 Jia xin Zhong1
Jia xin Zhong1 Yuan yuan Wang
Yuan yuan Wang Xiao li Du
Xiao li Du Yuan Wu
Yuan Wu Jin xing Lu
Jin xing Lu- 1State Key Laboratory of Infectious Disease Prevention and Control, Chinese Center for Disease Control and Prevention, National Institute for Communicable Disease Control and Prevention, Beijing, China
- 2Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Hangzhou, China
Clostridium perfringens (C. perfringens) is a significant foodborne pathogen and a common cause of intestinal diseases in both animals and humans. Our study investigated MLST, phenotypic antimicrobial resistance profiles, and resistance genes among isolates from human, animal and food. 186 C. perfringens isolates were obtained from nine provinces in China between 2013 and 2021. Additionally, some specific ST complexes were analyzed by cgMLST and cgSNP to investigate genetic relatedness. MLST indicated the most prevalent STs of C. perfringens of human and animal origin were as follows: ST221 (5/147), ST62 (4/147), ST408 (4/147), and ST493 (4/147) were predominant in humans, while ST479 (5/25) was the major type in animals. Within the same ST complex, genetically unrelated relationships or potential clustering/transmission events were further recognized by cgMLST and cgSNP, illustrating that these two methods are valuable in defining outbreaks and transmission events. All tested isolates were susceptible to vancomycin and meropenem. The rates of resistance to metronidazole, penicillin, cefoxitin, moxifloxacin, and chloramphenicol were low (metronidazole: 1.08%; penicillin: 9.68%; cefoxitin: 0.54%; moxifloxacin: 6.45%; and chloramphenicol: 3.76%). Interestingly, 49.66% of human origin were clindamycin-resistant, and 18.2% were penicillin-insensitive. Importantly, the portion of MDR isolates was significantly lower than in previous reports. The study provides an overview of the epidemiological characteristics of C. perfringens with different origins and hosts in China. C. perfringens demonstrated remarkable genetic diversity and distinct molecular features compared to antibiotic-resistance profiles from other studies.
Introduction
Clostridium perfringens (C. perfringens) is a Gram-positive anaerobic bacterium that is abundant in nature. The bacteria can cause gas gangrene, intestinal diseases, and food poisoning in both animals and humans (Brynestad and Granum, 2002; Kiu and Hall, 2018). The significant pathogenic mechanism of C. perfringens is the production of a wide variety of toxins and enzymes (Li et al., 2013). C. perfringens is classified into seven types: A (ɑ), B (ɑ, β, ƹ), C (ɑ, β), D (ɑ, ƹ), E (ɑ, L), F (ɑ, cpe), and G (ɑ, netB) by a toxin-based typing scheme (Li et al., 2013).
Antibiotic-associated diarrhea (AAD) continues to be a major healthcare issue, both in hospitalized patients and in the community (Kyne et al., 2002). Many investigations have revealed that C. perfringens played an important role in diarrheic patients with a history of antibiotic use (Carman, 1997; Azimirad et al., 2019). According to some studies, C. perfringens is responsible for 5–20% of all cases of AAD and sporadic non-foodborne diarrhea (Carman, 1997; Motamedi et al., 2021). Recently, an increase in C. perfringens antimicrobial resistance has been noted, and some investigations have indicated that most C. perfringens strains of animal origin are multidrug resistant (MDR) (Xu et al., 2021; Bendary et al., 2022). However, in China the research on C. perfringens antimicrobial resistance is limited.
The technique of MLST is widely applied for the identification of human, animal, and foodborne pathogens, and it has also been used to analyze bacterial population diversity and investigate C. perfringens outbreaks (Lacey et al., 2016). With the increasing application of whole-genome sequencing (WGS), high-resolution molecular subtyping approaches such as core genome MLST (cgMLST) based on WGS have gained popularity in epidemic investigation and bacterial tracing (Abdel-Glil et al., 2021a,b). Previous research has shown that C. perfringens from different sources had a large amount of genetic variation (Park et al., 2016; Álvarez-Pérez et al., 2017; Lacey et al., 2018), and human strains demonstrated more variation than animal strains (Nakano et al., 2017). However, the molecular characteristicistics of C. perfringens isolates from different sample origins and regions in China are limited. Therefore, this research aimed to provide valuable epidemiological data on genotype distribution, antibiotic resistance, toxinotype, genetic diversity, and phylogenetic characteristics of C. perfringens from distinct geographical regions in China. The findings can provide a deeper understanding of the molecular epidemiology of C. perfringens in China and offer a basis for the development of rapid detection and surveillance networks.
Materials and methods
Sample collection
A total of 186 C. perfringens strains were recovered and cultivated in our lab and characterized using 16S rRNA molecular technique. These isolates were obtained from people, including patients and healthy people, domestic animals (sheep, calves, and pigs), and food from nine different regions across China, from 2013 to 2021 (Figure 1). There were 47 isolates from Shijiazhuang, 81 isolates from Shandong, 16 isolates from Yunnan, three isolates from Sichuan, 11 isolates from Wenzhou, 14 isolates from Yulin, six isolates from Xi’an, two isolates from Hunan, and six isolates from Beijing (Figure 1). Among these 186 strains, 139 strains were from clinical sources; 24 strains were from animal sources and 14 were from food sources. Supplementary Table S1 contains a complete list of isolates background found in this study.
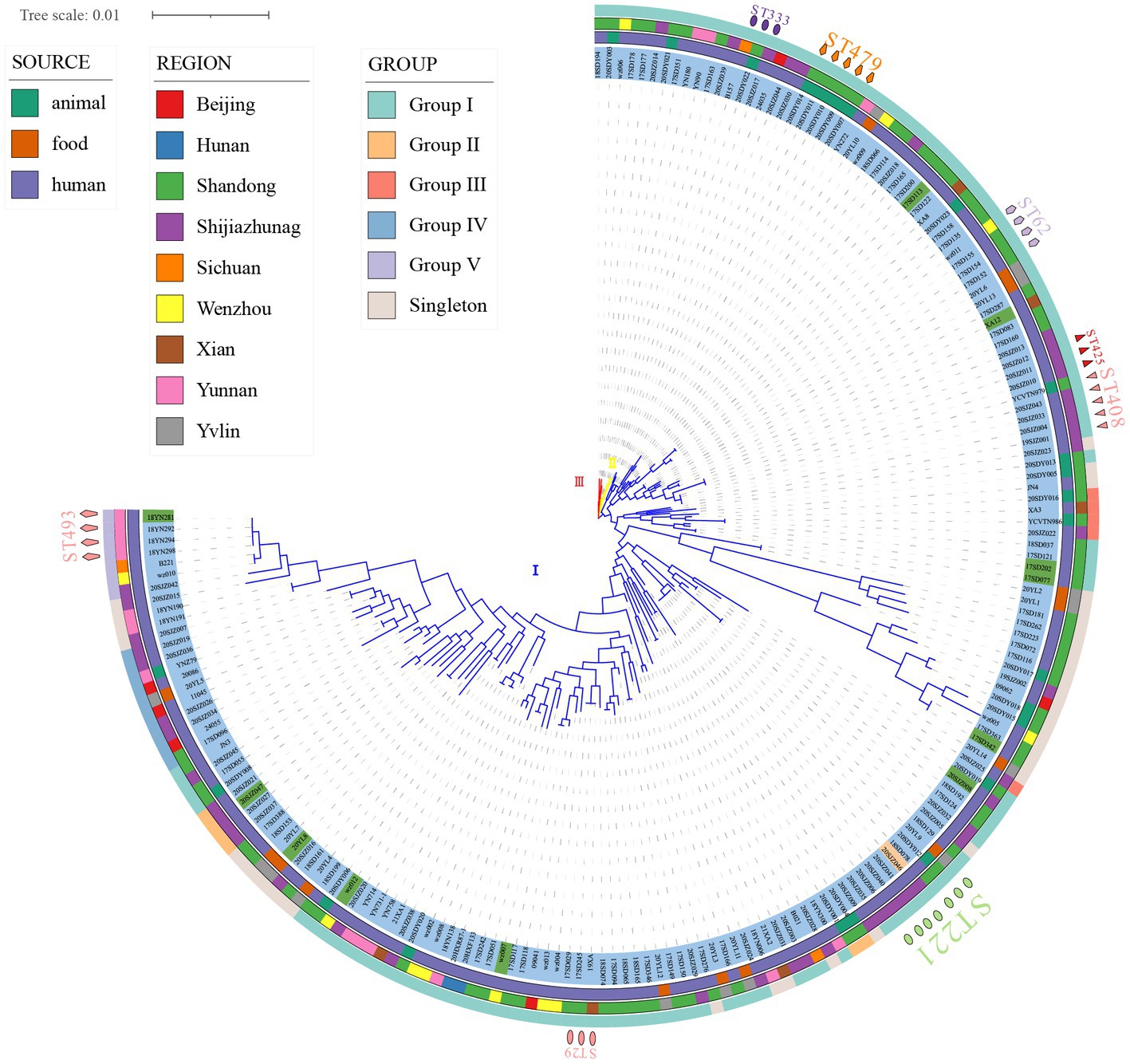
Figure 1. Phylogenetic maximum likelihood tree based on MLST identified in 186 strains of Clostridium perfringens (bootstrap test = 1,000 replicates) by fasttree. Three distinct branches are shown through the phylogenetic tree (blue, cluster I; yellow, cluster II; red, cluster III). The figure shows the population structure of the strains with toxinotype next to the ML tree (blue, type A; green, type F; and orange, type D), followed by the region, source, and ST of isolation as in the legend. The phylogenetic tree was visualized using iTOL.
Isolation and identification of C. perfringens
After ethanol shock treatment, all fecal specimens were inoculated with 5% egg yolk on selective tryptose sulfite cycloserine (TSC) agar (Oxoid, Basingstoke, UK) plates and incubated at 37°C for 48 h in an anaerobic jar (Mart, NL). Following vacuuming, an anaerobic atmosphere of 80% nitrogen, 10% hydrogen, and 10% carbon dioxide was injected. C. perfringens colonies were identified based on biochemical tests, colony and cell morphology, and other characteristics, and the 16S rRNA gene was used to identify the bacteria (short Gram-positive bacilli, dual hemolysis, and gelatinase and pectinase-producing bacteria) (Kikuchi et al., 2013).
DNA extraction
All isolates used in this study were maintained on BHI plates with 5% sheep agar under anaerobic conditions. The genomic DNA of these isolates was extracted using a bacterial genomic DNA extraction kit (Qiagen, Germany) according to the manufacturer’s instructions. DNA was then dissolved in 100 μL of TE (10mMTris-HCl1mM EDTA PH = 8.0) and stored at −20°C until use.
MLST and phylogenetic analysis
The eight gene loci (colA, groEL, sodA, plc, gyrB, sigK, pgk, and nadA) listed in the public C. perfringens MLST database1 were used in this study (Table 1) (Xiao et al., 2012). PCR assays were performed in final volumes of 25 μL containing 12.5 μL of 2 × PCR Taq Mastermix (MgCl, dNTP, Taq enzyme) (Takara Bio Inc., Japan); 0.5 μL of each primer (10 mmol/L); 1.5 μL of DNA template; and double-distilled water for a final volume of 25 μL. Reactions were performed with initial denaturation at 94°C for 2 min, followed by 35 cycles at 94°C for 30 s, at 55°C for 60 s and at 72°C for 60 s, and a final extension at 72°C for 8 min. Then, the PCR products were purified using a commercial kit (Omega Bio-Tek, United States) and were sent for Sanger nucleotide sequencing in both directions (Beijing DIA-UP BIOTECH Co., Ltd., China).
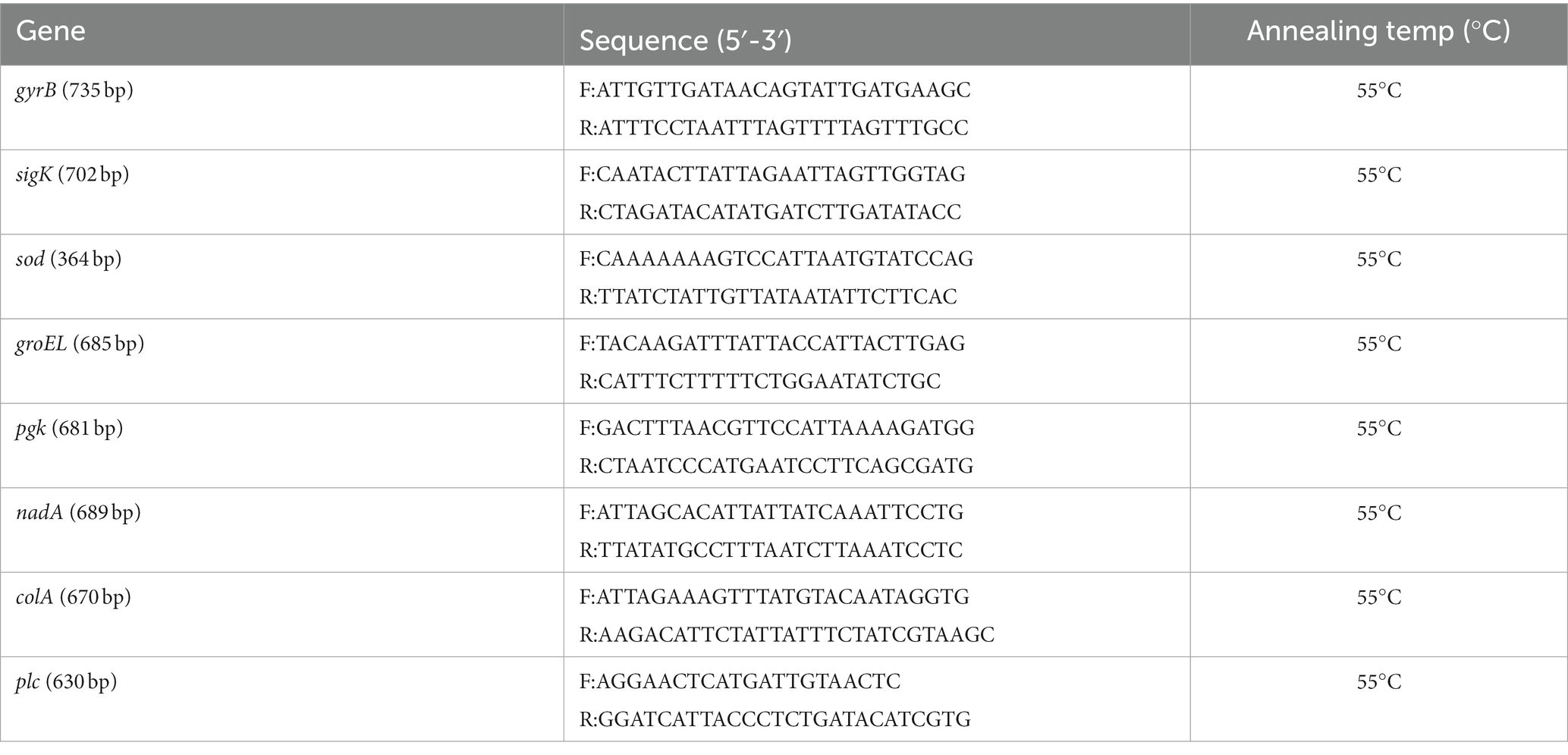
Table 1. The primer sequences of Clostridium perfringens housekeeping genes.
To determine the sequence type (ST) of all the tested 186 strains of C. perfringens, DNA sequences were submitted to a public C. perfringens MLST database (see text footnote 1) (Jolley et al., 2018). New alleles and STs were assigned in the C. perfringens MLST database. The minimum-spanning tree was constructed using BioNumerics software version 5.1. The tree in Figure 1 is based on nucleotide sequences, while the tree in Figure 2 is based on the allele matrix.
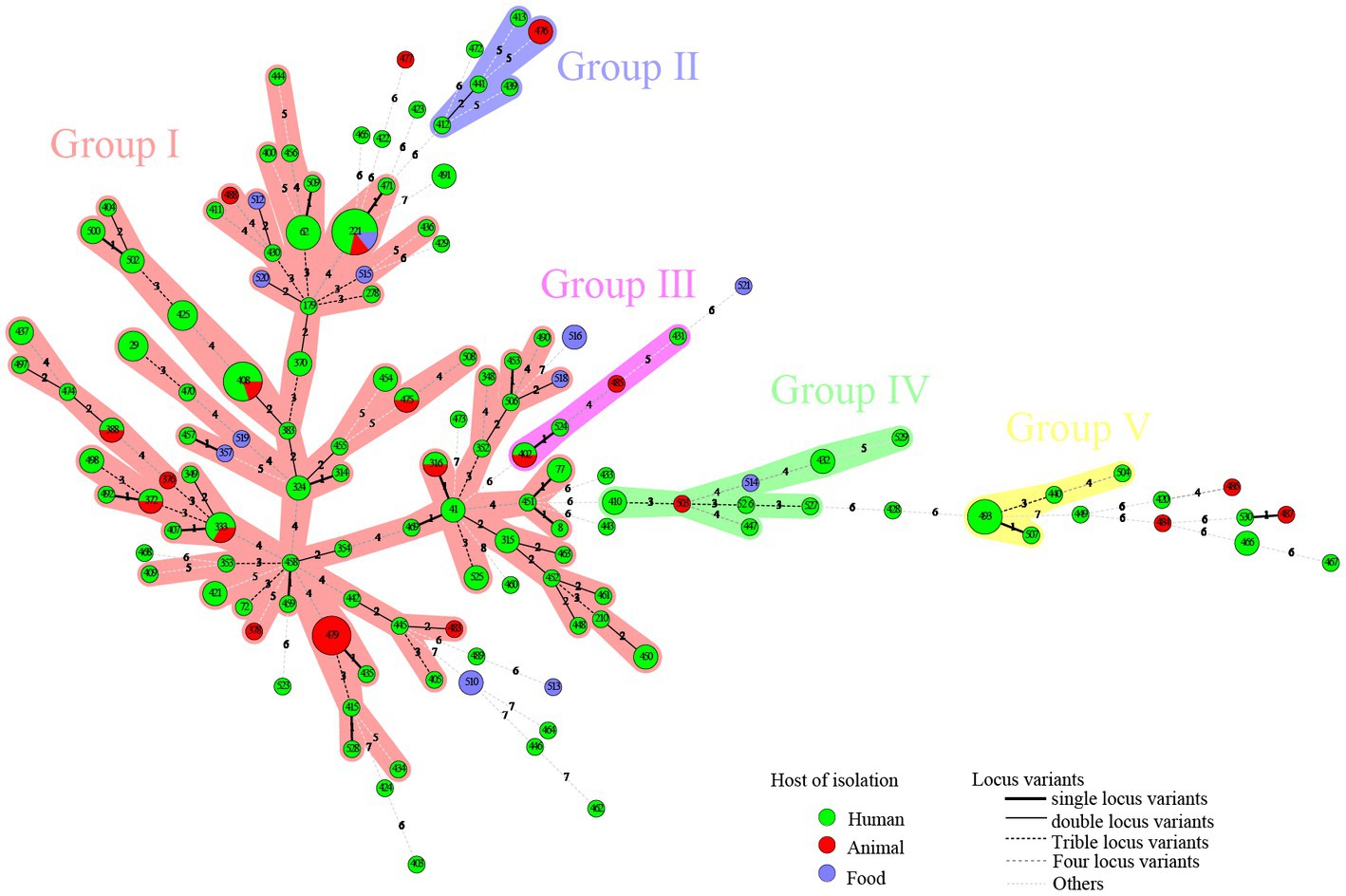
Figure 2. A total of 186 strains of C. perfringens from different sources were analyzed by constructing an MLST-minimal spanning tree. The minimum-spanning tree was constructed using the Bionumerics software (Bionumerics, version 5.1). The shaded section represents five clusters. The clusters were assigned for at least three types within five changes of neighbor distance. The area of the circle represents the number of strains; different colors represent different hosts; and the number on the branch represents the difference compared to housekeeping genes.
835 isolates were retrieved from public C. perfringens MLST database (see text footnote 1). Global C. perfringens MLST analysis was performed by online analysis on PubMLST.2
Toxinotype detection
Using previously described multiplex PCR assays, the isolates were examined for the presence of the cpa, cpb, etx, iap, cpb2, cpe, and netB genes (Yoo et al., 1997; Bailey et al., 2013; Hu et al., 2018; Rood et al., 2018). In this investigation, the multiplex PCR was carried out using the reference strains C. perfringens NCTC 528 (cpa), NCTC 3180 (cpb), NCTC 4989 (cpb, cpb2), NCTC 8346 (etx), NCTC 8084 (iap, cpe), and NetB-encoding plasmid (pNetB) as positive controls.
Sequencing and whole-genome sequences analysis
Genomic DNA was extracted and purified using the Wizard genomic DNA purification kit (Promega, United States). Whole-genome sequencing was carried out by Illumina HiSeq X 10 sequencing platform. Removing adapter sequences and low-quality sequences was performed using Trimmomatic version 0.39. Assembly was performed with SPAdes v3.11.1.
cgMLST and evolutionary relationship analysis
A C. perfringens cgMLST scheme established by Abdel-Glil et al. (2021b) comprising 1,431 target loci based on genomic sequences was used, and neighbor-joining was used to create dendrograms based on cgMLST information via the PubMLST website (see text footnote 1).
An alignment of SNPs in the core genome using ATCC 13124 as a reference was constructed using Snippy (version 4.6) and the maximum likelihood tree was constructed using fasttree (version 2.1.10).
Antimicrobial susceptibility test
The E-test method was applied for antimicrobial susceptibility analysis (Liofilchem s. r. l., Roseto degli Abruzzi, Italy) of the C. perfringens isolates. The following eight antimicrobials were tested: clindamycin, chloramphenicol, vancomycin, cefoxitin, meropenem, metronidazole, penicillin, and moxifloxacin. Penicillin and Clindamycin are the antibiotics of first choice for treating infections caused by C. perfringens. Other drugs are also common drugs in drug resistance studies of C. perfringens with powerful bactericidal action against gram-positive bacteria or anaerobes.
Isolates were suspended in Brucella broth to achieve a turbidity of 1.0 McFarland, and then inoculated onto Brucella agar supplemented with 5% sheep blood, 1 mg/L Vitamin K1, and 5 mg/L Hemin Chloride. Plates were incubated at 37°C for 18–24 h in anaerobic conditions. C. perfringens ATCC 13124 was used as a quality control. MDR was characterized as resistance to at least one agent in three or more antimicrobial classes (Magiorakos et al., 2012).
The interpretation criteria for Gram-positive anaerobic bacteria are available from the Clinical and Laboratory Standards Institute (CLSI) (Ferraro, 2001) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (Giske et al., 2022). MIC breakpoints for clindamycin (≥8 μg/mL), chloramphenicol (≥32 μg/mL), cefoxitin (≥64 μg/mL), meropenem (≥16 μg/mL), metronidazole (≥32 μg/mL), penicillin (≥2 μg/mL), moxifloxacin (≥8 μg/mL) (Ferraro, 2001), and vancomycin (>2 μg/mL) (Giske et al., 2022) were used to read the breakpoints.
Antibiotic resistant gene profiles
Antimicrobial resistance genes were screened using the ABRicate pipeline3 on the Comprehensive Antibiotic Resistance Database (CARD).4 Query genes with an identity higher than 80% and a coverage higher than 50% were considered potential antimicrobial resistance genes.
Results
Evolution and phylogenetic analysis of C. perfringens isolates in China by MLST
MLST analysis revealed that 186 C. perfringens isolates exhibited 135 sequence types (STs) with a high degree of genetic diversity, including 93 new STs. In the segments of colA (Kikuchi et al., 2013), groEL (Álvarez-Pérez et al., 2017), sodA (Jolley et al., 2018), plc (Rood et al., 2018), gyrB (Abdel-Glil et al., 2021b), sigK (Bendary et al., 2022), pgk (Xu et al., 2021), and nadA (Jolley et al., 2018), up to 129 new alleles were identified, resulting in new genotypes. Among the 135 STs, the most frequent ST was ST221 (7/186, 3.74%), followed by ST408 (5/186, 2.67%), ST479 (5/186, 2.67%), ST62 (4/186, 2.14%), and ST493 (4/186, 2.14%). There were 102 STs containing only one isolate. The 186 strains were from humans (n = 147), animals (n = 25) and food (n = 14) (Figure 2). The human strains comprised 110 STs, with ST221 (5/147), ST62 (4/147), ST408 (4/147), and ST493 (4/147) being the predominant STs. The animal strains contained 19 STs, with ST479 (5/25) and ST476 (2/25) being the predominant STs (Figure 2). Supplementary Table S1 contains a complete list of STs found in this study.
Of the 186 total examined isolates, 174 (93.55%) belonged to type A; 11 isolates (5.91%) were classified to type F; one isolate (0.54%) was classified to type D (Figure 1); and none of the investigated isolates were types B, C, and/or E. Clustering of the 186 genomes revealed that 91.94% (n = 171) belonged to cluster I, followed by cluster II (n = 10) and III (n = 5) (Figure 1). Cluster I, the most abundant subgroup, contained all predominant STs (ST221, ST408, ST479, ST62, and ST493), all toxinotypes (A, F, D), and all sources and all regions (Figure 1). As described, cluster I was associated with isolates from humans, food, and animals; and this cluster had a broad ecological distribution (Figure 1). C. perfringens type F and type D strains were also concentrated in cluster I (Figure 1). Cluster II contained ST333 (n = 3) as the predominant type and also included ST372 (n = 2) and five unique STs (Figure 1). Cluster III contained five isolates with different STs (Figure 1). Two animal isolates and eight human isolates were included in cluster II, while one animal isolate and four human isolates were included in cluster III (Figure 1). Strains in cluster II and cluster III were all type A.
There were no significant host, geographic distribution, or toxinotype connections among the strains involved (Figure 1). In the phylogenetic maximum likelihood tree, there were several STs, including isolates with different regions, hosts, or different toxinotypes within the same ST such as ST221, ST408, ST333, ST372, ST316, ST388, ST402, and ST475, indicating that isolates may have potentially spread across different hosts or regions (Figure 1). For example, ST221 contained seven strains from clinical diarrhea patients [Shijiazhuang (n = 4), Shandong (n = 1)], sheep [Shandong (n = 1)] and food [Yulin (n = 1)] (Figure 1). ST408 consisted of five strains originating from AAD patients [Shijiazhuang (n = 4)] and a calf [Shandong (n = 1)] (Figure 1).
A total of five clusters (cluster I–cluster V) were found in the minimal spanning tree (Figure 2) among the studied strains isolated in this investigation, accounting for 82.80% (154/186) of the strains. Thirty-two STs were determined to be singletons, with no observed cluster relationships. Cluster I, the most prolific cluster, included 80 STs (including advantage STs ST221, ST408, ST479, ST62, ST333, and ST493), with a total of 122 strains that accounted for 65.59% (122/186) of all detected strains. Cluster II contained four clinical origin strains (ST412, ST413, ST439, and ST441) and two animal origin strains (ST476), accounting for 3.23% (6/186) of the examined strains. Cluster III contained three clinical origin strains (ST402, ST431, and ST524) and two animal origin strains (ST402 and ST485), accounting for 2.69% (5/186) of the examined strains. Cluster IV contained eight human origin strains (ST410, ST432, ST447, ST526, ST527, and ST529), one animal origin strain (ST503), and one food origin strain (ST514), accounting for 5.38% (10/186) of the examined strains. Cluster V contained seven human origin strains (ST440, ST493, ST504, and ST507), accounting for 3.76% (7/186) of the examined strains; these showed genetic distance (≥6 allele differences) with other isolates in the minimum-spanning tree (Figure 2) and significant genetic distance (≥7 allele differences) with other clusters (Figure 2). Half of the food origin strains were dispersed as singletons, while most strains with animal origin clustered with strains of human origin, indicating the character of the zoonotic food-borne pathogen C. perfringens.
Interestingly, the distribution of isolates from multiple sources in the minimum-spanning tree (Figure 2) and in the phylogenetic tree (Figure 1) was not consistent. Due to different mathematical calculation models (the minimum-spanning tree was based on a matrix, while the phylogenetic tree was based on nucleic acid sequences), some strains belonging to similar branches in the phylogenetic tree did not form the same clusters in the minimum-spanning tree. For example, some isolates in cluster I were assigned to different clusters of the minimum-spanning tree (Figures 2, 3).
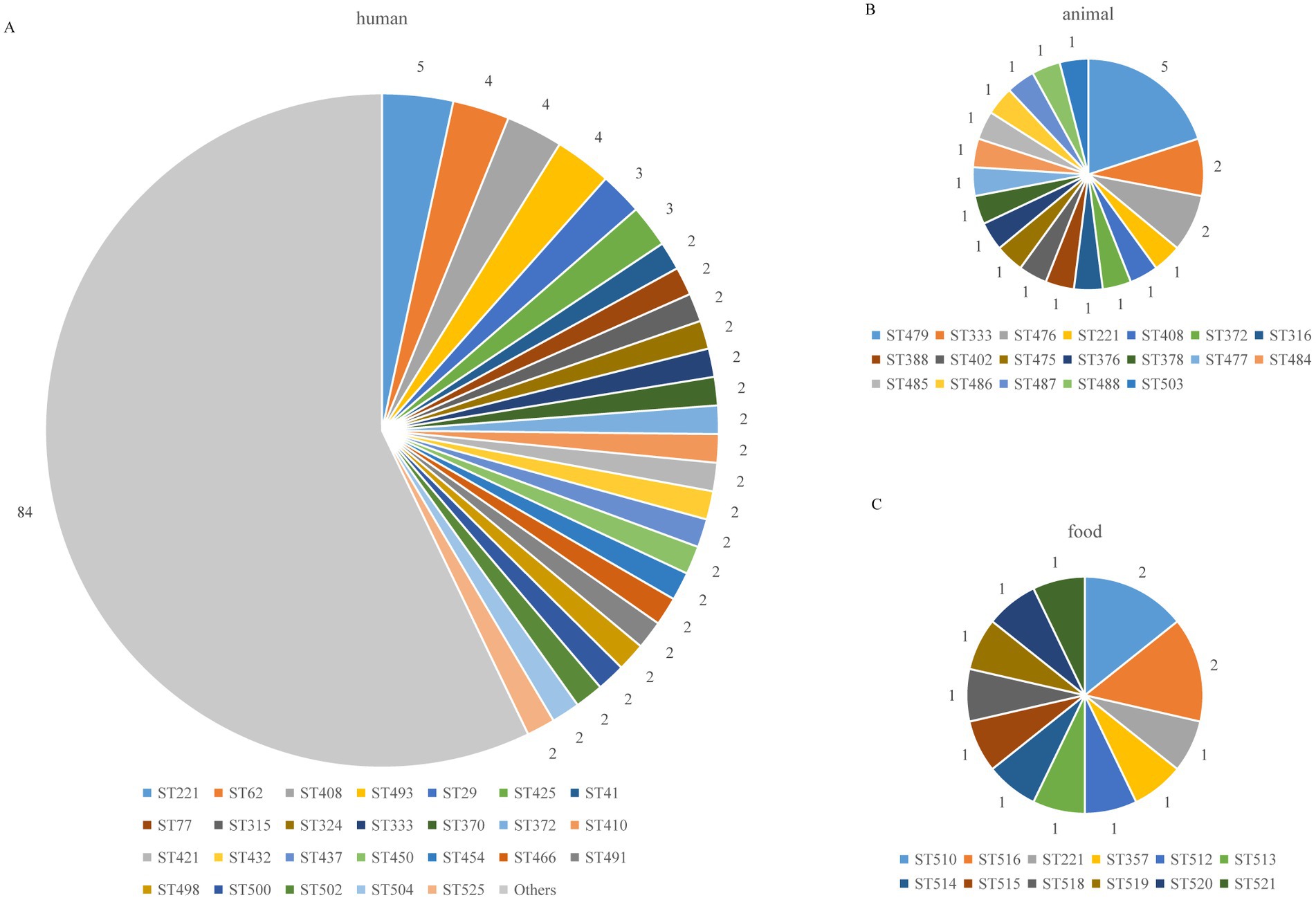
Figure 3. Distribution of C. perfringens ST types from different hosts. The “Others” include ST types with only 1 isolate; the number of other STs was 84: (A) human (B) animal (C) food.
Global C. perfringens MLST analysis
We performed MLST analysis on 835 strains isolated from different sources (clinical, animal, food and environment) (Figure 4). A total of 592 STs were identified (Figure 4). The global strains can be divided into three distinct clusters, and Chinese strains from database (n = 337), scattered among the three clusters (Figure 4). Japanese strains are mostly clustered in clusterI (Figure 4). The majority single ST types correlated to one isolate from one country (Figure 4). The main predominant types globally were ST80 (n = 24), ST248 (n = 15), ST251 (n = 11), ST73 (n = 11), ST221 (n = 10) (Figure 4). Among them, ST80, ST248, and ST73 are found in multiple nations (Figure 4). The molecular type ST80 strains came from the United States (n = 13), Canada (n = 9), and Switzerland (n = 2), the ST248 strains came from the United States (n = 13) and Finland (n = 2), and the ST73 strains came from the United States (n = 7), Australia (n = 3), and Canada (n = 1).

Figure 4. Minimum spanning tree (MST) of MLST for global C. perfringens as generated within PubMLST using GrapeTree. MST representing evolutionary relationships of C. perfringens sequence types (STs) between Chinese isolates and other 21 countries isolates. Isolates from one country were marked by one color. The size of the circle is corresponding to the number of isolates within that genotype. Numbers inside circles are STs and numbers over connecting lines are locus variant levels.
Cgmlst and cgSNP evolutionary relationship analysis of the same STs
Figure 3 shows the genetic relatedness of strains in the same STs on the cgMLST scheme from PubMLST based on the presence of the genes and sequences of 1,431 highly conserved core genes.
Isolates with the same ST221 were divided into seven cgMLST types, comprising five from clinical human isolates, one from food, and one from animals (Figure 5A). The seven genomes showed ≤100 allele differences (range 20–100). Furthermore, the food isolate cp2020YL9 (ST221) and sheep isolate cp2020SDY012 (ST221) showed 35 and 100 allele differences, respectively, compared to the human cp2018SD078 (ST221) isolate, indicating that these isolates with identical ST were genetically unrelated (Figure 5A).
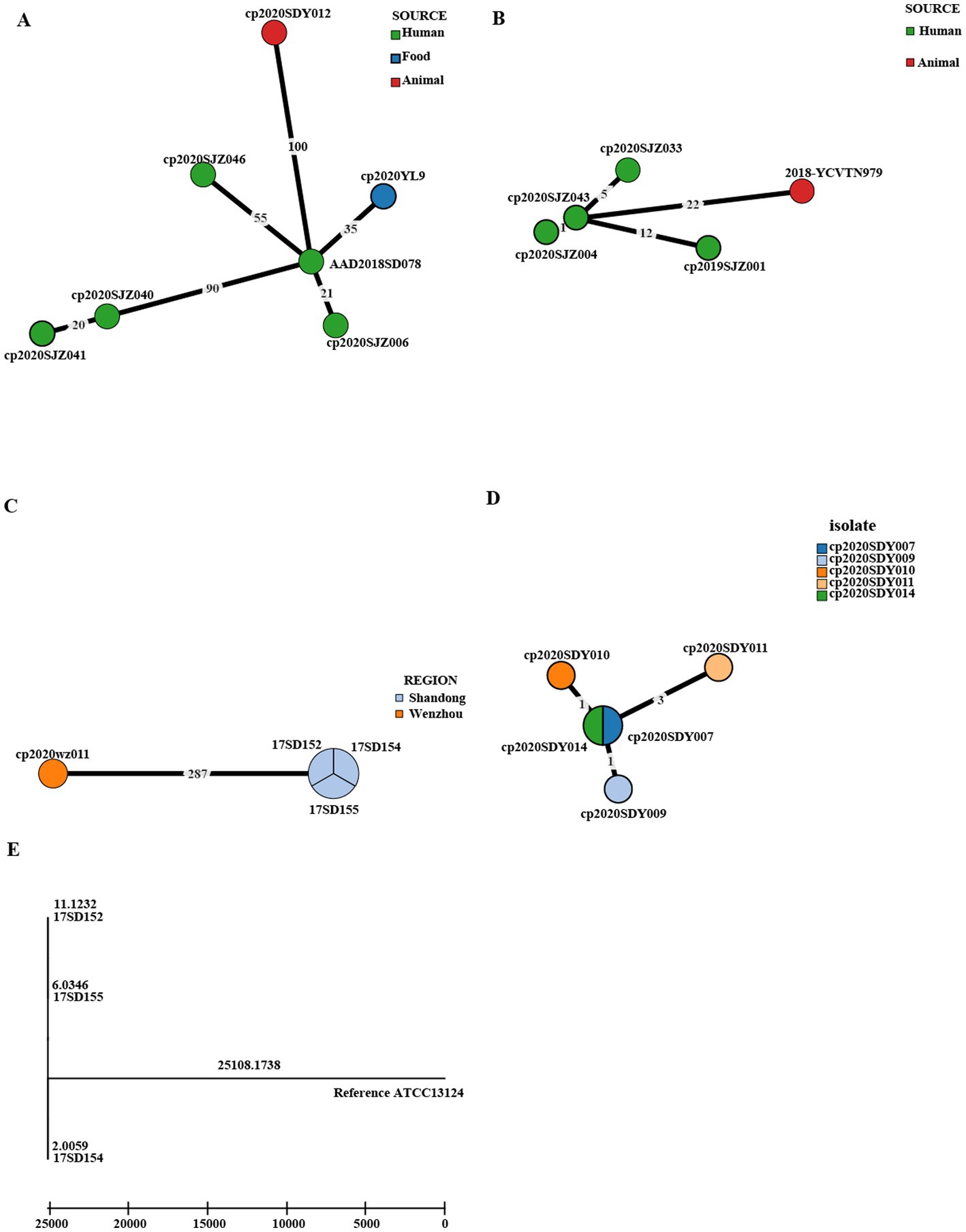
Figure 5. Phylogenetic tree of cgMLST and core SNP typing. (A–D) GrapeTree minimum-spanning tree of strains in the same STs based on the PubMLST cgMLST. The numbers on the branches represent the differences in alleles between neighboring nodes. (A) ST221, (B) ST408, (C) ST62, and (D) ST479. (E) A maximum likelihood (ML) phylogenetic tree of ST62 based on SNPs constructed by snippy and fasttree version 2.1.10 using ATCC 13124 as a reference.
In addition, within the ST408 complex, fetal cattle isolate 2018-YCVTN979 showed 22 allele differences compared to human isolate cp2020SJZ043, indicating that these isolates with identical cgMLST type were genetically heterogeneous (Figure 5B). However, the three human clinical isolates cp2020SJZ004, cp2020SJZ043, and cp2020SJZ033 showed high genetic relatedness with ≤5 allele differences.
Moreover, the three AAD strains with the same ST 62 isolated from the same hospital at Shandong in 2017 (17SD152, 17SD154 and 17SD155) were also classified to the same cgMLST type showing no allele differences (Figure 5C), strongly suggesting that they had arisen from a single source or were associated with the same transmission event. To confirm our hypothesis, cgSNP analysis was further performed for these three isolates (Figure 5E). The size of the core genome for C. perfringens isolates included in this study was 22,493 bp, and the average size of the C. perfringens genome for isolates sequenced in this study was 3.26 Mb. A phylogenetic tree was produced using the 6,914 bp core genome SNP alignment. Within 6,914 variant sites, pairwise SNP diversity ranged from 20 to 22 SNPs for the three genomes (Supplementary Table S1), illustrating that these three isolates had clear genetic differences. Notably, there was another strain (cp2020wz011) in the ST 62 complex that displayed 287 allele differences with the above three AAD strains (Figure 5C).
Furthermore, within the ST 479 complex, five isolates from sheep in Shandong were divided into four cgMLST types, including cp2020SDY007 and cp2020SDY014 that belonged to the same cgMLST type (Figure 5D). The five genomes showed ≤3 allele differences (range 0–3) below the cutoff threshold of seven allele differences.
Antibiotic-resistance profiles
Of the 186 strains tested, 96 were resistant to at least one antibiotic. All of the C. perfringens isolates showed 100% susceptibility to vancomycin and meropenem. The rates of resistance to metronidazole, penicillin, cefoxitin, moxifloxacin, and chloramphenicol were low (metronidazole: 1.08%; penicillin: 9.68%; cefoxitin: 0.54%; moxifloxacin: 6.45%; and chloramphenicol: 3.76%) (Figure 6A). However, these isolates displayed much higher rates of antibiotic resistance to clindamycin (47.85%) (Figure 6A).
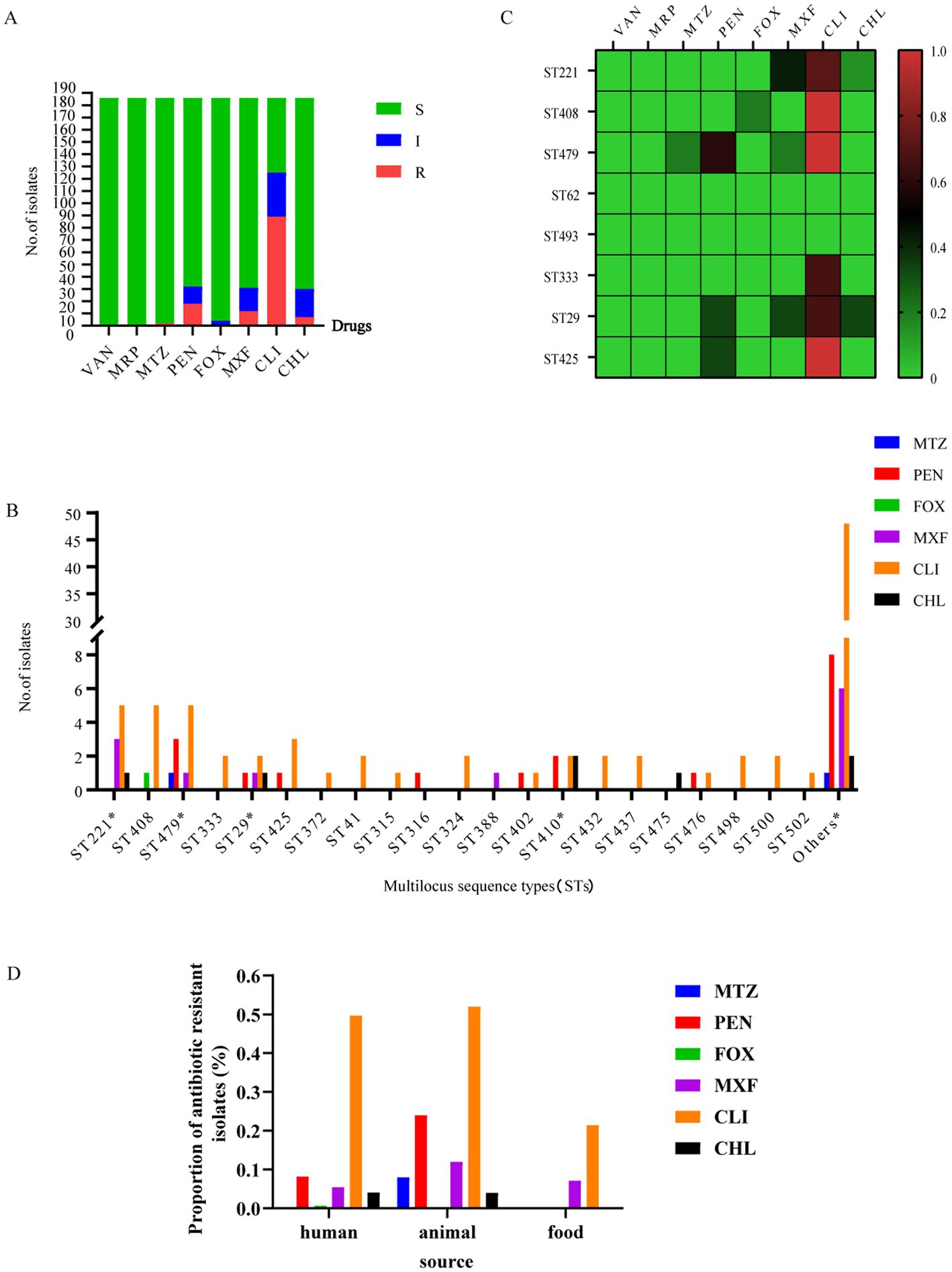
Figure 6. Antibiotic-resistance profiles of 186 C. perfringens strains. (A) Isolates sensitive, intermediate, or resistant to antimicrobial agents for 186 C. perfringens strains. S, sensitive; I, intermediate; and R, resistant. (B) Number of drug-resistant strains of different ST types of the 186 C. perfringens isolates.*, MDR strain. The “Others” group include resistance to antibiotics of ST types with only 1 strain; the number of other STs was 102. metronidazole (MTZ), penicillin (PEN), cefoxitin (FOX), moxifloxacin (MXF), clindamycin (CLI), chloramphenicol (CHL). (C) The heatmap correlating ST with antibiotic resistance. The percentages of resistance to antimicrobials are color-coded on the right side of the figure. Colors represent the proportion of ST-type resistance. Red indicates high resistance, brown indicates medium resistance and green indicates low resistance. The data analyzed here are from samples ≥3. (D) The percentages of resistant strains presented as the three different groups -human, animal and food.
These antibiotics (metronidazole, cefoxitin, moxifloxacin and chloramphenicol) with lower resistance rates, ST29 was the major molecular type of antibiotic-resistant C. perfringens (Figure 6B). Furthermore, there was an MDR strain (AAD2019XAzhou) in ST29 (Figure 6B). Moreover, genotypes ST408 and ST479 were the predominant types among clindamycin-resistant isolates, followed by ST221 and ST425 (Figure 6B). Overall, ST479, ST408, ST29, and ST410 were the most commonly identified genotypes that correlated with a high proportion of antibiotic resistance (Figure 6C). Moreover, the strains in cluster V were sensitive to all of the above antibiotics (Supplementary Tables S1, S2).
For isolates of human origin, all were susceptible to vancomycin, meropenem, and metronidazole. The rates of cefoxitin (0.68%), chloramphenicol (4.08%), moxifloxacin (5.44%), and penicillin (8.16%) resistance were low. The resistance rate to clindamycin (49.66%) was much higher (Figure 6D). For isolates of animal origin, all were susceptible to vancomycin, cefoxitin, and meropenem. The antibiotics to which the tested isolates were resistant were clindamycin (52%), penicillin (24%), moxifloxacin (12%), metronidazole (8%), and chloramphenicol (4%) (Figure 6D). For isolates of food origin, all isolates were susceptible to vancomycin, meropenem, metronidazole, penicillin, cefoxitin, and chloramphenicol (Figure 6D). The resistance rates of moxifloxacin and clindamycin were 7.14 and 21.43%, respectively (Supplementary Table S1).
The antimicrobial susceptibility testing showed that the multidrug resistance of isolates was low in this study (Figure 6B), with a proportion of multidrug-resistant isolates of 3.23% (6/186). There were two MDR strains in ST410, one MDR strain in ST29, one MDR strain in ST221, one MDR strain in ST443, and one MDR strain in ST479 (Figure 6B) isolated from sheep (n = 1) and humans (n = 5). The MDR proportion of human isolates was 3.40% (5/147). The MDR proportion of animal isolates was 4% (1/25). There were no multidrug-resistant strains of food origin in our study. Supplementary Table S1 contains a complete list of antibiotic-resistance profiles found in the study isolates.
Antibiotic resistant gene profiles
The carriage of resistance genes in C. perfringens is shown in Table 2, with specific information in Supplementary Table S3. The resistance genes with high carriage rates were as following: 100% C. perfringens carried the peptide resistance gene Clostridium_perfringens_mprF. The carriage rates of tetracycline resistance genes tetA(P), tetB(P), and tet44 were 97.19, 79.21, and 20.79%, respectively. The carriage rates of macrolide/lincosamide/streptomycin (MLS) resistance genes ErmQ, LnuP, and ErmB were 52.25, 33.71, and 8.99%, respectively. Antibiotics referred to as aminoglycosides, including ANT(6)-Ib and AAC(6′)-Ie-APH(2″)-Ia, had carriage rates of 21.35 and 20.79%, respectively.
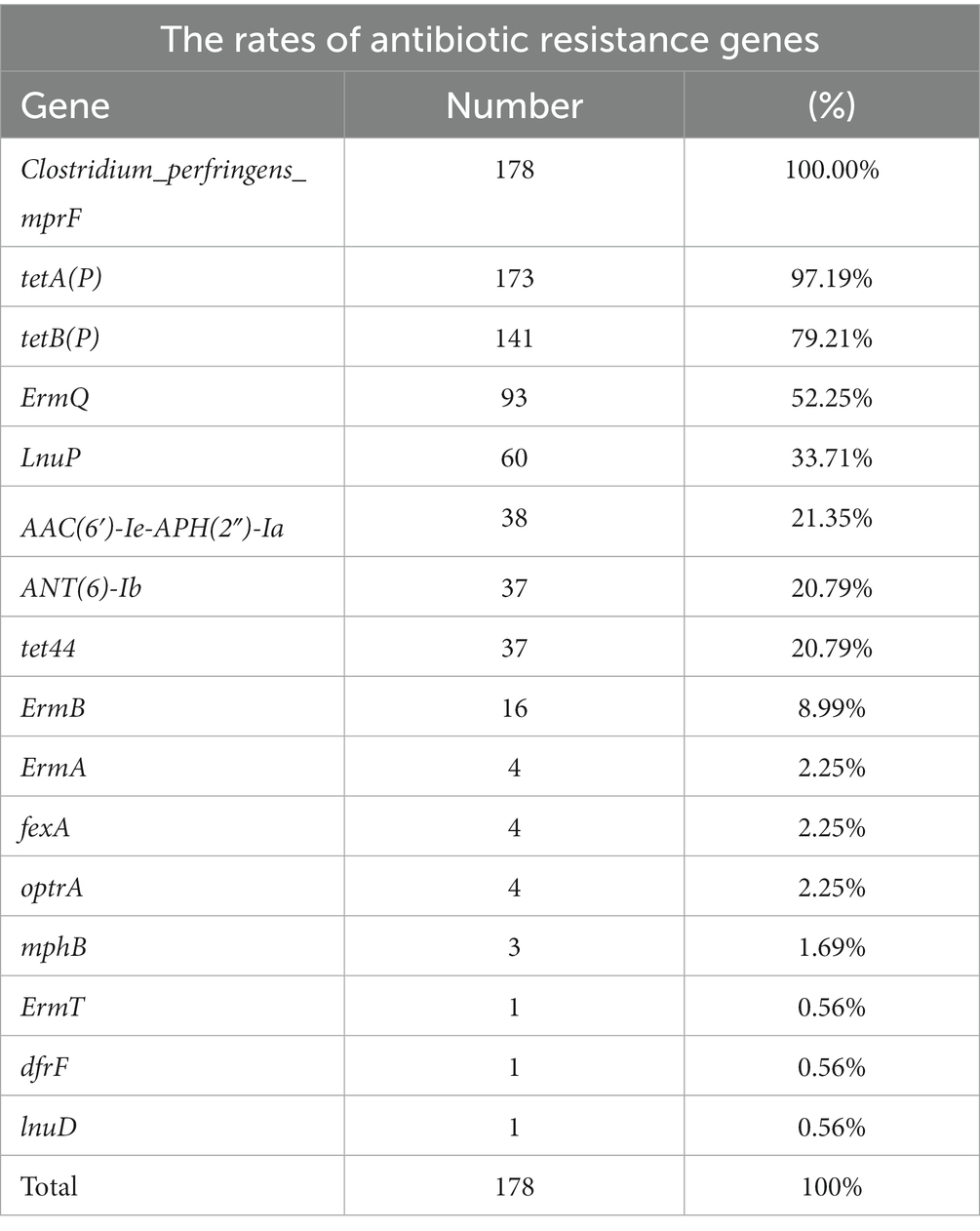
Table 2. The rates of antibiotic resistance genes.
Discussion
To the best of our knowledge, this is the first relatively comprehensive cross-sectional study concerning the molecular characterization of C. perfringens isolates in China. In our study, samples from patients, healthy humans, animals, and food were subjected to MLST analysis, toxinotype analysis, cgMLST analysis, and antibiotic susceptibility testing.
The MLST analysis of Chinese strains indicated a wide genetic diversity of C. perfringens, as previously reported (Verma et al., 2020). In our study, the predominant STs from human isolates (ST221, ST62, ST408) were significantly distinct from the predominant STs (ST479) from animal isolates, a result that was consistent with previous research on global C. perfringens strains, indicating a diverse distribution of C. perfringens among multiple sources (Verma et al., 2020; Wang et al., 2021). In global surveys, a clonal complex (CC) represented by three STs (ST98, ST41, and ST110) predominantly representing type F (18/20 strains) was mostly associated with human illnesses (Verma et al., 2020). Among the predominant STs, ST 54 was associated with enteritis cases in foals and dogs, and ST 58 was associated with necrotic enteritis in poultry (Verma et al., 2020). In our study, most type F strains (10/11 strains) were associated with human illnesses, different from the results of previous studies.
Global MLST analysis indicates a wide genetic diversity of C. perfringens. 835 isolates represented 563 STs. Among three predominant five STs, ST 80 represented maximum number of strains and belonged to three regions (United States, Canada and Switzerland). The same ST type can occur in geographically distant countries. In the minimum spanning tree, the distribution of strains of Japanese origin is relatively concentrated. It is probably due to the fact that they are all strains of clinical origin.
In terms of strains of human origin, the isolates recovered from human diarrhea cases (n = 111) were found to be of type A (n = 102), type F (n = 8), and type D (n = 1). Type A and type F were the major toxinotypes in the isolates, basically in accordance with earlier reports (Aung et al., 2021; Wang et al., 2021). Interestingly, there was one isolate of type D that carried cpe and/or cpb2 genes, indicating that it had the potential to produce the CPE toxin (Kiu and Hall, 2018). ST221 was the dominant genotype of C. perfringens among the isolates, with significant genetic diversity. Previous results revealed that ST41 and ST77 were associated with human illnesses (Verma et al., 2020; Wang et al., 2021). Consistent with these findings, we observed that the strains ST41 (2/2, type A) and ST77 (2/2, type F) were isolated from patients with diarrhea. The resistance rates of C. perfringens depend on the use of antibiotics in different regions. For example, some studies in Hungary, Slovenia, and northern Taiwan showed that cefoxitin, meropenem, and metronidazole were successful against C. perfringens. Only a minority were penicillin (2.6%) or clindamycin (3.8%) resistant (Sarvari and Schoblocher, 2020). However, in our study, we found high resistance (49.66%) to clindamycin in human origin samples in China. Similarly penicillin-resistant (10.2%) and penicillin-insensitive strains (18.2%) were much more frequent in this study than in previous studies. Moreover, recent data from Italy showed that clindamycin resistance was >20% for Clostridium spp. (Di Bella et al., 2022). In fact, penicillin and clindamycin have long been considered excellent drugs for Clostridial infections (Satyaprakash et al., 2010). In particular, penicillin is currently the drug of first choice for C. perfringens infections. Thus, our findings suggest that the two antibiotics should be used with caution as empirical therapies in China.
Among the strains of animal origin, all were found to be type A in this study. A relatively high prevalence of MDR (73.4%) was found among the isolates from different animals in a previous study (Xiaoting et al., 2021). The MDR profile was resistant against tetracycline (84%), erythromycin (72%), sulfamethoxazole (97.3%), trimethoprim (96.3%), and streptomycin (84.4%) (Xiaoting et al., 2021). Interestingly, the prevalence of MDR C. perfringens strains was rather low in this study (4%, 1/25 animals), possibly due to different choices of tested antimicrobials. The small scale of animal samples or low use of antibiotics in selected farms is also potential cause. Furthermore, the environment in which C. perfringens lives (e.g., the gut) may play a significant role in drug resistance. Therefore, in the future, close monitoring is necessary for a more thorough investigation of antibiotic resistance of C. perfringens in China. Rational antibiotic use may play an important role in reducing the drug resistance of C. perfringens.
C. perfringens is an important microorganism as a foodborne pathogen. Among the C. perfringens of food origin, in a previous study in Egypt, most of the food origin isolates (74%) (beef, chicken meat, and raw milk) exhibited MDR patterns. Strains isolated from raw camel milk in Isiolo County, Kenya, had high resistance to chloramphenicol (42.37%), kanamycin (40.68%), tetracycline (37.29%), and gentamycin (35.59%) (Aliwa et al., 2019). Another study in Korea reported that resistance to chloramphenicol (26/38, 68.4%) and metronidazole (13/38, 34.2%) was observed in C. perfringens from retail meats (Jang et al., 2020). Surprisingly, in our study, all of the isolates from retail meat products were susceptible to chloramphenicol and metronidazole, and none of the isolates showed an MDR pattern. Given that most of the samples in this study were of human origin, the selected antimicrobials were biased toward clinical use, a feature that was inconsistent with other studies using isolates of animal or food origin.
Antimicrobial resistance gene profiling among the recovered isolates revealed a higher presence of the mprF gene (100%) followed by tetA(P) (97.19%), tetB(P) (79.21%), ermQ (52.25%), and lnuP (33.71%) (Supplementary Table S3). These are common drug resistance genes in C. perfringens. All C. perfringens genomes had the mprF gene, which is known to impart resistance via altering the surface molecules of AMPs and is highly conserved in C. perfringens (Kiu et al., 2017). Since the 1980s, it has been known that the gene tetA(P) and tetB(P) are most frequently encoded in tetracycline efflux proteins of C. perfringens (Lyras and Rood, 1996). TetA(P) was found in 75% of the 56 strains in the prior genome investigation, which is more common than tetB(P) (42%) (Kiu et al., 2017). The high resistance of tetracycline (84%) was recorded out of the 50 animal origin isolates in previous study. In another study, clinical isolates (49%) as well as isolates from chickens (73%) and soil (63%) and food (42%) also were found to be tetracycline-resistant (Bendary et al., 2022). C. perfringens isolates from different sources varied in their tetracycline resistance levels and the frequency of the tetracycline-resistance genes tetA(P) and tetB(P) (Park et al., 2010). In this study, ErmQ was found to be the predominant macrolide-lincosamide-streptogramin (MLS) resistance gene of C. perfringens (Berryman et al., 1994). Also, lnuP gene conferring lincosamide resistance has been discovered in C. perfringens (Lyras et al., 2009). The isolates in this investigation were reported to be 52% resistant to the MLS drug clindamycin.
The three isolates (17SD152, 17SD154, and 17SD155) with the same ST62 isolated from the same hospital were assigned to the same cgMLST type (with no allele difference). Their visit times were close, no more than half a month apart. The three patients were 6 months old, 34 years old, and 62 years old. The three isolates collected at a single Shandong hospital inferred a potential transmission chain. The five isolates within the ST479 complex (cp2020SDY007, cp2020SDY009, cp2020SDY010, cp2020SDY011, and cp2020SDY014) of sheep origin in Shandong were divided into four cgMLST types with allele differences range 0–3, including cp2020SDY007 and cp2020SDY014 that belonged to the same cgMLST type (Figure 5D). Thus, we suspect that transmission of these C. perfringens strains in single farm took place. Genetic relatedness of strains isolated from three AAD patients from the same hospital and five sheep from the same farm in Shandong indicates that these isolates are originated from a single source or associated with the same transmission event and inter-host transfer of C. perfringens.
In our study, sequence type complex 221 was further divided into different cgMLST profiles. The STc221 strains across different provinces and hosts showed large allelic differences, indicating independent development and distribution. Meanwhile, this pattern showed that cgMLST had higher discrimination than the classical MLST method. Overall, cgMLST made it easy to clarify transmission routes, target the epidemiological investigation, and delineate infection control interventions. The cgSNP based on whole-genome sequencing (WGS) has advantages in understanding the dissemination of C. perfringens.
In conclusion, this study provides a glance at the molecular features and genetic characters of C. perfringens isolates in China. Using MLST analysis of 186 C. perfringens strains from diverse hosts, we found distinct differences in dominant STs among humans and animals. Similar characters were reported in other studies (Verma et al., 2020). Moreover, we gained insight into the antibiotic-resistance profiles in China of this significant pathogen. In our study, the percentage of MDR strains was much lower than previously reported. Interestingly, isolates of human origin were highly clindamycin-resistant and penicillin-insensitive, implying that their use as empirical therapies in China should be approached with caution. We analyzed the genetic relatedness of these specific complexes with the same STs to identify potential transmission links based on MLST, cgMLST, and cgSNP. Our results indicated that the isolates of the same ST may be genetically unrelated. cgMLST and cgSNP are more discriminant than traditional MLST and are suitable approaches for outbreak and transmission analyses. Further studies focused on C. perfringens of animal and food origin nationwide are needed to gain deep insights into prevention and control strategies, routine outbreak detection, and national surveillance in China.
Data availability statement
The genomic sequencing data set was deposited in the NCBI under submission: SUB12955969 and submission SUB12688567.
Author contributions
JZ, HZ, YW, LB and XD performed the experiments. JZ and HZ analyzed data and finished figures. JZ wrote the manuscript. YW designed the study and reviewed the manuscript. JL reviewed the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the National Key Research and Development Program of China (2021YFC2301000).
Acknowledgments
The authors thank Haijian Zhou, Yutong Kang, Jie Che, and Zhizhou Tan for their suggestions in both experiments and manuscript preparation. The authors thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1195083/full#supplementary-material
Footnotes
1. ^https://pubmlst.org/organisms/clostridium-perfringens
2. ^https://pubmlst.org/bigsdb?db=pubmlst_cperfringens_isolates&page=query&genomes=1
References
Abdel-Glil, M. Y., Thomas, P., Linde, J., Busch, A., Wieler, L. H., Neubauer, H., et al. (2021a). Comparative in silico genome analysis of Clostridium perfringens unravels stable phylogroups with different genome characteristics and pathogenic potential. Sci. Rep. 11:6756. doi: 10.1038/s41598-021-86148-8
Abdel-Glil, M. Y., Thomas, P., Linde, J., Jolley, K. A., Harmsen, D., Wieler, L. H., et al. (2021b). Establishment of a publicly available Core genome multilocus sequence typing scheme for Clostridium perfringens. Microbiol. Spectr. 9:e0053321. doi: 10.1128/Spectrum.00533-21
Aliwa, B. O., Dasel, K., and Mulwa, W. (2019). Antibiotic resistance of Clostridium perfringens isolated from raw camel Milk in Isiolo County, Kenya. Ann. Appl. Microbiol. Biotechnol. J. 3, 1–5. doi: 10.36876/aamb.1012
Álvarez-Pérez, S., Blanco, J. L., and García, M. E. (2017). Clostridium perfringens type a isolates of animal origin with decreased susceptibility to metronidazole show extensive genetic diversity. Microb. Drug Resist. 23, 1053–1058. doi: 10.1089/mdr.2016.0277
Aung, M. S., Matsuda, A., Urushibara, N., Kawaguchiya, M., Ohashi, N., Matsuda, N., et al. (2021). Clonal diversity of Clostridium perfringens human clinical isolates with various toxin gene profiles based on multilocus sequence typing and alpha-toxin (PLC) typing. Anaerobe 72:102473. doi: 10.1016/j.anaerobe.2021.102473
Azimirad, M., Gholami, F., Yadegar, A., Knight, D. R., Shamloei, S., Aghdaei, H. A., et al. (2019). Prevalence and characterization of Clostridium perfringens toxinotypes among patients with antibiotic-associated diarrhea in Iran. Sci. Rep. 9:7792. doi: 10.1038/s41598-019-44281-5
Bailey, M. A., Macklin, K. S., and Krehling, J. T. (2013). Use of a multiplex PCR for the detection of toxin-encoding Genes netB and tpeL in strains of clostridium perfringens. ISRN Vet. Sci. 2013, 1–4. doi: 10.1155/2013/865702
Bendary, M. M., Abd El-Hamid, M. I., El-Tarabili, R. M., Hefny, A. A., Algendy, R. M., Elzohairy, N. A., et al. (2022). Clostridium perfringens associated with foodborne infections of animal origins: insights into prevalence, antimicrobial resistance, toxin genes profiles, and Toxinotypes. Biology 11:551. doi: 10.3390/biology11040551
Berryman, D. I., Lyristis, M., and Rood, J. I. (1994). Cloning and sequence analysis of ermQ, the predominant macrolide-lincosamide-streptogramin B resistance gene in Clostridium perfringens. Antimicrob. Agents Chemother. 38, 1041–1046. doi: 10.1128/AAC.38.5.1041
Carman, R. J. (1997). Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microbiol. 8:S46. doi: 10.1097/00013542-199712001-00024
Di Bella, S., Antonello, R. M., Sanson, G., Maraolo, A. E., Giacobbe, D. R., Sepulcri, C., et al. (2022). Anaerobic bloodstream infections in Italy (ITANAEROBY): a 5-year retrospective nationwide survey. Anaerobe 75:102583. doi: 10.1016/j.anaerobe.2022.102583
Ferraro, M. J., et al. (2001). M100-S11, performance standards for antimicrobial susceptibility testing. Clin. Microbiol. Newsl. 23:49. doi: 10.1016/S0196-4399(01)88009-0
Giske, C. G., Turnidge, J., Cantón, R., Kahlmeter, G., Eucast, S. C., and Humphries, R. M. (2022). Update from the European committee on antimicrobial susceptibility testing (EUCAST). J. Clin. Microbiol. 60, –e0027621. doi: 10.1128/jcm.00276-21
Hu, W., Kim, H., and Koo, O. K. (2018). Molecular genotyping, biofilm formation and antibiotic resistance of enterotoxigenic Clostridium perfringens isolated from meat supplied to school cafeterias in South Korea. Anaerobe 52, 115–121. doi: 10.1016/j.anaerobe.2018.06.011
Jang, Y., Kim, D., Bae, D., Kim, S., Kim, H., Moon, J., et al. (2020). Prevalence, toxin-typing, and antimicrobial susceptibility of Clostridium perfringens from retail meats in Seoul, Korea. Anaerobe 64:102235. doi: 10.1016/j.anaerobe.2020.102235
Jolley, K. A., Bray, J. E., and Maiden, M. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Kikuchi, E., Miyamoto, Y., Narushima, S., and Itoh, K. (2013). Design of Species-Specific Primers to identify 13 species of Clostridium harbored in human intestinal tracts. Microbiol. Immunol. 46, 353–358. doi: 10.1111/j.1348-0421.2002.tb02706.x
Kiu, R., Caim, S., Alexander, S., Pachori, P., and Hall, L. J. (2017). Probing genomic aspects of the multi-host pathogen Clostridium perfringens reveals significant Pangenome diversity, and a diverse Array of virulence factors. Front. Microbiol. 8:2485. doi: 10.3389/fmicb.2017.02485
Kiu, R., and Hall, L. J. (2018). An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microb. Infect. 7, 1–15. doi: 10.1038/s41426-018-0144-8
Kyne, L., Hamel, M. B., Polavaram, R., and Kelly, C. P. (2002). Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin. Infect. Dis. 34, 346–353. doi: 10.1086/338260
Lacey, J. A., Allnutt, T. R., Vezina, B., Van, T. T. H., Stent, T., Han, X., et al. (2018). Whole genome analysis reveals the diversity and evolutionary relationships between necrotic enteritis-causing strains of Clostridium perfringens. BMC Genomics 19:379. doi: 10.1186/s12864-018-4771-1
Lacey, J. A., Johanesen, P. A., Lyras, D., and Moore, R. J. (2016). Genomic diversity of necrotic enteritis-associated strains of Clostridium perfringens: a review. Avian Pathol. 45, 302–307. doi: 10.1080/03079457.2016.1153799
Li, J., Adams, V., Bannam, T. L., Miyamoto, K., Garcia, J. P., Uzal, F. A., et al. (2013). Toxin plasmids of Clostridium perfringens. Microbiol. Mol. Biol. Rev. 77, 208–233. doi: 10.1128/MMBR.00062-12
Lyras, D., Adams, V., Ballard, S. A., Teng, W. L., Howarth, P. M., Crellin, P. K., et al. (2009). tISCpe8, an IS1595-family lincomycin resistance element located on a conjugative plasmid in Clostridium perfringens. J. Bacteriol. 191, 6345–6351. doi: 10.1128/JB.00668-09
Lyras, D., and Rood, J. I. (1996). Genetic organization and distribution of tetracycline resistance determinants in Clostridium perfringens. Antimicrob. Agents Chemother. 40, 2500–2504. doi: 10.1128/AAC.40.11.2500
Magiorakos, A. P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Motamedi, H., Fathollahi, M., Abiri, R., Kadivarian, S., Rostamian, M., and Alvandi, A. (2021). A worldwide systematic review and meta-analysis of bacteria related to antibiotic-associated diarrhea in hospitalized patients. PLoS One 16:e0260667. doi: 10.1371/journal.pone.0260667
Nakano, V., Ignacio, A., Llanco, L., Bueris, V., Sircili, M. P., and Avila-Campos, M. J. (2017). Multilocus sequence typing analyses of Clostridium perfringens type a strains harboring tpeL and netB genes. Anaerobe 44, 99–105. doi: 10.1016/j.anaerobe.2017.02.017
Park, M., Deck, J., Foley, S. L., Nayak, R., Songer, J. G., Seibel, J. R., et al. (2016). Diversity of Clostridium perfringens isolates from various sources and prevalence of conjugative plasmids. Anaerobe 38, 25–35. doi: 10.1016/j.anaerobe.2015.11.003
Park, M., Rooney, A. P., Hecht, D. W., Li, J., McClane, B. A., Nayak, R., et al. (2010). Phenotypic and genotypic characterization of tetracycline and minocycline resistance in Clostridium perfringens. Arch. Microbiol. 192, 803–810. doi: 10.1007/s00203-010-0605-5
Rood, J. I., Adams, V., Lacey, J., Lyras, D., Mcclane, B. A., Melville, S. B., et al. (2019). Pathogenesis and toxins expansion of the Clostridium perfringens toxin-based typing scheme.
Rood, J. I., Adams, V., Lacey, J., Lyras, D., McClane, B. A., Melville, S. B., et al. (2018). Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 53, 5–10. doi: 10.1016/j.anaerobe.2018.04.011
Sarvari, K. P., and Schoblocher, D. (2020). The antibiotic susceptibility pattern of gas gangrene-forming Clostridium spp. clinical isolates from south-eastern Hungary. Infect. Dis. (Lond) 52, 196–201. doi: 10.1080/23744235.2019.1696472
Satyaprakash, A. K., Ravanfar, P., and Tyring, S. K. (2010). “CHAPTER 49 - skin and soft-tissue infections” in Antibiotic and chemotherapy. eds. R. G. Finch, D. Greenwood, S. R. Norrby, and R. J. Whitley. 9th ed (London: W.B. Saunders), 617–632.
Brynestad, S., and Granum, E. (2002). Clostridium perfringens and foodborne infections. Int. J. Food Microbiol. 74, 195–202. doi: 10.1016/S0168-1605(01)00680-8
Verma, A. K., Abdel-Glil, M. Y., Madesh, A., Gupta, S., Karunakaran, A. C., Inbaraj, S., et al. (2020). Multilocus sequence typing of Clostridium perfringens strains from neonatal calves, dairy workers and associated environment in India. Anaerobe 63:102212. doi: 10.1016/j.anaerobe.2020.102212
Wang, B., Dong, W., Ma, L., Dong, Y., Wang, S., Yuan, Y., et al. (2021). Prevalence and genetic diversity of Clostridium perfringens isolates in hospitalized diarrheal patients from Central China. Infect. Drug Resist. 14, 4783–4793. doi: 10.2147/IDR.S338593
Xiao, Y., Wagendorp, A., Moezelaar, R., Abee, T., and Wells-Bennik, M. H. (2012). A wide variety of Clostridium perfringens type a food-borne isolates that carry a chromosomal cpe gene belong to one multilocus sequence typing cluster. Appl. Environ. Microbiol. 78, 7060–7068. doi: 10.1128/AEM.01486-12
Xiaoting, W., Chengcheng, N., Chunhui, J., Yan, L., Jing, L., Qingling, M., et al. (2021). Antimicrobial resistance profiling and molecular typing of ruminant-borne isolates of Clostridium perfringens from Xinjiang, China. J. Glob. Antimicrob. Resist. 27, 41–45. doi: 10.1016/j.jgar.2021.08.003
Xu, W., Wang, H., Chen, S., Chen, Y., Liu, L., and Wu, W. (2021). Tracing Clostridium perfringens strains along the chicken production chain from farm to slaughter by multilocus sequence typing. Zoonoses Public Health 68, 431–442. doi: 10.1111/zph.12831
Keywords: Clostridium perfringens, MLST (multilocus sequence typing), antimicrobial resistance (AMR), cgMLST, genomic
Citation: Zhong Jx, Zheng Hr, Wang Yy, Bai Ll, Du Xl, Wu Y and Lu Jx (2023) Molecular characteristics and phylogenetic analysis of Clostridium perfringens from different regions in China, from 2013 to 2021. Front. Microbiol. 14:1195083. doi: 10.3389/fmicb.2023.1195083
Edited by:
Alejandro Garrido-Maestu, International Iberian Nanotechnology Laboratory (INL), PortugalReviewed by:
Mostafa Y. Abdel-Glil, Friedrich Loeffler Institute, GermanyIman Mehdizadeh Gohari, University of Pittsburgh, United States
Copyright © 2023 Zhong, Zheng, Wang, Bai, Du, Wu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Wu, wuyuan@icdc.cn