 Lavanya Challagundla1
Lavanya Challagundla1 Jinnethe Reyes2
Jinnethe Reyes2 Iftekhar Rafiqullah3
Iftekhar Rafiqullah3 Daniel O. Sordelli4
Daniel O. Sordelli4 Gabriela Echaniz-Aviles5
Gabriela Echaniz-Aviles5 Maria E. Velazquez-Meza5
Maria E. Velazquez-Meza5 Santiago Castillo-Ramírez6Nahuel Fittipaldi7,8,9
Santiago Castillo-Ramírez6Nahuel Fittipaldi7,8,9 Michael Feldgarden10Sinéad B. Chapman11Michael S. Calderwood12
Michael Feldgarden10Sinéad B. Chapman11Michael S. Calderwood12 Lina P. Carvajal2
Lina P. Carvajal2 Sandra Rincon2
Sandra Rincon2 Blake Hanson13,14Paul J. Planet15Cesar A. Arias2,13,14
Blake Hanson13,14Paul J. Planet15Cesar A. Arias2,13,14 Lorena Diaz2
Lorena Diaz2 D. Ashley Robinson3*
D. Ashley Robinson3*- 1Department of Data Science, University of Mississippi Medical Center, Jackson, MS, United States
- 2Molecular Genetics and Antimicrobial Resistance Unit, International Center for Microbial Genomics, Universidad El Bosque, Bogota, Colombia
- 3Department of Microbiology and Immunology, University of Mississippi Medical Center, Jackson, MS, United States
- 4Instituto de Investigaciones en Microbiología y Parasitología Médica, Universidad de Buenos Aires and Consejo Nacional de Investigaciones Ciencias y Tecnicas, Buenos Aires, Argentina
- 5Department of Vaccine Evaluation, Instituto Nacional de Salud Pública, Cuernavaca, Mexico
- 6Programa de Genómica Evolutiva, Centro de Ciencias Génomicas, Universidad Nacional Autónoma de México, Cuernavaca, Mexico
- 7Public Health Ontario Laboratory, Toronto, ON, Canada
- 8Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
- 9Department of Cell and Systems Biology, University of Toronto, Toronto, ON, Canada
- 10National Center for Biotechnology Information, National Institutes of Health, Bethesda, MD, United States
- 11Broad Institute of MIT and Harvard, Cambridge, MA, United States
- 12Section of Infectious Disease and International Health, Dartmouth–Hitchcock Medical Center, Lebanon, NH, United States
- 13Division of Infectious Diseases and Center for Antimicrobial Resistance and Microbial Genomics, University of Texas Health Science Center, McGovern Medical School, Houston, TX, United States
- 14Center for Infectious Diseases, School of Public Health, University of Texas Health Science Center, Houston, TX, United States
- 15Children’s Hospital of Philadelphia, University of Pennsylvania, Philadelphia, PA, United States
Clonal complex 5 methicillin-resistant Staphylococcus aureus (CC5-MRSA) includes multiple prevalent clones that cause hospital-associated infections in the Western Hemisphere. Here, we present a phylogenomic study of these MRSA to reveal their phylogeny, spatial and temporal population structure, and the evolution of selected traits. We studied 598 genome sequences, including 409 newly generated sequences, from 11 countries in Central, North, and South America, and references from Asia and Europe. An early-branching CC5-Basal clade is well-dispersed geographically, is methicillin-susceptible and MRSA predominantly of ST5-IV such as the USA800 clone, and includes separate subclades for avian and porcine strains. In the early 1970s and early 1960s, respectively, two clades appeared that subsequently underwent major expansions in the Western Hemisphere: a CC5-I clade in South America and a CC5-II clade largely in Central and North America. The CC5-I clade includes the ST5-I Chilean/Cordobes clone, and the ST228-I South German clone as an early offshoot, but is distinct from other ST5-I clones from Europe that nest within CC5-Basal. The CC5-II clade includes divergent strains of the ST5-II USA100 clone, various other clones, and most known vancomycin-resistant strains of S. aureus, but is distinct from ST5-II strain N315 from Japan that nests within CC5-Basal. The recombination rate of CC5 was much lower than has been reported for other S. aureus genetic backgrounds, which indicates that recurrence of vancomycin resistance in CC5 is not likely due to an enhanced promiscuity. An increased number of antibiotic resistances and decreased number of toxins with distance from the CC5 tree root were observed. Of note, the expansions of the CC5-I and CC5-II clades in the Western Hemisphere were preceded by convergent gains of resistance to fluoroquinolone, macrolide, and lincosamide antibiotics, and convergent losses of the staphylococcal enterotoxin p (sep) gene from the immune evasion gene cluster of phage ϕSa3. Unique losses of surface proteins were also noted for these two clades. In summary, our study has determined the relationships of different clades and clones of CC5 and has revealed genomic changes for increased antibiotic resistance and decreased virulence associated with the expansions of these MRSA in the Western Hemisphere.
Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) is among the leading causes of antibiotic-resistant bacterial infections in hospital and community settings (Centers for Disease Control and Prevention, 2013; World Health Organization, 2014). These infections can range in severity from superficial skin infections to life-threatening invasive infections such as sepsis, infective endocarditis, and osteomyelitis (Boucher et al., 2010). Different MRSA clones predominate in different geographic regions and can differ in the rates and types of infections that they cause (Grundmann et al., 2010; David et al., 2014). Over the past 20 years, MRSA related to multilocus sequence type (ST) 5, which is classified into clonal complex (CC) 5, have been among the most prevalent clones causing hospital-associated infections in the Western Hemisphere (Aires De Sousa et al., 2001; Christianson et al., 2007; Rodriguez-Noriega et al., 2010; Diekema et al., 2014; Arias et al., 2017; Tickler et al., 2017). CC5-MRSA clones can differ in their Staphylococcal Chromosomal Cassette mec (SCCmec) genetic element, which carries the resistance determinant to anti-staphylococcal β-lactams, and in gene content (Christianson et al., 2007; Monecke et al., 2011). The identification of MRSA clones based on ST-SCCmec types (Robinson and Enright, 2004) has allowed for more precise transnational communication and tracking of these clones, and characterization of gene content has provided additional markers and insights into the lifestyles of these clones (Monecke et al., 2011).
Studies have identified the ST5-I Chilean/Cordobes clone (Sola et al., 2002), the ST5-II Canadian MRSA-2, New York/Japan, and USA100 clones (Roberts et al., 1998; McDougal et al., 2003; Christianson et al., 2007), and the ST5-IV USA800 clone (McDougal et al., 2003), as particularly common in the Western Hemisphere. For example, in the United States, ST5-II may have accounted for >40% of MRSA infections since the mid-1990s (Roberts et al., 1998; McDougal et al., 2003). In the southern cone of South America, ST5-I may have accounted for >60% of MRSA infections since the early 2000s (Sola et al., 2006). More recently in this region of South America, ST5-IV with pvl and sea toxin genes have emerged as a cause of community-associated infections and hospital infections especially among children (Sola et al., 2008; Egea et al., 2014). Other CC5-MRSA clones with SCCmec types I–VII have been reported elsewhere in the world (Monecke et al., 2011). In some cases, such as the ST225-II Rhine-Hesse clone in Central Europe (Nubel et al., 2010) and ST5-II in Western Australia (Coombs et al., 2007), the Western Hemisphere has been implicated as the origin of the clones.
Importantly, CC5 is the principal genetic background within S. aureus upon which full resistance to vancomycin—one of the drugs of choice for treating MRSA infections—repeatedly has arisen by acquisition of the vanA operon from Enterococcus spp. (Kos et al., 2012). The reasons why CC5 and not other S. aureus backgrounds has been the focal point for these acquisitions are not clear, but could include increased promiscuity, a genome that is conducive to vanA expression, or a shared niche with Enterococcus spp. In addition to the substantial disease caused by CC5-MRSA in human populations, the CC5 background has adapted to bird species and become a source of infection among broiler poultry (Lowder et al., 2009). The CC5 background also has been isolated from pigs (Frana et al., 2013), and with some traits that are present in other pig-adapted MRSA backgrounds (Hau et al., 2015).
The phylogenetic relationships between the numerous CC5-MRSA clones have not been determined, nor have the evolutionary events that led to their emergence. CC8 is the rival of CC5 for prominence among MRSA in the Western Hemisphere, and CC8-MRSA clone relationships and evolution have been studied extensively through genome sequencing (Uhlemann et al., 2014; Alam et al., 2015; Planet et al., 2015; Glaser et al., 2016; Challagundla et al., 2018). Relatively small samples of CC5-MRSA have been similarly studied (Kos et al., 2012; Aanensen et al., 2016; Arias et al., 2017). Here, we provide the first phylogenomic study of CC5-MRSA that is focused on large samples from the Western Hemisphere. Our goals are to resolve the phylogeny, place and time of origin of major CC5-MRSA clones, and to trace the evolution of their key traits.
Materials and Methods
Bacterial Samples and Genome Sequencing
The genome sequences of 598 strains of CC5 were studied here. Sequences were newly generated for 409 strains, and published sequences were downloaded from publicly available sources for 189 strains. The newly generated sequences were from three sequencing facilities: the Broad Institute (n = 297), the University of Mississippi Medical Center (UMMC, n = 74), and Universidad El Bosque (UEB, n = 38). Information on these sequences is provided in Supplementary Table S1. All new sequences were generated using Illumina instruments. The Broad Institute’s sequencing procedures are described in Supplementary Text S1, whereas previously published procedures were used at UMMC (Challagundla et al., 2018) and UEB (Arias et al., 2017). Eleven countries in the Western Hemisphere were sampled: Argentina, n = 30; Brazil, n = 23; Canada, n = 36; Chile, n = 23; Colombia, n = 56; Ecuador, n = 9; Guatemala, n = 21; Mexico, n = 40; Peru, n = 28; United States, n = 270; and Venezuela, n = 21. The US samples consisted of separate large collections: California, n = 40; Massachusetts, n = 40; Mississippi, n = 89; New York, n = 74; plus reference strains of USA100 and USA800, vancomycin-resistant S. aureus (VRSA), and avian and porcine strains. Other strains included completely sequenced reference strains JH1, N315, ED98, CF-Marseille, and 32 strains from Europe. Dates of isolation ranged from 1964 to 2017. Most strains were from clinical specimens.
Genome Assembly, Pseudoread Generation, and Alignment
Some of the published sequences were available only as assembled genomes. For those published sequences available as reads, and for the new sequences generated by UMMC, assemblies were made after filtering reads for minimum quality (base quality ≥ Q13, number of ambiguities ≤ 2, read length ≥ 15) using CLC Genomics Workbench v7 (Qiagen, Aarhus, Denmark). For those new sequences generated by the Broad Institute, assembly was carried out using the ALLPATHS-LG pipeline1, followed by automated assembly improvement using Pilon2. Quality measures of all assemblies are listed in Supplementary Table S1.
In order to align this heterogeneous sample of sequences, we first used wgsim v0.3.2 (Li et al., 2009) with each assembly to generate 500,000 paired-end pseudoreads of 100 bp, with a 200 bp distance between the ends. These pseudoreads were mapped to the complete reference sequence of CC5 strain JH1 (Mwangi et al., 2007) using bwa v0.7.12 (Li and Durbin, 2010), coordinate-sorted with Picard v1.141 (DePristo et al., 2011), and realigned around indels with GATK v2.8-1 (DePristo et al., 2011).
Variant Calling and Functional Effects
Single-nucleotide polymorphisms (SNPs) and short insertion–deletion polymorphisms (indels) were called with the UnifiedGenotyper walker of GATK. All self-similar sequences identified by pairwise megablast of the CC5 reference sequence against itself, plus five regions of mobile genetic elements, were excluded from variant calling. Mapping quality was >50 for all variants. The functional effects of the variants were determined using SnpEff v4 (Cingolani et al., 2012) with the annotation of the CC5 reference sequence.
Strain Typing and Prediction of Antibiotic Resistance
Multilocus sequence typing (MLST) was done by scanning the strains’ genome assemblies against the S. aureus MLST database3, using the mlst v2.10 program4 with default settings. Typing of the SCCmec element was done by mapping the strains’ pseudoreads to a custom-clustered database of ccr and mec gene complexes, plus SCCmec IV subtype-specific sequences (Kaya et al., 2018), using the SRST2 v0.2.0 program (Inouye et al., 2014) with the min_coverage 60 option. The validity of this approach was investigated using pseudoreads generated from reference sequences of SCCmec5 and by manually checking non-typeable elements with the SCCmecFinder web-based tool (Kaya et al., 2018). The detection of antibiotic resistance genes and mutations and the corresponding predicted antibiotic resistance was done with Mykrobe predictor v0.1.3, which has shown high (>99%) sensitivity and specificity for predicting resistance of S. aureus to a panel of 12 antibiotics (Bradley et al., 2015).
Detection of Selected Virulence Factors and Mobile Genetic Elements
To detect virulence factors, the full nucleotide dataset for S. aureus available at the Virulence Factors Database (Chen et al., 2016) was downloaded and then clustered into 265 groups with CD-HIT, using the tools available with SRST2 with default settings. The strains’ pseudoreads were mapped to this clustered database using SRST2 with the min_coverage 60 option. A similar approach was used to detect mobile genetic elements, specifically phages and integrative conjugative elements (ICEs) that integrate into the S. aureus chromosome. Phages were detected based on their integrase genes, using the 12 integrase groups described by Goerke et al. (2009) plus the integrase for ϕSPβ (Holden et al., 2010). ICEs were detected based on full-length sequences, using the seven subfamilies of ICE6013 and two subfamilies of Tn916 (Tn916 and Tn5801) described by Sansevere and Robinson (2017) and Sansevere et al. (2017). The validity of this approach was investigated using pseudoreads generated from complete sequences of reference strains of CC1, CC5, CC8, CC30, CC151, and reference sequences of the phages and ICEs.
Phylogenetic Inference and Recombination Detection
A maximum-likelihood (ML) phylogeny was constructed using PhyML v3.0 (Guindon et al., 2009) with the HKY+G model and five random and one BioNJ starting trees. For this analysis, an alignment representing biallelic SNPs and invariant core sequence present in all 598 genomes was used. Branch support was estimated with SH-like tests. The root of the tree was determined by first mapping reads from CC1, CC8, and ST72 outgroup sequences to the CC5 reference sequence, and then extracting the positions of the CC5 biallelic SNPs from the outgroups. These SNPs were then analyzed with distance-based (BioNJ) and maximum parsimony (MP) phylogenetic analysis. The outgroups consistently bisected a branch leading to a strain from Argentina, which was designated as the root. Recombination was detected using the PhyML tree with the method implemented in ClonalFrameML (CFML; Didelot and Wilson, 2015). Clade-specific recombination rates were examined on the recombination-corrected ML tree using R scripts.
Molecular Clock Analysis
The R package of Murray et al. (2016) was used to verify that no confounding existed between the path lengths on the recombination-corrected ML tree and the temporal distances of the genomes, and to verify a temporal phylogenetic signal in the positive relationship between the recombination-corrected ML tree root-to-tip distances and year of isolation of genomes. To estimate rates of evolution and dates of selected most recent common ancestors (MRCAs; tree nodes), the BEAST v1.7.5 program (Drummond et al., 2012) was used. We applied an HKY+G substitution model with empirical base frequencies, a Bayesian Skyline demographic model with default parameters, and both strict molecular clock and uncorrelated lognormal-relaxed molecular clock models using flat priors between 10−3 and 10−9 substitutions/site/year as informed by other S. aureus studies (Smyth et al., 2010). The basal strains from Argentina were constrained as outgroups, by forcing all other strains to be a monophyletic ingroup. Due to limitations in computational resources, the BEAST runs used three subsamples, each of 101 genomes. Each subsample consisted of 44 genomes chosen to represent early isolates and major nodes on the ML tree, plus 57 randomly selected genomes. Each subsample was run three times, and each run was 100,000,000 steps with sampling every 10,000 steps. Convergence and mixing of the MCMC chains, and the effective sample sizes of parameters (>2374 for the clock.rate parameter for every run of the selected strict clock model), was checked using Tracer v1.6. The first 10% of samples were removed as burn-in, the remaining samples were combined, and maximum clade credibility (MCC) trees were generated using LogCombiner v1.7.5 and TreeAnnotator v1.7.5. Since rate heterogeneity among branches was not extreme in any run of the relaxed clock model (i.e., the 95% credibility interval for the ucld.sd and coeeffvar parameters did not include 1.0), the strict clock model was selected.
Ancestral State Reconstruction
The evolution of selected traits was studied using ancestral state reconstruction under ML models. The ML analysis was done using the ace function of the ape R package (Paradis et al., 2004). Discrete, two-state characters were investigated including: geography (Central and North America vs. South America), predicted antibiotic resistance (resistant vs. susceptible), and the presence vs. absence of virulence factors, phages, ICEs, and predicted high-impact variants (i.e., frameshifts, start or stop codons gained or lost). Only those traits present in 5% (n = 30) to 95% (n = 568) of the genomes were investigated. The recombination-corrected ML tree was used as the estimate of the phylogeny. For each trait, both equal rates and all-rates-different models were fit to the data, and models were compared with a likelihood ratio test with one degree of freedom. In no case was the simpler, equal rates model rejected in favor of the more complicated, all-rates-different model.
Statistical Analysis and Tree Presentation
The basic stats package of R v3.4.2 (R Development Core Team, 2014) was used for two-sided tests of equal means (t-tests) and of no correlation between variables (Pearson’s coefficient). Tajima’s D, which examines the variance-normalized difference between two estimators of genetic diversity that are sensitive to different population dynamics, was calculated with the pegas (Paradis, 2010) R package. Tests of D = 0 were done assuming a beta distribution of the test statistic. Trees were visualized with iTOL (Letunic and Bork, 2016), and the ggtree (Yu et al., 2017) and phytools (Revell, 2012) R packages.
Results and Discussion
Outline of the CC5 Phylogeny
The alignment of 598 genomes of CC5 consisted of 1,081,440 bp present in all genomes, which included 11,961 biallelic SNPs. ML phylogenetic analysis with these sites, followed by correction of tree branch lengths for recombination and outgroup-rooting, resulted in the phylogeny presented in Figure 1. Several clades were defined by consideration of their ST-SCCmec types, geographic distribution, and statistical support. These clades are described below.
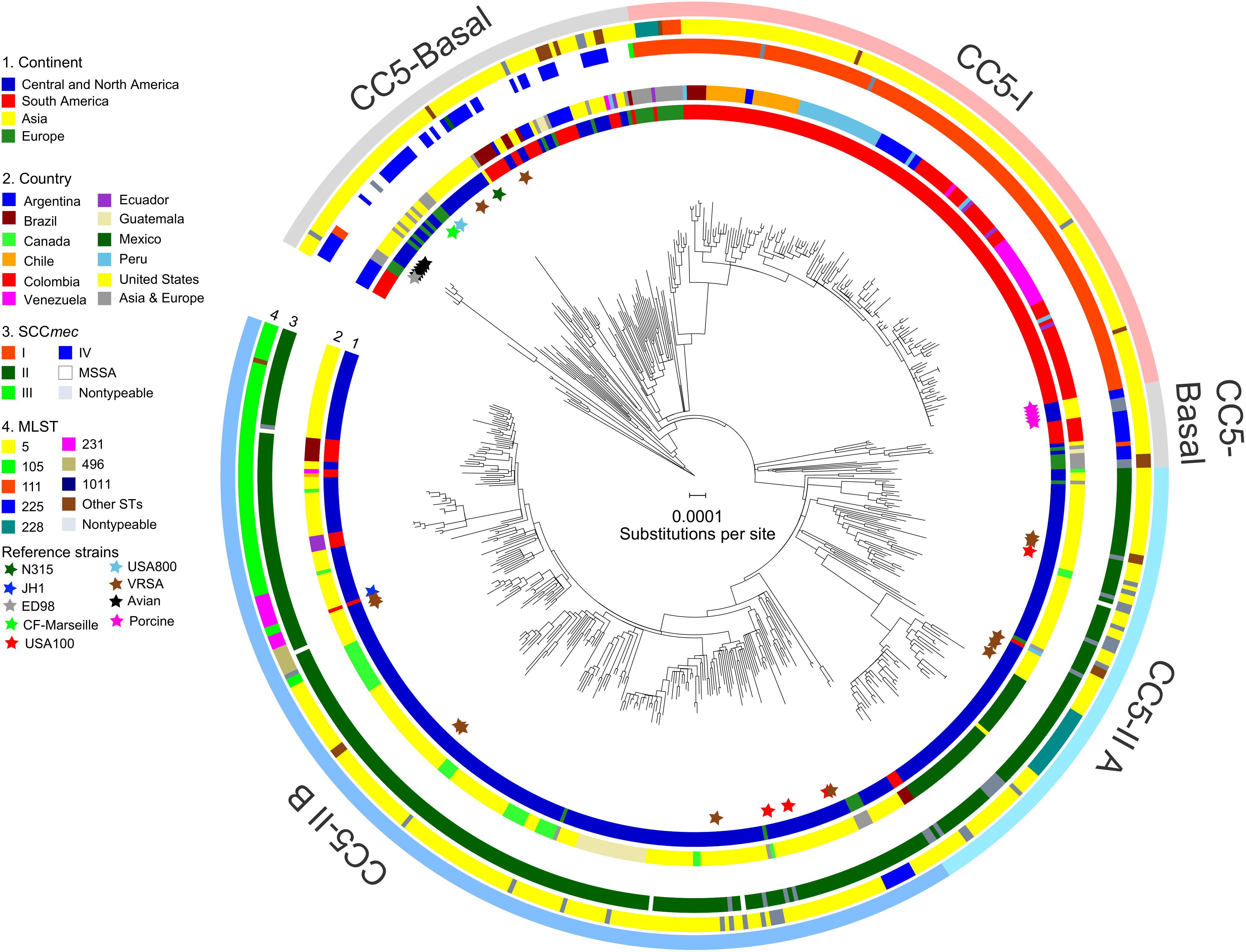
FIGURE 1. Phylogeny of CC5. The ML phylogeny has recombination-corrected branch lengths and is outgroup-rooted. The major clades of CC5-Basal, CC5-I, and CC5-II are indicated with shading. The statistical support for these clades is described in the text. The positions of reference strains are indicated with stars. The four rings in the direction of inner-outer, show continent (ring 1), country (ring 2), SCCmec type (ring 3), and multilocus ST (ring 4) for each genome. STs with less than three genomes were combined into “Other STs” for display purposes. Scale bar indicates number of substitutions per site.
CC5-Basal
CC5-Basal was defined as a large paraphyletic clade with a mixture of early-branching MSSA and MRSA predominantly of ST5-IV (SCCmec subtypes IVa, IVc, IVg, IVi) (Figure 1, light gray shading). This clade was well-dispersed across the Western and Eastern Hemispheres (Figure 1, ring 1). The most basal strain in our sample was an MSSA from Argentina, and the next most basal strains were a subclade of ST5-IVa (and one variant of ST5) from Argentina with the pvl and sea toxin genes that are characteristic of a recently emerged community-associated clone (Sola et al., 2008). Despite the exclusively South American source of these basal strains, ML ancestral state reconstruction did not reliably identify the continent of origin of CC5 (Supplementary Figure S1). The Bayesian phylogenetic analysis constrained these basal strains as an outgroup, resulting in maximal posterior probability (PP = 1) for the node, but the ML phylogenetic analysis was unconstrained and still strongly supported the node (SH-like support = 0.984). The Bayesian analysis estimated the MRCA of this sample of CC5 as the late 1930s (Table 1).
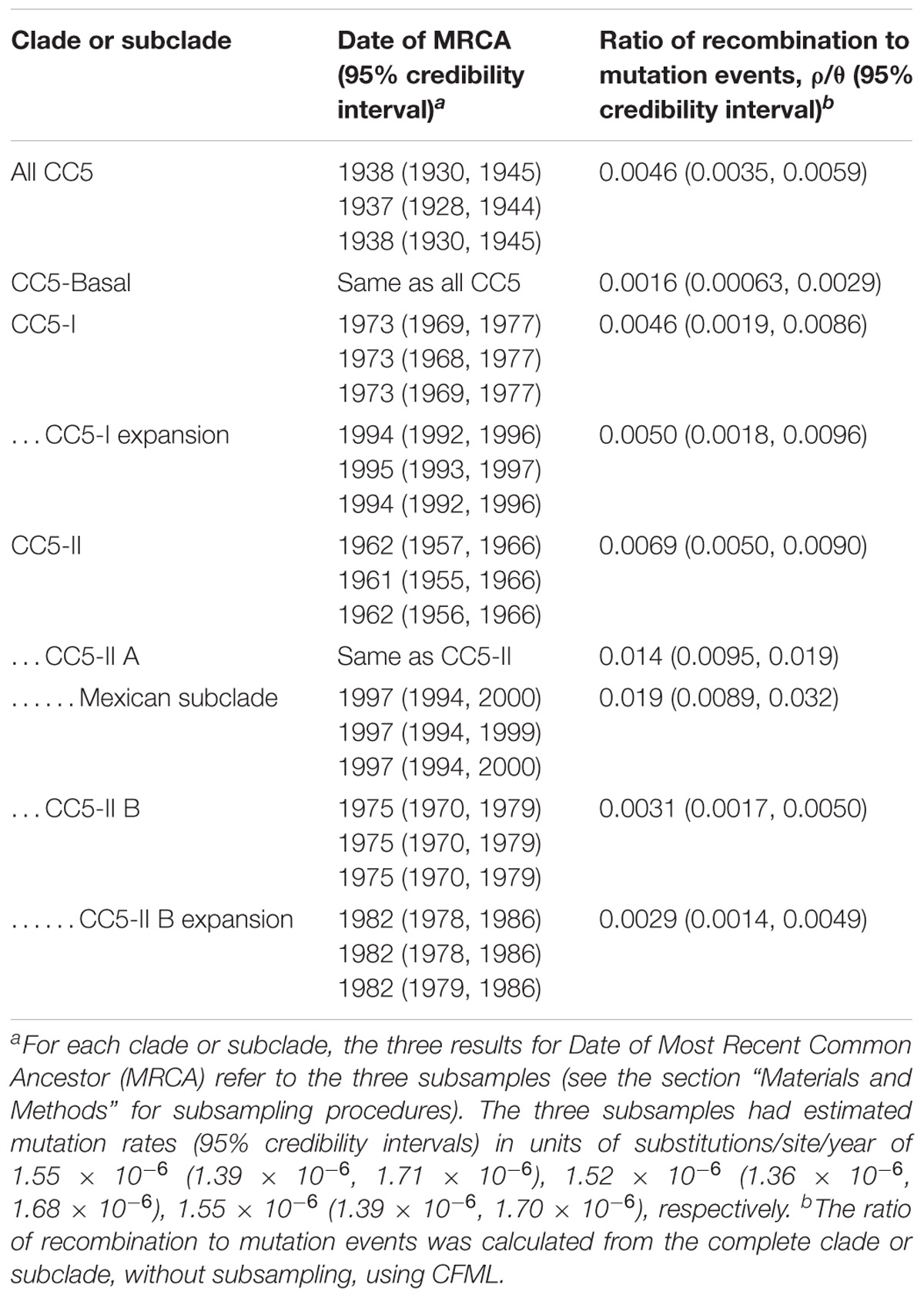
TABLE 1. Rates of evolution and dates of CC5 clades.
CC5-Basal included the ST5-IVc USA800 clone from the United States (McDougal et al., 2003), and the ST5-I Geraldine clone from France (Dauwalder et al., 2008). Also included are VRSA strains HOU1444-VR from Brazil and VRS3a from the United States, which represented independent acquisitions of vancomycin resistance and confirmed previous analysis (Panesso et al., 2015). CC5-Basal further included ST5-IVa reference strain CF-Marseille from France (Rolain et al., 2009), ST5-II reference strain N315 from Japan (Kuroda et al., 2001), and rare strains of ST5-I and ST5-III from Europe. The Bayesian and ML analyses provided maximal support (PP = 1, SH-like support = 1) for a previously described avian subclade, which included reference strain ED98 (Lowder et al., 2009), and for a separate subclade from porcine sources (Hau et al., 2017). The ML analysis presented a CC5-Basal subclade that included the porcine subclade (Figure 1, light gray shading), as a sister to the CC5-II clade described below. However, neither the Bayesian nor ML analyses provided statistical support for this arrangement, so this subclade should be considered to be part of the broader CC5-Basal clade.
CC5-I
CC5-I was defined as a large monophyletic clade with SCCmec I (Figure 1, light red shading). This clade represented one of three separate acquisitions of SCCmec I within CC5—the others occurred within the CC5-Basal clade. Most early-branching strains of CC5-I were from Europe, the rest were from South America. The ST228-I South German clone (Ghebremedhin et al., 2005) and related STs ST111 and ST1481 (Budimir et al., 2010) represented these early-branching European strains of CC5-I. The likelihood of a South American origin among CC5-I strains in the Western Hemisphere was >98% (Supplementary Figure S1). Bayesian and ML analyses provided maximal support for CC5-I, and its MRCA was estimated as the early 1970s (Table 1).
The terminal subclades of CC5-I represented an expansion of the ST5-I Chilean/Cordobes clone (Sola et al., 2002) throughout South America. The expansion node had maximal support in Bayesian and ML analyses, and its MRCA was estimated as the mid-1990s (Table 1). The initial branching events of the expansion were unresolved, which was consistent with a rapid expansion where the rate of geographic spread had outpaced the rate of mutation. Moreover, Tajima’s D for strains of the expansion was −2.75 (P = 3.46 × 10−5), which was consistent with a recent expansion because it indicated an excess of rare alleles that would be purged from an older equilibrium population. Subsequent subclades were strongly structured geographically by country within South America, and included strains from Brazil, Chile and nested Argentina strains, Peru, Argentina, Colombia, and nested Venezuela strains (Figure 1, ring 2).
CC5-II
CC5-II was defined as a very large monophyletic clade with SCCmec II (Figure 1, light blue shading). This clade represented one of two separate acquisitions of SCCmec II within CC5—the other occurred within the CC5-Basal clade on the branch leading to reference strain N315. CC5-II was subdivided into a paraphyletic early-branching subclade, CC5-II A, and a monophyletic terminal subclade, CC5-II B. In the Western Hemisphere, CC5-II strains were mostly from Central and North America, but some strains were from Brazil, Ecuador, and Venezuela. The likelihood of a Central or North American origin among CC5-II strains in the Western Hemisphere was >99% (Supplementary Figure S1). This clade had maximal support in Bayesian and ML analyses, and subclade CC5-II B had near maximal support (PP = 1, SH-like support = 0.997). The MRCAs of CC5-II and CC5-II B, respectively, were estimated as the early 1960s and mid-1970s (Table 1).
Of the four sampled reference strains of the ST5-II USA100 clone from the United States, one was within CC5-II A and three were within CC5-II B (Figure 1), which indicated that this clone was polyphyletic. Furthermore, the separation of ST5-II strains from New York in the United States and from Japan into distinct clades (CC5-II B and CC5-Basal, respectively), indicated that the New York/Japan clone was polyphyletic; in other words, there are separate New York and Japan clones, and not a single New York/Japan clone. Our study focused sampling on the Western Hemisphere, but it will be interesting to see if future studies place Asian SCCmec II-positive CC5 strains with strain N315 in CC5-Basal or with the diverse strains in CC5-II. Lastly, of the 11 sampled ST5-II VRSA strains from the United States, five were within CC5-II A and six were within CC5-II B (Figure 1, brown stars). Our analysis supported a previous analysis that indicated independent acquisitions of vancomycin resistance for most strains (Kos et al., 2012), but our analysis also highlighted the close relationships of strain pairs VRS1/VRS6, VRS5/VRS7, VRS9/VRS10, and VRS11a/VRS11b. Importantly, no evidence of further dissemination of these VRSA strains was obtained, as ML models of ancestral state reconstruction did not identify strains other than the known VRSAs to descend from vancomycin-resistant MRCAs (nodes). In the absence of vancomycin, the slight fitness burden reported for carriage of the vanA operon (Foucault et al., 2009) may be sufficient to impede the dissemination of these strains.
A major feature of CC5-II A was a divergent subclade that included all of the sampled strains from Mexico (Figure 1). Early-branching strains of this subclade were from the United States. This subclade had near maximal support (PP = 1, SH-like support = 0.999) in Bayesian and ML analyses, and its MRCA was estimated as the late 1990s (Table 1). This time period coincided with the replacement of CC30-MRSA in Mexico by CC5-MRSA, as previously described (Aires De Sousa et al., 2001; Velazquez-Meza et al., 2004; Echaniz-Aviles et al., 2006). This subclade was further subdivided into ST5-II and ST1011-II subclades that showed geographic structure by region within Mexico: ST5-II was from Central and Western Mexico, and ST1011-II was from Central and Northern Mexico.
The ST225-II Rhine-Hesse clone from Central Europe (Schulte et al., 2013) was an early offshoot of CC5-II B. The ST5-II reference strain JH1 from the United States (Mwangi et al., 2007) also nested within CC5-II B. The terminal subclades of CC5-II B represented an expansion of ST5-II, and subsequent proliferation of unnamed clones such as ST105-II, ST125-II, ST231-II, and ST496-II (Figure 1, ring 3), in Central and North America. The expansion node had maximal support in Bayesian and ML analyses, and dated to the early 1980s (Table 1). Several of the initial branching events of the expansion were unresolved, which was consistent with a rapid expansion. Also, Tajima’s D was −2.81 (P = 5.91 × 10−6), which was consistent with a recent expansion. CC5-II B showed some geographic structure at the country level, such as a subclade with 18 of 21 strains from Guatemala, two subclades with 10–11 strains each from Canada, and a subclade with 18 strains from California in the United States (Figure 1, ring 2).
Relatively Low Recombination Rates in CC5
Our estimate of the ratio of recombination to mutation events in this sample of CC5 (0.0046) (Table 1) was several orders of magnitude lower than previously estimated for CC5 (1.08) (Murray et al., 2017). However, the previous estimate detected a large proportion of recombination events in poultry strains and our sample included only six poultry strains for which no recombination events were detected. Sampling differences and the use of different recombination detection methods might explain the discrepancy between our recombination estimate and that of Murray et al. (2017). Our recombination estimate for CC5 was also lower than previously reported for the non-CC5 clones of ST239-III (Castillo-Ramirez et al., 2012) and ST8-IV USA300 (Challagundla et al., 2018); ranges of 0.05–0.29 and 0.12–0.29, respectively. The relatively low recombination rate estimated here using CFML was confirmed with an independent analysis using Gubbins (Croucher et al., 2015): 58 recombination events were detected by CFML and 29 were detected by Gubbins. In our sample, CC5-II A had a significantly higher ratio of recombination to mutation events compared to other CC5 clades (Table 1). This result was driven by a relatively large number of recombination events in the Mexican subclade. Thus, our analysis did not support the hypothesis that CC5 was generally more promiscuous than other S. aureus genetic backgrounds. Other explanations for why VRSA strains have repeatedly appeared in CC5 should be sought, such as whether CC5 has a unique gene regulatory environment that favors vanA expression or a unique ecological niche that favors interactions with Enterococcus spp.
Evidence for an Antibiotic Resistance-Toxicity Tradeoff in CC5
The full range of detected mutations and genes associated with antibiotic resistance, and selected virulence factors and mobile genetic elements, is provided in Supplementary Table S1. Our discussion is focused on those traits that are variably distributed among CC5 clades in the Western Hemisphere. Besides the high prevalence of resistance to β-lactams in all clades, CC5-I and CC5-II had a high prevalence of resistance to fluoroquinolones, macrolides, and lincosamides, and CC5-I also had a high prevalence of resistance to aminoglycosides (Table 2). The most common resistance mechanism to fluoroquinolones was the “double-serine mutations,” Ser84Leu in gyrA and Ser80Phe in grlA, that have been noted to occur in other successful MRSA clones (Fuzi et al., 2017). These double-serine mutations accounted for fluoroquinolone resistance in 141 of 147 (96%) resistant strains of CC5-I, and 107 of 239 (45%) resistant strains of CC5-II. Of note, CC5-II B had a lower prevalence of fluoroquinolone resistance compared to CC5-II A (58% vs. 98%, respectively) (Table 2), and the main mechanism of resistance in CC5-II B was the single mutation in gyrA that accounted for 117 of 141 (83%) resistant strains of CC5-II B. The most common resistance mechanism to macrolides and lincosamides was the ermA gene: alone accounting for 139 of 144 (97%) resistant strains of CC5-I, and 268 of 342 (78%) resistant strains of CC5-II. The ermA gene is known to be carried on Tn554 on the SCCmec II element (Kuroda et al., 2001), and it occurred on Tn554 elsewhere in the chromosome of the examined strains with SCCmec I elements. Aminoglycoside resistance, which occurred mostly in CC5-I, was exclusively attributed to the aacA–aphD bifunctional gene in our sample, and this gene occurred on composite transposon Tn4001 in examined strains. Mobile genetic elements also carried some rare antibiotic resistance genes. For example, ICE element (also known as conjugative transposon) Tn916 accounted for all eight strains with tetM-mediated tetracycline resistance. The other family of ICE elements known in S. aureus, ICE6013, was not observed to carry any antibiotic resistance genes and was common among the avian subclade of CC5-Basal (Table 2).
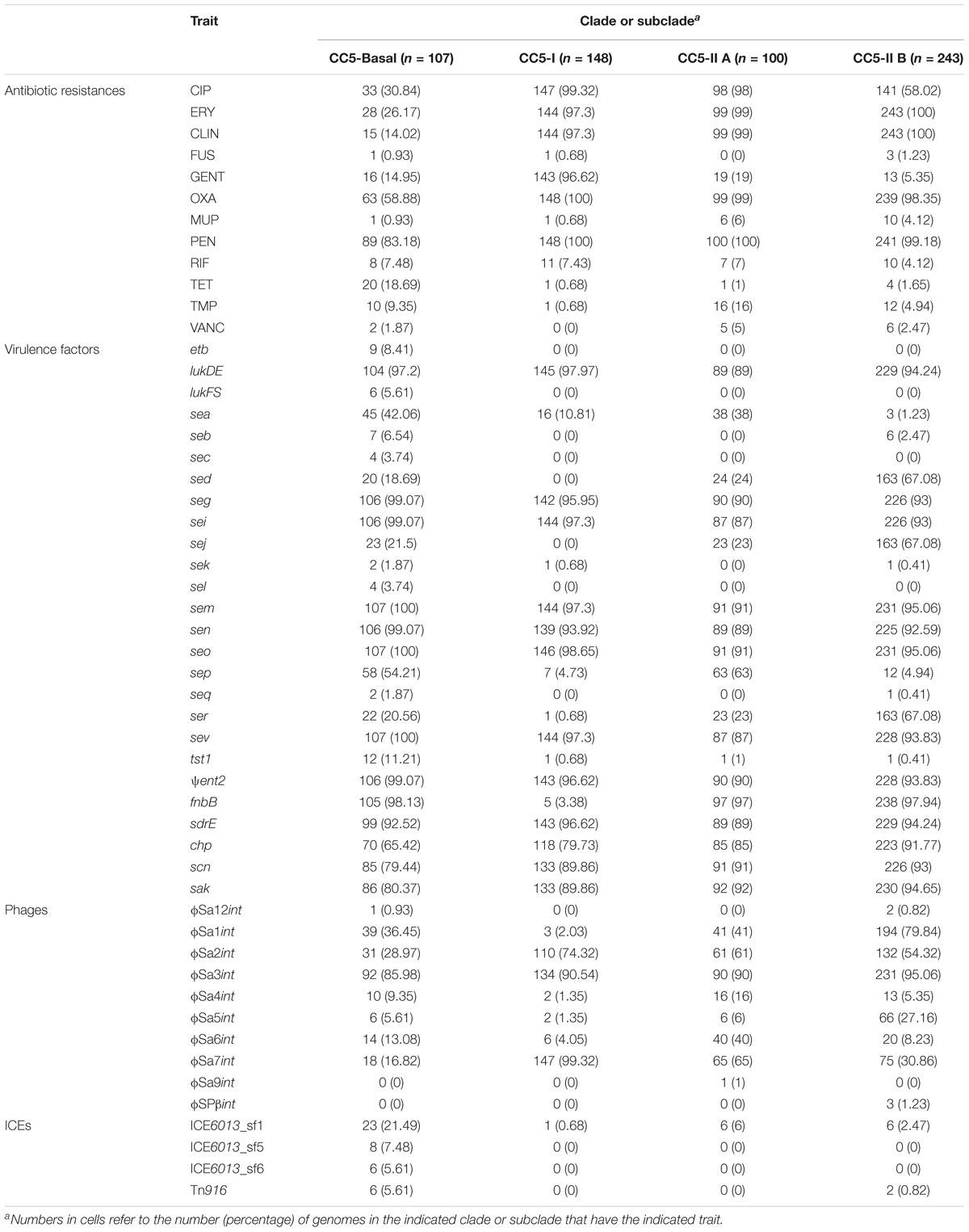
TABLE 2. Prevalence of selected traits in CC5 clades.
Common virulence factors of CC5 included the sak, scn, and chp genes of the immune evasion gene cluster (IEC) present on phage ϕSa3, and seg, sei, sem, sen, seo, and ψent of the enterotoxin gene cluster (EGC) as well as lukDE present on genomic island νSaβ (Table 2). CC5-Basal appeared to have a more diverse array of toxins, and the pvl (lukFS), sec, sel, and etb toxins occurred solely in this clade. CC5-I was unique in having a low prevalence of the fnbB adhesin gene; it was present in only 3% of CC5-I strains, and in >96% of strains of other clades (Table 2). On the other hand, the plasmid-borne sed, sej, ser toxins were most common in CC5-II B strains. Phages also showed some differences in CC5 clade distribution: phages ϕSa2 and ϕSa7 were most common in CC5-I strains, and phage ϕSa1 was most common in CC5-II B strains (Table 2).
We tested the hypothesis that antibiotic resistances and toxins were randomly distributed throughout CC5 and across time by comparing the average number of antibiotic resistances and toxins per genome in the various clades and by examining these traits with distance from the root of the CC5 tree. CC5-Basal had significantly fewer antibiotic resistances and more toxins than the other clades (Table 3). CC5-I had the opposite pattern, significantly more antibiotic resistances and fewer toxins than the other clades. CC5-II also had significantly more antibiotic resistances, but it had more toxins due to greater toxin acquisition in CC5-II B (e.g., sed, sej, ser) that offset the loss in CC5-II A (Table 3). Overall, the number of antibiotic resistances per genome increased with distance from root, and the number of toxins per genome decreased with distance from root (Table 3). A similar trend of increased number of antibiotic resistances with distance from root has been reported previously for CC8 (Strommenger et al., 2014). While these results highlight the importance of antibiotic resistance to the evolution of CC5, they are subject to a statistical caveat. Not all of the data points are independent because mobile genetic elements can carry multiple linked antibiotic resistance or toxin genes and affect multiple traits in a single evolutionary event; for example, ermA mediates both macrolide and lincosamide resistance, and the EGC encodes multiple enterotoxins.
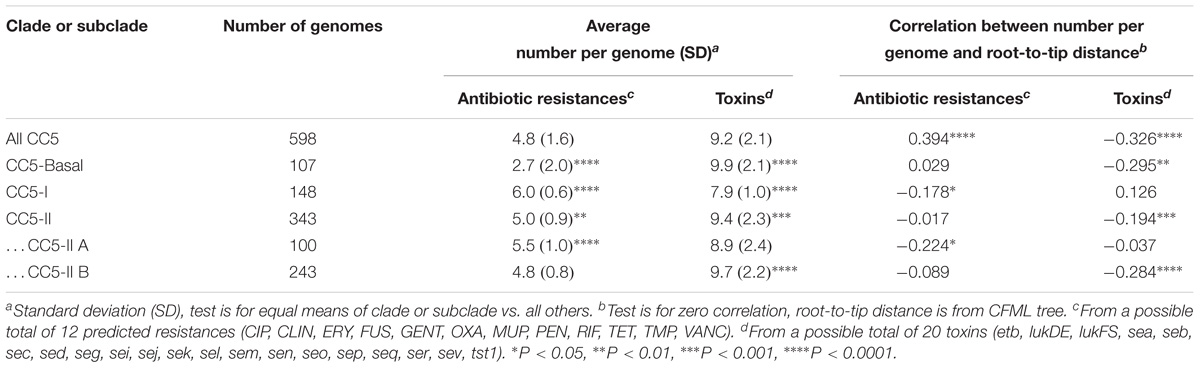
TABLE 3. Comparisons of antibiotic resistance and toxin traits in CC5 clades.
Convergent Genomic Changes Associated With Expansions of CC5
The evolution of selected traits was studied in more detail under an ML model of ancestral state reconstruction. A major finding of this analysis was that resistance to fluoroquinolones, macrolides, and lincosamides was gained, and the sep toxin gene was lost, independently, along the phylogenetic backbone leading to the expansion nodes of CC5-I and CC5-II B (Figure 2, pie-charts). The likelihood of the nodes that precede CC5-I and CC5-II B having these resistances was <5–6% for fluoroquinolones and <1% for macrolides and lincosamides, and >98% for the presence of sep. By the time of the expansion nodes of CC5-I and CC5-II B, the likelihood of having all three resistances was >99%, and <1% for the presence of sep (Figure 2, pie-charts).
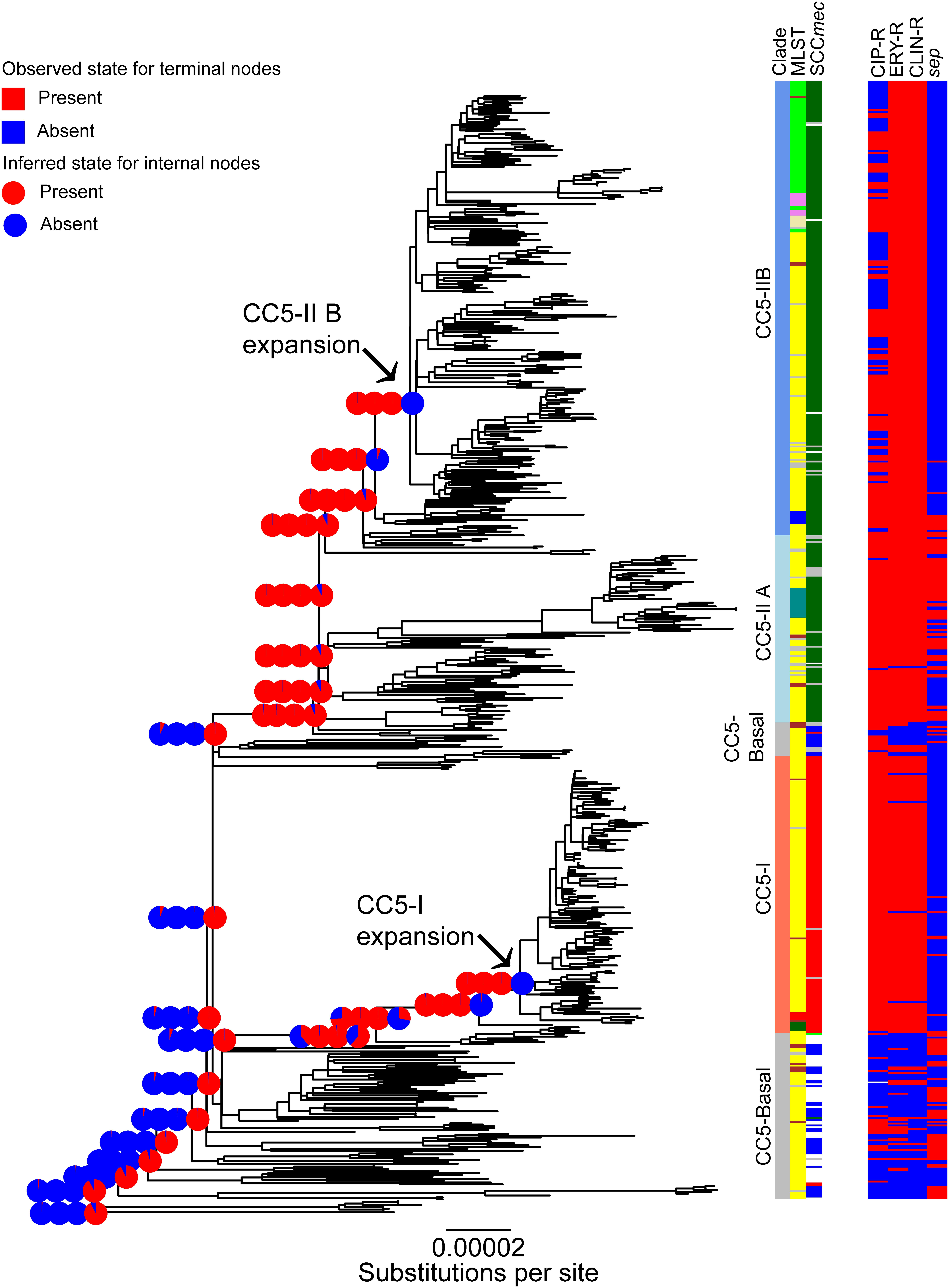
FIGURE 2. Convergent evolution within CC5. The ML phylogeny of Figure 1 is shown. The major clades, multilocus ST, and SCCmec type are shown for reference purposes and colored as in Figure 1. The CC5-I and CC5-II B expansion nodes are indicated with arrows. ML ancestral state reconstruction is shown along the phylogenetic backbone leading to the two expansion nodes; the four pie-charts at each of the backbone nodes indicate the likelihood of (in left-to-right order), fluoroquinolone, macrolide, and lincosamide resistance (red) or susceptibility (blue), and the presence (red) or absence (blue) of the sep toxin gene. Table adjacent to the tree indicates these traits in each genome. Scale bar indicates number of substitutions per site.
The independent evolution of the same traits from different starting points on the CC5 phylogeny (i.e., convergence), and the close timing of their evolution with independent expansions, suggests a causal relationship. Use of β-lactams, macrolides, and fluoroquinolones rank highly among antibiotics in the United States, Canada, and Brazil (Center for Disease Dynamics et al., 2015), and likely other countries in South America. Thus, the gain of these resistances may reflect a common selective pressure in the Western Hemisphere. As can be seen in Figure 2 (bar charts), some of the strains that appeared after the expansion of CC5-II B have subsequently lost fluoroquinolone resistance. As indicated above, many of the CC5-II B strains that retained fluoroquinolone resistance have retained the gyrA resistance allele but have lost the grlA resistance allele. Fluoroquinolone resistance mutations and especially the grlA mutation, have been shown to negatively impact the fitness of S. aureus strains in the absence of antibiotic (Horváth et al., 2012; Knight et al., 2012). However, these fitness effects may be mitigated in the presence of sub-inhibitory levels of the antibiotic (Gustave et al., 2018). These observations suggest that fluoroquinolone resistance may have an important role in the initial phase of epidemic spread CC5-MRSA clones, but less of a role in the subsequent phase of endemic residence in hospitals. Alternatively, there may have been efforts to reduce fluoroquinolone use in recent years, which in turn may have alleviated the selective pressure for clones to remain resistant. The CC5-II B expansion began approximately one decade earlier than the CC5-I expansion (Table 1), so it will be interesting to see if CC5-I begins to lose fluoroquinolone resistance and the grlA resistance allele over time as has happened in CC5-II B.
The association of the CC5-I and CC5-II B expansions with independent losses of the sep toxin gene was unexpected. This gene is present on the IEC of phage ϕSa3. Its loss preceding the CC5-I and CC5-II B expansions does not represent loss of the phage, since the majority of strains retained the phage and other virulence factors of the IEC such as sak, scn, and chp (Table 2). The completely sequenced reference strains N315 and JH1, respectively, provide full sequences of the IEC before and after loss of sep. A 1.75 kb region of the IEC that includes the sep gene was noted to be variably present in S. aureus by van Wamel et al. (2006). To our knowledge, the mechanism of sep excision from the IEC is unknown, and the function of sep is unstudied beyond its characterization as a superantigen with emetic activity (Omoe et al., 2005, 2013). In one study, the presence of sep was identified as the only S. aureus virulence factor among 30 tested, to associate with bacteremia in hospitalized MRSA carriers (Calderwood et al., 2014). While sep and other genome variations might influence the risk of bacteremia, those types of invasive infections are dead-ends for MRSA transmission.
The overall trend of decreased number of toxins per genome with distance from root, and parallel losses of sep in particular, suggests that the CC5 expansions occurred with strains that were less virulent than their precursors. Additional observations that support this notion come from an analysis of 35 high-frequency, high-impact mutations (listed in Supplementary Table S1). A total of 14 of these mutations occurred in CC5-I by the time of the expansion and three more occurred after the expansion. One of these was a frameshift mutation in the sasG adhesin gene, which also occurred in CC5-I strains that lacked the fnbB gene. Loss of sasG and fnbB function would be expected to result in reduced virulence especially in biofilm-associated infections (Vergara-Irigaray et al., 2009; Geoghegan et al., 2010). In CC5-II B, only two high-frequency, high-impact mutations occurred by the time of the expansion, but four occurred afterward. One of these mutations resulted in the loss of the stop codon of the srtA sortase gene, which is a virulence factor that attaches surface proteins with the LPXTG motif to S. aureus’ peptidoglycan cell wall. Loss of sortases function would be expected to result in reduced virulence (Mazmanian et al., 2000). One interesting gain-of-function change in CC5-II B was a frameshift mutation that restored the start codon of the toxin component of the axe1/txe1 toxin/antitoxin system. Expression of this system in CC8 strain Newman is increased after exposure to subinhibitory concentrations of erythromycin and tetracycline (Donegan and Cheung, 2009), but it has not been studied in CC5 to our knowledge.
Concluding Remarks
CC5-MRSA in the Western Hemisphere are highly diverse, even those strains that share the same ST-SCCmec type and circulate in the same country. More precise definitions for commonly sampled clones such as the USA100 and New York/Japan clones, and likely other clones such as USA800, are needed because different strains with those labels may have shared a MRCA > 50 years ago and may have divergent genomes. Our study provides the first systematic effort at organizing this diversity from a phylogenomic perspective and it provides a robust landmark for future genome studies of CC5-MRSA strains. The geographic structure of CC5 that is evident at the continent, country, and even region levels in some cases, suggests that a more precise delineation of the patterns of geographic spread of CC5-MRSA clones may be possible. MRSA clones of clinical relevance branch at nearly all time depths in the CC5 phylogeny; from the earliest-branching ST5-IV clones within the CC5-Basal clade that are an emerging problem in hospitals and communities in South America, to the latest-branching ST5-II clones within the CC5-II B clade that are a continuing problem in hospitals in Central and North America. Our analysis shows relatively low rates of recombination in CC5, which indicates that the propensity of CC5 to acquire vanA-mediated vancomycin resistance is not likely due to enhanced promiscuity and prompts study of other potential mechanisms. In tracing the evolution of selected traits of CC5, we discovered that some of the genomic changes that occurred prior to expansions of the CC5-I and CC5-II B clades represented instances of convergent evolution. While the convergent acquisition of resistance to widely prescribed antibiotics is a recurring theme in the history of successful MRSA clones, the convergent loss of the sep toxin and the trend of decreasing number of toxins with distance from the CC5 tree root and loss of other virulence factors is a unique finding. Taken together, our results suggest that more antibiotic-resistant and less virulent CC5-MRSA clones may be better able to spread geographically.
Data Availability
Sequence reads are available from NCBI Bioproject PRJNA224189, PRJNA454482, and PRJNA291213. Accession numbers for sequences are provided in Supplementary Table S1.
Data Deposition
NCBI Bioproject (http://www.ncbi.nlm.nih.gov/bioproject) PRJNA224189, PRJNA454482, and PRJNA291213.
Author Contributions
LC, MF, and DR conceived and designed the study. JR, DS, GE-A, MV-M, NF, CA, LD, and DR contributed bacterial strains or DNA. JR, MF, SC, LC, SR, BH, LD, and DR performed the genome sequencing. LC, JR, IR, MF, SC-R, MC, BH, PP, LD, and DR performed the analysis. LC and DR drafted the manuscript. All authors critically reviewed and approved the manuscript.
Funding
This work was supported in part by NIH grant R01-GM080602 to DR. The work of JR and LD, respectively, was supported by grants COL130871250417 and COL130874455850 from Colciencias. The work of DS was funded by grants ANPCyT PICT 2010-00941 and UBACyT 20020130100331BA. The work of MF was supported by the intramural research program of the National Library of Medicine, National Institutes of Health. CA was supported by NIH-NIAID award K24 AI121296. This project was funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No.: HHSN272200900018C. The work performed through the UMMC’s Molecular and Genomics Facility was supported in part by funds from the NIGMS, including Mississippi INBRE (P20GM103476), Center for Psychiatric Neuroscience (CPN)-COBRE (P30GM103328), Obesity, Cardiorenal and Metabolic Diseases-COBRE (P20GM104357), and Mississippi Center of Excellence in Perinatal Research (MS-CEPR)-COBRE (P20GM121334). The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
For provision of bacterial isolates, we gratefully acknowledge Rayo Morfin-Otero and Eduardo Rodriguez-Noriega from the Antiguo Hospital Civil “Fray Antonio Alcalde,” Guadalajara, Jalisco, Mexico; Elvira Garza-González from the Hospital Universitario “Dr. Jose Eleuterio Gonzalez,” Monterrey, Nuevo Leon, Mexico; Patricia Cornejo-Juárez and Patricia Volkow-Fernández, from the Instituto Nacional de Cancerologia, Mexico City, Mexico; The Canadian Nosocomial Infection Surveillance Program. For assistance with genome sequencing, we gratefully acknowledge Xiao Luo from UMMC; An Dinh from the University of Texas Health Science Center.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01901/full#supplementary-material
FIGURE S1 | Geographic structure within CC5.
TABLE S1 | Characteristics of CC5 genome sequences.
TEXT S1 | Genome sequencing procedures for the Broad Institute.
Footnotes
- ^ http://www.broadinstitute.org/software/allpaths-lg/blog
- ^ http://www.broadinstitute.org/software/pilon/
- ^ https://pubmlst.org/saureus/
- ^ https://github.com/tseemann/mlst
- ^ http://www.sccmec.org
References
Aanensen, D. M., Feil, E. J., Holden, M. T., Dordel, J., Yeats, C. A., Fedosejev, A., et al. (2016). Whole-genome sequencing for routine pathogen surveillance in public health: a population snapshot of invasive Staphylococcus aureus in Europe. mBio 7:e444-16. doi: 10.1128/mBio.00444-16
Aires De Sousa, M., Miragaia, M., Sanches, I. S., Avila, S., Adamson, I., Casagrande, S. T., et al. (2001). Three-year assessment of methicillin-resistant Staphylococcus aureus clones in Latin America from 1996 to 1998. J. Clin. Microbiol. 39, 2197–2205. doi: 10.1128/JCM.39.6.2197-2205.2001
Alam, M. T., Read, T. D., Petit, R. A., Boyle-Vavra, S., Miller, L. G., Eells, S. J., et al. (2015). Transmission and microevolution of USA300 MRSA in U.S. Households: evidence from whole-genome sequencing. mBio 6:e00054-15. doi: 10.1128/mbio.00054-15
Arias, C. A., Reyes, J., Carvajal, L. P., Rincon, S., Diaz, L., Panesso, D., et al. (2017). A prospective cohort multicenter study of molecular epidemiology and phylogenomics of Staphylococcus aureus bacteremia in nine Latin American Countries. Antimicrob. Agents Chemother. 61, e816–e817. doi: 10.1128/AAC.00816-17
Boucher, H., Miller, L. G., and Razonable, R. R. (2010). Serious infections caused by methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 51(Suppl. 2), S183–S197. doi: 10.1086/653519
Bradley, P., Gordon, N. C., Walker, T. M., Dunn, L., Heys, S., Huang, B., et al. (2015). Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 6:10063. doi: 10.1038/ncomms10063
Budimir, A., Deurenberg, R. H., Bosnjak, Z., Stobberingh, E. E., Cetkovic, H., and Kalenic, S. (2010). A variant of the Southern German clone of methicillin-resistant Staphylococcus aureus is predominant in Croatia. Clin. Microbiol. Infect. 16, 1077–1083. doi: 10.1111/j.1469-0691.2009.03042.x
Calderwood, M. S., Desjardins, C. A., Sakoulas, G., Nicol, R., Dubois, A., Delaney, M. L., et al. (2014). Staphylococcal enterotoxin P predicts bacteremia in hospitalized patients colonized with methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 209, 571–577. doi: 10.1093/infdis/jit501
Castillo-Ramirez, S., Corander, J., Marttinen, P., Aldeljawi, M., Hanage, W. P., Westh, H., et al. (2012). Phylogeographic variation in recombination rates within a global clone of methicillin-resistant Staphylococcus aureus. Genome Biol. 13:R126. doi: 10.1186/gb-2012-13-12-r126
Center for Disease Dynamics, Economics and Policy (2015). Antibiotic use in 2015. Available at: https://resistancemap.cddep.org/AntibioticUse.php [accessed May 1, 2018].
Centers for Disease Control and Prevention (2013). Antibiotic Resistance Threats in the United States, 2013. Available at: https://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf [accessed March 2, 2018].
Challagundla, L., Luo, X., Tickler, I. A., Didelot, X., Coleman, D. C., Shore, A. C., et al. (2018). Range expansion and the origin of USA300 North American epidemic methicillin-resistant Staphylococcus aureus. mBio 9:e02016-17. doi: 10.1128/mBio.02016-17
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Christianson, S., Golding, G. R., Campbell, J., Canadian Nosocomial Infection Surveillance Program, and Mulvey, M. R. (2007). Comparative genomics of Canadian epidemic lineages of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 45, 1904–1911. doi: 10.1128/JCM.02500-06
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92. doi: 10.4161/fly.19695
Coombs, G. W., Van Gessel, H., Pearson, J. C., Godsell, M. R., O’Brien, F. G., and Christiansen, K. J. (2007). Controlling a multicenter outbreak involving the New York/Japan methicillin-resistant Staphylococcus aureus clone. Infect. Control Hosp. Epidemiol. 28, 845–852. doi: 10.1086/518726
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Dauwalder, O., Lina, G., Durand, G., Bes, M., Meugnier, H., Jarlier, V., et al. (2008). Epidemiology of invasive methicillin-resistant Staphylococcus aureus clones collected in France in 2006 and 2007. J. Clin. Microbiol. 46, 3454–3458. doi: 10.1128/JCM.01050-08
David, M. Z., Daum, R. S., Bayer, A. S., Chambers, H. F., Fowler, V. G. Jr., Miller, L. G., et al. (2014). Staphylococcus aureus bacteremia at 5 US academic medical centers, 2008-2011: significant geographic variation in community-onset infections. Clin. Infect. Dis. 59, 798–807. doi: 10.1093/cid/ciu410
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comp. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Diekema, D. J., Richter, S. S., Heilmann, K. P., Dohrn, C. L., Riahi, F., Tendolkar, S., et al. (2014). Continued emergence of USA300 methicillin-resistant Staphylococcus aureus in the United States: results from a nationwide surveillance study. Infect. Control Hosp. Epidemiol. 35, 285–292. doi: 10.1086/675283
Donegan, N. P., and Cheung, A. L. (2009). Regulation of the mazEF toxin-antitoxin module in Staphylococcus aureus and its impact on sigB expression. J. Bacteriol. 191, 2795–2805. doi: 10.1128/JB.01713-08
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Echaniz-Aviles, G., Velazquez-Meza, M. E., Aires-de-Sousa, M., Morfin-Otero, R., Rodriguez-Noriega, E., Carnalla-Barajas, N., et al. (2006). Molecular characterisation of a dominant methicillin-resistant Staphylococcus aureus (MRSA) clone in a Mexican hospital (1999-2003). Clin. Microbiol. Infect. 12, 22–28. doi: 10.1111/j.1469-0691.2005.01283.x
Egea, A. L., Gagetti, P., Lamberghini, R., Faccone, D., Lucero, C., Vindel, A., et al. (2014). New patterns of methicillin-resistant Staphylococcus aureus (MRSA) clones, community-associated MRSA genotypes behave like healthcare-associated MRSA genotypes within hospitals, Argentina. Int. J. Med. Microbiol. 304, 1086–1099. doi: 10.1016/j.ijmm.2014.08.002
Foucault, M. L., Courvalin, P., and Grillot-Courvalin, C. (2009). Fitness cost of VanA-type vancomycin resistance in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53, 2354–2359. doi: 10.1128/AAC.01702-08
Frana, T. S., Beahm, A. R., Hanson, B. M., Kinyon, J. M., Layman, L. L., Karriker, L. A., et al. (2013). Isolation and characterization of methicillin-resistant Staphylococcus aureus from pork farms and visiting veterinary students. PLoS One 8:e53738. doi: 10.1371/journal.pone.0053738
Fuzi, M., Szabo, D., and Csercsik, R. (2017). Double-serine fluoroquinolone resistance mutations advance major international clones and lineages of various multi-drug resistant bacteria. Front. Microbiol. 8:2261. doi: 10.3389/fmicb.2017.02261
Geoghegan, J. A., Corrigan, R. M., Gruszka, D. T., Speziale, P., O’Gara, J. P., Potts, J. R., et al. (2010). Role of surface protein SasG in biofilm formation by Staphylococcus aureus. J. Bacteriol. 192, 5663–5673. doi: 10.1128/JB.00628-10
Ghebremedhin, B., Konig, W., and Konig, B. (2005). Heterogeneity of methicillin-resistant Staphylococcus aureus strains at a German university hospital during a 1-year period. Eur. J. Clin. Microbiol. Infect. Dis. 24, 388–398. doi: 10.1007/s10096-005-1339-1
Glaser, P., Martins-Simões, P., Villain, A., Barbier, M., Tristan, A., Bouchier, C., et al. (2016). Demography and intercontinental spread of the USA300 community-acquired methicillin-resistant Staphylococcus aureus lineage. mBio 7:e02183-15. doi: 10.1128/mbio.02183-15
Goerke, C., Pantucek, R., Holtfreter, S., Schulte, B., Zink, M., Grumann, D., et al. (2009). Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 191, 3462–3468. doi: 10.1128/JB.01804-08
Grundmann, H., Aanensen, D. M., van den Wijngaard, C. C., Spratt, B. G., Harmsen, D., Friedrich, A. W., et al. (2010). Geographic distribution of Staphylococcus aureus causing invasive infections in Europe: a molecular-epidemiological analysis. PLoS Med. 7:e1000215. doi: 10.1371/journal.pmed.1000215
Guindon, S., Delsuc, F., Dufayard, J.-F., and Gascuel, O. (2009). “Estimating maximum likelihood phylogenies with PhyML,” in Bioinformatics for DNA Sequence Analysis Methods in Molecular Biology, ed. D. Posada (Berlin: Springer Nature).
Gustave, C. A., Tristan, A., Martins-Simões, P., Stegger, M., Benito, Y., Andersen, P. S., et al. (2018). Demographic fluctuation of community-acquired antibiotic-resistant Staphylococcus aureus lineages: potential role of flimsy antibiotic exposure. ISME J. doi: 10.1038/s41396-018-0110-4 [Epub ahead of print].
Hau, S. J., Bayles, D. O., Alt, D. P., Frana, T. S., and Nicholson, T. L. (2017). Draft genome sequences of 63 swine-associated methicillin-resistant Staphylococcus aureus sequence type 5 isolates from the United States. Genome Announc. 5:e01081-17. doi: 10.1128/genomeA.01081-17
Hau, S. J., Sun, J., Davies, P. R., Frana, T. S., and Nicholson, T. L. (2015). Comparative prevalence of immune evasion complex genes associated with beta-hemolysin converting bacteriophages in MRSA ST5 isolates from swine, swine facilities, humans with swine contact, and humans with no swine contact. PLoS One 10:e0142832. doi: 10.1371/journal.pone.0142832
Holden, M. T., Lindsay, J. A., Corton, C., Quail, M. A., Cockfield, J. D., Pathak, S., et al. (2010). Genome sequence of a recently emerged, highly transmissible, multi-antibiotic- and antiseptic-resistant variant of methicillin-resistant Staphylococcus aureus, sequence type 239 (TW). J. Bacteriol. 192, 888–892. doi: 10.1128/JB.01255-09
Horváth, A., Dobay, O., Kardos, S., Ghidán,Á., Tóth,Á., Pászti, J., et al. (2012). Varying fitness cost associated with resistance to fluoroquinolones governs clonal dynamic of methicillin-resistant Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 31, 2029–2036. doi: 10.1007/s10096-011-1536-z
Inouye, M., Dashnow, H., Raven, L. A., Schultz, M. B., Pope, B. J., Tomita, T., et al. (2014). SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6:90. doi: 10.1186/s13073-014-0090-6
Kaya, H., Hasman, H., Larsen, J., Stegger, M., Johannesen, T. B., Allesoe, R. L., et al. (2018). SCCmecFinder, a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. mSphere 3, e00612-17. doi: 10.1128/mSphere.00612-17
Knight, G. M., Budd, E. L., Whitney, L., Thornley, A., Al-Ghusein, H., Planche, T., et al. (2012). Shift in dominant hospital-associated methicillin-resistant Staphylococcus aureus (HA-MRSA) clones over time. J. Antimicrob. Chemother. 67, 2514–2522. doi: 10.1093/jac/dks245
Kos, V. N., Desjardins, C. A., Griggs, A., Cerqueira, G., Van Tonder, A., Holden, M. T., et al. (2012). Comparative genomics of vancomycin-resistant Staphylococcus aureus strains and their positions within the clade most commonly associated with Methicillin-resistant S. aureus hospital-acquired infection in the United States. mBio 3:e00112-12. doi: 10.1128/mBio.00112-12
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., et al. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240. doi: 10.1016/S0140-6736(00)04403-2
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lowder, B. V., Guinane, C. M., Ben Zakour, N. L., Weinert, L. A., Conway-Morris, A., Cartwright, R. A., et al. (2009). Recent human-to-poultry host jump, adaptation, and pandemic spread of Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 106, 19545–19550. doi: 10.1073/pnas.0909285106
Mazmanian, S. K., Liu, G., Jensen, E. R., Lenoy, E., and Schneewind, O. (2000). Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. U.S.A. 97, 5510–5515. doi: 10.1073/pnas.080520697
McDougal, L. K., Steward, C. D., Killgore, G. E., Chaitram, J. M., McAllister, S. K., and Tenover, F. C. (2003). Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J. Clin. Microbiol. 41, 5113–5120. doi: 10.1128/jcm.41.11.5113-5120.2003
Monecke, S., Coombs, G., Shore, A. C., Coleman, D. C., Akpaka, P., Borg, M., et al. (2011). A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One 6:e17936. doi: 10.1371/journal.pone.0017936
Murray, G. G., Wang, F., Harrison, E. M., Paterson, G. K., Mather, A. E., Harris, S. R., et al. (2016). The effect of genetic structure on molecular dating and tests for temporal signal. Methods Ecol. Evol. 7, 80–89. doi: 10.1111/2041-210X.12466
Murray, S., Pascoe, B., Meric, G., Mageiros, L., Yahara, K., Hitchings, M. D., et al. (2017). Recombination-mediated host adaptation by avian Staphylococcus aureus. Genome Biol. Evol. 9, 830–842. doi: 10.1093/gbe/evx037
Mwangi, M. M., Wu, S. W., Zhou, Y., Sieradzki, K., de Lencastre, H., Richardson, P., et al. (2007). Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 104, 9451–9456. doi: 10.1073/pnas.0609839104
Nubel, U., Dordel, J., Kurt, K., Strommenger, B., Westh, H., Shukla, S. K., et al. (2010). A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog. 6:e1000855. doi: 10.1371/journal.ppat.1000855
Omoe, K., Hu, D. L., Ono, H. K., Shimizu, S., Takahashi-Omoe, H., Nakane, A., et al. (2013). Emetic potentials of newly identified staphylococcal enterotoxin-like toxins. Infect. Immun. 81, 3627–3631. doi: 10.1128/IAI.00550-13
Omoe, K., Imanishi, K., Hu, D. L., Kato, H., Fugane, Y., Abe, Y., et al. (2005). Characterization of novel staphylococcal enterotoxin-like toxin type P. Infect. Immun. 73, 5540–5546. doi: 10.1128/IAI.00550-13
Panesso, D., Planet, P. J., Diaz, L., Hugonnet, J. E., Tran, T. T., Narechania, A., et al. (2015). Methicillin-susceptible, vancomycin-resistant Staphylococcus aureus, Brazil. Emerg. Infect. Dis. 21, 1844–1848. doi: 10.3201/eid2110.141914
Paradis, E. (2010). pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420. doi: 10.1093/bioinformatics/btp696
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Planet, P. J., Diaz, L., Kolokotronis, S.-O., Narechania, A., Reyes, J., Xing, G., et al. (2015). Parallel epidemics of community-associated methicillin-resistant Staphylococcus aureus USA300 infection in North and South America. J. Infect. Dis. 212, 1874–1882. doi: 10.1093/infdis/jiv320
R Development Core Team (2014). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Revell, L. J. (2012). phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Roberts, R. B., de Lencastre, A., Eisner, W., Severina, E. P., Shopsin, B., Kreiswirth, B. N., et al. (1998). Molecular epidemiology of methicillin-resistant Staphylococcus aureus in 12 New York hospitals. MRSA Collaborative Study Group. J. Infect. Dis. 178, 164–171. doi: 10.1086/515610
Robinson, D. A., and Enright, M. C. (2004). Multilocus sequence typing and the evolution of methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 10, 92–97. doi: 10.1111/j.1469-0691.2004.00768.x
Rodriguez-Noriega, E., Seas, C., Guzman-Blanco, M., Mejia, C., Alvarez, C., Bavestrello, L., et al. (2010). Evolution of methicillin-resistant Staphylococcus aureus clones in Latin America. Int. J. Infect. Dis. 14, e560–e566. doi: 10.1016/j.ijid.2009.08.018
Rolain, J. M., François, P., Hernandez, D., Bittar, F., Richet, H., Fournous, G., et al. (2009). Genomic analysis of an emerging multiresistant Staphylococcus aureus strain rapidly spreading in cystic fibrosis patients revealed the presence of an antibiotic inducible bacteriophage. Biol. Direct 4:1. doi: 10.1186/1745-6150-4-1
Sansevere, E. A., Luo, X., Park, J. Y., Yoon, S., Seo, K. S., and Robinson, D. A. (2017). Transposase-mediated excision, conjugative transfer, and diversity of ICE6013 elements in Staphylococcus aureus. J. Bacteriol. 199:e00629-16. doi: 10.1128/JB.00629-16
Sansevere, E. A., and Robinson, D. A. (2017). Staphylococci on ICE: overlooked agents of horizontal gene transfer. Mob. Genet. Elements 7, 1–10. doi: 10.1080/2159256X.2017.1368433
Schulte, B., Bierbaum, G., Pohl, K., Goerke, C., and Wolz, C. (2013). Diversification of clonal complex 5 methicillin-resistant Staphylococcus aureus strains (Rhine-Hesse clone) within Germany. J. Clin. Microbiol. 51, 212–216. doi: 10.1128/JCM.01967-12
Smyth, D. S., McDougal, L. K., Gran, F. W., Manoharan, A., Enright, M. C., Song, J. H., et al. (2010). Population structure of a hybrid clonal group of methicillin-resistant Staphylococcus aureus, ST239-MRSA-III. PLoS One 5:e8582. doi: 10.1371/journal.pone.0008582
Sola, C., Cortes, P., Saka, H. A., Vindel, A., and Bocco, J. L. (2006). Evolution and molecular characterization of methicillin-resistant Staphylococcus aureus epidemic and sporadic clones in Cordoba, Argentina. J. Clin. Microbiol. 44, 192–200. doi: 10.1128/JCM.44.1.192-200.2006
Sola, C., Gribaudo, G., Vindel, A., Patrito, L., Bocco, J. L., and Cordoba MRSA Collaborative Study Group (2002). Identification of a novel methicillin-resistant Staphylococcus aureus epidemic clone in Cordoba, Argentina, involved in nosocomial infections. J. Clin. Microbiol. 40, 1427–1435. doi: 10.1128/JCM.40.4.1427-1435.2002
Sola, C., Saka, H. A., Cordoba MRSA Collaborative Study Group Vindel, A., and Bocco, J. L. (2008). Emergence and dissemination of a community-associated methicillin-resistant Panton-Valentine leucocidin-positive Staphylococcus aureus clone sharing the sequence type 5 lineage with the most prevalent nosocomial clone in the same region of Argentina. J. Clin. Microbiol. 46, 1826–1831. doi: 10.1128/JCM.01949-07
Strommenger, B., Bartels, M. D., Kurt, K., Layer, F., Rohde, S. M., Boye, K., et al. (2014). Evolution of methicillin-resistant Staphylococcus aureus towards increasing resistance. J. Antimicrob. Chemother. 69, 616–622. doi: 10.1093/jac/dkt413
Tickler, I. A., Goering, R. V., Mediavilla, J. R., Kreiswirth, B. N., Tenover, F. C., and Consortium, H. A. I. (2017). Continued expansion of USA300-like methicillin-resistant Staphylococcus aureus (MRSA) among hospitalized patients in the United States. Diagn. Microbiol. Infect. Dis. 88, 342–347. doi: 10.1016/j.diagmicrobio.2017.04.016
Uhlemann, A. C., Dordel, J., Knox, J. R., Raven, K. E., Parkhill, J., Holden, M. T. G., et al. (2014). Molecular tracing of the emergence, diversification, and transmission of S. aureus sequence type 8 in a New York community. Proc. Natl. Acad. Sci. U.S.A. 111, 6738–6743. doi: 10.1073/pnas.1401006111
van Wamel, W. J., Rooijakkers, S. H., Ruyken, M., van Kessel, K. P., and van Strijp, J. A. (2006). The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315. doi: 10.1128/JB.188.4.1310-1315.2006
Velazquez-Meza, M. E., Aires de Sousa, M., Echaniz-Aviles, G., Solorzano-Santos, F., Miranda-Novales, G., Silva-Sanchez, J., et al. (2004). Surveillance of methicillin-resistant Staphylococcus aureus in a pediatric hospital in Mexico City during a 7-year period (1997 to 2003): clonal evolution and impact of infection control. J. Clin. Microbiol. 42, 3877–3880. doi: 10.1128/JCM.42.8.3877-3880.2004
Vergara-Irigaray, M., Valle, J., Merino, N., Latasa, C., García, B., Ruiz de Los Mozos, I., et al. (2009). Relevant role of fibronectin-binding proteins in Staphylococcus aureus biofilm-associated foreign-body infections. Infect. Immun. 77, 3978–3991. doi: 10.1128/IAI.00616-09
World Health Organization (2014). Antimicrobial Resistance: Global Report on Surveillance. Available at: http://www.who.int/drugresistance/documents/surveillancereport/en/ [accessed May 1, 2018].
Keywords: methicillin-resistant Staphylococcus aureus, MRSA, phylogenomics, convergent evolution, local adaptation
Citation: Challagundla L, Reyes J, Rafiqullah I, Sordelli DO, Echaniz-Aviles G, Velazquez-Meza ME, Castillo-Ramírez S, Fittipaldi N, Feldgarden M, Chapman SB, Calderwood MS, Carvajal LP, Rincon S, Hanson B, Planet PJ, Arias CA, Diaz L and Robinson DA (2018) Phylogenomic Classification and the Evolution of Clonal Complex 5 Methicillin-Resistant Staphylococcus aureus in the Western Hemisphere. Front. Microbiol. 9:1901. doi: 10.3389/fmicb.2018.01901
Received: 01 May 2018; Accepted: 27 July 2018;
Published: 22 August 2018.
Edited by:
Stefan Monecke, Alere Technologies GmbH, GermanyReviewed by:
Ben Pascoe, University of Bath, United KingdomMiklos Fuzi, Semmelweis University, Hungary
Copyright © 2018 Challagundla, Reyes, Rafiqullah, Sordelli, Echaniz-Aviles, Velazquez-Meza, Castillo-Ramírez, Fittipaldi, Feldgarden, Chapman, Calderwood, Carvajal, Rincon, Hanson, Planet, Arias, Diaz and Robinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: D. Ashley Robinson, darobinson@umc.edu