Implications of GABAergic Neurotransmission in Alzheimer’s Disease
 Yanfang Li
Yanfang Li Hao Sun
Hao Sun Zhicai Chen
Zhicai Chen Huaxi Xu
Huaxi Xu Guojun Bu
Guojun Bu Hui Zheng
Hui Zheng- 1Fujian Provincial Key Laboratory of Neurodegenerative Disease and Aging Research, Institute of Neuroscience, College of Medicine, Xiamen University, Xiamen, China
- 2Neurodegenerative Disease Research Program, Sanford-Burnham Medical Research Institute, La Jolla, CA, USA
- 3Department of Neuroscience, Mayo Clinic, Jacksonville, FL, USA
- 4The Interdepartmental Program of Translational Biology and Molecular Medicine, Huffington Center on Aging, Baylor College of Medicine, Houston, TX, USA
Alzheimer’s disease (AD) is characterized pathologically by the deposition of β-amyloid peptides (Aβ) and the accumulation of neurofibrillary tangles (NFTs) composed of hyper-phosphorylated tau. Regardless of the pathological hallmarks, synaptic dysfunction is widely accepted as a causal event in AD. Of the two major types of synapses in the central nervous system (CNS): glutamatergic and GABAergic, which provide excitatory and inhibitory outputs respectively, abundant data implicate an impaired glutamatergic system during disease progression. However, emerging evidence supports the notion that disrupted default neuronal network underlies impaired memory, and that alterations of GABAergic circuits, either plays a primary role or as a compensatory response to excitotoxicity, may also contribute to AD by disrupting the overall network function. The goal of this review is to provide an overview of the involvement of Aβ, tau and apolipoprotein E4 (apoE4), the major genetic risk factor in late-onset AD (LOAD), in GABAergic neurotransmission and the potential of modulating the GABAergic function as AD therapy.
Introduction of Alzheimer’S Disease
Alzheimer’s disease (AD) is the most common age-associated neurodegenerative disorder, which is characterized by the deterioration of memory and cognition. About 10% of the population over the age of 65 and 30–50% of the population over the age of 85 suffer from AD (Querfurth and LaFerla, 2010). Despite significant research and drug development effort in the past decades, currently there are no effective therapies that can prevent, delay or stop the progression of AD, causing a severe burden for the patients, their families and the society.
A small subset (less than 2%) of AD cases result from dominantly inherited genetic mutations in genes encoding the β-amyloid precursor protein (APP) and presenilins (PSEN1 and PSEN2, Goate et al., 1991; Levy-Lahad et al., 1995; Sherrington et al., 1995). These AD cases usually develop disease before the age of 60, referring as early-onset familial AD (FAD). Sporadic or late-onset AD (LOAD) usually develops the disease later in life, representing the majority of AD cases (Kanekiyo et al., 2014). The pathological hallmarks of AD include widespread neuronal degeneration, senile plaques and intracellular neurofibrillary tangles (NFTs; Glenner and Wong, 1984; Querfurth and LaFerla, 2010; Tapia-Rojas et al., 2015).
β-Amyloid
The extracellular senile plaques are composed of accumulated small peptides called β-amyloid (Aβ) derived from the sequential cleavage of APP. There are three major isoforms of APP resulting from alternative splicing, named as APP695, APP751 and APP770 according to their number of amino acid residues. The isoform APP695 is predominantly expressed in neurons and lacks a 56 amino acid Kunitz Protease Inhibitor (KPI) domain at extracellular region (Goate et al., 1991; Zhang et al., 2012; Guzmán et al., 2014; Gautam et al., 2015). Full-length APP is a type I transmembrane protein and can undergo sequential proteolytic cleavage by distinct α-, β- or γ-secretase.
Depending on whether there’s toxic Aβ generation, the APP proteolytic cleavage is divided into two types: amyloidogenic processing and nonamyloidogenic processing. In amyloidogenic processing, APP is first cleaved by β-secretase (beta-site APP cleaving enzyme 1, BACE1), releasing a soluble ectodomain called sAPPβ. The remaining membrane associated carboxyl terminal fragment (βCTF) will be further cleaved by γ-secretase within the cell membrane, releasing neurotoxic Aβ peptides and amyloid intracellular domain (AICD; Li et al., 2014; Ohki et al., 2014; Jung et al., 2015; Neumann et al., 2015; Sadleir et al., 2015; Zhang et al., 2015). The exact cleavage site of γ-secretase may vary inside membrane, yielding Aβ peptides with 36–43 amino acids. Among them, Aβ40 is the major form while Aβ42 is the more amyloidogenic and toxic form (Querfurth and LaFerla, 2010; Zhang et al., 2012; Buggia-Prévot et al., 2014). The AICD tail released inside cytoplasm has been demonstrated to target the nucleus and regulate gene transcription activity (Querfurth and LaFerla, 2010).
As imbalance between production and clearance occurs, Aβ peptides could spontaneously self-aggregate into soluble oligomers, or further grow into insoluble fibers and finally amyloid plaques. The “amyloid hypothesis” is based on the idea that the accumulation of Aβ may be the initiating factor of AD pathogenesis. Multiple lines of evidence have indicated that accumulation of Aβ lead to a neurodegenerative cascade, resulting in synaptic dysfunction, NFT formation and eventually neuronal loss in vulnerable brain regions including cortex and hippocampus (Selkoe, 1998; Hu et al., 2014; Stancu et al., 2014). Compared to insoluble fibers, the soluble Aβ oligomers are more neurotoxic and confer the most deterious effect of Aβ (Querfurth and LaFerla, 2010; Zhang et al., 2012; Tu et al., 2014; Xu et al., 2015).
The nonamyloidogenic processing of APP initiates from the proteolytic cleavage by α-secretase, releasing a soluble sAPPα (Postina, 2012; Jiang et al., 2014; Wang et al., 2014). It is 16 amino acids bigger than sAPPβ because the cleavage site of α-secretase is within the Aβ domain, therefore excluding the possibility of Aβ generation. The membrane remaining αCTF could be further cleaved by γ-secretase, releasing the shorter P83 peptide and AICD. In contrast to Aβ, sAPPα showed important protective roles in neuronal survival and synaptic plasticity against Aβ (Mattson et al., 1993; Goodman and Mattson, 1994; Yamamoto et al., 1994; Furukawa and Mattson, 1998).
Tau
The major component of AD hallmark NFTs was revealed to be abnormally hyperphosphorylated microtubule-associated protein tau (MAPT), which is essential for assembly and stabilization of microtubules (Spillantini and Goedert, 1998; Querfurth and LaFerla, 2010). The encoding gene of MAPT is located on chromosome 17 in human and expresses six isoforms by alternative splicing in central nervous system (CNS). As a result, the six tau isoforms possess variable N-terminal repeats (0, 1 or 2N) and C-terminal microtubule-binding domains (3 or 4R; Kolarova et al., 2012; Caillet-Boudin et al., 2015; Song et al., 2015).
In physiological conditions, tau is very soluble and mainly located in neuronal axons, where it binds microtubules and regulates the axonal transportation for vesicles and organelles. Since many amino acid residues of the tau protein are potential phosphorylation sites (Ser, Thr, or Tyr), tau is highly phosphorylation-labile (Hasegawa et al., 1992; Hanger et al., 2009). It dynamically switches between phosphorylated and dephosphorylated state during each cell cycle (Pedersen and Sigurdsson, 2015; Wang et al., 2015). It has been demonstrated that increasing tau phosphorylation reduces its affinity for microtubules (Iqbal et al., 2005). The imbalance between tau kinase and phosphatase activities under phathological conditions could lead to tau hyperphosphorylation, which makes tau insoluble and self-aggregate into paired helical filament structure of NFTs. In NFTs, at least 7–8 residues were phosphorylated (Hasegawa et al., 1992; Hanger et al., 2009; Mandelkow and Mandelkow, 2012; Marttinen et al., 2015). Hyperphosporylated tau lacks the affinity for microtubules, therefore making microtubules unstable and impairing their critical function in axonal transportation, eventually resulting in synaptic dysfunction.
The specific tau pathology was reported to correlate well with cognitive abilities. In the cerebrospinal fluid (CSF) of AD patients, the levels of both total tau and phosphorylated tau were found increased (Jack et al., 2013). Besides AD, NFTs composed of hyperphosphorylated tau was found to be a common pathological feature in a number of neurodegenerative disorders including Parkinson’s disease, frontotemporal dementia and progressive supranuclear palsy (PSP), referring as a class of neurodegenerative diseases called tauopathies (Spillantini and Goedert, 1998; Lonskaya et al., 2014; Golovyashkina et al., 2015; Yamada et al., 2015). Considering the critical contribution of tau to the pathological progression of AD, a tau based hypothesis for AD has received wide notice. It emphasizes that the intracellular aggregation of hyperphosphorylated tau leads to the disassembly of microtubules, collapse of synapses, and eventually the cell death in AD (Pedersen and Sigurdsson, 2015).
Introduction of GABAergic Neurotransmitter System
γ-aminobutyric acid (GABA) is the principle inhibitory neurotransmitter in mammalian CNS. The inhibitory effect of GABA can be conferred through three distinct receptor subfamilies named GABAA, GABAB and GABAC receptors. Both GABAA and GABAC receptors are ligand-gated chloride (Cl−) channels, whereas GABAB receptors are G-protein coupled metabotropic receptors (Chebib and Johnston, 1999; Bormann, 2000). In the vertebrate brain, GABAA receptors mediate the majority of fast inhibition in the brain. They are composed of five distinct subunits pentamerically assembled, forming a ligand-gated Cl− ion channel. According to their gene identity, the identified GABAA receptor subunits are classified as α1–6, β1–3, γ1–3, ρ1–3, θ, δ, π and ε. In the mammalian brain, the most common combination of GABAA receptor contains two α, two β and one γ subunits. GABAC receptors are composed of ρ1–3 subunits, form homomeric or heteromeric channels, making them distinct from GABAA receptors in pharmacology and function (Johnston, 1994; Lüscher and Keller, 2004). GABAA receptors are widely expressed in all the CNS, while GABAC receptors are highly enriched in retina. Since both GABAA and GABAC receptors are ligand-gated Cl− channels, sometimes we also consider GABAC receptors as a minor subgroup in GABAA receptors (Barnard et al., 1998). GABAB receptor is a metabotropic receptor coupling with Gi/o protein. It regulates neuronal activity by either opening the K+ channel or inhibiting Ca2+ channel via the Gi/o protein-dependent signaling cascade (Bowery, 1989; Marshall et al., 1999).
In CNS, the inhibitory action of GABA can be broadly divided into two classes: phasic inhibition and tonic inhibition (Farrant and Nusser, 2005; McQuail et al., 2015). In GABAergic interneurons, neurotransmitter GABA is synthesized from glutamate by the enzyme glutamic acid decarboxylase (GAD). Synthesized GABA is transported along the axon to presynaptic terminals and recruited into vesicles by the vesicular GABA transporter (vGAT; Glykys and Mody, 2007; Gonzalez-Burgos et al., 2009). Upon membrane depolarization induced by action potential, neurotransmitter GABA can be released from presynaptic vesicles into the synaptic cleft, resulting in the burst increase of GABA concentration in the cleft. Most of the released neurotransmitter GABA transiently activates specific GABA receptors on postsynaptic membrane and results in the phasic inhibition of postsynaptic neurons. The phasic inhibition has been demonstrated to be mainly mediated by GABAA receptors contains γ2 subunits postsynaptically (Schweizer et al., 2003; Farrant and Nusser, 2005). Tonic inhibition refers to the sustained form of inhibition upon neuronal cells. Beyond binding to postsynaptic receptors, released neurotransmitter GABA also spills over from synaptic cleft and activates GABA receptors at extrasynaptic area. Tonic inhibition has been demonstrated to be mainly mediated by extrasynaptic GABAA receptors containing π subunit in most brain regions, and GABAA receptors containing α5 subunit especially in hippocampus (Glykys et al., 2008). Recently evidence also indicates the participation of astrocytes and GABAB receptors in tonic inhibition. Especially under pathological conditions, reactive astrocytes release GABA through Bestrophin 1 (Best1) channel. The released GABA could activate both GABAA and GABAB receptors at extrasynaptic area and confer inhibitory effect (Wu et al., 2014).
GABAergic Neurotransmission in the Pathogenesis of AD
Alteration of GABAergic Neurotransmission in AD
In the past, considerable research has focused on the mechanisms of calcium permeable excitatory acetylcholine receptor or glutamate receptors including NMDA and AMPA receptors in AD. Compared to the marked deficits seen in excitatory cholinergic and glutamatergic systems, much less consistent results were revealed for GABAergic system, the main inhibitory neurotransmission in brain. Early studies in postmortem human brains or using animal models concluded that GABAergic neurons and receptors appear more resistant to AD pathology, with only modest loss in AD (Rossor et al., 1982). However, during recent years this statement has been challenged with accumulating evidence, indicating that GABAergic neurotransmission also undergoes profound pathological changes in AD and may be a promising therapeutic target for this neurodegenerative disorder (Figure 1).
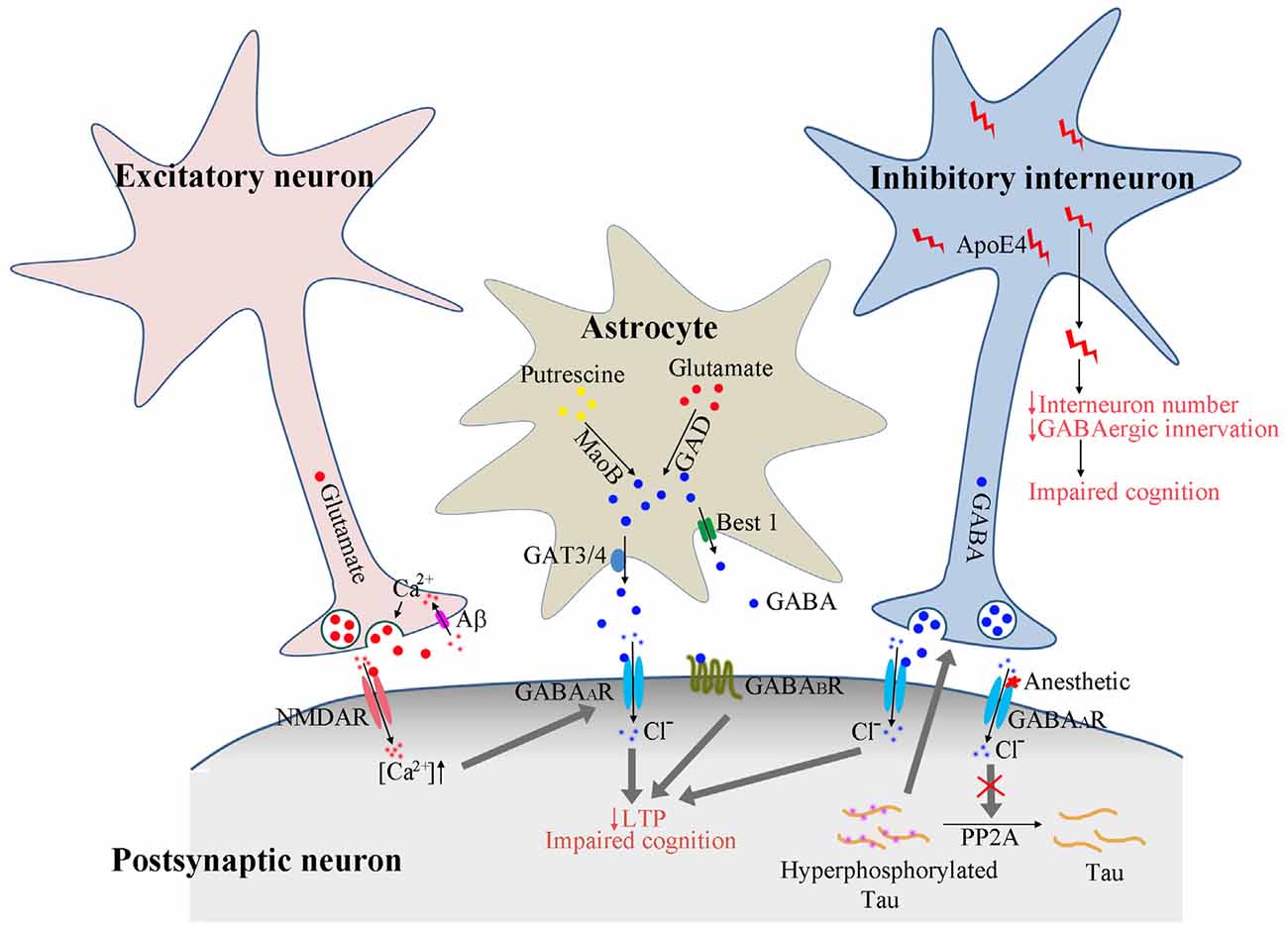
Figure 1. Proposed model of GABAergic signaling in AD pathogenesis. Calcium enters the presynaptic terminal via Aβ formed pores on cell membrane. The increased calcium concentration triggers presynaptic glutamate neurotransmitter release and activates postsynaptic receptors. The activated NMDA receptors further enhance the GABAA receptor activation to dampen the overexcitation. In astrocytes, GABA could be synthesized from putresine or glutamate and released via GAT3/4 or Best1 channel. The release of GABA from astrocytes may be enhanced under AD conditions to activate the extrasynaptic GABAA and GABAB receptors, resulting in suppressed long-term potentiation (LTP) and impaired cognition. The activation of GABAA receptors by anesthetics reduces PP2A binding with tau protein, resulting in tau hyperphosphorylation, which feeds back and enhances the activation of GABAergic interneurons and GABA release. ApoE4 secreted from GABAergic interneurons results in reduced interneuron number and reduced GABAergic innervation to other neurons, eventually leading to the disruption of neuronal circuitry and impaired cognition.
By using HPLC, the concentrations of different neurotransmitters were measured in the brain of AD patient samples and age-matched control subjects. In the temporal cortex of AD patients, significantly lower levels of GABA and glutamate neurotransmitters were observed, indicating deficient synaptic function and neuronal transmission in AD (Gueli and Taibi, 2013). The decreased GABA neurotransmitter levels were also observed in the CSF of AD patients and normal humans with aging (Bareggi et al., 1982; Zimmer et al., 1984; Grouselle et al., 1998). An immunocytochemistry study showed diminished perisomatic GABAergic terminals in brain sections from both AD patients and APP/PS1 transgenic mice, especially on cortical neurons adjacent to amyloid plaques, implicating the loss of GABAergic neuronal function in AD (Garcia-Marin et al., 2009; Ramos-Miguel et al., 2015). However, the alteration of synaptic function in AD appears more complicated during the disease progression. By using 4-month old tgCRND8 and 18-month old APP/PS1 transgenic mice respectively, both glutamatergic and GABAergic presynaptic terminals were found elevated at early stage, but declined at late time point in the distinct AD mice models (Bell et al., 2003, 2006; Bell and Claudio Cuello, 2006; Marttinen et al., 2015).
In the nervous system, maintaining a proper dynamic balance between the excitatory glutamate and inhibitory GABA neurotransmitters is critical for neuronal function. Altered synaptic balance was found to be one of the pathological factors that contribute to neuronal disorders including AD, Huntington’s disease and schizophrenia (Kehrer et al., 2008; Cummings et al., 2009; Sun et al., 2009). Aβ the most well-studied neurotoxic factor in AD pathogenesis, has been demonstrated to be a pore-forming molecule. Similar to other pore-forming neurotoxins, Aβ treatment induced perforation in cell membrane, causing a rapid increase of calcium influx in cultured hippocampal neurons (Parodi et al., 2010; Sepulveda et al., 2010). The increased intracellular calcium concentration triggered the presynaptic neurotransmitter release, leading to disrupted neuronal excitation. In the presence of low concentration of Aβ, the frequency of electrophysiological recorded miniature currents increased quickly, but decreased gradually after a couple of hours, indicating the presynaptic vesicular depletion caused by Aβ (Parodi et al., 2010).
A subset of AD patients have been reported to suffer from epilepsy, which is a typical disorder resulting from imbalanced neuronal excitation. In a study with transgenic human APP (hAPP) mice, Aβ was demonstrated to cause aberrant neuronal overexcitation and spontaneous nonconvulsive seizure activity in cortical and hippocampal networks, the most vulnerable brain regions in AD. The increased epileptic activities in turn triggered downstream alterations including GABAergic sprouting and increased synaptic inhibition in hippocampal circuits. These alterations are characterized as compensatory inhibitory mechanisms to ameliorate neuronal overexcitation and keep the normal neuronal excitation (Palop et al., 2007). The finding of direct crosstalk between postsynaptic glutamate NMDA receptors and GABAA receptors further supported the neuronal circuit remodeling mechanisms. In hypothalamic neurosecretory neurons, the NMDA receptor activation by endogenous glutamate was observed to evoke a transient and reversible enhancement of postsynaptic GABAA receptors (Potapenko et al., 2013). The inter-receptor crosstalk between NMDA receptor and GABAA receptor was considered as a compensatory mechanism for dampening the overexcitation commonly observed in pathological conditions. While on the other hand, the increased inhibitory function mediated by GABAergic synapses may interfere with processes required for learning and memory, as indicated by long-term potentiation (LTP) deficits in dentate gyrus (Palop et al., 2007). Consistent with this result, application of GABAA receptor antagonist picrotoxin was demonstrated to prevent such LTP deficits observed in animal model of AD (Kleschevnikov et al., 2004).
Alteration in postsynaptic GABAA receptors was also observed along with AD pathology. Immunohistochemistry study indicates that the β2/3 subunit was markedly preserved, while α1 and γ subunits were upregulated in human AD subjects (Mizukami et al., 1998; Iwakiri et al., 2009). By microtransplating cell membrane isolated from temporal cortices of control and AD patients into Xenopus oocytes, the level of transplanted GABAA receptors were determined by electrophysiological recording. The whole-cell currents mediated by transplanted GABAA receptors were recorded (Limon et al., 2012). A reduction of GABA-evoked currents was observed in cells transplanted with GABAA receptors from AD brains. In particular, the mRNA and protein levels of α1 and γ2 subunits were found down-regulated, whereas α2, β1 and γ1 subunits were up-regulated in AD brains, indicating that GABAergic neurotransmission undergoes a functional remodeling in the cortex of AD patients.
Consistent results for the impact of Aβ on GABAA receptors were obtained in cerebellum. In cultured rat cerebellar granule neurons (CGNs), treatment of recombinant Aβ40, rather than Aβ42, significantly increased the expression level of α6 subunit containing GABAA receptors and their functional recorded currents. In addition, the expression level of α6 protein in APP knockout mice was significantly lower than in WT CGNs. Further investigation demonstrated that Aβ could induce the phosphorylation of ERK and mTOR, resulting in the increased translation of GABAA receptor α6 subunit (Zhan et al., 2014).
The elevated inhibition mediated by GABAergic neurotransmission found in AD mice models do not appear to be consistent with results obtained from AD patients. Even though transgenic mice models have been widely used for AD mechanism investigation, it should be noted that they may not completely represent the complex pathologic characteristics of AD. It should also be noticed that the transgenic mice used in these studies were with various ages, between 4– to 11-month old, at which age even the amyloid plaques were observed, the neuronal death was not detectable (Jo et al., 2014). AD is by far one of the most complicated progressive neurodegenerative disorders. The differential results indicate that at various stages of the disease, GABAergic system might undergo dynamic remodeling and play different roles in AD pathology.
Alteration of GABAergic Gliotransmission in AD Mice Models
It’s well known that astrocytes are important for uptake and recycling of specific neurotransmitters including GABA and glutamate. In CNS, not only neurons, but also astrocytes were found to be able to produce and release GABA, activating GABAA and GABAB receptors in nearby neurons (Yoon et al., 2012; Yoon and Lee, 2014). Recently, several studies indicate that astrocytes activated by Aβ could release GABA and participate in AD pathology (Mitew et al., 2013; Jo et al., 2014; Wu et al., 2014).
In APP/PS1 and 5× FAD mice, significantly more astrocytes were found activated in hippocampus. Normally astrocytes in wild type mice show minimal GABA immunoreactivity. While in AD mice models, reactive astrocytes were found to abundantly produce and release inhibitory GABA gliotransmitter. As a consequence, HPLC analysis with collected interstitial fluid samples from dentate gyrus revealed significantly elevated GABA level in APP/PS1 mice than wild-type littermates (Jo et al., 2014). In the hippocampus of 5× FAD, the GABA, glutamate and GAD immunostaining intensity were dramatically elevated in astrocytes (Wu et al., 2014). In the synaptosomes isolated from cortex of aged APP/PS1 mice with high amyloid load, the protein level of GAD was found significantly higher than in wild type control and plaque-free region cerebellum. Further study revealed that the increased GAD activity was localized in isolated glial synaptosome, rather than neuronal synaptosome, suggesting that in APP/PS1 transgenic mice, Aβ plaques stimulate the astrocytic GABA synthesis and release (Mitew et al., 2013).
In astrocytes, there may exist more than one pathways for the synthesis and release of GABA. In APP/PS1 mice, the GABA gliotransmitter in astrocytes was demonstrated to be synthesized from putrescine by enzyme monoamine oxidase-B (MaoB) and was released from astrocytes via Best1 channel. The immunoactivity of GABA and MaoB in astrocytes were abnormally and strongly upregulated in the dentate gyrus of APP/PS1 mice and the postmortem brain of AD patients, especially around Aβ plaques (Jo et al., 2014). While in the study with 5× FAD mice, the increased GABA gliotransmitter was synthesized from glutamate by enzyme GAD, and released from astrocytes via the specific GABA transporter GAT3/4 (Wu et al., 2014). The upregulated GABA release from astrocytes could bind to extrasynaptic GABAA and GABAB receptors, strongly inhibit synaptic function and finally leads to the memory and cognitive deficits in AD (Jo et al., 2014).
Linkage between Tau and GABAA Receptors
GABAA receptor is the most well-known pharmacological target for anesthetics including isoflurane, pentobarbital, propofol and chloral hydrate. In recent years, studies indicate that general anesthesia may contribute to the development and exacerbation of AD (Whittington et al., 2013). Besides Aβ plaques, NFTs composed of hyperphosphorylated tau protein is the most important pathological hallmark of AD. Both pre-clinical and clinical studies have found that anesthesia significantly increase the phosphorylation of tau protein. (Le Freche et al., 2012; Whittington et al., 2013). Since GABAA receptor is the major pharmacological target of most anesthetics, the activation of GABAA receptors was assumed and later confirmed to participate in the anesthesia-induced tau hyperphosphorylation.
By using a live cell reporter system, the direct protein interaction between tau and peptidyl-prolyl cis-transisomerase 1 (Pin1) was identified (Nykänen et al., 2012). Pin1 controls the access of phosphatases to serine-proline or threonine-proline (SP/TP) sites of tau, and therefore promotes dephosphorylation of tau via protein phosphatase 2A (PP2A). Interestingly, with pharmaceutical chemical library screening, several GABAA receptor modulators including anesthetics benzodiazepines and barbiturates were found to increase the interaction between tau and Pin1, but significantly promoted the phosphorylation of tau. Further study revealed that these GABAA receptor modulators do not directly inhibit the activity of PP2A, but recruited more PP2A to cell surface for GABAA receptor β3 subunit dephosphorylation and receptor desensitization, therefore reduced the availability of PP2A for tau dephosphorylation. GABAA receptor activation significantly increased tau phosphorylation at AT8 epitope (Ser199/Ser202/Thr205) in cultured cortical neurons (Nykänen et al., 2012).
On the other hand, the hyperphosphorylated tau also has influence on GABAergic synapses. In tau P301L transgenic mice, in which the extent of tau phosphorylation was remarkably upregulated, the GABAergic interneurons were observed hyperactivated, leading to increased GABA neurotransmitter level in the brain (Nilsen et al., 2013).
Overall, GABAA receptor activation could enhance tau phosphorylation by reducing the association of PP2A with tau, consequently increase the intracellular NFTs in neurons and contribute to the development of AD. Vice versa, the hyperphosphorylated tau could enhance GABAergic neurotransmission. There might be a feedback loop between GABAA receptor activation and tau phosphorylation in nervous system (Figure 1).
Contribution of GABAergic Interneurons in apoE4-Induced Deleterious Effect
Apolipoprotein E4 (apoE4), the major genetic risk factor for AD, accounts for 60–75% of all AD cases, increasing significantly the risk of AD and lowering the age of onset of this disorder (Hu et al., 2015; Liu et al., 2015). By using mice model knockout of endogenous Apoe and knockin with various human APOE alleles, the neurogenesis was found reduced in both apoE knockout and human apoE4 knockin mice, leading to impaired learning and memory. In apoE4 knockin mice, the GABAergic interneuron number and presynaptic GABAergic input to newly born neurons both decreased, which was associated with increased tau phosphorylation and neurotoxic apoE4 fragments. Treatment with GABAA receptor agonist pentobarbital restored the neurogenesis deficit in apoE4 knockin mice. Consistently, treatment of apoE3 knockin mice with GABAA receptor antagonist picrotoxin decreased the neurogenesis in hippocampus (Li et al., 2009). These findings suggest that the activation of GABAA receptors and GABAergic signaling pathway could be targeted to mitigate the deleterious effects of apoE4 on neurogenesis.
ApoE is expressed in various cell types. In the brain, apoE is mainly released from astrocytes, increasing during aging. It could also be released from neurons, increasing with stress and injury (Huang, 2010; Huang and Mucke, 2012; Mahley and Huang, 2012). Dr. Huang’s group generated specific human APOE allele knockin mice models, in which the human APOE gene in knockin mice was conditionally deleted in astrocytes, neurons or specific GABAergic interneurons. Deleting apoE4 in neurons, but not in astrocytes, rescued the apoE4-induced deficits including GABAergic interneuron loss and impaired learning and memory. In addition and importantly, conditionally deleting apoE4 in GABAergic interneurons was sufficient to have similar effect and completely prevented the apoE4-induced deficits (Knoferle et al., 2014), suggesting that the apoE4 sourced from GABAergic interneurons is responsible for the deleterious effect of apoE4 on neuronal loss and cognitive deficit.
In human apoE4-positive AD individuals were found often associated with elevated Aβ levels, and Aβ has been shown to impair GABAergic neurotransmission (Huang and Mucke, 2012; Verret et al., 2012). Mice expressing human apoE4 knockin and human APP FAD transgene (apoE4-KI/hAPP-FAD) exhibited high Aβ level and severe cognitive deficit (Palop et al., 2007; Verret et al., 2012). Embryonic interneuron progenitors transplanted into the hilus of apoE4 knockin and apoE4-KI/hAPP-FAD mice successfully developed into mature interneurons that release inhibitory neurotransmitter GABA. In addition, the recovered hippocampal circuitry functionally restored normal learning and memory (Tong et al., 2014), highlighting the importance of GABAergic interneuron and GABAergic neurotransmission in AD pathogenesis.
Interestingly apoE4 also showed sex-dependent characteristics. The risk of developing AD is significantly higher in APOE4 carrying females than males. Studies found that in female apoE4 knockin mice, the GAD67 or somatostatin positive GABAergic interneuron number decreased in an age-dependent manner, accompanied by spatial learning deficits. The ratio of hilar inhibitory GABAergic interneurons to excitatory mossy cells also decreased. However, in male apoE knockin mice, such ratio was kept consistent, regardless of various apoE genotype and age. Furthermore, in aged male apoE knockin mice, the number of hilar GABAergic interneurons even increased, independent of APOE genotype (Leung et al., 2012). These findings suggest that the sex-dependent effect of apoE4 on AD developing risk is at least partially mediated by its differential effects on GABAergic function.
All together, the reported data strongly suggest that GABAergic interneuron plays critical roles in the deleterious effect of apoE4. ApoE4 expressed in GABAergic interneurons may result in apoptosis of these interneurons, leading to reduced GABAergic innervation to other neurons, and disruption of the inhibitory/excitatory balance and neuronal network (Figure 1). The expression of apoE4 was found to cause hyperactivity in human hippocampus (Filippini et al., 2009), which is consistent with the disrupted inhibitory signaling system.
Potential GABAergic Therapies for AD
Intense therapeutic effort has been taken in the AD field, but the outcomes have been disappointing. As outlined in this review, multiple lines of evidence have strongly suggested that GABAergic neurotransmission plays very important roles in AD pathogenesis. There’s close linkage between GABAergic signaling system and various aspect of AD pathology including Aβ toxicity, tau hyperphosphorylation and apoE4 effect. Accordingly GABAA and GABAB receptor modulators have been investigated in preclinical or clinical tests (Table 1).
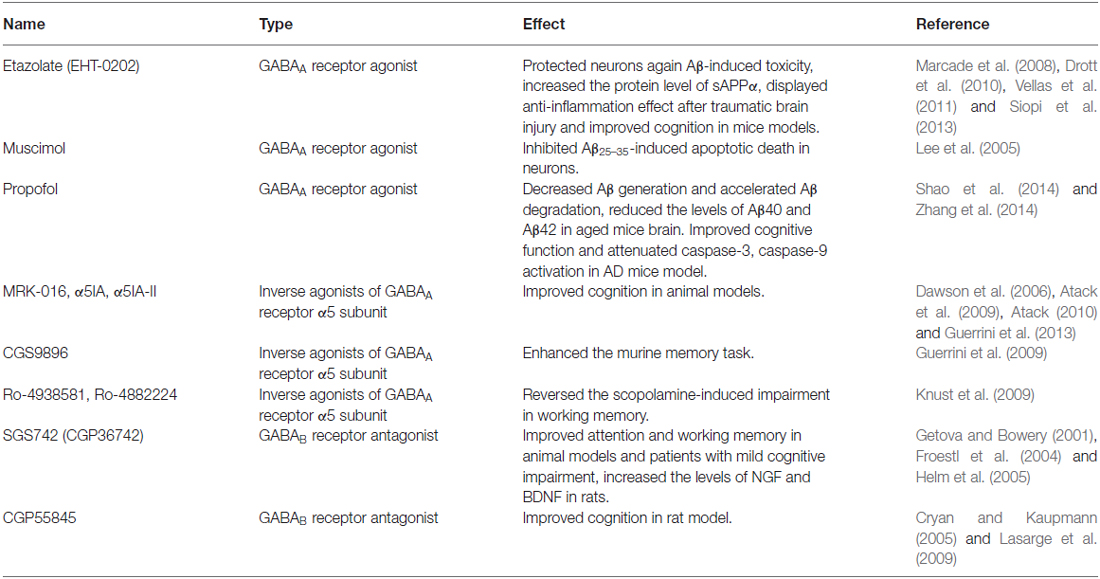
Table 1. Effect of GABAergic chemicals in AD models.
GABAA Receptor Agonists
Hyperexcitation of neuronal activity has been observed in AD brain and was considered one of the toxic factors leading to neuronal death. GABAergic neurotransmission was found upregulated in the hippocampus of AD mice models before general cell death (Jo et al., 2014; Wu et al., 2014), which is possibly a neuronal mechanism to neutralize the abnormal hyperexcitation. Some GABAA receptor agonists have been tested and displayed promising effect.
Etazolate, the GABAA receptor modulator, has been shown to exert neuroprotective effect against Aβ toxicity, anti-inflammation after traumatic brain injury and improvement of cognition (Marcade et al., 2008; Drott et al., 2010). Further investigation revealed that etazolate exerted its neuroprotective effect by activating GABAA receptor and stimulating α-secretase cleavage of APP. The neuroprotective effect of etazolate could be fully blocked by GABAA receptor antagonist. Both in rat cortical neurons in vitro and in guinea pigs in vivo, treatment with etazolate significantly increased the protein level of sAPPα, whose neuroprotective effect has been well demonstrated (Marcade et al., 2008). There might be a relationship between GABAA receptor signaling and the α-secretase cleavage pathway of APP. Etazolate (EHT-0202) has entered Phase II clinical trial for the treatment of AD, with an encouraging result on safety and patient tolerance (Vellas et al., 2011).
Beneficial results have also been obtained with two GABAA receptor agonists, muscimol and propofol (Shao et al., 2014; Zhang et al., 2014). In cultured rat cortical neurons, pretreatment of muscimol significantly inhibited Aβ25–35-induced neuronal apoptotic death. GABAA receptor antagonist bicuculine completely blocked the neuroprotective effect of muscimol (Lee et al., 2005). Chronic treatment of aged mice (18-month old) with propofol reduced the levels of Aβ40 and Aβ42 in brain tissue. In addition, decreased expression of BACE1, the critical enzyme for Aβ generation, and increased level of neprilysin, the primary enzyme for Aβ degradation, were both observed after propofol treatment (Zhang et al., 2014), indicating that chronic activation of GABAA receptor by propofol plays neuroprotective role against Aβ by decreasing Aβ generation and accelerating Aβ degradation. Further investigation showed that propofol treatment also improved cognitive function and attenuated caspase-3, -9 activation in both WT and APP/PS1 mice (Shao et al., 2014). All these results support the notion that GABAA receptor activation might have neuroprotective function against Aβ and could effectively improve cognitive function.
Inverse Agonists of GABAA Receptor α5 Subunit
GABAA receptor is a pentamer containing various allosteric binding sites. It has been suggested that different subunits of GABAA receptor may exert relatively distinct function. For example, α1 subunit is mainly responsible for the sedative action of diazepam, α2 subunit mediates the anxiolytic-like action, whereas α5 subunit may be associated with cognition and memory (Gabriella and Giovanna, 2010). It’s reported that α5 subunit deficiency enhanced hippocampus-dependent memory and spatial learning ability in mice (Collinson et al., 2002; Crestani et al., 2002). In addition, GABAA receptors containing α5 subunit was found upregulated in the dentate gyrus of 5× FAD mice, and has been suggested to mediate the tonic inhibition in CNS. A series of compounds have been developed, serving as inverse agonists of GABAA receptor α5 subunit, which bind to α5 subunit with much higher affinity than other subunits, but negatively regulate receptor activity.
In the early 2000s, the Merck Sharp and Dohme identified a series of benzothiophene derivatives with a notable binding selectivity for GABAA receptor α5 subunit. Among them the ligands MRK-016, α5IA and α5IA-II, all of which displayed encouraging effect on cognition in animal models. The compound α5IA has further advanced to preclinical and clinical studies (Dawson et al., 2006; Atack et al., 2009; Atack, 2010; Guerrini et al., 2013).
Another series of pyrazolo [5, 1-c] [1, 2, 4] benzotriazine 5-oxide (CGS9896) derivatives that are closely correlated to α5IA-II also showed important activity in enhancing the murine memory task (Guerrini et al., 2009). Other two compounds Ro-4938581 and Ro-4882224 from Hoffmann-La Roche Company were shown to significantly reverse the scopolamine-induced working memory impairment. Supported by this result, these two compounds Ro-4938581 and Ro-4882224 have been selected as candidates for further clinical studies (Knust et al., 2009).
GABAB Receptor Antagonists
In AD mice models and human AD patients, GABA released from activated astrocytes was significantly increased. The released GABA could bind to neuronal GABAB receptors at extrasynaptic area, and in turn participate in the inhibition of synaptic release in APP/PS1 mice (Jo et al., 2014). To alleviate the inhibition of synaptic function and improve the cognition deficit in AD, several compounds served as GABAB receptor antagonists have been tested.
SGS742 (CGP36742) is the first GABAB receptor antagonist tested in clinical trials for AD treatment. In rodents and Rhesus monkeys, SGS742 displayed pronounced cognition enhancing effects in various cognitive and learning tasks. It blocked the inhibitory postsynaptic potential (IPSP) and paired-pulse inhibition (PPI) in hippocampus both in vitro and in vivo. It also increased the mRNA and protein levels of NGF (nerve growth factor) and BDNF (brain derived neurotrophic factor) in cortex and hippocampus of rats (Froestl et al., 2004). In addition, SGS742 was well tolerated in both experimental animals and human volunteers. In a Phase II study, oral administration of SGS742 for 8 weeks significantly improved attention and working memory in patients with mild cognitive impairment (Getova and Bowery, 2001; Froestl et al., 2004; Helm et al., 2005). These encouraging findings make SGS742 a promising candidate for dementia treatment and pushed it for further clinical test.
CGP55845 is another GABAB receptor antagonist undergoing preclinical study. In an aged rat model with impaired cognition, treatment with CGP55845 completely reversed its olfactory discrimination learning deficits and restored its performance (Cryan and Kaupmann, 2005; Lasarge et al., 2009). These results supported the potential importance of GABAB receptor as the pharmaceutical target in cognition enhancing activities.
Outlook and Conclusions
Although tremendous understanding of AD pathogenesis has been achieved since last decade, there is still no effective therapy to prevent, delay or stop the disease progression. For a long time, inhibitory GABAergic interneurons and GABA receptors were considered generally preserved in AD, compared to the more vulnerable excitatory glutamate and acetylcholine neurotransmission systems. However, in recent years, abundant evidence has emerged to support the notion that GABAergic signaling system undergoes pathological alterations and contribute to AD pathogenesis. Accordingly, targeting GABAergic neurotransmission is being explored as a potential therapy for AD treatment. However, inconsistent and controversial results have been reported, and these are likely attributed by the complex pathological processes, limitations of the animal models, and differences in the timing and duration of the experimental design. Overall, we hope that this review provides an overall of the current understanding of the role of GABAergic inhibitory neurons in AD and calls for the need of further investigating the GABAergic system in AD pathogenesis using more sophisticated models, rigorous methods and advanced technology. In this review, we present multiple lines of evidence that there is significant GABAergic derangement in AD and that Aβ, tau and apoE4 all mediate GABAergic dysfunction.
Author Contributions
YL wrote the manuscript. HZ reviewed and edited the manuscript. ZC contributed to the references organization. All authors have read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81301105).
References
Atack, J. R. (2010). Preclinical and clinical pharmacology of the GABAA receptor α5 subtype-selective inverse agonist α5IA. Pharmacol. Ther. 125, 11–26. doi: 10.1016/j.pharmthera.2009.09.001
Atack, J. R., Maubach, K. A., Wafford, K. A., O’connor, D., Rodrigues, A. D., Evans, D. C., et al. (2009). In vitro and in vivo properties of 3-tert-butyl-7–(5-methylisoxazol-3-yl)-2(1-methyl-1H-1,2,4-triazol-5-ylmethoxy)-pyrazolo[1,5-d]-[1,2,4]triazine (MRK-016), a GABAA receptor α5 subtype-selective inverse agonist. J. Pharmacol. Exp. Ther. 331, 470–484. doi: 10.1124/jpet.109.157636
Bareggi, S. R., Franceschi, M., Bonini, L., Zecca, L., and Smirne, S. (1982). Decreased CSF concentrations of homovanillic acid and γ-aminobutyric acid in Alzheimer’s disease. Age- or disease-related modifications? Arch. Neurol. 39, 709–712. doi: 10.1001/archneur.1982.00510230035010
Barnard, E. A., Skolnick, P., Olsen, R. W., Mohler, H., Sieghart, W., Biggio, G., et al. (1998). International union of pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 50, 291–313.
Bell, K. F., and Claudio Cuello, A. (2006). Altered synaptic function in Alzheimer’s disease. Eur. J. Pharmacol. 545, 11–21. doi: 10.1016/j.ejphar.2006.06.045
Bell, K. F., de Kort, G. J., Steggerda, S., Shigemoto, R., Ribeiro-Da-Silva, A., and Cuello, A. C. (2003). Structural involvement of the glutamatergic presynaptic boutons in a transgenic mouse model expressing early onset amyloid pathology. Neurosci. Lett. 353, 143–147. doi: 10.1016/j.neulet.2003.09.027
Bell, K. F., Ducatenzeiler, A., Ribeiro-Da-Silva, A., Duff, K., Bennett, D. A., and Cuello, A. C. (2006). The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol. Aging 27, 1644–1657. doi: 10.1016/j.neurobiolaging.2005.09.034
Bormann, J. (2000). The ‘ABC’ of GABA receptors. Trends Pharmacol. Sci. 21, 16–19. doi: 10.1016/s0165-6147(99)01413-3
Bowery, N. (1989). GABAB receptors and their significance in mammalian pharmacology. Trends Pharmacol. Sci. 10, 401–407. doi: 10.1016/0165-6147(89)90188-0
Buggia-Prévot, V., Fernandez, C. G., Riordan, S., Vetrivel, K. S., Roseman, J., Waters, J., et al. (2014). Axonal BACE1 dynamics and targeting in hippocampal neurons: a role for Rab11 GTPase. Mol. Neurodegener. 9:1. doi: 10.1186/1750-1326-9-1
Caillet-Boudin, M. L., Buée, L., Sergeant, N., and Lefebvre, B. (2015). Regulation of human MAPT gene expression. Mol. Neurodegener. 10:28. doi: 10.1186/s13024-015-0025-8
Chebib, M., and Johnston, G. A. (1999). The ‘ABC’ of GABA receptors: a brief review. Clin. Exp. Pharmacol. Physiol. 26, 937–940. doi: 10.1046/j.1440-1681.1999.03151.x
Collinson, N., Kuenzi, F. M., Jarolimek, W., Maubach, K. A., Cothliff, R., Sur, C., et al. (2002). Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J. Neurosci. 22, 5572–5580.
Crestani, F., Keist, R., Fritschy, J. M., Benke, D., Vogt, K., Prut, L., et al. (2002). Trace fear conditioning involves hippocampal α5 GABAA receptors. Proc. Natl. Acad. Sci. U S A 99, 8980–8985. doi: 10.1073/pnas.142288699
Cryan, J. F., and Kaupmann, K. (2005). Don’t worry ‘B’ happy!: a role for GABAB receptors in anxiety and depression. Trends Pharmacol. Sci. 26, 36–43. doi: 10.1016/j.tips.2004.11.004
Cummings, D. M., André, V. M., Uzgil, B. O., Gee, S. M., Fisher, Y. E., Cepeda, C., et al. (2009). Alterations in cortical excitation and inhibition in genetic mouse models of Huntington’s disease. J. Neurosci. 29, 10371–10386. doi: 10.1523/JNEUROSCI.1592-09.2009
Dawson, G. R., Maubach, K. A., Collinson, N., Cobain, M., Everitt, B. J., Macleod, A. M., et al. (2006). An inverse agonist selective for α5 subunit-containing GABAA receptors enhances cognition. J. Pharmacol. Exp. Ther. 316, 1335–1345. doi: 10.1124/jpet.105.092320
Drott, J., Desire, L., Drouin, D., Pando, M., and Haun, F. (2010). Etazolate improves performance in a foraging and homing task in aged rats. Eur. J. Pharmacol. 634, 95–100. doi: 10.1016/j.ejphar.2010.02.036
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Filippini, N., Macintosh, B. J., Hough, M. G., Goodwin, G. M., Frisoni, G. B., Smith, S. M., et al. (2009). Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc. Natl. Acad. Sci. U S A 106, 7209–7214. doi: 10.1073/pnas.0811879106
Froestl, W., Gallagher, M., Jenkins, H., Madrid, A., Melcher, T., Teichman, S., et al. (2004). SGS742: the first GABAB receptor antagonist in clinical trials. Biochem. Pharmacol. 68, 1479–1487. doi: 10.1016/j.bcp.2004.07.030
Furukawa, K., and Mattson, M. P. (1998). Secreted amyloid precursor protein alpha selectively suppresses N-methyl-D-aspartate currents in hippocampal neurons: involvement of cyclic GMP. Neuroscience 83, 429–438. doi: 10.1016/s0306-4522(97)00398-9
Gabriella, G., and Giovanna, C. (2010). γ-Aminobutyric acid type A GABAA receptor subtype inverse agonists as therapeutic agents in cognition. Methods Enzymol. 485, 197–211. doi: 10.1016/B978-0-12-381296-4.00011-7
Garcia-Marin, V., Blazquez-Llorca, L., Rodriguez, J. R., Boluda, S., Muntane, G., Ferrer, I., et al. (2009). Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front. Neuroanat. 3:28. doi: 10.3389/neuro.05.028.2009
Gautam, V., D’avanzo, C., Berezovska, O., Tanzi, R. E., and Kovacs, D. M. (2015). Synaptotagmins interact with APP and promote Aβ generation. Mol. Neurodegener. 10:31. doi: 10.1186/s13024-015-0028-5
Getova, D. P., and Bowery, N. G. (2001). Effects of high-affinity GABAB receptor antagonists on active and passive avoidance responding in rodents with γ-hydroxybutyrolactone-induced absence syndrome. Psychopharmacology 157, 89–95. doi: 10.1007/s002130100766
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease and down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135. doi: 10.1016/0006-291x(84)91209-9
Glykys, J., Mann, E. O., and Mody, I. (2008). Which GABAA receptor subunits are necessary for tonic inhibition in the hippocampus? J. Neurosci. 28, 1421–1426. doi: 10.1523/JNEUROSCI.4751-07.2008
Glykys, J., and Mody, I. (2007). The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol. 582, 1163–1178. doi: 10.1113/jphysiol.2007.134460
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Golovyashkina, N., Penazzi, L., Ballatore, C., Smith, A. B., III, Bakota, L., and Brandt, R. (2015). Region-specific dendritic simplification induced by Aβ, mediated by tau via dysregulation of microtubule dynamics: a mechanistic distinct event from other neurodegenerative processes. Mol. Neurodegener. 10:60. doi: 10.1186/s13024-015-0049-0
Gonzalez-Burgos, G., Rotaru, D. C., Zaitsev, A. V., Povysheva, N. V., and Lewis, D. A. (2009). GABA transporter GAT1 prevents spillover at proximal and distal GABA synapses onto primate prefrontal cortex neurons. J. Neurophysiol. 101, 533–547. doi: 10.1152/jn.91161.2008
Goodman, Y., and Mattson, M. P. (1994). Secreted forms of β-amyloid precursor protein protect hippocampal neurons against amyloid β-peptide-induced oxidative injury. Exp. Neurol. 128, 1–12. doi: 10.1006/exnr.1994.1107
Grouselle, D., Winsky-Sommerer, R., David, J. P., Delacourte, A., Dournaud, P., and Epelbaum, J. (1998). Loss of somatostatin-like immunoreactivity in the frontal cortex of Alzheimer patients carrying the apolipoprotein epsilon 4 allele. Neurosci. Lett. 255, 21–24. doi: 10.1016/s0304-3940(98)00698-3
Gueli, M. C., and Taibi, G. (2013). Alzheimer’s disease: amino acid levels and brain metabolic status. Neurol. Sci. 34, 1575–1579. doi: 10.1007/s10072-013-1289-9
Guerrini, G., Ciciani, G., Cambi, G., Bruni, F., Selleri, S., Guarino, C., et al. (2009). Synthesis, in vivo evaluation and molecular modeling studies of new pyrazolo[5,1-c][1,2,4]benzotriazine 5-oxide derivatives. Identification of a bifunctional hydrogen bond area related to the inverse agonism. J. Med. Chem. 52, 4668–4682. doi: 10.1021/jm801599a
Guerrini, G., Ciciani, G., Costanzo, A., Daniele, S., Martini, C., Ghelardini, C., et al. (2013). Synthesis of novel cognition enhancers with pyrazolo[5,1-c][1,2,4]benzotriazine core acting at gamma-aminobutyric acid type A GABAA receptor. Bioorg. Med. Chem. 21, 2186–2198. doi: 10.1016/j.bmc.2013.02.027
Guzmán, E. A., Bouter, Y., Richard, B. C., Lannfelt, L., Ingelsson, M., Paetau, A., et al. (2014). Abundance of Aβ5-x like immunoreactivity in transgenic 5XFAD, APP/PS1KI and 3xTG mice, sporadic and familial Alzheimer’s disease. Mol. Neurodegener. 9:13. doi: 10.1186/1750-1326-9-13
Hanger, D. P., Anderton, B. H., and Noble, W. (2009). Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112–119. doi: 10.1016/j.molmed.2009.01.003
Hasegawa, M., Morishima-Kawashima, M., Takio, K., Suzuki, M., Titani, K., and Ihara, Y. (1992). Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J. Biol. Chem. 267, 17047–17054.
Helm, K. A., Haberman, R. P., Dean, S. L., Hoyt, E. C., Melcher, T., Lund, P. K., et al. (2005). GABAB receptor antagonist SGS742 improves spatial memory and reduces protein binding to the cAMP response element (CRE) in the hippocampus. Neuropharmacology 48, 956–964. doi: 10.1016/j.neuropharm.2005.01.019
Hu, J., Liu, C. C., Chen, X. F., Zhang, Y. W., Xu, H., and Bu, G. (2015). Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Aβ metabolism in apoE4-targeted replacement mice. Mol. Neurodegener. 10:6. doi: 10.1186/s13024-015-0001-3
Hu, X., Li, X., Zhao, M., Gottesdiener, A., Luo, W., and Paul, S. (2014). Tau pathogenesis is promoted by Aβ1–42 but not Aβ1–40. Mol. Neurodegener. 9:52. doi: 10.1186/1750-1326-9-52
Huang, Y. (2010). Mechanisms linking apolipoprotein E isoforms with cardiovascular and neurological diseases. Curr. Opin. Lipidol. 21, 337–345. doi: 10.1097/MOL.0b013e32833af368
Huang, Y., and Mucke, L. (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. doi: 10.1016/j.cell.2012.02.040
Iqbal, K., Alonso Adel, C., Chen, S., Chohan, M. O., El-Akkad, E., Gong, C. X., et al. (2005). Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 1739, 198–210. doi: 10.1016/j.bbadis.2004.09.008
Iwakiri, M., Mizukami, K., Ikonomovic, M. D., Ishikawa, M., Abrahamson, E. E., DeKosky, S. T., et al. (2009). An immunohistochemical study of GABAA receptor γ subunits in Alzheimer’s disease hippocampus: relationship to neurofibrillary tangle progression. Neuropathology 29, 263–269. doi: 10.1111/j.1440-1789.2008.00978.x
Jack, C. R., Jr., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., et al. (2013). Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/s1474-4422(12)70291-0
Jiang, S., Li, Y., Zhang, X., Bu, G., Xu, H., and Zhang, Y. W. (2014). Trafficking regulation of proteins in Alzheimer’s disease. Mol. Neurodegener. 9:6. doi: 10.1186/1750-1326-9-6
Jo, S., Yarishkin, O., Hwang, Y. J., Chun, Y. E., Park, M., Woo, D. H., et al. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896. doi: 10.1038/nm.3639
Johnston, G. A. (1994). Rand lecture, ASCEPT. GABA receptors: as complex as ABC? Australaisian society for clinical and experimental pharmacologists and toxicologists. Clin. Exp. Pharmacol. Physiol. 21, 521–526. doi: 10.1111/j.1440-1681.1994.tb02550.x
Jung, J. I., Price, A. R., Ladd, T. B., Ran, Y., Park, H. J., Ceballos-Diaz, C., et al. (2015). Cholestenoic acid, an endogenous cholesterol metabolite, is a potent γ-secretase modulator. Mol. Neurodegener. 10:29. doi: 10.1186/s13024-015-0021-z
Kanekiyo, T., Xu, H., and Bu, G. (2014). ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754. doi: 10.1016/j.neuron.2014.01.045
Kehrer, C., Maziashvili, N., Dugladze, T., and Gloveli, T. (2008). Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front. Mol. Neurosci. 1:6. doi: 10.3389/neuro.02.006.2008
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of down syndrome. J. Neurosci. 24, 8153–8160. doi: 10.1523/jneurosci.1766-04.2004
Knoferle, J., Yoon, S. Y., Walker, D., Leung, L., Gillespie, A. K., Tong, L. M., et al. (2014). Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci. 34, 14069–14078. doi: 10.1523/JNEUROSCI.2281-14.2014
Knust, H., Achermann, G., Ballard, T., Buettelmann, B., Gasser, R., Fischer, H., et al. (2009). The discovery and unique pharmacological profile of RO4938581 and RO4882224 as potent and selective GABAA α5 inverse agonists for the treatment of cognitive dysfunction. Bioorg. Med. Chem. Lett. 19, 5940–5944. doi: 10.1016/j.bmcl.2009.08.053
Kolarova, M., García-Sierra, F., Bartos, A., Ricny, J., and Ripova, D. (2012). Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012:731526. doi: 10.1155/2012/731526
Lasarge, C. L., Bañuelos, C., Mayse, J. D., and Bizon, J. L. (2009). Blockade of GABAB receptors completely reverses age-related learning impairment. Neuroscience 164, 941–947. doi: 10.1016/j.neuroscience.2009.08.055
Lee, B. Y., Ban, J. Y., and Seong, Y. H. (2005). Chronic stimulation of GABAA receptor with muscimol reduces amyloid β protein (25–35)-induced neurotoxicity in cultured rat cortical cells. Neurosci. Res. 52, 347–356. doi: 10.1016/j.neures.2005.04.008
Le Freche, H., Brouillette, J., Fernandez-Gomez, F. J., Patin, P., Caillierez, R., Zommer, N., et al. (2012). Tau phosphorylation and sevoflurane anesthesia: an association to postoperative cognitive impairment. Anesthesiology 116, 779–787. doi: 10.1097/ALN.0b013e31824be8c7
Leung, L., Andrews-Zwilling, Y., Yoon, S. Y., Jain, S., Ring, K., Dai, J., et al. (2012). Apolipoprotein E4 causes age- and sex-dependent impairments of hilar GABAergic interneurons and learning and memory deficits in mice. PLoS One 7:e53569. doi: 10.1371/journal.pone.0053569
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H., et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977. doi: 10.1126/science.7638622
Li, G., Bien-Ly, N., Andrews-Zwilling, Y., Xu, Q., Bernardo, A., Ring, K., et al. (2009). GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 5, 634–645. doi: 10.1016/j.stem.2009.10.015
Li, Y., Bohm, C., Dodd, R., Chen, F., Q11mar, S., Schmitt-Ulms, G., et al. (2014). Structural biology of presenilin 1 complexes. Mol. Neurodegener. 9:59. doi: 10.1186/1750-1326-9-59
Limon, A., Reyes-Ruiz, J. M., and Miledi, R. (2012). Loss of functional GABAA receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. U S A 109, 10071–10076. doi: 10.1073/pnas.1204606109
Liu, D. S., Pan, X. D., Zhang, J., Shen, H., Collins, N. C., Cole, A. M., et al. (2015). APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol. Neurodegener. 10:7. doi: 10.1186/s13024-015-0002-2
Lonskaya, I., Hebron, M., Chen, W., Schachter, J., and Moussa, C. (2014). Tau deletion impairs intracellular β-amyloid-42 clearance and leads to more extracellular plaque deposition in gene transfer models. Mol. Neurodegener. 9:46. doi: 10.1186/1750-1326-9-46
Lüscher, B., and Keller, C. A. (2004). Regulation of GABAA receptor trafficking, channel activity and functional plasticity of inhibitory synapses. Pharmacol. Ther. 102, 195–221. doi: 10.1016/j.pharmthera.2004.04.003
Mahley, R. W., and Huang, Y. (2012). Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron 76, 871–885. doi: 10.1016/j.neuron.2012.11.020
Mandelkow, E. M., and Mandelkow, E. (2012). Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2:a006247. doi: 10.1101/cshperspect.a006247
Marcade, M., Bourdin, J., Loiseau, N., Peillon, H., Rayer, A., Drouin, D., et al. (2008). Etazolate, a neuroprotective drug linking GABAA receptor pharmacology to amyloid precursor protein processing. J. Neurochem. 106, 392–404. doi: 10.1111/j.1471-4159.2008.05396.x
Marshall, F. H., White, J., Main, M., Green, A., and Wise, A. (1999). GABAB receptors function as heterodimers. Biochem. Soc. Trans. 27, 530–535. doi: 10.1042/bst0270530
Marttinen, M., Kurkinen, K. M., Soininen, H., Haapasalo, A., and Hiltunen, M. (2015). Synaptic dysfunction and septin protein family members in neurodegenerative diseases. Mol. Neurodegener. 10:16. doi: 10.1186/s13024-015-0013-z
Mattson, M. P., Cheng, B., Culwell, A. R., Esch, F. S., Lieberburg, I., and Rydel, R. E. (1993). Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron 10, 243–254. doi: 10.1016/0896-6273(93)90315-i
McQuail, J. A., Frazier, C. J., and Bizon, J. L. (2015). Molecular aspects of age-related cognitive decline: the role of GABA signaling. Trends Mol. Med. 21, 450–460. doi: 10.1016/j.molmed.2015.05.002
Mitew, S., Kirkcaldie, M. T., Dickson, T. C., and Vickers, J. C. (2013). Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 34, 2341–2351. doi: 10.1016/j.neurobiolaging.2013.04.010
Mizukami, K., Grayson, D. R., Ikonomovic, M. D., Sheffield, R., and Armstrong, D. M. (1998). GABAA receptor β2 and β3 subunits mRNA in the hippocampal formation of aged human brain with Alzheimer-related neuropathology. Brain Res. Mol. Brain Res. 56, 268–272. doi: 10.1016/s0169-328x(97)00347-1
Neumann, U., Rueeger, H., Machauer, R., Veenstra, S. J., Lueoend, R. M., Tintelnot-Blomley, M., et al. (2015). A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of Aβ and neuroinflammation in APP transgenic mice. Mol. Neurodegener. 10:44. doi: 10.1186/s13024-015-0033-8
Nilsen, L. H., Rae, C., Ittner, L. M., Götz, J., and Sonnewald, U. (2013). Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J. Cereb. Blood Flow Metab. 33, 684–691. doi: 10.1038/jcbfm.2012.212
Nykänen, N. P., Kysenius, K., Sakha, P., Tammela, P., and Huttunen, H. J. (2012). gamma-Aminobutyric acid type A GABAA receptor activation modulates tau phosphorylation. J. Biol. Chem. 287, 6743–6752. doi: 10.1074/jbc.m111.309385
Ohki, Y., Shimada, N., Tominaga, A., Osawa, S., Higo, T., Yokoshima, S., et al. (2014). Binding of longer Abeta to transmembrane domain 1 of presenilin 1 impacts on Abeta42 generation. Mol. Neurodegener. 9:7. doi: 10.1186/1750-1326-9-7
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Parodi, J., Sepúlveda, F. J., Roa, J., Opazo, C., Inestrosa, N. C., and Aguayo, L. G. (2010). β-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J. Biol. Chem. 285, 2506–2514. doi: 10.1074/jbc.m109.030023
Pedersen, J. T., and Sigurdsson, E. M. (2015). Tau immunotherapy for Alzheimer’s disease. Trends Mol. Med. 21, 394–402. doi: 10.1016/j.molmed.2015.03.003
Postina, R. (2012). Activation of alpha-secretase cleavage. J. Neurochem. 120(Suppl. 1), 46–54. doi: 10.1111/j.1471-4159.2011.07459.x
Potapenko, E. S., Biancardi, V. C., Zhou, Y., and Stern, J. E. (2013). Astrocytes modulate a postsynaptic NMDA-GABAA-receptor crosstalk in hypothalamic neurosecretory neurons. J. Neurosci. 33, 631–640. doi: 10.1523/jneurosci.3936-12.2013
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Ramos-Miguel, A., Hercher, C., Beasley, C. L., Barr, A. M., Bayer, T. A., Falkai, P., et al. (2015). Loss of Munbc18–1 long splice variant in GABAergic terminals is associated with cognitive decline and increased risk of dementia in a community sample. Mol. Neurodegener. 10:65. doi: 10.1186/s13024-015-0061-4
Rossor, M. N., Garrett, N. J., Johnson, A. L., Mountjoy, C. Q., Roth, M., and Iversen, L. L. (1982). A post-mortem study of the cholinergic and GABA systems in senile dementia. Brain 105, 313–330. doi: 10.1093/brain/105.2.313
Sadleir, K. R., Eimer, W. A., Cole, S. L., and Vassar, R. (2015). Aβ reduction in BACE1 heterozygous null 5XFAD mice is associated with transgenic APP level. Mol. Neurodegener. 10:1. doi: 10.1186/1750-1326-10-1
Schweizer, C., Balsiger, S., Bluethmann, H., Mansuy, I. M., Fritschy, J. M., Mohler, H., et al. (2003). The γ 2 subunit of GABAA receptors is required for maintenance of receptors at mature synapses. Mol. Cell. Neurosci. 24, 442–450. doi: 10.1016/s1044-7431(03)00202-1
Selkoe, D. J. (1998). The cell biology of β-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 8, 447–453. doi: 10.1016/s0962-8924(98)01363-4
Sepulveda, F. J., Parodi, J., Peoples, R. W., Opazo, C., and Aguayo, L. G. (2010). Synaptotoxicity of Alzheimer β-amyloid can be explained by its membrane perforating property. PLoS One 5:e11820. doi: 10.1371/journal.pone.0011820
Shao, H., Zhang, Y., Dong, Y., Yu, B., Xia, W., and Xie, Z. (2014). Chronic treatment with anesthetic propofol improves cognitive function and attenuates caspase activation in both aged and Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 41, 499–513. doi: 10.3233/JAD-132792
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. doi: 10.1038/375754a0
Siopi, E., Llufriu-Dabén, G., Cho, A. H., Vidal-Lletjós, S., Plotkine, M., Marchand-Leroux, C. et al. (2008). Etazolate, an α-secretase activator, reduces neuroinflammation and offers persistent neuroprotection following traumatic brain injury in mice. Neuropharmacology 67, 183–192. doi: 10.1016/j.neuropharm.2012.11.009
Song, L., Lu, S. X., Ouyang, X., Melchor, J., Lee, J., Terracina, G., et al. (2015). Analysis of tau post-translational modifications in rTg4510 mice, a model of tau pathology. Mol. Neurodegener. 10:14. doi: 10.1186/s13024-015-0011-1
Spillantini, M. G., and Goedert, M. (1998). Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 21, 428–433. doi: 10.1016/s0166-2236(98)01337-x
Stancu, I. C., Vasconcelos, B., Terwel, D., and Dewachter, I. (2014). Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol. Neurodegener. 9:51. doi: 10.1186/1750-1326-9-51
Sun, B., Halabisky, B., Zhou, Y., Palop, J. J., Yu, G., Mucke, L., et al. (2009). Imbalance between GABAergic and Glutamatergic transmission impairs adult neurogenesis in an animal model of Alzheimer’s disease. Cell Stem Cell 5, 624–633. doi: 10.1016/j.stem.2009.10.003
Tapia-Rojas, C., Lindsay, C. B., Montecinos-Oliva, C., Arrazola, M. S., Retamales, R. M., Bunout, D., et al. (2015). Is L-methionine a trigger factor for Alzheimer’s-like neurodegeneration?: changes in Aβ oligomers, tau phosphorylation, synaptic proteins, Wnt signaling and behavioral impairment in wild-type mice. Mol. Neurodegener. 10:62. doi: 10.1186/s13024-015-0057-0
Tong, L. M., Djukic, B., Arnold, C., Gillespie, A. K., Yoon, S. Y., Wang, M. M., et al. (2014). Inhibitory interneuron progenitor transplantation restores normal learning and memory in ApoE4 knock-in mice without or with Aβ accumulation. J. Neurosci. 34, 9506–9515. doi: 10.1523/JNEUROSCI.0693-14.2014
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 9:48. doi: 10.1186/1750-1326-9-48
Vellas, B., Sol, O., Snyder, P. J., Ousset, P. J., Haddad, R., Maurin, M., et al. (2011). EHT0202 in Alzheimer’s disease: a 3-month, randomized, placebo-controlled, double-blind study. Curr. Alzheimer Res. 8, 203–212. doi: 10.2174/156720511795256053
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Wang, L., Cheng, S., Yin, Z., Xu, C., Lu, S., Hou, J., et al. (2015). Conditional inactivation of Akt three isoforms causes tau hyperphosphorylation in the brain. Mol. Neurodegener. 10:33. doi: 10.1186/s13024-015-0030-y
Wang, X., Huang, T., Bu, G., and Xu, H. (2014). Dysregulation of protein trafficking in neurodegeneration. Mol. Neurodegener. 9:31. doi: 10.1186/1750-1326-9-31
Whittington, R. A., Bretteville, A., Dickler, M. F., and Planel, E. (2013). Anesthesia and tau pathology. Prog. Neuropsychopharmacol. Biol. Psychiatry 47, 147–155. doi: 10.1016/j.pnpbp.2013.03.004
Wu, Z., Guo, Z., Gearing, M., and Chen, G. (2014). Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat. Commun. 5:4159. doi: 10.1038/ncomms5159
Xu, Z., Xiao, N., Chen, Y., Huang, H., Marshall, C., Gao, J., et al. (2015). Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 10:58. doi: 10.1186/s13024-015-0056-1
Yamada, K., Patel, T. K., Hochgräfe, K., Mahan, T. E., Jiang, H., Stewart, F. R., et al. (2015). Analysis of in vivo turnover of tau in a mouse model of tauopathy. Mol. Neurodegener. 10:55. doi: 10.1186/s13024-015-0052-5
Yamamoto, K., Miyoshi, T., Yae, T., Kawashima, K., Araki, H., Hanada, K., et al. (1994). The survival of rat cerebral cortical neurons in the presence of trophic APP peptides. J. Neurobiol. 25, 585–594. doi: 10.1002/neu.480250510
Yoon, B. E., and Lee, C. J. (2014). GABA as a rising gliotransmitter. Front. Neural Circuits 8:141. doi: 10.3389/fncir.2014.00141
Yoon, B. E., Woo, J., and Lee, C. J. (2012). Astrocytes as GABA-ergic and GABA-ceptive cells. Neurochem. Res. 37, 2474–2479. doi: 10.1007/s11064-012-0808-z
Zhan, X. Q., Yao, J. J., Liu, D. D., Ma, Q., and Mei, Y. A. (2014). Aβ40 modulates GABAA receptor α6 subunit expression and rat cerebellar granule neuron maturation through the ERK/mTOR pathway. J. Neurochem. 128, 350–362. doi: 10.1111/jnc.12471
Zhang, H., Ma, Q., Zhang, Y.-W., and Xu, H. (2012). Proteolytic processing of Alzheimer’s β-amyloid precursor protein. J. Neurochem. 120, 9–21. doi: 10.1111/j.1471-4159.2011.07519.x
Zhang, Y., Shao, H., Dong, Y., Swain, C. A., Yu, B., Xia, W., et al. (2014). Chronic treatment with anesthetic propofol attenuates β-amyloid protein levels in brain tissues of aged mice. Transl. Neurodegener. 3:8. doi: 10.1186/2047-9158-3-8
Zhang, X., Yu, C. J., and Sisodia, S. S. (2015). The topology of pen-2, a γ-secretase subunit, revisited: evidence for a reentrant loop and a single pass transmembrane domain. Mol. Neurodegener. 10:39. doi: 10.1186/s13024-015-0037-4
Keywords: GABAergic neurotransmission, amyloid beta-peptides, tau proteins, apolipoproteins E, neuronal inhibition
Citation: Li Y, Sun H, Chen Z, Xu H, Bu G and Zheng H (2016) Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front. Aging Neurosci. 8:31. doi: 10.3389/fnagi.2016.00031
Received: 02 December 2015; Accepted: 08 February 2016;
Published: 23 February 2016.
Edited by:
Xiongwei Zhu, Case Western Reserve University, USAReviewed by:
Gopal Thinakaran, The University of Chicago, USAYueming Li, Memorial Sloan Kettering Cancer Center, USA
Copyright © 2016 Li, Sun, Chen, Xu, Bu and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanfang Li, yfli@xmu.edu.cn