 Hunter D. Whittington
Hunter D. Whittington Mahatam Singh2
Mahatam Singh2 M. Andrea Azcárate-Peril
M. Andrea Azcárate-Peril José M. Bruno-Bárcena
José M. Bruno-Bárcena- 1Department of Plant and Microbial Biology, North Carolina State University, Raleigh, NC, United States
- 2BASF Corporation, Research Triangle Park, NC, United States
- 3Department of Medicine, Division of Gastroenterology and Hepatology, and UNC Microbiome Core, Center for Gastrointestinal Biology and Disease, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
Pesticide-resistant plant pathogens are an increasing threat to the global food supply and have generated a need for novel, efficacious agrochemicals. The current regulatory process for approving new agrochemicals is a tedious but necessary process. One way to accelerate the safety evaluation process is to utilize in vitro systems to demonstrate pesticide degradation by soil microbes prior to ex vivo soil evaluations. This approach may have the capability to generate metabolic profiles free of inhibitory substances, such as humic acids, commonly present in ex vivo soil systems. In this study, we used a packed-bed microbial bioreactor to assess the role of the natural soil microbial community during biodegradation of the triazolopyrimidine fungicide, ametoctradin. Metabolite profiles produced during in vitro ametoctradin degradation were similar to the metabolite profiles obtained during environmental fate studies and demonstrated the degradation of 81% of the parent compound in 72 h compared to a half-life of 2 weeks when ametoctradin was left in the soil. The microbial communities of four different soil locations and the bioreactor microbiome were compared using high throughput sequencing. It was found that biodegradation of ametoctradin in both ex vivo soils and in vitro in the bioreactor correlated with an increase in the relative abundance of Burkholderiales, well characterized microbial degraders of xenobiotic compounds.
Introduction
The human population has rapidly increased since the development of farming; however, the current growth rate is unsustainable and will eventually become limited by food demands that cannot persist using our current farming practices (Alexandratos, 1999; Tilman et al., 2002). These demands are being tackled by the scientific and agricultural communities through a collaboration set up to improve crop production, plant breeding, and disease mitigation (Tilman et al., 2002). In particular, disease management is important for maintaining food security from pests that are responsible for an annual crop loss of 5–10% (Oerke, 2006). To help combat these losses farmers have spent billions of dollars on industrialized genetically-modified (GM) plants and agrochemicals with marginal success (Klümper and Qaim, 2014; Oristep Consulting, 2015; Atwood and Paisley-Jones, 2017), as common plant pests have adapted to resist both GM plants and agrochemicals, setting in motion the need for development and testing of novel agrochemicals, which require crucial environmental fate studies to guarantee safety prior obtaining regulatory certifications.
The Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA) is the most relevant regulatory statute and the compliance basis for all agrochemicals used in the United States. This regulation states that for safety before an agrochemical product can be approved for use it must be demonstrated that its residues, products of its degradation, and/or any other ingredients in the formulation do not cause adverse effects on human health and the environment (Federal Insecticide, Fungicide, and Rodenticide Act, 1910). These requirements present a unique challenge to agrochemical companies which must retrieve the agrochemical residues and products of their degradation from the soil for characterization and to show that these residues are not unreasonably harmful. This process can be costly and time-consuming, with environmental fate studies for single compounds lasting nearly a year (McDonald et al., 2006).
Currently, radiolabeling techniques coupled with advanced chemical analyses are used to determine the fate of agrochemicals in soils and plants (Soeda et al., 1972; McDonald et al., 2006). However, working with radiolabeled compounds is not always feasible, limiting residue analysis to more conventional methods such as UV detection combined with high performance liquid chromatography (HPLC-UV). An additional challenge encountered when working with soil samples arises from the presence of humic substances, which must be removed prior to the quantification of agrochemical residues, effectually raising the number of steps and the limit of detection (Masqué et al., 1998; Hogendoorn et al., 1999). Additionally, humic acids have been shown to reduce degradation efficiency in vitro (Zhao et al., 2018; Singh et al., 2019). Thus, ex vivo environmental fate and residue quantification studies could be improved by obtaining samples free of humic materials while still maintaining the sample’s biology.
While it is accepted that microorganisms provide the critical driving force behind degradation of xenobiotic compounds most studies are executed using in vivo (i.e., samples taken from agrochemical-treated soils at a research farm), ex vivo (i.e., soil cores removed from the environment and treated in the lab), and monoculture in vitro degradation experiments. All of these approaches present challenges such as environmental contamination, lengthy degradation times, or incomplete generation of metabolite fingerprints (Warhurst et al., 1994; van Ginkel, 1996; McDonald et al., 2006). One solution would be to use a controlled in vitro system to culture complex microbial consortia from soils. This strategy would facilitate the elimination of background signals coming from humic materials, allowing the rapid generation of “clean” agrochemical degradation profiles and providing detailed information regarding the specific community members responsible for actively participating in the degradation of the compound of interest. Although such a system cannot act as a replacement for field-based environmental fate studies, the metabolite profiles generated would allow for targeted downstream analyses.
In this study, we used a traditional ex vivo soil system as a comparison control to investigate the response of soil microbiome communities from four different locations challenged with the EPA-approved fungicide ametoctradin. Next, we validated an in vitro dynamic packed-bed bioreactor system (PBBR) to select for and culture complex microbial consortia capable of degrading the fungicide. This culturing technique generated a “clean” metabolite profile that was similar to the profile found in ex vivo soil experiments. Furthermore, we identified the microbial community members that were likely responsible for degradation of ametoctradin, subsequently making these members useful biomarkers for future field studies.
Results
Ametoctradin Biodegradation in an ex vivo Soil System
Soil samples were collected from four different locations previously utilized for ametoctradin environmental fate testing and regularly used for agricultural studies: California (CA), New Jersey (NJ), and Germany (LUFA 2.2 and LUFA 2.3) (Table 1). Roughly 1 kg of each soil sample was placed in two replicate containers linked to a CO2-scrubbed air source. Each sample was then either treated with 2.4 μg mL–1 of ametoctradin or remained untreated. After 14 days, the soil samples were harvested and analyzed in triplicate using HPLC-MS/MS. In each case, degradation of ametoctradin was confirmed along with the identification of four major metabolites: M650F01, M650F02, M650F03, and M650F04 (Figure 1). These compounds appeared to be the product of degradation by soil microorganisms capable of metabolizing the long aliphatic chain on the ametoctradin molecule while the amine ring structures remain largely unaltered. The NJ soil sample displayed the highest and most complete level of ametoctradin degradation, with only 13.6% of the original amount of agrochemical remaining after the 14-day period. The M650F03 metabolite appeared to be the main degradation product in every soil type.

Table 1. Soil samples used in this study.
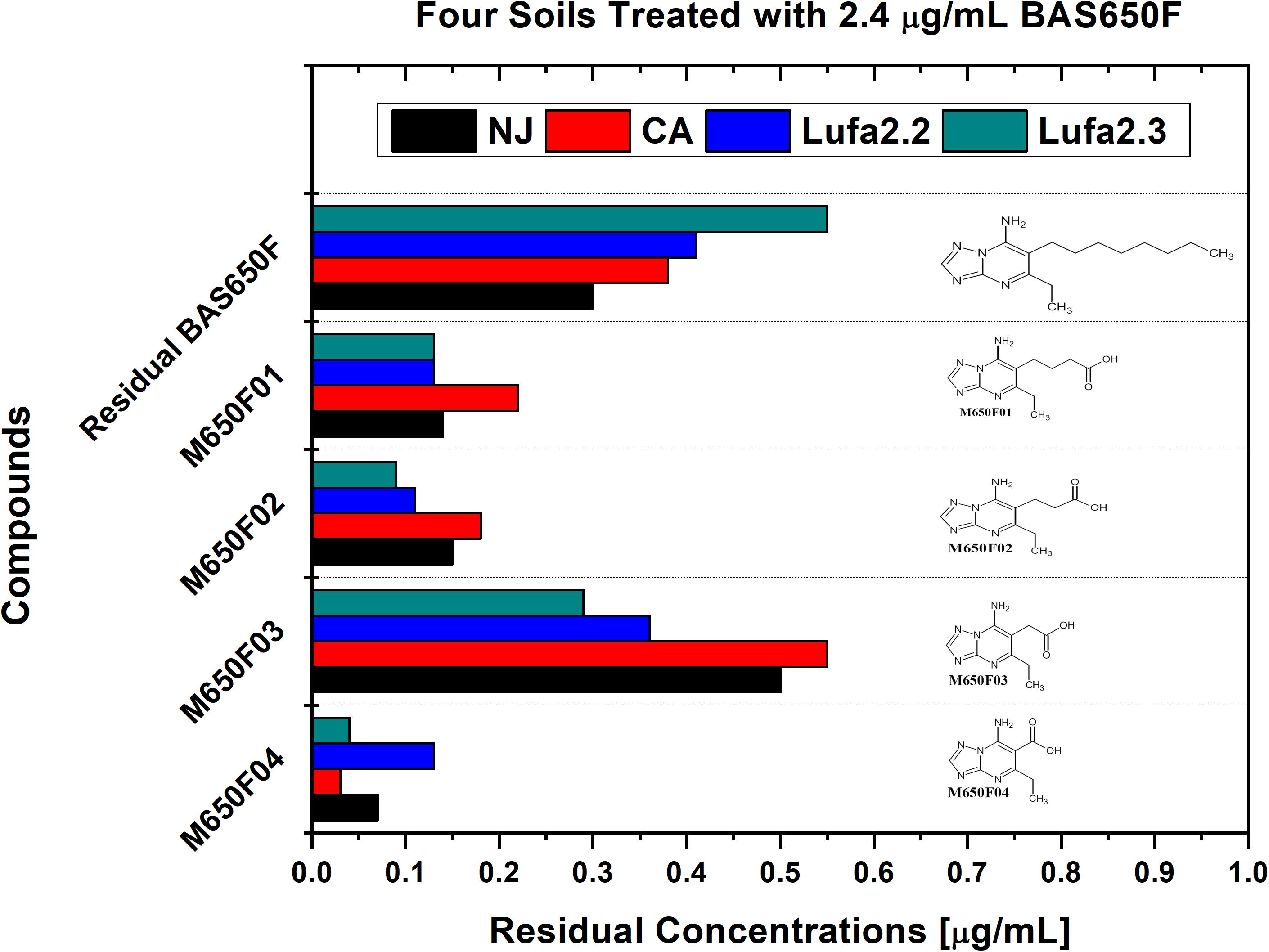
Figure 1. Residual concentrations of ametoctradin (BAS650F) and metabolites in soil. Residual concentrations of ametoctradin and its major metabolites in four treated soil samples. Concentrations of each compound were determined using HPLC-MS/MS. The chemical structure of each compound is shown to highlight the oxidized structure of the aliphatic side chain in the parent compound.
Microbial Community Composition of ex vivo Systems
Microbiome analysis of treated and untreated soils by 16S (Figure 2 and Supplementary Table S1) and 16S/18S (Supplementary Figure S2 and Supplementary Table S2) rRNA amplicon sequencing showed that Proteobacteria were the dominant bacterial phylum present in both the treated and untreated soils, representing up to 54% of the reads in untreated soils and 78% of the reads in treated soils. The Acidobacteria were also commonly identified, comprising up to 39% of the reads in untreated soils and 28% in treated soils. Within Eukaryotes, ametoctradin treatment only revealed significant reduction in the Oomycetes, particularly Phytophthora infestans, which are the target organisms for ametoctradin (Supplementary Table S2).
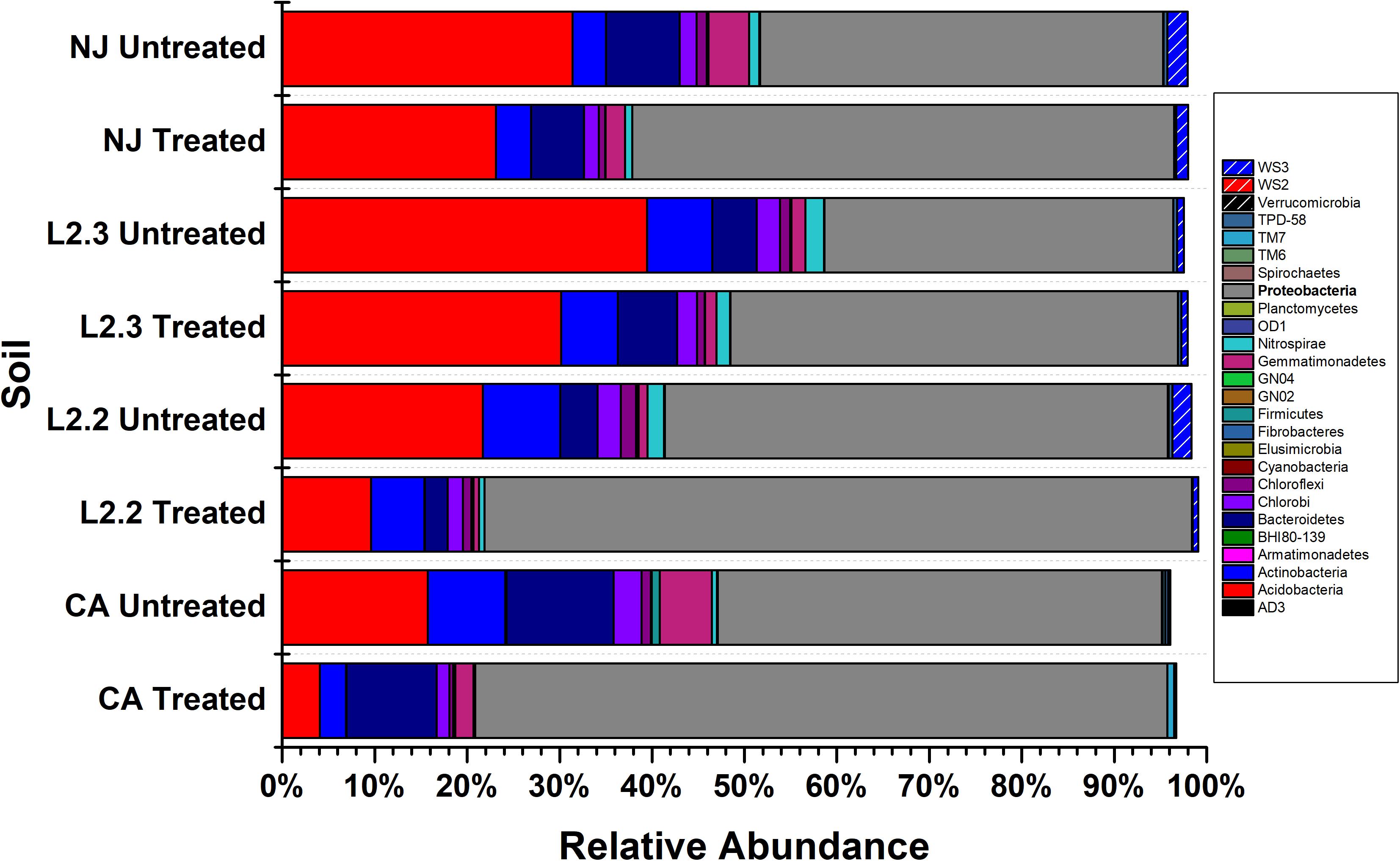
Figure 2. Relative abundance of bacterial phyla across soil samples. Treated soils were exposed to 2.4 μg mL–1 ametoctradin for 14 days. L2.2 and L2.3 represent LUFA 2.2 and LUFA 2.3 soil samples, respectively. Percent relative abundance is expressed as the mean relative abundance of three samples for each treatment.
Species richness varied between the four soil samples, while the most diverse soils were NJ and LUFA 2.3, with Shannon-Wiener Index (SWI) values around 8. The CA and LUFA 2.2 soils had SWI values of 7.75 and 7.3, respectively (Table 1). All soils showed a significant decrease in diversity when treated with ametoctradin (CA, NJ, LUFA 2.2: p < 0.0001, LUFA 2.3 p = 0.053). Moreover, as indicated in Supplementary Table S1, several OTUs increased in relative abundance after treatment. Those OTUs that increased in relative abundance include known degraders of xenobiotic compounds such as Mycobacterium, Rhodococcus, Comamonas, Burkholderia, Variovorax, Methylibium, and Pseudomonas. In fact, many of these genera were below the limit of detection in untreated soils. This increase in relative abundance after soil treatment suggested the presence of resistant organisms and may be connected to microbially mediated degradation (Berg et al., 2012; Jia et al., 2015).
Soil microbial community similarities and differences have been highlighted by beta diversity analysis (Figure 6A). Although the number of samples did not allow for statistical analysis, the data showed that untreated soils tended to cluster together while treated soil sample communities tended to diverge from each other, suggesting that ametoctradin treatment on soil microbial communities varied according to the location of the soil.
Configuration of the Packed-Bed Bioreactor System
Analysis of ex vivo systems showed that M650F01, M650F02, M650F03, and M650F04 were the main products of degradation of BASF650F and identified several species including Mycobacterium, Rhodococcus, Comamonas, Burkholderia, Variovorax, Methylibium, and Pseudomonas as relevant participants in the degradation process. We next implemented an in vitro continuous up-flow PBBR system to culture the complex microbial communities present in the soils in an attempt to confirm and reproduce data from ex vivo systems. The PBBR system consisted of a water-jacketed glass column packed with expanded glass beads. Mixing was achieved using a recirculation loop and a peristaltic pump operated at a recycling ratio of 50:1. ametoctradin was added to the medium at a maximum soluble concentration of 148 μg L–1. Finally, continuous addition of fresh medium containing ametoctradin was performed at five dilution rates over the course of the experiment (see section “Materials and Methods”).
Degradation of Ametoctradin in the Packed-Bed Bioreactor
Since NJ soil samples contained the highest number of taxa and the highest diversity values, the ametoctradin-treated NJ soil was chosen to inoculate the PBBR. After inoculation, the reactor was maintained under continuous culture conditions for 1 month to permit enrichment and selection of taxa. After this maturation period, the reactor was subjected to five different dilution rates (0.03, 0.05, 0.06, 0.07, and 0.08 h–1) over the course of the experiment. Sequencing confirmed that no eukaryotic taxa remained in the reactor after maturation, indicating that any ametoctradin degradation observed in the reactor was entirely due to prokaryotic organisms.
Once the culture reached steady-state conditions, liquid samples were taken at retention time intervals and residual levels of ametoctradin, and its degradation products, were monitored by LC-MS/MS. The community structure was also analyzed at those times by 16S rRNA amplicon sequencing. Following this period of maturation, the reactor was sequentially exposed to five sequentially increasing dilution rates (0.03, 0.05, 0.06, 0.07, and 0.08 h–1) of ametoctradin-containing medium over the course of the experiment. Sequencing of rRNA amplicons from samples obtained at each dilution rate confirmed the washout and absence of eukaryotic OTUs in the reactor after maturation. Nearly complete degradation of ametoctradin was observed in the reactor due to prokaryotic activity. At the lower dilution rates (0.03, 0.05, and 0.06 h–1), residual levels of ametoctradin were low (4.3 ± 0.89 μg L–1). When the dilution rate was increased to 0.07 and 0.08 h–1, the residual levels of ametoctradin also increased, reaching a maximum concentration of 15.2 μg L–1 (Figure 3). These variations in residual levels of ametoctradin matched shifts in the reactor microbial community, with the Burkholderiales, particularly Comamonas, dominating the community at the lower dilution rates (0.03, 0.04, and 0.05 h–1). A shift towards members of the Clostridiales, specifically Clostridium, was observed at the higher dilution rates (0.06 h–1 and above) (Figure 3 and Supplementary Table S3). These results indicated that Burkholderiales may be key to the ametoctradin biodegradation process.
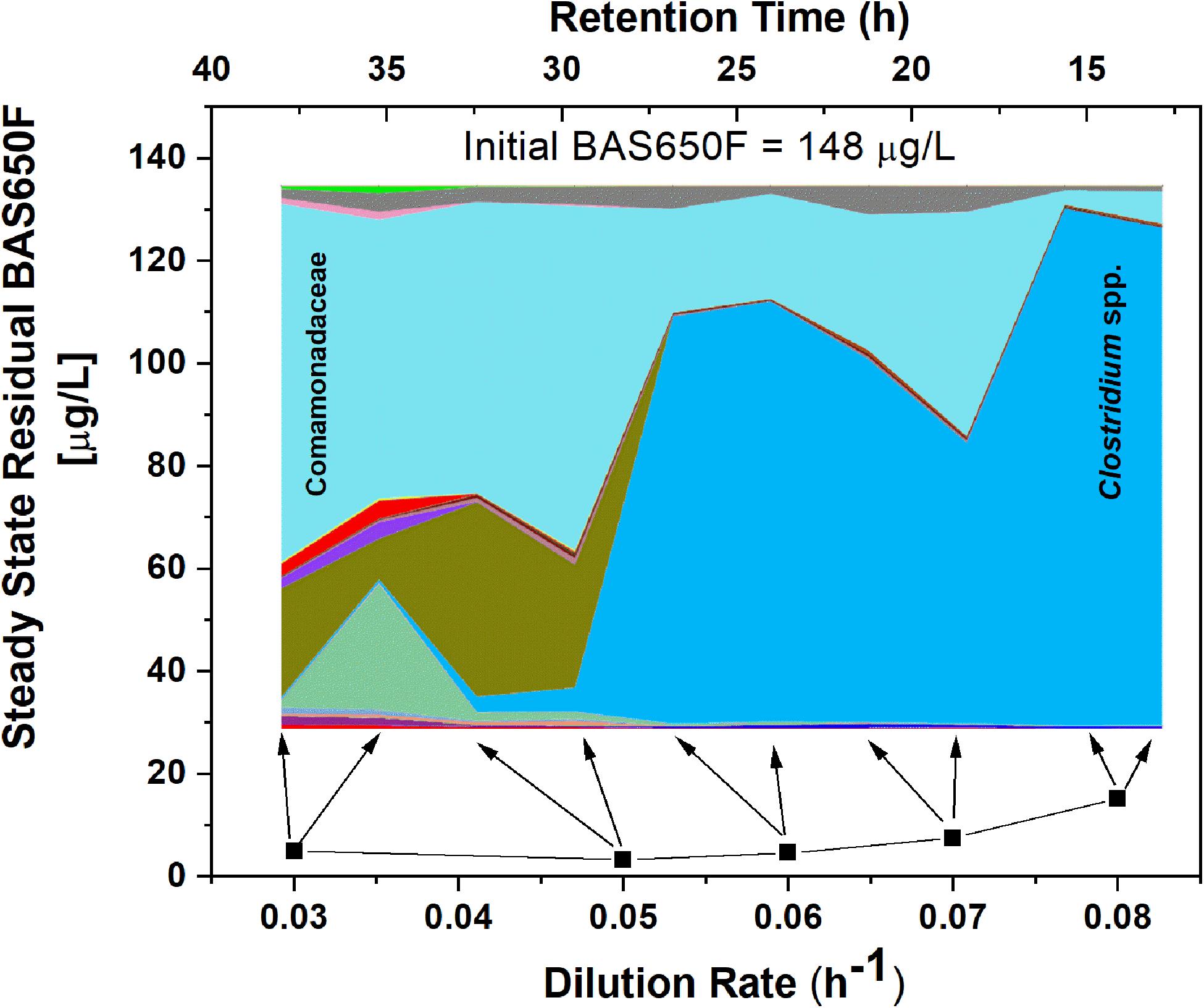
Figure 3. OTU relative abundance and residual ametoctradin (BAS650F) at steady-state. The area chart was generated in QIIME using OTU relative abundance data, with colors representing the relative abundance of a particular OTU. The highly abundant Comamonadaceae and Clostridium spp. OTUs are labeled as a reference. Residual ametoctradin levels shown are the average of two samples taken at steady-state for each respective dilution rate. Arrows indicate the two points on the area chart corresponding to these two samples at steady-state.
To confirm that the low residual levels of ametoctradin found in the reactor were due to microbial degradation, we performed a pulse experiment in which we quantified the levels of ametoctradin and its metabolites using LC-MS/MS at various sampling points over a period of 83 h. This data was then compared against a model for theoretical washout in which the compound would slowly be removed from the system at predictable intervals based on the dilution rate. It is important to note that the pulse of ametoctradin into the reactor was well above its limit of solubility in water, and the rapid decrease in ametoctradin levels in the first 8 h is an artifact of this limitation (Figure 4). However, increased levels of metabolites M650F01, M650F03, and M650F04 were detected in the samples, indicating active biodegradation. Additionally, the levels of ametoctradin remained relatively stable around 0.148 μg mL–1 (limit of solubility in water) for a period of 42 h, suggesting that the additional insoluble portion from the pulse was being solubilized as degradation of the parent compound was occurring.
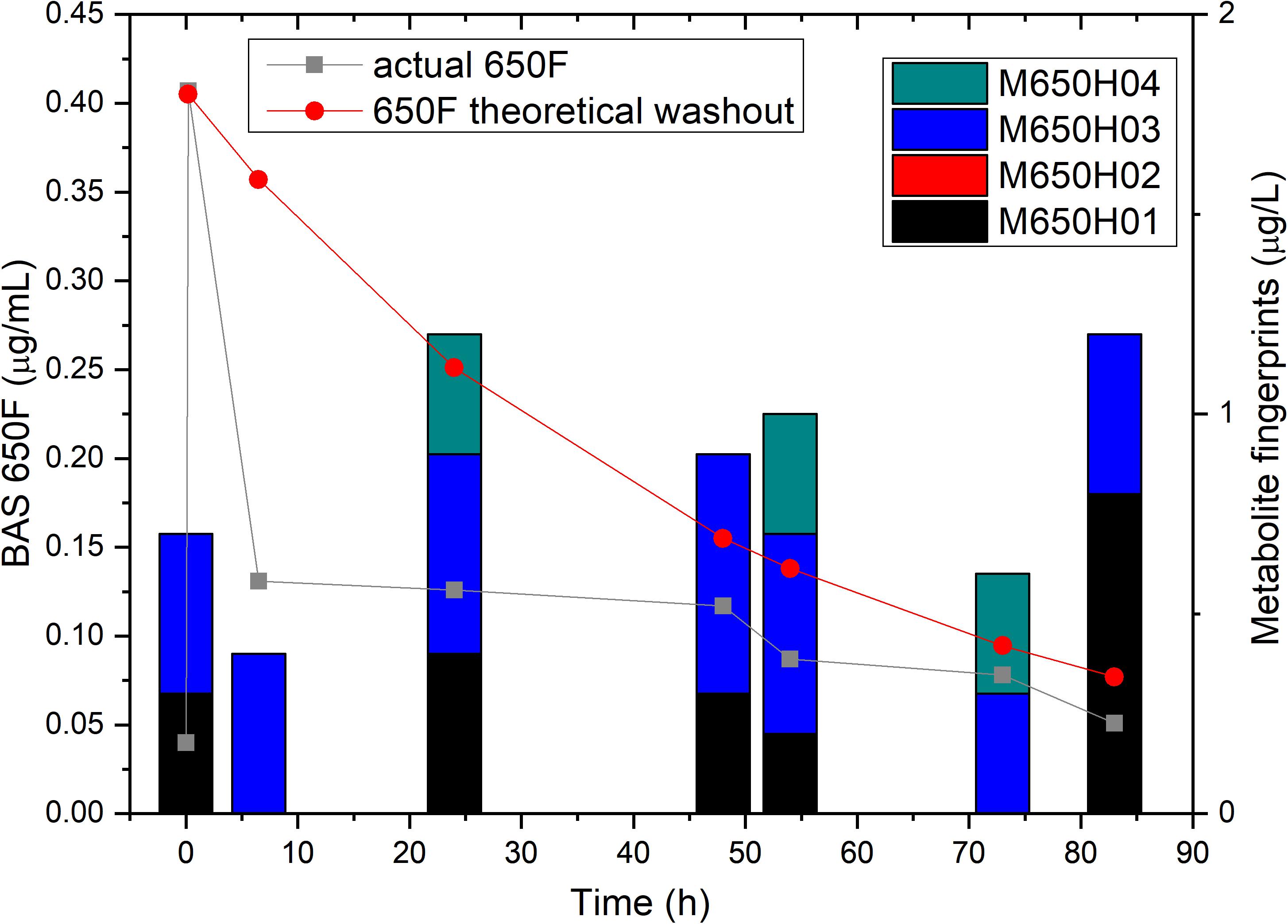
Figure 4. Ametoctradin (BAS650F) pulse experiment. A pulse of 0.41 μg mL–1 ametoctradin was performed at time 0, with samples taken immediately before and after the pulse to confirm initial levels of parent and metabolites. Samples were taken at various intervals up to 83 h post pulse. At each sampling point, levels of ametoctradin and its four major metabolites were analyzed using HPLC-MS/MS. A theoretical washout curve based on reactor parameters was calculated and plotted to confirm degradation. Residual ametoctradin levels are expressed in μg mL–1, while metabolite levels are expressed in μg L–1.
In vitro Composition of the New Jersey Soil-Derived Microbial Community
Microbiome analysis of the soil-derived community showed that abundance of members of the Betaproteobacteria continued to increase up to a dilution rate of 0.06 h–1 after which they begin to wash out of the system (Figure 5 and Supplementary Table S3). A Comamonas sp. was particularly abundant at these dilution rates, where it comprised nearly 60% of the reads. At higher dilution rates (0.07 and 0.08 h–1), it is apparent that members of the Firmicutes, particularly Clostridium spp., begin to dominate the microbial community in the bioreactor. Beta diversity analysis of the soil-derived communities sampled at different dilution rates showed clear differences between soil and soil-derived populations, with treated and untreated soil samples clustering separately from the PBBR samples. Likewise, samples collected from low dilution rate conditions were grouped separately from high dilution rate condition samples illustrating the dynamics of the microbial community (Figure 6). Community shifts were likely due to the changes in relative abundance in Betaproteobacteria and Clostridia detailed above. We also observed that the disappearance of Acidobacteria, a highly abundant phylum in the soil samples, in the PBBR did not hinder the ability of the consortium to degrade ametoctradin, supporting our hypothesis that Burkholderiales were the major drivers of ametoctradin degradation.
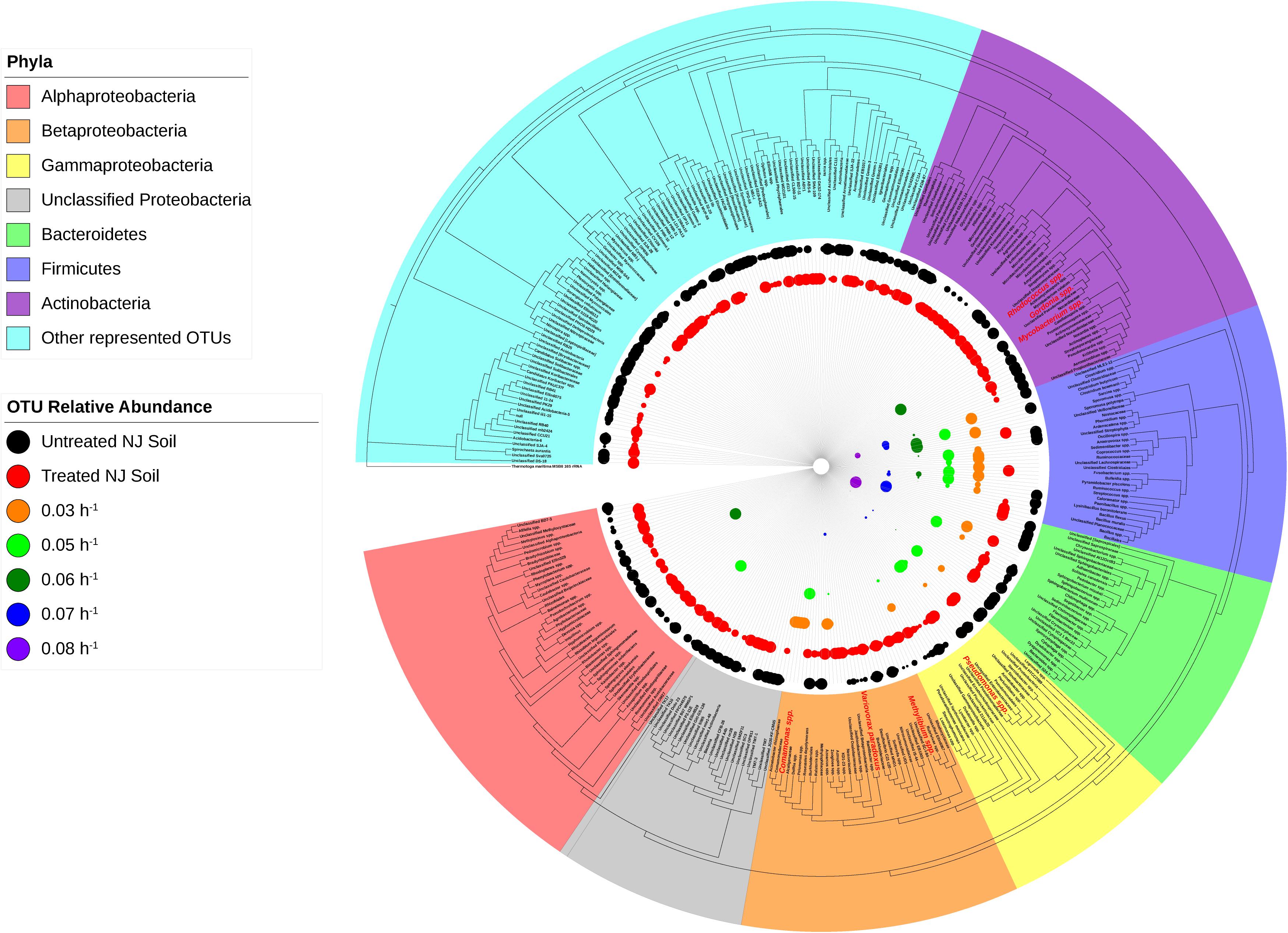
Figure 5. Phylogeny and relative abundance in the bioreactor and in New Jersey soil sample. Phylogenetic tree was generated in Geneious using the PHYML algorithm from the set of representative sequences generated by QIIME. The diameter of each circle is directly correlated to the percent relative abundance of the OTU, with larger circles representing higher relative abundance. OTUs with a relative abundance of <0.3% have circles of a diameter too small to be displayed. OTUs highlighted in red are well-known degraders of xenobiotic compounds.
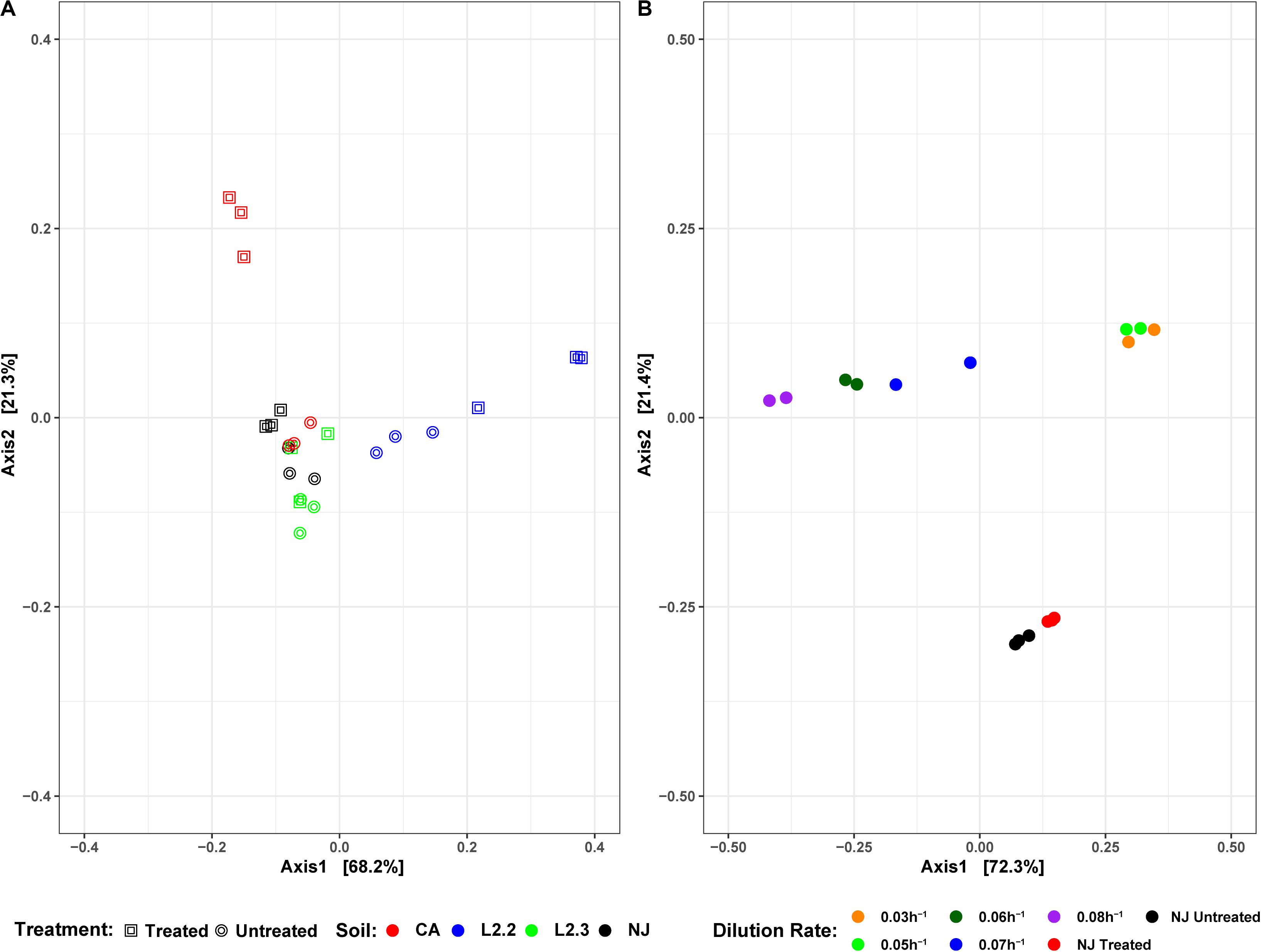
Figure 6. DPCoA plots for each soil sample, treatment, and reactor dilution rate. Panel (A) presents both the untreated and treated samples for each soil, while panel (B) presents the samples from each reactor dilution rate, as well as the NJ Treated and Untreated samples. Plots were generated using the vegan package in R 3.5.1. Axis 1 and Axis 2 represent principal components 1 and 2, respectively.
Predicted Metabolic Pathways Impacted by Ametoctradin
Using the predictive software PICRUSt, which permits the determination of metabolic potential from 16S rRNA amplicon sequencing data, we analyzed the KEGG gene pathways associated with degradation of xenobiotic compounds in ex vivo and in vitro systems (Figure 7). The only pathway that showed a significant difference in relative abundance between treatments in all three soils was caprolactam degradation. This pathway contains several enzymes that may participate in the oxidation of the aliphatic side chain present on ametoctradin, including alkane 1-monoxygenase (EC 1.14.15.3), NADP-dependent aldehyde dehydrogenase (EC 1.2.1.4), and NADP-alcohol dehydrogenase (EC 1.1.1.2). The LUFA 2.2 soil showed the largest number of changes in metabolic potential between non-treated and treated samples, with 15 out of 18 pathways examined showing significantly higher gene counts in the treated soil. Specifically, gene predictions related to bisphenol, chloroalkane, fluorobenzene, naphthalene, and styrene degradation only showed significant increases in the LUFA 2.2 soil. Several of these pathways include decyclizing enzymes such as catechol dioxygenase (ECs 1.13.11.1 and 1.13.11.2) which might participate in the degradation of the ring structure in ametoctradin.
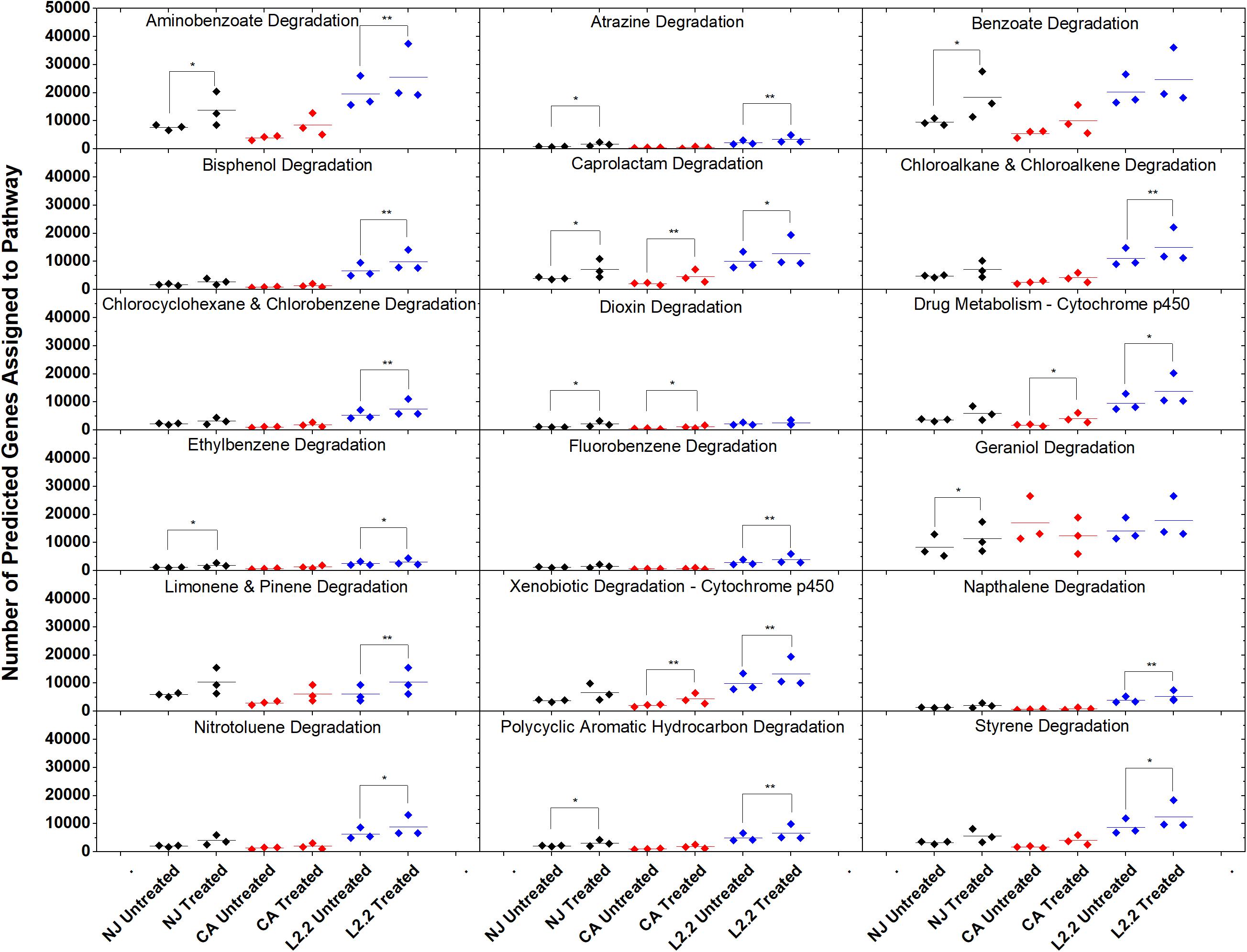
Figure 7. Gene counts in KEGG pathways associated with xenobiotic degradation in soils. Gene counts are determined in PICRUSt by first finding sequenced representatives for each OTU, summing the number of genes involved in a pathway for that OTU, multiplying this number by the relative abundance of the OTU, and finally by summing across OTUs. Boxplots were created in Origin 9.1 by pooling three replicates for each soil and treatment. Statistics on metagenome profiles were performed in STAMP. Each sample is represented with a diamond. Colored lines represent the mean of the three samples. Brackets with a single asterisk correspond to a p-value of <0.05, while two asterisks correspond to a p-value of <0.01.
In vitro analysis of changes in metabolic potential showed that the dilution rates impacted a number of genes for each category that were directly correlated with changes in the relative abundance of Betaproteobacteria, specifically a Comamonas species (Figure 8). Upon examination of the Comamonas genomes in the KEGG database, we found that all five annotated genomes contained decyclizing dioxygenases such as catechol dioxygenase (ECs 1.13.11.1 and 1.13.11.2) and protocatechuate dioxygenase (EC 1.13.11.8) (Kanehisa and Goto, 2000). This abundance of ring-opening enzymes may help to explain the low amounts of ametoctradin and its metabolites in the reactor effluent.
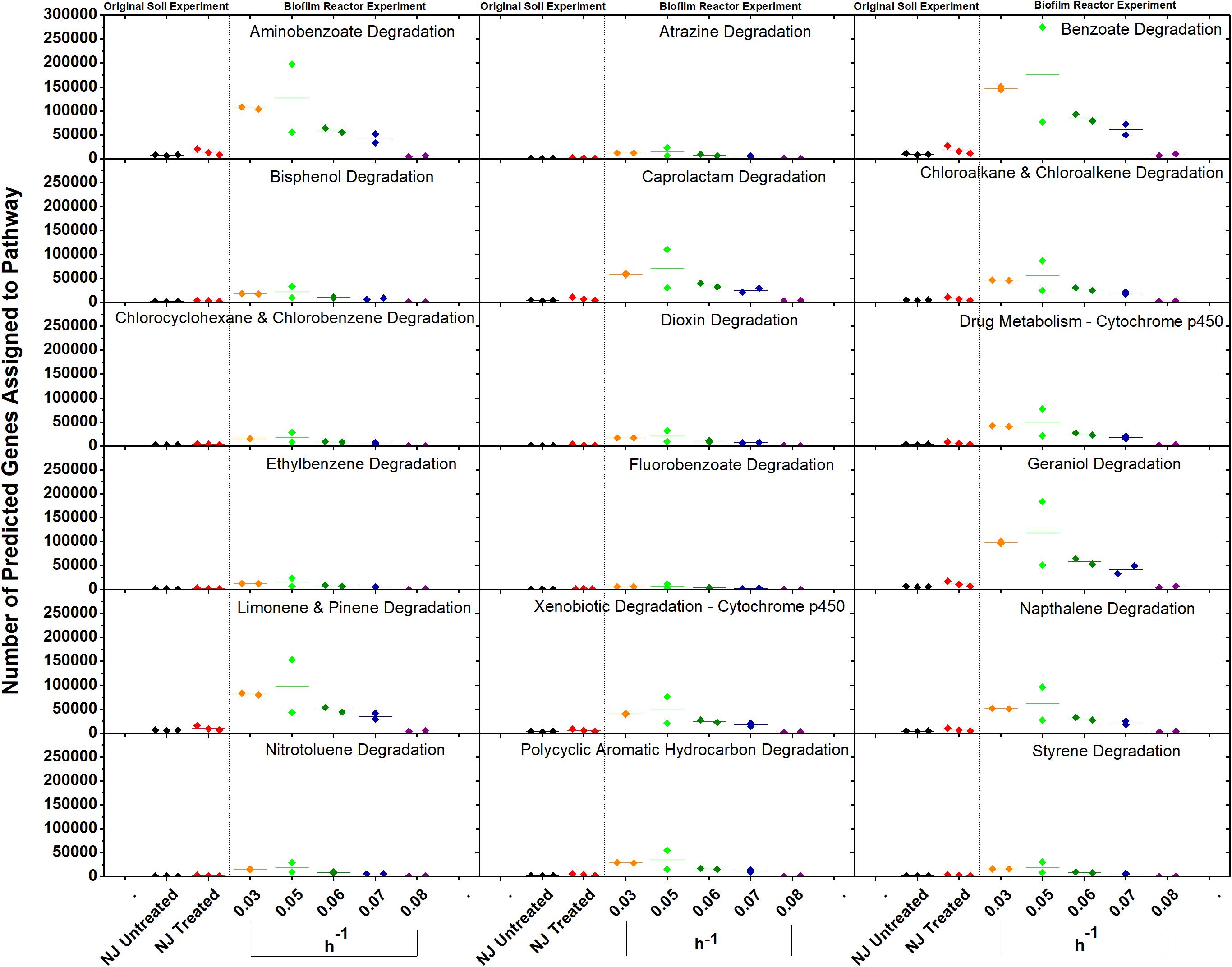
Figure 8. Gene counts in KEGG pathways associated with xenobiotic degradation in the bioreactor. Gene counts are determined in PICRUSt by first finding sequenced representatives for each OTU, summing the number of genes involved in a pathway for that OTU, multiplying this number by the relative abundance of the OTU, and finally by summing across OTUs. Boxplots were created in Origin 9.1 by pooling three replicates for each soil and two replicates for each dilution rate. A dashed line separates the soil and bioreactor experiments in each panel to stress the fact that each graph contains data from two separate experiments. Dilution rates are expressed in h–1.
Discussion
This study examined the ability of an in vitro dynamic PBBR to simulate agrochemical metabolite degradation profiles found in ex vivo soil experiments. Additionally, we analyzed the microbiome of both soil and bioreactor samples to identify the members of the microbial community who responded to treatment with an EPA-approved agrochemical, ametoctradin, and potentially participated in its degradation.
Notably, the packed-bed reactor system was able to accelerate the degradation of ametoctradin. Packed-bed bioreactors have been used for decades for microbial biodegradation of environmental contaminants and compounds of interest, as well as for the production of value-added products such as ethanol and lactic acid (Shiotani and Yamané, 1981; Strandberg et al., 1989; Bruno-Bárcena et al., 1999; Alfonso-Gordillo et al., 2016; Geed et al., 2017; Mavriou et al., 2020). It has been shown previously that sessile microbial cells are more resistant to toxic substances than planktonic cells, making packed-bed bioreactors attractive for studying the degradation of xenobiotic compounds (Parkin and Speece, 1983). Additionally, packed-bed bioreactors have several advantages over stirred-tank bioreactors including compactness, resistance to temperature fluctuations, and limited biomass washout (Sagberg et al., 1992). In the soil, this compound exhibits a half-life of approximately 14 days, whereas 81% of the compound had degraded in 72 h in the bioreactor. Furthermore, 3 of the 4 major metabolites were detected in small amounts in each of the samples, indicating rapid assimilation or mineralization to CO2 (Figure 4). Additionally, the culturing medium was free of compounds that may interfere with the analysis of ametoctradin and its metabolites, allowing for a simple and rapid downstream analysis.
The degradative ability of the members of the Burkholderiales has been well documented in the literature (Pérez-Pantoja et al., 2012). Our data demonstrated that members of the Burkholderiaceae and Comamonadaceae, including Comamonas, Variovorax, and Methylibium, are highly abundant in samples that showed evidence of ametoctradin degradation. These species have been well studied and are commonly associated with xenobiotic degradation (Vallaeys et al., 1998; Nakatsu et al., 2006; Satola et al., 2012; Chen et al., 2016a, b; Öztürk et al., 2020; Zhang et al., 2020). In the packed-bed reactor, Comamonas spp. account for nearly 60% of sequence reads at the dilution rates 0.03 and 0.05 h–1 (Supplementary Table S3). The high levels of Comamonas spp. at these dilution rates also coincides with lower residual levels of ametoctradin in the reactor effluent, likely indicating that Comamonas is largely responsible for ametoctradin degradation in the reactor (Figure 3). Additionally, several Actinomycetes, including Gordonia, Mycobacterium, and Rhodococcus were identified in soil and bioreactor samples via their 16S rRNA gene sequences (Figure 5 and Supplementary Tables S1, S3). These genera have been extensively studied and have been shown to be excellent degraders of various xenobiotic compounds (Burback and Perry, 1993; Arenskötter et al., 2004; Chen et al., 2016b). Interestingly, an unclassified OTU from the family Lachnospiraceae, a part of the Clostridia, was highly abundant in the 0.03 and 0.05 h–1 conditions, comprising 25 and 30% of reads, respectively (Supplementary Table S3). There are reports of Lachnospiraceae degrading hydrocarbons (Schouw et al., 2016), however, this family are mainly known for their cellulolytic activity in ruminants (Zhang et al., 2017). Considering that cellulosic compounds are present in the culture medium and that ruminant manure is a common fertilizer in agriculture, this Lachnospiraceae group is likely not contributing to ametoctradin degradation.
Furthermore, we examined the impact of ametoctradin treatment on archaea and eukaryotes, since it is well understood that fungi play a noteworthy role in the biodegradation of xenobiotic compounds (Bending et al., 2002; Gupta et al., 2017; Manasfi et al., 2020). Overall, these populations were quite stable, with significant changes only occurring in the oomycetes, which are the target organisms of ametoctradin (Supplementary Figure S2 and Supplementary Table S2). No archaeal or eukaryotic sequences were detected in the bioreactor, indicating that bacteria mediated all degradation occurring therein.
Our PICRUSt-generated metagenomes also indicated the possible presence of genes associated with xenobiotic degradation, including monooxygenases and dioxygenases, hydrolases, cytochromes p450 and more, many of which are produced by microorganisms found in our packed-bed reactor, such as Comamonas (Rhee et al., 2004). Due to the predicted presence of many of these genes, this system would likely be useful in examining the microbial degradation of many different classes of xenobiotic compounds. A packed-bed system would also be ideal for examining the ability of a defined microbial consortium to degrade various other xenobiotic compounds in a controlled environment while also allowing for complete compound accountability.
Here, we demonstrated the ability of a naturally sourced microbial consortium to degrade the triazolopyrimidine fungicide ametoctradin and generate metabolite profiles in vitro. Additionally, we presented a novel usage of a PBBR that facilitated cleaner downstream analysis of agrochemical degradation. This system may also provide researchers with future opportunities for a more intimate analysis of microbial community dynamics in the degradation of xenobiotic compounds.
Materials and Methods
Collection and Treatment of Soil Samples
Two soil samples from the United States and two samples from Germany were collected for ex vivo degradation analysis (Table 1). Each of the four soil samples were treated with 2.4 mg kg–1 soil of ametoctradin and allowed to react for a total of 14 days. After this incubation period, the soils were analyzed following established methods for residual ametoctradin and its associated metabolites (M650F01, M650F02, M650F03, M650F04) using HPLC-UV and HPLC-MS/MS, respectively (European Food Safety Authority, 2012).
Sequencing of Soil and Reactor Samples
Amplification of the V1–V2 and V3–V4 regions of the bacterial 16S rRNA gene were performed on total DNA from soil samples. Master mixes for these reactions used the Qiagen HotStar Hi-Fidelity Polymerase Kit (Qiagen, Valencia, CA, United States) with a forward primer composed of the Roche Titanium Fusion Primer A (5′-CCATCTCATCCCTGCG TGTCTCCGACTCAG-3′), a 10 bp Multiplex Identifier (MID) sequence (Roche, Indianapolis, IN, United States) unique to each of the samples. The gene-specific part of the primer used the universal bacterial primer 8F (5′-AGAGTTTGATCCTGGC TCAG-3′) to amplify V1–V2 region or 515 F bacterial primer (5′-GTGCCAGCMGCCGCGGTAA-3′) to amplify V3–V4 region (Edwards et al., 1989; Fierer et al., 2008; Caporaso et al., 2011; Walters et al., 2016). The reverse primer was composed of the Roche Titanium Primer B (5′-CCTATCCCCTGT GTGCCTTGGCAGTCTCAG-3′), the identical 10 bp MID sequence as the forward primer and the reverse bacterial primer 338R (5′-GCTGCCTCCCGTAGGAGT-3′), which span the V1–V2 hyper variable region of the bacterial 16S or 806R bacterial primer (5′-GGACTACHVGGGTWTCTAAT-3′) for the amplification of V3–V4 region. The thermal profile for the amplification of both V1–V2 and V3–V4 regions had an initial denaturing step at 94°C for 5 min, followed by a cycling of denaturing of 94°C for 45 s, annealing at 50°C for 30 s and a 1 min 30 s extension at 72°C (35 cycles), a 10 min extension at 72°C and a final hold at 4°C. Each sample was individually gel purified using the Qiaquick Gel Extraction Kit (Qiagen, Valencia, CA, United States) and E-Gel Electrophoresis System (Life Technologies, Invitrogen division), and standardized prior to pooling. Pooled barcoded libraries were sequenced on a Roche 454 Genome Sequencer FLX Titanium instrument (Microbiome Core Facility, Chapel Hill, NC, United States) using the GS FLX Titanium XLR70 sequencing reagents and protocols. Initial data analysis, base pair calling and trimming of each sequence to yield high quality reads were performed by Research Computing at the University of North Carolina at Chapel Hill (Chapel Hill, NC, United States). Sequence data were cleaned and analyzed using Version 1.9 of QIIME software (Caporaso et al., 2010). Sequences are available on the NCBI GenBank under SRA accession number PRJNA625701.
Building and Annotating Phylogenetic Trees
Redundant sequences were removed from the set of representative sequences using the PhyloToAST (Dabdoub et al., 2016) plugin for QIIME. The trimmed set of representative sequences was aligned using the MUSCLE algorithm in Geneious 5.5.9 (Biomatters Inc., Newark, NJ, United States). Maximum likelihood phylogenetic trees were generated using the PHYML plugin for Geneious. Sequence ID numbers on leaves were replaced with taxon information using PhyloToAST (Dabdoub et al., 2016). Trees were annotated with relative abundance information using the Interactive Tree of Life (iToL) software (Letunic and Bork, 2011). Relative abundance data were generated by QIIME and normalized using the cumulative sum scaling (CSS) method and the normalize_table.py command in QIIME for each project (Paulson et al., 2013). Mean relative abundance was calculated from the mean of the triplicate data points for each sample and repeated for each of the different treatments using PhyloToAST.
Construction and Analysis of Metagenomic Profiles
In order to obtain a rough metagenome for each soil treatment and bioreactor condition, we ran PICRUSt analysis (Langille et al., 2013). The input file used in PICRUSt was a closed-reference OTU table generated for both the soil and bioreactor experiments in QIIME. Predicted metagenomes were statistically analyzed in STAMP (Parks et al., 2014).
Bioreactor Enrichment Experiment
The bioreactor enrichment experiment was performed in a packed-bed reactor consisting of a single water-jacketed glass column (6 cm diameter by 13 cm height) filled with porous foam glass particles having a mean diameter of 4–8 mm with a porosity of 55%, and a density of ∼0.2 g cm–3 (A generous gift from Poraver GmbH). The column had an external loop that consisted of an input for the growth medium, a peristaltic pump controlling the dilution rate, a port from which to take samples, and a harvest tank and associated peristaltic pump. A schematic of the PBBR configuration can be found in the Supplementary Figure S3. The reactor was fed with medium containing 148 μg L–1 ametoctradin (maximum solubility in water); a full description of medium composition can be found in the Cultivation Medium section “Materials and Methods” below. This medium was recycled within the reactor at ratio of 50:1. The final active volume for the duration of the experiment was 220 mL. The temperature during the experiment was controlled via water jacket at 24°C. The pH was monitored but not controlled and varied between 4.8 and 6.7. Inoculum for the experiment consisted of 1 g of ametoctradin treated NJ soil. After inoculation, the in vitro microbial community was allowed to mature and stabilize for 1 month at a dilution rate of 0.03 h–1 before adjustments were made to the dilution rate. Over the next 3 weeks the reactor’s microbial community was subjected to five different sequential dilution rates (0.03, 0.05, 0.06, 0.07, and 0.08 h–1). For each dilution rate, performed for at least a three retention times (RT) (period of adaptation), three samples were taken and stored for sequencing and HPLC-UV or HPLC-MS/MS analysis. The samples represent the mature microbial community that has been influenced by the presence of ametoctradin.
Ametoctradin Pulse Experiment
To determine if residual levels of ametoctradin and products of its degradation in the bioreactor were microbially mediated degradation and not washout, an experiment injecting 0.41 μg mL–1 ametoctradin was performed while maintaining the reactor at a dilution rate of 0.03 h–1. Samples were taken at 0.2, 6.5, 24, 48, 54, 73, and 83 h to monitor levels of ametoctradin and its metabolites. Actual data analyzed by HPLC-UV or HPLC-MS/MS were compared to theoretical values obtained from a mathematical model for washout at a given dilution rate. The formula for theoretical substrate concentration at time t [(Y)t] is as follows:
where C0 and V0 represent the initial pulse concentration and volume, VR is the total reactor volume, and D is the reactor dilution rate.
Culture Media
The medium used during these experiments contained (g L–1) yeast extract 1, ammonium acetate 2, carboxymethyl cellulose 2, dextrose 1, cellobiose 0.1, xylose 0.771, K2HPO4 0.8, KH2PO4 0.2, CaCl2⋅H2O 0.09, FeSO4⋅7H2O 0.0028, MgSO4⋅7H2O 0.2, MnSO4⋅H2O 0.003, NaCl 0.1, NaMoO4⋅2H2O 0.00025, ZnSO4⋅7H2O 0.00025, Biotin 0.0001, Pyridoxol⋅HCl 0.0002, and 0.01 mM EDTA.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA625701.
Author Contributions
HW, MA-P, and JB-B wrote and interpreted the data in the manuscript. JB-B and MA-P designed and executed the experiments. CT and MS analyzed using HPLC-UV or HPLC-MS/MS the samples presented in the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project was partially supported by a generous gift from BASF Corporation and by the North Carolina State University Department of Plant and Microbial Biology. HW was supported by the NC State Graduate Student Support Plan. BASF funds were utilized to conduct the experiments, HPLC-MS/MS sample analysis at BASF, and the microbiome analysis by the UNC Microbiome Core Facility (grant no. NIH P30 DK34987). The funder bodies were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
CT and MS were employed by the BASF Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Shareef M. Dabdoub for his help with Python and PhyloToAST. The authors would also like to thank Drs. Harald Seulberger and Laura Sears of BASF for the partial financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01898/full#supplementary-material
FIGURE S1 | Phylogeny and relative abundance of bacterial sequences in each of the four soils examined. Phylogenetic tree was generated in Geneious using the PHYML algorithm from the set of representative sequences generated by QIIME. The diameter of each circle is directly correlated to the percent relative abundance of the OTU, with larger circles representing higher relative abundance. OTUs with a relative abundance of <0.3% have circles of a diameter too small to be displayed. OTUs highlighted in red are well-known degraders of xenobiotic compounds.
FIGURE S2 | Phylogeny and relative abundance of archaeal and fungal sequences in each of the four soil samples examined. Phylogenetic tree was generated in Geneious using the PHYML algorithm from the set of representative sequences generated by QIIME. The diameter of each circle is directly correlated to the percent relative abundance of the OTU, with larger circles representing higher relative abundance. OTUs with a relative abundance of <0.3% have circles of a diameter too small to be displayed. The Oomycetes, highlighted by the black box, are the target organisms of ametoctradin.
FIGURE S3 | A schematic of the packed-bed bioreactor. Arrows indicate the direction of flow through the system. The speed of the harvest pump and the recirculation pump were constant, while the speed of the feed pump was varied according to the dilution rate examined.
References
Alexandratos, N. (1999). World food and agriculture: Outlook for the medium and longer term. Proc. Natl. Acad. Sci. U.S.A. 96, 5908–5914. doi: 10.1073/pnas.96.11.5908
Alfonso-Gordillo, G., Flores-Ortiz, C. M., Morales-Barrera, L., and Cristiani-Urbina, E. (2016). Biodegradation of methyl tertiary butyl ether (MTBE) by a microbial consortium in a continuous up-flow packed-bed biofilm reactor: kinetic study, metabolite identification and toxicity bioassays. PLoS One 11:e0167494. doi: 10.1371/journal.pone.0167494
Arenskötter, M., Bröker, D., and Steinbüchel, A. (2004). Biology of the Metabolically Diverse Genus Gordonia. Appl. Environ. Microbiol. 70, 3195–3204. doi: 10.1128/AEM.70.6.3195-3204.2004
Atwood, D., and Paisley-Jones, C. (2017). Pesticides Industry Sales and Usage: 2008-2012 Market Estimates. Available online at: https://www.epa.gov/sites/production/files/2017-01/documents/pesticides-industry-sales-usage-2016_0.pdf (accessed April 18, 2017).
Bending, G. D., Friloux, M., and Walker, A. (2002). Degradation of contrasting pesticides by white rot fungi and its relationship with ligninolytic potential. FEMS Microbiol. Lett. 212, 59–63. doi: 10.1111/j.1574-6968.2002.tb11245.x
Berg, J., Brandt, K. K., Al-Soud, W. A., Holm, P. E., Hansen, L. H., Sørensen, S. J., et al. (2012). Selection for Cu-tolerant bacterial communities with altered composition, but unaltered richness, via long-term Cu exposure. Appl. Environ. Microbiol. 78, 7438–7446. doi: 10.1128/AEM.01071-12
Bruno-Bárcena, J. M., Ragout, A. L., Cordoba, P. R., and Sineriz, F. (1999). Continuous production of L (+)-lactic acid by Lactobacillus casei in two-stage systems. Appl. Microbiol. Biotechnol. 51, 316–324. doi: 10.1007/s002530051397
Burback, B. L., and Perry, J. J. (1993). Biodegradation and biotransformation of groundwater pollutant mixtures by Mycobacterium vaccae. Appl. Environ. Microbiol. 59, 1025–1029. doi: 10.1128/aem.59.4.1025-1029.1993
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chen, K., Xu, X., Zhang, L., Gou, Z., Li, S., Freilich, S., et al. (2016a). Comparison of four Comamonas catabolic plasmids reveals the evolution of pBHB to catabolize haloaromatics. Appl. Environ. Microbiol. 82, 1401–1411. doi: 10.1128/AEM.02930-15
Chen, Y.-L., Wang, C.-H., Yang, F.-C., Ismail, W., Wang, P.-H., Shih, C.-J., et al. (2016b). Identification of Comamonas testosteroni as an androgen degrader in sewage. Sci. Rep. 6, 1–13. doi: 10.1038/srep35386
Dabdoub, S. M., Fellows, M. L., Paropkari, A. D., Mason, M. R., Huja, S. S., Tsigarida, A. A., et al. (2016). PhyloToAST: bioinformatics tools for species-level analysis and visualization of complex microbial datasets. Sci. Rep. 6:29123. doi: 10.1038/srep29123
Edwards, U., Rogall, T., Blöcker, H., Emde, M., and Böttger, E. C. (1989). Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 17, 7843–7853.
European Food Safety Authority (2012). Conclusion on the peer review of the pesticide risk assessment of the active substance ametoctradin (BAS 650 F). EFSA J. 10, 2921–3005. doi: 10.2903/j.efsa.2012.2921
Federal Insecticide, Fungicide, and Rodenticide Act (1910). Federal Insecticide, Fungicide, and Rodenticide Act. 7 U.S.C. ch. 6 § 136a. Washington, DC: 61st United States Congress.
Fierer, N., Hamady, M., Lauber, C. L., and Knight, R. (2008). The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 17994–17999. doi: 10.1073/pnas.0807920105
Geed, S. R., Kureel, M. K., Giri, B. S., Singh, R. S., and Rai, B. N. (2017). Performance evaluation of Malathion biodegradation in batch and continuous packed bed bioreactor (PBBR). Bioresour. Technol. 227, 56–65. doi: 10.1016/j.biortech.2016.12.020
Gupta, S., Wali, A., Gupta, M., and Annepu, S. K. (2017). “Fungi: An Effective Tool for Bioremediation,” in Plant-Microbe Interactions in Agro-Ecological Perspectives. Singapore: Springer, 593–606. doi: 10.1007/978-981-10-6593-4_24
Hogendoorn, E. A., Dijkman, E., Baumann, B., Hidalgo, C., Sancho, J.-V., and Hernandez, F. (1999). Strategies in using analytical restricted access media columns for the removal of humic acid interferences in the trace analysis of acidic herbicides in water samples by coupled column liquid chromatography with UV detection. Anal. Chem. 71, 1111–1118. doi: 10.1021/ac980918x
Jia, S., Shi, P., Hu, Q., Li, B., Zhang, T., and Zhang, X.-X. (2015). Bacterial community shift drives antibiotic resistance promotion during drinking water chlorination. Environ. Sci. Technol. 49, 12271–12279. doi: 10.1021/acs.est.5b03521
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Khan, F. (2012). Review of Soil Taxonomy Information for Submitted Environmental Fate Studies of Ametroctradin. Washington, D.C: U.S. Environmental Protection Agency.
Klümper, W., and Qaim, M. (2014). A meta-analysis of the impacts of genetically modified crops. PLoS One 9:e111629. doi: 10.1371/journal.pone.0111629
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Letunic, I., and Bork, P. (2011). Interactive tree of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478. doi: 10.1093/nar/gkr201
Manasfi, R., Chiron, S., Montemurro, N., Perez, S., and Brienza, M. (2020). Biodegradation of fluoroquinolone antibiotics and the climbazole fungicide by Trichoderma species. Environ. Sci. Pollut. Res. 27, 23331–23341. doi: 10.1007/s11356-020-08442-8
Masqué, N., Marcé, R. M., and Borrull, F. (1998). Chemical removal of humic substances interfering with the on-line solid-phase extraction—Liquid chromatographic determination of polar water pollutants. Chromatographia 48:231. doi: 10.1007/BF02467676
Mavriou, Z., Alexandropoulou, I., Melidis, P., Karpouzas, D. G., and Ntougias, S. (2020). Biotreatment and bacterial succession in an upflow immobilized cell bioreactor fed with fludioxonil wastewater. Environ. Sci. Pollut. Res. doi: 10.1007/s11356-020-09231-z [Epub ahead of print].
McDonald, J. A., Gaston, L. A., Jackson, S. H., Locke, M. A., and Zablotowicz, R. M. (2006). Degradation kinetics assessment for the fungicide BAS 505 in intact soil cores versus batch soils. Soil Sci. 171, 239–248. doi: 10.1097/01.ss.0000187375.38649.5b
Nakatsu, C. H., Hristova, K., Hanada, S., Meng, X.-Y., Hanson, J. R., Scow, K. M., et al. (2006). Methylibium petroleiphilum gen. nov., sp. nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. Int. J. Syst. Evol. Microbiol. 56, 983–989. doi: 10.1099/ijs.0.63524-0
Oerke, E.-C. (2006). Crop losses to pests. J. Agric. Sci. 144, 31–43. doi: 10.1017/S0021859605005708
Oristep Consulting (2015). Global Pesticide Market - By Regions and Vendors - Market Size, Demand Forecasts, Industry Trends and Updates, Supplier Market Shares 2014-2020. Available online at: http://www.researchandmarkets.com/publication/mdxunlv/global_pesticide_market_by (accessed April 20, 2017).
Öztürk, B., Werner, J., Meier-Kolthoff, J. P., Bunk, B., Spröer, C., and Springael, D. (2020). Comparitive genomics suggests mechanisms of genetic adaptation toward the catabolism of the phenylurea herbicide linuron in Variovorax. Genome Biol. Evol. 12, 827–841. doi: 10.1093/gbe/evaa085
Parkin, G. F., and Speece, R. E. (1983). Attached versus suspended growth anaerobic reactors: response to toxic substances. Water Sci. Technol. Lond. 15, 261–289. doi: 10.2166/wst.1983.0171
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Paulson, J. N., Stine, O. C., Bravo, H. C., and Pop, M. (2013). Robust methods for differential abundance analysis in marker gene surveys. Nat. Methods 10, 1200–1202. doi: 10.1038/nmeth.2658
Pérez-Pantoja, D., Donoso, R., Agulló, L., Córdova, M., Seeger, M., Pieper, D. H., et al. (2012). Genomic analysis of the potential for aromatic compounds biodegradation in Burkholderiales. Environ. Microbiol. 14, 1091–1117. doi: 10.1111/j.1462-2920.2011.02613.x
Rhee, S.-K., Liu, X., Wu, L., Chong, S. C., Wan, X., and Zhou, J. (2004). Detection of genes involved in biodegradation and biotransformation in microbial communities by using 50-Mer oligonucleotide microarrays. Appl. Environ. Microbiol. 70, 4303–4317. doi: 10.1128/AEM.70.7.4303-4317.2004
Sagberg, P., Dauthuille, P., and Hamon, M. (1992). “Biofilm reactors: a compact solution for the upgrading of waste water treatment plants,” in Water Science & Technology (London: International Water Association), 733–742. doi: 10.2166/wst.1992.0454
Satola, B., Wübbeler, J. H., and Steinbüchel, A. (2012). Metabolic characteristics of the species Variovorax paradoxus. Appl. Microbiol. Biotechnol. 97, 541–560. doi: 10.1007/s00253-012-4585-z
Schouw, A., Leiknes Eide, T., Stokke, R., Pedersen, R. B., Steen, I. H., and Bødtker, G. (2016). Abyssivirga alkaniphila gen. nov., sp. nov., an alkane-degrading, anaerobic bacterium from a deep-sea hydrothermal vent system, and emended descriptions of Natranaerovirga pectinivora and Natranaerovirga hydrolytica. Int. J. Syst. Evol. Microbiol. 66, 1724–1734. doi: 10.1099/ijsem.0.000934
Shiotani, T., and Yamané, T. (1981). A horizontal packed-bed bioreactor to reduce CO2 gas holdup in the continuous production of ethanol by immobilized yeast cells. Eur. J. Appl. Microbiol. Biotechnol. 13, 96–101. doi: 10.1007/BF00499695
Singh, S., Kumar, V., Singh, S., and Singh, J. (2019). Influence of humic acid, iron and copper on microbial degradation of fungicide Carbendazim. Biocatal. Agric. Biotechnol. 20:101196. doi: 10.1016/j.bcab.2019.101196
Soeda, Y., Kosaka, S., and Noguchi, T. (1972). The fate of thiophanate-methyl fungicide and its metabolites on plant leaves and glass plates. Agric. Biol. Chem. 36, 931–936. doi: 10.1080/00021369.1972.10860358
Strandberg, G. W., Donaldson, T. L., and Farr, L. L. (1989). Degradation of trichloroethylene and trans-1,2-dichloroethylene by a methanotrophic consortium in a fixed-film, packed-bed bioreactor. Environ. Sci. Technol. 23, 1422–1425. doi: 10.1021/es00069a016
Tilman, D., Cassman, K. G., Matson, P. A., Naylor, R., and Polasky, S. (2002). Agricultural sustainability and intensive production practices. Nature 418, 671–677. doi: 10.1038/nature01014
Vallaeys, T., Albino, L., Soulas, G., Wright, A. D., and Weightman, A. J. (1998). Isolation and characterization of a stable 2,4-dichlorophenoxyacetic acid degrading bacterium, Variovorax paradoxus, using chemostat culture. Biotechnol. Lett. 20, 1073–1076. doi: 10.1023/A:1005438930870
van Ginkel, C. G. (1996). Complete degradation of xenobiotic surfactants by consortia of aerobic microorganisms. Biodegradation 7, 151–164. doi: 10.1007/BF00114627
Walters, W., Hyde, E. R., Berg-Lyons, D., Ackermann, G., Humphrey, G., Parada, A., et al. (2016). Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1:e00009-15. doi: 10.1128/mSystems.00009-15
Warhurst, A. M., Clarke, K. F., Hill, R. A., Holt, R. A., and Fewson, C. A. (1994). Metabolism of styrene by Rhodococcus rhodochrous NCIMB 13259. Appl. Environ. Microbiol. 60, 1137–1145. doi: 10.1128/aem.60.4.1137-1145.1994
Zhang, L., Chung, J., Jiang, Q., Sun, R., Zhang, J., Zhong, Y., et al. (2017). Characteristics of rumen microorganisms involved in anaerobic degradation of cellulose at various pH values. RSC Adv. 7, 40303–40310. doi: 10.1039/C7RA06588D
Zhang, X., Zhang, L., Wu, M., Tang, Q., Song, Z., Zhou, H., et al. (2020). Comparative characterization and functional genomic analysis of two Comamonas sp. strains for biodegradation of quinoline. J. Chem. Technol. Biotechnol. 95, 2017–2026. doi: 10.1002/jctb.6390
Keywords: biodegradation, fungicide, ametoctradin, microbial consortium, packed-bed reactor
Citation: Whittington HD, Singh M, Ta C, Azcárate-Peril MA and Bruno-Bárcena JM (2020) Accelerated Biodegradation of the Agrochemical Ametoctradin by Soil-Derived Microbial Consortia. Front. Microbiol. 11:1898. doi: 10.3389/fmicb.2020.01898
Received: 17 April 2020; Accepted: 20 July 2020;
Published: 25 August 2020.
Edited by:
Guining Lu, South China University of Technology, ChinaCopyright © 2020 Whittington, Singh, Ta, Azcárate-Peril and Bruno-Bárcena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José M. Bruno-Bárcena, jbbarcen@ncsu.edu