 Jing Cong1,2
Jing Cong1,2 Hua Zhu1Dong Liu1,2Tianjun Li1Chuantao Zhang1Jingjuan Zhu1Hongying Lv1Kewei Liu1Chenxing Hao1Zibin Tian3Jianli Zhang4
Hua Zhu1Dong Liu1,2Tianjun Li1Chuantao Zhang1Jingjuan Zhu1Hongying Lv1Kewei Liu1Chenxing Hao1Zibin Tian3Jianli Zhang4 Xiaochun Zhang1,2*
Xiaochun Zhang1,2*- 1Department of Medical Oncology, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, China
- 2Qingdao Cancer Institute, Qingdao, China
- 3Department of Gastroenterology, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, China
- 4Department of General Surgery, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, China
Colorectal cancer (CRC) is a growing health problem throughout the world. Strong evidences have supported that gut microbiota can influence tumorigenesis; however, little is known about what happens to gut microbiota following surgical resection. Here, we examined the changes of gut microbiota in CRC patients after the surgical resection. Using the PCoA analysis and dissimilarity tests, the microbial taxonomic compositions and diversities of gut microbiota in post-surgery CRC patients (A1) were significantly different from those in pre-surgery CRC patients (A0) and healthy individuals (H). Compared with A0 and H, the Shannon diversity and Simpson diversity were significantly decreased in A1 (P < 0.05). Based on the LEfSe analysis, the relative abundance of phylum Proteobacteria in A1 was significantly increased than that in A0 and H. The genus Klebsiella in A1 had higher proportions than that in A0 (P < 0.05). Individual variation was distinct; however, 90% of CRC patients in A1 had more abundances of Klebsiella than A0. The Klebsiella in A1 was significantly associated with infectious diseases (P < 0.05), revealed by the correlation analysis between differentiated genera and metabolic pathway. The Klebsiella (Proteobacteria, Gammaproteobacteria, Enterobacteriales, Enterobacteriaceae) in A1 was significantly linked with lymphatic invasion (P < 0.05). Furthermore, the PCA of KEGG pathways indicated that gut microbiota with a more scattered distribution in A1 was noticeably different from that in A0 and H. The nodes, the links, and the kinds of phylum in each module in A1 were less than those in A0 and H, indicating that gut microbiota in A1 had a relatively looser ecologcial interaction network. To sum up, this pilot study identified the changes of gut microbiota in post-surgery CRC patients, and highlights future avenues in which the gut microbiota is likely to be of increasing importance in the care of surgical patients.
Introduction
Colorectal cancer (CRC) is the third leading cause of cancer mortality in the world (Jemal et al., 2011), and is influenced by heredity, diet, lifestyle, gut microbiota, and other factors (Berstad et al., 2015; Sun and Kato, 2016). The human intestinal tract is a nutrient-rich environment housing the largest microbial communities (Zheng et al., 2016). The gut microbiota has garnered great attentions because of its important role in influencing CRC risk by metabolites and immunity in the host (Saleh and Trinchieri, 2011). For example, some bacteria producing hydrogen sulfide, acetaldehyde and secondary bile acids can contribute to the risk of CRC (Huycke and Gaskins, 2004; Bernstein et al., 2009). Simultaneously, some bacteria, including the orders Clostridiales, Lactobacillales, Bifidobacteriales, and Actinomycetales (Devillard et al., 2007), may reduce CRC risk by producing butyrate and conjugated linoleic acids (Scharlau et al., 2009). Therefore, understanding the role of gut microbiota contributes to improving CRC patients’ care.
Human gut microbiota is considered as an essential “organ,” which plays a key role in providing nourishment, regulating epithelial development, and modulating immunity (Eckburg et al., 2005). In recent years, many researchers have attempted to understand the differences in gut microbiota by comparing the microbial community structures of CRC patients and healthy individuals and identify reliable microbial markers for CRC precursors (Zeller et al., 2014). Previously, it has been reported that the abundances of Enterococcus faecalis (Balamurugan et al., 2008) and Desulfovibrio sp. (Scanlan et al., 2009) were observably higher in CRC patients than that in healthy individuals, whereas Bacteroides/Prevotella levels were significantly lower (Sobhani et al., 2011). Differences were also observed in the overall structure of gut microbiota between CRC patients and healthy individuals (Sobhani et al., 2011). Despite of advances in understanding the connections between gut microbiota and CRC, little is known about how surgery resection influences the gut microbiota. Current studies have indicated that many patients who undergo treatment may experience recurrence and even die within several years (Ryuk et al., 2014). Therefore, identifying valid methods to evaluate post-surgery patients’ condition is vital in reducing mortality and healthcare costs.
Gut microbiota is strongly influenced by the surgical removal of lesions, and influences the intestinal healing, particularly with respect to anastomotic tissues in colorectal surgery (Bachmann et al., 2017). In this pilot study, we analyzed fecal samples of CRC patients and healthy individuals by 16S rRNA gene sequencing and real-time quantitative PCR. First, we used the classical community analysis and statistical tests to compare the gut microbial community structure and composition between CRC patients and healthy individuals. Next, we generated functional discrepancy prediction and molecular ecological network to further examine the differences among them. Finally, we discussed the correlates of gut microbiota and clinical variables for the probabilities to assess whether gut microbiota played a key role in identifying the condition for post-surgery CRC patients.
Materials and Methods
Study Design
Our pilot study subjects comprised 10 CRC patients and 11 healthy individuals (Supplementary Figure S1). The CRC patients, aged 34–63 years, were from the affiliated hospital of Qingdao University (Qingdao, China, Table 1 and Supplementary Tables S1, S2). The pilot study selected from untreated CRC patients and excluded those (N = 6) who had previously undergone surgery, chemotherapy, radiation, or targeted therapies before samples collection. Fecal samples from these pre-surgery CRC patients were collected prior to a colonoscopy (O’Brien et al., 2013). The lesion location of all the selected CRC patients was in rectum. These CRC patients were treated with palliative surgery or radical surgery, such as Dixon, Miles and Hartmann (Supplementary Table S2). Following up samples were obtained in approximately 1 month after the surgery. The healthy individuals, aged 49–64 years, were selected as controls (Table 1 and Supplementary Table S2). During a routine physical examination, none had any recorded antibiotics usage or gastrointestinal tract disorders within 3 months preceding the sample collection. All of the participants have been local residents of Qingdao city. This pilot study was approved by the Affiliated Hospital of Qingdao University Institutional Review Board, and all pilot study participants signed the informed consent before participation. All fecal samples were collected within 3 h after defecation in the morning. The collected samples from the healthy individuals, pre-surgery CRC patients and post-surgery CRC patients were named by H, A0, and A1, respectively. Fresh fecal samples were put into 5 ml tubes and immediately stored at -80°C until the day of analysis.
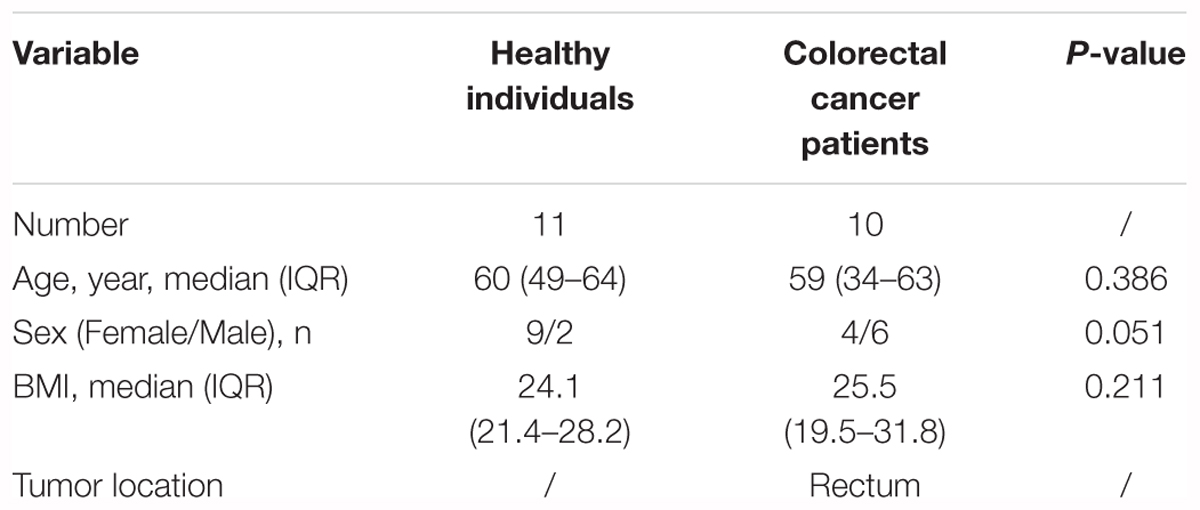
TABLE 1. Baseline characteristics in healthy individuals and colorectal cancer patients.
DNA Extraction, Purification, Sequencing and Data Processing
Extraction of bacterial DNA was performed from fecal samples using a QIAamp Fast DNA Stool Mini Kit as previously reported (Wu et al., 2016). The freshly extracted DNA was purified by 0.5% melting point agarose gel followed by phenolchloroform-butanol extraction. The V3-V4 region of the bacterial 16S rRNA gene from each DNA sample was amplified using the bacterial universal primers (forward primer: 5′-ACTCCTACGGGRSGCAGCAG-3′; reverse primer: 5′-GGACTACVVGGGTATCTAATC-3′). PCR amplification was performed in a 30 μl reaction, containing 15 μl of 2 × KAPA HiFi Hotstart ReadyMix, 1 μl of each primer (forward and reverse primer), 10 ng of template DNA, and the remaining volume of ddH2O. The reaction mixtures were denatured at 95°C for 1 min; followed by 12 cycles of 98°C for 15 s, 72°C for 10 s, 94°C for 20 s, 65°C for 10 s and 72°C for 10 s; then 11 cycles of 94°C for 20 s, 58°C for 30 s, 72°C for 30 s; and a final extension at 72°C for 150 s. The PCR amplification products were purified with an AxyPrep DNA Gel Extraction Kit (Axygen, United States), eluted in 30 μl water, and aliquoted into three PCR tubes. DNA quality and quantity were assessed by the ratios of 260/280 nm and 260/230 nm, and final DNA contents were quantified with a Qubit® dsDNA HS Assay Kit (Invitrogen, United States). Finally, we used bacterial DNA amplicons from each fecal sample for 2 × 250 bp paired-end sequencing based on the Illumina Hiseq 2500.
Raw sequences were separated into samples by barcodes using the Galaxy Illumina sequencing pipeline1. Adapters, ambiguous and low-quality reads (“N”) were trimmed by Btrim (Kong, 2011). Forward and reverse reads were incorporated into a whole sequence by FLASH (Magoč and Salzberg, 2011). After quality control of the raw data, the clean reads were clustered into operational taxonomic units (OTUs) at 97% similarity level by using UCLUST (Edgar, 2010). Each OTU was considered to represent a species (Deng et al., 2012). Rarefaction analysis was performed using the original detected OTUs (Supplementary Figure S2). The ribosomal database project (RDP) classifier was used to determine the taxonomic assignment (Wang et al., 2007). Random resampling was conducted on 48,360 sequences per fecal sample.
Real-Time Quantitative PCR
Three specific primers were used for real-time quantitative PCR (qPCR), including 16S rRNA universal primer for bacteria, primer for Fusobacterium nucleatum (Castellarin et al., 2012), primer for Klebsiella pneumonia (Sun et al., 2010) (Supplementary Table S3). The primers were synthesized at Shanghai Sangon Company (China). The PCR program was as follows: 95°C for 3 min; 35 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 30 s; 72°C for 8 min. The PCR products were purified with gel extraction kit (Sangon SK8131), quantified using Micro-spectrophotometer SMA4000 (Merinton), and used to construct standard curves. The copy number of PCR products was calculated based on the formula: Copy number/μL = 6.02 × 1014 × C/(M × W). The C (unit, ng/μL) represents the concentration of PCR products, M (unit, bp) represents the length of PCR products, and W (660 Da/bp) represents the constant. The PCR products were diluted from 107 to 1010 copies/μL and amplified to construct standard curves. The real-time qPCR was run with LightCycler480 II (Roche, German). The reaction mixture contained 5 μL SybrGreen qPCR Master Mix (Roche, German), 1 μL of template DNA, and 0.2 μL forward/reverse primer (10 μM), and ddH2O was added to reach a total volume of 10 μL. The real-time qPCR program was as follows: 95°C for 3 min; 45 cycles of 95°C for 15 s, 57°C for 20 s, and 72°C for 30 s. The melting curves for the amplicons were measured while monitoring fluorescence. The amplification efficiencies of three primers were between 80 and 110%, and the melting curves all showed a single peak (Supplementary Figure S3), indicating that the results were credible. The copies of each sample based on 16S rRNA universal primer were considered as the bacterial biomass per gram. The following formula was used to calculate the relative abundance: Relative abundance = Ci/C0 × 100%, where Ci represents the copies of the species and C0 represents the bacterial biomass (Xiao et al., 2018).
Network Analysis
Gut microbial ecological networks were constructed and analyzed by random matrix theory (RMT) methods by the online MENA pipeline2. OTUs detected in less than 70% from each group were removed to ensure reliable correlations. For comparisons with different networks, the same cutoff of 0.77 was applied to construct ecological networks for gut microbial communities. Each ecological network was separated into modules by the fast greedy modularity optimization to characterize the modularity property. Furthermore, a network developed from OTU abundance data represented the ecological co-occurrence (links) of different OTU markers (nodes) in a microbial community, and different nodes played distinct roles (Guimerà et al., 2007).
Statistical Analysis
The common OTUs mean that the OTUs present in three groups (A1, A0 and H). Principal coordinate analysis (PCoA) was used to identify overall gut microbial composition between CRC patients and healthy individuals based on Bray-Curtis dissimilarity index. Principal components analysis (PCA) was used to determine the changes of KEGG pathways between CRC patients and healthy individuals. Alpha diversity was calculated using the observed species (richness), phylogenetic diversity, Shannon index and Simpson index. The significant differences referred to the multiple response permutation procedure (MRPP) algorithms and analysis of similarity (Anosim). Significant P-values associated with microbial clades and functions were identified by Linear Discriminant Analysis with Effect Size (LEfSe). Characteristics with an LDA score cut-off of 2.0 were considered as being different. Community analysis and differential abundance of OTUs were performed using the STAMP 2.0.8 (Parks et al., 2014). According to the Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology, functional profiling of microbial communities was predicted using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) (Langille et al., 2013). Gut microbial metabolic and other pathway differences were predicted by the correlations between the PICRUSt-generated functional profiles and STAMP-generated genus level bacterial abundance. Mantel test was used to evaluate the linkages between gut microbial structure and environmental attributes. The R software package (v3.4.1) was used for all statistical analysis, except for two-tailed unpaired t-tests by Microsoft Excel 2010, and Analysis of variance (ANOVA) and Pearson correlation by IBM SPSS statistic 19.0 to determine the significance of the differences and the clinical correlates.
Results
Taxonomic Composition and Diversity of Gut Microbiota
A total of 1,819,210 quality-filtered 16S rRNA gene sequences were acquired from 31 samples, with an average of 58,684 ± 2602 reads per sample (Supplementary Table S4). A total of 648 OTUs were generated at the 97% similarity level, with an average of 189 ± 60 OTUs per sample (Supplementary Table S4). We compared the microbial alpha diversity (richness, phylogenetic diversity, Shannon diversity and Simpson diversity) between CRC patients and healthy individuals (Table 2). The results demonstrated that the Shannon diversity and Simpson diversity were significantly decreased in A1 compared with the A0 and H (P < 0.05, Table 2). However, no statistically significant differences were identified in the richness and phylogenetic diversity for CRC patients and healthy individuals (P > 0.05, Table 2). The dissimilarity tests showed that A1 was significantly different from A0 and H based on the multiple response permutation procedure (MRPP) algorithms and analysis of similarity (ANOSIM) (P < 0.05, Supplementary Table S5). PCoA based on Bray-Curtis dissimilarity index revealed overall gut microbial composition in CRC patients was well separated from each other, but partly overlapping with healthy individuals (Figure 1). Furthermore, A1 presented a more scattered distribution and had a distance from the A0 and H (Figure 1). Therefore, A1 had significantly different community structure with A0 and H.
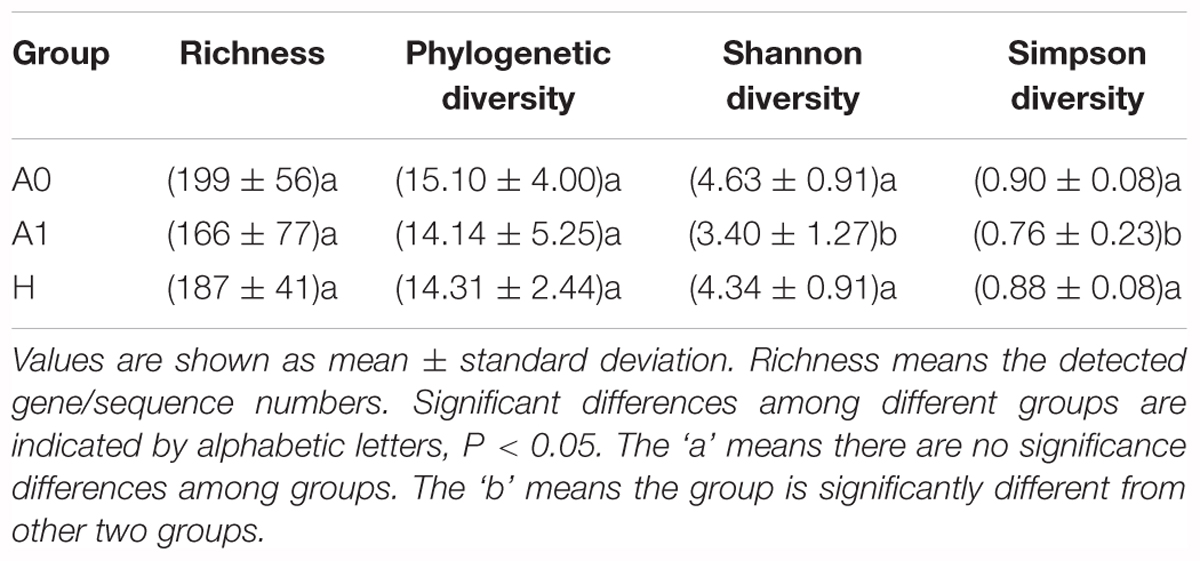
TABLE 2. Comparison of alpha diversity indices of gut microbiota between the healthy volunteers (H) and CRC patients before and after surgery (A0 and A1).
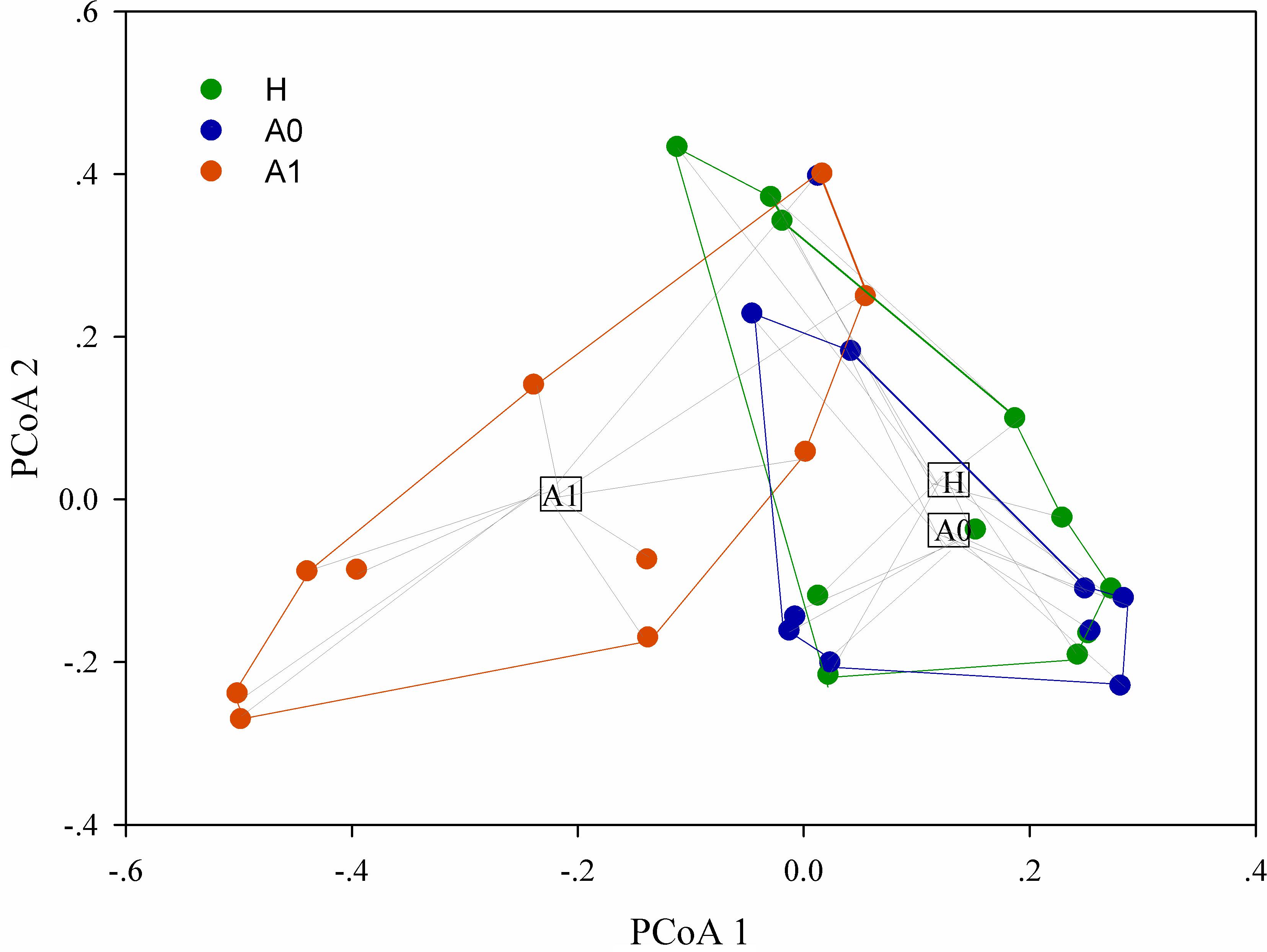
FIGURE 1. Principal coordinates analysis (PCoA) ordination (operational taxonomic units = 97% 16S rRNA sequence similarity) showing distinctly different microbial composition between CRC patients and healthy individuals based on the Bray-Curtis dissimilarity matrix.
Taxonomy-Based Comparisons of the Gut Microbiota
The gut microbial taxa and their relative abundance were significantly different among H, A0, and A1. At the phylum level, H was mainly characterized by Firmicutes and Bacteroidetes, whereas A0 and A1 had a very complicated community composition, especially for A1 (Supplementary Figure S4). Compared with H, the relative abundance of Proteobacteria was significantly increased by 12.90%, and Bacteroidetes was significantly decreased by 23.06% in A1 (P < 0.05, Supplementary Table S6). At the genus level, the members of Faecalibacterium, Roseburia, Ruminococcus, and Lachnospiracea_incertae_sedis were significantly lower in A1 than those in H (P < 0.05, Supplementary Figure S5 and Supplementary Table S7). To identify gut microbial responses associated with surgery at the taxonomical level, we determined microbial clade differences using LEfSe analysis (Figure 2). At the phylum level, higher proportions of Proteobacteria were observed in A1 than that in A0 and H (Figure 2A). At the genus level, greater proportions of Klebsiella were detected in A1 than that in A0 (Supplementary Figure S6). The genus Fusobacteria was significantly enriched in A0 than that in A1 and H (Figure 2B). The members of Clostridium XlVa, Fusobacterium, Parvimonas, and Peptostreptococcus were more abundant on A0 than that on A1 and H (Figure 2A).
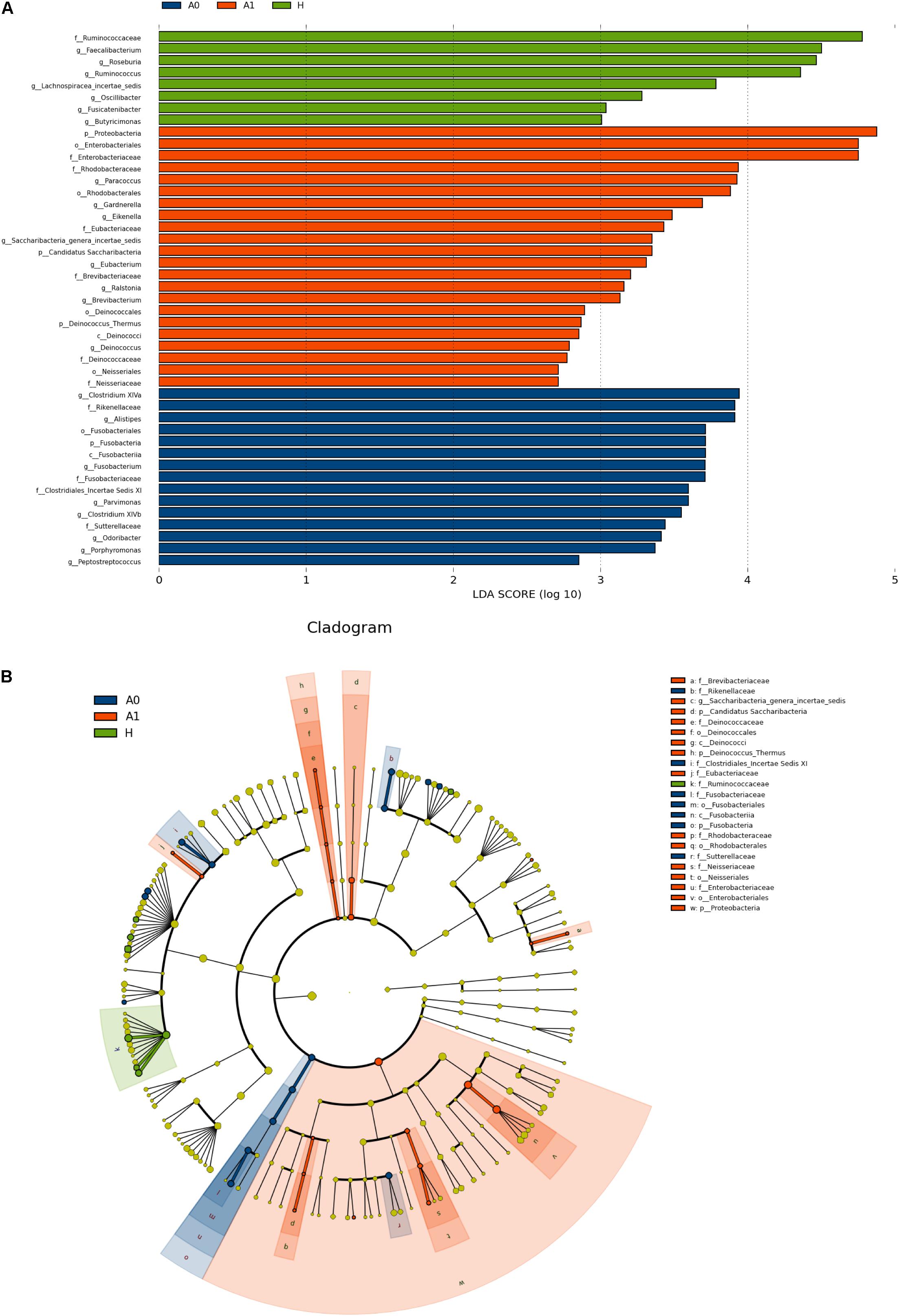
FIGURE 2. Microbial biomarkers among healthy volunteers (H) and CRC patients (A0 and A1). (A) LEfSe analysis shows differentially abundant taxa as biomarkers using Kruskal–Wallis test (P < 0.05) with LDA score > 2.0. (B) Cladogram representation of the differentially abundant taxa. The root of the cladogram represents the domain bacteria. The size of each node represents their relative abundance. No significantly different taxa are labeled by yellow. Significant different taxa are labeled by following the color of each group.
Sequencing data suggested that gut microbiota made changes in response to surgery, and we further used real-time qPCR to help validate changes observed and detected with 16S sequencing data. We selected Fusobacterium nucleatum and Klebsiella pneumoniae to examine the variations, which frequently present in CRC patients (Supplementary Figure S7). The results showed that the relative abundance of Fusobacterium nucleatum in A0 was significantly higher than that in H (P < 0.05). However, the relative abundance of Klebsiella pneumoniae was not significantly changed. In addition, individual differences were evident showed in Supplementary Figure S8. Notably, the relative abundance of Fusobacterium increased markedly in 8/10 patients of A0 compared with A1. The members of Klebsiella were distinctly higher in 9/10 patients of A1 than that in A0.
Functional and Metabolic Discrepancy of the Gut Microbiota
Further studies were required to understand the dynamics of gut microbiota following surgical treatment to evaluate the role of microbiota. The PCA of KEGG pathways indicated that A1 was notably different from A0 and H, which had a more scattered distribution based on the STAMP analysis (Figure 3A). In terms of KEGG pathways (L2, Figure 3B), the functions of ‘replication and repair,’ ‘folding, sorting and degradation,’ and ‘cell growth and death,’ which belong to the first level ‘Genetic information processing’ and ‘Cellular processes,’ respectively, were significantly enriched in A0 (P < 0.05) compared with H and A1 based on the LEfSe analysis. The results indicated that gut microbiota in pre-surgery patients was enriched in more conservative housekeeping functions. The ‘infectious diseases,’ ‘xenobiotics biodegradation and metabolism,’ ‘metabolism of other amino acids,’ ‘neurodegenerative diseases,’ and ‘metabolism’ were significant function hallmarks of gut microbiota in A1. Furthermore, we found that the genus Klebsiella in A1 was significantly and closely associated with infectious diseases, such as bacterial invasion of epithelial cells and Staphylococcus aureus infection (P < 0.05, Figure 4).
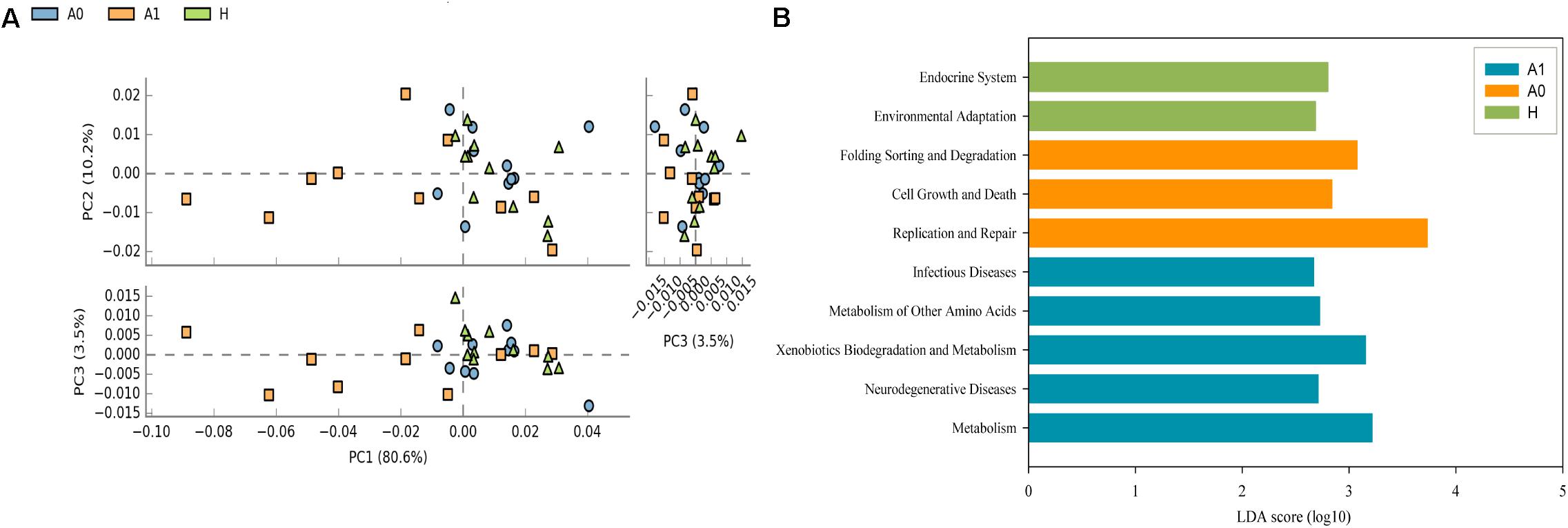
FIGURE 3. Functional and metabolic discrepancy of the gut microbiota between CRC patients and healthy individuals. (A) Principal component analysis (PCA) plot of the KEGG pathway (L2) shows that the post-surgery CRC patients were noticeably different from the pre-surgery CRC patients and healthy individuals based on the STAMP analysis. Characteristics with an LDA score cut-off of 2.0 were considered as being different. The LDA scores (log10) > 2 are listed; (B) Discriminatory functional pathways (KEGG L2) shows the significantly different between CRC patients and healthy individuals based on the LDA score using the LEfSe analysis.
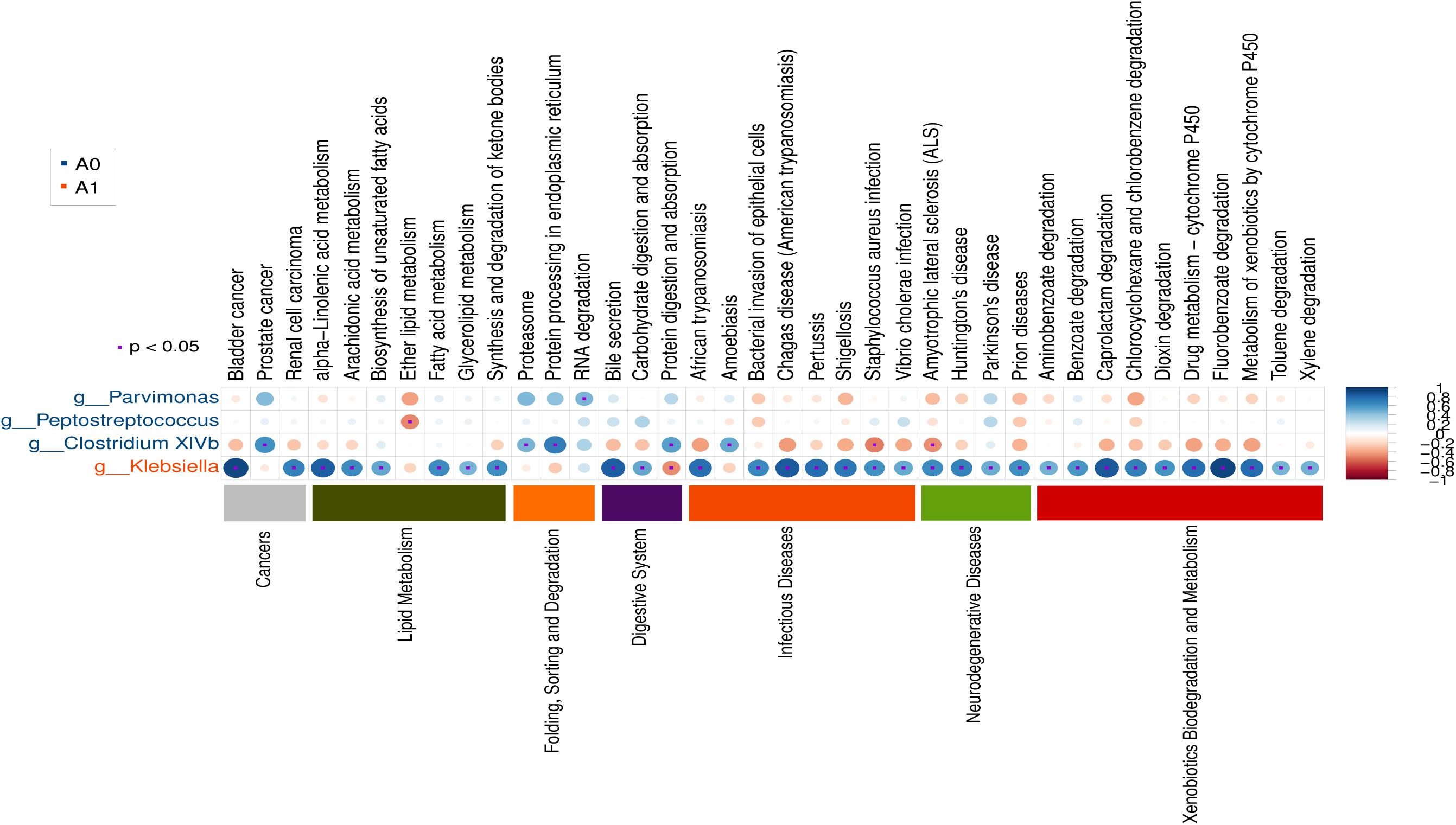
FIGURE 4. Gut microbial metabolic and other pathway differences in pre-surgery CRC patients (A0) and post-surgery CRC patients (A1). Correlations between the PICRUSt-generated functional profiles and STAMP-generated genus level bacterial abundance are calculated and plotted.
Modularity Analysis in Gut Microbiota
Microbes rarely live in isolation, but instead interact in complex ecological networks. To identify the assemblages that potentially interact within the intestinal tract, we focused on representative networks from CRC patients and healthy individuals. We selected more than five nodes to construct the modules and visualized the phylogeny for modules with at least two kinds of phyla (Figure 5). Overall, OTU tended to co-occur (positive correlations, gray lines) rather than co-exclude (negative correlations, pink lines). However, there were more negative correlations between gut microbes in A0 than that in A1 and H (Figure 5). The modules in A1 became smaller and less connected, which had fewer nodes and links (35, 37) than that in A0 (60, 113) and in H (91, 231). Furthermore, the kinds of phylum in each module were only two in A1, which were less than that in A0 and H.
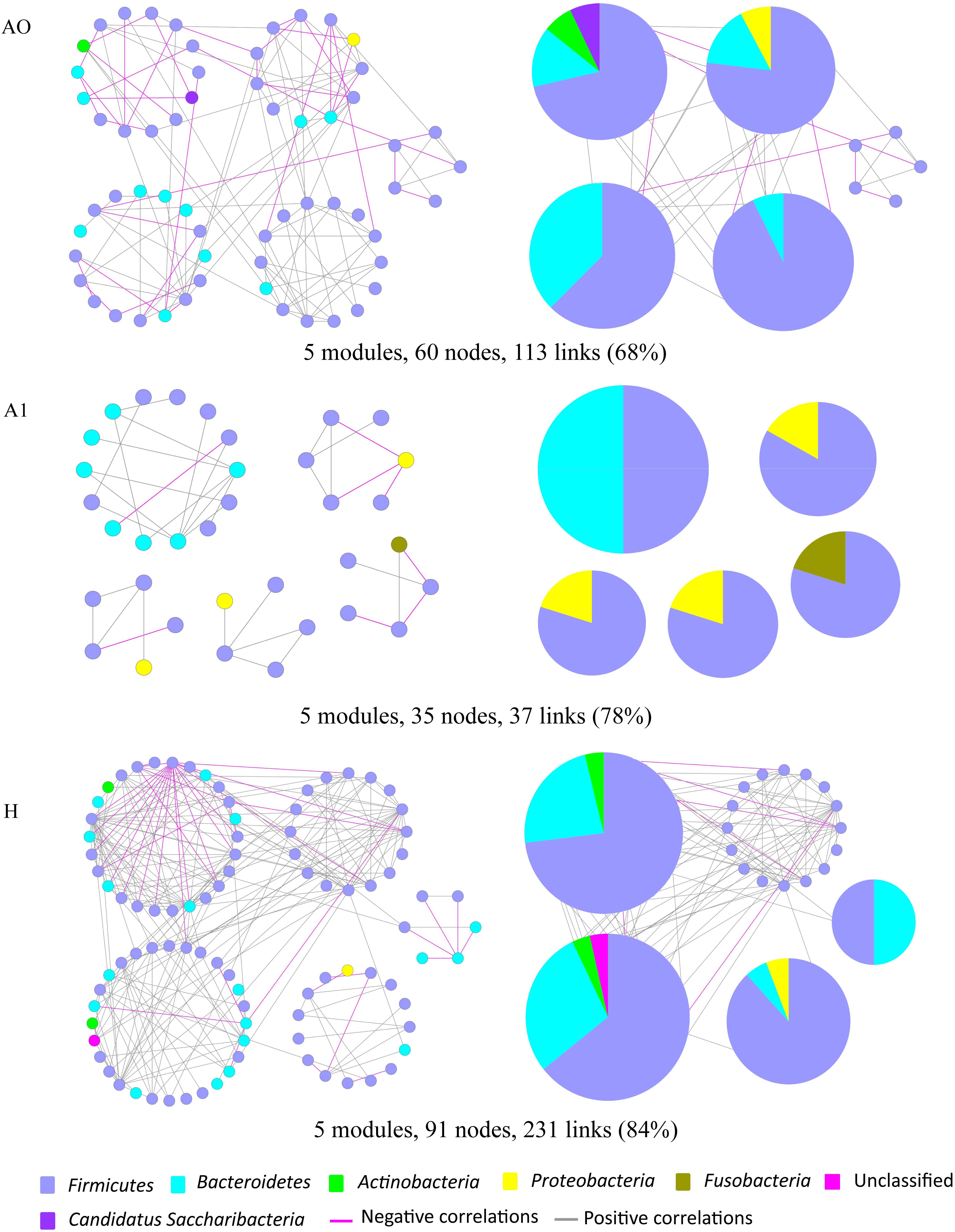
FIGURE 5. Highly connected modules of gut microbial networks within CRC patients before and after surgery (A0, A1) and healthy individuals (H). The colors of nodes indicate different major phyla; pie charts represent the composition of modules with ≥ 2 phyla. A pink link indicates negative correlations between two individual nodes, whereas a gray link indicates positive correlations. The percentage in parentheses indicates the ratio of positive correlations.
Correlates of Gut Microbiota and Clinical Variables
Based on the mantel test, we explored the clinical correlates of gut microbiota and patients age, sex, BMI, and bowel treatment. The results showed that the whole gut microbiota and key phyla had no significant correlations with these clinical variables (Supplementary Table S8). Then, we analyzed the histopathological correlates of gut microbiota and tumor stage, grade of tumor differentiation, lymphatic invasion, perineural invasion, number of metastasis lymph nodes and tumor markers (CEA and CA199) (Supplementary Table S9). We found that the Fusobacterium (Fusobacteria, Fusobacteriia, Fusobacteriales, Fusobacteriaceae) in A0 and Klebsiella (Proteobacteria, Gammaproteobacteria, Enterobacteriales, Enterobacteriaceae) in A1 were significantly correlated with lymphatic invasion (P < 0.05). The Fusobacterium (Fusobacteria, Fusobacteriia, Fusobacteriales, Fusobacteriaceae) in A0 was significantly correlated with CA199 (P < 0.05).
Discussion
The gut microbiota interacts extensively with the host by the co-metabolism of substrates and metabolic exchanges to maintain a healthy status and normal functions of the body (Nicholson et al., 2005). Previous studies have highlighted the significance of gut microbiota in the progression of intestinal diseases such as Crohn’s disease, ulcerative colitis (Willing et al., 2010), celiac disease in children (Nadal et al., 2007), allergic inflammation in infants (Kalliomäki and Isolauri, 2003), and CRC (Bachmann et al., 2017). In this pilot study, we focused on exploring the feedback of gut microbiota of CRC patients in response to the surgical removal of lesions.
The gut microbial community composition and diversity in post-surgery CRC patients significantly differ from that in pre-surgery CRC patients and healthy individuals (P < 0.05, Figure 1, Table 2, and Supplementary Table S5). However, the gut microbial alpha diversity in A0 was not significantly different from that in H (Table 2). The findings may be explained by previous report that the fecal microbiota only partially reflected mucosal microbiota in CRC (Flemer et al., 2017a). The alpha diversity in A1 was significantly lower than that in H (P < 0.05). High diversity is always linked to health and temporal stability (Flores et al., 2014). Conversely, a relative lack of diversity is often observed in the gut microbiota of CRC patients (Gao et al., 2015). Antibiotics use causes a dramatic reduction in the diversity of gut microbiota (Dethlefsen and Relman, 2011), which was similar to our results that the gut microbial diversity was significantly decreased in post-surgery CRC patients, potentially weakening the community’s ability to resist pathogens.
Flemer et al. (2017b) had previously confirmed that the microbiota of CRC patients differed from that of controls (Flemer et al., 2017b), and similar results had been found in our study that gut microbial taxa and their relative abundance of CRC patients, especially for post-surgery CRC patients, significantly differed from that of healthy individuals (P < 0.05). Generally, the gut microbiota in healthy individuals is dominated by the phyla Firmicutes and Bacteroidetes (Consortium, 2012), whereas Proteobacteria, Actinobacteria, Verrucomicrobia, and Fusobacteria are less abundant (Bäckhed et al., 2005). The relative abundance of phylum Proteobacteria was significantly higher in A1 than that in A0 and H (P < 0.05, Figure 2A and Supplementary Table S6). Imbalanced gut microbiota often results from a sustained increase in Proteobacteria, and human gut microbiota normally contains a minor proportion of this phylum (Shin et al., 2015). A bloom of Proteobacteria in the gut often reflects an unstable structure of the gut microbial community, which was often observed in the disease states (Morgan et al., 2012). Proteobacteria was often associated with dysbiosis and was a potential diagnostic criterion for disease (Shin et al., 2015). The relative abundance of phylum Fusobacteria was significantly increased in A0 than that in A1 and H (P < 0.05, Figure 2B). At the genus level, Klebsiella played more significantly key roles in A1 than that in A0 (P < 0.05, Supplementary Figure S6). However, the relative abundance of Klebsiella pneumoniae was not significantly changed based on the real-time qPCR (Supplementary Figure S7). The reasons were probably ascribed to the small sample size, followed by the low abundance in sample itself, DNA degradation of human feces, the sensitivity and specificity of primer, reaction condition of qPCR. In addition, Clostridium XlVa, Parvimonas, Peptostreptococcus, and Fusobacterium played significantly important roles in A0 (P < 0.05, Figure 2A). Parvimonas was frequently and significantly increased in stools from CRC patients (Shah et al., 2017). Peptostreptococcus played a key role in the dysbiosis of mucosa-associated microbiota in CRC patients (Shah et al., 2017). It was reported that Fusobacterium species led the development of CRC (Kostic et al., 2012). The qPCR results also showed that Fusobacterium nucleatum in A0 was significantly higher than that in H (P < 0.05, Supplementary Figure S7). Fusobacterium nucleatum infection is prevalent in human CRC (Castellarin et al., 2012). Furthermore, we found that Fusobacterium (Fusobacteria, Fusobacteriia, Fusobacteriales, Fusobacteriaceae) in A0 was significantly correlated with lymphatic invasion and tumor marker CA199 (P < 0.05, Supplementary Table S9), similar to Castellarin et al. (2012) results that indicated that high relative abundance of Fusobacterium was more likely to have regional lymph node metastases. Therefore, gut microbiota from fecal samples was increasingly possible to be considered as potential diagnostic biomarkers of dysbiosis for CRC patients (Zackular et al., 2014).
Potential function and metabolism of gut microbiota in CRC patients were greatly changed in response to the surgery. Although great differences were observed in the taxonomic composition of gut microbiota in different individuals, the metabolic pathways are considerably more consistent across people (Consortium, 2012). A healthy microbiota may contain specific microbial combinations, metabolic modules, and regulatory pathways that together maintain a stable host-associated ecology (Martiny et al., 2015). The housekeeping functions are necessary for all microbial life, such as ‘replication and repair’ and ‘cell growth and death’ (Consortium, 2012). Our results demonstrated that the housekeeping functions were significantly associated with gut microbiota in A0, whereas the gut microbiota in A1 was more closely involved with the functions of infectious diseases and xenobiotics biodegradation and metabolism (Figure 3). The Klebsiella in A1 was determined to be closely associated with infectious diseases (Figure 4). Klebsiella is an opportunistic pathogen routinely found in human gut that causes diarrhea, pneumonia, and bloodstream infections (Yan et al., 2017). It was reported that overgrowth of Klebsiella often foreshadowed gut flora dysbiosis (Garrett et al., 2010) and markedly increased the rates of treatment failure and death (Yan et al., 2017). Furthermore, Klebsiella (Proteobacteria, Gammaproteobacteria, Enterobacteriales, Enterobacteriaceae) in A1 was significantly correlated with lymphatic invasion (P < 0.05, Supplementary Table S9). Therefore, the gut microbiota in post-surgery CRC patients maybe presented a weaker robustness when random and specific perturbations influence its functional stability.
Molecular ecological networks of gut microbiota in CRC patients were also greatly changed in response to the surgery. In network biology, a group of microbial species strongly interacting with one another constructs a module, which may reflect physical contact, divergent selection, functional association, and/or the phylogenetic clustering of closely related species (Olesen et al., 2007). Considering the characteristic of a smaller and looser network module in A1 (Figure 5), it implied that A1 had a weaker coupling between gut microbes than A0 and H. This could partially be explained by some sharply growing pathogenic bacterium, such as the phylum Proteobacteria, influencing the microbial community structure (Figure 2 and Supplementary Table S6). Therefore, the gut microbiota in post-surgery patients probably had a higher sensitivity in response to external changes.
The age, sex, BMI, bowel treatment and diet were considered as the important influencing factors in changing the gut microbiota of CRC patients. However, we found that there were no significant correlations between these factors (age, sex, BMI, and bowel treatment) and gut microbiota based on the mantel test (Supplementary Table S8). Comprehensive information on microbial species across a great number of samples is essential in identifying the changes among microbial communities (Barberán et al., 2012). Sample sets should ideally be ample to achieve sufficient variability (Barberán et al., 2012). However, the number of patients in the treatment group was relatively small and individual differences were evident (Supplementary Figure S8). Therefore, there was not statistical power to adequately examine these relationships between these factors (age, sex, BMI, and bowel treatment) and gut microbiota. Therefore, we could not scale the results to all the situations with only a few samples. In addition, samples from the post-surgery patients were also influenced by the colonoscopy. More attentions should be paid on these correlative factors in the future. Yet, this pilot study would provide better understanding of the responses of gut microbiota to the surgical removal of lesions for CRC patients (Supplementary Figure S9). The gut microbiota probably plays a key role in identifying the condition for post-surgery CRC patients. Additional sampling efforts, colonoscopy effects, and diet records combined with clinical follow-up are required to further obtain unique insight into gut microbial changes in post-surgery CRC patients to predict disease states and develop therapies to correct dysbiosis.
Conclusion
In summary, this pilot study explored the changes of gut microbiota in CRC patients following the surgery. The gut microbial taxonomic compositions in post-surgery CRC patients were significantly different from those in pre-surgery CRC patients and healthy individuals. The gut microbiota in post-surgery CRC patients had a significantly lower alpha diversity and a looser ecological interaction network. Most post-surgery CRC patients had more abundances of Klebsiella. The Klebsiella in post-surgery CRC patients was significantly associated with lymphatic invasion. These results indicated that gut microbiota was probably considered to be the valuable biomarkers in evaluating the condition of post-surgery CRC patients. More attentions should be paid to advance our understanding of the role of gut microbiota in recovering the intestinal health of post-surgery CRC patients.
Availability of Data and Material
Sequencing data are accessible in NCBI SRA database with Accession No. SRP133809 (https://www.ncbi.nlm.nih.gov/sra/SRP133809).
Ethics Statement
The Affiliated Hospital of Qingdao University Institutional Review Board approved this study, and all study subjects provided informed consent.
Author Contributions
All authors were involved in the design of the study. DL, HZ, and CH collected fecal samples. JjZ, TL, CZ, HL, KL, ZT, and JlZ analyzed the data. All authors interpreted the data. JC participated in all the work including writing the manuscript. All authors reviewed and revised the manuscript and read and approved the final manuscript.
Funding
This study was supported by funding from Project funded by China Postdoctoral Science Foundation (2016M602094), Qingdao Postdoctoral Application Research Project (2016047), Youth Research Fund Project, and Taishan Scholar Foundation (201502061).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the CRC patients and healthy volunteers for providing the fecal samples that were used in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02777/full#supplementary-material
Footnotes
References
Bachmann, R., Leonard, D., Delzenne, N., Kartheuser, A., and Cani, P. D. (2017). Novel insight into the role of microbiota in colorectal surgery. Gut 66, 739–749. doi: 10.1136/gutjnl-2016-312569
Bäckhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A., and Gordon, J. I. (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920. doi: 10.1126/science.1104816
Balamurugan, R., Rajendiran, E., George, S., Samuel, G. V., and Ramakrishna, B. S. (2008). Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, Desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J. Gastroenterol. Hepatol. 23, 1298–1303. doi: 10.1111/j.1440-1746.2008.05490.x
Barberán, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351. doi: 10.1038/ismej.2011.119
Bernstein, H., Bernstein, C., Payne, C. M., and Dvorak, K. (2009). Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. 15, 3329–3340. doi: 10.3748/wjg.15.3329
Berstad, P., Løberg, M., Larsen, I. K., Kalager, M., Holme,Ø, Botteri, E., et al. (2015). Long-term lifestyle changes after colorectal cancer screening: randomized controlled trial. Gut 64, 1268–1276. doi: 10.1136/gutjnl-2014-307376
Castellarin, M., Warren, R. L., Freeman, J. D., Dreolini, L., Krzywinski, M., Strauss, J., et al. (2012). Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306. doi: 10.1101/gr.126516.111
Consortium, H. M. P. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Deng, Y., Jiang, Y. H., Yang, Y., He, Z., Luo, F., and Zhou, J. (2012). Molecular ecological network analyses. BMC Bioinformatics 13:113. doi: 10.1186/1471-2105-13-113
Dethlefsen, L., and Relman, D. A. (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4554–4561. doi: 10.1073/pnas.1000087107
Devillard, E., Mcintosh, F. M., Duncan, S. H., and Wallace, R. J. (2007). Metabolism of linoleic acid by human gut bacteria: different routes for biosynthesis of conjugated linoleic acid. J. Bacteriol. 189, 2566–2570. doi: 10.1128/JB.01359-06
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., et al. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638. doi: 10.1126/science.1110591
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Flemer, B., Lynch, D. B., Brown, J. M. R., Jeffery, I. B., Ryan, F. J., Claesson, M. J., et al. (2017a). Original article: tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 66, 633–643. doi: 10.1136/gutjnl-2015-309595
Flemer, B., Lynch, D. B., Brown, J. M. R., Jeffery, I. B., Ryan, F. J., Claesson, M. J., et al. (2017b). Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 66, 633–643. doi: 10.1136/gutjnl-2015-309595
Flores, G. E., Caporaso, J. G., Henley, J. B., Rideout, J. R., Domogala, D., Chase, J., et al. (2014). Temporal variability is a personalized feature of the human microbiome. Genome Biol. 15, 1–13. doi: 10.1186/s13059-014-0531-y
Gao, Z., Guo, B., Gao, R., Zhu, Q., and Qin, H. (2015). Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 6:20. doi: 10.3389/fmicb.2015.00020
Garrett, W. S., Gallini, C. A., Yatsunenko, T., Michaud, M., Dubois, A., Delaney, M. L., et al. (2010). Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300. doi: 10.1016/j.chom.2010.08.004
Guimerà, R., Sales-Pardo, M., and Amaral, L. A. N. (2007). Classes of complex networks defined by role-to-role connectivity profiles. Nat. Phys. 3, 63–69. doi: 10.1038/nphys489
Huycke, M. M., and Gaskins, H. R. (2004). Commensal bacteria, redox stress, and colorectal cancer: mechanisms and models. Exp. Biol. Med. 229, 586–597. doi: 10.1177/153537020422900702
Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., and Forman, D. (2011). Global cancer statistics. Cancer J. Clin. 61, 69–90. doi: 10.3322/caac.20107
Kalliomäki, M., and Isolauri, E. (2003). Role of intestinal flora in the development of allergy. Curr. Opin. Allergy Clin. Immunol. 3, 15–20. doi: 10.1097/00130832-200302000-00003
Kong, Y. (2011). Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98, 152–153. doi: 10.1016/j.ygeno.2011.05.009
Kostic, A. D., Gevers, D., Pedamallu, C. S., Michaud, M., Duke, F., Earl, A. M., et al. (2012). Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298. doi: 10.1101/gr.126573.111
Langille, M. G., Zaneveld, J., Caporaso, J. G., Mcdonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Martiny, J. B., Jones, S. E., Lennon, J. T., and Martiny, A. C. (2015). Microbiomes in light of traits: a phylogenetic perspective. Science 350:aac9323. doi: 10.1126/science.aac9323
Morgan, X. C., Tickle, T. L., Sokol, H., Gevers, D., Devaney, K. L., Ward, D. V., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79. doi: 10.1186/gb-2012-13-9-r79
Nadal, I., Donat, E., Ribes-Koninckx, C., Calabuig, M., and Sanz, Y. (2007). Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 56, 1669–1674. doi: 10.1099/jmm.0.47410-0
Nicholson, J. K., Holmes, E., and Wilson, I. D. (2005). Gut microorganisms, mammalian metabolism and personalized health care. Nat. Rev. Microbiol. 3, 431–438. doi: 10.1038/nrmicro1152
O’Brien, C. L., Allison, G. E., Grimpen, F., and Pavli, P. (2013). Impact of colonoscopy bowel preparation on intestinal microbiota. PLoS One 8:e62815. doi: 10.1371/journal.pone.0062815
Olesen, J. M., Bascompte, J., Dupont, Y. L., and Jordano, P. (2007). The modularity of pollination networks. Proc. Natl. Acad. Sci. U.S.A. 104, 19891–19896. doi: 10.1073/pnas.0706375104
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Ryuk, J. P., Choi, G. S., Park, J. S., Kim, H. J., Park, S. Y., Yoon, G. S., et al. (2014). Predictive factors and the prognosis of recurrence of colorectal cancer within 2 years after curative resection. Ann. Surg. Treat. Res. 86, 143–151. doi: 10.4174/astr.2014.86.3.143
Saleh, M., and Trinchieri, G. (2011). Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat. Rev. Immunol. 11, 9–20. doi: 10.1038/nri2891
Scanlan, P. D., Shanahan, F., and Marchesi, J. R. (2009). Culture-independent analysis of desulfovibrios in the human distal colon of healthy, colorectal cancer and polypectomized individuals. FEMS Microbiol. Ecol. 69, 213–221. doi: 10.1111/j.1574-6941.2009.00709.x
Scharlau, D., Borowicki, A., Habermann, N., Hofmann, T., Klenow, S., Miene, C., et al. (2009). Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat. Res. 682, 39–53. doi: 10.1016/j.mrrev.2009.04.001
Shah, M. S., Desantis, T. Z., Weinmaier, T., Mcmurdie, P. J., Cope, J. L., Altrichter, A., et al. (2017). Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut 67, 882–891. doi: 10.1136/gutjnl-2016-313189
Shin, N. R., Whon, T. W., and Bae, J. W. (2015). Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503. doi: 10.1016/j.tibtech.2015.06.011
Sobhani, I., Sobhani, J., Roudot-Thoraval, F., Roperch, J. P., Letulle, S., Letulle, P., et al. (2011). Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One 6:e16393. doi: 10.1371/journal.pone.0016393
Sun, F., Wu, D., Qiu, Z., Jin, M., Wang, X., and Li, J. (2010). Development of real-time PCR systems based on SYBR Green for the specific detection and quantification of Klebsiella pneumoniae in infant formula. Food Control 21, 487–491. doi: 10.1016/j.foodcont.2009.07.014
Sun, J., and Kato, I. (2016). Gut microbiota, inflammation and colorectal cancer. Annu. Rev. Microbiol. 3, 130–143. doi: 10.1016/j.gendis.2016.03.004
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Willing, B. P., Dicksved, J., Halfvarson, J., Andersson, A. F., Lucio, M., Zheng, Z., et al. (2010). A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139, 1844–1854. doi: 10.1053/j.gastro.2010.08.049
Wu, X., Zhang, H., Chen, J., Shang, S., Wei, Q., Yan, J., et al. (2016). Comparison of the fecal microbiota of dholes high-throughput Illumina sequencing of the V3–V4 region of the 16S rRNA gene. Appl. Microbiol. Biotechnol. 100, 3577–3586. doi: 10.1007/s00253-015-7257-y
Xiao, Y., Liu, X., Meng, D., Tao, J., Gu, Y., Yin, H., et al. (2018). The role of soil bacterial community during winter fallow period in the incidence of tobacco bacterial wilt disease. Appl. Microbiol. Biotechnol. 102, 2399–2412. doi: 10.1007/s00253-018-8757-3
Yan, Q., Gu, Y., Li, X., Yang, W., Jia, L., Chen, C., et al. (2017). Alterations of the gut microbiome in hypertension. Front. Cell. Infect. Microbiol. 7:381. doi: 10.3389/fcimb.2017.00381
Zackular, J. P., Rogers, M. A., and Schloss, P. D. (2014). The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev. Res. 7, 1112–1121. doi: 10.1158/1940-6207.CAPR-14-0129
Zeller, G., Tap, J., Voigt, A. Y., Sunagawa, S., Kultima, J. R., Costea, P. I., et al. (2014). Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 10:766. doi: 10.15252/msb.20145645
Keywords: gut microbiota, colorectal cancer, surgery, high-throughput sequencing, real-time quantitative PCR
Citation: Cong J, Zhu H, Liu D, Li T, Zhang C, Zhu J, Lv H, Liu K, Hao C, Tian Z, Zhang J and Zhang X (2018) A Pilot Study: Changes of Gut Microbiota in Post-surgery Colorectal Cancer Patients. Front. Microbiol. 9:2777. doi: 10.3389/fmicb.2018.02777
Received: 08 May 2018; Accepted: 30 October 2018;
Published: 20 November 2018.
Edited by:
Nezar Al-hebshi, Temple University, United StatesReviewed by:
Stéphanie Olivier-Van Stichelen, National Institutes of Health (NIH), United StatesElizabeth P. Ryan, Colorado State University, United States
Copyright © 2018 Cong, Zhu, Liu, Li, Zhang, Zhu, Lv, Liu, Hao, Tian, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaochun Zhang, zhangxiaochun9670@126.com