Ferroptosis in Neurological Diseases
 Jia-Xin Ren1,2†
Jia-Xin Ren1,2†  Xin Sun
Xin Sun Xiu-Li Yan
Xiu-Li Yan Zhen-Ni Guo
Zhen-Ni Guo Yi Yang
Yi Yang- 1Department of Neurology, The First Hospital of Jilin University, Changchun, China
- 2School of Clinical Medicine, Jilin University, Changchun, China
- 3Clinical Trial and Research Center for Stroke, Department of Neurology, The First Hospital of Jilin University, Changchun, China
Ferroptosis is mechanism for non-apoptotic, iron-dependent, oxidative cell death that is characterized by glutathione consumption and lipid peroxides accumulation. Ferroptosis is crucially involved in neurological diseases, including neurodegeneration, stroke and neurotrauma. This review provides detailed discussions of the ferroptosis mechanisms in these neurological diseases. Moreover, it summarizes recent drugs that target ferroptosis for neurological disease treatment. Furthermore, it compares the differences and relationships among the various cell death mechanisms involved in neurological diseases. Elucidating the ferroptosis role in the brain can improve the understanding of neurological disease mechanism and provide potential prevention and treatment interventions for acute and chronic neurological diseases.
Ferroptosis
Ferroptosis is a recently identified type of regulated cell death (Dixon, 2017). Specifically, it is a non-apoptotic, iron-dependent, oxidative cell death mechanism that was proposed by Dixon et al. (2012). Ferroptosis, which is classified as “cell sabotage” (Green and Victor, 2012), differs from apoptosis, necrosis, and autophagy in terms of morphology, biochemistry, and heredity (Cao and Dixon, 2016). Ferroptosis is mainly characterized glutathione (GSH) consumption and lipid peroxidation, which involves specific oxidation of phosphatidylethanolamine containing arachidonic and adrenal acids (D’Herde and Krysko, 2017). The following unique gene set is required for erastin-induced ferroptosis: ribosomal protein L8, iron response element-binding protein 2, F0-F1-ATPase subunit C3, citrate synthase, tetratricopeptide repeat domain 35, and acetyl-CoA synthetase family member (Speer et al., 2013).
Ferroptosis is studied through step-wise in vivo and in vitro experiments using various activators and inhibitors (Xie et al., 2016). Previous cell experiments (Dixon et al., 2012; Yang et al., 2014; Do Van et al., 2016) have shown that ferroptosis is triggered by the oncogenic RAS-selective lethal small molecule erastin, RAS-selective lethal 3, buthionine sulfoximine and sulfasalazine. Ferroptosis induced by the aforementioned compounds and drugs has been detected in cancer cells (Yagoda et al., 2007), kidney tubule cells (Friedmann Angeli et al., 2014), neurons (Guiney et al., 2017), and T cells (Matsushita et al., 2015). Consequently, there are various types of ferroptosis inhibitors with different targets including iron chelators (Ward et al., 2014), antioxidants (Dixon et al., 2012), ferrostatins-1 (Fer-1) (Skouta et al., 2014), glutathione peroxidase 4 (GPX4) (Yang et al., 2014), hypoxia-inducible factor prolyl hydroxylase inhibitors (Speer et al., 2013) and other Chinese medicine (Andersen et al., 2019). Trials on these drugs have shown that the ferroptosis mechanism involve lipid reactive oxygen species (ROS) production, plasma membrane polyunsaturated fatty acid (PUFA) oxidation, iron metabolism, and antioxidant GSH metabolism (Ward et al., 2014; Kagan et al., 2017; Wu et al., 2018). Notably, ferroptosis is not cell suicide; rather it involves cell sabotage that occurs normal life activities to adapt to stimuli and body changes (Green and Victor, 2012; Dixon, 2017). Therefore, ferroptosis is only observed in multiple physiological and pathological processes, including cancer cell death, neurotoxicity, neurodegenerative diseases, acute renal failure, drug-induced hepatotoxicity, hepatic and heart ischemia/reperfusion injury, and T-cell immunity (Xie et al., 2016), when it is misregulated or unbalanced where it induces other cell death mechanisms and immunoreactions (Wenzel et al., 2017; Martin-Sanchez et al., 2018; Proneth and Conrad, 2019).
Ferroptosis plays an important role in the brain and neurological diseases (Kenny et al., 2019). The brain has the highest levels of PUFAs, which are recognized as lipid peroxide precursors, in the human body (Bazinet and Layé, 2014). Moreover, there is a close to correlation of GSH depletion and lipid peroxidation with neurological diseases, including neurotrauma, stroke, and neurodegeneration (Bayir et al., 2006; Weiland et al., 2019). Studies have reported an increase in the ferroptosis phosphatidylethanolamine (PE) marker after traumatic brain injury (TBI) (Wu et al., 2019). Moreover, cellular ferroptosis has been observed in dopaminergic neurons in Parkinson’s disease (PD) and other neurodegenerative diseases (Do Van et al., 2016). Similarly, ferroptosis inhibitors, including Fer-1 and liproxstatin-1 (Lip-1), have been shown to significantly reduce functional deficits in ischemic stroke mouse models. This indicates that ferroptosis is involved in stroke and ischemia-reperfusion injury (Tuo et al., 2017). Further, GPX4 genetic defects have been observed in in vivo and in vitro neuronal death (Yoo et al., 2012; Chen et al., 2015). Taken together, ferroptosis is a potential therapeutic target for neurological diseases given the complexity and fine regulation of the central nervous system (CNS), as well as the unclear changes in neurological diseases.
The targeted ferroptotic mechanism depends on the pathogenesis of the neurological disease; however, lipid peroxidation is generally regarded as the driving force of ferroptosis (Yang and Stockwell, 2016). Hence, we first introduce lipid peroxidation followed by a summary of the main ferroptotic mechanisms and drugs targeting ferroptosis to treat neurodegenerative diseases, stroke, and brain trauma.
Lipid Peroxidation Is the Driving Force of Ferroptosis
Lipid peroxidation products are recognized as the most potent inducers of ferroptosis (Kagan et al., 2017). Lipid peroxide biosynthesis can be performed by enzymes, including 12/15-lipoxygenase (12/15-LOX) and 5-lipoxygenase (5-LOX), or through non-enzymatic processes known as Fenton-type chemistry (Gaschler and Stockwell, 2017). Lipid peroxidation is preferential for PUFAs, which are long-chain fatty acids with more than one double bond, including linoleic, arachidonic, and docosahexaenoic acids. They are highly enriched in the brain where they increase membrane fluidity and plasticity to promote neurotransmitter release and neural network development and migration (Gaschler and Stockwell, 2017; Ingold et al., 2018).
Lipid peroxidation is inhibited by GPX enzymes, especially GPX4, which reduces phospholipid hydroperoxide to lipid alcohols (Brigelius-Flohé and Maiorino, 2013) and restricts the formation of reactive lipid alkoxy groups (Seiler et al., 2008). Lipid peroxide-induced ferroptosis involves the following three steps shown in Figure 1. First, as a key regulator, acyl-CoA synthetase long-chain family member 4 (ACSL4) catalyzes the esterification of arachidonoyl (AA) or adrenoyl (AdA) into PE. Second, lysophosphatidylcholine acyltransferase 3 (LPCAT3) is specific for PE-based substrates and generates PUFA-PE. Finally, 15-LOX oxidizes AA-PE and AdA-PE into ferroptotic signals, including PE-AdA-OOH and PE-AA-OH (D’Herde and Krysko, 2017). For high lipid peroxide levels, lipid peroxide aggregates have been reported in the endoplasmic reticulum (Doll et al., 2017). Moreover, other studies have shown that lipid peroxidation accumulation on the mitochondrial membrane is a key factor for ferroptosis (Friedmann Angeli et al., 2014; Krainz et al., 2016). Here, CRISPR-based genome-wide genetic screening and microarray analysis have demonstrated ACSL4 and LPCAT3 as key ferroptosis regulators (Dixon et al., 2015; Doll et al., 2017). Notably, GPX4 depletion induces ferroptosis only when ACSL4 and LPCAT3 are expressed and involved in arachidonic acid modification with subsequent insertion into membrane phospholipids (Cheng and Li, 2007; Shindou and Shimizu, 2009). Downstream events of lipid peroxides include PUFA fragmentation and membrane lipid damage, as well as toxic reactive lipid intermediate 4-hydroxynonenal production. This leads to cellular ferroptosis promotion through covalent modification and inactivation of essential intracellular proteins (Schneider et al., 2008).
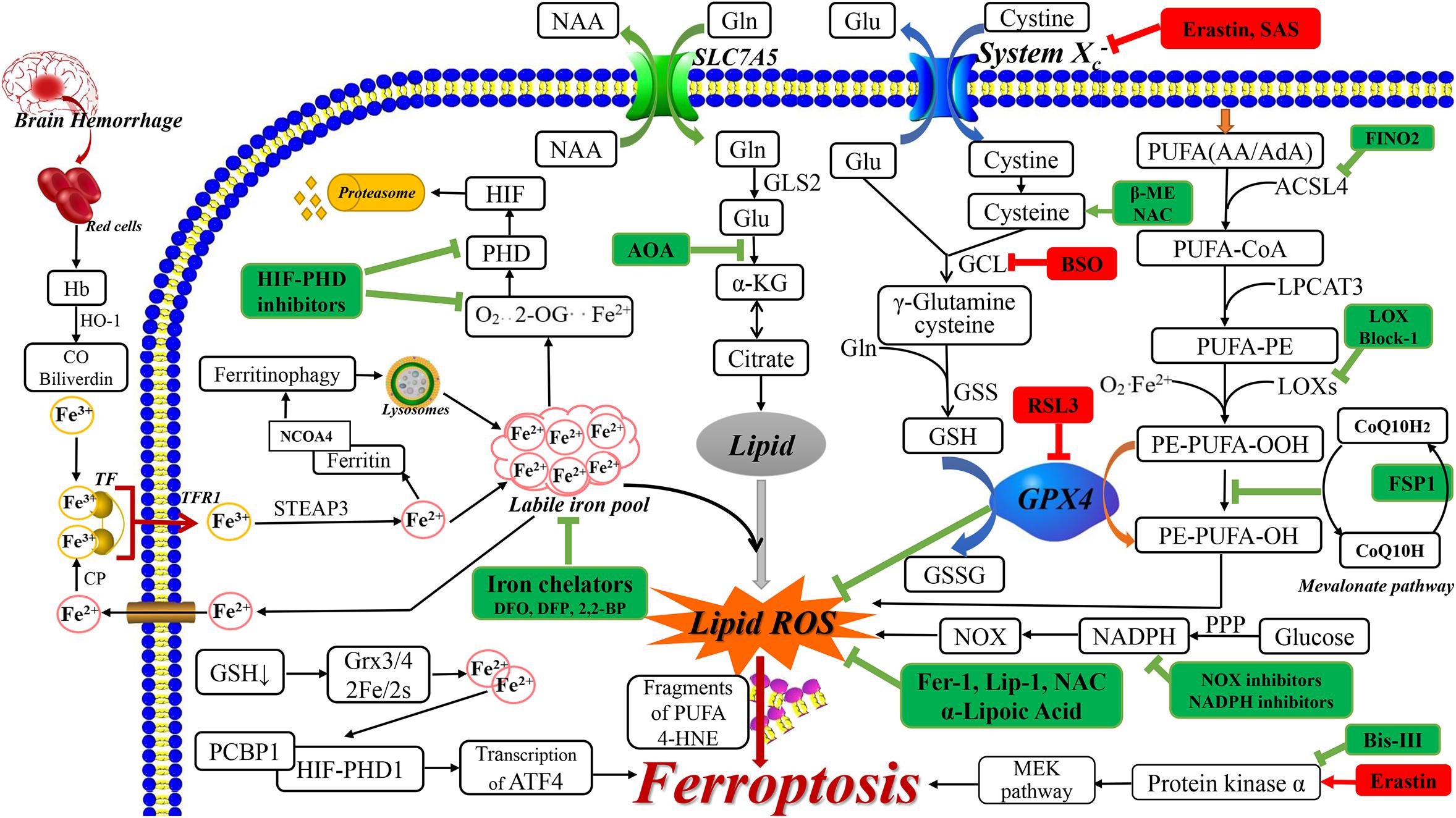
Figure 1. The related ferroptosis mechanisms in neurological diseases. The drugs with red box are ferroptosis inducer and the drugs with green box are ferroptosis inhibitor. AA, arachidonic acid; AdA, adrenic acid; ACSL4, acyl-CoA synthetase long-chain family member 4; AOA, amino acetate; α-KG, α ketoglutarate; BSO, buthionine sulfoximine; 2,2-BP, 2,2-bipyridyl; β-ME, β-mercaptoethanol; CO, carbon monoxide; CoQ 10, Coenzyme Q10; CP, ceruloplasmin; DMT1, divalent metal transporter 1; FAC, ferric citrate; FINO2, 1, 2-dioxolane; FSP-1, Ferroptosis-suppressor-protein 1; GCL, glutamate cysteine ligase; GLS2, glutaminase 2; Glu, glutamate; GSS, glutathione synthetase; GSSG, oxidized GSH; Hb, hemoglobin; HIF, hypoxia-inducible factor; 4-HNE, 4-hydroxynonenal; HO-1, heme oxygenase 1; Lip-1, liproxstatin-1; LPCAT3, lysophosphatidylcholine acyltransferase 3; NAA, neutral amino acids; NAC, N-acetylcysteine; NOX, nitrogen oxide; 2-OG, 2-oxoglutarate; PPP, pentose phosphate pathway; RSL3, RAS-selective lethal 3; SAS, sulfasalazine; STEAP3, 6-transmembrane epithelial antigen of the prostate 3.
Studies using erastin have shown that system Xc–, which is a transmembrane cystine/glutamate reverse transporter, plays an important role in ferroptosis (Dixon et al., 2012). System Xc– is comprised of the 12-pass transmembrane transporter SLC7A11 (Sato et al., 1999), which promotes cysteine-dependent GSH synthesis (Dixon et al., 2012). The following steps lead to ferroptosis. First, erastin inhibits system Xc– and competitively inhibits cystine uptake, which leads to cysteine depletion (the rate-limiting precursor for GSH synthesis). Subsequently, this causes GSH depletion, which leads to imbalanced cellular oxidants and antioxidants, and oxidative, which induces cell death (Siddiq et al., 2005; Figure 1). Furthermore, A Gln- and citrate synthase-dependent lipid synthesis pathway can supply specific lipid precursors required for ferroptosis (Dixon et al., 2012).
Ferroptosis in Neurodegeneration
The Ferroptosis Mechanism in Neurodegeneration: Free Iron Accumulation
Neurodegenerative diseases include complex and elaborate cell death mechanisms and multiple pathways, which involve excessive accumulation of iron and lipid peroxidation in different brain regions (Guiney et al., 2017). An important feature of nervous system dysfunction is the metabolic and nutritional coupling between glial cells (astroglial cells, oligodendrocytes, and microglia) and neurons, which can lead to neuronal death, especially in ferroptosis (Ratan, 2020). Ferroptosis is dependent on excessive iron accumulation, which is a crucial component of lipid oxidation and is derived from iron metabolism disorder (Cao and Dixon, 2016). Under normal CNS conditions, iron is mainly combined with ferritin and neuromelanin. Iron, which is an important cofactor in the CNS (Moreau et al., 2018), participates in several important processes, including oxygen transportation, oxidative phosphorylation, myelin production, and neurotransmitter synthesis and metabolism (Ward et al., 2014). Through the transferrin-transferrin receptor 1 (TF-TFR1) system, iron is released into the cytoplasmic after crossing the blood-brain barrier. Subsequently, it continuously moves among neurons, oligodendrocytes, and astrocytes. The inactive iron (Fe3+) form is recognized by TF and moved into the cell by TFR1. Subsequently, Fe3+ is converted into free iron (Fe2+) by 6-transmembrane epithelial antigen of the prostate 3 (STEAP3) (Ke and Qian, 2007; Wu et al., 2019). Free iron is transported by divalent metal transporter 1 (DMT1) from the endosome and exported by ferroportin. Free iron can be stored in ferritin or the labile iron pool with the latter being involved in lipid ROS generation. Fe2+ is a cofactor for metalloenzymes in many oxidation reactions, including those modulated by lipoxygenase and hypoxia-inducible factor prolyl hydroxylase (HIF-PHD) (Karuppagounder and Ratan, 2012; Kagan et al., 2017; Yan and Zhang, 2019). Further, free iron can be released to the labile iron pool by lysosomes, which degrade ferritin via nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy (Torii et al., 2016). Therefore, brain iron homeostasis is maintained through the regulation of iron movement between blood and brain tissue; intracellular and extracellular; and among different iron pools (Ke and Qian, 2007; Garton et al., 2016). Protein-bound iron is safe; contrastingly, abnormal iron homeostasis involving excessive free iron generates molecules that can destroy proteins and nucleotides through Fenton chemical reactions, which leads to lipid peroxidation (Ke and Qian, 2007; Wu et al., 2019). Therefore, iron is intricately involved in the onset and progression of neurodegenerative diseases (Siddiq et al., 2005; Ward et al., 2014). The involvement of excessive iron accumulation in neurodegenerative diseases includes oxidative stress induction, mitochondrial function interference, excessive ROS production, and destruction of nuclear DNA and mitochondria (Ke and Qian, 2007). Daniel I Orellana et al. (2016) have shown that in neurodegenerative diseases, pantothenate kinase-associated neurodegeneration may disrupt lipid homeostasis, change the mitochondrial iron-dependent biosynthetic pathway and cytoplasmic iron homeostasis and produce harmful ROS. Moreover, iron accumulation in neurodegenerative diseases could involve gene mutations related to iron transport and binding, changes in the TF transport system, increased divalent metal transporter 1 expression (Cheah et al., 2006), and blood-brain barrier dysfunction (Ward et al., 2014; Figure 1).
Ferroptosis Inhibitors in PD
Ferroptosis Induction by Excess Iron in PD
PD is characterized by neuronal loss in multiple brain regions, especially dopaminergic neurons in the substantia nigra. Moreover, the causative factor of PD is α-synuclein protein (Guiney et al., 2017). Based on these PD traits, the key treatment target is restoring brain dopamine levels and preventing neuron degeneration. Iron-dependent ferroptosis is a crucial cell death pathway for dopaminergic neurons (Do Van et al., 2016). In PD, dopaminergic neurons have abundant iron, which is an integral part of enzymatic and non-enzymatic reactions involved in dopamine metabolism (Moreau et al., 2018). In addition, free iron induces dopamine oxidation and α-synuclein aggregation (Duce et al., 2017) which leads to increased dopaminergic neuron loss in PD; moreover, it mutates TF and causes excessive iron uptake (Ward et al., 2014). The possible underlying mechanism could involve excess free iron being able to reduce GPX4 activity, which, in turn, leads to GSH depletion. Subsequently, there is a reduced ROS clearance ability of cells, which leads to excessive ROS, membrane oxidation, and eventual ferroptosis (Imai et al., 2017; Stockwell et al., 2017; Wang et al., 2017). Moreover, α-synuclein oligomers significantly increase the rate of active oxygen production, and induce lipid peroxidation and the increase of iron-dependent free radicals in neurons and astrocytes, such aggregates will be incorporated into the cell membrane through mutual action leading to ferroptosis, which may play an important role in the cellular mechanism of neuronal cell loss in Parkinson’s disease (Angelova et al., 2015, 2020). Experiments show that PUFA is more resistant to lipid peroxidation through site-specific deuteration, which can prevent α-Syn-induced lipid peroxidation and α-Syn-induced cell death (Angelova et al., 2015).
Iron Chelators in PD
Iron chelators are ferroptosis inhibitors, that target free iron in labile iron pools and prevent iron from supplying electrons to oxygen to form ROS (Dixon and Stockwell, 2014). There are two categories of iron chelators, membrane-permeable (lipophilic iron chelators), and membrane-impermeable (Cao and Dixon, 2016). Lipophilic iron chelators, which include ciclopirox and 2,2-bipyridyl, penetrate the cell membrane and blood-brain barrier, target iron accumulation areas, and remove chelatable iron from labile iron pools or transport it to other proteins (Boddaert et al., 2007). For ferroptosis inhibition, lipophilic iron chelators prevent PUFA fragment oxidation and lipid ROS formation (Gao et al., 2015). Moreover, lipophilic iron chelators can directly inactivate ferrous enzymes that promote membrane lipid oxidation (Cao and Dixon, 2016), as well as hypoxia-inducible factor-prolyl hydroxylases (Siddiq et al., 2005). This exerts neuroprotective effects by simulating hypoxia, stabilizing HIF-1α, and activating hypoxia-adaptive genetic programs (Karuppagounder and Ratan, 2012). In contrast, membrane-impermeable iron chelators include deferoxamine (DFO) and deferiprone (DFP), which accumulates in lysosomes through endocytosis from where they capture excess free iron (Barradas et al., 1989).
Numerous in vivo and in vitro experiments have shown that iron chelators play an essential role in PD (Do Van et al., 2016). DFP reduces oxidative stress, increases cellular dopamine utilization, improves existing motor symptom, and prevents worsening in MPTP mouse models (Devos et al., 2014). A phase III clinical trial on iron chelators treatment in patients with PD reported reduced dyskinesia and increased GSH peroxide activity in the cerebrospinal fluid within 6 months (Guiney et al., 2017; Martin-Bastida et al., 2017). Notably, most patients with neurodegenerative disease present normal systemic iron homeostasis during iron chelators application (Ward et al., 2014). Excessive systemic iron chelation therapy is unsuitable for patients with PD and may cause iatrogenic iron depletion and anemia. Therefore, low-dose iron chelation therapy should be used to allow neuroprotective effects with minimal side effects (Moreau et al., 2018). A novel strategy based on iron clearance and redeployment targets has been proposed to allow targeted and differential specificity (Cabantchik et al., 2013). A study conducted by Devos et al. (2014) administered 30 mg/10 g/day DFP to patients with PD. Clinical benefits were observed at 6 months, dyskinesia was significantly reduced at 12 months, and a normal iron index was restored at 18–24 months. This indicated that a lower DFP dose was safe with a risk for reversible neutropenia of 1–3% (Devos et al., 2014). Moreover, a study by Zhang et al. (2020) reported that ferroptosis occurred at a relatively low ferric citrate concentration, while apoptosis occurred with an increase in the iron dose. This is indicative of the role of early death in PD and shows that ferroptosis can result in apoptosis caused by iron overload. Furthermore, the specific ferroptosis inhibitor Fer-1 and the iron chelator DFO inhibit iron-induced cell death and apoptosis in the early PD stages (Zhang et al., 2020). In addition, a recent study shows that in the rodent Parkinson’s disease model, Clioquinol has the effect of an iron chelator. By reducing the iron content in substantia nigra, it inhibits the production of lipid peroxides and ferroptosis, and produce a therapeutic effect (Shi et al., 2020).
Ferroptosis Inhibitors in Alzheimer’s Disease (AD)
The Relationship Between Ferroptosis and AD
AD is characterized by progressive cortical and hippocampal neuronal dysfunction and death. Its main pathological changes include hyperphosphorylation, abnormal Tau aggregation, and synapses and neurons degeneration (Dugger and Dickson, 2017; Roubroeks et al., 2017); synapses are lost and neuronal death is the underlying AD cause (Hambright et al., 2017). Ferroptosis characteristics (iron dysregulation, lipid peroxidation, and inflammation) are considered as important preclinical signs of AD and cognitive impairment (Chen et al., 2015; Hambright et al., 2017). Lipid peroxidation is considered an early event in AD pathogenesis (He et al., 2003). Specifically, excess iron aggravates toxic amyloid β peptide and hyperphosphorylated tau aggregation; moreover, it directly induces oxidative neuronal damage (Thirupathi and Chang, 2019). Iron interacts with amyloid β and tau through peptide-heme complex formation, and thus contributes to ROS generation, which might be involved in the ferroptosis pathway (Derry et al., 2020). Moreover, brain tissues from patients with AD have increased levels of 4-hydroxynonenal, a lipid oxidation by-product, which contributes to β-amyloid accumulation (Yoo et al., 2010).
Lipid Peroxidation Inhibitors in AD
A study by Zhang Y. H. et al. (2018) reported that an AD mouse model with Tau overexpression and hyperphosphorylation presented with excess iron. Studies have proposed treatment with α-lipoic acid (LA), which has natural enzyme cofactors with antioxidants and iron chelator properties; moreover, it easily penetrates the blood-brain barrier. LA administration has been shown to down-regulate TFR expression and up-regulate ferroportin 1expression, which reduces the iron overload. LA regulates iron redistribution through iron chelation; moreover, it acts as a direct free radical scavenger and an indirect antioxidant. Therefore, it reduces ROS levels and increases antioxidant enzymes expressions (GPX4 and Xc–) to play the neuroprotective role in AD (Persson et al., 2003). Furthermore, GPX4-knockout mice present cognitive impairment and neurodegenerative changes, which are associated with lipid peroxidation, extracellular signal-regulated kinase activation, and markedly increased neuroinflammation (Hambright et al., 2017). Therefore, GPX4 protects cortical neurons from oxidative damage and amyloid toxicity (Ran et al., 2006; Yoo et al., 2010). Notably, hydroxylated chalcone inhibits Aβ aggregation and ferroptosis by exerting a pharmacological effect on factors for iron-induced death (including GPX4 or Xc– inhibition); therefore, it could be a candidate treatment for AD (Cong et al., 2019).
Others
Bersuker et al. (2019) used synthetic lethal CRISPR/Cas9 screening technology, and the results showed that myristoylation recruited ferroptosis-suppressor-protein 1(FSP1) to the plasma membrane, which acts as an oxidoreductase, reducing coenzyme Q10, producing a lipophilic free radical trapping antioxidant to prevent the spread of lipid peroxides. The FSP1-CoQ 10 -NAD(P)H pathway exists as an independent parallel system and can be used in conjunction with GPX4 and glutathione to inhibit phospholipid peroxidation and ferroptosis (Bersuker et al., 2019; Doll et al., 2019). Virus-inactivated and heat-treated human platelet lysate has a strong neuroprotective effect against erastin-induced ferroptosis (Nebie et al., 2019) and has been developed as a novel and effective neurodegenerative therapy. Human platelet lysates can inhibit ferroptosis and reduce neuronal loss in cellular models of PD and amyotrophic lateral sclerosis (ALS) (Gouel et al., 2017). Third, as another type of inhibitor, Mithramycin, DNA-binding drugs, can reduce c-Myc expression by binding to the Sp1 site in its promoter, thereby playing a role in inhibiting iron death and treating animal models of HD and AD (Sleiman et al., 2011; Ratan, 2020). Fourth, in mouse hippocampal neuron cells, gastrodin inhibits glutamate-induced ferroptosis through the Nrf2/HO-1 signaling pathway (Jiang et al., 2020). Besides, Nrf-2 activators (such as Tecfidera) are now used in clinical applications to reduce the exacerbation of multiple sclerosis involving ferroptosis and see hope in other neurodegenerative diseases (Sejbaek et al., 2019). Fifth, it is worth noting that diacetyl-bis (4-methyl-3-thiosemicarbazonato) copper II [Cu II (atsm)], by inhibiting ferroptosis, delays the progression of ALS and PD. Compared with lipoxstatin-1 (which has similar potency in vitro), Cu II has better properties including oral bioavailability and entry into the brain (Southon et al., 2020). Moreover, Vitamin E/α-tocopherol could act as a 12/15-LOX inhibitor in neurodegenerative diseases (Matsushita et al., 2015). Finally, it was also found that the transglutaminase inhibitor Cystamine can eliminate glutamate-induced ferroptosis (Farrelly et al., 2019) and the histone deacetylase inhibitor PCI-34051 is an effective neuroprotection and has a role in preventing neuronal ferroptosis (Sleiman et al., 2014).
Ferroptosis in Stroke
Ferroptosis in Ischemic Stroke
The Mechanism of Ferroptosis in Ischemic Stroke
Hypoxia-inducible factor prolyl hydroxylases (HIF-PHD) is considered as a neuroprotective target for “antioxidant” metal chelators. PHD, which is the main oxygen sensor, promotes HIF decomposition and inhibition during the normoxic state. However, PHD inhibition during the hypoxia condition maintains HIF stability (Karuppagounder and Ratan, 2012). On the one hand, HIF-PHD is involved in iron-chelator-related mechanism. Specifically, iron chelator inhibits HIF-PHD activity, which inhibits hydroxyl group production. Consequently, unhydroxylated HIF-1α does not bind ubiquitin-protein ligase and is not degraded. As a result, the [HIF-1α]-[HIF-1β] heterodimer activates protective gene expression, including vascular endothelial growth factor and erythropoietin. This exerts further neuroprotective effects and improves acute stroke recovery (Semenza et al., 2000; Bruick, 2003; Siddiq et al., 2005). On the other hand, the possible process of HIF-PHD causing ferroptosis is as follows: when glutathione is depleted, iron released from the disruption of the glutathione-Grx3/Grx4 Fe/S cluster complex could then be taken up into iron chaperones (PCB1) for enzymes such as the HIF PHDs, and then drive the expression of iron-promoting ATF4 gene, thereby causing ferroptosis (Ratan, 2020).
Ferroptosis Inhibitors in Ischemic Stroke
Ischemic stroke is characterized by rapid neuronal death and dysfunction. Moreover, post-ischemic stroke complications cause further neuronal damage. Therefore, there is a need for further research to further improve stroke prevention and treatment (Tuo et al., 2017). HIF-PHD inhibitors have been shown to reduce neuronal damage caused by permanent focal ischemia and can be clinically used as neuroprotective agents in cases with an imminent risk of ischemic or oxidative neuron damage (Siddiq et al., 2005). In addition, dimethyloxalylglycine significantly reduced the infarct volumes and improved behavior at 24 h and 8 days. Moreover, it improved regional cerebral blood flow after 24 h in a rat model with permanent and transient middle cerebral artery occlusion (Nagel et al., 2011).
A feasible strategy for first-line stroke treatment 12/15-LOX is inhibition using LOXBlock-1, which protects neuronal HT22 cells from oxidative stress. In a mouse model of transient focal ischemia, LOXBlock-1 reduced the infarct size and improved behavioral parameters at both 24 h and 14 days after stroke. This protective effect was observed even with delayed treatment until at least 4 h after the ischemic attack. Additionally, since LOXBlock-1 showed no adverse effects in the collagenase-induced bleeding model, it could be used as an adjuvant for tissue plasminogen activator thrombolysis (Yigitkanli et al., 2013). Notably, intraperitoneal Tat Selpep injection to induce GPX4 expression can reduce the post-focal ischemia infarct size. Moreover, in the mouse ischemic model, it drives the transcription reaction to offset reactive lipid formation and cell death, as well as improves important nerve function within clinically relevant time windows (Alim et al., 2019). Intranasal Lip-1/Fer-1 administration to middle cerebral artery occlusion mice significantly alleviated MCAO-induced functional defects and reduced the infarct size after 14 days (Tuo et al., 2017). Analysis of the drug structure indicated that “amines” are essential for inhibition of cellular ferroptosis; further, Fer-1 analog synthesis can develop more efficient ferroptosis inhibitors, including SRS11-92, SRS12-45, etc. (Skouta et al., 2014).
Ferroptosis in Hemorrhagic Stroke
The Mechanism of Ferroptosis in Hemorrhagic Stroke
Post-hemorrhagic stroke damage is triggered by red blood cells lysis; hemoglobin, heme, and iron release; and coagulation cascade activation. This leads to irreversible destruction of neurovascular unit components with subsequent blood-brain barrier destruction, extensive neuronal death, and fatal brain edema (Qureshi et al., 2009; Keep et al., 2012; Zhao et al., 2018). After intracranial hemorrhage (Garton et al., 2016), heme is degraded by heme-oxygenase into carbon monoxide, biliverdin, and free iron, which induces ferroptosis (Wu et al., 2019; Figure 1).
Ferroptosis Inhibitors in Hemorrhagic Stroke
In the hemorrhagic stroke model, Tat Selpep effectively drives GPX4 expression in the brain, protects and protect neurons, and improves behavior (Alim et al., 2019). Adaptaquin reduces cell death and enhances functional recovery in intracerebral hemorrhage (ICH) rodent models (Karuppagounder et al., 2016). Through toxic lipids neutralizations, the synergistic effect of clinically approved thiol-containing redox-regulating N-acetylcysteine and prostaglandins E inhibits heme-induced ferroptosis and improve the prognosis of hemorrhagic stroke in mice (Karuppagounder et al., 2018).
Others
Naotaifang reduces TFR1 expression, ROS and iron accumulation, and neurobehavioral scores in a rat model of acute cerebral artery occlusion (Lan et al., 2020). Moreover, Carvacrol reduces the tissue lipid peroxide levels to protect gerbils from ischemia-reperfusion injury (Guan et al., 2019).
Ferroptosis in Neurotrauma
The Mechanism of Ferroptosis in Neurotrauma
Post-TBI cell death is a cause of neurological deficits and death (Stoica and Faden, 2010; Anthonymuthu et al., 2018). After brain trauma, there is overexpression of lipid peroxidase, including cyclooxygenase and LOX. One of the earliest mechanical injury events is excessive free fatty acid release. There was a significant increase in free fatty acids in the brain within 5 min after TBI, which remained high at 24 h after the injury (Dhillon et al., 1994). There is a post-TBI increase in the biomarkers of iron-induced death. Further, LOX inhibition in ferroptosis can protect HT22 neurons from in vitro TBI and promote tissue neuroprotection and PE oxidation after control cortex injury (Kenny et al., 2019). Therefore, ferroptosis prevention in brain trauma strengthens the defense system, removes oxidized membrane lipids, prevents 15LOX/PEBP1 complex formation, and reduces the membrane lipid dioxide oxidation rate (Anthonymuthu et al., 2018).
Ferroptosis Inhibitors in Neurotrauma
First, the PEBP1/15LOX complex in cortical and hippocampal neurons is a potential treatment target for brain trauma. This is based on the balance between 15-LOX-produced PE-PUFA-OOH metabolites and GPX4 reduction to hydroxyl metabolites in ferroptosis (Wenzel et al., 2017). Therefore, specific inhibitors targeting the PEBP1/15-LOX complex could serve as a novel treatment strategy for ferroptosis (Jameson et al., 2014). Similarly, Baicalein inhibits ferroptosis and improves post-TBI behavioral outcomes (Andersen et al., 2019; Kenny et al., 2019). Additionally, N-acetylcysteine has a protective effect on GSH in adult, but not pediatric, patients with TBI (Amen et al., 2011; Hoffer et al., 2013).
Differences and Relationships Among Various Cell Death Mechanisms in Neurological Diseases (Table 1)
Different cell death mechanisms, apoptosis, necrosis, autophagy and ferroptosis occur in neurological diseases (Guiney et al., 2017). Each mechanism has typical morphology, regulators, and inhibitors (Table 1). Assessment of intracellular proteases in neurological diseases by Yagami et al. (2019) indicated that neuronal cell death is induced by the generation of ROS, including H2O2; increased intracellular calcium levels; activation proteases (caspase, calpain, cathepsin), and inactivation/activation of kinases.
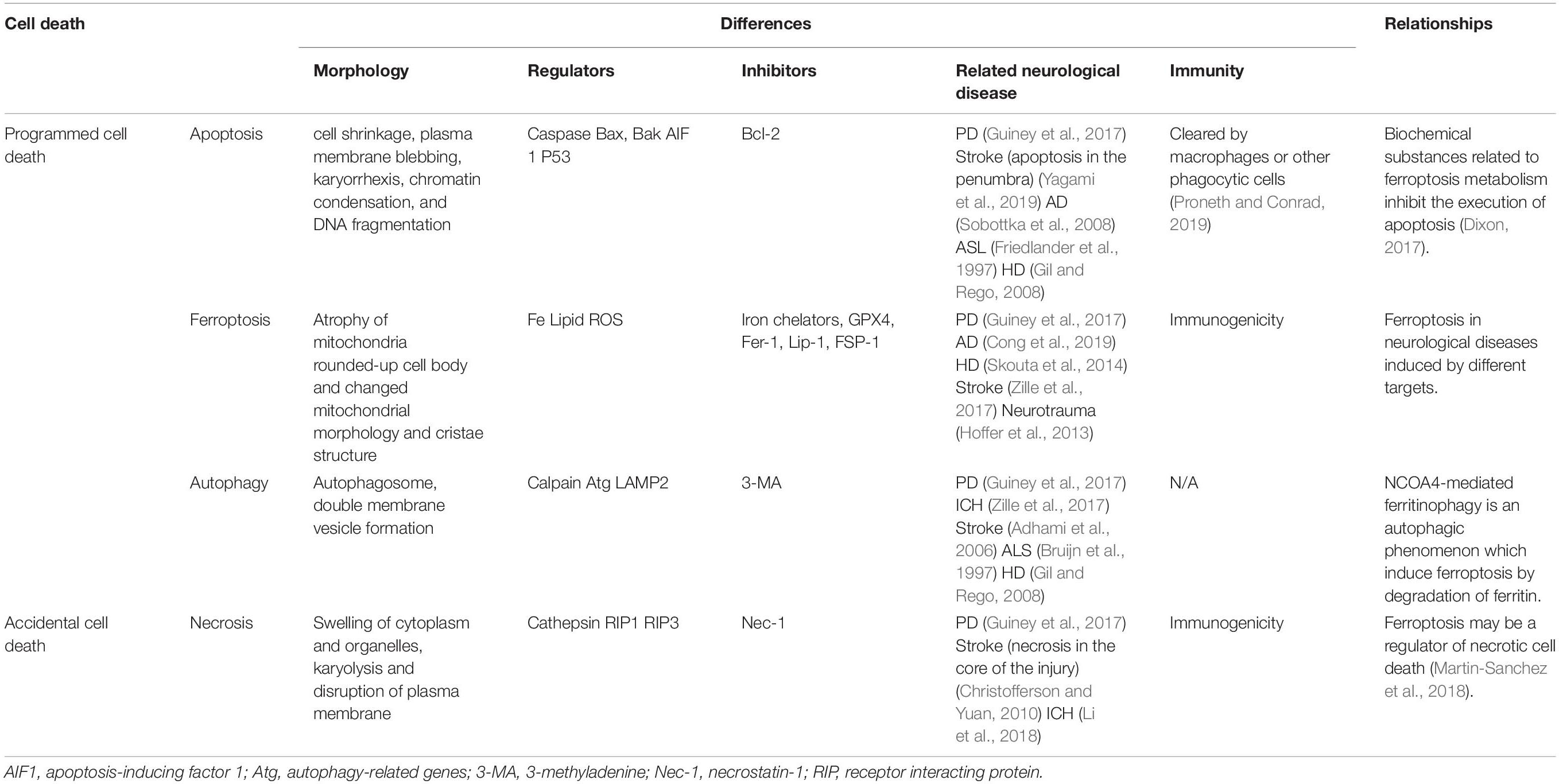
Table 1. Differences and relationships among various cell death mechanisms in neurological diseases.
First, the morphological characteristics of apoptotic neuron cells include cell shrinkage, plasma membrane blebbing, karyorrhexis, chromatin condensation, and DNA fragmentation. Apoptosis, which is a common programmed cell death mechanism, has been demonstrated in the penumbra of patients with stroke (Yagami et al., 2019) and neurodegenerative diseases, including PD, AD (Sobottka et al., 2008), ALS (Friedlander et al., 1997), and Huntington disease (HD) (Gil and Rego, 2008). Further, patients with PD present with elevated levels of proapoptotic protein Bax and activated caspase in dopaminergic neurons (Guiney et al., 2017). In vitro cell experiments have shown that caspase-activated upstream apoptotic mechanisms are not involved in cellular ferroptosis; moreover, GSH-depleted cells cannot properly activate caspase (Dixon et al., 2012). Accordingly, Dixon (2017) suggested that ferroptosis-related biochemical substances inhibit apoptosis.
Second, necrosis is characterized by the swelling of cytoplasm and organelles, karyolysis and plasma membrane disruption in collagenase-induced mouse ICH models observable using transmission electron microscope (Li et al., 2018). Necrosis, which refers to accidental cell death (Yagami et al., 2019), is observed in early stage stroke injury (Christofferson and Yuan, 2010). In addition, necroststin-1, the necrosis inhibitor, reduces the toxicity of 6-hydroxydopamine (6-OHDA) in the PD model (Guiney et al., 2017). Ferroptosis may be a regulator of necrotic cell death (Proneth and Conrad, 2019); specifically, it may induce secondary upregulation of necrotic mechanical components and exacerbate tissue damage (Martin-Sanchez et al., 2018).
Third, autophagy has been observed in neurological diseases. Three days after ICH, autophagosome and double-membrane vesicle formation have been observed in the mouse ICH model (Li et al., 2018). Moreover, the morphological characteristics of autophagy have been observed in patients with PD (Guiney et al., 2017). In adult mice with carotid artery occlusion and hypoxia, stroke causes reperfusion deficits and autophagic/lysosomal cell death in the brain (Adhami et al., 2006). In addition, autophagosomes appear in the neurons of patients with ALS (Bruijn et al., 1997) and HD (Gil and Rego, 2008). Notably, it induces ferroptosis, which leads to ferritinophagy (Vanden Berghe et al., 2014). NCOA4-mediated ferritinophagy is an autophagic phenomenon that induces ferroptosis via ferritin degradation (Hou et al., 2016). Under ferroptotic conditions, ferritin is degraded by autophagy, which preserves the labile iron pool and makes the cell more sensitive to ferroptosis (Gao et al., 2016). Further, autophagy may share key regulators with ferroptosis (Santana-Codina and Mancias, 2018).
Taken together, the relationship among apoptosis, necrosis, autophagy and ferroptosis is delicate and complex in neuronal and surrounding glial cells, with each mechanism having its corresponding time windows. In addition, cell death forms vary across different brain regions with different neuronal sensitivity. For example, the ventral region is more sensitive to PD neurodegeneration in the substantia nigra area (Guiney et al., 2017). In addition, given that pathology caused by tissue damage and degeneration is related to inflammation, the immune response and involved cells could be used to distinguish among the different cell death mechanisms (Green et al., 2009). Apoptotic cells are cleared by macrophages or other phagocytic cells based on the plasma membrane integrity (Proneth and Conrad, 2019). Contrastingly, in necroptosis and ferroptosis, the release of damage-associated molecular patterns through the ruptured plasma membrane triggers the innate immune system in brain tissues with high immunogenicity (Dixon, 2017; Proneth and Conrad, 2019). Regarding therapy, Fer-1 combined with different cell death inhibitors allows better prevention of hemoglobin-induced cell death in organotypic hippocampal slice cultures and human pluripotent stem cell-derived neurons than any of the inhibitors alone. In a collagenase-induced mouse ICH model, a combination of Fer-1, caspase3 inhibitor, and necrostatin-1 can reduce cell death more than any of the inhibitors alone (Li et al., 2017).
Summary
The related ferroptosis mechanisms in neurological diseases are summarized in Figure 1 and the recent drugs that target ferroptosis for neurological disease treatment are summarized in Table 2. There are several prospects regarding ferroptosis in the nervous system. First, there is a need for chemical probes or biomarkers capable of elucidating the ferroptosis mechanism to determine its role in the nervous system given the quick cell death occurrence and difficulty in obtaining appropriate neurons (Guiney et al., 2017). In contrast with other cell death mechanisms, mitochondrial atrophy (Zille et al., 2017), as well as immunity-related cells and factors (Proneth and Conrad, 2019), may be potential markers. Moreover, cyclooxygenase (Li et al., 2017) and lipid peroxides can be assessed as potential chemical probes or biomarkers (Dixon, 2017; Stockwell et al., 2017). It is showed in the latest report that nanoparticles with adjustable size, charge, and targeting ligands show high specificity for the brain, which may provide a reference for the design of new anti- ferroptotic nanomedicines in the future (Xu et al., 2020). Second, since ferroptosis is a cell sabotage mechanism, it should be assessed as a potential adaptive change in the body’s response to stimuli (Dixon, 2017). Third, redox phosphatidyl liposomes in the ferroptosis process involve, PE- and PUFA-PEox as substrates and redox reactions products and catalytic or regulatory proteins, such as 15-LOX, PEBP1, and GPX4. The interactions among these liposome components could elucidate relevant pathogeneses and they could be assessed as potential targets for drug therapy (Wenzel et al., 2017). Fourth, the use of LOXBlock-1 with tissue plasminogen activator significantly reduces bleeding, which suggests that this combination could be a useful treatment strategy for ischemic stroke. Further research is required to determine whether simultaneous administration is required, as well as the appropriate time window (Yigitkanli et al., 2013). Fifth, while assessing the role of the various ferroptosis inhibitors, attention should be paid to the combined use of other cell deaths inhibitors and different drug delivery methods.
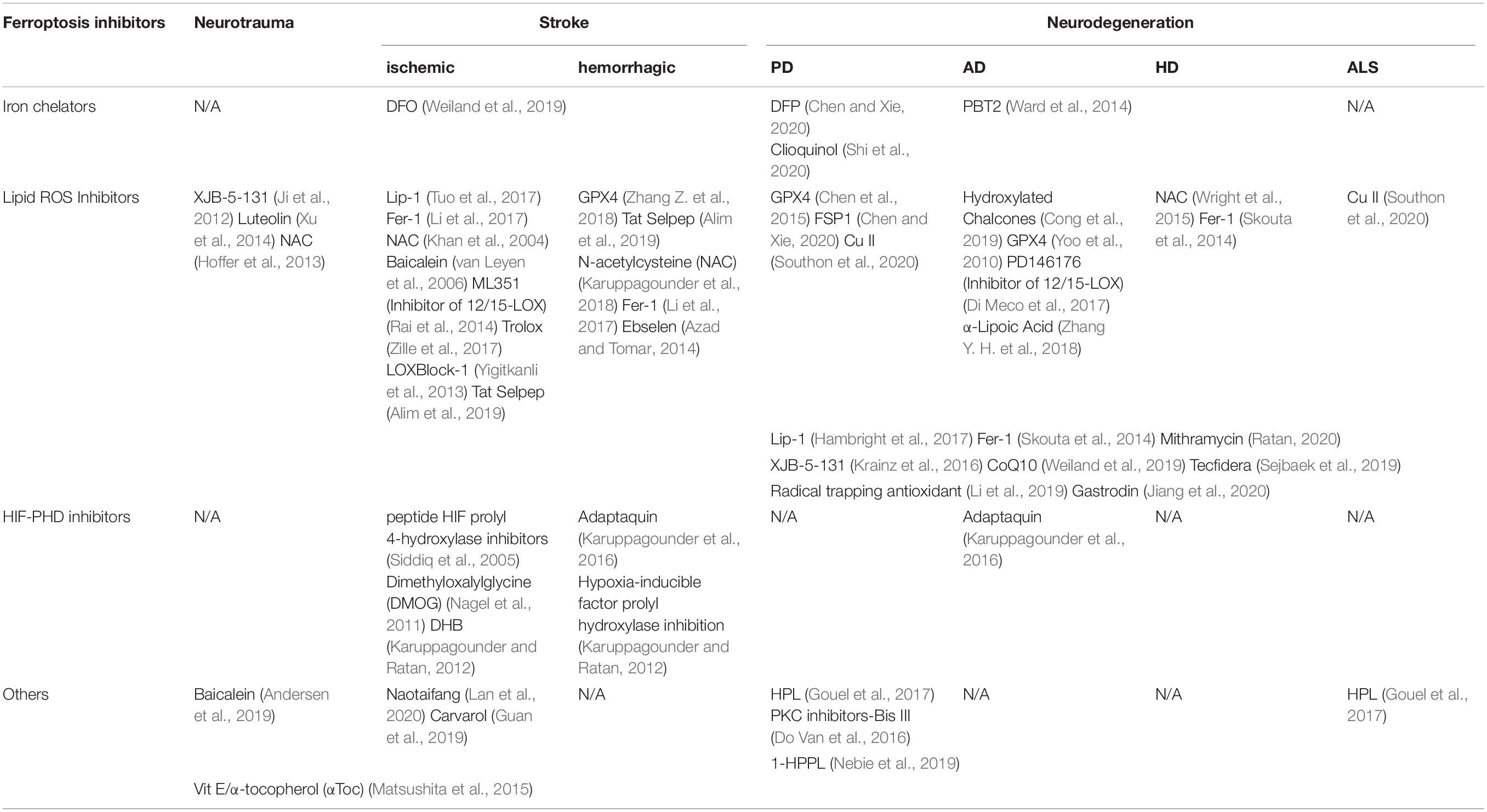
Table 2. Potential drugs that target ferroptosis for neurological disease treatment.
Author Contributions
J-XR and XS searched the literature and drafted the manuscript. X-LY, Z-NG, and YY critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81771243), the Program for JLUSTIRT (2017TD-12), and the Jilin Provincial Key Laboratory (20190901005JC) to YY.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Editage for English language editing.
Abbreviations
AD, Alzheimer’s disease; CNS, Central nervous system; DFO, Deferoxamine; DFP, Deferiprone; Fer-1, ferrostatin-1; Gln, glutamine; GPX4, Glutathione peroxidase-4; GSH, glutathione; HD, Huntington’s disease; HIF, hypoxia-inducible factor; ICH, intracerebral hemorrhage; Lip-1, liproxstatin-1; 12/15-LOX, 12/15-lipoxygenases; NCOA4, Nuclear receptor coactivator 4; PD, Parkinson’s disease; PE, phosphatidylethanolamines; PHDs, prolyl-hydroxylases; PUFA, polyunsaturated fatty acid; ROS, reactive oxygen species; TBI, traumatic brain injury; TF, transferrin; TFR1, transferrin receptor 1.
References
Adhami, F., Liao, G., Morozov, Y. M., Schloemer, A., Schmithorst, V. J., Lorenz, J. N., et al. (2006). Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. 169, 566–583. doi: 10.2353/ajpath.2006.051066
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262.e25–1279.e25. doi: 10.1016/j.cell.2019.03.032
Amen, D. G., Wu, J. C., Taylor, D., and Willeumier, K. (2011). Reversing brain damage in former NFL players: implications for traumatic brain injury and substance abuse rehabilitation. J. Psychoactive Drugs 43, 1–5. doi: 10.1080/02791072.2011.566489
Andersen, C. R., Hawkins, B. E., and Prough, D. S. (2019). Finding the hidden (Statistical) platform. Crit. Care Med. 47, 480–483. doi: 10.1097/ccm.0000000000003611
Angelova, P. R., Choi, M. L., Berezhnov, A. V., Horrocks, M. H., Hughes, C. D., De, S., et al. (2020). Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death Differ. [Epub ahead of print]. doi: 10.1038/s41418-020-0542-z
Angelova, P. R., Horrocks, M. H., Klenerman, D., Gandhi, S., Abramov, A. Y., and Shchepinov, M. S. (2015). Lipid peroxidation is essential for α-synuclein-induced cell death. J. Neurochem. 133, 582–589. doi: 10.1111/jnc.13024
Anthonymuthu, T. S., Kenny, E. M., Lamade, A. M., Kagan, V. E., and Bayır, H. (2018). Oxidized phospholipid signaling in traumatic brain injury. Free Radic. Biol. Med. 124, 493–503. doi: 10.1016/j.freeradbiomed.2018.06.031
Azad, G. K., and Tomar, R. S. (2014). Ebselen, a promising antioxidant drug: mechanisms of action and targets of biological pathways. Mol. Biol. Rep. 41, 4865–4879. doi: 10.1007/s11033-014-3417-x
Barradas, M. A., Jeremy, J. Y., Kontoghiorghes, G. J., Mikhailidis, D. P., Hoffbrand, A. V., and Dandona, P. (1989). Iron chelators inhibit human platelet aggregation, thromboxane A2 synthesis and lipoxygenase activity. FEBS Lett. 245, 105–109. doi: 10.1016/0014-5793(89)80201-7
Bayir, H., Kochanek, P. M., and Kagan, V. E. (2006). Oxidative stress in immature brain after traumatic brain injury. Dev. Neurosci. 28, 420–431. doi: 10.1159/000094168
Bazinet, R. P., and Layé, S. (2014). Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 15, 771–785. doi: 10.1038/nrn3820
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi: 10.1038/s41586-019-1705-2
Boddaert, N., Le Quan Sang, K. H., Rötig, A., Leroy-Willig, A., Gallet, S., Brunelle, F., et al. (2007). Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood 110, 401–408. doi: 10.1182/blood-2006-12-065433
Brigelius-Flohé, R., and Maiorino, M. (2013). Glutathione peroxidases. Biochim. Biophys. Acta 1830, 3289–3303. doi: 10.1016/j.bbagen.2012.11.020
Bruick, R. K. (2003). Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev. 17, 2614–2623. doi: 10.1101/gad.1145503
Bruijn, L. I., Becher, M. W., Lee, M. K., Anderson, K. L., Jenkins, N. A., Copeland, N. G., et al. (1997). ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338. doi: 10.1016/s0896-6273(00)80272-x
Cabantchik, Z. I., Munnich, A., Youdim, M. B., and Devos, D. (2013). Regional siderosis: a new challenge for iron chelation therapy. Front. Pharmacol. 4:167. doi: 10.3389/fphar.2013.00167
Cao, J. Y., and Dixon, S. J. (2016). Mechanisms of ferroptosis. Cell Mol. Life. Sci. 73, 2195–2209. doi: 10.1007/s00018-016-2194-1
Cheah, J. H., Kim, S. F., Hester, L. D., Clancy, K. W., Patterson, S. E. III, Papadopoulos, V., et al. (2006). NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 51, 431–440. doi: 10.1016/j.neuron.2006.07.011
Chen, L., Hambright, W. S., Na, R., and Ran, Q. (2015). Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 290, 28097–28106. doi: 10.1074/jbc.M115.680090
Chen, L., and Xie, J. (2020). Ferroptosis-suppressor-protein 1: a potential neuroprotective target for combating ferroptosis. Mov. Disord. 35:400. doi: 10.1002/mds.27990
Cheng, Z., and Li, Y. (2007). What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem. Rev. 107, 748–766. doi: 10.1021/cr040077w
Christofferson, D. E., and Yuan, J. (2010). Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 22, 263–268. doi: 10.1016/j.ceb.2009.12.003
Cong, L., Dong, X., Wang, Y., Deng, Y., Li, B., and Dai, R. (2019). On the role of synthesized hydroxylated chalcones as dual functional amyloid-β aggregation and ferroptosis inhibitors for potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 166, 11–21. doi: 10.1016/j.ejmech.2019.01.039
Derry, P. J., Hegde, M. L., Jackson, G. R., Kayed, R., Tour, J. M., Tsai, A. L., et al. (2020). Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog. Neurobiol. 184:101716. doi: 10.1016/j.pneurobio.2019.101716
Devos, D., Moreau, C., Devedjian, J. C., Kluza, J., Petrault, M., Laloux, C., et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox. Signal. 21, 195–210. doi: 10.1089/ars.2013.5593
D’Herde, K., and Krysko, D. V. (2017). Ferroptosis: oxidized PEs trigger death. Nat. Chem. Biol. 13, 4–5. doi: 10.1038/nchembio.2261
Dhillon, H. S., Donaldson, D., Dempsey, R. J., and Prasad, M. R. (1994). Regional levels of free fatty acids and Evans blue extravasation after experimental brain injury. J. Neurotrauma 11, 405–415. doi: 10.1089/neu.1994.11.405
Di Meco, A., Li, J. G., Blass, B. E., Abou-Gharbia, M., Lauretti, E., and Pratico, D. (2017). 12/15-lipoxygenase inhibition reverses cognitive impairment, brain amyloidosis, and tau pathology by stimulating autophagy in aged triple transgenic mice. Biol. Psychiatry 81, 92–100. doi: 10.1016/j.biopsych.2016.05.023
Dixon, S. J. (2017). Ferroptosis: bug or feature? Immunol. Rev. 277, 150–157. doi: 10.1111/imr.12533
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., and Stockwell, B. R. (2014). The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17. doi: 10.1038/nchembio.1416
Dixon, S. J., Winter, G. E., Musavi, L. S., Lee, E. D., Snijder, B., Rebsamen, M., et al. (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609. doi: 10.1021/acschembio.5b00245
Do Van, B., Gouel, F., Jonneaux, A., Timmerman, K., Gelé, P., Pétrault, M., et al. (2016). Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 94, 169–178. doi: 10.1016/j.nbd.2016.05.011
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. doi: 10.1038/s41586-019-1707-0
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. doi: 10.1038/nchembio.2239
Duce, J. A., Wong, B. X., Durham, H., Devedjian, J. C., Smith, D. P., and Devos, D. (2017). Post translational changes to α-synuclein control iron and dopamine trafficking; a concept for neuron vulnerability in Parkinson’s disease. Mol. Neurodegener. 12:45. doi: 10.1186/s13024-017-0186-8
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9:a028035. doi: 10.1101/cshperspect.a028035
Farrelly, L. A., Thompson, R. E., Zhao, S., Lepack, A. E., Lyu, Y., Bhanu, N. V., et al. (2019). Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539. doi: 10.1038/s41586-019-1024-7
Friedlander, R. M., Brown, R. H., Gagliardini, V., Wang, J., and Yuan, J. (1997). Inhibition of ICE slows ALS in mice. Nature 388:31. doi: 10.1038/40299
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi: 10.1038/ncb3064
Gao, M., Monian, P., Pan, Q., Zhang, W., Xiang, J., and Jiang, X. (2016). Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032. doi: 10.1038/cr.2016.95
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell. 59, 298–308. doi: 10.1016/j.molcel.2015.06.011
Garton, T., Keep, R. F., Hua, Y., and Xi, G. (2016). Brain iron overload following intracranial haemorrhage. Stroke Vasc. Neurol. 1, 172–184. doi: 10.1136/svn-2016-000042
Gaschler, M. M., and Stockwell, B. R. (2017). Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 482, 419–425. doi: 10.1016/j.bbrc.2016.10.086
Gil, J. M., and Rego, A. C. (2008). Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 27, 2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x
Gouel, F., Do Van, B., Chou, M. L., Jonneaux, A., Moreau, C., Bordet, R., et al. (2017). The protective effect of human platelet lysate in models of neurodegenerative disease: involvement of the Akt and MEK pathways. J. Tissue Eng. Regen. Med. 11, 3236–3240. doi: 10.1002/term.2222
Green, D. R., Ferguson, T., Zitvogel, L., and Kroemer, G. (2009). Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363. doi: 10.1038/nri2545
Green, D. R., and Victor, B. (2012). The pantheon of the fallen: why are there so many forms of cell death? Trends Cell. Biol. 22, 555–556. doi: 10.1016/j.tcb.2012.08.008
Guan, X., Li, X., Yang, X., Yan, J., Shi, P., Ba, L., et al. (2019). The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 235:116795. doi: 10.1016/j.lfs.2019.116795
Guiney, S. J., Adlard, P. A., Bush, A. I., Finkelstein, D. I., and Ayton, S. (2017). Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 104, 34–48. doi: 10.1016/j.neuint.2017.01.004
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi: 10.1016/j.redox.2017.01.021
He, Q., Xu, R. Z., Shkarin, P., Pizzorno, G., Lee-French, C. H., Rothman, D. L., et al. (2003). Magnetic resonance spectroscopic imaging of tumor metabolic markers for cancer diagnosis, metabolic phenotyping, and characterization of tumor microenvironment. Dis. Markers 19, 69–94. doi: 10.1155/2004/424395
Hoffer, M. E., Balaban, C., Slade, M. D., Tsao, J. W., and Hoffer, B. (2013). Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One 8:e54163. doi: 10.1371/journal.pone.0054163
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi: 10.1080/15548627.2016.1187366
Imai, H., Matsuoka, M., Kumagai, T., Sakamoto, T., and Koumura, T. (2017). Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr. Top. Microbiol. Immunol. 403, 143–170. doi: 10.1007/82_2016_508
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2018). Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409.e21–422.e21. doi: 10.1016/j.cell.2017.11.048
Jameson, J. B. II, Kantz, A., Schultz, L., Kalyanaraman, C., Jacobson, M. P., Maloney, D. J., et al. (2014). A high throughput screen identifies potent and selective inhibitors to human epithelial 15-lipoxygenase-2. PLoS One 9:e104094. doi: 10.1371/journal.pone.0104094
Ji, J., Kline, A. E., Amoscato, A., Samhan-Arias, A. K., Sparvero, L. J., Tyurin, V. A., et al. (2012). Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat. Neurosci. 15, 1407–1413. doi: 10.1038/nn.3195
Jiang, T., Cheng, H., Su, J., Wang, X., Wang, Q., Chu, J., et al. (2020). Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol. In Vitro 62:104715. doi: 10.1016/j.tiv.2019.104715
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P., Doll, S., Croix, C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi: 10.1038/nchembio.2238
Karuppagounder, S. S., Alim, I., Khim, S. J., Bourassa, M. W., Sleiman, S. F., John, R., et al. (2016). Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci. Transl. Med. 8:328ra329. doi: 10.1126/scitranslmed.aac6008
Karuppagounder, S. S., Alin, L., Chen, Y., Brand, D., Bourassa, M. W., Dietrich, K., et al. (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 84, 854–872. doi: 10.1002/ana.25356
Karuppagounder, S. S., and Ratan, R. R. (2012). Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J. Cereb. Blood Flow Metab. 32, 1347–1361. doi: 10.1038/jcbfm.2012.28
Ke, Y., and Qian, Z. M. (2007). Brain iron metabolism: neurobiology and neurochemistry. Prog. Neurobiol. 83, 149–173. doi: 10.1016/j.pneurobio.2007.07.009
Keep, R. F., Hua, Y., and Xi, G. (2012). Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 11, 720–731. doi: 10.1016/s1474-4422(12)70104-7
Kenny, E. M., Fidan, E., Yang, Q., Anthonymuthu, T. S., New, L. A., Meyer, E. A., et al. (2019). Ferroptosis contributes to neuronal death and functional outcome after traumatic brain injury. Crit. Care Med. 47, 410–418. doi: 10.1097/ccm.0000000000003555
Khan, M., Sekhon, B., Jatana, M., Giri, S., Gilg, A. G., Sekhon, C., et al. (2004). Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J. Neurosci. Res. 76, 519–527. doi: 10.1002/jnr.20087
Krainz, T., Gaschler, M. M., Lim, C., Sacher, J. R., Stockwell, B. R., and Wipf, P. (2016). A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent. Sci. 2, 653–659. doi: 10.1021/acscentsci.6b00199
Lan, B., Ge, J. W., Cheng, S. W., Zheng, X. L., Liao, J., He, C., et al. (2020). Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J. Integr. Med. [Epub ahead of print]. doi: 10.1016/j.joim.2020.01.008
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:e90777. doi: 10.1172/jci.insight.90777
Li, Q., Weiland, A., Chen, X., Lan, X., Han, X., Durham, F., et al. (2018). Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: coexistence of ferroptosis, autophagy, and necrosis. Front. Neurol. 9:581. doi: 10.3389/fneur.2018.00581
Li, Y. Z., Zhou, X. L., Huo, B. Q., Chen, D. Z., Liu, Z. H., and Sheng, X. H. (2019). Reactions of the lipid hydroperoxides with aminic antioxidants: the influence of stereoelectronic and resonance effects on hydrogen atom transfer. Front. Chem. 7:850. doi: 10.3389/fchem.2019.00850
Martin-Bastida, A., Ward, R. J., Newbould, R., Piccini, P., Sharp, D., Kabba, C., et al. (2017). Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 7:1398. doi: 10.1038/s41598-017-01402-2
Martin-Sanchez, D., Fontecha-Barriuso, M., Carrasco, S., Sanchez-Niño, M. D., Mässenhausen, A. V., Linkermann, A., et al. (2018). TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl. Acad. Sci. U.S.A. 115, 4182–4187. doi: 10.1073/pnas.1716578115
Matsushita, M., Freigang, S., Schneider, C., Conrad, M., Bornkamm, G. W., and Kopf, M. (2015). T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212, 555–568. doi: 10.1084/jem.20140857
Moreau, C., Duce, J. A., Rascol, O., Devedjian, J. C., Berg, D., Dexter, D., et al. (2018). Iron as a therapeutic target for Parkinson’s disease. Mov. Disord. 33, 568–574. doi: 10.1002/mds.27275
Nagel, S., Papadakis, M., Chen, R., Hoyte, L. C., Brooks, K. J., Gallichan, D., et al. (2011). Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 31, 132–143. doi: 10.1038/jcbfm.2010.60
Nebie, O., Devos, D., Vingtdeux, V., Barro, L., Devedjian, J. C., Jonneaux, A., et al. (2019). The neuroprotective activity of heat-treated human platelet lysate biomaterials manufactured from outdated pathogen-reduced (amotosalen/UVA) platelet concentrates. J. Biomed. Sci. 26:89. doi: 10.1186/s12929-019-0579-9
Orellana, D. I., Santambrogio, P., Rubio, A., Yekhlef, L., Cancellieri, C., Dusi, S., et al. (2016). Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration. EMBO Mol. Med. 8, 1197–1211. doi: 10.15252/emmm.201606391
Persson, H. L., Yu, Z., Tirosh, O., Eaton, J. W., and Brunk, U. T. (2003). Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radic. Biol. Med. 34, 1295–1305. doi: 10.1016/s0891-5849(03)00106-0
Proneth, B., and Conrad, M. (2019). Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death. Diff. 26, 14–24. doi: 10.1038/s41418-018-0173-9
Qureshi, A. I., Mendelow, A. D., and Hanley, D. F. (2009). Intracerebral haemorrhage. Lancet 373, 1632–1644. doi: 10.1016/s0140-6736(09)60371-8
Rai, G., Joshi, N., Jung, J. E., Liu, Y., Schultz, L., Yasgar, A., et al. (2014). Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. J. Med. Chem. 57, 4035–4048. doi: 10.1021/jm401915r
Ran, Q., Gu, M., Van Remmen, H., Strong, R., Roberts, J. L., and Richardson, A. (2006). Glutathione peroxidase 4 protects cortical neurons from oxidative injury and amyloid toxicity. J. Neurosci. Res. 84, 202–208. doi: 10.1002/jnr.20868
Ratan, R. R. (2020). The chemical biology of ferroptosis in the central nervous system. Cell Chem. Biol. 27, 479–498. doi: 10.1016/j.chembiol.2020.03.007
Roubroeks, J. A. Y., Smith, R. G., van den Hove, D. L. A., and Lunnon, K. (2017). Epigenetics and DNA methylomic profiling in Alzheimer’s disease and other neurodegenerative diseases. J. Neurochem. 143, 158–170. doi: 10.1111/jnc.14148
Santana-Codina, N., and Mancias, J. D. (2018). The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals 11:114. doi: 10.3390/ph11040114
Sato, H., Tamba, M., Ishii, T., and Bannai, S. (1999). Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 274, 11455–11458. doi: 10.1074/jbc.274.17.11455
Schneider, C., Porter, N. A., and Brash, A. R. (2008). Routes to 4-hydroxynonenal: fundamental issues in the mechanisms of lipid peroxidation. J. Biol. Chem. 283, 15539–15543. doi: 10.1074/jbc.R800001200
Seiler, A., Schneider, M., Förster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell. Metab. 8, 237–248. doi: 10.1016/j.cmet.2008.07.005
Sejbaek, T., Nielsen, H. H., Penner, N., Plavina, T., Mendoza, J. P., Martin, N. A., et al. (2019). Dimethyl fumarate decreases neurofilament light chain in CSF and blood of treatment naïve relapsing MS patients. J. Neurol. Neurosurg. Psychiatry 90, 1324–1330. doi: 10.1136/jnnp-2019-321321
Semenza, G. L., Agani, F., Feldser, D., Iyer, N., Kotch, L., Laughner, E., et al. (2000). Hypoxia, HIF-1, and the pathophysiology of common human diseases. Adv. Exp. Med. Biol. 475, 123–130. doi: 10.1007/0-306-46825-5_12
Shi, L., Huang, C., Luo, Q., Xia, Y., Liu, W., Zeng, W., et al. (2020). Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson’s disease through AKT/mTOR pathway. Aging 12, 9515–9533. doi: 10.18632/aging.103225
Shindou, H., and Shimizu, T. (2009). Acyl-CoA:lysophospholipid acyltransferases. J. Biol. Chem. 284, 1–5. doi: 10.1074/jbc.R800046200
Siddiq, A., Ayoub, I. A., Chavez, J. C., Aminova, L., Shah, S., LaManna, J. C., et al. (2005). Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J. Biol. Chem. 280, 41732–41743. doi: 10.1074/jbc.M504963200
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556. doi: 10.1021/ja411006a
Sleiman, S. F., Langley, B. C., Basso, M., Berlin, J., Xia, L., Payappilly, J. B., et al. (2011). Mithramycin is a gene-selective Sp1 inhibitor that identifies a biological intersection between cancer and neurodegeneration. J. Neurosci. 31, 6858–6870. doi: 10.1523/jneurosci.0710-11.2011
Sleiman, S. F., Olson, D. E., Bourassa, M. W., Karuppagounder, S. S., Zhang, Y. L., Gale, J., et al. (2014). Hydroxamic acid-based histone deacetylase (HDAC) inhibitors can mediate neuroprotection independent of HDAC inhibition. J. Neurosci. 34, 14328–14337. doi: 10.1523/jneurosci.1010-14.2014
Sobottka, B., Reinhardt, D., Brockhaus, M., Jacobsen, H., and Metzger, F. (2008). ProNGF inhibits NGF-mediated TrkA activation in PC12 cells. J. Neurochem. 107, 1294–1303. doi: 10.1111/j.1471-4159.2008.05690.x
Southon, A., Szostak, K., Acevedo, K. M., Dent, K. A., Volitakis, I., Belaidi, A. A., et al. (2020). Cu(II) (atsm) inhibits ferroptosis: implications for treatment of neurodegenerative disease. Br. J. Pharmacol. 177, 656–667. doi: 10.1111/bph.14881
Speer, R. E., Karuppagounder, S. S., Basso, M., Sleiman, S. F., Kumar, A., Brand, D., et al. (2013). Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by “antioxidant” metal chelators: from ferroptosis to stroke. Free Radic. Biol. Med. 62, 26–36. doi: 10.1016/j.freeradbiomed.2013.01.026
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Stoica, B. A., and Faden, A. I. (2010). Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 7, 3–12. doi: 10.1016/j.nurt.2009.10.023
Thirupathi, A., and Chang, Y. Z. (2019). Brain iron metabolism and CNS diseases. Adv. Exp. Med. Biol. 1173, 1–19. doi: 10.1007/978-981-13-9589-5_1
Torii, S., Shintoku, R., Kubota, C., Yaegashi, M., Torii, R., Sasaki, M., et al. (2016). An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 473, 769–777. doi: 10.1042/bj20150658
Tuo, Q. Z., Lei, P., Jackman, K. A., Li, X. L., Xiong, H., Li, X. L., et al. (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530. doi: 10.1038/mp.2017.171
van Leyen, K., Kim, H. Y., Lee, S. R., Jin, G., Arai, K., and Lo, E. H. (2006). Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke 37, 3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5
Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., and Vandenabeele, P. (2014). Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 15, 135–147. doi: 10.1038/nrm3737
Wang, N., Jin, X., Guo, D., Tong, G., and Zhu, X. (2017). Iron chelation nanoparticles with delayed saturation as an effective therapy for parkinson disease. Biomacromolecules 18, 461–474. doi: 10.1021/acs.biomac.6b01547
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/s1474-4422(14)70117-6
Weiland, A., Wang, Y., Wu, W., Lan, X., Han, X., Li, Q., et al. (2019). Ferroptosis and its role in diverse brain diseases. Mol. Neurobiol. 56, 4880–4893. doi: 10.1007/s12035-018-1403-3
Wenzel, S. E., Tyurina, Y. Y., Zhao, J., St Croix, C. M., Dar, H. H., Mao, G., et al. (2017). PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628.e6–641.e6. doi: 10.1016/j.cell.2017.09.044
Wright, D. J., Renoir, T., Smith, Z. M., Frazier, A. E., Francis, P. S., Thorburn, D. R., et al. (2015). N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 5:e492. doi: 10.1038/tp.2014.131
Wu, J. R., Tuo, Q. Z., and Lei, P. (2018). Ferroptosis, a recent defined form of critical cell death in neurological disorders. J. Mol. Neurosci. 66, 197–206. doi: 10.1007/s12031-018-1155-6
Wu, Y., Song, J., Wang, Y., Wang, X., Culmsee, C., and Zhu, C. (2019). The potential role of ferroptosis in neonatal brain injury. Front. Neurosci. 13:115. doi: 10.3389/fnins.2019.00115
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: process and function. Cell Death Diff. 23, 369–379. doi: 10.1038/cdd.2015.158
Xu, J., Wang, H., Ding, K., Zhang, L., Wang, C., Li, T., et al. (2014). Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic. Biol. Med. 71, 186–195. doi: 10.1016/j.freeradbiomed.2014.03.009
Xu, Y., Qin, Z., Ma, J., Cao, W., and Zhang, P. (2020). Recent progress in nanotechnology based ferroptotic therapies for clinical applications. Eur. J. Pharmacol. 880:173198. doi: 10.1016/j.ejphar.2020.173198
Yagami, T., Yamamoto, Y., and Koma, H. (2019). Pathophysiological roles of intracellular proteases in neuronal development and neurological diseases. Mol. Neurobiol. 56, 3090–3112. doi: 10.1007/s12035-018-1277-4
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. doi: 10.1038/nature05859
Yan, N., and Zhang, J. J. (2019). The emerging roles of ferroptosis in vascular cognitive impairment. Front. Neurosci. 13:811. doi: 10.3389/fnins.2019.00811
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26, 165–176. doi: 10.1016/j.tcb.2015.10.014
Yigitkanli, K., Pekcec, A., Karatas, H., Pallast, S., Mandeville, E., Joshi, N., et al. (2013). Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann. Neurol. 73, 129–135. doi: 10.1002/ana.23734
Yoo, M. H., Gu, X., Xu, X. M., Kim, J. Y., Carlson, B. A., Patterson, A. D., et al. (2010). Delineating the role of glutathione peroxidase 4 in protecting cells against lipid hydroperoxide damage and in Alzheimer’s disease. Antioxid. Redox. Signal. 12, 819–827. doi: 10.1089/ars.2009.2891
Yoo, S. E., Chen, L., Na, R., Liu, Y., Rios, C., Van Remmen, H., et al. (2012). Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biol. Med. 52, 1820–1827. doi: 10.1016/j.freeradbiomed.2012.02.043
Zhang, P., Chen, L., Zhao, Q., Du, X., Bi, M., Li, Y., et al. (2020). Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic. Biol. Med. 152, 227–234. doi: 10.1016/j.freeradbiomed.2020.03.015
Zhang, Y. H., Wang, D. W., Xu, S. F., Zhang, S., Fan, Y. G., Yang, Y. Y., et al. (2018). α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 14, 535–548. doi: 10.1016/j.redox.2017.11.001
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125. doi: 10.1016/j.brainres.2018.09.012
Zhao, H., Chen, Y., and Feng, H. (2018). P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr. Neuropharmacol. 16, 1282–1295. doi: 10.2174/1570159x16666180516094500
Keywords: ferroptosis, neurodegeneration, stroke, inhibitors, iron
Citation: Ren J-X, Sun X, Yan X-L, Guo Z-N and Yang Y (2020) Ferroptosis in Neurological Diseases. Front. Cell. Neurosci. 14:218. doi: 10.3389/fncel.2020.00218
Received: 22 April 2020; Accepted: 19 June 2020;
Published: 13 July 2020.
Edited by:
Pier Giorgio Mastroberardino, Erasmus University Rotterdam, NetherlandsReviewed by:
Senthilkumar Rajagopal, Rayalaseema University, IndiaAndrey Y. Abramov, University College London, United Kingdom
Copyright © 2020 Ren, Sun, Yan, Guo and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen-Ni Guo, zhen1ni2@163.com; Yi Yang, doctoryangyi@163.com; doctor_yangyi@hotmail.com
†These authors have contributed equally to this work