoV-2 receptor binding domain (RBD, aa319-550, based on Wuhan-Hu-1 GenBank: MN908947.3) including a C-terminal stop codon and EcoRI and SpeI restriction sites at the 5’- and 3’-end, respectively, were synthesized (GenScript Biotech Corp) and cloned in frame into the previous used pHW2000 neuraminidase (NA) plasmid encoding a fusion of the N-terminal region of PR8 NA with enhanced green fluorescent protein (eGFP) (36). Thereby, eGFP was replaced by TBEV NS1 or the SARS-CoV-2 RBD as non-TBEV insert. By using reverse genetics based on the 8 plasmid system (37), recombinant PR8 (rPR8), IAV-NS1 and IAV-RBD (vector control virus) were rescued. For this, HEK293T cells were transfected with plasmids encoding the respective chimeric or wildtype NA and the remaining PR8 gene segments by using TransIT®-LT1 (Mirus Bio LLC). Infection medium was supplemented with exogenous Vibrio cholerae neuraminidase (eNA, 1:60,000, Sigma). After 48 h, HEK293T supernatant was transferred to MDCK cells for virus amplification. To generate IAV stocks, MDCK cells were infected with multiplicity of infection (MOI) 0.01 of the respective virus in presence of eNA. 48 hours post infection (hpi), supernatant was harvested and clarified by low-speed centrifugation. Stocks for animal experiments were subsequently concentrated 120x by ultracentrifugation through a 25% sucrose cushion (28,000 rpm, 2 h, 4°C). Virus pellets were resuspended in 1x PBS (Gibco™). Viral titers were determined by plaque assay as described before with avicel overlay (38).
Non-recombinant MVA F6 (wtMVA) and MVA-GFP (expression of GFP under transcriptional control of VACV promotor P11 in deletion site III) were obtained from the Institute for Infectious Diseases and Zoonoses, Ludwig Maximilian University Munich, Munich, Germany. Plasmid encoding for the Kozak sequence followed by the TBEV E gene signal peptide and entire TBEV NS1 were synthesized (based on TBEV Neudoerfl, UniProtKB: P14336; GenScript Biotech Corp) and cloned into MVA transfer plasmid pIIIsynIIred under transcriptional control of VACV late promotor psynII (39). pIIIsynIIred contains mCherry as marker gene which is flanked by short repetitive regions. MVA-NS1 was produced as described previously (39) (Figure 1A). Virus stocks were propagated on primary CEF cells and virus was concentrated by ultracentrifugation at 38,400 rcf through 36% sucrose cushion. Virus pellets were resuspended in tris-buffered saline (120 mM NaCl/10 mM Tris-HCl, pH 7.4). MVA-specific immune peroxidase staining after slightly modified standard protocol (39) including overlay consisting of 2.5% Avicel in 2X MEM, 2% FBS, 1% Pen/Strep and 1% MEM NEAA solution was performed to determine viral titers.
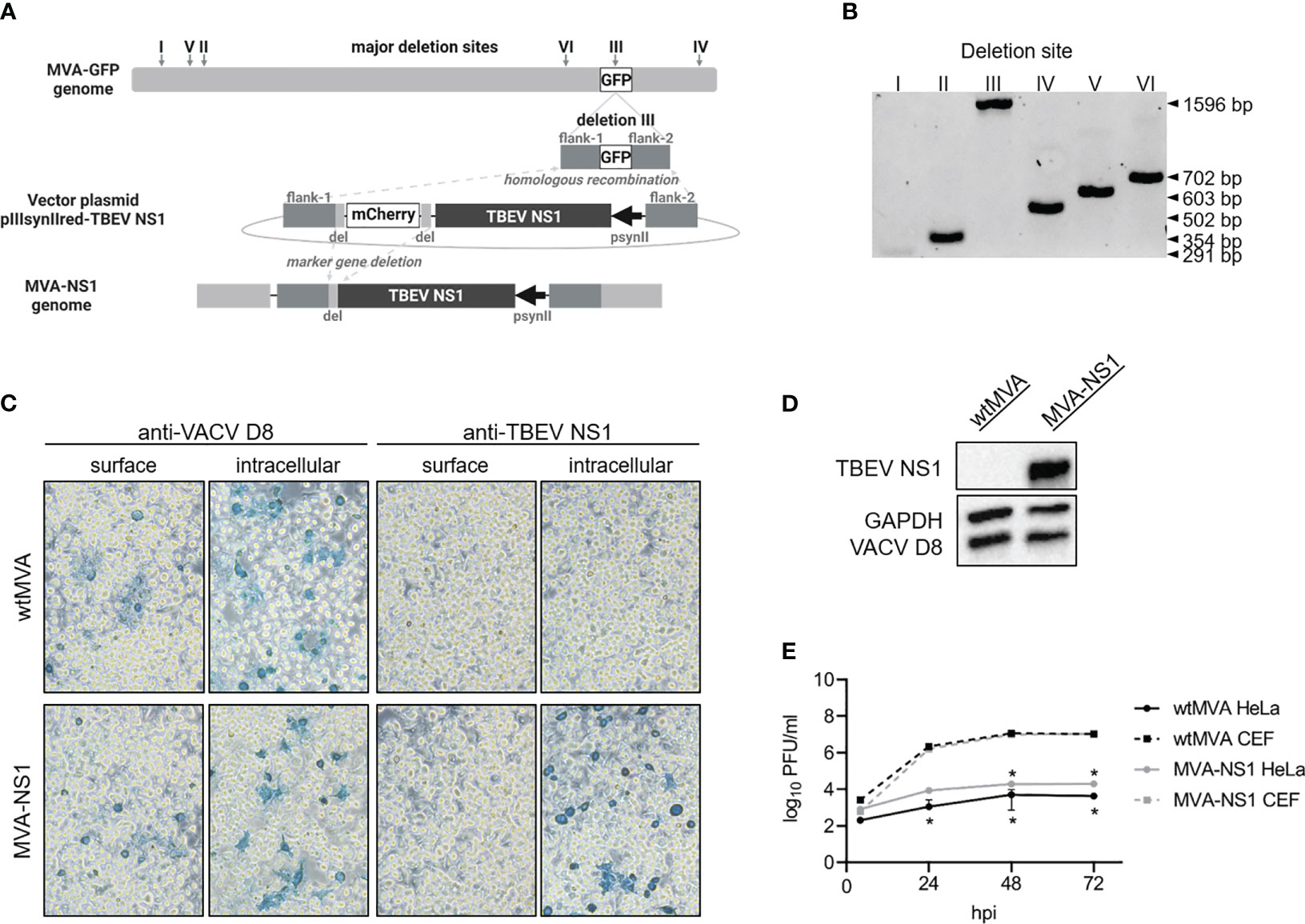
Figure 1 In vitro characterization of MVA-NS1. (A) Homologous and intragenomic homologous (marker gene deletion) recombinations lead to production of MVA-NS1 with TBEV NS1 expression under transcriptional control of VACV late promotor psynII. (B) PCR products specific for the six major deletion sites inside the MVA genome performed on MVA-NS1 (1% agarose TBE gel) (I: 291 bp, II: 354 bp, III: 447 bp, IV: 502 bp, V: 603 bp, VI: 702 bp). Integration of the NS1 gene in deletion site III was confirmed (III: 1596 bp). (C) Immunostaining of wtMVA or MVA-NS1 infected HeLa cells (MOI 1). 24 hpi, cells were fixed with 4% PFA, for intracellular staining permeabilized with TritonX®-100 and immunostained against VACV D8 and TBEV NS1 (20x magnification). (D) Western blot analysis of whole cell lysate from HeLa cells infected with wtMVA or MVA-NS1 for 24 h (MOI 5). Blots were stained against TBEV NS1. For control, antibodies against VACV D8 and GAPDH were included. (E) Growth curves of wtMVA (black) and MVA-NS1 (gray) on primary CEF cells (dotted line) and HeLa cells (solid line) (MOI 0.05). Mann-Whitney test was used for statistical comparison between CEF and HeLa cells (* p<0.05).
All viruses were stored at -80°C and tested negative for mycoplasma before use (MycoStrip™–Mycoplasma detection Kit, InvivoGen).
2.3 In vitro characterization of vector constructs
2.3.1 Integration of NS1 and sequence analysis
IAV RNA was isolated using the QIAmp® Viral RNA Mini Kit (Qiagen) following manufacturer’s instructions and reverse transcribed into cDNA using SuperScript III reverse transcriptase (Invitrogen) with the Uni12 primer (5’-AGCAAAAGCAGG-3’) (40). Chimeric NA cDNA regions were amplified using AmpliTag Gold DNA polymerase (Applied Biosystems) with primers NA int for (5´-ATCTGTCTGGTAGTCGGA-3´) and NA int rev (5´-GGCCAAGACCAATCTACA-3´). For amplification of the hemagglutinin (HA) gene segment, primers HA for (5’-AGCAAAAGCAGGGG-3’) and HA rev (5’-AGTAGAAACAAGGGTGTTTT-3’) were used. PCR products were separated on 0.8% agarose TBE gel and sequence identity of NA and HA was confirmed by sequencing (Microsynth Seqlab).
NS1 gene sequence integration into deletion site III of the MVA genome was verified by PCRs specific for the six major deletion sites of MVA as described previously (39). Purified PCR products were separated on 1% agarose TBE gel and analyzed with imaging system (ChemiDoc, ImageLab v6.0.1, Bio-Rad Laboratories, Inc.). For sequencing of NS1, deletion site III-specific PCR was performed (39) and purified PCR product was sequenced (Microsynth Seqlab). For PCRs, GoTaq® DNA polymerase (Promega) and for DNA purification, GeneJET Gel Extraction Kit (Thermo Scientific™) were used.
2.3.2 Western blot and immunostaining
To show expression of TBEV NS1 by the IAV vector system, MDCK cells were infected with MOI 0.01 of the respective IAV in the absence of eNA. For MVA vector system, HeLa cells were infected with MOI 5 of the respective MVA. For Western blot analysis, cells were lysed 24 hpi using RIPA buffer with 1% Halt™ Protease-Inhibitor-Cocktail (100x) (Thermo Fisher Scientific). Lysates were separated on 10% SDS-PAGE gels (Bio-Rad Laboratories, Inc.) and blotted on Cytiva Amersham™ Hybond™ P 0.45μm PVDF Membrane (VWR). Proteins were stained by using the mouse anti-TBEV NS1 monoclonal antibody (mAb) (M838, The Native Antigen Company), rabbit anti-GAPDH (D16H11) XP® mAb (Cell Signaling Technology®), mouse anti-influenza NP mAb (ATCC, clone HB65) or polyclonal rabbit anti-Cell Surface-Binding Protein (D8L) antibody (Biozol), respectively. After incubation with goat anti-rabbit IgG (H+L) HRP (Invitrogen) or goat anti-mouse IgG (H+L) HRP (Invitrogen) as secondary antibodies, blots were developed using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific™) and ChemiDoc Imaging System (Bio-Rad Laboratories, Inc.). For immunostaining, infected cells (IAV: MDCK cells, MOI 0.01; MVA: HeLa cells, MOI 1) were fixed 24 hpi with 4% ice-cold paraformaldehyde (PFA, Roth). For intracellular staining, cells were permeabilized using 0.5% Triton X®-100 (Roth). Viral proteins were detected by using the above mentioned antibodies. For visualization, stained cells were incubated with HRP-substrate TrueBlue™ Peroxidase Substrate (SeraCare) supplemented with 1:1000 H2O2 (Roth) and imaged with Leica DM IL LED microscope.
2.3.3 Growth kinetics
MDCK cells were infected in the presence or absence of eNA with MOI 0.001 of the respective IAV. For MVA, primary CEF and HeLa cells were infected with MOI 0.05. Supernatant was taken 2/4, 24, 48 and 72 hpi and viral titers were determined by plaque assay as described above.
2.4 Animal experiments
2.4.1 Ethical statement
All animal experiments were conducted at the University of Veterinary Medicine Hannover, Foundation, Hannover, Germany in strict compliance with European guidelines (EU directive on animal testing 2010/63/EU) and German Animal Welfare Law. The study protocol was approved by the Lower Saxony State Office for Consumer Protection and Food Safety (LAVES, Approval No. 33.8-42502-04-20/3437).
2.4.2 Mice
Female C57BL/6JOlaHsd (BL6) mice were purchased from the commercial breeder Envigo RMS. Depending on the required biosafety level, mice were housed in individually ventilated cages type Sealsafe Plus GM500 or IsoCage N Biocontainment System (Tecniplast), respectively. Sterilized food pellets and water were provided ad libitum. All experiments started after at least one week of habituation and acclimatization of mice. Treatment of mice was done under isoflurane anesthesia.
2.4.3 Immunogenicity study
Six to eight weeks old BL6 mice (n=4/group) were vaccinated twice in a 4-week interval with 107 plaque-forming units (PFU) of the IAV [subcutaneous, dorsally in the neck region (s.c.) as described previously (41)] or MVA [intramuscular, left hind limb (i.m.)] vector constructs. 170 µl of FSME-IMMUN® 0.5 mL (Pfizer, lot number EM2898) was administered as positive control (0.816 µg/mouse; s.c.). 100 µl PBS (s.c.) was administered as negative (mock) control. To minimize the number of experimental animals and to comply with the 3R principle (replacement, reduction and refinement), data of wtMVA-vaccinated mice (empty vector control group) were shared with an experiment performed in parallel under identical experimental conditions (same approval number). This was deemed justified because many studies failed to demonstrate any effect of MVA vector control induced immunity on immune responses to the pathogen of interest and protective efficacy (e.g (42–46). Mice were bled on day 0 and 28 before the first and second vaccination, respectively, by puncturing Vena facialis and on day 56 by retrobulbar sinus puncture. Blood was collected in MiniCollect® CAT Serum Sep Clot Activator tubes (Greiner Bio-One GmbH), incubated 30 min at room temperature (RT) and centrifuged at 3000 xg for 10 min to collect serum. After final blood drawing, mice were euthanized and spleens from individual mice were collected for the generation of single-cell suspensions through cell strainers followed by erythrocyte lysis using ACK Lysing buffer (Gibco™). Splenocytes were resuspended in RPMI 1640 (1X) with 10% FBS, Pen/Strep and 5 mM ß-mercaptoethanol (R10F) and directly used for ELISpot and FACS assays.
2.4.4 TBEV challenge infection
All challenge infection experiments were done under biosafety level 3**. Mice (n=12/group) were vaccinated as described above. On day 56 post prime immunization, blood was taken at Vena facialis and mice were infected with 5.4x103 TCID50 TBEV strain Neudoerfl (100 µl, s.c.). Upon challenge infection, mice were scored daily based on the clinical score sheet including the categories outer appearance, activity, movement, body weight and neurological signs. Half of the mice (n=6/group) were euthanized 8 days post infection (dpi) for determination of viral loads, remaining mice (n=6/group) were taken out of the experiment according to humane endpoint (HEP) or study endpoint (SEP, 16 dpi). On day of sacrifice, mice were bled by retrobulbar sinus puncture and euthanized. Organs (left brain hemisphere, cervical part of spinal cord, spleen, rice-corn sized part of ileum and colon) were collected in 1 ml PBS, homogenized with a stainless steel bead using the TissueLyser II (Qiagen) with 30 Hz for 1 min and stored at -80°C. The right brain hemisphere and remaining gastrointestinal tract were fixed in ROTI®Histofix 4% (Roth, for at least 48 h) for histopathological analysis.
2.5 Serology
2.5.1 Enzyme-linked immunosorbent assay
To detect TBEV NS1-specific IgG antibodies, Mouse Anti-Tick Borne Encephalitis Virus Non-Structural Protein 1 IgG Elisa Kit (Alpha Diagnostic International) was used according to manufacturer’s instructions. Serum was heat-inactivated before use (30 min, 56°C). Concentration of specific anti-NS1 antibodies was measured in arbitrary units (U/ml).
2.5.2 Luciferase immunoprecipitation systems assay
Luciferase Immunoprecipitation System (LIPS) assay for TBEV NS1 was performed as described previously (47) with 1:100 diluted, heat-inactivated mouse sera (30 min, 56°C). LIPS plasmids were kindly provided by Imke Steffen (Institute for Biochemistry and Research Center for Emerging Infections and Zoonoses, University of Veterinary Medicine Hannover, Foundation, Hannover, Germany). Luminescence was measured using the microplate reader infinite 200Pro (Tecan) and Tecan i-control software (version 2.0.10.0, Tecan). Average of triplicate measurements was determined and expressed in relative light units (RLU). RLU values higher than the average of negative samples plus five times standard deviation are considered positive.
2.5.3 Virus neutralization assay
To test mouse sera for TBEV-neutralizing antibodies, heat-inactivated sera (30 min, 56°C) were two-fold serially diluted in A549 infection medium, starting with a 1:10 dilution. 100 TCID50/well TBEV Neudoerfl was added and incubated for 1 h at 37°C. Subsequently, virus-serum mix was transferred in triplicates to 80% confluent A549 cells which were incubated for 5-6 days at 37°C, 5% CO2. Reduction of the cytopathic effect (CPE) by 100% compared to the negative serum control was considered as virus neutralization. Virus neutralizing titers (VNT100) were defined microscopically as the reciprocal of the highest serum dilution still resulting in complete inhibition of CPE.
2.5.4 Hemagglutination inhibition assay
To demonstrate IAV-specific antibodies, heat-inactivated (30 min, 56°C) mouse sera were tested in hemagglutination inhibition (HI) assay as described before (48). In short, serum was pre-incubated for 16 h at 37°C with Vibrio cholerae filtrate (generously provided by Ron Fouchier, Erasmus Medical Center, Rotterdam, Netherlands) and subsequently heat-inactivated at 56°C for 1 h. HI assay was then performed after standard protocol with 4 hemagglutination units of rPR8 and 1% chicken erythrocytes.
2.6 Splenocytes
2.6.1 Restimulation of splenocytes
TBEV-specific peptide pools based on TBEV Neudoerfl NS1 (UniProtKB: P14336) were used for the ex vivo restimulation of splenocytes. Lyophilized 15-mer peptides with 11 amino acid overlaps (≥ 75% purity, GenScript Biotech Corp) were dissolved in DMSO and two pools were generated (NS11-183: 43 peptides; NS1173-352: 42 peptides). For ex vivo restimulation, mouse splenocytes were incubated overnight at 37°C/5% CO2 with a final peptide concentration of 1 µg/ml, 30 ng/ml Phorbol 12-myrisate 13-acetate (PMA; Cayman Chemical) and 0.5 µg/ml Ionomycin (Cayman Chemical) (positive control), MOI 1 of live IAV or MOI 3 of live MVA, and DMSO/R10F (negative control), respectively.
2.6.2 IFN-γ ELISpot
IFN-γ ELISpot assays were performed with Mouse IFN-γ ELISpotPLUS kit (ALP) (Mabtech) according to manufacturer’s standard protocol. Mouse splenocytes were restimulated as described above on pre-coated 96-well ELISpot plates in the presence of antigen/control for 20 h at 37°C, 5% CO2. After staining, spots were scanned and counted by using the ImmunoSpot® S6 Ultimate Reader and the ImmunoSpot® software (Version 7.0.20.1, Immunospot, CTL). Triplicates were averaged and data expressed as spot-forming cells (SFC) per 106 splenocytes after background subtraction (DMSO/R10F stimulation).
2.6.3 Flow cytometry
Mouse splenocytes (1x106 cells/well) were restimulated as described above. For the final 4 h of restimulation, Brefeldin A (10 µg/ml, Sigma-Aldrich) was added to the medium. Cells were stained with LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit for 633 or 635 nm excitation (Invitrogen™) for 20 min in the dark followed by Fc receptor blocking with anti-Mouse CD16-CD32 (Clone: 93) for 15 min at RT in the dark. Surface staining was performed for 20 min at 4°C in the dark. Cells were then permeabilized and fixed with BD Cytofix/Cytoperm™ (BD Biosciences) for 20 min at 4°C in the dark, intracellular staining was performed for 30 min at 4°C in the dark. After resuspension in PBS, cells were acquired by BD LSR Fortessa X-20 (BD Biosciences) using BD FACSDiva (version 9.0, BD Biosciences). Used antibodies are listed in Table 1 and were used at 1:200 dilution.
Table 1 Antibody panel used in flow cytometry experiments.
2.7 Determination of live virus in serum and organs by tissue culture infectious dose 50%
Tissue samples were thawed and tissue debris was removed by centrifugation (3000 xg, 10 min). 80% confluent A549 cells were inoculated in quadruplicates with 10-fold serial dilutions of sera or organ samples prepared in A549 infection medium. After 5-6 days at 37°C, 5% CO2, cells were screened for the presence/absence of CPE. TCID50 values for individual samples were determined using the Reed and Muench method (49) and calculated to 1 g tissue (TCID50/g tissue) or 1 ml (TCID50/ml). Detection limit for each organ was defined by dividing the lowest dilution (101) by the respective averaged organ weight.
2.8 RNA isolation and real time reverse transcriptase quantitative PCR
Total RNA was extracted from sera and clarified organ homogenate supernatants using the QIAmp® Viral RNA Mini Kit (Qiagen) following manufacturer’s protocol. Real time reverse transcriptase quantitative PCR (RT-qPCR) was performed with Luna® Universal One-Step RT-qPCR Kit (New England BioLabs® GmbH) based on the modified protocol by Schwaiger and Cassinotti (50) including a TBEV Neudoerfl RNA standard kindly provided by Stefanie Becker (Institute for Parasitology and Research Center for Emerging Infections and Zoonoses at University of Veterinary Medicine Hannover, Foundation, Hannover, Germany). Real time RT-qPCR was run in duplicates using AriaMx Real-time PCR System (Agilent Technologies) with Agilent Aria software (version 1.5, Agilent Technologies). Absolute copy numbers were calculated based on the standard curve and expressed as TBEV copies/ml or g tissue.
2.9 Histology
Two longitudinal sections of the right brain hemisphere and representative sections of duodenum, jejunum, ileum, caecum, colon and rectum were embedded in paraffin wax followed by cutting 2-3 µm thick sections using a microtome. Sections were stained with hematoxylin and eosin (H&E) and analyzed as described previously (51). Briefly, three brain regions (olfactory bulb, cerebral cortex, hippocampus) were selected for histopathological analysis and evaluated with respect to microscopic lesions including perivascular as well as vascular inflammation, vascular lesions including perivascular edema, hemorrhage and fibrinoid necrosis, microgliosis characterized by hyperplasia and/or hypertrophy of microglia/macrophages as well as cellular necrosis characterized by karyorrhexis, karyolysis, pyknosis and triangularly shaped, hypereosinophilic and shrunken neurons. To assess histological changes in the intestine of mice, submucosal and myenteric plexus were investigated with respect to microscopic lesions including necrosis of ganglion neurons characterized by karyorrhexis, karyolysis and pyknosis, hypereosinophilia and shrinkage of neurons as well as presence of inflammatory cell infiltrates/hypercellularity in plexus.
2.10 Immunohistochemistry
For immunohistochemistry (IHC), the avidin-biotin-peroxidase (ABC) complex method was applied as previously published (52). For immunohistochemical detection of TBEV antigen, a mouse anti-TBEV E mAb (clone 19/1493, diluted 1:2000, kindly provided by Matthias Niedrig) was used. Sections of brain and intestine were examined with respect to the presence of TBEV-antigen positive cells as described previously (51).
2.11 Statistics
GraphPad Prism software (version 9.0.0, GraphPad Software Inc.) was used for statistical analysis. Statistical test used for analyses are stated in the respective figure legends. A p value <0.05 was considered as significant.
3 Results
3.1 TBEV NS1 is expressed by the highly attenuated IAV- and MVA-vectors
Using reverse genetics (37), the PR8-based virus IAV-NS1 harboring a chimeric NA gene segment was rescued. Most of the open reading frame of the IAV NA gene was replaced by the NS1 gene of the TBEV strain Neudoerfl terminated by a stop codon. Essential IAV packaging signals located in the 3’ and 5’ ends were maintained (53). This way, IAV-NS1 expresses TBEV NS1 as a fusion protein with the N-terminal region of the NA of IAV PR8 (Figure 2A). Accordingly, the vector control virus IAV-RBD expressing a non-TBEV gene was designed and rescued. Correct insertion into NA was confirmed by RT-PCR specific for the NA gene (Figure 2B). Nucleotide sequences of the HA and chimeric NA genes and absence of mutations were confirmed by Sanger sequencing (data not shown). MVA served as a second viral vector system delivering TBEV NS1 (Figure 1A). Correct integration of the NS1 gene in deletion site III of the MVA genome was verified by PCRs specific for the six major MVA deletion sites (Figure 1B). Additionally, nucleotide sequencing confirmed complete insertion of the correct NS1 sequence (data not shown).
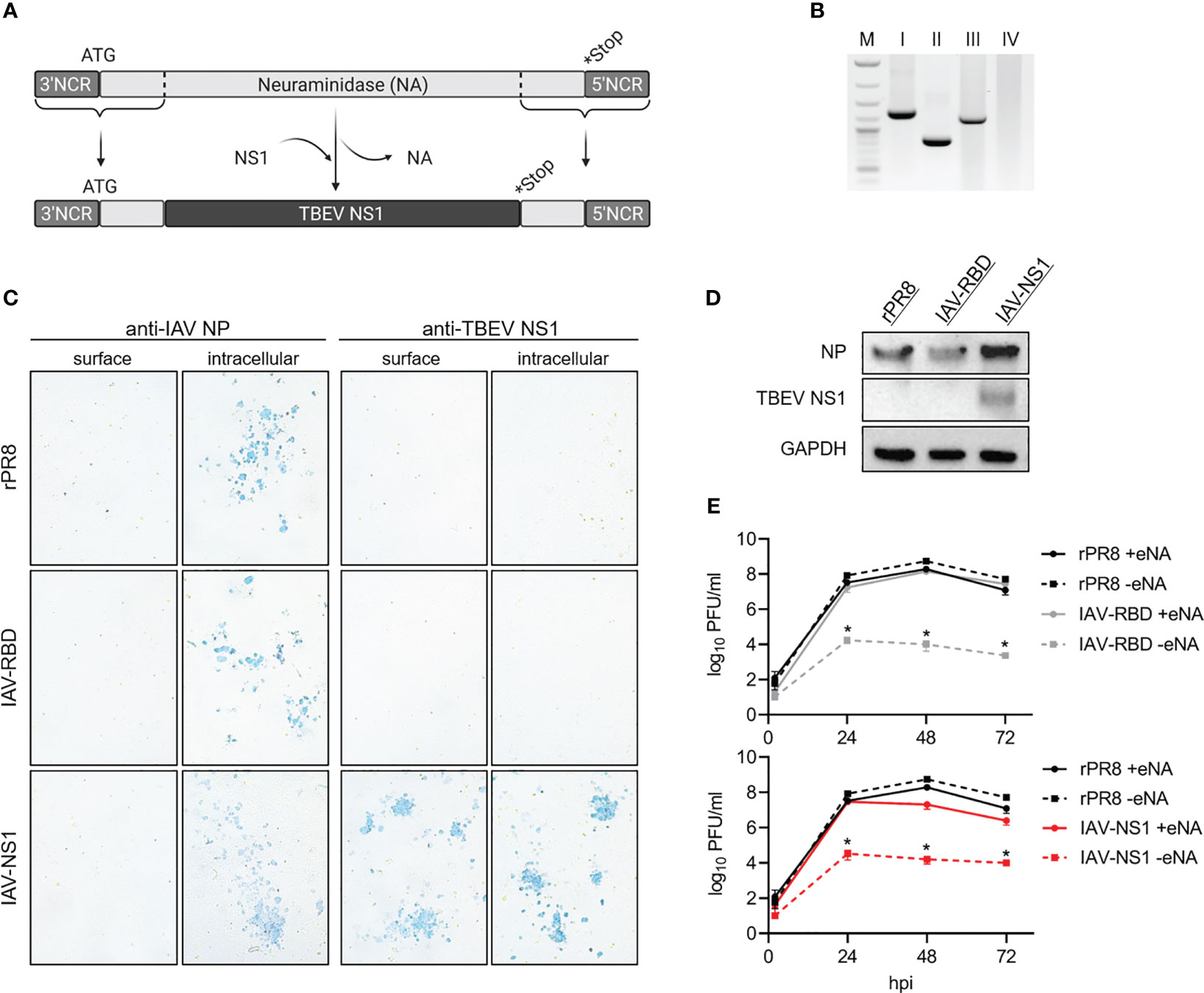
Figure 2 In vitro characterization of IAV-NS1. (A) Schematic representation of the NA-TBEV NS1 gene segment, NCR = non-coding region, ATG = start codon, *Stop = stop codon. (B) PCR products specific for IAV NA performed on I = rPR8, II = IAV-RBD, III = IAV-NS1 and IV = negative control (1% agarose TBE gel, M = 1 kb ladder). (C) Immunostaining of rPR8, IAV-RBD or IAV-NS1 infected MDCK cells (MOI 0.01). 24 hpi, cells were fixed with 4% PFA, for intracellular staining permeabilized with TritonX®-100 and immunostained against IAV NP or TBEV NS1 (10x magnification). (D) Western blot analysis of whole cell lysate from MDCK cells infected with rPR8, IAV-RBD or IAV-NS1 for 24 h (MOI 0.01). Blots were stained against TBEV NS1. For control, antibodies against IAV nucleoprotein (NP) and GAPDH were included. (E) Growth curves for rPR8 (black), IAV-RBD (gray) and IAV-NS1 (red) in the presence (solid line) or absence (dotted line) of eNA on MDCK cells (MOI 0.001). Mann-Whitney test was used for statistical comparison between rPR8 –eNA and IAV-RBD –eNA or IAV-NS1 –eNA, respectively (* p<0.05).
Expression of TBEV NS1 was shown by Western blot analysis of lysates of MDCK cells infected with IAV-NS1 or HeLa cells infected with MVA-NS1 (Figures 1D, 2D). Immunostaining of respective cells infected with IAV-NS1 or MVA-NS1 confirmed the intracellular expression of TBEV NS1 (Figures 1C, 2C). In addition, cell surface expression of NS1 in non-permeabilized cells was shown for IAV-NS1 (Figure 2C).
Insertion of foreign genes should not affect the attenuated phenotype of vector-based vaccine constructs. Therefore, growth characteristics of the vector controls, IAV-NS1 and MVA-NS1 were analyzed by performing multi-step growth kinetics (Figures 1E, 2E). The vector control IAV-RBD and IAV-NS1 displayed a highly attenuated growth phenotype compared to the recombinant wildtype virus rPR8 in the absence of eNA. However, addition of eNA to the cell culture medium restored the in vitro replicative capacity of IAV-RBD and IAV-NS1. Viral titers peaked at 48 hpi and declined thereafter. rPR8 replicated independently of eNA to high titers (Figure 2E). Similarly, wtMVA and MVA-NS1 replicated well in permissive primary CEF cells up to titers of 107 PFU/ml. However, in non-permissive human HeLa cells, replication-deficiency for wtMVA and MVA-NS1 was confirmed (Figure 1E).
Thus, two attenuated vector-constructs based on IAV and MVA were constructed that drive the expression of the TBEV NS1 gene. Subsequently, we tested the tolerability and immunogenicity in mice.
3.2 IAV-NS1 and MVA-NS1 are immunogenic in mice
To test the immunogenicity of the respective NS1 vector constructs, mice were vaccinated twice at a 4-weeks interval with 107 PFU of IAV-NS1 or MVA-NS1. For heterologous prime/boost vaccination regimens, we immunized mice with MVA-NS1 followed by an IAV-NS1 booster immunization (MVA-NS1/IAV-NS1) and vice versa (IAV-NS1/MVA-NS1). As controls, mice were vaccinated twice with 0.816 µg FSME-IMMUN®, PBS or vector controls (IAV-RBD, wtMVA).
All vaccine preparations and regimens were well tolerated and all mice continued to gain weight over the course of immunization without displaying any clinical signs (Supplementary Figure S1). Eight weeks after the first immunization, serum samples and splenocytes were collected to analyze TBEV-specific immune responses.
None of the mock or vector control-vaccinated mice developed TBEV-specific antibodies as measured by NS1-specific ELISA, LIPS assay and VN assay (Figure 3, Supplementary Figure S2). In contrast, all mice vaccinated with IAV-NS1 or MVA-NS1 developed TBEV NS1-specific antibodies after a single immunization. These antibody levels were boosted by a second immunization (Figure 3B). Two doses with MVA-NS1 induced higher antibody levels than two doses of IAV-NS1. With the heterologous prime/boost regimens, significantly higher TBEV NS1 antibody levels were achieved (Figures 3A, B). Vaccination with FSME-IMMUN® did not induce TBEV NS1-specific antibodies (Figure 3), but resulted in the induction of high VN antibody titers (Supplementary Figure S2). The use of NS1-based vaccine preparations did not result in the induction of VN antibodies (Supplementary Figure S2).
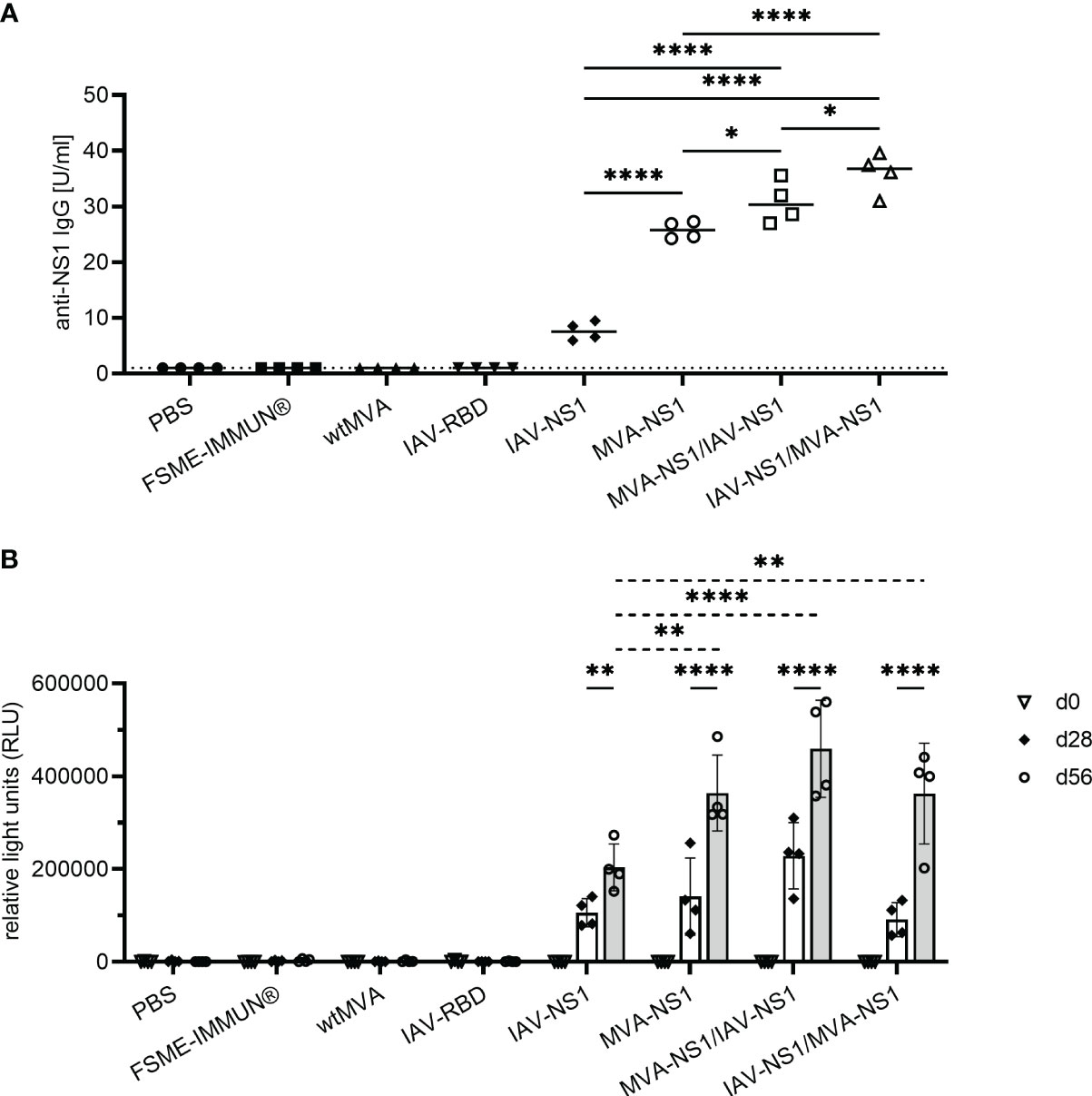
Figure 3 Antibody response against TBEV NS1. (A) Quantitative measurement of TBEV NS1-specific IgG by NS1 ELISA of sera samples collected 56 days post prime immunization. Results are reported as arbitrary units per ml (U/ml). Cut-off values were calculated according to the manufacturer’s instructions (1 U/ml). One-way ANOVA with Tukey’s multiple-comparison test was used for statistical analysis. Significances are shown for all NS1-specific vaccine groups (* p<0.05, **** p<0.0001). (B) Semi-quantitative measurement of TBEV NS1-specific antibodies by LIPS assay with mouse sera from day 0, 28 and 56 post prime immunization in relative light units (RLU). Mean values with SD are shown from 2-5 independent experiments (n=4 mice/group). Two-way ANOVA with Tukey’s multiple-comparison test was used for statistical analysis. Significances are shown for RLU values on d28 vs. d56 (solid line) and for all NS1-specific vaccine groups compared at d56 (dotted line) (** p<0.01, **** p<0.0001).
Vaccination with the IAV-vector induced high titers of PR8-specific antibodies as measured by HI assay. Mice receiving IAV-NS1 prime and MVA-NS1 boost developed slightly lower IAV-specific HI titers compared to the other groups immunized with IAV-based vector constructs. Mice of all other treatment groups were negative in the HI assay (Supplementary Figure S3).
NS1-specific T cell responses were analyzed by IFN-γ ELISpot assay using splenocytes restimulated with peptide pools of TBEV NS1 (NS11-183 and NS1173-352). IFN-γ SFC were detectable in all mice that received IAV-NS1 and/or MVA-NS1 (Figure 4A). Splenocytes from all NS1-vaccinated mice responded to both peptide pools, however, the response to NS1173-352 peptide pool was in general higher than the response to NS11-183. Mice that received heterologous prime/boost regimens displayed significantly higher combined numbers of IFN-γ positive spots than the mice that received IAV-NS1 or MVA-NS1 only (Figure 4B). In FSME-IMMUN®-vaccinated and control mice, NS1-specific IFN-γ SFC were not detected. Further flow cytometric analysis revealed that both antigen-specific CD4+ and CD8+ T cells were the source of IFN-γ. In agreement with the ELISpot data, frequencies of NS1173-352-specific IFN-γ+ cells were higher than those specific for NS11-183 (Figures 4D, F). Moreover, CD8+ T cells positive for Granzyme B were detected in mice that received two doses of MVA-NS1 or heterologous prime/boost regimens (Figure 4G). The induction of NS1-specific CD4+ and CD8+ T cells was further confirmed by the detection of T cells expressing the early activation marker CD69 (Figures 4C, E). The respective viral vectors also induced vector-specific CD4+IFN-γ+, CD8+IFN-γ+ and CD8+Granzyme B+ T cells. In general, the frequencies of these cells were higher after homologous prime/boost regimens with the respective vectors than after heterologous vaccination (Supplementary Figure S4).
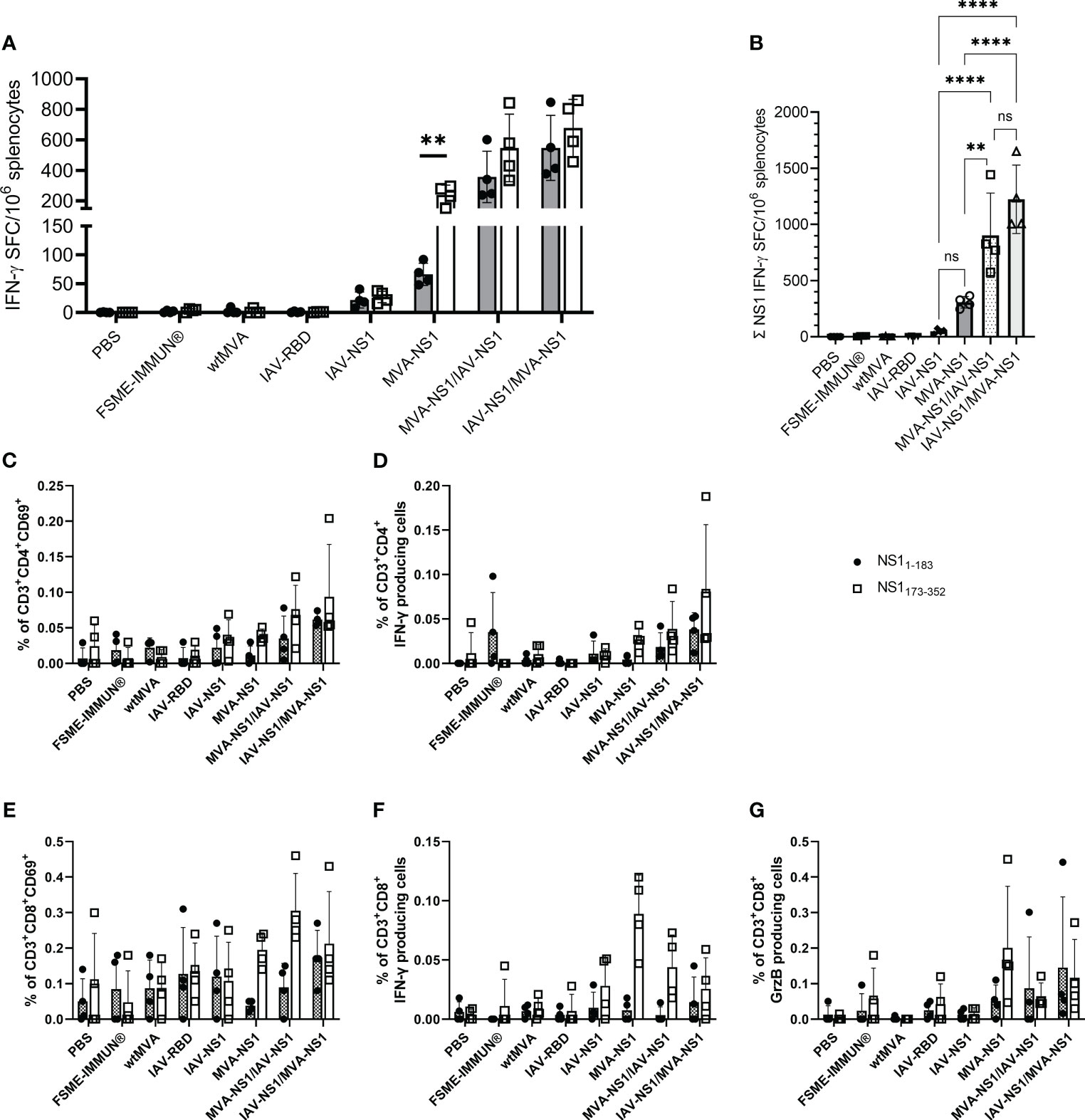
Figure 4 T cell response against TBEV NS1. (A) For IFN-γ ELISpot, mouse splenocytes were restimulated overnight with TBEV NS1 peptide pools NS11-183 and NS1173-352. Mean values with SD are shown as IFN-γ spot-forming cells (SFC) per one million splenocytes after background subtraction. For statistical analysis, unpaired t-test was used (**p<0.01). (B) Calculated IFN-γ SFC per million splenocytes for total NS1 (NS11-183 + NS1173-352). One-way ANOVA with Tukey’s multiple-comparison test was used for statistical analysis. Significances are only shown for NS1-vaccine groups (ns = not significant, ** p<0.01, **** p<0.0001). (C–G) Flow cytometric analysis of mouse splenocytes. Frequency of CD3+ subpopulations gated on CD4+ (C, D) and CD8+ (E–G) T cells positive for CD69, IFN-γ and Granzyme B (GrzB) upon restimulation with NS11-183 (black circle) or NS1173-352 (unfilled square) (n = 4). Bars represent mean with SD. Data is shown after background subtraction.
3.3 Heterologous prime/boost regimens with IAV-NS1 and MVA-NS1 partly protect against TBEV challenge infection
Next, the protective efficacy of IAV-NS1 and MVA-NS1 immunization against a lethal challenge with TBEV Neudoerfl was assessed. All mock and vector control-vaccinated mice started to lose body weight 8 dpi and developed severe signs of TBEV infection reaching the HEP at 10-13 dpi (Figures 5A, B). All FSME-IMMUN®-vaccinated mice, which served as positive controls, survived until the SEP (16 dpi) without severe clinical scores (Figures 5A, B).
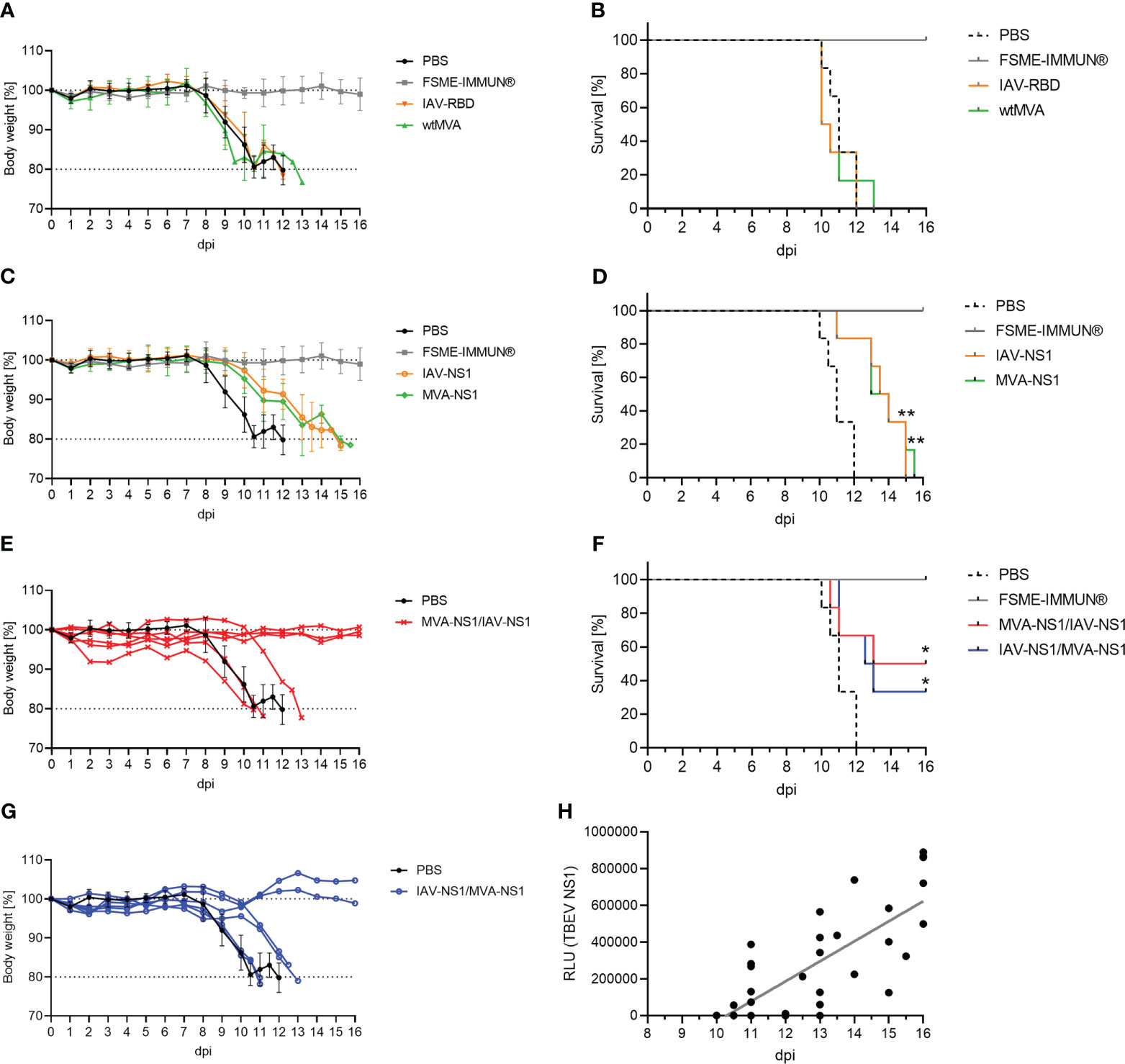
Figure 5 Protective efficacy of TBEV NS1-based vector constructs. Body weights and survival of BL6 mice immunized with controls (A, B), IAV-NS1 or MVA-NS1 (C, D), MVA-NS1/IAV-NS1 (E, F) or IAV-NS1/MVA-NS1 (G, F) after challenge infection with TBEV Neudoerfl. In (A) and (C) mean body weights are shown (n = 6). For (E) and (G) body weights from individual mice are shown. Weights of PBS mice are shown as mean (n = 6). (B, D, F) Kaplan-Meier curves were analyzed by log-rank test (*p<0.05, **p<0.01). (H) Pearson correlation of TBEV NS1-specific antibodies on day 56 before challenge infection measured by LIPS with days of survival post TBEV infection for all groups (FSME-IMMUN® group was excluded). Linear regression is depicted by gray line (r = 0.7964, p<0.0001).
Mice that had received two doses of either IAV-NS1 or MVA-NS1 displayed a delayed onset of body weight loss starting at 10 dpi and a statistically significant prolonged survival up to 15 dpi compared to the PBS group (Figures 5C, D). Interestingly, 50% of the MVA-NS1/IAV-NS1- (3/6) and 33% of the IAV-NS1/MVA-NS1- (2/6) vaccinated mice were fully protected against lethal TBEV challenge (Figures 5E–G). These mice maintained their body weight and did not show clinical signs until SEP. Of note, prolonged survival post challenge infection correlated with levels of NS1-specific antibodies prior to infection (r=0.7964; p<0.0001) (Figure 5H).
3.4 Heterologous prime/boost vaccination reduces viral loads and histopathological changes in the central nervous system and intestine
To assess whether NS1 vaccination against TBEV has an effect on viral replication, half of the mice from each group (n = 6) were taken out of the challenge experiment 8 dpi to examine TBEV titers in different organs by TCID50 and TBEV RNA copy numbers by real time RT-qPCR. At 8 dpi, high viral loads were observed in the spleens of mock-vaccinated mice with RNA copy numbers of around 108 per gram of tissue, which were similar in the vector control groups. FSME-IMMUN® vaccination afforded partial protection and prevented virus replication in 4 out of 6 mice (Figure 6A). Interestingly, vaccination with MVA-NS1/IAV-NS1 or IAV-NS1/MVA-NS1 reduced the virus loads in the spleen approximately 100-fold compared to mock-vaccinated mice. None of the serum samples were tested positive for viral RNA 8 dpi (Figure 6B). Except for one FSME-IMMUN®-vaccinated mouse, no infectious virus was seen in any spleen and serum samples (Supplementary Figures S5A, B).
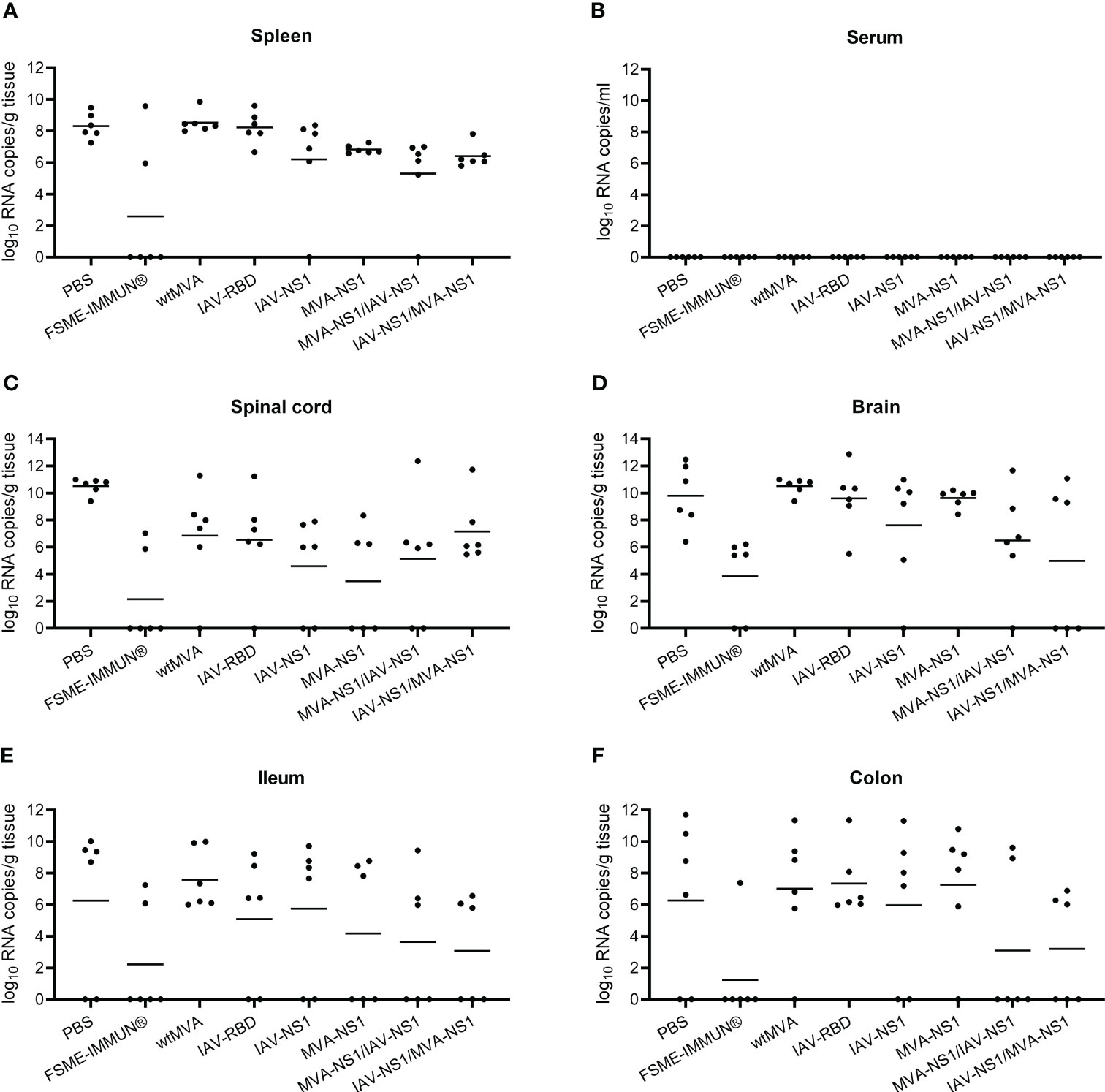
Figure 6 Viral loads in the periphery, CNS and GIT. Presence of TBEV RNA in spleen (A), serum (B), spinal cord (C), brain (D), ileum (E) and colon (F) was determined by performing real time RT-qPCR on cleared organ homogenates or serum from TBEV challenged mice sacrificed at 8 dpi (n = 6). Bars depict geometric means.
To assess the viral spread into the central nervous system (CNS), the cervical part of the spinal cord and brain tissues were tested for the presence of virus. Additionally, the olfactory bulbs of selected mice were histologically and immunohistochemically analyzed (Figure 7). In the spinal cords and brains of mock-vaccinated mice, high TBEV RNA copy numbers were detected 8 dpi (Figures 6C, D). In the brains of these mice also high titers of infectious virus were detected (Supplementary Figure S5D). This was accompanied by microscopic lesions in the brains of these mice consisting of cellular necrosis, microgliosis, perivascular inflammation and vasculitis 8 dpi. Cellular necrosis was characterized by shrunken and hypereosinophilic cells with karyorrhectic, karyolytic and pyknotic nuclei. Shrunken, hypereosinophilic, triangular shaped necrotic neurons are shown representatively for the olfactory bulb of a selected mock-vaccinated mouse (Figure 7A). Accordingly, IHC for TBEV E protein revealed high numbers of antigen-positive cells characterized by a cytoplasmic staining as shown for the olfactory bulb in Figure 7I.
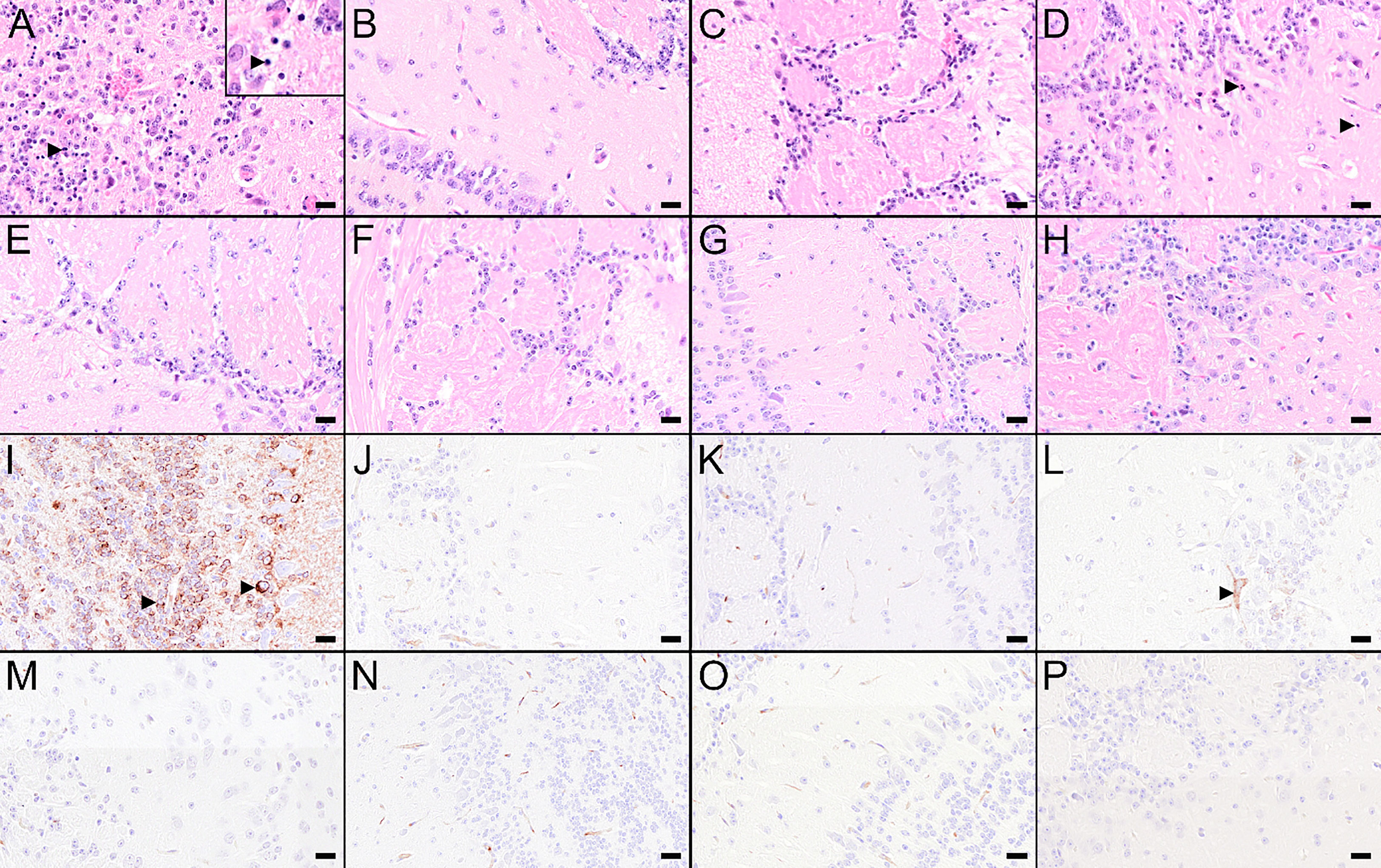
Figure 7 Histological and immunohistochemical changes in the olfactory bulb at 8 dpi. (A–H) Hematoxylin and eosin (H&E) stained sections of the olfactory bulb of a mouse treated with PBS (A) or vaccinated with FSME-IMMUN® (B), wtMVA (C), IAV-RBD (D), IAV-NS1 (E), MVA-NS1 (F), MVA-NS1/IAV-NS1 (G) or IAV-NS1/MVA-NS1 (H), respectively. (A) Olfactory bulb of the PBS-treated mouse displays marked cellular necrosis with karyorrhectic, karyolytic and pyknotic cells (insert, arrowhead) and shrunken, hypereosinophilic, triangular shaped necrotic neurons as well as inflammatory cell infiltrates. (B–H) In FSME-IMMUN®- (B), wtMVA- (C), IAV-NS1- (E), MVA-NS1- (F), MVA-NS1/IAV-NS1- (G) or IAV-NS1/MVA-NS1- (H) vaccinated mice, no significant microscopic lesions within the olfactory bulb are visible. In the IAV-RBD (D) vaccinated mouse, single necrotic cells are present. (I–P) Immunohistochemistry for TBEV E antigen of the olfactory bulb of a mouse treated with PBS (I) or vaccinated with FSME-IMMUN® (J), wtMVA (K), IAV-RBD (L), IAV-NS1 (M), MVA-NS1 (N), MVA-NS1/IAV-NS1 (O) or IAV-NS1/MVA-NS1 (P), respectively. (I) Immunohistochemically, a cytoplasmic TBEV immunoreactivity (arrowhead) is present in multiple cells of the olfactory bulb from the PBS-treated mouse. (J–P) Olfactory bulbs of FSME-IMMUN®- (J), wtMVA- (K), IAV-NS1- (M), MVA-NS1- (N), MVA-NS1/IAV-NS1- (O) or IAV-NS1/MVA-NS1- (P) vaccinated mice do not show immunoreactivity for TBEV, while the IAV-RBD- (N) vaccinated mouse shows single TBEV-positive cells (arrowhead). Scale bars: 20µm.
Vaccination with FSME-IMMUN® reduced TBEV replication considerably, but not completely. In 2 and 4 out of six mice low levels of viral RNA were still detectable in spinal cords and brains, respectively (Figures 6C, D). This is in accordance with findings of histological and immunohistochemical analysis (Figures 7B, J).
Of special interest, in mice that received heterologous prime/boost regimens MVA-NS1/IAV-NS1 or IAV-NS1/MVA-NS1, a clear reduction in viral titers (Supplementary Figure S5D) and RNA copy numbers (Figure 6D) was observed in the brains compared to mock or vector control-vaccinated mice. In the brains of these mice no major histopathological changes nor TBEV antigen positive cells using IHC were observed (Figures 7G, H, O, P). Mice that received two doses of IAV-NS1 or MVA-NS1, respectively, showed some reduction in viral loads in the CNS (Figures 6C, D, Supplementary Figures S5C, D). However, the histopathological changes in the CNS of these mice were less prominent than in the CNS of mock-vaccinated animals (Figures 7E, F, M, N).
Upon dissection at 8 dpi, 100% of the mock-vaccinated and 67% of the vector control mice displayed macroscopically visible acute distension and segmental dilation of the gastrointestinal tract as described previously (54). In contrast, only 50% of mice vaccinated with two doses of IAV-NS1 or MVA-NS1 displayed such lesions. In mice that received the heterologous prime/boost vaccination regimens, this proportion was only 16.7%. No macroscopic abnormalities were observed in FSME-IMMUN®-vaccinated mice (data not shown).
In the majority of the mock and vector control-vaccinated mice, high TBEV RNA levels were observed in ileum and colon at 8 dpi with RNA copy numbers ranging from 106-1012 per gram of tissue (Figures 6E, F). Infectious virus was mainly detectable in the colon of infected animals (Supplementary Figures S5E, F). In FSME-IMMUN®-vaccinated mice, geometric mean titers were considerably reduced and viral RNA was detected in two and one animals in ileum and colon, respectively (Figures 6E, F). In these mice infectious virus was not detected in any of the organs tested 8 dpi (Supplementary Figures S5E, F) or 16 dpi (data not shown). Of interest, also in mice that received MVA-NS1/IAV-NS1 or IAV-NS1/MVA-NS1 viral copy numbers, especially in the colon, were reduced >1000-fold compared to mock and vector control-vaccinated mice (Figures 6E, F).
Histopathological examination of the intestine revealed histopathological lesions characterized by ganglioneuritis of the myenteric and submucosal plexus in mock and vector control-vaccinated mice 8 dpi as described previously (54). Ganglioneuritis of the myenteric and submucosal plexus are representatively shown in the colon (Figure 8). Ganglia show signs of neuronal necrosis and an infiltration with inflammatory cells and/or hyperplasia of resident immune cells. In concordance with the viral loads that were measured, only minor histopathological changes were observed in mice that received heterologous prime/boost NS1 vaccination (Figures 8G, H, O, P). In line with the low numbers of mice with infectious virus in the intestine (Supplementary Figure S5F), IHC showed no significant portion of antigen-positive cells in the plexus of FSME-IMMUN®- or NS1-immunized mice (Figures 8J, M–P), whereas TBEV antigen positive cells were shown for mock and vector control-vaccinated mice (Figures 8I, K, L).
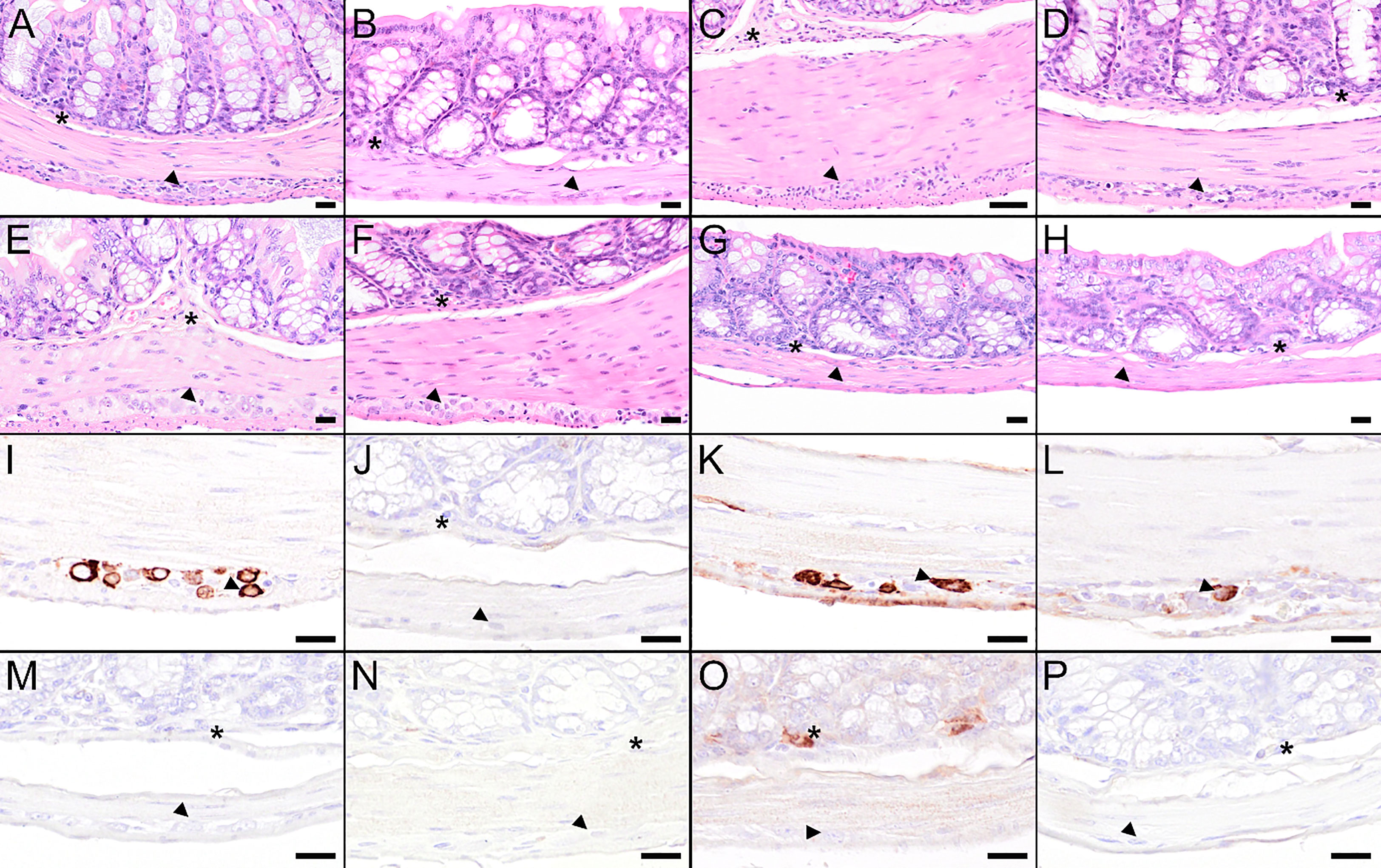
Figure 8 Histological and immunohistochemical analysis of colon at 8 dpi. (A–H) Hematoxylin and eosin (H&E) stained sections of the colon of a mouse treated with PBS (A) or vaccinated with FSME-IMMUN®, (B), wtMVA (C), IAV-RBD (D), IAV-NS1 (E), MVA-NS1 (F), MVA-NS1/IAV-NS1 (G) or IAV-NS1/MVA-NS1 (H), respectively. (A) Colon of the PBS-treated mouse displays minor to mild hypercellularity and cellular necrosis in the myenteric (arrowhead) and submucosal plexus (asterisk). (B–F) Colon of the FSME-IMMUN®- (B), wtMVA- (C), IAV-RBD- (D), and MVA-NS1- (F) vaccinated mice show minimal to moderate histopathological changes mainly characterized by hypercellularity/inflammatory cell infiltrates in the myenteric and submucosal plexus. (E–H) In the IAV-NS1- (E), MVA-NS1/IAV-NS1- (G) and IAV-NS1/MVA-NS1- (H) vaccinated mice, no histopathological changes in the plexi of the colon are observed. (I–P) Immunohistochemistry for TBEV E antigen of the colon of a mouse treated with PBS (I) or vaccinated with FSME-IMMUN® (J), wtMVA (K), IAV-RBD (L), IAV-NS1 (M), MVA-NS1 (N), MVA-NS1/IAV-NS1 (O) or IAV-NS1/MVA-NS1 (P), respectively. (I, K, L) Immunohistochemically, a cytoplasmic TBEV immunoreactivity is present in cells of the myenteric plexus (arrowhead) of the colon from the PBS-treated mouse (I), the wtMVA-vaccinated mouse (K) and the IAV-RBD-vaccinated mouse (L). A single TBEV-antigen positive cell is visible in the submucosal plexus of the colon from the MVA-NS1/IAV-NS1-vaccinated mouse (O, asterisk). No TBEV-antigen positive cells are detectable in the submucosal (asterisk) or myenteric plexus (arrowhead) of FSME-IMMUN®- (J), IAV-NS1- (M), MVA-NS1- (N) and IAV-NS1/MVA-NS1-vaccinated (P) mice. Scale bars (A, B, D–H): 20µm; Scale bar: (C, I–P): 50µm.
For 3/5 of the surviving mice receiving the heterologous NS1-based vaccinations, neither infectious virus (data not shown) nor virus RNA were detectable in all tested organs at study endpoint (Supplementary Figure S6). The other two mice showed low TBEV RNA copy numbers in brain (105 RNA copies/g tissue) and colon (106 RNA copies/g tissue) (MVA-NS1/IAV-NS1) or colon only (IAV-NS1/MVA-NS1). Histopathological examination and IHC for TBEV of brain and intestine showed no major lesions or TBEV antigen staining in those mice as shown for representative mice and organ sections (Supplementary Figure S7).
4 Discussion
In the present study, we tested the efficacy of two TBEV NS1-carrying viral vectors to induce protective immunity against TBEV. We showed that the use of IAV- and MVA-based viral vectors were highly immunogenic, especially in heterologous prime/boost regimens, resulting in the induction of TBEV NS1-specific antibodies and T cells. The vector-induced NS1-specific immunity afforded mice partial protection against a lethal challenge infection with TBEV.
First, the two recombinant vectors were characterized in vitro and it was shown that they both drive the expression of TBEV NS1 intracellularly. However, cell surface expression was only observed with IAV-NS1. Since TBEV NS1 lacks a transmembrane domain, it interacts with the plasma membrane via hydrophobic protrusions and is associated with lipid rafts [reviewed in (55)]. However, in the IAV-NS1 construct, NS1 was fused to the N-terminal part of the IAV NA which contains its transmembrane domain facilitating insertion of the NA-NS1 fusion in the cell membrane and expression on the cell surface. This was further supported by the fact that NS1 secretion was only observed from cells infected with MVA-NS1 but not from cells infected with IAV-NS1 (data not shown).
Insertion of the NS1 gene did not affect the attenuated phenotype of both recombinant vectors. In the absence of exogenous NA, NA-deficient IAV-RBD and IAV-NS1 replicated only to low titers, which is in concordance with previous studies (56, 57). Attenuation is achieved by the replacement of a large part of the IAV NA, which as receptor destroying enzyme plays an important role in the IAV replication cycle and release of newly budded IAV virions. Lack of a functional NA results in aggregation of virions at the cell surface as demonstrated by electron microscopy for a NA-deficient IAV expressing a NA-GFP fusion protein (56). As shown previously, trans-complementation by addition of NA from V. cholerae restored the replicative capacity of NA-deficient IAVs (58). Attenuation of MVA was achieved by extensive passaging in avian cells, resulting in the loss of genes involved in virus-host interactions and replication-deficiency in most mammalian cells [reviewed in (34)]. Due to their highly attenuated phenotype both IAV-NS1 and MVA-NS1 were well tolerated by mice.
Upon vaccination with the NS1-expressing vectors, NS1-specific antibodies were readily induced in all mice. As expected, these antibodies fail to display neutralizing activity because NS1 is not a component of TBEV virions. In contrast, vaccination with FSME-IMMUN®, which was included as a positive control in our study design, induced high levels of VN antibodies but failed to induce TBEV NS1-specific antibodies. These findings are in line with a recent publication showing that a high number of FSME-IMMUN® vaccine doses is required to induce measurable quantities of NS1-specific antibodies in mice (18). Two doses of MVA-NS1 proved to be more immunogenic than two doses of IAV-NS1. MVA and IAV are two different viral expression systems with their own unique biological properties. Thus, the level of NS1 gene expression from these viral vectors may differ. Furthermore, as mentioned above, NS1 was secreted from MVA-NS1-infected cells, whereas IAV-NS1 drove the expression of a membrane NA-NS1 fusion protein that was not secreted. To increase NS1 expression and immunogenicity by the IAV vector, the exchange of 5´- and 3´-end NA packaging signals with those of the HA gene might be considered (59). For both IAV-NS1 and MVA-NS1, the respective homologous booster vaccination significantly increased NS1-specific antibody levels induced after the first immunization. This indicates that vector-immunity induced by the prime immunization did not prevent boosting of the NS1-specific antibody response as has been shown previously (60–63). It is well known that prime/boost vaccination regimens with heterologous vectors or antigen delivery systems can improve immune responses significantly [(64–66), reviewed in (35)]. The use of IAV-NS1 and MVA-NS1 for heterologous prime/boost vaccination increased the NS1-specific antibody and T cell responses significantly compared to prime/boost with the same vectors. The order of immunization with the two vectors did not make a big difference, which is in contrast to a previous vaccination study with recombinant IAV and VACV expressing malaria antigens (67). The superior immunogenicity of heterologous prime/boost vaccination with IAV-NS1 or MVA-NS1 translated also in superior protection of mice against infection with TBEV Neudoerfl. In comparison to PBS and vector control groups, a delayed onset of body weight loss and significant prolonged survival was observed in mice that received two doses of the same vector. No difference in disease progression and survival between IAV-NS1 and MVA-NS1 immunized mice was observed, although MVA-NS1 was more immunogenic. In contrast, heterologous prime/boost vaccination resulted in partial survival of 33% and 50% of mice vaccinated with IAV-NS1/MVA-NS1 or MVA-NS1/IAV-NS1, respectively. In general, these results are in agreement with previous studies which demonstrated that with NS1-based vaccine preparations partial protection against TBEV infection can be achieved (21–27). Reduced viral loads in the periphery, CNS and intestine suggest that induction of immunity to NS1 favors restriction of virus replication and associated pathological changes in infected tissues.
Although the exact mode of protective immunity is unclear, adoptive transfer experiments with TBEV NS1-specific serum or B cells indicated that antibodies are involved as a correlate of protection (22, 25). Also in our study, the magnitude of the NS1-specific antibody response correlated with the duration of survival. Several studies identified antibody-dependent complement-mediated cytolysis (CMC) as a possible mechanism involved in protection [reviewed in (19)]. However, NS1-immunized complement-deficient mice were still protected upon TBEV challenge infection indicating that protective immunity is not exclusively mediated by CMC (21). As shown for other flaviviruses, NS1-specific antibodies can also contribute to complement-independent Fc-mediated effector functions like antibody-dependent cellular cytotoxicity and antibody-dependent cellular phagocytosis (68–73). Therefore, the mechanism of how TBEV NS1-specific antibodies exert their protective effect should be subject of further investigation. Since the homology of the NS1 amino acid sequence of European and Far-Eastern TBEV subtypes is between 92-98.9% (74) and cross-reactivity of NS1-specific antibodies was shown between these subtypes (20), we speculate that NS1-specific immunity offers a certain degree of cross-protection against other TBEV subtypes.
The use of NS1 as vaccine antigen may offer the advantage over the use of the E protein that NS1 vaccination will not result in the induction of virion-reactive antibodies. Consequently, the risk of ADE of infection is reduced (28). Although clear evidence for ADE of TBEV infection in vivo is lacking [reviewed in (15)], ADE was shown in vitro (29–32). Furthermore, flavivirus NS1, like that of Dengue virus (DENV), may contribute to pathogenesis by causing plasma leakage, thrombocytopenia and hemorrhages, all characteristic of severe dengue disease. Moreover, autoantibodies elicited by DENV NS1 can cross-react with host-antigens on e.g. endothelial cells or coagulation factors by molecular mimicry [reviewed in (75, 76)]. In our studies, we did not observe any obvious detrimental effects of TBEV NS1 expression or NS1-specific antibodies in mice. NS1-immunized animals developing TBE displayed no signs of increased TBE severity compared to infected control mice. NS1 of the respective flaviviruses may play distinct roles during infection and in pathogenesis which is underscored by the low sequence homology (37%) between TBEV and DENV NS1 (25).
Apart from NS1-specific antibodies, NS1-specific T cells can contribute to protective immunity against TBEV infection as was demonstrated in early adoptive transfer experiments (22). For Zika virus (ZIKV) it was shown that functional CD4+ and CD8+ T cell responses were required to control and clear infection in ZIKV NS1-vaccinated mice, despite the presence of high anti-NS1 antibody levels (77). In our study, mice that received a heterologous prime/boost vaccination had stronger NS1-specific IFN-γ+ T cell responses than those received two doses of the same viral vector. CD4+ and CD8+ T cells contributed to this response of which the CD8+ T cells also were Granzyme B positive. The induction of these improved T cell responses may have been the basis for the partial protection against TBEV challenge infection. In addition to Fc-mediated viral clearance of infected cells via NS1-specific antibodies, CD4+ and CD8+ cytotoxic T cells may have contributed to restricted viral replication in spleen, CNS and intestine in mice that received heterologous prime/boost vaccination. Although it has been demonstrated in TBEV mouse models that CD8+ T cells can have detrimental effects and contribute to the pathogenesis of infection (78), we did not observe such effects. Examination of representative H&E stained tissue sections of the CNS and intestine did not reveal more severe histopathological changes in mice that received the heterologous prime/boost vaccination than in those that received two doses of the same vector. However, to obtain a better understanding of the role of TBEV NS1-specific T cells in the pathogenesis of TBEV infection, further studies are required like adoptive T cell transfer experiments with selected T cell populations obtained from NS1-vaccinated mice.
In conclusion, we have shown that with the viral vectors IAV-NS1 and MVA-NS1 potent NS1-specific antibody and T cell responses can be induced in mice using heterologous prime/boost vaccination regimens. This NS1-specific immunity afforded partial protection against a lethal challenge infection with the Neudoerfl strain of TBEV. Therefore, the inclusion of a NS1 component in improved next generation TBEV vaccines seems desirable as has been demonstrated for other flaviviruses, like Japanese encephalitis virus and ZIKV (79, 80). Ideally, in such vaccines NS1 would be combined with the E or prM/E proteins, the major target for the induction of VN antibodies.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Lower Saxony State Office for Consumer Protection and Food Safety (LAVES, Approval No. 33.8-42502-04-20/3437).
Author contributions
Conceptualization: JBe, MK, TG, CKP, and GR. Formal analysis: JBe and MK. Investigation: JBe, MK, JBi, and CKP performed the animal experiments and analyzed the samples. IZ, CP, and WB performed and analyzed histology and immunohistochemistry data. Resources: GS. Writing-original draft preparation: JBe and MK. Writing-review and editing: IZ, CP, JBi, TG, WB, AO, GS, CKP, and GR. Visualization: JBe and MK. Supervision: TG, CKP, and GR. Funding acquisition: GR. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - 398066876/GRK 2485/1 and the Alexander von Humboldt Foundation in the framework of the Alexander von Humboldt Professorship endowed by the German Federal Ministry of Education and Research to GR. This Open Access publication was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - 491094227 “Open Access Publication Funding” and the University of Veterinary Medicine Hannover, Foundation. The funders played no role in study design, data collection, analysis and interpretation of data or the writing of this manuscript.
Acknowledgments
We thank L. Engels, S. Stelz, A. Molle and J. Friese for their excellent technical support. In addition, we thank S. Pfeifer for her excellent technical support in the animal facility. We thank Prof. Dr. S. Becker and Dr. I. Steffen for TBEV RNA standard and LIPS plasmids, respectively, and Prof. R. Fouchier for the Vibrio cholerae filtrate. Moreover, we thank Dr. G. Dobler for TBEV strain Neudoerfl, Prof. Dr. R. Webby and Prof. Dr. R. Webster for providing the reverse genetics pHW2000 plasmids and Prof. Dr. A. Volz and Dr. A. Tscherne for advice on the MVA vector system.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1177324/full#supplementary-material
References
1. Wondim MA, Czupryna P, Pancewicz S, Kruszewska E, Groth M, Moniuszko-Malinowska A. Epidemiological trends of trans-boundary tick-borne encephalitis in Europe, 2000–2019. Pathogens (2022) 11:1–10. doi: 10.3390/pathogens11060704
2. Dai X, Shang G, Lu S, Yang J, Xu J. A new subtype of eastern tick-borne encephalitis virus discovered in qinghai-Tibet plateau, China article. Emerg Microbes Infect (2018) 7:1–9. doi: 10.1038/s41426-018-0081-6
3. Kovalev SY, Mukhacheva TA. Reconsidering the classification of tick-borne encephalitis virus within the Siberian subtype gives new insights into its evolutionary history. Infect Genet Evol (2017) 55:159–65. doi: 10.1016/j.meegid.2017.09.014
4. Adelshin RV, Sidorova EA, Bondaryuk AN, Trukhina AG, Sherbakov DY, White RA, et al. “886-84-like” tick-borne encephalitis virus strains: intraspecific status elucidated by comparative genomics. Ticks Tick Borne Dis (2019) 10:1168–72. doi: 10.1016/j.ttbdis.2019.06.006
5. Lindquist L, Vapalahti O. Tick-borne encephalitis. Lancet (2008) 371:1861–71. doi: 10.1016/S0140-6736(08)60800-4
6. Ruzek D, Avšič Županc T, Borde J, Chrdle A, Eyer L, Karganova G, et al. Tick-borne encephalitis in Europe and Russia: review of pathogenesis, clinical features, therapy, and vaccines. Antiviral Res (2019) 164:23–51. doi: 10.1016/j.antiviral.2019.01.014
7. Růžek D, Dobler G, Mantke OD. Tick-borne encephalitis: pathogenesis and clinical implications. Travel Med Infect Dis (2010) 8:223–32. doi: 10.1016/j.tmaid.2010.06.004
8. Loew-Baselli A, Poellabauer E-M, Pavlova BG, Koska M, Bobrovsky R, Konior R, et al. Seropersistence of tick-borne encephalitis antibodies, safety and booster response to FSME-IMMUN® 0.5 ml in adults aged 18–67 years. Hum Vaccin (2009) 5:551–6. doi: 10.4161/hv.5.8.8571
9. Lotrič-Furlan S, Bogovič P, Avšič-Županc T, Jelovšek M, Lusa L, Strle F. Tick-borne encephalitis in patients vaccinated against this disease. J Intern Med (2017) 282:142–55. doi: 10.1111/joim.12625
10. Lotric-Furlan S, Avšič-Županc T, Strle F. Tick-borne encephalitis after active immunization. Int J Med Microbiol (2008) 298:309–13. doi: 10.1016/j.ijmm.2008.03.006
11. Sendi P, Hirzel C, Pfister S, Ackermann-Gäumann R, Grandgirard D, Hewer E, et al. Fatal outcome of European tick-borne encephalitis after vaccine failure. Front Neurol (2017) 8:119. doi: 10.3389/fneur.2017.00119
12. Lenhard T, Ott D, Jakob NJ, Martinez-Torres F, Grond-Ginsbach C, Meyding-Lamadé U. Clinical outcome and cerebrospinal fluid profiles in patients with tick-borne encephalitis and prior vaccination history. Ticks Tick Borne Dis (2018) 9:882–8. doi: 10.1016/j.ttbdis.2018.02.021
13. Andersson CR, Vene S, Insulander M, Lindquist L, Lundkvist Å, Günther G. Vaccine failures after active immunisation against tick-borne encephalitis. Vaccine (2010) 28:2827–31. doi: 10.1016/j.vaccine.2010.02.001
14. Dobler G, Kaier K, Hehn P, Böhmer MM, Kreusch TM, Borde JP. Tick-borne encephalitis virus vaccination breakthrough infections in Germany - a retrospective analysis from 2001-2018. Clin Microbiol Infect (2019) 26:1090.e7–1090.e13. doi: 10.1016/j.cmi.2019.12.001
15. Kubinski M, Beicht J, Gerlach T, Volz A, Sutter G, Rimmelzwaan GF. Tick-borne encephalitis virus: a quest for better vaccines against a virus on the rise. Vaccines (2020) 8:451. doi: 10.3390/VACCINES8030451
16. Salat J, Mikulasek K, Larralde O, Formanova PP, Chrdle A, Haviernik J, et al. Tick-borne encephalitis virus vaccines contain non-structural protein 1 antigen and may elicit NS1-specific antibody responses in vaccinated individuals. Vaccines (2020) 8:1–13. doi: 10.3390/vaccines8010081
17. Stiasny K, Leitner A, Holzmann H, Heinz FX. Dynamics and extent of non-structural protein 1-antibody responses in tick-borne encephalitis vaccination breakthroughs and unvaccinated patients. Viruses (2021) 13:1–9. doi: 10.3390/v13061007
18. Salat J, Strakova P, Ruzek D. Dynamics of whole virus and non-structural protein 1 (NS1) IgG response in mice immunized with two commercial tick-borne encephalitis vaccines. Vaccines (2022) 10:1–6. doi: 10.3390/vaccines10071001
19. Muller DA, Young PR. The flavivirus NS1 protein: molecular and structural biology, immunology, role inpathogenesis and application as a diagnostic biomarker. Antiviral Res (2013) 98:192–208. doi: 10.1016/j.antiviral.2013.03.008
20. Crooks AJ, Lee JM, Easterbrook LM, Timofeev AV, Stephenson JR. The NS1 protein of tick-borne encephalitis virus forms multimeric species upon secretion from the host cell. J Gen Virol (1994) 75:3453–60. doi: 10.1099/0022-1317-75-12-3453
21. Jacobs SC, Stephenson JR, Wilkinson GWG. Protection elicited by a replication-defective adenovirus vector expressing the tick-borne encephalitis virus non-structural glycoprotein NS1. J Gen Virol (1994) 75:2399–402. doi: 10.1099/0022-1317-75-9-2399
22. Timofeev AV, Ozherelkov SV, Pronin AV, Deeva AV, Karganova GG, Elbert LB, et al. Immunological basis for protection in a murine model of tick-borne encephalitis by a recombinant adenovirus carrying the gene encoding the NS1 non-structural protein. J Gen Virol (1998) 79:689–95. doi: 10.1099/0022-1317-79-4-689
23. Khoretonenko MV, Vorovitch MF, Zakharova LG, Pashvykina GV, Ovsyannikova NV, Stephenson JR, et al. Vaccinia virus recombinant expressing gene of tick-borne encephalitis virus non-structural NS1 protein elicits protective activity in mice. Immunol Lett (2003) 90:161–3. doi: 10.1016/j.imlet.2003.09.002
24. Aleshin SE, Timofeev AV, Khoretonenko MV, Zakharova LG, Pashvykina GV, Stephenson JR, et al. Combined prime-boost vaccination against tick-borne encephalitis (TBE) using a recombinant vaccinia virus and a bacterial plasmid both expressing TBE virus non-structural NS1 protein. BMC Microbiol (2005) 5:1–5. doi: 10.1186/1471-2180-5-45
25. Volpina OM, Volkova TD, Koroev DO, Ivanov VT, Ozherelkov SV, Khoretonenko MV, et al. A synthetic peptide based on the NS1 non-structural protein of tick-borne encephalitis virus induces a protective immune response against fatal encephalitis in an experimental animal model. Virus Res (2005) 112:95–9. doi: 10.1016/j.virusres.2005.03.026
26. Volkova TD, Koroev DO, Titova MA, Oboznaya MB, Filatova MP, Vorovich MF, et al. Synthetic fragments of the NS1 protein of the tick-borne encephalitis virus exhibiting a protective effect. Russ J Bioorganic Chem (2007) 33:213–7. doi: 10.1134/S1068162007020021
27. Jacobs SC, Stephenson JR, Wilkinson GW. High-level expression of the tick-borne encephalitis virus NS1 protein by using an adenovirus-based vector: protection elicited in a murine model. J Virol (1992) 66:2086–95. doi: 10.1128/jvi.66.4.2086-2095.1992
28. Gibson CA, Schlesinger JJ, Barrett ADT. Prospects for a virus non-structural protein as a subunit vaccine. Vaccine (1988) 6:7–9. doi: 10.1016/0264-410X(88)90004-7
29. Haslwanter D, Blaas D, Heinz FX, Stiasny K. A novel mechanism of antibody-mediated enhancement of flavivirus infection. PloS Pathog (2017) 13:1–27. doi: 10.1371/journal.ppat.1006643
30. Kreil TR, Eibl MM. Pre- and postexposure protection by passive immunoglobulin but no enhancement of infection with a flavivirus in a mouse model. J Virol (1997) 71:2921–7. doi: 10.1128/jvi.71.4.2921-2927.1997
31. Phillpotts RJ, Stephenson JR, Porterfield JS. Antibody-dependent enhancement of tick-borne encephalitis virus infectivity. J Gen Virol (1985) 66:1831–7. doi: 10.1099/0022-1317-66-8-1831
32. Kopecký J, Grubhoffer L, Tomková E. Interaction of tick-borne encephalitis virus with mouse peritoneal macrophages. the effect of antiviral antibody and lectin. Acta Virol (1991) 35:218–25.
33. Gerlach T, Elbahesh H, Saletti G, Rimmelzwaan GF. Recombinant influenza a viruses as vaccine vectors. Expert Rev Vaccines (2019) 18:379–92. doi: 10.1080/14760584.2019.1582338
34. Volz A, Sutter G. Modified vaccinia virus Ankara: history, value in basic research, and current perspectives for vaccine development. 1st ed. Elsevier Inc (2017) p. 187–243. doi: 10.1016/bs.aivir.2016.07.001
35. Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol (2009) 21:346–51. doi: 10.1016/j.coi.2009.05.016
36. Rimmelzwaan GF, Nieuwkoop NJ, de Mutsert G, Boon ACM, Kuiken T, Fouchier RAM, et al. Attachment of infectious influenza a viruses of various subtypes to live mammalian and avian cells as measured by flow cytometry. Virus Res (2007) 129:175–81. doi: 10.1016/j.virusres.2007.07.007
37. Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza a virus from eight plasmids. Proc Natl Acad Sci (2000) 97:6108–13. doi: 10.1073/pnas.100133697
38. Matrosovich M, Matrosovich T, Garten W, Klenk HD. New low-viscosity overlay medium for viral plaque assays. Virol J (2006) 3:1–7. doi: 10.1186/1743-422X-3-63
39. Kremer M, Volz A, Kreijtz JHCM, Fux R, Lehmann MH, Sutter G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. Methods Mol Biol (2012), 890:59–92. doi: 10.1007/978-1-61779-876-4
40. Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza a viruses. Arch Virol (2001) 146:2275–89. doi: 10.1007/s007050170002
41. Martina BEE, van den Doel P, Koraka P, van Amerongen G, Spohn G, Haagmans BL, et al. A recombinant influenza a virus expressing domain III of West Nile virus induces protective immune responses against influenza and West Nile virus. PloS One (2011) 6:e18995. doi: 10.1371/journal.pone.0018995
42. Volz A, Lim S, Kaserer M, Lülf A, Marr L, Jany S, et al. Immunogenicity and protective efficacy of recombinant modified vaccinia virus Ankara candidate vaccines delivering West Nile virus envelope antigens. Vaccine (2016) 34:1915–26. doi: 10.1016/j.vaccine.2016.02.042
43. Veit S, Jany S, Fux R, Sutter G, Volz A. CD8+ T cells responding to the middle east respiratory syndrome coronavirus nucleocapsid protein delivered by vaccinia virus MVA in mice. Viruses (2018) 10:1–18. doi: 10.3390/v10120718
44. van den Doel P, Volz A, Roose JM, Sewbalaksing VD, Pijlman GP, van Middelkoop I, et al. Recombinant modified vaccinia virus Ankara expressing glycoprotein E2 of chikungunya virus protects AG129 mice against lethal challenge. PloS Negl Trop Dis (2014) 8:1–12. doi: 10.1371/journal.pntd.0003101
45. Pérez P Q, Marín M, Lázaro-Frías A, Jiménez de Oya N, AB Blázquez, Escribano-Romero E, et al. A vaccine based on a modified vaccinia virus Ankara vector expressing zika virus structural proteins controls zika virus replication in mice. Sci Rep (2018) 8:1–15. doi: 10.1038/s41598-018-35724-6
46. Kreijtz JHCM, Suezer Y, de Mutsert G, van Amerongen G, Scwantes A, van den Brand JMA, et al. MVA-based H5N1 vaccine affords cross-clade protection in mice against influenz a A/H5N1 viruses at low doses and after single immunization. PloS One (2009) 4:1–8. doi: 10.1371/journal.pone.0007790
47. Könenkamp L, Ziegler U, Naucke T, Groschup MH, Steffen I. Antibody ratios against NS1 antigens of tick-borne encephalitis and West Nile viruses support differential flavivirus serology in dogs. Transbound Emerg Dis (2022) 69:e2789–99. doi: 10.1111/tbed.14630
48. Kaufmann L, Syedbasha M, Vogt D, Hollenstein Y, Hartmann J, Linnik JE, et al. An optimized hemagglutination inhibition (HI) assay to quantify influenzaspecific antibody titers. J Vis Exp (2017) 2017:1–10. doi: 10.3791/55833
49. Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Epidemiol (1938) 27:493–7. doi: 10.1093/oxfordjournals.aje.a118408
50. Schwaiger M, Cassinotti P. Development of a quantitative real-time RT-PCR assay with internal control for the laboratory detection of tick borne encephalitis virus (TBEV) RNA. J Clin Virol (2003) 27:136–45. doi: 10.1016/S1386-6532(02)00168-3
51. Kubinski M, Beicht J, Zdora I, Saletti G, Kircher M, Petry-Gusmag M, et al. Cross-reactive antibodies against langat virus protect mice from lethal tick-borne encephalitis virus infection. Front Immunol (2023) 14:1134371. doi: 10.3389/fimmu.2023.1134371
52. Leitzen E, Raddatz BB, Jin W, Goebbels S, Nave K-A, Baumgärtner W, et al. Virus-triggered spinal cord demyelination is followed by a peripheral neuropathy resembling features of Guillain-Barré syndrome. Sci Rep (2019) 9:4588. doi: 10.1038/s41598-019-40964-1
53. Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y. Selective incorporation of influenza virus RNA segments into virions. Proc Natl Acad Sci U.S.A. (2003) 100:2002–7. doi: 10.1073/pnas.0437772100
54. Boelke M, Puff C, Becker K, Hellhammer F, Gusmag F, Marks H, et al. Enteric ganglioneuritis, a common feature in a subcutaneous tbev murine infection model. Microorganisms (2021) 9:1–14. doi: 10.3390/microorganisms9040875
55. Carpio KL, Barrett ADT. Flavivirus NS1 and its potential in vaccine development. Vaccines (2021) 9:622. doi: 10.3390/vaccines9060622
56. Shinya K, Fujii Y, Ito H, Ito T, Kawaoka Y. Characterization of a neuraminidase-deficient influenza a virus as a potential gene delivery vector and a live vaccine. J Virol (2004) 78:3083–8. doi: 10.1128/jvi.78.6.3083-3088.2004
57. Kalthoff D, Röhrs S, Höper D, Hoffmann B, Bogs J, Stech J, et al. Truncation and sequence shuffling of segment 6 generate replication-competent neuraminidase-negative influenza H5N1 viruses. J Virol (2013) 87:13556–68. doi: 10.1128/jvi.02244-13
58. Hughes MT, Matrosovich M, Rodgers ME, McGregor M, Kawaoka Y. Influenza a viruses lacking sialidase activity can undergo multiple cycles of replication in cell culture, eggs, or mice. J Virol (2000) 74:5206–12. doi: 10.1128/jvi.74.11.5206-5212.2000
59. Zheng A, Sun W, Xiong X, Freyn AW, Peukes J, Strohmeier S, et al. Enhancing neuraminidase immunogenicity of influenza a viruses by rewiring RNA packaging signals. J Virol (2020) 94:1–12. doi: 10.1128/jvi.00742-20
60. Brault AC, Domi A, McDonald EM, Talmi-Frank D, McCurley N, Basu R, et al. A zika vaccine targeting NS1 protein protects immunocompetent adult mice in a lethal challenge model. Sci Rep (2017) 7:1–11. doi: 10.1038/s41598-017-15039-8
61. Koch T, Dahlke C, Fathi A, Kupke A, Krähling V, Okba NMA, et al. Safety and immunogenicity of a modified vaccinia virus Ankara vector vaccine candidate for middle East respiratory syndrome: an open-label, phase 1 trial. Lancet Infect Dis (2020) 20:827–38. doi: 10.1016/S1473-3099(20)30248-6
62. Kreijtz JHCM, Goeijenbier M, Moesker FM, van den Dries L, Goeijenbier S, De Gruyter HLM, et al. Safety and immunogenicity of a modified-vaccinia-virus-Ankara-based influenza a H5N1 vaccine: a randomised, double-blind phase 1/2a clinical trial. Lancet Infect Dis (2014) 14:1196–207. doi: 10.1016/S1473-3099(14)70963-6
63. de Vries RD, Altenburg AF, Nieuwkoop NJ, de Bruin E, van Trierum SE, Pronk MR, et al. Induction of cross-clade antibody and T-cell responses by a modified vaccinia virus Ankara–based influenza A(H5N1) vaccine in a randomized phase 1/2a clinical trial. J Infect Dis (2018) 218:614–23. doi: 10.1093/infdis/jiy214
64. Gherardi MM, Nájera JL, Pérez-Jiménez E, Guerra S, García-Sastre A, Esteban M. Prime-boost immunization schedules based on influenza virus and vaccinia virus vectors potentiate cellular immune responses against human immunodeficiency virus env protein systemically and in the genitorectal draining lymph nodes. J Virol (2003) 77:7048–57. doi: 10.1128/jvi.77.12.7048-7057.2003
65. Ba L, Yi CE, Zhang L, Ho DD, Chen Z. Heterologous MVA-s prime Ad5-s boost regimen induces high and persistent levels of neutralizing antibody response against SARS coronavirus. Appl Microbiol Biotechnol (2007) 76:1131–6. doi: 10.1007/s00253-007-1073-y
66. Ratto-Kim S, Currier JR, Cox JH, Excler JL, Valencia-Micolta A, Thelian D, et al. Heterologous prime-boost regimens using rAd35 and rMVA vectors elicit stronger cellular immune responses to HIV proteins than homologous regimens. PloS One (2012) 7:1–10. doi: 10.1371/journal.pone.0045840
67. Li S, Rodrigues M, Rodriguez D, Rodriguez JR, Esteban M, Palese P, et al. Priming with recombinant influenza virus followed by administration of recombinant vaccinia virus induces CD8+ T-cell-mediated protective immunity against malaria. Proc Natl Acad Sci U.S.A. (1993) 90:5214–8. doi: 10.1073/pnas.90.11.5214
68. García G, Arango M, Pérez AB, Fonte L, Sierra B, Rodríguez-Roche R, et al. Antibodies from patients with dengue viral infection mediate cellular cytotoxicity. J Clin Virol (2006) 37:53–7. doi: 10.1016/j.jcv.2006.04.010
69. Chung KM, Thompson BS, Fremont DH, Diamond MS. Antibody recognition of cell surface-associated NS1 triggers fc-γ receptor-mediated phagocytosis and clearance of West Nile virus-infected cells. J Virol (2007) 81:9551–5. doi: 10.1128/jvi.00879-07
70. Wan S-W, Chen P-W, Chen C-Y, Lai Y-C, Chu Y-T, Hung C-Y, et al. Therapeutic effects of monoclonal antibody against dengue virus NS1 in a STAT1 knockout mouse model of dengue infection. J Immunol (2017) 199:2834–44. doi: 10.4049/jimmunol.1601523
71. Bailey MJ, Duehr J, Dulin H, Broecker F, Brown JA, Arumemi FO, et al. Human antibodies targeting zika virus NS1 provide protection against disease in a mouse model. Nat Commun (2018) 9:1–11. doi: 10.1038/s41467-018-07008-0
72. Bailey MJ, Broecker F, Duehr J, Arumemi F, Krammer F, Palese P, et al. Antibodies elicited by an NS1-based vaccine protect mice against zika virus. MBio (2019) 10:1–16. doi: 10.1128/mBio.02861-18
73. Yu L, Liu X, Ye X, Su W, Zhang X, Deng W, et al. Monoclonal antibodies against zika virus ns1 protein confer protection via fccreceptor-dependent and-independent pathways. MBio (2021) 12:1–18. doi: 10.1128/mBio.03179-20
74. Jacobs SC, Stephenson JR, Wilkinson GWG. Sequence of the NS1 gene of the K 23 isolate of tick-borne encephalitis virus and identification of conserved motifs. Arch Virol (1993) 129:279–85. doi: 10.1007/BF01316902
75. Reyes-Sandoval A, Ludert JE. The dual role of the antibody response against the flavivirus non-structural protein 1 (NS1) in protection and immuno-pathogenesis. Front Immunol (2019) 10:1651. doi: 10.3389/fimmu.2019.01651
76. Wilken L, Rimmelzwaan GF. Adaptive immunity to dengue virus: slippery slope or solid ground for rational vaccine design? Pathogens (2020) 9:E470. doi: 10.3390/pathogens9060470
77. Grubor-Bauk B, Wijesundara DK, Masavuli M, Abbink P, Peterson RL, Prow NA, et al. NS1 DNA vaccination protects against zika infection through T cell – mediated immunity in immunocompetent mice. Sci Adv (2019) 5:1–15. doi: 10.1126/sciadv.aax2388
78. Růžek D, Salát J, Palus M, Gritsun TS, Gould EA, Dyková I, et al. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology (2009) 384:1–6. doi: 10.1016/j.virol.2008.11.023
79. Ishikawa T, Wang G, Widman DG, Infante E, Winkelmann ER, Bourne N, et al. Enhancing the utility of a prM/E-expressing chimeric vaccine for Japanese encephalitis by addition of the JEV NS1 gene. Vaccine (2011) 29:7444–55. doi: 10.1016/j.vaccine.2011.07.058
Keywords: TBEV, IAV, MVA, NS1, protection, vaccination, virus-neutralizing antibodies, T cells
Citation: Beicht J, Kubinski M, Zdora I, Puff C, Biermann J, Gerlach T, Baumgärtner W, Sutter G, Osterhaus ADME, Prajeeth CK and Rimmelzwaan GF (2023) Induction of humoral and cell-mediated immunity to the NS1 protein of TBEV with recombinant Influenza virus and MVA affords partial protection against lethal TBEV infection in mice. Front. Immunol. 14:1177324. doi: 10.3389/fimmu.2023.1177324
Received: 01 March 2023; Accepted: 20 June 2023;
Published: 07 July 2023.
Edited by:
Leyi Wang, University of Illinois at Urbana-Champaign, United StatesReviewed by:
Jianqiang Ye, Yangzhou University, ChinaElizaveta Starodubova, Engelhardt Institute of Molecular Biology (RAS), Russia
Copyright © 2023 Beicht, Kubinski, Zdora, Puff, Biermann, Gerlach, Baumgärtner, Sutter, Osterhaus, Prajeeth and Rimmelzwaan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guus F. Rimmelzwaan, Guus.Rimmelzwaan@tiho-hannover.de
†These authors have contributed equally to this work and share first authorship