 Peixin Fan1,2†
Peixin Fan1,2† Miju Kim1,2†
Miju Kim1,2† Grace Liu1,2Yuting Zhai1,2Ting Liu1,2Joseph Danny Driver2
Grace Liu1,2Yuting Zhai1,2Ting Liu1,2Joseph Danny Driver2 Kwangcheol C. Jeong1,2*
Kwangcheol C. Jeong1,2*- 1Emerging Pathogens Institute, University of Florida, Gainesville, FL, United States
- 2Department of Animal Sciences, University of Florida, Gainesville, FL, United States
Calf diarrhea is one of the most concerning challenges facing both the dairy and beef cattle industry. Maintaining healthy gut microbiota is essential for preventing gastrointestinal disorders. Here, we observed significantly less bacterial richness in the abnormal feces with watery or hemorrhagic morphology compared to the normal solid feces. The normal solid feces showed high relative abundances of Osllospiraceae, Christensenellaceae, Barnesiella, and Lactobacillus, while the abnormal feces contained more bacterial taxa of Negativicutes, Tyzzerella, Parasutterella, Veillonella, Fusobacterium, and Campylobacter. Healthy calves had extensive bacterial-bacterial correlations, with negative correlation between Lactobacillus and potential diarrheagenic Escherichia coli-Shigella, but not in the abnormal feces. We isolated Lactobacillus species (L. reuteri, L. johnsonii, L. amylovorus, and L. animalis), with L. reuteri being the most abundant, from the healthy gut microbiota. Isolated Lactobacillus strains inhibited pathogenic strains including E. coli K88 and Salmonella Typhimurium. These findings indicate the importance of a diverse gut microbiota in newborn calf’s health and provide multiple potential probiotics that suppress pathogen colonization in the gastrointestinal tract to prevent calf diarrhea.
Introduction
Newborn calves are susceptible to diarrhea, especially during the first month of their life (Bendali et al., 1999). The US Department of Agriculture estimated that between 4 and 25% of calves die from diarrhea each year in the US, leading to tremendous economic losses in the cattle industry (USDA, 2017). Calf diarrhea is challenging to mitigate because of its multifactorial etiology, which comprises of both infectious and noninfectious factors (Whon et al., 2021). Antibiotics are commonly used as a treatment for calf diarrhea, but the efficacy of antibiotic treatment is questionable (Kim et al., 2021). Furthermore, antibiotic use is nontargeted that can negatively impact calf gut microbial composition (Ianiro et al., 2016; Ramirez et al., 2020).
A healthy gut microbiota prevent colonization of foreign pathogens (Pickard et al., 2017; Fan et al., 2019, 2020, 2021; Kim et al., 2021) and enhance host immune systems through interactions between antigens and immune cells during early stage of life (Zhao and Elson, 2018; Fan et al., 2020). The increased intestinal permeability and disturbance of gut microbiota are key factors leading to the pathogen-induced diarrhea. The association of calf diarrhea with both microbial composition and functional dysbiosis shed light to ameliorate diarrhea through modulating gut microbial ecosystems. A study attempted to perform fecal microbiota transplantation (FMT) from healthy donors to six Korean brown cattle calves exhibiting diarrhea and showed promising results in which incidence of diarrhea was reduced significantly (Kim et al., 2021). In addition, administration of oral probiotics can be possible preventative treatments or supportive therapies for calf diarrhea. Duration of calf diarrhea has been shown to be diminished through administration of multispecies probiotics and yeast bolus (Renaud et al., 2019). To develop more efficient probiotics for diarrhea mitigation strategies, it is necessary to characterize the gut microbiota in healthy and diarrheic calves. A few studies have investigated the difference in gut microbial composition between healthy and diarrheic dairy calves and indicated the critical role of microbiota development in regulating gut health during early stage (Gomez et al., 2017; Hennessy et al., 2021). However, the important gut commensals that contribute to preventing diarrhea in newborn beef cattle, especially raised on pasture remain poorly defined.
In this study, we seek to elucidate the role of gut microbiota in the progression of diarrhea in one-month-old calves and identified several potential probiotics that may contribute to the gut health of young calves through suppressing diarrheagenic bacteria. The findings will aid in the future development of dietary interventions that may resolve or prevent enteric and diarrheal disease in young calves.
Materials and Methods
Animal Management
Preweaning calves enrolled in this study were from one generation of the multibreed Angus-Brahman (MAB) herd of the University of Florida. The calves were naturally born on pasture during the delivery season. The birthweight of newborns was measured within 24h after delivery. The calves were raised together with dams during preweaning stage on pasture on the cow-calf operation in Santa Fe River Ranch Beef Unit, FL, United States.
Fecal Sample Collection, Morphology Categorization, and Sample Processing
Fecal samples were collected from 91 beef calves ranging in age from 21 to 35days. Briefly, each fecal sample was collected from the rectal-anal junction (RAJ) using two sterile cotton swabs. During sample collection, the morphology of fecal samples was observed and recorded based on criteria of Bristol stool scale, especially the color, consistency, and presence of blood (Koppen et al., 2016). Swabs with fecal samples were placed in a 15ml conical tube on ice and were transported to the laboratory within 1h for further processing. Each swab sample was resuspended in mixture of 2ml of Luria-Bertani (LB) broth and 2ml of 30% glycerol as previously described (Fan et al., 2020). The fecal solution was then split into four 2ml tubes and frozen in an ultra-low freezer at −80°C.
16S rRNA Gene Amplicon Sequencing
Fecal samples were thawed on ice and homogenized. Then 1ml of each sample was used for DNA extraction using the QIAamp PowerFecal DNA kit according to the manufacturer’s instructions (Qiagen, United States). To understand the bacterial community, a dual-index sequencing strategy was applied (Kozich et al., 2013). Briefly, the V4 region of the 16S rRNA gene was amplified using the PCR with dual-index primers (Kozich et al., 2013). The PCR amplification reaction included 1μl forward index primer (10mM), 1μl reverse index primer (10mM), 1μl 10ng/μl DNA template, and 17μl Pfx AccuPrime master mix (Invitrogen, United States). Amplification was initiated with denaturation for 5min at 95°C, followed by 30cycles of 95°C for 30s, annealing at 55°C for 30s and extension at 72°C for 1min, with a final elongation for 5min at 72°C. The amplicons were then purified and normalized using the SequalPrep plate normalization kit (Invitrogen, United States). The same amount of barcoded V4 amplicons from each sample was pooled to construct the DNA library.
Analysis for 16S rRNA Gene Amplicon Sequencing
The sequencing data of 16S rRNA gene were analyzed using version 2 of the Quantitative Insights into Microbial Ecology (QIIME 2) pipeline (Bolyen et al., 2019). Briefly, paired-end raw reads were imported, and the quality of the initial bases was evaluated according to the Interactive Quality Plot. The sequence quality control was performed with the Divisive Amplicon Denoising Algorithm (DADA2) pipeline implemented in QIIME 2, including steps for filtering low quality reads, denoising reads, merging the paired-end reads, and removing chimeric reads. The phylogenetic tree was generated using the align-to-tree-mafft-fasttree pipeline from the q2-phylogeny plugin of QIIME 2. The sequencing depth was normalized to 10,800 sequences per sample. The number of amplicon sequence variants (ASVs), Shannon index, and Bray-Curtis distance were calculated by the core-metrics-phylogenetic method. All ASVs were classified into the bacterial taxonomy using the q2-feature-classifier plugin of QIIME 2 and the SILVA 138 database.1
Co-occurrence Network Analysis
To investigate bacterial-bacterial interactions in the gut bacterial community of one-month-old calves, co-occurrence events of core bacterial genera that were present in at least 50% of normal and abnormal fecal samples were evaluated in the network interface, respectively, using pairwise Spearman’s rank correlations (rs) based on the relative abundance according to the previous study (Fan et al., 2020). The Spearman rank correlation was analyzed using Hmisc within RStudio (version 1.1456). A significant rank correlation between two bacterial genera (rs>0.2 or rs<−0.2, FDR-adjusted p value<0.05) was considered as a co-occurrence event. The network was visualized using the Force Atlas algorithm in the interactive platform Gephi 0.9.2.2 In the network, nodes represented different genera, and edges indicated significant correlations between nodes. The size of the nodes represented the degree of connection, and the thickness of edges indicated the strength of the correlation.
Microbial Function Prediction
Functional capacity of the gut microbial community was predicted using the online Galaxy version of phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt; http://galaxy.morganlangille.com/; Langille et al., 2013). The closed reference operational taxonomic unit (OTU) table was generated by picking OTU against the 13 August 2013 Greengenes database using QIIME (version 1.9.0). Normalization of copy numbers, metagenome prediction, and function categorization based on Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were conducted according to a standard analysis process.
Isolation of Lactobacillus spp. Strains and Antimicrobial Activities
The normal and abnormal feces were plated after 10-fold serial dilution in phosphate-buffered saline (PBS) on de Man, Rogosa, and Sharpe (MRS) agar (Difco, United States) to determine the concentrations of lactic acid bacteria (LAB), then plates were anaerobically incubated at 37°C for 48h using the GasPak EZ Anaerobe System (BD, United States). To isolate Lactobacillus strains from the normal feces, colonies with different morphologies were randomly selected and purified on the same solid media to isolate pure colonies. A total of 79 colonies were isolated from fecal samples of healthy calves. Genomic DNA of the isolated colonies was extracted by bead beating method, as previously described (Ma et al., 2021). Then, to speciate the isolates, the 16S gene was amplified using the universal primer pair KCP812 (5'-CAG GCC TAA CAC ATG CAA GTC-3') and KCP813 (5'-GGG CGG WGT GTA CAA GGC-3'; Marchesi et al., 1998). Amplified PCR products were purified using the QIAquick PCR purification kit (Qiagen, United States). Purified PCR products were sequenced in Genewiz (South Plainfield, United States), and the 16S rRNA gene sequences were identified by BLAST searching against National Center for Biotechnology Information (NCBI) 16S ribosomal RNA sequences database.
Antimicrobial activity of the Lactobacillus isolates was assessed against Escherichia coli KCJ2K2616, E. coli K88 KCJ4567, and Salmonella enterica Typhimurium KCJ187. For antimicrobial activity of the isolates, the agar well diffusion method was used with slight modification (Espeche et al., 2009; Boranbayeva et al., 2020). Briefly, antimicrobial activities of filter (0.22μm) sterilized supernatants of 18h cultures of Lactobacillus spp. were tested against pathogens described above. Overnight cultures of pathogens (107CFU) were mixed with soft LB agar medium (0.75% agar) and solidified. Then, 100μl of the supernatants was loaded in the agar well. After overnight incubation at 37°C, the diameters of inhibition zones were measured. Lactoplantibacillus plantarum (formerly known as Lactobacillus plantarum) KCJ4051 was used as a positive control (Yu et al., 2021), and each strain was tested in triplicate.
Statistical Analysis
Differences in alpha diversity and antimicrobial activity were analyzed using Student’s t-test or a one-way ANOVA followed by an F-test and Tukey’s HSD test for pairwise comparison of multiple means using the GraphPad prism. Differences between Bray-Curtis distances were analyzed using a permutational multivariate ANOVA (PERMANOVA) with the beta-group-significance command in QIIME 2 pipeline.
A Linear discriminant analysis Effect Size (LEfSe) analysis was applied to identify unique bacteria in normal and abnormal feces based on the non-parametric factorial Kruskal–Wallis sum-rank test. An effect size of 2 was considered as significant difference.
Results
High Prevalence of Abnormal Feces in One-Month-Old Beef Calves
One-month-old calves had high prevalence of abnormal feces. Among the 91 fecal samples, 74 (81.3%) were firm and brownish, which were classified as normal feces, whereas 17 (18.7%) were abnormal, including six mild watery, two extremely watery, eight with slight blood observed, and one extremely hemorrhagic (Figure 1A; Supplementary Table 1). Twelve out of 17 abnormal feces were from heifer. Interestingly, the birthweight of heifer with abnormal feces were significantly lower than those with normal feces (31.86±2.69kg vs. 35.26±4.50kg, p=0.0178, Figure 1B), suggesting birth weight might be associated with gut health. However, the pattern was not observed in bulls (38.44±8.13kg vs. 38.14±5.17kg; Figure 1C).
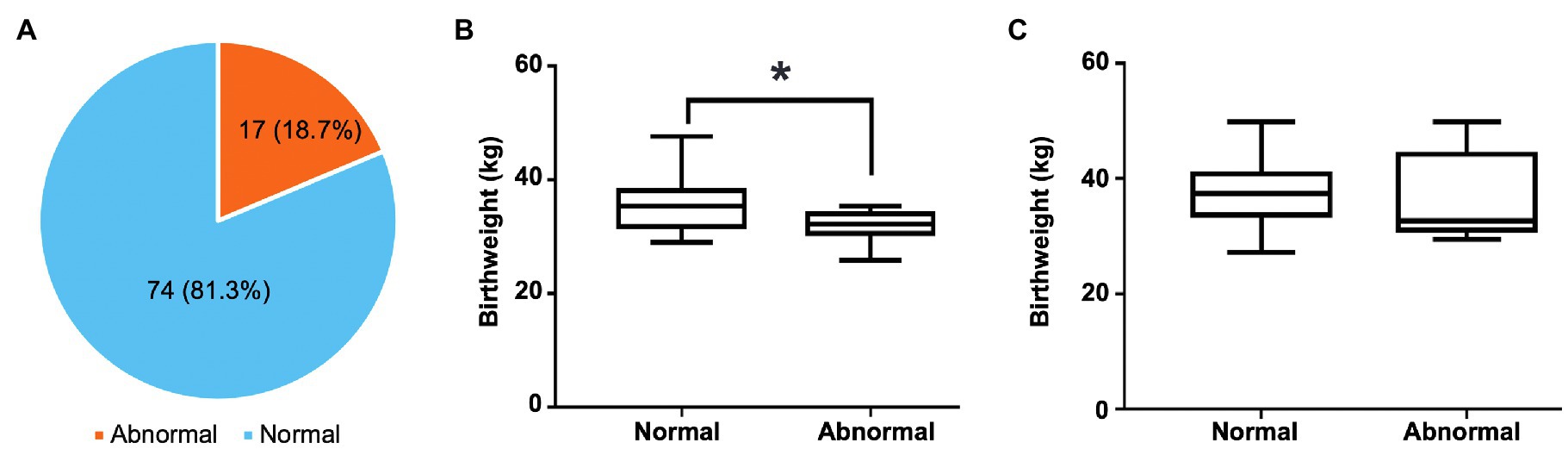
Figure 1. The distribution of normal and abnormal feces and its relationship with birthweight of preweaning calves. (A) The prevalence of normal and abnormal feces of preweaning calves. (B) The difference in birthweight between heifers with normal feces and those with abnormal feces. (C) The difference in birthweight between bulls with normal feces and those with abnormal feces. In the box plot, the asterisk reflects that they are significantly different (p<0.05) based on the Student’s t-test.
Normal and Abnormal Feces in Young Calves Carried Distinctive Microbiota
To compare the microbiota composition of normal and abnormal feces, the sequencing of the V4 region of the 16S gene was conducted. A total of 2,172,004 (23,868±455) raw reads were generated and after removing substitution and chimera errors using DADA2 pipeline, 1,703,949 (18,724±367) high-quality ASVs were yielded (Supplementary Table 2).
Alpha diversity analysis was first conducted to compare the richness and diversity of bacterial community between the normal and abnormal feces using the alpha diversity metrics, the number of ASVs, and the Shannon index, respectively. The abnormal feces had significantly lower number of ASVs compared with normal feces (p=0.017, Figure 2A), indicating a lower gut bacterial richness in calves with abnormal feces. However, we did not observe an apparent difference in the Shannon Index between groups (p=0.2841, Figure 2B), denoting comparable levels of species diversity. Moreover, visualization of the differences in gut microbiota structure between the abnormal and normal fecal groups via a principal coordinates analysis (PCoA) plot showed no significant difference (p=0.214) between groups based on Bray-Curtis distance evaluated using PERMANOVA (Figure 2C). Both feces were dominated by Firmicutes, Bacteroidota, and Proteobacteria phyla (Figure 2D; Supplementary Table 3). To differentiate significant bacterial taxa between the fecal groups, a cladogram was generated based on LefSe. The abnormal feces harbored higher relative abundance of Negativicutes, while normal feces had higher relative abundance of commensals, including Rikenellaceae, Tannerellaceae, Erysipelatoclostridiaceae, Christensenellaceae, Oscillospiraceae, and Anaerovoraceae families (Figure 2E). To understand the influence of different microbiota on microbial functional properties, the PICRUSt was applied to predict the relative abundance of bacterial genes. The bacterial genes involved in metabolism of cofactors and vitamins, and cellular processes and signaling were enriched in abnormal feces, whereas those participate in cell motility had higher relative abundance in normal feces (Figure 2F).
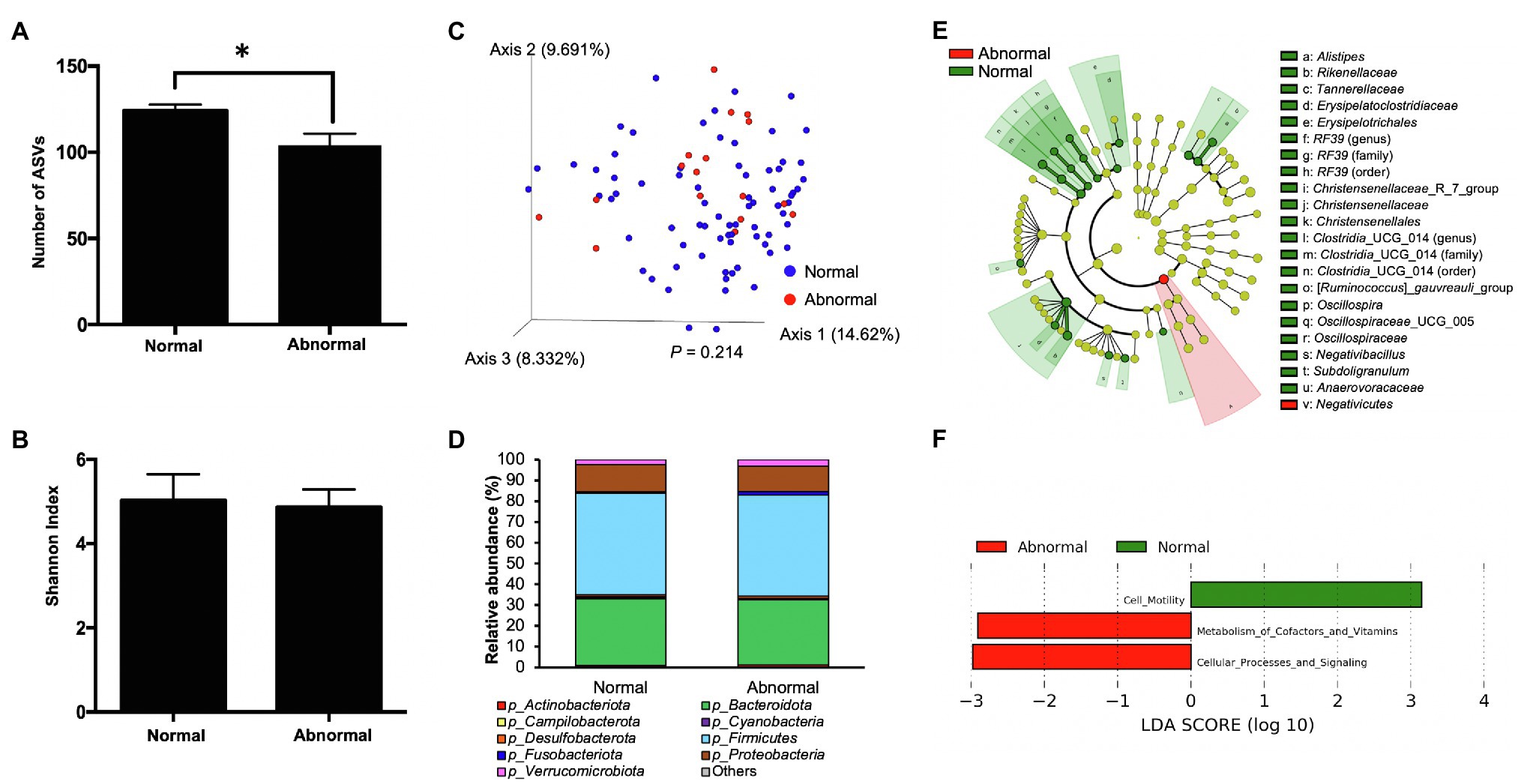
Figure 2. The microbiota characteristics of normal and abnormal feces. (A) The number of amplicon sequence variants (ASVs) of normal and abnormal feces. (B) The Shannon index of normal and abnormal feces. (C) The PCoA plot based on Bray-Curtis distances comparing microbiota composition between normal and abnormal feces. The difference in Bray-Curtis distance was analyzed by the permutational multivariate ANOVA (PERMANOVA). (D) Distribution of abundant bacterial phyla in normal and abnormal feces. (E) The taxonomic cladogram obtained from the linear discriminant analysis effect size (LEfSe) analysis showing the unique bacterial taxa in the normal and abnormal feces. (F) Results of the LEfSe analysis showing microbial function that were significantly different between normal and abnormal feces.
The Variation in Microbiota of Abnormal Feces With Different Morphologies
After comparing gut microbiota composition between calves with abnormal feces and calves with normal feces, the abnormal fecal group was subcategorized based on differing morphologies (mild watery, severe watery, mild hemorrhagic, and severe hemorrhagic) for further analysis. The number of ASVs was lower in watery feces and was dependent on severity of the watery property (Figure 3A). Average number of ASVs for normal, mild watery, and severe watery feces was 124.3, 102.2, and 78.00, respectively. The feces with presence of slight blood contained slightly lower level of ASVs (114.4) compared to normal feces, but the severe hemorrhagic feces harbored much fewer ASVs (81.0). Similar but less obvious pattern was observed for the Shannon index (Figure 3B). The results show an incremental decrease in bacterial species richness and diversity with increased levels of severity of both hemorrhagic and watery feces. However, no significant difference was observed for overall microbiota structure between severe watery feces and severe hemorrhagic feces with the normal feces according to the PCoA plot based on Bray-Curtis distance (Figure 3C).
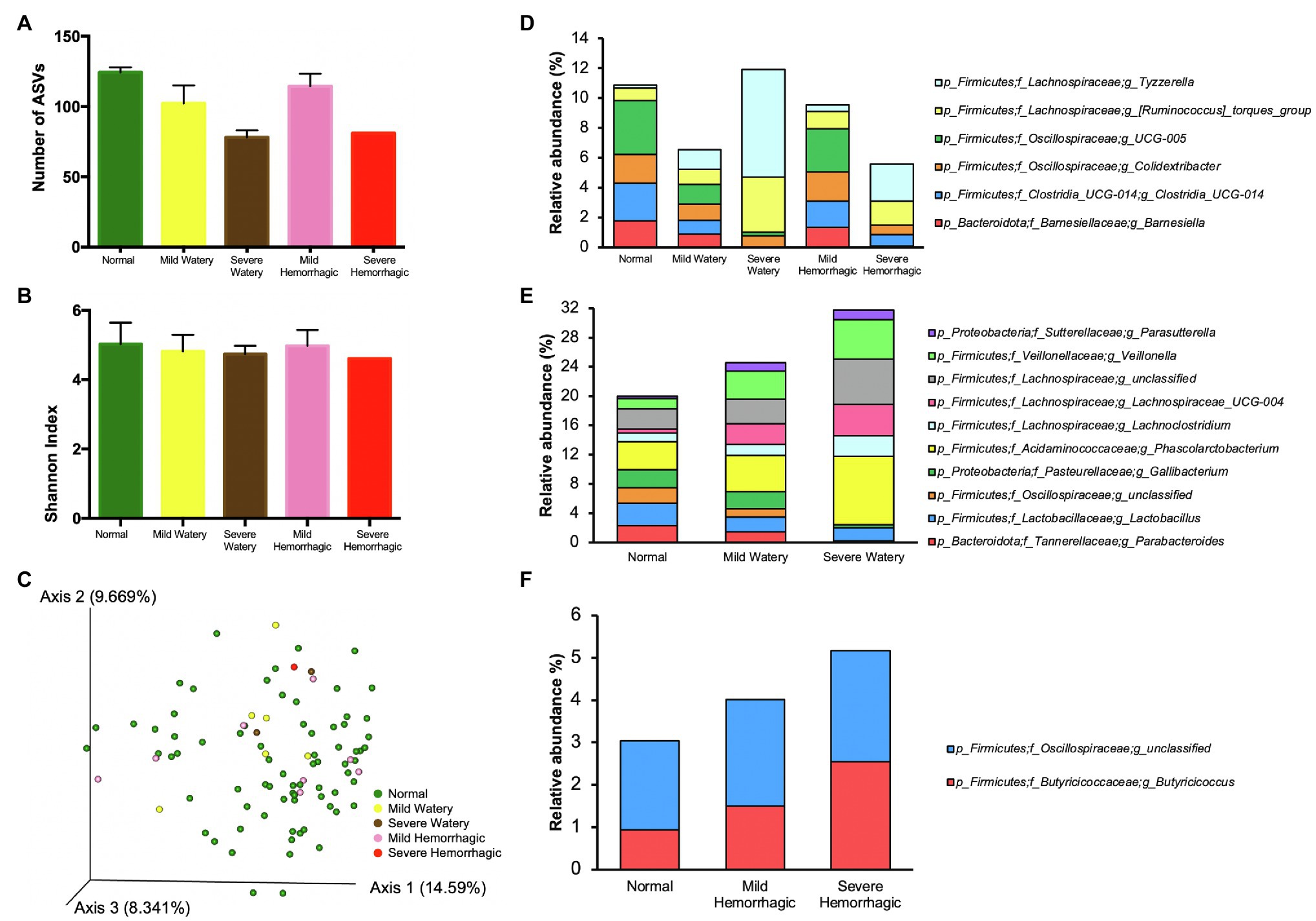
Figure 3. The microbiota characteristics of feces with different morphologies. (A) The number of ASVs of feces with different morphologies. (B) The Shannon index of feces with different morphologies. (C) The PCoA plot based on Bray-Curtis distances comparing microbiota composition between feces with different morphologies. (D) The bacterial genera that either showed highest relative abundance or lowest relative abundance in normal feces compared with other morphologies. (E) The bacterial genera that were gradually changed with the severity of fecal watery morphology. (F) The bacterial genera that were gradually changed with the severity of hemorrhagic morphology.
We further pinpointed key differences in microbiota composition of feces with varied morphologies. The microbiota at phylum level was similar among normal feces, mild watery feces, and mild hemorrhagic feces, while severe watery feces contained 20 folds more Fusobacteriota compared with normal feces, and severe hemorrhagic feces had much less level of Verrucomicrobiota (Supplementary Figure 1). At genus level, normal feces harbored highest relative abundance of Barnesiella and Clostridia_UCG-014, Colidextribacter and Oscillospiraceae_UCG_005 but lowest relative abundance of [Ruminococcus]_torques_group and Tyzzerella compared to feces with other morphologies (Figure 3D; Supplementary Table 4). Moreover, the relative abundances of Parabacteroides, Lactobacillus, unclassified Oscillospiraceae, and Gallibacterium were gradually decreased with the increase of severity of the watery morphology, whereas the relative abundances of Phascolarctobacterium, Lachnoclostridium, Lachnospiraceae_UCG-004, Veillonella, and Parasutterella were elevated, indicating these bacteria were sensitive to fecal watery morphology (Figure 3E; Supplementary Table 5). For the hemorrhagic morphology, Butyricicoccus and unclassified Oscillospiraceae were gradually enriched in feces with increase of severity (Figure 3F; Supplementary Table 6).
The Microbiota Profile of Severe Watery and Hemorrhagic Feces
We further extracted the individual severe watery and hemorrhagic fecal samples and characterized their microbiota structure, as they would pose greater risk to the intestinal health of calves. All severe abnormal fecal samples had much less bacterial richness according to the number of ASVs (Figure 4A) and slightly lower bacterial diversity according to the Shannon index (Figure 4B). For the microbiota composition, one of the severe watery feces (Severe Watery #1) harbored more than 5-fold higher relative abundance of Fusobacterium (26.56%), Peptostreptococcus (3.23%), Campylobacter (2.96%), Parasutterella (2.25%), and Tyzzerella (1.18%) compared to normal feces (0.61, 0.01, 0.34, 0.36, and 0.22%), and the other one (Severe Watery #2) contained more Tyzzerella (13.27%), Veillonella (10.52%), Lachnospiraceae_UCG-004 (6.94%), [Ruminococcus]_torques_group (4.53%) than normal feces (0.22, 1.37, 0.57, and 0.8%; Figure 4C). For the severe hemorrhagic feces, it shed more than 10-fold higher relative abundance of Tyzzerella (2.5%) and approximately 5-fold more Veillonella (6.06%) than normal feces (0.22 and 1.37%; Figure 4C).
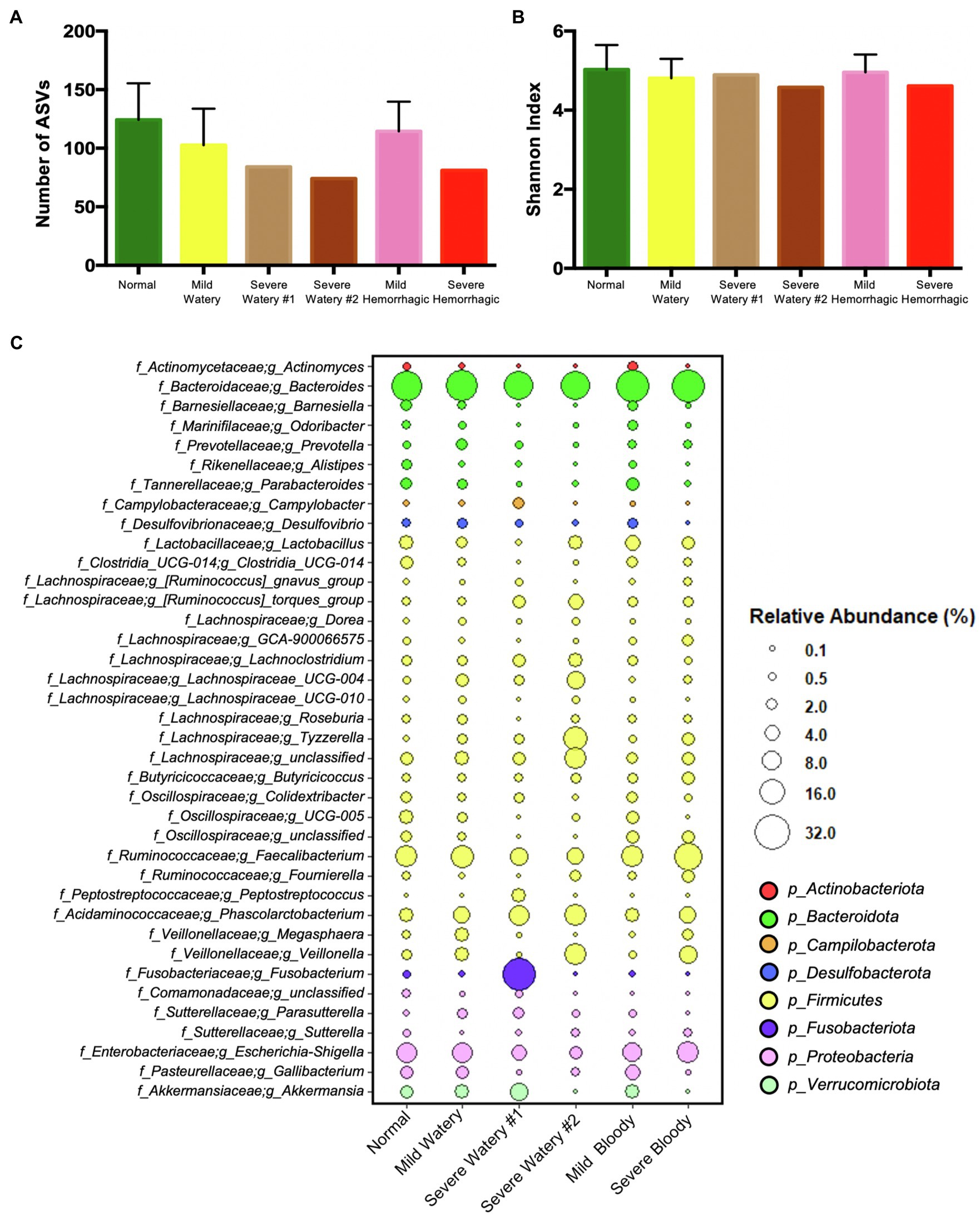
Figure 4. The microbiota characteristics of severe watery or hemorrhagic feces. (A) The number of ASVs of subgroup feces. (B) The Shannon index of subgroup feces. (C) The dotplot that shows the relative abundance of genera in subgroup feces.
Bacterial-Bacterial Interactions Predicted by Co-occurrence Network
To investigate whether there are differences in bacterial-bacterial interactions in the gut microbial ecosystems, the co-occurrence networks were analyzed using the 59 and 54 core bacterial genera that were present in more than 50% of the normal and abnormal fecal samples, respectively. The normal feces had eight unique core bacterial genera including Actinomyces, Christensenellaceae R-7 group, Collinsella, Coprococcus, Erysipelatoclostridiaceae UCG-004, Incertae Sedis, Oscillospiraceae NK4A214 group, RF39, and Rhodospirillales uncultured, whereas the abnormal feces had only four unique core bacterial genera including Coprobacter, Intestinimonas, Streptococcus, and Tyzzerella. The normal feces had more bacteria-bacteria interactions (n=656) compared to the abnormal feces (n=272) based on Spearman correlations (Padjust<0.05, rs>0.2, or rs<−0.2). For the normal feces, the network was grouped into three modules according to the inner interactions among the nodes (Figure 5A). In module 1, Oscillospira, which had higher relative abundance in normal feces (Figure 2E), was the hub among the 22 core bacterial genera, having 38 significant correlations with other bacteria (Figure 5A). Notably, Oscillospira was negatively associated with Campylobacter, which showed high relative abundance in the severe watery feces, and Escherichia-Shigella, which contain multiple pathogenic species that can cause diarrhea in young calves. In module 2, Oscillospiraceae UCG-005 was the hub among the 20 core bacteria, having 38 correlations with other bacteria. Oscillospiraceae UCG-005 showed negative associations with Veillonella, which was enriched in watery feces (Figure 3E), as well as Campylobacter in Module 1. In module 3, Lachnospiraceae UCG-004 associated with watery feces was the hub among the 17 core bacteria and it was negatively correlated with the Christensenellaceae R-7 group in module 1, which was belonged to the unique core bacteria of the normal feces. Importantly, a significant negative correlation was also observed between beneficial bacteria Lactobacillus and Escherichia-Shigella.
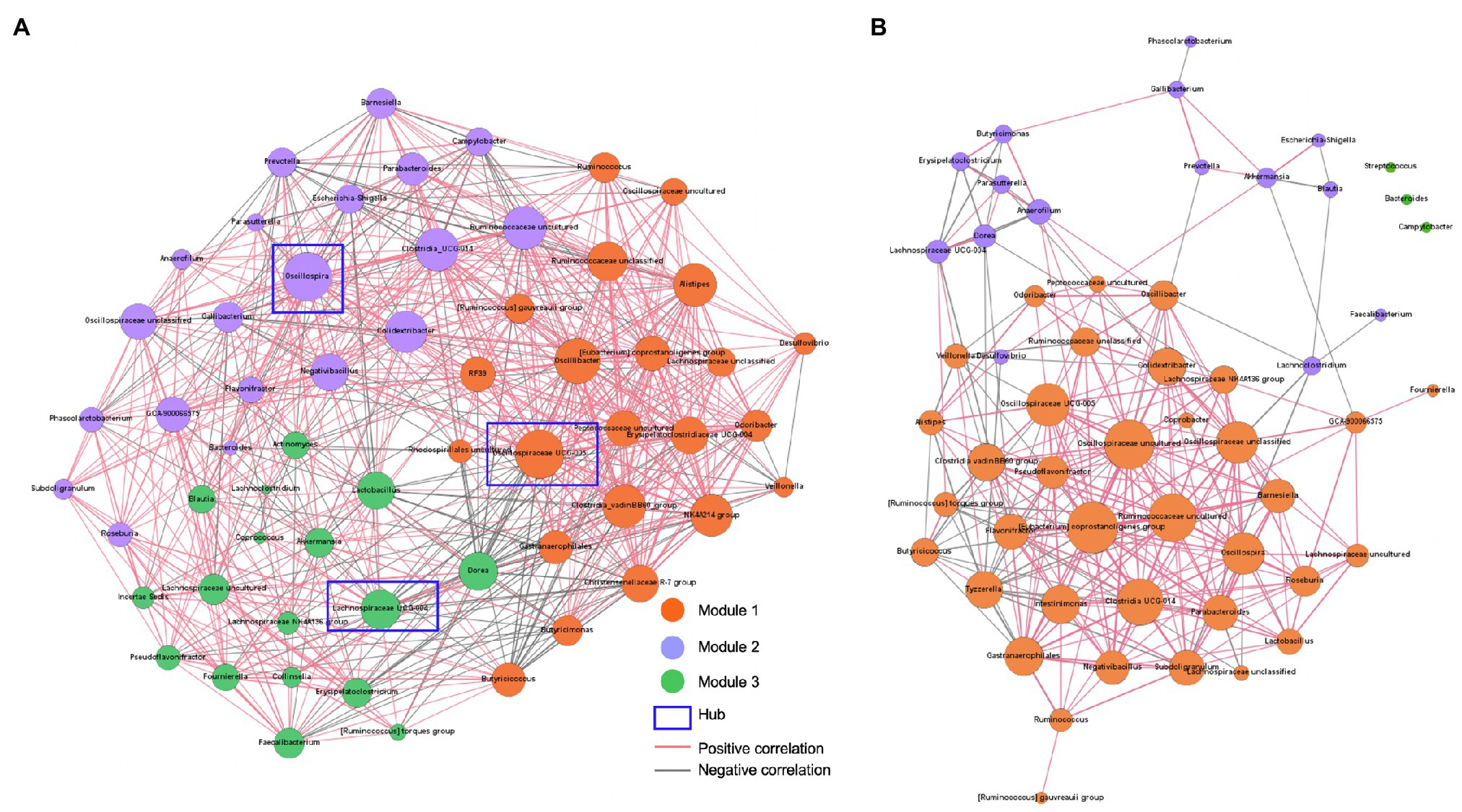
Figure 5. Bacterial-bacterial interactions predicted by co-occurrence networks. (A) Bacterial co-occurrence network in normal feces. (B) Bacterial co-occurrence network in abnormal feces. Co-occurrence networks predicting the bacteria-bacteria interactions among core bacterial genera (present in more than 50% of normal or abnormal fecal samples) in calf feces. Connections were detected based on Spearman’s rank correlations (rs>0.2 or rs<−0.2, FDR adjusted p value<0.05). Dot sizes represent number of connections with other taxa. Thickness of lines represents strength of relatedness. The modules were designated based on a community detection algorithm.
For the abnormal feces, the bacterial-bacterial interaction network was also grouped to three modules, with module 1 being the dominant, but the number of nods and correlations were significantly less compared to normal feces (Figure 5B). The [Eubacterium] coprostanoligenes group was the hub among the 38 core bacteria, with 26 significant correlations with other bacteria. Notably, the negative associations between potential beneficial bacteria, Oscillospira and Lactobacillus, and opportunistic pathogens, Campylobacter and Escherichia-Shigella, which were detected in the normal feces, were not observed in the abnormal feces, suggesting that beneficial bacteria may suppress diarrheagenic pathogens in normal healthy gut microbiota.
Identification of Lactobacillus spp. Strains and Their Antimicrobial Activities
As we discovered higher abundance of Lactobacillus in normal feces (3.12%) compared to mild watery feces (2.02%) and severe watery feces (1.82%), as well as negative correlations between Lactobacillus and Escherichia-Shigella in normal feces, we isolated Lactobacillus strains and evaluated their antimicrobial activity against pathogens to test if these strains may contribute to the gastrointestinal tract health. The number of LAB in normal feces was significantly greater than that of abnormal feces (p=0.0198; Figure 6A), suggesting the concentrations of LAB might be associated with the gut health in the calves. In addition, 79 Lactobacillus spp. strains were isolated from the normal feces, and they were identified as Limosilactobacillus reuteri (formerly known as Lactobacillus reuteri), which was the most predominant species in the normal feces (Figure 6B). We also identified Lactobacillus johnsonii, Lactobacillus amylovorus, and Ligilactobacillus animalis (formerly known as Lactobacillus animalis) in the normal feces (Figure 6B). To test if these strains may suppress pathogenic strains as suggested in Figure 5, we conducted antimicrobial activity of these strains against E. coli and S. Typhimurium, which can cause calf diarrhea (Boranbayeva et al., 2020). All Lactobacillus spp. strains inhibited the growth of three pathogens (Figures 6C–E). Notably, L. reuteri and L. johnsonii showed clear inhibition that was similar to that of well-known probiotic strain L. plantarum against all test pathogens. Lactobacillus reuteri and L. johnsonii were the most and second most abundant Lactobacillus species in healthy calves (Figure 6B), suggesting these Lactobacillus spp. may affect gut health positively by suppressing the colonization of pathogenic bacteria in the gut of preweaning calves.
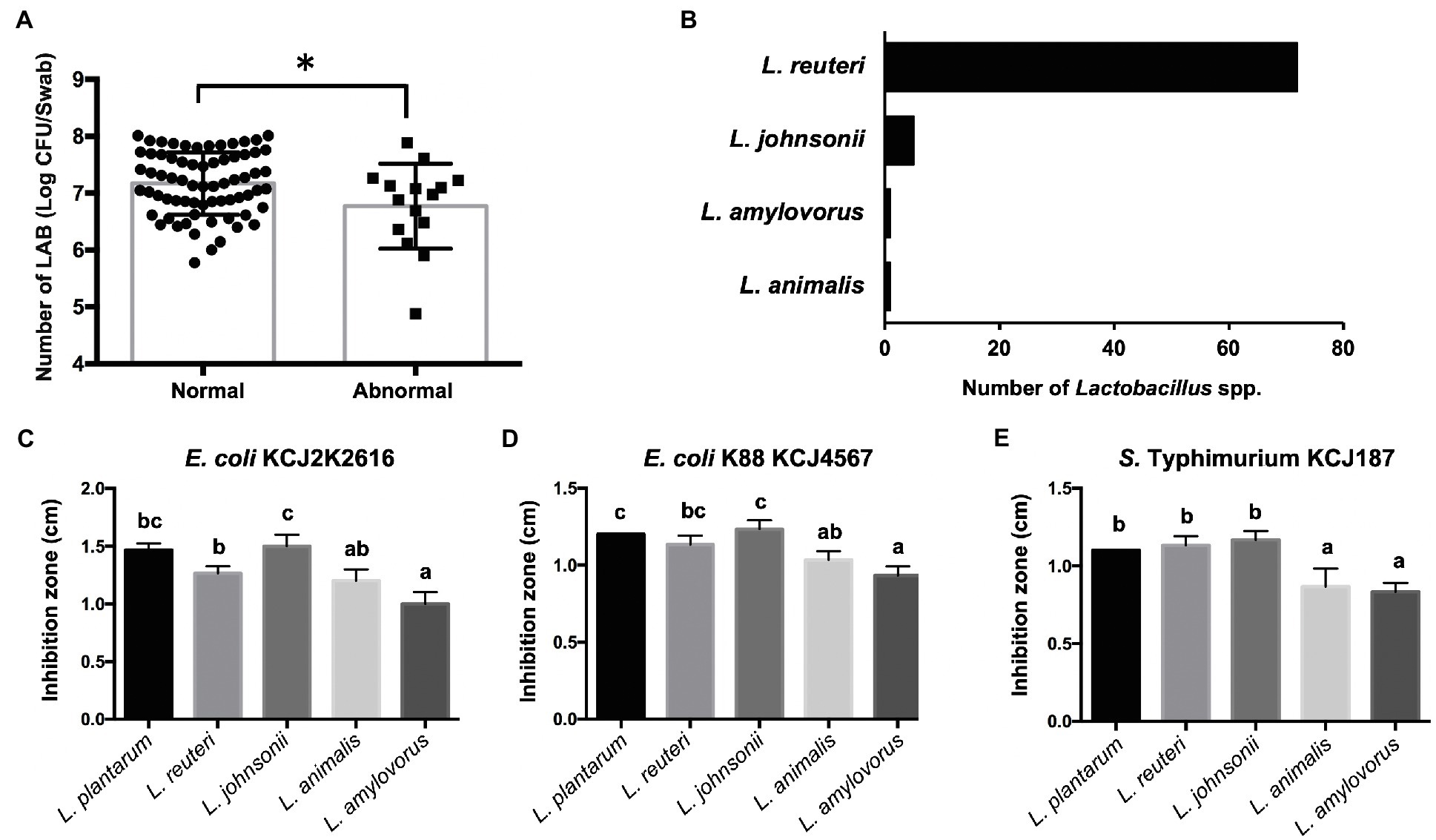
Figure 6. Isolation of Lactobacillus strains and their antimicrobial activity. (A) The concentrations of lactic acid bacteria (LAB) in normal and abnormal feces. In the bar graph, the asterisk reflects they are significantly different (p<0.05) based on the Student’s t-test. (B) Number of Lactobacillus spp. isolated from healthy calves identified by 16S rRNA gene sequencing. (C-E) Antimicrobial activity of Lactobacillus spp. against pathogenic strains. Bars with different letters indicate significant difference (p<0.05) based on one-way ANOVA followed by an F-test and Tukey’s HSD test for pairwise comparison of multiple means.
Discussion
Diarrhea is a leading cause of death for beef and dairy calves in their first month of life (Caffarena et al., 2021). The homeostasis of gut microbial ecosystems is of central importance for maintaining the functions of the gastrointestinal tract of early preweaning calves (Virginio Junior and Bittar, 2021). However, there are limited studies reporting relationships between gut microbiota and diarrhea in young beef cattle. In this study, we investigated the fecal microbiota composition of one-month-old beef calves raised on pasture based on their fecal morphology. Many bacteria were identified to distinguish the normal solid feces and abnormal watery or hemorrhagic feces that had negative correlations with diarrheagenic pathogens.
The most apparent pattern for the abnormal fecal microbiota is the low bacterial richness. The number of bacterial types in feces was dramatically decreased with the severity of the watery or hemorrhagic phenomenon. A diverse gut microbiome is composed of 100 times more genes than host and serves as a functional expansion of host genomes (Zhu et al., 2010). The reduction in gut bacterial richness could result in limitation of microbiota function, including digestion, educating immune system, and inhibiting pathogens (Nicholson et al., 2012; Pickard et al., 2017; Schluter et al., 2020). The loss of microbial richness could be due to the great proliferation of pathogenic bacteria, such as Fusobacterium and Campylobacter. It is noteworthy that we found a significant lower birthweight of heifers with abnormal feces compared with those that had normal feces, suggesting the fetal development likely affects the gut microbiota maturation of young calves.
In this study, we screened for potential pathogenic and beneficial bacteria according to the fecal morphology. The bacteria enriched in abnormal feces include Negativicutes, Tyzzerella, [Ruminococcus]_torques_group, Parasutterella, Veillonella, Lachnospiraceae_UCG-004, Lachnoclostridium, Butyricicoccus, Fusobacterium, and Campylobacter. Campylobacter, including C. jejuni, C. hyointestinalis, and C. fetus are capable to cause irritation in the intestinal tract of calves and induce diarrhea (Firehammer and Myers, 1981; Diker et al., 1990). Previous studies reported that Negativicutes, Veillonella, and Fusobacterium were more abundant in diarrheic piglets compared to healthy weaned piglets (Huang et al., 2019; Karasova et al., 2021). In human studies, Tyzzerella contribute to neuroinflammation and its increase is associated with diarrhea (Nakajima et al., 2020). Parasutterella is in association with the development and progression of IBS (Chen et al., 2018). However, a few studies report the role of these bacteria in calf diarrhea.
We also identified several potential beneficial bacteria that showed higher relative abundance in normal feces, including Christensenellaceae, Oscillospiraceae (Colidextribacter and Oscillospira), Barnesiella, Parabacteroides, and Lactobacillus. Although many studies showed the beneficial effects of oral administration of Lactobacillus strains on reducing the diarrhea in swine and calves, only a few studies measured the change of Lactobacillus abundance between diarrheic and healthy beef cattle (Hou et al., 2015; Fernandez et al., 2020). In this study, we further isolated Lactobacillus strains from the healthy calves with normal feces, including L. reuteri, L. johnsonii, L. amylovorus, and L. animalis. In particular, the most abundant Lactobacillus species from healthy calves in our analysis, L. reuteri and L. johnsnii, are being administrated as potential probiotics and improved gastrointestinal health in pre-weaned calves (Fernandez et al., 2020). Jia et al. (2021) proposed L. animalis with high antibacterial activity against intestinal pathogens as anti-diarrheal probiotic for pig (Jia et al., 2021). Interestingly, the four Lactobacillus species isolated from the calves in this study all exerted antibacterial activities against pathogenic bacteria that commonly induce diarrhea in cattle, suggesting the high abundance of commensal Lactobacillus in healthy calves are beneficial in preventing colonization of pathogens.
In a previous study, Kim et al. (2021) applied FMT to effectively ameliorate calf diarrhea and found an increase in relative abundance in Christensenellaceae and Parabacteroides (Kim et al., 2021). Another study showed a significant decrease of Oscillospira in feedlot cattle with hemorrhagic diarrhea compared to healthy cattle (Zeineldin et al., 2018). Our study in line with other findings indicates the essential role of these bacteria in maintaining the cattle gut health. Although the function of Barnesiella in cattle is largely unknown, it showed impact in alleviating DSS-induced colitis in mice (Yang et al., 2020). Moreover, as we detected strong correlations between these potential beneficial bacteria with pathogenic bacteria, future studies will be directed to look into their competitive interactions.
Data Availability Statement
The 16S rRNA gene amplicon sequencing data generated during the current study were submitted to NCBI under BioProject PRJNA751859.
Ethics Statement
The animal study was reviewed and approved by University of Florida Institutional Animal Care and Use Committee.
Author Contributions
PF and KJ designed the study. JD managed the animals. PF, GL, MK, YZ, and TL collected and processed the samples. PF, MK, and GL analyzed the data. PF, MK, GL, and KJ drafted the manuscript. PF, MK, and KJ finalized this manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Southern Sustainable Agriculture Research & Education Graduate Student Grant, GS19-206, supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture to PF and KJ. MK was partly supported by Basic Research Program by the National Research Foundation of Korea, The Ministry of Education (2020R1A6A3A03038510).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.772863/full#supplementary-material
Footnotes
References
Bendali, F., Sanaa, M., Bichet, H., and Schelcher, F. (1999). Risk factors associated with diarrhoea in newborn calves. Vet. Res. 30, 509–522.
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Boranbayeva, T., Karahan, A. G., Tulemissova, Z., Myktybayeva, R., and Ozkaya, S. (2020). Properties of a new probiotic candidate and Lactobacterin-TK(2) against diarrhea in calves. Probiotics Antimicrob. Proteins 12, 918–928. doi: 10.1007/s12602-020-09649-4
Caffarena, R. D., Casaux, M. L., Schild, C. O., Fraga, M., Castells, M., Colina, R., et al. (2021). Causes of neonatal calf diarrhea and mortality in pasture-based dairy herds in Uruguay: a farm-matched case-control study. Braz. J. Microbiol. 52, 977–988. doi: 10.1007/s42770-021-00440-3
Chen, Y. J., Wu, H., Wu, S. D., Lu, N., Wang, Y. T., Liu, H. N., et al. (2018). Parasutterella, in association with irritable bowel syndrome and intestinal chronic inflammation. J. Gastroenterol. Hepatol. 33, 1844–1852. doi: 10.1111/jgh.14281
Diker, K. S., Diker, S., and Ozlem, M. B. (1990). Bovine diarrhea associated with campylobacter hyointestinalis. Zentralbl. Veterinarmed. B 37, 158–160. doi: 10.1111/j.1439-0450.1990.tb01040.x
Espeche, M. C., Otero, M. C., Sesma, F., and Nader-Macias, M. E. F. (2009). Screening of surface properties and antagonistic substances production by lactic acid bacteria isolated from the mammary gland of healthy and mastitic cows. Vet. Microbiol. 135, 346–357. doi: 10.1016/j.vetmic.2008.09.078
Fan, P., Bian, B. L., Teng, L., Nelson, C. D., Driver, J., Elzo, M. A., et al. (2020). Host genetic effects upon the early gut microbiota in a bovine model with graduated spectrum of genetic variation. ISME J. 14, 302–317. doi: 10.1038/s41396-019-0529-2
Fan, P., Nelson, C. D., Driver, J. D., Elzo, M. A., and Jeong, K. C. (2019). Animal breed composition is associated with the hindgut microbiota structure and beta-lactam resistance in the multibreed Angus-Brahman herd. Front. Microbiol. 10:1846. doi: 10.3389/fmicb.2019.01846
Fan, P., Nelson, C. D., Driver, J. D., Elzo, M. A., Penagaricano, F., and Jeong, K. C. (2021). Host genetics exerts lifelong effects upon hindgut microbiota and its association with bovine growth and immunity. ISME J. 15, 2306–2321. doi: 10.1038/s41396-021-00925-x
Fernandez, S., Fraga, M., Castells, M., Colina, R., and Zunino, P. (2020). Effect of the administration of lactobacillus spp. strains on neonatal diarrhoea, immune parameters and pathogen abundance in pre-weaned calves. Benefic. Microbes 11, 477–488. doi: 10.3920/BM2019.0167
Firehammer, B. D., and Myers, L. L. (1981). Campylobacter fetus subsp jejuni: its possible significance in enteric disease of calves and lambs. Am. J. Vet. Res. 42, 918–922.
Gomez, D. E., Arroyo, L. G., Costa, M. C., Viel, L., and Weese, J. S. (2017). Characterization of the fecal bacterial microbiota of healthy and diarrheic dairy calves. J. Vet. Intern. Med. 31, 928–939. doi: 10.1111/jvim.14695
Hennessy, M., Indugu, N., Vecchiarelli, B., Redding, L., Bender, J., Pappalardo, C., et al. (2021). Short communication: comparison of the fecal bacterial communities in diarrheic and nondiarrheic dairy calves from multiple farms in southeastern Pennsylvania. J. Dairy Sci. 104, 7225–7532. doi: 10.3168/jds.2020-19108
Hou, C. L., Zeng, X. F., Yang, F. J., Liu, H., and Qiao, S. Y. (2015). Study and use of the probiotic lactobacillus reuteri in pigs: a review. J. Anim. Sci. Biotechnol. 6:14. doi: 10.1186/s40104-015-0014-3
Huang, A., Cai, R., Wang, Q., Shi, L., Li, C., and Yan, H. (2019). Dynamic change of gut microbiota during porcine epidemic diarrhea virus infection in suckling piglets. Front. Microbiol. 10:322. doi: 10.3389/fmicb.2019.00322
Ianiro, G., Tilg, H., and Gasbarrini, A. (2016). Antibiotics as deep modulators of gut microbiota: between good and evil. Gut 65, 1906–1915. doi: 10.1136/gutjnl-2016-312297
Jia, D., Wang, Y., Wang, J. H., Liu, J. L., Li, H. H., Liu, A. H., et al. (2021). Lactobacillus animalis pZL8a: a potential probiotic isolated from pig feces for further research. 3 Biotech. 11:132. doi: 10.1007/s13205-021-02681-3
Karasova, D., Crhanova, M., Babak, V., Jerabek, M., Brzobohaty, L., Matesova, Z., et al. (2021). Development of piglet gut microbiota at the time of weaning influences development of postweaning diarrhea—A field study. Res. Vet. Sci. 135, 59–65. doi: 10.1016/j.rvsc.2020.12.022
Kim, H. S., Whon, T. W., Sung, H., Jeong, Y. S., Jung, E. S., Shin, N. R., et al. (2021). Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 12:161. doi: 10.1038/s41467-020-20389-5
Koppen, I. J. N., Velasco-Benitez, C. A., Benninga, M. A., Di Lorenzo, C., and Saps, M. (2016). Using the Bristol stool scale and parental report of stool consistency as part of the Rome III criteria for functional constipation in infants and toddlers. J. Pediatr. 177, 44–48. doi: 10.1016/j.jpeds.2016.06.055
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Ma, Z., Lee, S., Fan, P., Zhai, Y., Lim, J., Galvao, K. N., et al. (2021). Diverse beta-lactam antibiotic-resistant bacteria and microbial community in milk from mastitic cows. Appl. Microbiol. Biotechnol. 105, 2109–2121. doi: 10.1007/s00253-021-11167-4
Marchesi, J. R., Sato, T., Weightman, A. J., Martin, T. A., Fry, J. C., Hiom, S. J., et al. (1998). Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Environ. Microbiol. 64, 795–799. doi: 10.1128/AEM.64.2.795-799.1998
Nakajima, H., Takewaki, F., Hashimoto, Y., Kajiyama, S., Majima, S., Okada, H., et al. (2020). The effects of metformin on the gut microbiota of patients with type 2 diabetes: A two-center, quasi-experimental study. Lifestyles 10:195. doi: 10.3390/life10090195
Nicholson, J. K., Holmes, E., Kinross, J., Burcelin, R., Gibson, G., Jia, W., et al. (2012). Host-gut microbiota metabolic interactions. Science 336, 1262–1267. doi: 10.1126/science.1223813
Pickard, J. M., Zeng, M. Y., Caruso, R., and Nunez, G. (2017). Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 279, 70–89. doi: 10.1111/imr.12567
Ramirez, J., Guarner, F., Fernandez, L. B., Maruy, A., Sdepanian, V. L., and Cohen, H. (2020). Antibiotics as major disruptors of gut microbiota. Front. Cell. Infect. Microbiol. 10:572912. doi: 10.3389/fcimb.2020.572912
Renaud, D. L., Kelton, D. F., Weese, J. S., Noble, C., and Duffield, T. F. (2019). Evaluation of a multispecies probiotic as a supportive treatment for diarrhea in dairy calves: a randomized clinical trial. J. Dairy Sci. 102, 4498–4505. doi: 10.3168/jds.2018-15793
Schluter, J., Peled, J. U., Taylor, B. P., Markey, K. A., Smith, M., Taur, Y., et al. (2020). The gut microbiota is associated with immune cell dynamics in humans. Nature 588, 303–307. doi: 10.1038/s41586-020-2971-8
Virginio Junior, G. F., and Bittar, C. M. M. (2021). Microbial colonization of the gastrointestinal tract of dairy calves—a review of its importance and relationship to health and performance. Anim. Health Res. Rev. 1–12. doi: 10.1017/S1466252321000062 [Epub ahead of print]
Whon, T. W., Kim, H. S., Shin, N. R., Sung, H., Kim, M. S., Kim, J. Y., et al. (2021). Calf diarrhea caused by prolonged expansion of autochthonous gut Enterobacteriaceae and their lytic bacteriophages. msystems 6, e00816–e00820. doi: 10.1128/mSystems.00816-20
Yang, L., Lin, Q., Han, L., Wang, Z., Luo, M., Kang, W., et al. (2020). Soy hull dietary fiber alleviates inflammation in BALB/C mice by modulating the gut microbiota and suppressing the TLR-4/NF-kappaB signaling pathway. Food Funct. 11, 5965–5975. doi: 10.1039/D0FO01102A
Yu, H., Ma, Z., Meng, S., Qiao, S., Zeng, X., Tong, Z., et al. (2021). A novel nanohybrid antimicrobial based on chitosan nanoparticles and antimicrobial peptide microcin J25 with low toxicity. Carbohydr. Polym. 253:117309. doi: 10.1016/j.carbpol.2020.117309
Zeineldin, M., Aldridge, B., and Lowe, J. (2018). Dysbiosis of the fecal microbiota in feedlot cattle with hemorrhagic diarrhea. Microb. Pathog. 115, 123–130. doi: 10.1016/j.micpath.2017.12.059
Zhao, Q., and Elson, C. O. (2018). Adaptive immune education by gut microbiota antigens. Immunology 154, 28–37. doi: 10.1111/imm.12896
Keywords: gut microbiota, diarrhea, newborn calves, Lactobacillus, probiotics
Citation: Fan P, Kim M, Liu G, Zhai Y, Liu T, Driver JD and Jeong KC (2021) The Gut Microbiota of Newborn Calves and Influence of Potential Probiotics on Reducing Diarrheic Disease by Inhibition of Pathogen Colonization. Front. Microbiol. 12:772863. doi: 10.3389/fmicb.2021.772863
Edited by:
Si Hong Park, Oregon State University, United StatesReviewed by:
Hyun Joong Kim, Mokpo National University, South KoreaRaies Mir, United States Department of Agriculture (USDA), United States
Copyright © 2021 Fan, Kim, Liu, Zhai, Liu, Driver and Jeong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kwangcheol C. Jeong, kcjeong@ufl.edu
†These authors have contributed equally to this work