Perspectives on Systems Modeling of Human Peripheral Blood Mononuclear Cells
 Partho Sen
Partho Sen Esko Kemppainen1
Esko Kemppainen1 - 1Turku Centre for Biotechnology, University of Turku and Åbo Akademi University, Turku, Finland
- 2School of Medical Sciences, Örebro University, Örebro, Sweden
Human peripheral blood mononuclear cells (PBMCs) are the key drivers of the immune responses. These cells undergo activation, proliferation and differentiation into various subsets. During these processes they initiate metabolic reprogramming, which is coordinated by specific gene and protein activities. PBMCs as a model system have been widely used to study metabolic and autoimmune diseases. Herein we review various omics and systems-based approaches such as transcriptomics, epigenomics, proteomics, and metabolomics as applied to PBMCs, particularly T helper subsets, that unveiled disease markers and the underlying mechanisms. We also discuss and emphasize several aspects of T cell metabolic modeling in healthy and disease states using genome-scale metabolic models.
Introduction
Human peripheral blood mononuclear cells (PBMCs) are peripheral blood cells carrying a single round nuclei. PBMCs are comprised of several classes of immune cells, including T cells (~70%), B cells (~15%), monocytes (~5%), dendritic cells (~1%) and natural killer (NK) cells (~10%) (Autissier et al., 2010; Kleiveland, 2015). The T cell co-receptor (CD3+ expressing T lymphocytes) can be divided into CD4+ and CD8+ cytotoxic cells, which are present in PBMCs in approximately 2:1 ratio (Kleiveland, 2015). Activated CD4+ T cells are further divided into Th1, Th2, Th17, Th9, Th22, follicular helper (Tfh) cell and regulatory T cell (Treg) subsets, based on the panel of cytokines produced, transcription factors and surface markers expressed (Stockinger and Veldhoen, 2007; Sakaguchi et al., 2008; Broere et al., 2011; Crotty, 2011; Akdis et al., 2012; Luckheeram et al., 2012; Tan and Gery, 2012; Kleiveland, 2015; Golubovskaya and Wu, 2016). Treg cells can be natural cells (nTreg) generated in the thymus or inducible Treg cells (iTreg) when activated in the periphery (Wing and Sakaguchi, 2010). Likewise, activated CD8+ T cells (cytotoxic T cells) can be divided into Tc1 or Tc2 subsets based on their signature cytokines (Croft et al., 1994). Different subsets of T cells, their mechanisms of activation, differentiation and their functions have been extensively reviewed (Broere et al., 2011; Luckheeram et al., 2012).
B cells or B lymphocytes are bone marrow derived cells, which express the B cell receptor and bind to specific antigens against which they initiate antibody responses, thus forming the core of the adaptive humoral immune system (Cooper, 2015). B cells mature into plasmablasts and plasma cells, memory B cells, follicular B cells, marginal zone B cells, B regulatory and B-1 cells. The cytotoxic natural killer cells (NK cells), unlike T and B cells, are critical components of the innate immune system and can directly destroy pathogen infected cells. In addition, NK cells secrete lymphokines and interact with other immune cells and thus participate in immune responses by means other than direct cytotoxicity (Yuan et al., 1994).
Systems Approaches Applied to PBMCs
Systems biology together with bioinformatics has begun to emerge as an essential tool in immunological research. Integration of complex multi-omics datasets has unveiled several biomarkers and elucidated their physiological role (Buonaguro et al., 2011; Li et al., 2013, 2014b, 2017b; Olafsdottir et al., 2016). PBMCs, a large complement of inflammatory cells which is easy and inexpensive to acquire, can provide a more comprehensive overview of the immune system status than circulating serum or plasma markers. PBMCs have been used extensively to study several autoimmune disorders such as type 1 diabetes mellitus (T1DM) (Foss-Freitas et al., 2008), asthma (Iikura et al., 2011; Falcai et al., 2015), numerous allergies and cancer (Payne et al., 2013). Below we provide examples of omics and systems based approaches as applied to PBMCs, particularly to T helper cells (Figure 1).
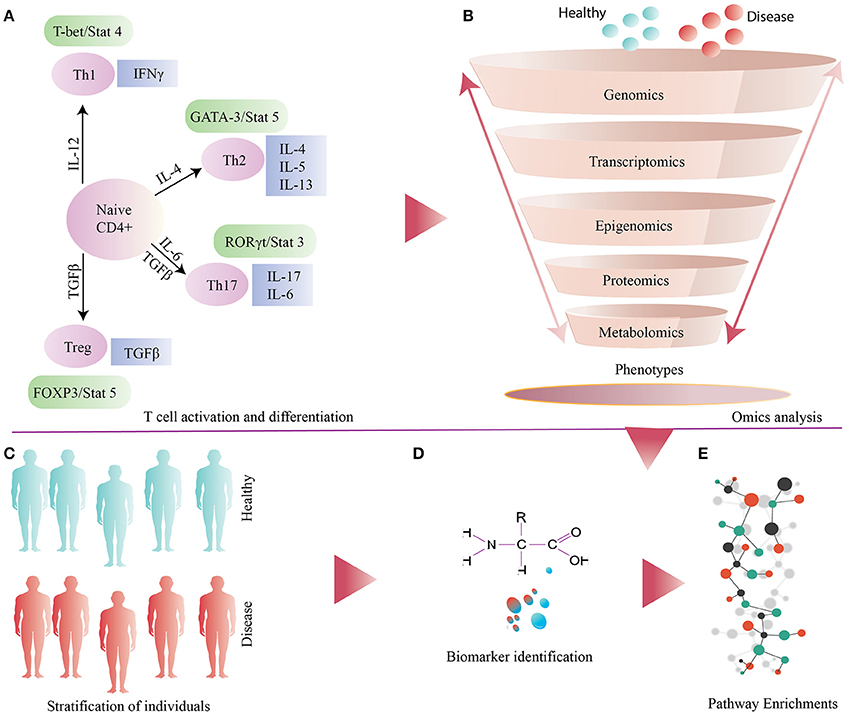
Figure 1. (A) General illustration of T cell activation and differentiation. (B) Several omics based approaches applied to samples obtained from disease and healthy individuals (controls). (C) Stratification of individuals based on metabolic phenotype. (D) Identification and validation of biomarkers. (E) Down-stream analysis of omics datasets for identification and enrichments of differential pathways.
Transcriptomics
Global transcriptomics analyses of PBMCs have been successfully used in elucidating the inflammatory mechanisms underlying different autoimmune diseases (Bennett et al., 2003; Crow et al., 2003; Greenberg et al., 2005; Achiron et al., 2007; Edwards et al., 2007). A proinflammatory transcriptional signature of interleukin-1 cytokine family was marked in patients with recent-onset of T1DM (Wang et al., 2008; Levy et al., 2012). Gene expression profiling of PBMCs using oligonucleotide array was used to identify 330 transcripts that were differentially expressed in rheumatoid arthritis (RA) patients as compared to the healthy controls (Edwards et al., 2007).
Transcriptomics data from PBMCs across multiple studies were used to characterize multiple types of diabetes, which revealed that gestational and T1DM were related at the transcriptome level (Collares et al., 2013). Meta-analysis of PBMC based microarray datasets was used to identify dysregulated pathways in patients with systemic lupus erythematosus (SLE). The study revealed that toll-like receptor (TLR) signaling, oxidative phosphorylation, diapedesis and adhesion regulatory networks were differentially regulated in the PBMCs of affected individuals (Kröger et al., 2016).
Transcriptomes from PBMCs have also been used to characterize HIV phenotypes. Distinct transcriptomics signatures with several dysregulated genes involved in apoptosis were identified in rapid HIV progressors. The expression of five miRNAs (miR-31, 200c, 526a, 99a, and 503) were also found to be altered (Zhang et al., 2013). In another study, gene expression profiling of PBMCs obtained from smokers exhibited a signature of chronic obstructive pulmonary disease (COPD) and emphysema characterized by multiple differentially regulation of genes FOXP1, TCF7, and ASAH1 involved in sphingolipid (ceramide) metabolism. Plasma metabolomics validated the identity of glycoceramide as a marker of emphysema (Bahr et al., 2013).
In addition, integration of transcriptomics and protein expression profiles of PBMCs obtained from a large study cohort suggested an association between decreased IL-16 and emphysema; it also identified IL-16 cis-eQTL as a novel disease biomarker (Bowler et al., 2013). PBMCs have also been analyzed in the context of cancer. Whole genome cDNA microarray analysis study of PBMC samples from 26 patients with pancreatic cancer and 33 matched healthy controls identified an eight-gene predictor set comprising SSBP2, Ube2b-rs1, CA5B, F5, TBC1D8, ANXA3, ARG1, and ADAMTS20 (Baine et al., 2011). Similarly, significant differences were observed in the PBMC transcriptomes as obtained from renal cell carcinoma patients and normal volunteers (Twine et al., 2003; Burczynski et al., 2005).
RNA-Seq and microarray based transcriptomics datasets have been used to characterize different subsets of T helper cells. Transcriptomics of the differentiated subsets (Ciofani et al., 2012; Hu et al., 2013) characterized differences between Th17 and Th0 cells (TCR stimulated CD4+ T cells), while functional analysis inspired by these transcriptomes suggested differences in the control of cell cycle regulation (Simeoni et al., 2015). In another study, transcriptome analysis of cord blood-derived naïve T cell precursors was used to identify several lineage-specific genes involved in the early differentiation of Th1 and Th2 subsets (Kanduri et al., 2015). Moreover, comparative transcriptomics of mouse and human Th17 cells marked novel transcripts related to Th17 polarization. Several human long non-coding RNAs were identified in response to cytokines stimulating Th17 cell differentiation (Tuomela and Lahesmaa, 2013; Tuomela et al., 2016).
Epigenomics
Epigenetics play a pivotal role in the regulation of gene expression and inheritance of genetic information. Epigenome-wide association studies of three human immune cell types (CD14+ monocytes, CD16+ neutrophils and naïve CD4+ T cells) obtained from 197 subjects were performed to assess the impact of cis-genetic and epigenetic factors. The major outcome of this study was the identification of 345 molecular trait QTLs (quantitative trait loci) which co-localized with immune disease specific loci (Chen et al., 2016). Epigenetic mechanisms in naïve CD4+ T cell have been extensively reviewed (Lee et al., 2006; Sanders, 2006; Aune et al., 2009; Hirahara et al., 2011; Oestreich and Weinmann, 2012).
Proteomics
Proteome profiling of PBMCs has been carried out primarily for two purposes: (a) to identify protein biomarker(s) associated with specific pathophysiological processes, and (b) to characterize different subsets of immune cells based on their proteomes. Recently, comparative proteomics using tandem mass spectrometry (MS) was applied to PBMC samples obtained from kidney biopsies of 40 kidney allograft recipients, either with healthy transplants or those suffering acute rejection. A total of 344 proteins were identified, cataloged and mapped to 2905 proteoforms (Savaryn et al., 2016). Comparative proteome analysis also revealed differences between untreated and inflammatory activated human PBMCs (T cells and monocytes) using 2D-PAGE and LC–MS/MS. Several cell specific proteomic signatures of activation and inflammation were identified as NAMPT and PAI2 (PBMCs), IRF-4 and GBP1 (T cells), PDCD5, IL1RN, and IL1B (monocytes) (Haudek-Prinz et al., 2012).
Proteome profiling of the Th1 cells induced from naïve T cells by stimulating with interleukin 12 (IL-12) was used to identify 42 IL-12 regulated genes, among which 22 were up- and 20 were down-regulated. Functional characterization of the up-regulated proteins helped to identify a multifunctional cytokine macrophage migration inhibitory factor and a novel IL-12 target gene (Rosengren et al., 2005). In another study, MS (stable isotope labeling by amino acids in cell culture, SILAC) based profiling of cell surface proteome was used to identify differentially expressed proteins between human Th1 and Th2 cells. Among the differentially expressed proteins, BST2 (bone marrow stromal protein 2) and TRIM (T cell receptor interacting molecule) were found to be significantly differently regulated (Loyet et al., 2005). Moreover, global analysis of highly purified primary naïve T and Th1 cell proteomes using LC-MS/MS revealed differential regulation of ubiquitination pathway upon T cell differentiation (Pagani et al., 2015). Quantitative proteomics of Th cells using ICAT labeling and LC MS/MS have identified (557) and quantified (304) IL-4-regulated proteins from the microsomal fractions of CD4+ cells extracted from umbilical cord blood. Among these, small GTPases, mainly GIMAP1 and GIMAP4, were down-regulated by IL-4 during Th2 differentiation (Filén et al., 2009).
Metabolomics
Circulating PBMCs are a complex mixture of different subsets of immune cells in highly variable stages of their lifespan. In addition to the natural genetic variation and immune challenges, this heterogeneity is shaped by the myriad of environmental conditions around them. In the light of the current understanding, the key role of the cell metabolism in immune cell function also underscores the potential impact of metabolites in regulating immune system directly or indirectly (Buck et al., 2015). For example, external perturbations to key metabolic processes such as glycolysis, energy metabolism, fatty acid and amino acid metabolism are known to affect and impair T cell activation and differentiation (Berod et al., 2014; Almeida et al., 2016; Geiger et al., 2016; Ma et al., 2017).
Metabolomics of PBMCs obtained from affected or healthy mice and humans have been used to identify metabolic markers in various pathological conditions. For example, gas chromatography coupled to MS (GC-MS) based targeted metabolomics was used to quantify glucose derived metabolites in PBMCs of healthy controls, schizophrenia and major depressions. Most of these metabolites were found to be significantly altered particularly in schizophrenic subjects. In addition, ribose 5-phosphate showed a high diagnostic performance for first-episode drug-naïve schizophrenia subjects (Liu et al., 2015). Similarly, GC–MS was used to identify metabolites such as malic acid, ornithine, L-lysine, stigmasterol, oleic acid, adenosine and N-acetyl-D-glucosamine which were significantly altered in resilient rats while statistical analysis of metabolic pathways showed aberrant energy metabolism (Li et al., 2017a).
Fatty acid composition of PBMCs phospholipids obtained from 150 subjects were estimated and linked with immune cell functions. The proportions of total polyunsaturated fatty acids (PUFAs) in PBMC phospholipids were positively correlated with phagocytosis by neutrophils and monocytes, neutrophil oxidative burst, lymphocyte proliferation, and interferon-γ production. The study also suggested that variations in the fatty acid composition of PBMCs phospholipids might induce subtle variations in immune cell functions as seen in healthy individuals (Kew et al., 2003). Since the phospholipids are primarily incorporated into cellular membranes, this effect may be mediated by the altered membrane properties such as fluidity and lateral pressure, due to their altered phospholipid composition (Mouritsen, 2011).
High-resolution MS was recently used to generate dynamic metabolome and proteome profiles of human primary naïve T cells upon activation. The study reported a dramatic decrease in intracellular L-arginine concentration which has impact on metabolic fitness and survival capacity of T cells related to anti-tumor responses (Geiger et al., 2016). Metabolism of T cells during naïve, activated, proliferative and differentiated states have been extensively reviewed (Gerriets and Rathmell, 2012; MacIver et al., 2013; Pearce and Pearce, 2013; Pearce et al., 2013; Buck et al., 2015; Dimeloe et al., 2017).
Gut Microbes and Immune Cells
The link between diet, gut microbiota and the immune response is currently well recognized. It is known that the immune system plays a significant role in the regulation of gut microbiota and in turn microbiota contribute to the development, training and tuning of the immune responses (Round and Mazmanian, 2009; Belkaid and Hand, 2014). Imbalances in the microbial composition or host specific interactions have been linked to inflammatory and autoimmune diseases (Brugman et al., 2006; Wen et al., 2008; Roesch et al., 2009; Kostic et al., 2015). It has been demonstrated that composition of the gut microbiota may be altered in individuals at risk of developing T1DM (Brown et al., 2011; Giongo et al., 2011; de Goffau et al., 2013; Murri et al., 2013). The phenomenon was first observed in a cohort of Finnish children at high HLA-associated risk of developing T1DM, where fecal samples from individuals seropositive with multiple pancreatic islet antigen specific autoantibodies were compared to seronegative healthy controls (Giongo et al., 2011; Kostic et al., 2015). Furthermore, Kostic et al., examined the relationship between dynamics of human gut microbiome throughout the infancy in a cohort of 33 infants genetically predisposed to T1DM. The study showed a decline in alpha-diversity in T1DM progressors between seroconversion and T1DM diagnosis; followed by an increase in microbial species which promote in inflammation, altered gene functions and stool metabolites (Kostic et al., 2015). Links between diet, gut microbiota and T cell associated disorders have been reviewed elsewhere (Kosiewicz et al., 2014; Mejía-León and Barca, 2015; Knip and Siljander, 2016).
A comprehensive list of omic approaches applied to PBMCs and T helper subsets is provided in (Table 1).
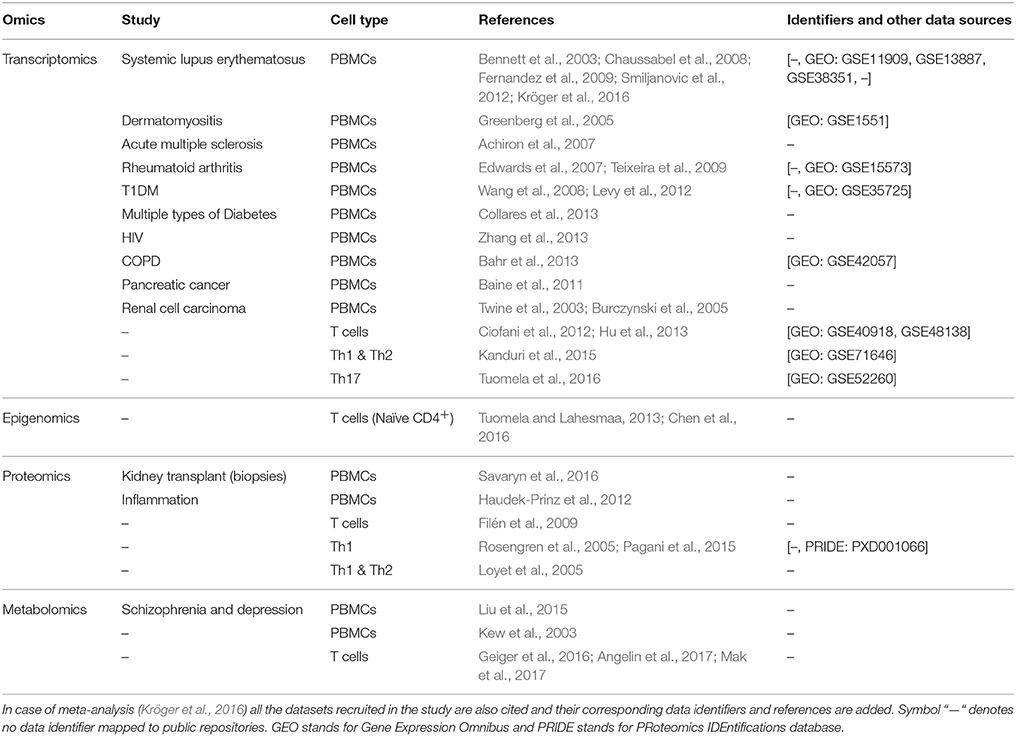
Table 1. List of studies performed by using PBMCs and T cells as model systems.
Genome-scale Metabolic Models as a Tool to Study Metabolism
With the rapid advancement of cutting-edge technologies in PBMC research, there is a growing need for development of integrative methods and computational models to cope with the increasing amounts of data. These approaches when applied at the systems level could mechanistically relate entities like gene, proteins and metabolites that might unveil the disease markers and related processes at the systems level (Sen et al., 2016).
Genome-scale metabolic modeling (GSMM) is a constraint-based mathematical modeling approach that integrates biochemical, genetic and genomic informations within a computational framework (Price et al., 2004; Orth et al., 2010; Bordbar et al., 2014; O'Brien et al., 2015). It is used to study metabolic genotype-phenotype relationship of an organism. GSMM have been continuously evolving over the past 30 years. Genome-scale metabolic models (GEMs) have been used in in silico metabolic engineering for designing studies such as essentiality of the reaction/gene (Patil et al., 2005; Suthers et al., 2009), relevance of foreign pathway(s) (Pharkya et al., 2004) and over expression or suppression of metabolites and metabolic pathways (Pharkya and Maranas, 2006). They are efficient tools for prediction of growth in living cells/tissues exposed to different nutrients (Förster et al., 2003; O'Brien et al., 2013).
Over the past years, the components and functionalities of GEMs have been extended to study metabolism in human. The first in silico global reconstruction of human metabolic network Recon 1 (1,905 genes, 3,742 reactions, and 2,766 metabolites) was built with a vision to integrate and analyze biological datasets (Duarte et al., 2007). Subsequently, the Edinburgh Human Metabolic Network (EHMN) (2,322 genes, 2,823 reactions, and 2,671 metabolites) (Ma et al., 2007) was developed, these models were parsimonious and provided partial knowledge about human metabolism. Thereafter, Recon 2 (2,194 genes, 7,440 reactions, and 5,063 metabolites) (Thiele et al., 2013), Recon 2.2 (1,675 genes, 7,785 reactions, and 5,324 metabolites) (Swainston et al., 2016), a community-driven consensus human metabolic reconstruction, and Human Metabolic Reaction (HMR) (3,668 genes, 8,181 reactions, and 9,311 metabolites) (Mardinoglu et al., 2013, 2014) were designed that comprehensively captured human metabolism. The human metabolic reconstructions have been used to study cell, tissue and organ specific metabolism (Agren et al., 2012; Wang et al., 2012) in the context of various diseases such as cancer (Yizhak et al., 2015), non-alcoholic fatty liver disease (NAFLD) (Mardinoglu et al., 2014; Hyötyläinen et al., 2016), diabetes (Väremo et al., 2016). Furthermore, GEMs as an integrative tool has been used to model diet-tissue (Sen et al., 2017) and multi-tissue interactions in humans (Bordbar et al., 2011).
The structure of GEM provides scaffolds for integration of different types of omics data such as transcriptome, proteome and metabolome/fluxome (Blazier and Papin, 2012). Several algorithms were designed that allow integration and contextualization of GEMs based on expression datasets. GIMME designed by Becker and Palsson considers a single gene expression dataset and compares it to a certain threshold, it subsequently lists active and inactive reactions within a GEM model (Becker and Palsson, 2008). On the other hand, iMAT discretize expression dataset to low, moderate and highly expressed genes and categorize GEM reactions into low, moderate and active sets (Shlomi et al., 2008; Zur et al., 2010). MADE allows integration of multiple expression datasets, it was devised to overcome the user supplied expression threshold that might be unrealistic (Jensen and Papin, 2011). MADE decomposes gene expression data into a binary state and determines sets of low or highly active reactions. E-flux is a threshold based method that does not reduce the expression data into binary states, rather it converts the expression data to some suitable constraints that sets upper and lower limits to the reactions (Colijn et al., 2009). INIT (Integrative Network Inference for Tissues) algorithm uses cell specific protein abundances to generate genome-scale active metabolic networks (Agren et al., 2012).
GEMs have been used to model cataloged human gut microbes (Qin et al., 2010; Li et al., 2014a) based on their metabolic functions (El-Semman et al., 2014; Shoaie and Nielsen, 2014; Bauer et al., 2015; Magnúsdóttir et al., 2016). Magnúsdóttir et al., introduced AGORA (Assembly of Gut Organisms through Reconstruction and Analysis) that includes semi-automatically reconstructed GEMs of 773 human gut bacteria (205 genera, 605 species). The reconstruction can accommodate metagenomics or 16S rRNA sequencing datasets that can be used to study metabolic diversities among microbial communities (Magnúsdóttir et al., 2016). Furthermore, GEMs derived from human gut microbiome were used to decipher microbe-microbe, diet-microbe and microbe-host interactions. Another GEM based comprehensive computational platform, CASINO (Community And Systems-level INteractive Optimization) was designed to study the effect of diet on microbial communities (Shoaie et al., 2015).
Genome-scale Metabolic Models Applied to PBMCs and Concluding Remarks
The availability of genome sequences of human cell lines together with the existing human metabolic reconstructions (Duarte et al., 2007; Agren et al., 2012; Wang et al., 2012; Mardinoglu et al., 2013, 2014; Thiele et al., 2013; Swainston et al., 2016; Väremo et al., 2016) and large volume of PBMC data, provides an opportunity to develop the PBMC-specific GEMs (Figure 2). These metabolic networks could be refined by the experimental data such as metabolite intensities, fluxes, enzyme abundances, and gene/transcripts expression. Network refinement adds more confidence to the metabolic reactions and their associated entities, and thus eliminates the false positives (Becker et al., 2007; Schellenberger et al., 2011). Integration of omics data with these networks makes it condition-specific, on which different analyses could be performed. One such analysis is the identification of reporter metabolites (RMs), i.e., metabolite within a metabolic network around which significant transcriptional changes occurs (Patil and Nielsen, 2005). RMs are actively involved in one or more metabolic reactions regulated by gene expression and/or enzyme abundances. RMs could also inform about the regulation of a metabolic pathway(s)/subsystem(s) (for e.g., glycolysis).
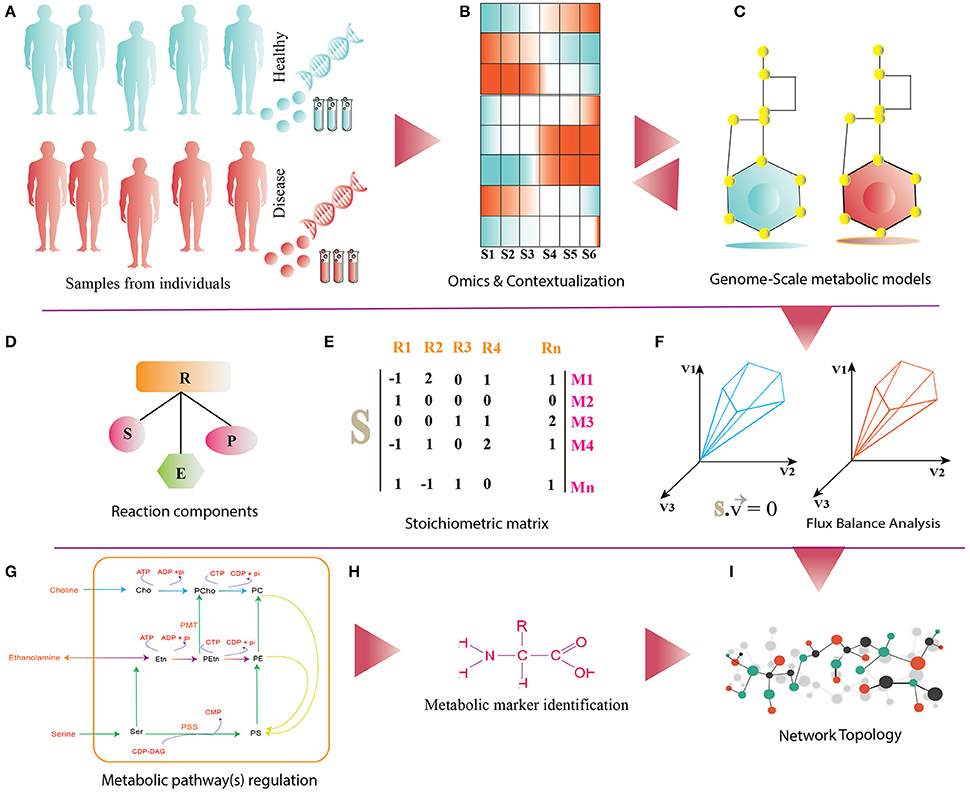
Figure 2. (A) It shows disease and healthy individuals (controls) from which PBMCs samples are obtained for omics analysis. (B) Differential omics expression and analysis for contextualization. (C) Reconstruction and contextualization of condition specific genome-scale metabolic models. (D) Reaction components (R) of Genome-Scale metabolic models: S, substrates; E, enzymes; P, products. (E) Stoichiometric matrix (S) of Mn metabolites and Rn reactions, directionality of each metabolites consumed (−1) or produced (+1) or not involved in the reaction (0). (F) Flux-Balance Analysis (FBA) for model simulation, optimization and estimation of flux (v) phenotype at the steady state. (G–I) The panel shows functionalities of genome-scale metabolic models such as regulations of metabolic pathway, metabolic marker identification and identification of differential pathways.
Likewise, omics data can be used to contextualize PBMC-specific networks under healthy and disease states. RM analysis can identify metabolic hotspots, modules and subnetworks, which might enhance our knowledge and understanding of immunometabolism under specific conditions. Moreover, integration of metabolomics data could help to characterize reporter reaction(s), i.e., reactions marked by significant and coordinated changes in the surrounding metabolites following the environmental/genetic perturbations. By combining transcriptome data, it is possible to infer whether the reactions are hierarchically or metabolically regulated (Cakir et al., 2006). Furthermore, fluxes estimated by PBMC-specific GEMs using Flux Balance Analysis (FBA) (Orth et al., 2010) could guide to understand the relevance of multiple pathways involved in glucose, energy, arginine and serine metabolism and ubiquinone biosynthesis with higher proficiency than previously possible (Liu et al., 2015; Almeida et al., 2016; Ma et al., 2017).
Similarly, GEMs can be reconstructed for specific immune cells. RAW 264.7 cell line, a GEM for macrophage have been developed by integration of transcriptomics, proteomics, and metabolomics datasets (Bordbar et al., 2012). The model was used to assess metabolic features that are critical for macrophage activation. It was also used to determine the metabolic modulators of the cellular activation. In another study, GEMs for naïve T cells (CD4T1670) were reconstructed by integrating transcriptomics and metabolomics datasets. This model was used to study carbohydrate metabolism, fatty acid metabolism and glutaminolysis (Han et al., 2016). Availability of the omics data for immune cell subsets, particularly CD4+ T helper cells (Th1, Th2, Th17) (Kanduri et al., 2015; Tuomela et al., 2016) provides an opportunity to reconstruct T helper specific GEMs, that could be used to characterize metabolic phenotypes of Th subsets and predict differences between them.
There is growing evidence suggesting metabolism could be regulated by epigenetic modifications (Lu and Thompson, 2012). This is facilitated by perturbation of metabolic gene(s) under suitable conditions (Colyer et al., 2012; Yun et al., 2012). Salehzadeh-Yazdi et al., incorporated epigenetic constraints in GEMs to show the impact of the mutated histone tails on metabolic reactions, thereby estimating its overall impact on yeast metabolism. The network topology was analyzed with an assumption that down-regulated metabolic genes are presumably under epigenetic control and thus affecting the metabolism of the entire organism (Salehzadeh-Yazdi et al., 2014). Similar strategy can be adopted when modeling the effect of epigenetic modification on T cell metabolism. The estimated epigenetic constraints for the down-regulated genes (presumably under epigenetic control) can be added as an additional constraint (reaction score or weight) to the associated metabolic reaction(s) within GEMs.
GEMs can be used to model and study metabolic interactions between immune cells and gut microbes on a genome-scale. This enables the identification of key regulators (metabolites/substrates, genes and enzymes) that modulate immune responses. They could also be used to identify resident microbe(s) which perform specialized metabolic functions. Moreover, GEMs can provide mechanistic overview of substrate allocation, microbe-microbe competition for resources and microbe-assisted modulation of the host immune responses. Modeling metabolic interactions among cells- and tissue-specific GEMs using a cellular compartment and/or metabolic intermediates have been previously possible (Bordbar et al., 2011; Shoaie et al., 2015; Magnúsdóttir et al., 2016; Bauer et al., 2017).
While GEMs mechanistically link metabolic genotypes and phenotypes, at the same time they handle multitude of constraints and variables which could in turn enhance uncertainty of predictions. Therefore, clear standards for GEM reconstruction, solver integration and usability has to be decided prior to the modeling (Orth et al., 2010; Chindelevitch et al., 2014; Ebrahim et al., 2015; Ravikrishnan and Raman, 2015). Availability of experimental data can help to refine GEMs to higher quality and thus lead to more accurate predictions. It is important that the predictions of GEMs are iteratively validated with the experimental data.
As indicated in this review, transcriptome, proteome, epigenome and signaling of PBMCs and Th subsets, have been well studied. In comparison, the metabolism of Th subsets and its underlying regulations is so far poorly studied. It is known that metabolism of circulating T cell undergoes dramatic changes under the environmental stress which drives the immunity (Gerriets and Rathmell, 2012; Pearce and Pearce, 2013; Pearce et al., 2013; Buck et al., 2015). We are currently making several efforts to characterize metabolic phenotype and regulations of PBMCs as obtained from pre-diabetic children at risk of developing T1DM. We believe that the congruence of GEMs based predictions and experimental data could bridge the gaps in “Big data” generated from PBMCs research. Furthermore, GEMs of PBMCs could enhance our knowledge of immune cell metabolism and allow one to better characterize PBMCs as a model system for studying immune responses under metabolically aberrant conditions.
Author Contributions
PS: drafted the manuscript; EK and MO: provided critical comments and edits to the manuscript; All authors approved the final version of the manuscript.
Funding
This work was supported by the Academy of Finland (Centre of Excellence in Molecular Systems Immunology and Physiology Research 2012–2017, Decision No. 250114, to MO) and the Juvenile Diabetes Research Foundation (2-SRA-2014-159-Q-R to MO).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank to Alex Dickens, Santosh Lamichhane, and Riitta Lahesmaa for helpful discussions related to the metabolism of Th cells and the development of T1DM.
References
Achiron, A., Feldman, A., Mandel, M., and Gurevich, M. (2007). Impaired expression of peripheral blood apoptotic-related gene transcripts in acute multiple sclerosis relapse. Ann. N. Y. Acad. Sci. 1107, 155–167. doi: 10.1196/annals.1381.017
Agren, R., Bordel, S., Mardinoglu, A., Pornputtapong, N., Nookaew, I., and Nielsen, J. (2012). Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 8:e1002518. doi: 10.1371/journal.pcbi.1002518
Akdis, M., Palomares, O., van de Veen, W., van Splunter, M., and Akdis, C. A. (2012). TH17 and TH22 cells: a confusion of antimicrobial response with tissue inflammation versus protection. J. Allergy Clin. Immunol. 129, 1438–1449. doi: 10.1016/j.jaci.2012.05.003
Almeida, L., Lochner, M., Berod, L., and Sparwasser, T. (2016). Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 28, 514–524. doi: 10.1016/j.smim.2016.10.009
Angelin, A., Gil-de-Gómez, L., Dahiya, S., Jiao, J., Guo, L., Levine, M. H., et al. (2017). Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 25, 1282–1293. e1287. doi: 10.1016/j.cmet.2016.12.018
Aune, T. M., Collins, P. L., and Chang, S. (2009). Epigenetics and T helper 1 differentiation. Immunology 126, 299–305. doi: 10.1111/j.1365-2567.2008.03026.x
Autissier, P., Soulas, C., Burdo, T. H., and Williams, K. C. (2010). Evaluation of a 12-color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans. Cytometry A. 77, 410–419. doi: 10.1002/cyto.a.20859
Bahr, T. M., Hughes, G. J., Armstrong, M., Reisdorph, R., Coldren, C. D., Edwards, M. G., et al. (2013). Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 49, 316–323. doi: 10.1165/rcmb.2012-0230OC
Baine, M. J., Chakraborty, S., Smith, L. M., Mallya, K., Sasson, A. R., Brand, R. E., et al. (2011). Transcriptional profiling of peripheral blood mononuclear cells in pancreatic cancer patients identifies novel genes with potential diagnostic utility. PLoS ONE 6:e17014. doi: 10.1371/journal.pone.0017014
Bauer, E., Laczny, C. C., Magnusdottir, S., Wilmes, P., and Thiele, I. (2015). Phenotypic differentiation of gastrointestinal microbes is reflected in their encoded metabolic repertoires. Microbiome 3:55. doi: 10.1186/s40168-015-0121-6
Bauer, E., Zimmermann, J., Baldini, F., Thiele, I., and Kaleta, C. (2017). BacArena: individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 13:e1005544. doi: 10.1371/journal.pcbi.1005544
Becker, S. A., and Palsson, B. O. (2008). Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 4:e1000082. doi: 10.1371/journal.pcbi.1000082
Becker, S. A., Feist, A. M., Mo, M. L., Hannum, G., Palsson, B. O., and Herrgard, M. J. (2007). Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat. Protoc. 2, 727–738. doi: 10.1038/nprot.2007.99
Belkaid, Y., and Hand, T. W. (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. doi: 10.1016/j.cell.2014.03.011
Bennett, L., Palucka, A. K., Arce, E., Cantrell, V., Borvak, J., Banchereau, J., et al. (2003). Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197, 711–723. doi: 10.1084/jem.20021553
Berod, L., Friedrich, C., Nandan, A., Freitag, J., Hagemann, S., Harmrolfs, K., et al. (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333. doi: 10.1038/nm.3704
Blazier, A. S., and Papin, J. A. (2012). Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 3:299. doi: 10.3389/fphys.2012.00299
Bordbar, A., Feist, A. M., Usaite-Black, R., Woodcock, J., Palsson, B. O., and Famili, I. (2011). A multi-tissue type genome-scale metabolic network for analysis of whole-body systems physiology. BMC Syst. Biol. 5:180. doi: 10.1186/1752-0509-5-180
Bordbar, A., Mo, M. L., Nakayasu, E. S., Schrimpe-Rutledge, A. C., Kim, Y. M., Metz, T. O., et al. (2012). Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol. Syst. Biol. 8:558. doi: 10.1038/msb.2012.21
Bordbar, A., Monk, J. M., King, Z. A., and Palsson, B. O. (2014). Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120. doi: 10.1038/nrg3643
Bowler, R. P., Bahr, T. M., Hughes, G., Lutz, S., Kim, Y.-I., Coldren, C. D., et al. (2013). Integrative omics approach identifies interleukin-16 as a biomarker of emphysema. Omics 17, 619–626. doi: 10.1089/omi.2013.0038
Broere, F., Apasov, S. G., Sitkovsky, M. V., and van Eden, W. (2011). “A2 T cell subsets and T cell-mediated immunity,” in Principles of Immunopharmacology: 3rd Revised and Extended Edition, eds F. P. Nijkamp and M. J. Parnham (Basel: Birkhäuser Basel), 15–27.
Brown, C. T., Davis-Richardson, A. G., Giongo, A., Gano, K. A., Crabb, D. B., Mukherjee, N., et al. (2011). Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 6:e25792. doi: 10.1371/journal.pone.0025792
Brugman, S., Klatter, F., Visser, J., Wildeboer-Veloo, A., Harmsen, H., Rozing, J., et al. (2006). Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 49, 2105–2108. doi: 10.1007/s00125-006-0334-0
Buck, M. D., O'Sullivan, D., and Pearce, E. L. (2015). T cell metabolism drives immunity. J. Exp. Med. 212, 1345–1360. doi: 10.1084/jem.20151159
Buonaguro, L., Wang, E., Tornesello, M. L., Buonaguro, F. M., and Marincola, F. M. (2011). Systems biology applied to vaccine and immunotherapy development. BMC Syst. Biol. 5:146. doi: 10.1186/1752-0509-5-146
Burczynski, M. E., Twine, N. C., Dukart, G., Marshall, B., Hidalgo, M., Stadler, W. M., et al. (2005). Transcriptional profiles in peripheral blood mononuclear cells prognostic of clinical outcomes in patients with advanced renal cell carcinoma. Clin. Cancer Res. 11, 1181–1189.
Cakir, T., Patil, K. R., Onsan, Z., Ulgen, K. O., Kirdar, B., and Nielsen, J. (2006). Integration of metabolome data with metabolic networks reveals reporter reactions. Mol. Syst. Biol. 2:50. doi: 10.1038/msb4100085
Chaussabel, D., Quinn, C., Shen, J., Patel, P., Glaser, C., Baldwin, N., et al. (2008). A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity 29, 150–164. doi: 10.1016/j.immuni.2008.05.012
Chen, L., Ge, B., Casale, F. P., Vasquez, L., Kwan, T., Garrido-Martin, D., et al. (2016). Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167, 1398–1414 e1324. doi: 10.1016/j.cell.2016.10.026
Chindelevitch, L., Trigg, J., Regev, A., and Berger, B. (2014). An exact arithmetic toolbox for a consistent and reproducible structural analysis of metabolic network models. Nat. Commun. 5:4893. doi: 10.1038/ncomms5893
Ciofani, M., Madar, A., Galan, C., Sellars, M., Mace, K., Pauli, F., et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. doi: 10.1016/j.cell.2012.09.016
Colijn, C., Brandes, A., Zucker, J., Lun, D. S., Weiner, B., Farhat, M. R., et al. (2009). Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 5:e1000489. doi: 10.1371/journal.pcbi.1000489
Collares, C., Evangelista, A., Xavier, D., Takahashi, P., Almeida, R., Macedo, C., et al. (2013). Transcriptome meta-analysis of peripheral lymphomononuclear cells indicates that gestational diabetes is closer to type 1 diabetes than to type 2 diabetes mellitus. Mol. Biol. Rep. 40, 5351–5358. doi: 10.1007/s11033-013-2635-y
Colyer, H. A., Armstrong, R. N., and Mills, K. I. (2012). Microarray for epigenetic changes: gene expression arrays. Methods Mol. Biol. 863, 319–328. doi: 10.1007/978-1-61779-612-8_20
Cooper, M. D. (2015). The early history of B cells. Nat. Rev. Immunol. 15, 191–197. doi: 10.1038/nri3801
Croft, M., Carter, L., Swain, S. L., and Dutton, R. W. (1994). Generation of polarized antigen-specific CD8 effector populations: reciprocal action of interleukin (IL)-4 and IL-12 in promoting type 2 versus type 1 cytokine profiles. J. Exp. Med. 180, 1715–1728. doi: 10.1084/jem.180.5.1715
Crotty, S. (2011). Follicular helper CD4 T cells (Tfh). Annu. Rev. Immunol. 29, 621–663. doi: 10.1146/annurev-immunol-031210-101400
Crow, M. K., Kirou, K. A., and Wohlgemuth, J. (2003). Microarray analysis of interferon-regulated genes in SLE. Autoimmunity 36, 481–490. doi: 10.1080/08916930310001625952
de Goffau, M. C., Luopajärvi, K., Knip, M., Ilonen, J., Ruohtula, T., Härkönen, T., et al. (2013). Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes 62, 1238–1244. doi: 10.2337/db12-0526
Dimeloe, S., Burgener, A. V., Grahlert, J., and Hess, C. (2017). T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology 150, 35–44. doi: 10.1111/imm.12655
Duarte, N. C., Becker, S. A., Jamshidi, N., Thiele, I., Mo, M. L., Vo, T. D., et al. (2007). Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. U.S.A. 104, 1777–1782. doi: 10.1073/pnas.0610772104
Ebrahim, A., Almaas, E., Bauer, E., Bordbar, A., Burgard, A. P., Chang, R. L., et al. (2015). Do genome-scale models need exact solvers or clearer standards? Mol. Syst. Biol. 11:831. doi: 10.15252/msb.20156157
Edwards, C. J., Feldman, J. L., Beech, J., Shields, K. M., Stover, J. A., Trepicchio, W. L., et al. (2007). Molecular profile of peripheral blood mononuclear cells from patients with rheumatoid arthritis. Mol. Med. 13, 40–58. doi: 10.2119/2006-00056.Edwards
El-Semman, I. E., Karlsson, F. H., Shoaie, S., Nookaew, I., Soliman, T. H., and Nielsen, J. (2014). Genome-scale metabolic reconstructions of Bifidobacterium adolescentis L2-32 and Faecalibacterium prausnitzii A2-165 and their interaction. BMC Syst. Biol. 8:41. doi: 10.1186/1752-0509-8-41
Falcai, A., Soeiro-Pereira, P., Kubo, C., Aranda, C., Solé, D., and Condino-Neto, A. (2015). Peripheral blood mononuclear cells from severe asthmatic children release lower amounts of IL-12 and IL-4 after LPS stimulation. Allergol. Immunopathol. 43, 482–486. doi: 10.1016/j.aller.2014.10.005
Fernandez, D. R., Telarico, T., Bonilla, E., Li, Q., Banerjee, S., Middleton, F. A., et al. (2009). Activation of mammalian target of rapamycin controls the loss of TCRζ in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J. Immunol. 182, 2063–2073. doi: 10.4049/jimmunol.0803600
Filén, J.-J., Filén, S., Moulder, R., Tuomela, S., Ahlfors, H., West, A., et al. (2009). Quantitative proteomics reveals GIMAP family proteins 1 and 4 to be differentially regulated during human T helper cell differentiation. Mol. Cell. Proteomics 8, 32–44. doi: 10.1074/mcp.M800139-MCP200
Förster, J., Famili, I., Fu, P., Palsson, B. Ø., and Nielsen, J. (2003). Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 13, 244–253. doi: 10.1101/gr.234503
Foss-Freitas, M. C., Foss, N. T., Rassi, D. M., Donadi, E. A., and Foss, M. C. (2008). Evaluation of cytokine production from peripheral blood mononuclear cells of type 1 diabetic patients. Ann. N.Y. Acad. Sci. 1150, 290–296. doi: 10.1196/annals.1447.053
Geiger, R., Rieckmann, J. C., Wolf, T., Basso, C., Feng, Y., Fuhrer, T., et al. (2016). L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842 e813. doi: 10.1016/j.cell.2016.09.031
Gerriets, V. A., and Rathmell, J. C. (2012). Metabolic pathways in T cell fate and function. Trends Immunol. 33, 168–173. doi: 10.1016/j.it.2012.01.010
Giongo, A., Gano, K. A., Crabb, D. B., Mukherjee, N., Novelo, L. L., Casella, G., et al. (2011). Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 5, 82–91. doi: 10.1038/ismej.2010.92
Golubovskaya, V., and Wu, L. (2016). Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers 8:36. doi: 10.3390/cancers8030036
Greenberg, S. A., Pinkus, J. L., Pinkus, G. S., Burleson, T., Sanoudou, D., Tawil, R., et al. (2005). Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann. Neurol. 57, 664–678. doi: 10.1002/ana.20464
Han, F., Li, G., Dai, S., and Huang, J. (2016). Genome-wide metabolic model to improve understanding of CD4+ T cell metabolism, immunometabolism and application in drug design. Mol. Biosyst. 12, 431–443. doi: 10.1039/C5MB00480B
Haudek-Prinz, V. J., Klepeisz, P., Slany, A., Griss, J., Meshcheryakova, A., Paulitschke, V., et al. (2012). Proteome signatures of inflammatory activated primary human peripheral blood mononuclear cells. J. Proteomics 76 Spec No., 150–162. doi: 10.1016/j.jprot.2012.07.012
Hirahara, K., Vahedi, G., Ghoreschi, K., Yang, X. P., Nakayamada, S., Kanno, Y., et al. (2011). Helper T-cell differentiation and plasticity: insights from epigenetics. Immunology 134, 235–245. doi: 10.1111/j.1365-2567.2011.03483.x
Hu, G., Tang, Q., Sharma, S., Yu, F., Escobar, T. M., Muljo, S. A., et al. (2013). Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat. Immunol. 14, 1190–1198. doi: 10.1038/ni.2712
Hyötyläinen, T., Jerby, L., Petäjä, E. M., Mattila, I., Jäntti, S., Auvinen, P., et al. (2016). Genome-scale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat. Commun. 7:8994. doi: 10.1038/ncomms9994
Iikura, K., Katsunuma, T., Saika, S., Saito, S., Ichinohe, S., Ida, H., et al. (2011). Peripheral blood mononuclear cells from patients with bronchial asthma show impaired innate immune responses to rhinovirus in vitro. Int. Arch. Allergy Immunol. 155(Suppl. 1), 27–33. doi: 10.1159/000327262
Jensen, P. A., and Papin, J. A. (2011). Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 27, 541–547. doi: 10.1093/bioinformatics/btq702
Kanduri, K., Tripathi, S., Larjo, A., Mannerstrom, H., Ullah, U., Lund, R., et al. (2015). Identification of global regulators of T-helper cell lineage specification. Genome Med. 7:122. doi: 10.1186/s13073-015-0237-0
Kew, S., Banerjee, T., Minihane, A. M., Finnegan, Y. E., Williams, C. M., and Calder, P. C. (2003). Relation between the fatty acid composition of peripheral blood mononuclear cells and measures of immune cell function in healthy, free-living subjects aged 25–72 y. Am. J. Clin. Nutr. 77, 1278–1286.
Kleiveland, C. R. (2015). “Peripheral blood mononuclear cells,” in The Impact of Food Bioactives on Health, eds K. Verhoeckx, P. Cotter, I. López-Expósito, C. Kleiveland, T. Lea, A. Mackie, T. Requena, D. Swiatecka, H. Wichers (Springer), 161–167.
Knip, M., and Siljander, H. (2016). The role of the intestinal microbiota in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 12, 154–167. doi: 10.1038/nrendo.2015.218
Kosiewicz, M. M., Dryden, G. W., Chhabra, A., and Alard, P. (2014). Relationship between gut microbiota and development of T cell associated disease. FEBS Lett. 588, 4195–4206. doi: 10.1016/j.febslet.2014.03.019
Kostic, A. D., Gevers, D., Siljander, H., Vatanen, T., Hyötyläinen, T., Hämäläinen, A.-M., et al. (2015). The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 17, 260–273. doi: 10.1016/j.chom.2015.01.001
Kröger, W., Mapiye, D., Entfellner, J.-B. D., and Tiffin, N. (2016). A meta-analysis of public microarray data identifies gene regulatory pathways deregulated in peripheral blood mononuclear cells from individuals with systemic lupus erythematosus compared to those without. BMC Med. Genomics 9:66. doi: 10.1186/s12920-016-0227-0
Lee, G. R., Kim, S. T., Spilianakis, C. G., Fields, P. E., and Flavell, R. A. (2006). T helper cell differentiation: regulation by cis elements and epigenetics. Immunity 24, 369–379. doi: 10.1016/j.immuni.2006.03.007
Levy, H., Wang, X., Kaldunski, M., Jia, S., Kramer, J., Pavletich, S. J., et al. (2012). Transcriptional signatures as a disease-specific and predictive inflammatory biomarker for type 1 diabetes. Genes Immun. 13, 593–604. doi: 10.1038/gene.2012.41
Li, J., Jia, H., Cai, X., Zhong, H., Feng, Q., Sunagawa, S., et al. (2014a). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32, 834–841. doi: 10.1038/nbt.2942
Li, J., Zhang, S. X., Wang, W., Cheng, K., Guo, H., Rao, C. L., et al. (2017a). Potential antidepressant and resilience mechanism revealed by metabolomic study on peripheral blood mononuclear cells of stress resilient rats. Behav. Brain Res. 320, 12–20. doi: 10.1016/j.bbr.2016.11.035
Li, S., Nakaya, H. I., Kazmin, D. A., Oh, J. Z., and Pulendran, B. (2013). “Systems biological approaches to measure and understand vaccine immunity in humans,” in Seminars in Immunology (Elsevier), 209–218.
Li, S., Rouphael, N., Duraisingham, S., Romero-Steiner, S., Presnell, S., Davis, C., et al. (2014b). Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 15, 195–204. doi: 10.1038/ni.2789
Li, S., Sullivan, N. L., Rouphael, N., Yu, T., Banton, S., Maddur, M. S., et al. (2017b). Metabolic phenotypes of response to vaccination in humans. Cell 169, 862–877.e17. doi: 10.1016/j.cell.2017.04.026
Liu, M. L., Zhang, X. T., Du, X. Y., Fang, Z., Liu, Z., Xu, Y., et al. (2015). Severe disturbance of glucose metabolism in peripheral blood mononuclear cells of schizophrenia patients: a targeted metabolomic study. J. Transl. Med. 13, 226. doi: 10.1186/s12967-015-0540-y
Loyet, K. M., Ouyang, W., Eaton, D. L., and Stults, J. T. (2005). Proteomic profiling of surface proteins on Th1 and Th2 cells. J. Proteome Res. 4, 400–409. doi: 10.1021/pr049810q
Lu, C., and Thompson, C. B. (2012). Metabolic regulation of epigenetics. Cell Metab. 16, 9–17. doi: 10.1016/j.cmet.2012.06.001
Luckheeram, R. V., Zhou, R., Verma, A. D., and Xia, B. (2012). CD4+T cells: differentiation and functions. Clin. Dev. Immunol. 2012:12. doi: 10.1155/2012/925135
Ma, E. H., Bantug, G., Griss, T., Condotta, S., Johnson, R. M., Samborska, B., et al. (2017). Serine is an essential metabolite for effector T cell expansion. Cell Metab. 25, 345–357. doi: 10.1016/j.cmet.2016.12.011
Ma, H., Sorokin, A., Mazein, A., Selkov, A., Selkov, E., Demin, O., et al. (2007). The Edinburgh human metabolic network reconstruction and its functional analysis. Mol. Syst. Biol. 3:135. doi: 10.1038/msb4100177
MacIver, N. J., Michalek, R. D., and Rathmell, J. C. (2013). Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283. doi: 10.1146/annurev-immunol-032712-095956
Magnúsdóttir, S., Heinken, A., Kutt, L., Ravcheev, D. A., Bauer, E., Noronha, A., et al. (2016). Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89. doi: 10.1038/nbt.3703
Mak, T. W., Grusdat, M., Duncan, G. S., Dostert, C., Nonnenmacher, Y., Cox, M., et al. (2017). Glutathione primes T cell metabolism for inflammation. Immunity 46, 1089–1090. doi: 10.1016/j.immuni.2017.06.009
Mardinoglu, A., Agren, R., Kampf, C., Asplund, A., Nookaew, I., Jacobson, P., et al. (2013). Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 9:649. doi: 10.1038/msb.2013.5
Mardinoglu, A., Agren, R., Kampf, C., Asplund, A., Uhlen, M., and Nielsen, J. (2014). Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 5:3083. doi: 10.1038/ncomms4083
Mejía-León, M. E., and Barca, A. M. (2015). Diet, microbiota and immune system in type 1 diabetes development and evolution. Nutrients 7, 9171–9184. doi: 10.3390/nu7115461
Mouritsen, O. G. (2011). Lipidology and lipidomics–quo vadis? A new era for the physical chemistry of lipids. Phys. Chem. Chem. Phys. 13, 19195–19205. doi: 10.1039/c1cp22484k
Murri, M., Leiva, I., Gomez-Zumaquero, J. M., Tinahones, F. J., Cardona, F., Soriguer, F., et al. (2013). Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 11:46. doi: 10.1186/1741-7015-11-46
O'Brien, E. J., Monk, J. M., and Palsson, B. O. (2015). Using genome-scale models to predict biological capabilities. Cell 161, 971–987. doi: 10.1016/j.cell.2015.05.019
O'Brien, E. J., Lerman, J. A., Chang, R. L., Hyduke, D. R., and Palsson, B. O. (2013). Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 9:693. doi: 10.1038/msb.2013.52
Oestreich, K. J., and Weinmann, A. S. (2012). Encoding stability versus flexibility: lessons learned from examining epigenetics in T helper cell differentiation. Curr. Top. Microbiol. Immunol. 356, 145–164. doi: 10.1007/82_2011_141
Olafsdottir, T. A., Lindqvist, M., Nookaew, I., Andersen, P., Maertzdorf, J., Persson, J., et al. (2016). Comparative systems analyses reveal molecular signatures of clinically tested vaccine adjuvants. Sci. Rep. 6:39097. doi: 10.1038/srep39097
Orth, J. D., Thiele, I., and Palsson, B. Ø. (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248. doi: 10.1038/nbt.1614
Pagani, M., Rockstroh, M., Schuster, M., Rossetti, G., Moro, M., Crosti, M., et al. (2015). Reference proteome of highly purified human Th1 cells reveals strong effects on metabolism and protein ubiquitination upon differentiation. Proteomics 15, 3644–3647. doi: 10.1002/pmic.201400139
Patil, K. R., and Nielsen, J. (2005). Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. U.S.A. 102, 2685–2689. doi: 10.1073/pnas.0406811102
Patil, K. R., Rocha, I., Förster, J., and Nielsen, J. (2005). Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinformatics 6:308. doi: 10.1186/1471-2105-6-308
Payne, K. K., Zoon, C. K., Wan, W., Marlar, K., Keim, R. C., Kenari, M. N., et al. (2013). Peripheral blood mononuclear cells of patients with breast cancer can be reprogrammed to enhance anti-HER-2/neu reactivity and overcome myeloid-derived suppressor cells. Breast Cancer Res. Treat. 142, 45–57. doi: 10.1007/s10549-013-2733-5
Pearce, E. L., and Pearce, E. J. (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643. doi: 10.1016/j.immuni.2013.04.005
Pearce, E. L., Poffenberger, M. C., Chang, C.-H., and Jones, R. G. (2013). Fueling immunity: insights into metabolism and lymphocyte function. Science 342:1242454. doi: 10.1126/science.1242454
Pharkya, P., and Maranas, C. D. (2006). An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab. Eng. 8, 1–13. doi: 10.1016/j.ymben.2005.08.003
Pharkya, P., Burgard, A. P., and Maranas, C. D. (2004). OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 14, 2367–2376. doi: 10.1101/gr.2872004
Price, N. D., Reed, J. L., and Palsson, B. Ø. (2004). Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol. 2, 886–897. doi: 10.1038/nrmicro1023
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalog established by metagenomic sequencing. Nature 464:59. doi: 10.1038/nature08821
Ravikrishnan, A., and Raman, K. (2015). Critical assessment of genome-scale metabolic networks: the need for a unified standard. Brief. Bioinformatics 16, 1057–1068. doi: 10.1093/bib/bbv003
Roesch, L. F., Lorca, G. L., Casella, G., Giongo, A., Naranjo, A., Pionzio, A. M., et al. (2009). Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J. 3, 536–548. doi: 10.1038/ismej.2009.5
Rosengren, A. T., Nyman, T. A., and Lahesmaa, R. (2005). Proteome profiling of interleukin-12 treated human T helper cells. Proteomics 5, 3137–3141. doi: 10.1002/pmic.200401151
Round, J. L., and Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323. doi: 10.1038/nri2515
Sakaguchi, S., Yamaguchi, T., Nomura, T., and Ono, M. (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. doi: 10.1016/j.cell.2008.05.009
Salehzadeh-Yazdi, A., Asgari, Y., Saboury, A. A., and Masoudi-Nejad, A. (2014). Computational analysis of reciprocal association of metabolism and epigenetics in the budding yeast: a genome-scale metabolic model (GSMM) approach. PLoS ONE 9:e111686. doi: 10.1371/journal.pone.0111686
Sanders, V. M. (2006). Epigenetic regulation of Th1 and Th2 cell development. Brain Behav. Immun. 20, 317–324. doi: 10.1016/j.bbi.2005.08.005
Savaryn, J. P., Toby, T. K., Catherman, A. D., Fellers, R. T., LeDuc, R. D., Thomas, P. M., et al. (2016). Comparative top down proteomics of peripheral blood mononuclear cells from kidney transplant recipients with normal kidney biopsies or acute rejection. Proteomics 16, 2048–2058. doi: 10.1002/pmic.201600008
Schellenberger, J., Que, R., Fleming, R. M., Thiele, I., Orth, J. D., Feist, A. M., et al. (2011). Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 6, 1290–1307. doi: 10.1038/nprot.2011.308
Sen, P., Mardinogulu, A., and Nielsen, J. (2017). Selection of complementary foods based on optimal nutritional values. Sci. Rep. 7:5413. doi: 10.1038/s41598-017-05650-0
Sen, P., Vial, H. J., and Radulescu, O. (2016). Mathematical modeling and omic data integration to understand dynamic adaptation of Apicomplexan parasites and identify pharmaceutical targets. Compr. Anal. Parasite Biol. 7:457. doi: 10.1002/9783527694082.ch20
Shlomi, T., Cabili, M. N., Herrgard, M. J., Palsson, B. O., and Ruppin, E. (2008). Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 26, 1003–1010. doi: 10.1038/nbt.1487
Shoaie, S., and Nielsen, J. (2014). Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 5:86. doi: 10.3389/fgene.2014.00086
Shoaie, S., Ghaffari, P., Kovatcheva-Datchary, P., Mardinoglu, A., Sen, P., Pujos-Guillot, E., et al. (2015). Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 22, 320–331. doi: 10.1016/j.cmet.2015.07.001
Simeoni, O., Piras, V., Tomita, M., and Selvarajoo, K. (2015). Tracking global gene expression responses in T cell differentiation. Gene 569, 259–266. doi: 10.1016/j.gene.2015.05.061
Smiljanovic, B., Grün, J. R., Biesen, R., Schulte-Wrede, U., Baumgrass, R., Stuhlmüller, B., et al. (2012). The multifaceted balance of TNF-α and type I/II interferon responses in SLE and RA: how monocytes manage the impact of cytokines. J. Mol. Med. 90, 1295–1309. doi: 10.1007/s00109-012-0907-y
Stockinger, B., and Veldhoen, M. (2007). Differentiation and function of Th17 T cells. Curr. Opin. Immunol. 19, 281–286. doi: 10.1016/j.coi.2007.04.005
Suthers, P. F., Zomorrodi, A., and Maranas, C. D. (2009). Genome-scale gene/reaction essentiality and synthetic lethality analysis. Mol. Syst. Biol. 5:301. doi: 10.1038/msb.2009.56
Swainston, N., Smallbone, K., Hefzi, H., Dobson, P. D., Brewer, J., Hanscho, M., et al. (2016). Recon 2.2: from reconstruction to model of human metabolism. Metabolomics 12:109. doi: 10.1007/s11306-016-1051-4
Tan, C., and Gery, I. (2012). The unique features of Th9 cells and their products. Crit. Rev. Immunol. 32, 1–10. doi: 10.1615/CritRevImmunol.v32.i1.10
Teixeira, V. H., Olaso, R., Martin-Magniette, M. L., Lasbleiz, S., Jacq, L., Oliveira, C. R., et al. (2009). Transcriptome analysis describing new immunity and defense genes in peripheral blood mononuclear cells of rheumatoid arthritis patients. PLoS ONE 4:e6803. doi: 10.1371/journal.pone.0006803
Thiele, I., Swainston, N., Fleming, R. M., Hoppe, A., Sahoo, S., Aurich, M. K., et al. (2013). A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 31, 419–425. doi: 10.1038/nbt.2488
Tuomela, S., and Lahesmaa, R. (2013). Early T helper cell programming of gene expression in human. Semin. Immunol. 25, 282–290. doi: 10.1016/j.smim.2013.10.013
Tuomela, S., Rautio, S., Ahlfors, H., Oling, V., Salo, V., Ullah, U., et al. (2016). Comparative analysis of human and mouse transcriptomes of Th17 cell priming. Oncotarget 7, 13416–13428. doi: 10.18632/oncotarget.7963
Twine, N. C., Stover, J. A., Marshall, B., Dukart, G., Hidalgo, M., Stadler, W., et al. (2003). Disease-associated expression profiles in peripheral blood mononuclear cells from patients with advanced renal cell carcinoma. Cancer Res. 63, 6069–6075.
Väremo, L., Scheele, C., Broholm, C., Mardinoglu, A., Kampf, C., Asplund, A., et al. (2016). Proteome-and transcriptome-driven reconstruction of the human myocyte metabolic network and its use for identification of markers for diabetes. Cell Rep. 14:1567. doi: 10.1016/j.celrep.2015.04.010
Wang, X., Jia, S., Geoffrey, R., Alemzadeh, R., Ghosh, S., and Hessner, M. J. (2008). Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J. Immunol. 180, 1929–1937. doi: 10.4049/jimmunol.180.3.1929
Wang, Y., Eddy, J. A., and Price, N. D. (2012). Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst. Biol. 6:153. doi: 10.1186/1752-0509-6-153
Wen, L., Ley, R. E., Volchkov, P. Y., Stranges, P. B., Avanesyan, L., Stonebraker, A. C., et al. (2008). Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455, 1109–1113. doi: 10.1038/nature07336
Wing, K., and Sakaguchi, S. (2010). Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 11, 7–13. doi: 10.1038/ni.1818
Yizhak, K., Chaneton, B., Gottlieb, E., and Ruppin, E. (2015). Modeling cancer metabolism on a genome scale. Mol. Syst. Biol. 11:817. doi: 10.15252/msb.20145307
Yuan, D., Koh, C., and Wilder, J. (1994). Interactions between B lymphocytes and NK cells. FASEB J. 8, 1012–1018.
Yun, J., Johnson, J. L., Hanigan, C. L., and Locasale, J. W. (2012). Interactions between epigenetics and metabolism in cancers. Front. Oncol. 2:163. doi: 10.3389/fonc.2012.00163
Zhang, Z.-N., Xu, J.-J., Fu, Y.-J., Liu, J., Jiang, Y.-J., Cui, H.-L., et al. (2013). Transcriptomic analysis of peripheral blood mononuclear cells in rapid progressors in early HIV infection identifies a signature closely correlated with disease progression. Clin. Chem. 59, 1175–1186. doi: 10.1373/clinchem.2012.197335
Keywords: systems biology, multi-omics, peripheral blood mononuclear cells, PBMCs, immune system, metabolomics, genome-scale metabolic models, pathways
Citation: Sen P, Kemppainen E and Orešič M (2018) Perspectives on Systems Modeling of Human Peripheral Blood Mononuclear Cells. Front. Mol. Biosci. 4:96. doi: 10.3389/fmolb.2017.00096
Received: 09 September 2017; Accepted: 21 December 2017;
Published: 09 January 2018.
Edited by:
Wolfram Weckwerth, University of Vienna, AustriaReviewed by:
Atsushi Fukushima, Riken, JapanFabien Jourdan, Institut National de la Recherche Agronomique, France
Copyright © 2018 Sen, Kemppainen and Orešič. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Partho Sen, partho.sen@utu.fi