 Evangeline M. Foster
Evangeline M. Foster Adrià Dangla-Valls
Adrià Dangla-Valls Simon Lovestone
Simon Lovestone Elena M. Ribe
Elena M. Ribe Noel J. Buckley
Noel J. Buckley- Department of Psychiatry, University of Oxford, Oxford, United Kingdom
Clusterin (CLU) or APOJ is a multifunctional glycoprotein that has been implicated in several physiological and pathological states, including Alzheimer’s disease (AD). With a prominent extracellular chaperone function, additional roles have been discussed for clusterin, including lipid transport and immune modulation, and it is involved in pathways common to several diseases such as cell death and survival, oxidative stress, and proteotoxic stress. Although clusterin is normally a secreted protein, it has also been found intracellularly under certain stress conditions. Multiple hypotheses have been proposed regarding the origin of intracellular clusterin, including specific biogenic processes leading to alternative transcripts and protein isoforms, but these lines of research are incomplete and contradictory. Current consensus is that intracellular clusterin is most likely to have exited the secretory pathway at some point or to have re-entered the cell after secretion. Clusterin’s relationship with amyloid beta (Aβ) has been of great interest to the AD field, including clusterin’s apparent role in altering Aβ aggregation and/or clearance. Additionally, clusterin has been more recently identified as a mediator of Aβ toxicity, as evidenced by the neuroprotective effect of CLU knockdown and knockout in rodent and human iPSC-derived neurons. CLU is also the third most significant genetic risk factor for late onset AD and several variants have been identified in CLU. Although the exact contribution of these variants to altered AD risk is unclear, some have been linked to altered CLU expression at both mRNA and protein levels, altered cognitive and memory function, and altered brain structure. The apparent complexity of clusterin’s biogenesis, the lack of clarity over the origin of the intracellular clusterin species, and the number of pathophysiological functions attributed to clusterin have all contributed to the challenge of understanding the role of clusterin in AD pathophysiology. Here, we highlight clusterin’s relevance to AD by discussing the evidence linking clusterin to AD, as well as drawing parallels on how the role of clusterin in other diseases and pathways may help us understand its biological function(s) in association with AD.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia, accounting for over 60% of the 46.8 million cases worldwide. Due to an increasing aging population, this number is predicted to rise to over 130 million cases by 2050 (Prince et al., 2015; Alzheimer Association, 2016). Currently, there are no treatments that prevent or slow the progression of AD, and this is in part explained by the lack of mechanistic understanding of the processes underlying the disease. Although the etiology is unknown, AD is considered a multifactorial disease with age, lifestyle, and genetics as main contributing factors.
Mutations in genes such as PSEN1, PSEN2, and APP result in the rare, familial, early onset forms of AD, while over 20 genes have been identified that influence the risk of the more common, sporadic, late onset AD (LOAD) (Van Cauwenberghe et al., 2016). In 2009, two large independent Genome Wide Association Studies (GWAS) identified clusterin (CLU) as a novel LOAD-risk gene (Harold et al., 2009; Lambert et al., 2009) and numerous single nucleotide polymorphisms (SNPs) were identified as susceptibility loci in these and subsequent studies (Seshadri et al., 2010; Tan et al., 2016). CLU is now considered the third greatest genetic risk factor for LOAD, after APOE and BIN1. From histopathological to biomarker studies, numerous lines of evidence also suggest a link between clusterin and AD, such as the observation that clusterin is upregulated in the hippocampus and cortex of the AD brain, colocalizing with amyloid beta (Aβ) plaques (May et al., 1990). Or later, it was demonstrated that clusterin is upregulated in AD cerebrospinal fluid (CSF) (Nilselid et al., 2006). Recently, CSF clusterin levels were used in an endophenotype-based approach to try to identify novel loci that might be linked to AD pathogenesis through an alteration of clusterin in CSF (Deming et al., 2016). Additionally, higher plasma clusterin levels have been associated with increased hippocampal atrophy and increased rate of clinical progression (Thambisetty et al., 2010, 2011), suggestive of clusterin as a promising biomarker. However, although a multitude of genetic, biomarker, and post-mortem evidence suggests a role for clusterin in AD, it is unclear as to whether clusterin is a causal factor leading to AD development or is a contributing factor to disease progression. Either way, it is important to identify clusterin’s mechanism of action. We anticipate that the groundswell of CRISPR-based studies aimed at introducing and correcting specific variants will be pivotal in this regard.
Clusterin was traditionally referred to as an extracellular chaperone (Humphreys et al., 1999; reviewed in Satapathy, 2017) and a number of binding partners have been identified. Clusterin’s ability to interact and bind to Aβ appears to alter aggregation and promote Aβ clearance, suggesting a neuroprotective role (DeMattos et al., 2004; Bell et al., 2007; Nuutinen et al., 2007; Yerbury and Wilson, 2010; Cascella et al., 2013; Narayan et al., 2014; Merino-Zamorano et al., 2016; Yeh et al., 2016; Zandl-Lang et al., 2017). However, other studies show that clusterin may in fact reduce the clearance of Aβ (Oda et al., 1995; Lambert et al., 1998; DeMattos et al., 2002; Nielsen et al., 2010; Mulder et al., 2014) and may be a key mediator regulating Aβ-induced neurotoxicity (Killick et al., 2014; Robbins et al., 2018). Finally, it has been argued that the nature of the interaction between Aβ and clusterin is dependent on the clusterin:Aβ ratio (Yerbury et al., 2007) and the factor in excess might determine whether clusterin exhibits neuroprotective or neurotoxic properties.
As can be readily appreciated, many previous attempts have been made to understand the contribution that clusterin plays in a number of diseases including AD (Nuutinen et al., 2009; Bertram and Tanzi, 2010; Li et al., 2014; Rohne et al., 2016), and yet this role has not been fully elucidated. In this review, we discuss the complexity of clusterin and the importance of this protein in the context of neurodegenerative diseases while drawing parallels from other fields, particularly, oncology. We discuss the different roles played by clusterin in other diseases and how these may enable us to better understand the role of clusterin in neurodegeneration and AD.
Clusterin Complexity: From Gene to Protein
Clusterin is a ubiquitously and constitutively expressed protein found in a wide range of tissues and bodily fluids (De Silva et al., 1990; Rizzi et al., 2009). Its wide expression is accompanied by a number of attributed functions including inhibition of the complement system (Murphy et al., 1988; Jenne and Tschopp, 1989), chaperone function (Humphreys et al., 1999), lipid transport (Wang and Eckel, 2014), and regulation of cell survival and cell death pathways (Scaltriti et al., 2004a,b; Zhang et al., 2005; Takase et al., 2008; Trougakos et al., 2009; Kim et al., 2012).
Originally, the 85 kDa protein isolated from ram rete testis fluid with an aggregating or ‘clustering’ effect on Sertoli cells was identified as clusterin protein (Blaschuk et al., 1983). Over the years, however, clusterin has been re-identified numerous times and given several names based on its location of identification and function, including: testosterone repressed prostate messenger-2 (TRPM-2) (Leger et al., 1987), serum protein-40,40 (SP-40,40) (Murphy et al., 1988), complement cytolysis inhibitor (CLI) (Jenne and Tschopp, 1989), sulfated glycoprotein 2 (SGP-2) (Purrello et al., 1991), and apolipoprotein J (APOJ) (De Silva et al., 1990; James et al., 1991). It was eventually determined that all these proteins were in fact produced from the same gene (Wong et al., 1993) and the name CLU was decided on at the Workshop on Clusterin held in Cambridge in 1992 (Fritz and Murphy, 1993). Despite extensive links between clusterin and both physiological and pathological processes, the exact role of this protein is unclear. One fundamental reason for this is the complexity of CLU and the lack of clarity in its mRNA and protein structures.
CLU Gene Structure and Regulation
CLU is a single copy gene located at the p21-p12 locus on chromosome 8. CLU encodes nine exons and the majority of clusterin protein is produced from the CLU mRNA transcript NM_001831.3 (Prochnow et al., 2013). Translation is initiated at the start site located in exon 2. Although alternative start codons have been described in exons 1, 2, and 3 (Reddy et al., 1996; Rizzi and Bettuzzi, 2010; Prochnow et al., 2013), their functional importance has not been shown (Reddy et al., 1996; Yang et al., 2000; Leskov et al., 2003; Prochnow et al., 2013). The N-terminal endoplasmic reticulum (ER)-signal peptide located within exon 2 ensures the production of secreted protein (Wong et al., 1993), and two nuclear localization signals (NLS) are located in exon 3 and exons 8–9 (Leskov et al., 2003) (Figure 1).
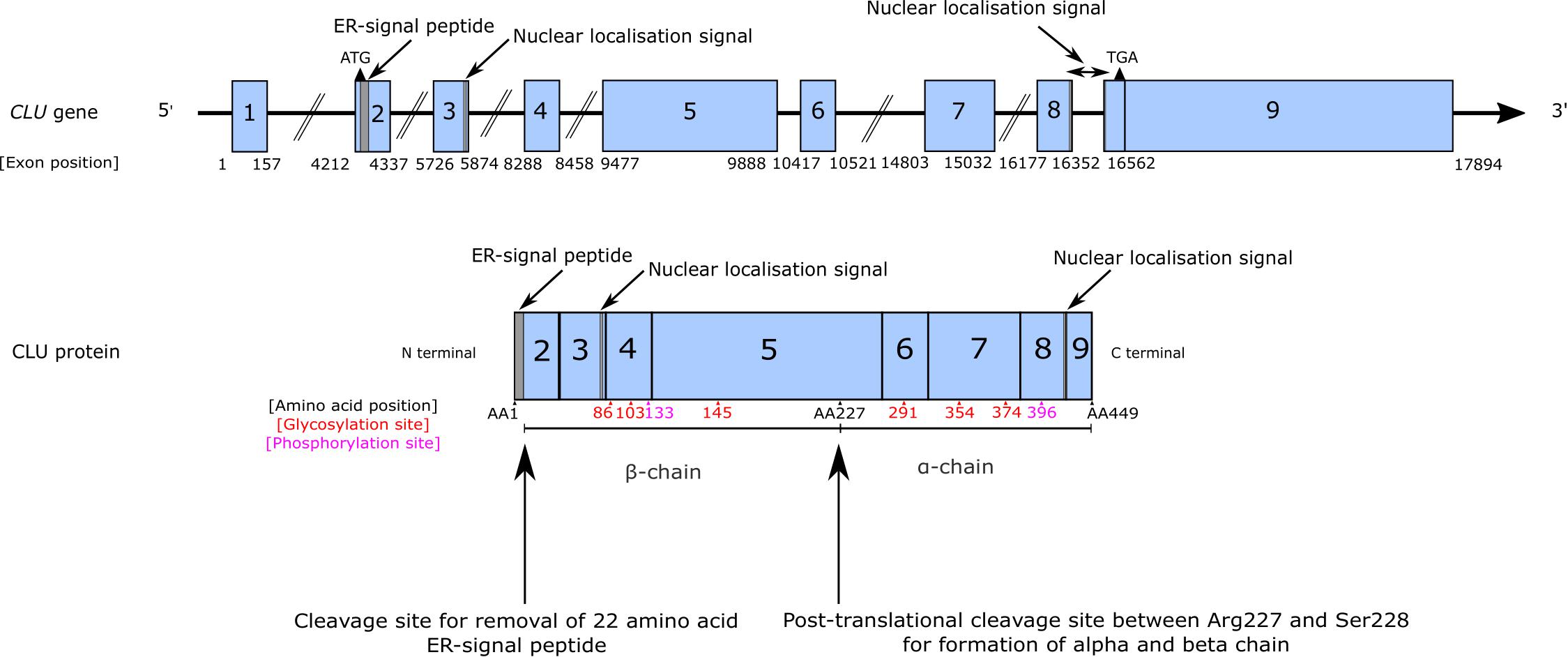
Figure 1. CLU gene and protein structure. CLU is a single copy gene containing nine exons. Exon 1 is a non-coding exon and two translational start sites have been identified, located in exons 2 and 3. An additional ATG has been predicted to be found in exon 1 of alternative variants of CLU, but their biological relevance is unclear (Prochnow et al., 2013). The main CLU mRNA transcript is transcript NM_001831.3 and typically translation begins at the translational start site located in exon 2. This produces an immature preproprotein (NP_001822.3) that contains exons 2–8 and the coding portion of exon 9 and includes an endoplasmic reticulum (ER)-signal peptide located in exon 2 that enables this immature protein to be processed, modified and cleaved in the traditional secretory pathway to produce mature sCLU. During the production of mature sCLU, the ER-signal peptide is cleaved and removed, and an additional cleavage event takes place between amino acid residues 227 and 228 resulting in the formation of an α-chain and a β-chain linked by disulphide bonds. Glycosylation occurs at 6 sites (indicated in red) on both the β-chain (sites 86, 103, and 145) and the α-chain (sites 291, 354, and 374). Two phosphorylation sites are also known (indicated in pink) at residues 133 and 396. Due to discrepancies in the literature, positioning of the α-chain and a β-chain, amino acid annotations, and length of the ER-signal peptide have been drawn based on current annotations provided by NCBI for the clusterin preproprotein NP_001822.3.
Although the regulation of CLU is complex and incompletely characterized, it is apparent that there is a need for tight control of CLU expression. siRNA-knockdown of CLU in cancer cells increases apoptosis (Trougakos and Gonos, 2006) while overexpression of CLU in L929 cells potentiates the toxicity induced by TGF-β while protecting the same cells from TNF-α cytotoxicity (Humphreys et al., 1997). CLU mRNA was first observed to increase in the rat ventral prostate after castration, an observation initially attributed to androgen repression but is now thought to be due to castration-induced apoptosis (Montpetit et al., 1986; Leger et al., 1987). Nevertheless, CLU intron 1 does contain putative androgen response elements and treatment with androgens increases both clusterin mRNA and protein expression via the androgen receptor (AR) (Cochrane et al., 2007). A number of factors have now been observed to regulate CLU expression including NF-κB, growth factors, lipopolysaccharide, and several apoptosis-inducing agents such as ionizing radiation and oxidative stress (Saura et al., 2003; Trougakos et al., 2009; Zoubeidi and Gleave, 2012).
The CLU promoter is highly conserved in mammals (Steve Jones and Jomary, 2002) and contains a number of regulatory elements that may contribute to the control of CLU expression. These elements include activator protein-1 (AP-1), activator protein-2 (AP-2), specificity protein 1 (SP-1) motifs (Wong et al., 1993), androgen response elements (AREs) (Cochrane et al., 2007), and cyclic-AMP response elements (CREs) (Rosemblit and Chen, 1994). Cell death and apoptotic signals also regulate CLU. CLU mRNA expression was observed to be upregulated in rat hippocampus after both neocortical and hippocampal lesioning (May et al., 1990) and after ischaemia (May et al., 1992). Ionizing radiation increases CLU promoter activity in cultured cancer cells, an effect mediated via EGR-1 and EGR-1 consensus sites (Criswell et al., 2005). Apoptotic activator p53 represses CLU promoter activity and transcription in the MCF-7 breast cancer and the HCT 116 colon cancer cell lines, resulting in reduced levels of secreted clusterin (Criswell et al., 2003). The increase in CLU expression combined with clusterin’s extracellular chaperone activity has resulted in comparisons being made between heat shock proteins and clusterin (Poon et al., 2000; Wilson and Easterbrook-Smith, 2000; Nizard et al., 2007). The CLU promoter contains a 14 bp clusterin element (CLE), which differs from the heat shock element (HSE) by only a single base pair (Michel et al., 1997). During stress, this element becomes bound by HSF-1 (Michel et al., 1997) and by HSF-2 during proteasome inhibition (Loison et al., 2006), which causes an induction of CLU expression (Balantinou et al., 2009). Clusterin enhances HSF-1-mediated transcriptional activity and suppresses stress-induced apoptosis (Lamoureux et al., 2011). Heat shock conditions have been shown to not influence CLU expression in mouse astrocytes or motor neurons (Zinkie et al., 2013), suggesting the influence of heat shock signals on CLU expression may be cell- and/or tissue-dependent.
CLU is highly influenced by stress; the AP-1 binding site responds to several stress-related transcription factors including TGF-β (Herault et al., 1992; Wong et al., 1993; Jin and Howe, 1997), which reduces CLU expression (Jin and Howe, 1997) through the induction of c-fos (Marti et al., 1994; Jin and Howe, 1997, 1999; Itahana et al., 2007). TGF-β also down-regulates CLU in a number of cell types including porcine smooth muscle cells (Thomas-Salgar and Millis, 1994) and in rat astrocyte monotypic cultures (Morgan et al., 1995). Tissue-specific regulation has been reported in the case of TGF-β; an upregulation of CLU is in fact observed in epithelial and endothelial cells through the AP-1 binding site (Jin and Howe, 1997). CLU upregulation by TGF-β is also observed in rat astrocytes when co-cultured with microglia and oligodendrocytes (Laping et al., 1994; Morgan et al., 1995). In contrast, in fibroblasts, TGF-β1 downregulates CLU expression (Peix et al., 2018). Stress-activated transcription factor Y box binding protein 1 (YB-1) binds to the CLU promoter resulting in an upregulation of CLU expression (Shiota et al., 2011). Overexpression of CLU or YB-1 results in increased resistance of cancer cells to taxane, a drug used in the treatment of prostate cancer. Taxane, like many anti-cancer drugs, works by promoting stress-induced apoptosis, but this is reduced in the presence of elevated clusterin levels. These observations suggest that this upregulation of CLU during cellular stress is a protective mechanism, enabling cells to survive and adapt to stress through anti-apoptotic pathways. A variety of signals have been shown to induce CLU expression during stress, but downstream pathways influenced by this induction have not been fully characterized. Stress-induced rise in clusterin is predicted to be protective considering the chaperone function of clusterin and its comparisons to heat shock proteins.
CLU also appears to be controlled epigenetically. Several CpG islands have been identified in the CLU promoter (Rosemblit and Chen, 1994). Aging affects both DNA methylation and histone acetylation status, and epigenetic regulation may have an important role in CLU expression during aging (Gemenetzi and Lotery, 2014). CLU expression appears highly influenced by epigenetic factors in retinal pigment epithelial cells where valproic acid, a histone deacetylase (HDAC) inhibitor, induces a significant increase in clusterin protein expression and secretion (Suuronen et al., 2007). Suuronen et al. (2007) observed an increase in both clusterin mRNA and protein expression after treatment of retinal cells with the HDAC inhibitor, Trichostatin A. Additionally, clusterin secretion was increased after the cells were treated HDAC and DNA methyltransferase inhibitors. Hepatitis delta virus increases CLU expression by histone acetylation in human carcinoma cells (Liao et al., 2009). In comparison, histone deacetylation of the CLU promoter and histone methylation via the histone methyltransferase EZH2 in tumor cells results in CLU silencing (Hellebrekers et al., 2007; Wang et al., 2012). DNA demethylation by 5-aza-2′-deoxycytidine increases expression of CLU in prostate cancer cell lines (Rauhala et al., 2008). In human colon cancer cell lines, CLU is regulated predominantly by histone modifications such as histone 3 lysine 9 trimethylation (H3K9me3) and histone 3 lysine 4 trimethylation (H3K4me3) (Deb et al., 2015). Both H3K9me3 and H3K4me3 enrichment may result in altered expression of clusterin in the nucleus of colon cells (Deb et al., 2015). Furthermore, treatment of human astrocytes with valproic acid and the anti-cancer/HDAC inhibitor Vorinostat, resulted in the induction of CLU expression and an increase in clusterin secretion under therapeutic conditions in both cases (Nuutinen et al., 2010). All these observations indicate that the regulation of CLU is cell- and tissue-specific, and that this regulation is complex, involving a diverse array of intracellular and extracellular signals.
Secreted Clusterin (sCLU) – Biogenesis
Mature, secreted clusterin or sCLU is derived from mRNA transcript NM_001831.3, containing exons 1–9 (Figure 1). The ATG located in exon 2 is used for the majority of clusterin translation (Prochnow et al., 2013) and results in the synthesis of the preproprotein (NP_001822.3), which is targeted to the ER and subsequently undergoes extensive post-translational modifications (reviewed in Rohne et al., 2016). Cleavage of the N-terminal ER-signal peptide within the ER produces an immature proprotein of 50 kDa, which is then modified by phosphorylation and glycosylation in the ER and the Golgi (Urban et al., 1987; Wong et al., 1993; Kapron et al., 1997; Lakins et al., 1998; Yang et al., 2000; Sabatte et al., 2011; Rohne et al., 2014), as indicated in Figure 1. Within the Golgi, cleavage between residues 227 and 228 results in the formation of the α- and β-chains linked by five disulphide bonds (Urban et al., 1987; Wong et al., 1993; Kapron et al., 1997; Yang et al., 2000). The resulting secreted protein is a highly glycosylated heterodimer (MW 75–80 kDa) consisting of two chains of 40 kDa each (Jenne and Tschopp, 1989; Kirszbaum et al., 1992; Choi-Miura and Oda, 1996) (Figure 1).
Intracellular Clusterin – Biogenesis
Previously, clusterin was considered to be essentially a secreted protein, but subsequently intracellular clusterin species have also been described (Reddy et al., 1996; Yang et al., 2000; Nizard et al., 2007). Unlike sCLU’s biogenesis, the origin of intracellular clusterin is not well characterized and is still debated. Initially, intracellular clusterin was thought to arise from use of alternative exon 1s and splicing of CLU mRNA (Wong et al., 1993; Reddy et al., 1996; Leskov et al., 2003; Rizzi et al., 2009). However, the reports of multiple CLU mRNA transcripts (outlined in Table 1) were interpreted as a differential transcriptomic origin of intracellular and secreted clusterin proteins (Reddy et al., 1996; Schepeler et al., 2007). A variety of transcripts have been described, but they are not consistently observed in different cell types and/or are only expressed in cells after stress and at very low abundance (Reddy et al., 1996; Leskov et al., 2003; Rizzi et al., 2009; Prochnow et al., 2013), calling into question their physiological relevance in cells. The current consensus is that transcript NM_001831.3 is translated to produce the majority of clusterin protein and that non-secreted clusterin isoforms produced from other transcripts are rare (Prochnow et al., 2013; Rohne et al., 2016).

Table 1. Summary of described CLU transcripts and relevant notes regarding their discovery, annotation, and expression.
The loss of exon 2 and absence of the ER-signal peptide is predicted to produce a single-chain intracellular clusterin that does not undergo cleavage or glycosylation (Reddy et al., 1996; Yang et al., 2000; Leskov et al., 2003; Moretti et al., 2007; Nizard et al., 2007; Trougakos et al., 2009). However, the seminal work on this line of research only demonstrated that exogenous expression of a low-abundance transcript lacking exon 2 led to cytosolic localization of a GFP-fusion protein, which was also found in the nuclei of apoptotic cells (Leskov et al., 2003). Although endogenous expression of this protein was observed to increase after treatment of cells with ionizing radiation in MCF-7 cells (Yang et al., 2000), its transcriptional origin has never been investigated, and others have not observed any nuclear localization in other cell types (Prochnow et al., 2013). Additionally, there is little evidence for the existence of a specific precursor protein in the synthesis of intracellular clusterin. The ∼49 kDa protein described by Leskov et al. (2003), which was detected in stressed cells and predicted to be a nCLU precursor, may in fact be a modified version of secreted clusterin with altered subcellular localization, given that deglycosylated, mature, secreted clusterin has a molecular weight of around ∼50 kDa (Stewart et al., 2007).
There have been numerous reports of glycosylated clusterin with altered cellular localization, particularly subsequent to induction of cellular stressors, including treatment with Nerve Growth Factor (O’Sullivan et al., 2003) and ER stress (Nizard et al., 2007; Li et al., 2013; Gregory et al., 2017). ER stress resulted in the cytoplasmic accumulation of clusterin in a Drosophila model of amyotrophic lateral sclerosis (ALS), reducing accumulation of TDP-43 protein inclusions and partially rescuing the ALS-like phenotype (Gregory et al., 2017). Treatment with autophagy-inducing stressors in cancer cell lines resulted in the upregulation of both clusterin mRNA and protein, which co-localized with LC3 puncta on autophagosomes’ membrane, promoting their biogenesis and increasing cell survival (Zhang F. et al., 2014). Multiple strands of evidence suggest that intracellular clusterin is a modified form of secreted clusterin. There are several ways in which this could occur, including: (i) the impaired secretion of clusterin after cellular stress resulting in the intracellular accumulation of mature, secreted clusterin; (ii) reuptake of secreted, mature clusterin after release from cells (Ghiso et al., 1993; Zlokovic et al., 1996; Kang et al., 2005); and (iii) improper trafficking of clusterin through the secretory pathway and premature escape resulting in the intracellular accumulation of incompletely modified or unglycosylated clusterin (Prochnow et al., 2013). Furthermore, nuclear compartmentalization of clusterin is increased after the direct inhibition of clusterin secretion (Sansanwal et al., 2015) and rare AD-mutations found in CLU have been shown to alter clusterin trafficking, resulting in intracellular accumulation and a loss of secreted clusterin (Bettens et al., 2015). Additionally, our group also observed that treatment of rodent primary neurons with Aβ leads to reduced clusterin secretion and increased intracellular clusterin (Killick et al., 2014). Collectively, these studies indicate a tight relationship between intracellular and secreted clusterin and highlight a potential role of altered clusterin trafficking in AD pathogenesis.
When considering the brain, it is important to note that astrocytes are the main source of clusterin, although subpopulations of neurons also do express clusterin (Pasinetti et al., 1994; Morgan et al., 1995). In the healthy brain, astrocytes synthesize and release clusterin into the extracellular space. After injury, in inflammatory states, and in neurodegenerative diseases like AD, both neuronal and astrocytic CLU expression is increased (Johnson et al., 1992; Zoli et al., 1993; Liu et al., 1999; Wiggins et al., 2003), although whether this rise is protective or toxic is unknown. It will be important for AD research to identify the source of neuronal intracellular clusterin. As we will discuss later, sCLU plays a role in cell survival pathways, a role which requires it to interact with intracellular proteins such as Ku70. Earlier studies have suggested that clusterin is taken up by cells subsequent to release (Ghiso et al., 1993; Zlokovic et al., 1996; Kang et al., 2005), a process that would enable sCLU to participate in intracellular pathways. This process may be enhanced in cells during damage or stress, as a protective mechanism against cell death (Pasinetti et al., 1994; Morgan et al., 1995).
Clusterin in Health and Disease
The main focus of this review is to explore the role of clusterin in AD. However, understanding the role of clusterin in other pathologies, tissues, and cells lends further insight into clusterin mechanism of action that may further illuminate its role in AD. Here, we focus on clusterin’s role in other neurological disorders, cancer, and cardiovascular diseases.
Clusterin and Neurological Disorders
Increased levels of clusterin have been observed not only in the AD brain but also in other neurodegenerative diseases, including ALS (Grewal et al., 1999), multiple sclerosis (Ingram et al., 2014), transmissible spongiform encephalopathies (Sasaki et al., 2002a), and Huntington’s disease (Labadorf et al., 2015). Similarly to the AD pathology, where clusterin co-localizes with Aβ in the senile plaques (reviewed in section “Clusterin in Aβ Metabolism”), in the case of alpha-synucleinopathies, clusterin co-localizes with cortical Lewy bodies (LBs) (Sasaki et al., 2002b). Interestingly, cortical LBs with a stronger clusterin immunostaining show a reduction in alpha-synuclein, perhaps indicating a role for clusterin in modifying its aggregation. This is in line with the well-known function of clusterin as an extracellular chaperone (Humphreys et al., 1999), although the different locations of the intracellular LBs and of mature, secreted clusterin pose the questions of how secreted clusterin could associate with LBs inside the cell, or whether clusterin could abandon the secretory pathway and, if that was the case, whether non-secreted clusterin species would still retain a chaperone function. Nevertheless, given that protein aggregation is a common pathological hallmark across neurodegenerative diseases, it is not surprising that the chaperone function of CLU has been studied in several proteinopathies. A recent study using N2a mouse neuroblastoma cultures and in vivo Drosophila models of ALS showed that CLU overexpression reduces TDP-43 protein aggregation and toxicity (Gregory et al., 2017). These findings were replicated in the same study using Drosophila photoreceptor neurons expressing pathogenic Huntingtin-Q128 and mutant (R406W) human tau proteins, where co-expression of CLU protected cells from proteotoxicity. Importantly, clusterin-mediated neuroprotection was only observed under ER stress conditions, which had previously been shown to induce clusterin translocation from the ER to the cytosol (Nizard et al., 2007; Li et al., 2013). However, once again the origin of clusterin interacting with intracellular protein aggregates remains unsolved and, if clusterin translocation from the ER occurred in this context, then it is uncertain whether the chaperone function of mature secreted clusterin would be present in these non-secreted forms.
Clusterin is also upregulated in some neurodevelopmental disorders that are not associated with proteotoxicity, such as schizophrenia and Rett syndrome. In schizophrenia, increased CLU mRNA levels are found in cortical neuronal subpopulations (Pietersen et al., 2014a,b), and sCLU is upregulated in prefrontal cortex homogenates (Athanas et al., 2015). The authors hypothesize that clusterin could be playing a neuroprotective role against oxidative stress mediated by TGF-β-signaling (Athanas et al., 2015), which is associated with schizophrenia pathophysiology (Bitanihirwe and Woo, 2011). CLU is also upregulated in the frontal cortex of Rett syndrome (RTT), a neurodevelopmental disorder mainly caused by mutations in the methyl CpG binding protein 2 (MECP2) gene (Gibson et al., 2010). MECP2 siRNA knockdown led to CLU upregulation and furthermore, a direct interaction between MECP2 and the CLU promoter was shown, indicating that loss-of-function mutations in MECP2 might lead to the overexpression of CLU (Gibson et al., 2010). It is worth noting that links between RTT and oxidative stress have also been established, with oxidative stress measurements correlating with pathogenicity of MECP2 mutations and clinical severity of the disease (Leoncini et al., 2011; Filosa et al., 2015). Therefore, the reported CLU upregulation in RTT could again be a stress response partially driven by oxidative stress.
Hypoxia-ischaemia brain insult also triggers CLU expression (Zoli et al., 1993; Yasuhara et al., 1994; Walton et al., 1996; Kurian and McGuone, 2012). Some studies found a protective effect of CLU against hypoxia-ischaemia and showed that recovery from middle cerebral artery occlusion (MCAO) was better in wild-type (WT) than in CLU-KO mice, which had a larger inflammatory response around the scar area (Imhof et al., 2006; Wehrli et al., 2001), while overexpression of human CLU reduced the presence of inflammatory cells and cell death (Wehrli et al., 2001). However, clusterin’s role in hypoxia-ischaemia is debated, as some studies report a detrimental function of clusterin. In a neonatal hypoxia-ischaemia mouse model, in which tissue loss was accompanied by accumulation of clusterin in dying neurons, CLU-KO animals had reduced brain damage in an allele-dose-dependent manner (Han et al., 2001). At the cellular level, addition of exogenous clusterin potentiated neuronal death in mixed cultures of primary cortical neurons and astrocytes in an oxygen/glucose deprivation environment, while having no effect under normoxic conditions (Han et al., 2001). Similarly, CLU-KO mouse hippocampal slices showed higher tolerance to oxygen/glucose deprivation compared to WT brain slices, and the addition of clusterin increased their sensitivity to that of WT (Hakkoum et al., 2008). At this point it is not clear what determines the neuroprotective or neurodegenerative role of clusterin in hypoxia-ischaemia conditions, since there are numerous confounds in the various experimental systems including differences in the animal models, their developmental stage, over-expression versus gene KO and the measurements used to assess ischaemic brain damage (Holtzman et al., 2001; Wehrli et al., 2001). Importantly, hypoxia-ischaemia is a known risk factor in AD (Zhang and Le, 2010), so a better understanding of clusterin’s function in this context could provide valuable insight into the contribution of clusterin to AD pathology. In light of our preceding discussion it is also clear that we need to know more about which clusterin species are found in the disease models. In this sense, CLU-KO is a blunt tool since both secreted and intracellular clusterin is abolished, thereby blinding us to any function(s) performed by specific species. This concept is explored further in our discussion of the role of clusterin in cancer (see section “Clusterin and Cancer”).
Clusterin and Cancer
Much of what we currently understand about clusterin arises from oncology research. Clusterin is overexpressed in a variety of cancers including breast (Ranney et al., 2007; Yom et al., 2009), ovarian (Xie D. et al., 2005; Wei et al., 2009), and prostate cancer (July et al., 2002), where it promotes tumorigenesis and contributes to chemoresistance (Cao et al., 2005; Flanagan et al., 2010; Kususda et al., 2012; Tang et al., 2012; Xiu et al., 2013). Oncology research has highlighted the opposing roles of clusterin proteins in cell death and survival, presenting the same apparent contradiction as seen in neurodegeneration. Here, we will discuss the current knowledge of clusterin’s role in these pathways, and how research from oncology may lend insight into the role of clusterin in AD.
Initial suggestions gave rise to the idea that sCLU provides a pro-cell survival role while intracellular clusterin proteins are pro-apoptotic (Leskov et al., 2011). This apparent functional dichotomy was thought to arise from the existence of different clusterin proteins with different cellular localizations. An overall increase in CLU expression may not be the critical factor in cancer, instead it is thought that an altered ratio of intracellular: secreted CLU proteins may be the key factor, such that cancer is accompanied by an increase in CLU secretion and a reduction in intracellular CLU (Pucci et al., 2004). Most oncology research in clusterin focuses on the role of sCLU, since it is believed to promote chemoresistance (Cao et al., 2005; Flanagan et al., 2010; Kususda et al., 2012; Tang et al., 2012; Xiu et al., 2013) and survival of cancer cells through both anti-apoptotic and pro-survival mechanisms (Ammar and Closset, 2008; Shim et al., 2011; Wang et al., 2013). Numerous studies have shown that the knockdown of CLU increases cell sensitivity to chemotherapy drugs and enhances cell death (Viard et al., 1999; Lee et al., 2002; Trougakos et al., 2004; So et al., 2005; Redondo et al., 2007). Although these studies have not specifically knocked down expression of either secreted or intracellular clusterin, it is likely, given the postulated origins of intracellular clusterin, that knockdown of CLU will result in a reduction of secreted clusterin and thus, also intracellular clusterin. The potential therapeutic advantage of silencing CLU in cancer has been examined in clinical trials. Custirsen (OGX-011) is an antisense oligonucleotide that targets CLU (Chi et al., 2005; Zielinksi and Chi, 2012). The antisense design strategy was aimed at specifically targeting secreted clusterin and had no reported effect on nuclear clusterin expression (Cao et al., 2005), but nevertheless, from the oligonucleotide design it would not be able to distinguish between sCLU and intracellular forms of CLU unless altered translation initiation start sites are used. However, addition of Custirsen to the current treatment regime for prostate cancer does promote sensitization of cancer cells during chemo-therapy (He et al., 2009; Saad et al., 2011; Kususda et al., 2012; Laskin et al., 2012), but effects were no better than the current regime alone (Beer et al., 2017; Chi et al., 2017).
Clusterin and Cardiovascular Disease
As discussed above, clusterin possesses both pro-survival and pro-apoptotic functions and is cytoprotective in tumor cells. Clusterin’s role in the heart appears more straightforward and is considered primarily protective in cardiac cells during damage and disease. CLU expression is frequently observed to rise in the heart after damage (Swertfeger et al., 1996; Ishikawa et al., 1998; McLaughlin et al., 2000; Miyata et al., 2015; Liu et al., 2018). This rise in clusterin is a potential prognosis marker for heart damage: post-heart transplant patients that recover show increased plasma clusterin compared to those who do not recover (Hollander et al., 2014). Strong evidence suggests that clusterin’s protective role in the heart is attributed to its function as a lipid and fat transporter. Firstly, clusterin is only expressed in damaged arteries in the early stages of atherosclerosis, where it may be acting to increase fat and lipid transport, and not in healthy arteries (Ishikawa et al., 1998); secondly, clusterin and APOE form HDL particles in blood plasma together with APOA-I and paraoxonase (Baralla et al., 2015), promoting their transport and processing in the liver (Rizzi et al., 2009); thirdly, clusterin removes cholesterol from macrophage-foam cells, which are a key cell type in atherosclerotic lesions (Gelissen et al., 1998). This is not to say that the anti-apoptotic and anti-oxidant functions of clusterin do not also contribute to its protective role. Clusterin has been shown to inhibit apoptosis and to protect cardiac cells from ischaemic-reperfusion after heart transplantation (Liu et al., 2018), and it has been associated with a reduction in pro-inflammatory signals such as TNF-α and pro-apoptotic BAX, and with an increase in anti-apoptotic gene BCL-XL (Jun et al., 2011; Foglio et al., 2015; Liu et al., 2018). Clusterin may also act as an anti-oxidant in the heart (Mackness et al., 1997), a function that appears dependent on the phosphorylation of Akt/GSK-β and PI3K pathways (Jun et al., 2011; Liu et al., 2015).
So, do these observations of diverse disease states provide any gestalt perspective of clusterin function? Clearly clusterin is involved in regulating cell death in neurological disease, cardiovascular disease and cancer but whether clusterin is protective or pro-apoptotic requires a more nuanced answer. It is clear from both oncology and cardiovascular research that clusterin has a protective function. In the heart, clusterin’s protective function is attributed to its ability to bind lipids and fats, combined with its anti-apoptotic and anti-oxidant properties. However, these same ‘protective’ functions give clusterin a pathological role in oncology, contributing to both tumorigenesis and treatment resistance. The older view that a duality of function may reflect the actions of different extracellular and intracellular clusterin species, each with distinct structure, function, and location has given way to the realization that sCLU is likely the source of intracellular clusterin. Whether this is due to stress-induced translocation of secreted clusterin, or immature/improperly modified versions of secreted clusterin diverting the protein from the secretory pathway resulting in its accumulation within the cell (and a subsequent loss of secreted clusterin being released from the cell) or whether extracellular sCLU becomes internalized is still unclear. So, for instance, in cancer it may not be the overall rise in clusterin levels that determines its role but the altered location of clusterin proteins resulting in an increased ratio of sCLU to intracellular CLU (Pucci et al., 2004). Interestingly, something similar has been observed in AD, whereby clusterin trafficking has been altered by both Aβ treatment (Killick et al., 2014) and CLU AD-mutations (Bettens et al., 2015), resulting in increased intracellular clusterin and a reduction in secreted clusterin. These observations suggest that an alteration in the distribution of clusterin inside and outside of cells may contribute to AD pathogenesis. Additionally, the demonstration that CLU silencing provides protection from Aβ-induced neurotoxicity in rodent neurons and iPSC-neurons (Killick et al., 2014; Robbins et al., 2018) also points to the importance of altered distribution of clusterin across the cell and clusterin’s pathological role in mediating Aβ toxicity in neurons. This brings into sharp focus the relevance of understanding how intracellular clusterin arises in cells, the structural differences and similarities between secreted and intracellular clusterin, and how these contribute to pathophysiological events. Resolving these issues is critical not only to understanding the role of clusterin in AD but also to exploiting the therapeutic potential unveiled by CLU knockdown while preventing unwanted off-target effects.
Clusterin and Disease Mechanisms
Given the wide distribution of clusterin in the body and its varied functions, it is not surprising that clusterin is implicated in several diseases. Even though clusterin might affect specific pathways in each of these diseases, its dysregulation appears to be driven by common underlying processes, prominently cell death and survival, and oxidative stress.
Cell Death and Survival
Both secreted and intracellular clusterin have been reported to interact with common proteins in survival and apoptosis pathways, including BAX and Ku70, mediators of BAX-dependent apoptosis (Figure 2). BAX is a key regulator of apoptosis and translocates from the cytosol to the mitochondria during cellular stress whereupon it stimulates pore formation in the mitochondria membrane, enabling the release of pro-apoptotic proteins, thereby promoting cell death (Green and Kroemer, 2004; Moldoveanu et al., 2014). In healthy cells, cytosolic Ku70, a DNA repair protein, binds to BAX to form a complex that inhibits BAX movement to the mitochondria. sCLU can alter this interaction by binding to the complex of Ku70-BAX to stabilize it, thereby reducing the amount of free BAX that can translocate to the mitochondria to induce apoptosis (Zhang et al., 2005; Trougakos et al., 2009). Depletion of clusterin activity has been shown to reduce the binding of Ku70-BAX and increase BAX mitochondrial levels (Trougakos et al., 2009) with the resulting accumulation of pro-apoptotic proteins, increased release of cytochrome C, and increased caspase 9 expression. This is accompanied by a reduction in anti-apoptotic proteins, including Bcl-2, resulting in cells becoming more vulnerable to the pro-apoptotic effects of BAX (Trougakos et al., 2009). Since sCLU is usually located in the extracellular space, the mechanism enabling this intracellular interaction remains unclear. However, internalization of clusterin may occur after binding to receptors such as the megalin receptor (Ghiso et al., 1993; Zlokovic et al., 1996), and several groups have reported glycosylated clusterin protein in the cytosol during cell stress (Nizard et al., 2007), which supports the feasibility of this intracellular interaction between sCLUi, Ku70, and BAX to reduce cell death (Zhang et al., 2005). In contrast, there is some indication that intracellular clusterin may promote apoptosis. Previously Leskov et al. (2003) described nuclear clusterin as a cell death signal and it is thought that nuclear clusterin binds directly to Ku70 (Yang et al., 2000; Leskov et al., 2003), blocking the interaction between Ku70 and BAX, therefore promoting BAX translocation to the mitochondria where it can promote cell death. The interaction and binding with Ku70 were observed with a 60 kDa clusterin protein but not with mature, secreted clusterin and therefore was suggested to be specific to intracellular clusterin (Yang et al., 1999, 2000). The structure or identity of this 60 kDa clusterin protein was not determined and may in fact represent immature secreted clusterin or a modified version of the mature secreted protein. The origin of these differences in these protein interactions of the clusterin proteins has not been explored but may be in part explained by structural differences, such as the level of glycosylation of clusterin, which is known to influence chaperone activity (Rohne et al., 2014). Structural differences between these proteins may therefore be key in explaining the dysfunctionality of clusterin in apoptosis and cell death pathways, as opposed to the existence of different clusterin proteins with separate cellular localizations.
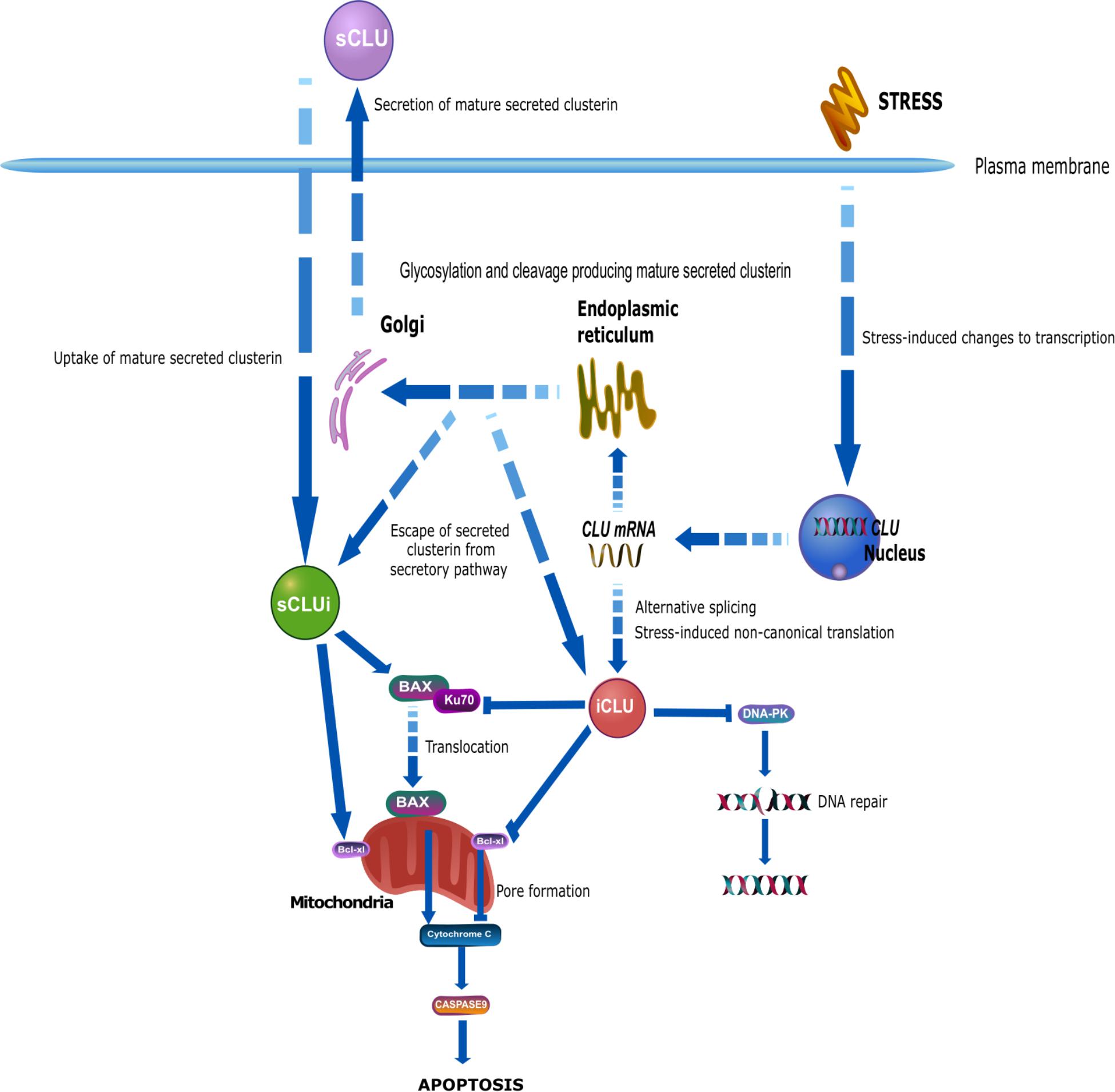
Figure 2. The biogenesis of mature, secreted clusterin involves the traditional secretory pathway common to other secreted proteins. CLU is transcribed into the mRNA NM_001831.3, containing exons 1–9. Within exon 2 is a 22-amino acid signal peptide that targets the preproprotein (NP_001822.3) to the endoplasmic reticulum (ER) and the Golgi apparatus. This immature preproprotein undergoes a series of modifications including phosphorylation, cleavage, and extensive glycosylation. Finally, mature secreted clusterin (sCLU) is produced and secreted from cells. Although traditionally referred to as a secreted protein, a number of reports have shown the existence of clusterin inside the cell, referred to as intracellular clusterin. In comparison to secreted clusterin, which has a well described biogenesis, the origin and production of intracellular clusterin is much less clear. Although originally believed that intracellular clusterin arose from a distinct CLU mRNA transcript to sCLU, it is now extensively believed that these two proteins share the common mRNA transcript NM_001831.3 and that intracellular clusterin may exist due to altered localization of secreted clusterin. Stress may impair a cell’s ability to secrete sCLU, resulting in its localization within the cell. Alternatively, sCLU may not undergo canonical biogenesis, resulting in incomplete glycosylation and/or cleavage, and this may lead immature sCLU to escape the secretory pathway and remain inside the cell. Additionally, secreted clusterin may be taken into cells by uptake mechanisms resulting in the presence of mature, glycosylated sCLU within the cell. Alternative theories suggest that CLU mRNA may undergo stress-induced alternative splicing and non-canonical translation to produce a truncated, non-glycosylated clusterin protein that lacks exon 2 and therefore does not become secreted, but instead accumulates within the cell. Research from oncology indicates a pro-survival function of secreted clusterin and a pro-apoptotic function of intracellular clusterin. Secreted clusterin interacts with the BAX-Ku70 complex, stabilizing it thereby inhibiting the translocation of BAX to the mitochondria where it would promote apoptotic pathways. In contrast, intracellular clusterin competes with BAX for binding with Ku70 and thereby inhibiting their complex formation, resulting in increased free BAX that can translocate to the mitochondria. Secreted clusterin is also thought to interact with Bcl-xl proteins promoting its anti-apoptotic function, whereas intracellular clusterin interacts with this same protein to reduce its anti-apoptotic function. Intracellular clusterin has also been shown to interact with DNA-PK complexes and thereby inhibit DNA repair, resulting in DNA damage and cell stress and death. This figure was produced using icons in the icon library of the Reactome database that are free for download and modification: (https://reactome.org/icon-lib) Accessed August 28, 2018; (Sidiropoulos et al., 2017).
Other pro-apoptotic roles of clusterin in promoting cell death are mediated via interaction with Bcl-xl, an anti-apoptotic protein located on the mitochondrial membrane which is believed to contribute to the regulation of the permeability of the outer mitochondrial membrane (Kelekar and Thompson, 1998; Youle and Strasser, 2008) and the release of pro-apoptotic proteins including cytochrome C, resulting in cell death (Kim et al., 2012). In the heart, cardiomyocytes are protected from H2O2-induced apoptosis if they are treated with clusterin; cells show increased expression of Bcl-2 proteins and reduced BAX expression, and clusterin-treated cells also show increased protection from H2O2-induced cell death (Jun et al., 2011). After heart transplantation, clusterin expression reduces the expression of inflammatory proteins including TNF-α and BAX, additionally increasing Bcl-xl expression both in vivo and in vitro. This reduces cell death and ischaemia reperfusion injury after heart transplantation, a key source of heart damage (Liu et al., 2018). Overexpression of intracellular clusterin cDNA fragments reduces cell viability, which can be rescued by overexpression of Bcl-xl and inhibition of caspases (Debure et al., 2003). nCLU has also been implicated in DNA repair pathways. In fact, nCLU was the first stress-inducible protein shown to be associated with the DNA-dependent protein kinase complex (DNA-PK) during double-stranded break repair of DNA, an association that prevents DNA repair resulting in cell death (Leskov et al., 2001). Currently the contribution of nCLU to cell death and DNA repair mechanisms is not fully understood. Additionally, cytosolic clusterin inhibits NF-κB-dependent Bcl-xl expression and induces arrest of the cell cycle promoting cell death (Scaltriti et al., 2004a,b; Zhang et al., 2005; Takase et al., 2008; Kim et al., 2012). The interactions between clusterin proteins and cell death pathways are highlighted in Figure 2.
Not all clusterin proteins have been observed to influence BAX-mediated apoptosis. Prochnow et al. (2013) observed that non-secreted clusterin proteins observed after proteasomal inhibition did not influence BAX apoptosis. In addition to interactions with apoptotic pathways, there is some evidence supporting interaction of clusterin with pro-survival pathways. PI3K activates and phosphorylates Akt where it localizes to the plasma membrane and can influence several downstream effectors such as mTOR. In several cell lines, clusterin has been observed to interact with this pathway and promote cell survival. In the heart, clusterin protects H9c2 cardiomyocytes from oxidative stress-induced cell death, possibly by regulation of PI3K-Akt and GSK-3β signaling (Jun et al., 2011). This pathway is also active in retinal cells (ARPE-19) where cell death induced by H2O2 was reduced by clusterin, an effect that that was blocked by inhibition of the PI3K-Akt pathway (Kim et al., 2010). This interaction is also observed in the induced prostate cancer cell line, MLLTet-sClu, where the same dose of doxycycline that was enough to induce clusterin synthesis, increases Akt and Bad phosphorylation, and reduces cytochrome C release, thus promoting cell survival (Ammar and Closset, 2008). Although not much is known about the role of clusterin in mediating cell survival pathways directly, the combinatory role of clusterin in both an anti-apoptotic role and in the enhancement of pro-survival pathways suggests a key role for clusterin in promoting cell survival during cellular stress.
Oxidative Stress
Oxidative stress is the result of an imbalance between the production of reactive oxygen species (ROS) and the body’s defenses to remove them, and is implicated in many disorders including cancer (Campisi and Fleshner, 2003), cardiovascular diseases (Stocker, 2004), and numerous neurodegenerative diseases including Parkinson’s disease (Lin and Beal, 2006) and AD (Praticò et al., 2002; Honda et al., 2004; Mattson, 2004). In the aging brain, oxidative stress is increased (Finkel and Holbrook, 2000; Golden et al., 2002) and the AD brain is characterized by increased oxidative stress markers including DNA damage and lipid peroxidation (Praticò et al., 2002), which can result in cell death (Butterfield et al., 2002). ROS are produced naturally in the body by the interaction of oxygen with redox metal ions such as copper and zinc. Aβ produces a large amount of ROS through activation of NMDA receptors (Parks et al., 2001; De Felice et al., 2007; Shelat et al., 2008), which may contribute to synaptic dysfunction (Hu et al., 2009; Rönicke et al., 2011). Interaction between Aβ and redox metal ions is also observed; Cu2+ and Aβ produce H2O2 (Huang et al., 1999; Cuajungco et al., 2000; Opazo et al., 2002) and Aβ toxicity is enhanced in the presence of Cu2+ (Opazo et al., 2002). Clusterin is thought to be a sensitive biosensor of oxidative stress (Trougakos and Gonos, 2006) since a number of sites in the CLU promoter are responsive to changes in oxidative stress (Herault et al., 1992; Wong et al., 1993; Michel et al., 1995; Jin and Howe, 1997), including proteolytic stress-mediated increases in CLU expression via binding of both HSF-1 and HSF-2 to a CLE in the CLU promoter (Chondrogianni et al., 2003; Loison et al., 2006). Oxidants such as H2O2 can alter CLU mRNA expression and sCLU protein levels (Frippiat et al., 2002) whilst ionizing radiation-induced oxidative stress leads to the accumulation of sCLU at low doses and nCLU at higher, toxic doses (Leskov et al., 2003; Criswell et al., 2005). Numerous studies in various cell types suggest a protective role of sCLU against oxidative stress. For instance, in the heart, sCLU accumulates in arterial walls during the early stages of atherosclerosis and is thought to be a protective mechanism against oxidative stress (Mackness et al., 1997). sCLU also protects cancer cells from H2O2 and UVA radiation (Viard et al., 1999; Dumont et al., 2002; Miyake et al., 2005). Time-dependent increases in CLU mRNA and protein are also observed in neuroblastoma cells under conditions of lipid peroxidation and production of ROS, suggested to be a protective mechanism inhibiting cellular damage during oxidative stress (Strocchi et al., 2006). The silencing of CLU in human cancer cells increases apoptosis, reduces growth and increases vulnerability to oxidative stress (Trougakos et al., 2004).
Overall, clusterin appears to be protective against oxidative stress and provides both short term resistance to oxidative stress-induced damage and also long-term survival. In common with the manifold functions of clusterin the exact contributions of sCLU and intracellular clusterin to protection against oxidative stress remain unclear. Oxidative stress is a critical factor observed both during normal aging and in neurodegenerative diseases like AD. A loss of sCLU’s protective role against damage induced by ROS may underwrite increased vulnerability of neurons to insults such as Aβ, which produces both H2O2 and other ROS. If sCLU is the critical player then the observed reduction in sCLU after Aβ treatment in neurons may result in a loss of protection against oxidative stress and cell death.
Clusterin (dys)Function in AD
Clusterin and Aβ Metabolism
The relationship between clusterin and Aβ has been reported for over two decades (Ghiso et al., 1993), with pioneering work already showing that clusterin prevented Aβ aggregation in an in vitro acellular system (Matsubara et al., 1996). Around that time, LRP2 (also known as megalin/gp330) was identified as an endocytic receptor for clusterin (Kounnas et al., 1995) and was shown to mediate CLU-Aβ transport through the blood–brain barrier (BBB) and through the blood-CSF barrier (Zlokovic et al., 1996). Despite these initial observations, the anti-amyloidogenic nature of clusterin continues to be a matter of controversy fuelled by the confounds arising from use of numerous and diverse biological and disease models.
Effects on Amyloid Toxicity
A common way of studying the effect of clusterin on Aβ aggregation and resulting toxicity has been to incubate amyloid preparations with or without clusterin in different biological systems. The use of different Aβ species, aggregation protocols and incubation strategies has undoubtedly led to the variable downstream biological effects that have been observed. Nevertheless, a common finding is that clusterin interferes with Aβ aggregation, in agreement with observations from Narayan et al. (2012) showing that clusterin is able to bind a wide range of Aβ oligomers (from dimers to 50-mers), and consequently to prevent further aggregation into fibrils. However, this interference with Aβ aggregation has been observed to influence toxicity of the amyloid products in differing ways. On the one hand, clusterin’s cytoprotective effects were detected in a study with SH-SY5Y cells treated with Aβ-supplemented AD CSF, where the addition of a mix of extracellular chaperones including clusterin into the CSF preserved cell viability (Yerbury and Wilson, 2010). Similarly, co-culture experiments on rat hippocampal astrocytes and neurons showed that clusterin incubation prevents Aβ-induced astrocytic calcium uptake, resulting in decreased ROS generation and caspase 3 activation; additionally, clusterin blocked Aβ-induced inhibition of LTP on hippocampal slices (Narayan et al., 2014). In another study, incubation of Aβ aggregates with clusterin before injection into rat brains improved performance in the Morris water maze test and decreased glial activation and neuronal degeneration compared to that observed in rats injected with Aβ42 alone or clusterin and Aβ42 with no previous incubation (Cascella et al., 2013). On the other hand, reports show an increase in the amount of more diffusible, soluble oligomeric species and reduced fibril formation resulting in increased cellular oxidative stress (Oda et al., 1995) and neurotoxicity in organotypic mice brain slices (Lambert et al., 1998). In a conciliatory study, the duality of clusterin’s effect on Aβ toxicity was postulated to be dependent on the molar ratio of clusterin and Aβ (Yerbury et al., 2007). Clusterin reduced Aβ aggregation and toxicity but, when Aβ was present in an excess compared to clusterin, there was an increase in amyloid formation. In the latter, clusterin was also incorporated into these amyloid aggregates, which were more toxic than aggregates containing Aβ42 alone. Given that concentrations, timescale, and tissue location of the clusterin-Aβ interaction may vary significantly between the AD brain and these simplified experimental models, the relevance of these observations to AD pathology remains to be proven.
Effects on Amyloid Clearance
Several mechanisms of brain Aβ clearance have been described, including intracellular uptake and transport across the BBB (Tarasoff-Conway et al., 2015), both of which have implications for the role of clusterin in AD.
Astrocytes and microglia are both implicated in capturing and eliminating Aβ from the brain interstitial fluid (ISF) (Tarasoff-Conway et al., 2015). Again, the role of clusterin on these interactions is far from clear. Treatment of human astrocytes with Aβ results in a simultaneous increase of intracellular clusterin, decrease of secreted clusterin, and appearance of vacuoles containing fibrillary Aβ, arguing in favor of clusterin’s role in promoting Aβ clearance (Nuutinen et al., 2007). However, in other studies, incubation of pre-aggregated Aβ with clusterin resulted in reduced amyloid uptake from oligomer-enriched preparations in cultures of human primary astrocytes (Nielsen et al., 2010) and from fibril-enriched preparations by microglia (Mulder et al., 2014). Interestingly, these studies showed that, despite both cell types being more efficient at taking up amyloid from oligomer-enriched rather than from fibril-enriched preparations, microglia had a higher capacity for binding and internalizing amyloid from a fibril-enriched preparation than astrocytes, which suggests different roles and mechanisms of Aβ clearance by microglia and astrocytes (Mulder et al., 2014). Another study observed that incubation of AD CSF with clusterin actually increased the removal of Aβ42 from the supernatant by macrophage-like U937 cells (Yerbury and Wilson, 2010). More recently, it was described that mouse primary microglia and human monocyte-derived macrophages use TREM2 as a receptor to bind and take up lipoproteins and apolipoproteins including clusterin, and that internalization of Aβ is more efficient when complexed with LDL or clusterin (Yeh et al., 2016). TREM2, like CLU, is an AD-risk locus (Guerreiro et al., 2012; Jonsson et al., 2012). All of this underlies the potential importance of functional links between multiple risk genes in disease mechanism (see section “Relationships to Other AD Genes”).
Clusterin also regulates Aβ transport through the BBB, and Aβ clearance is significantly increased when complexed with clusterin in vivo, an effect mediated by LRP2 (Bell et al., 2007). More recent studies have used in vitro systems to model a simplified version of the BBB by culturing endothelial cells on a permeable support that allows for separation between apical and basolateral compartments simulating blood and brain, respectively. In a study with mouse primary cerebral endothelial cells, trafficking of fluorescently labeled Aβ40 from basolateral to apical compartment was enhanced when complexed with clusterin, and blockage of LRP1/LRP2 reduced crossing of clusterin between compartments (Merino-Zamorano et al., 2016). In a similar system using porcine brain capillary endothelial cells, exogenous addition of clusterin to the culture increased intracellular protein levels of APP and trafficking of radiolabeled Aβ40 (Zandl-Lang et al., 2017).
These studies indicate that interaction between clusterin and Aβ is potentially important for amyloid clearance. However, these findings also underscore the importance of context when studying this interaction. Different cell-types, varying levels of model complexity, and conditions that represent distinct physiological situations often lead to differing conclusions. Sifting through these confounds to identify the factors that contribute to AD pathology remains a challenge.
Effects on AD-Pathology in AD Animal Models
The use of CLU-knockout (KO) animal models of amyloidosis has enabled the study of the relationship between clusterin and Aβ in vivo. These studies, unsurprisingly, have not provided clear answers on the anti-amyloidogenic nature of clusterin. In seminal work by DeMattos et al. (2002), plaque formation was reduced in CLU-KO PDAPP mice compared to CLU-expressing PDAPP mice. Plaques formed in the absence of CLU displayed decreased surrounding neuritic dystrophy, which argued in favor of a pro-amyloidogenic role of clusterin in this mouse model (DeMattos et al., 2002). However, subsequent work by the same group showed that knocking out both CLU and APOE in PDAPP mice resulted in increased Aβ production and amyloid deposition and earlier onset of AD-pathology than the APOE-KO alone (DeMattos et al., 2004). These findings suggest that clusterin and APOE may work cooperatively in this mouse model to reduce amyloid deposition and illustrate the complexity of understanding Aβ-clusterin interactions in vivo. A more recent study in APP/PS1 mice showed that CLU-KO shifts deposition of Aβ from plaques to accumulation in the cerebrovasculature, resulting in increased amyloid angiopathy but, surprisingly, reduced hemorrhage and inflammation (Wojtas et al., 2017). These varied findings across CLU-KO mice with different genetic backgrounds illustrate the need to consider potential compensatory mechanisms and parallel pathways when drawing conclusions from a KO model, and offer a cautionary note on the even greater distance that might exist in clusterin function between animal models and humans.
Clusterin in DKK1-Driven Altered Wnt Signaling
In addition to its direct interactions with Aβ and the resulting downstream effects, clusterin has been implicated in Wnt signaling, a pathway that has been intensely scrutinized in relation to AD. Aβ treatment of neurons gives rise to a neurotoxic response and an associated upregulation of dickkopf 1 (DKK1), an antagonist of canonical Wnt signaling (Killick et al., 2014), which leads to an upregulation of GSK-3β, increased tau phosphorylation (Caricasole et al., 2004) and synapse loss (Purro et al., 2012). CLU knockdown in cultures of rat primary cortical neurons prevents the Aβ-driven increase in DKK1 expression levels and protects cells from Aβ-induced neurotoxicity (Killick et al., 2014). Furthermore, treatment of cells with Aβ increases intracellular levels of clusterin while decreasing release of extracellular, secreted clusterin, similarly to what has also been described in astrocytes (Nuutinen et al., 2007). These changes took place within 30 min and in the absence of any change in CLU mRNA expression (Killick et al., 2014), suggesting that changes in clusterin are post-transcriptional and primarily result from changes in clusterin secretion. Further characterization of this pathway led to the postulation of a switch in Wnt signaling to the non-canonical Wnt-PCP-JNK pathway, and to the subsequent activation of downstream transcription factors upregulated by both Aβ and DKK1 – an upregulation prevented by CLU-knockdown. When individually silenced, these genes protected cultures from Aβ-induced neuronal cell death (EGR1 and KLF10) and/or restored phosphorylated tau (p-tau) down to basal unstimulated levels (EGR1 and NAB2) (Killick et al., 2014). Moreover, a very recent study from our lab performed in human induced pluripotent stem cell (iPSC)-derived neurons gives further support to clusterin mediating Aβ toxicity (Robbins et al., 2018), showing neurite length after Aβ-insult is preserved in CLU-KO cells. Interestingly, previous reports had demonstrated co-localization of clusterin and Aβ plaques surrounded by p-tau-positive dystrophic neurites and with p-tau deposits within the neurites in the temporal cortex of AD patients (Martin-Rehrmann et al., 2005). Furthermore, in the AD entorhinal cortex, clusterin co-localized with neurofibrillary tangles (NFTs), and neurons containing NFTs showed increased expression of CLU (Dunckley et al., 2006). Both mouse primary cortical neurons treated with clusterin and rats intracerebrally injected with clusterin showed an increase in tau and p-tau levels (Martin-Rehrmann et al., 2005). These studies are in disagreement with initial observations describing an increased proportion of NFT-free neurons expressing CLU in AD entorhinal, temporal, and frontal cortices, and clusterin localization in NFT-containing neurons was only rarely observed (Giannakopoulos et al., 1998). Further exploration of the effectors responsible for this neuroprotection is urgently needed, as these hold promise for developing new AD therapeutics.
Clusterin in Immunomodulation
Changes in the immunological mechanisms in the brain are considered a key component of AD pathogenesis, whereby proteotoxic aggregates trigger an innate immune response that exacerbates disease progression (Heneka et al., 2015). Moreover, recent GWAS have identified several immune genes as risk factors for AD, with a particular enrichment in complement components, highlighting even further the relevance of the innate immune system in AD pathogenesis (Lambert et al., 2009, 2013; Seshadri et al., 2010; Hollingworth et al., 2011; Guerreiro et al., 2013; Sims et al., 2017). The complement system (CS) is part of the innate immune system and recognizes a variety of potentially harmful elements ranging from invading microorganisms to toxic protein aggregates such as amyloid deposits. Through an activation cascade that includes over 30 factors operating in a network of three integrated pathways (classical, alternative, and lectin), the CS is involved in neuro-inflammatory signaling and culminates in assembly of the membrane attack complex (MAC), a multi-component structure that generates pores in the membrane of the targeted cells leading to lysis (reviewed in Veerhuis et al., 2011). The CS is overactive in AD brain, where components of the MAC and several complement factors (C1q, C4, C3) are found not only in association with amyloid aggregates but also with tau inclusions and damaged neurons (McGeer et al., 1989; Shen et al., 2001, 2013). Within its immunomodulatory functions, clusterin is best known for regulating the CS. Clusterin was found to regulate the formation of the MAC, specifically as an inhibitor of C5b-6, the first step in MAC formation (Jenne and Tschopp, 1989; Murphy et al., 1989). Soon after, immunohistochemistry data showed similarities in the staining pattern of clusterin and MAC in brain tissue, both of which localized in dystrophic neurites and neuropil threads in AD but not in control brains, suggesting a potential increase in clusterin levels as a protective response to MAC formation (McGeer et al., 1992). Later on, it was argued that CS-inhibition is not possible at the physiological concentrations at which clusterin is found (Hochgrebe et al., 1999), but additional studies have shown more examples of local clusterin up-regulation leading to CS-inhibition (Urbich et al., 2000; Hallström et al., 2015). Nevertheless, a mechanistic relationship of CS-inhibition by clusterin in the context of AD remains to be investigated. Links between clusterin and other inflammatory processes have been studied, including modulation of NF-κβ signaling (Santilli et al., 2003) and multiple cytokines including TGF-β (Morgan et al., 1995), TNF-α (Shim et al., 2012), and IL-2 (Sonn et al., 2010). In a study with rat microglia, clusterin treatment induced cell morphological activation both in vitro and in vivo (Xie Z. et al., 2005); additionally, clusterin-activated microglia secreted more reactive nitrogen intermediates and TNF-α, and boosted neurotoxicity when co-cultured with rat primary cortical neurons. The inflammatory component of AD is an area under intense study, and it is clear that glial responses to protein aggregation contribute to AD pathogenesis (Heneka et al., 2015); however, the potential neuro-inflammation regulatory role of clusterin in AD has not been studied in depth. Given the multiple links that have been established between clusterin and several immunomodulatory actors, clusterin could offer some therapeutic potential as a mediator of the boosted immune response observed in the AD brain.
Clusterin and Copper Homeostasis
Dysregulation of copper homeostasis is a known pathophysiological event occurring in AD (reviewed in Greenough et al., 2016). Copper has a role in limiting the amyloidogenic processing of APP (Borchardt et al., 1999), and accumulation of copper in amyloid deposits, as well as brain copper deficiencies, have been described in AD (Deibel et al., 1996; Lovell et al., 1998). Copper-transporting P-type ATPases (copper-ATPases) ATP7A and ATP7B regulate brain copper transport so that levels are sufficient for copper-associated proteins but also ensuring that toxic copper excess is removed (Telianidis et al., 2013). Experiments performed in the context of Menkes and Wilson diseases, in which mutations in ATP7A and ATP7B lead to copper deficiency and copper toxicity disorders, respectively, showed that clusterin participates in the degradation of ATP7A and ATP7B (Materia et al., 2011) via the lysosomal pathway (Materia et al., 2012), an observation that tallies with the reported function of intracellular clusterin in facilitating autophagy (Zhang F. et al., 2014). Interaction between clusterin and the copper-ATPases increases under conditions of oxidative stress and by mutations in ATP7B, suggesting that oxidative stress caused by a dysregulation of copper levels might be driving clusterin-associated degradation of ATP7A and ATP7B (Materia et al., 2011). Interestingly, two SNPs in high linkage disequilibrium within ATP7B (rs1061472 and rs732774) have been linked with increased risk of AD (Bucossi et al., 2012), being the haplotype located in the ATP7B regions encoding for functionally important transmembrane and transduction domains (Squitti et al., 2013). Clearly further investigation is needed to fully unravel the potential role of CLU in copper dyshomeostasis observed in AD, but nevertheless these studies do point to an alternative pathway that could be used as a novel therapeutic target.
CLU Genetics and Alzheimer’s Disease
Mutations in several genes have been identified to cause familial AD, but these are rare. Most cases of AD are sporadic and arise due to complex interactions between the environment and risk genes. Heritability of LOAD is estimated to be between 60 and 80%, and the exact contribution that genetics play in LOAD is still unclear (Gatz et al., 2006). Extensive genetic studies have identified genes that alter AD risk and numerous SNPs have also been identified associated with these genes (Bertram et al., 2007; Bertram and Tanzi, 2009; Harold et al., 2009; Lambert et al., 2009, 2013; Seshadri et al., 2010).
GWAS and CLU
Repeatedly the APOE genotype has been identified as the greatest common genetic risk factor for LOAD and until 2009 was the only one collectively recognized by the AD community (Corder et al., 1993; Saunders et al., 1993). However, APOE genotype only accounts for an estimated 27% of LOAD heritability (Gatz et al., 2006; Lambert et al., 2013) suggesting other genes must also contribute to LOAD risk. GWAS have now identified several genes that alter LOAD risk including, among others, bridging integrator 1 (BIN1) and CLU, which are the second and third most common genetic risk factors after APOE1. Two large GWAS independently identified a number of SNPs located at the CLU locus associated with altered AD risk (Figure 3). Lambert et al. (2009) identified 36 SNPs in CLU in control individuals, and 3 (rs11136000, rs93318888, and rs2279590) were associated with altered AD risk. Additionally, these SNPs were observed in a linkage disequilibrium (LD) block, which only encompassed CLU (Lambert et al., 2009). Harold et al. (2009) similarly observed a significant association between CLU SNP rs11136000 and AD, and this was further replicated in another GWAS (Seshadri et al., 2010) and in smaller Caucasian and European cohorts (Carrasquillo et al., 2010; Naj et al., 2011; Shuai et al., 2015; Zhang S. et al., 2015, 2016; Zhu et al., 2017). Rs11136000 was considered to be the main CLU SNP that altered AD risk (Harold et al., 2009; Lambert et al., 2009). However, replication in Asian cohorts has been less consistent (Yu et al., 2010; Ma et al., 2011; Lin et al., 2012; Ohara et al., 2012; Chung et al., 2013; Miyashita et al., 2013; Liu et al., 2014; Du et al., 2016), suggesting other SNPs contribute to AD risk in Asian cohorts. Similarly, less consistent associations have been observed in Turkish populations (Alaylloglu et al., 2016).
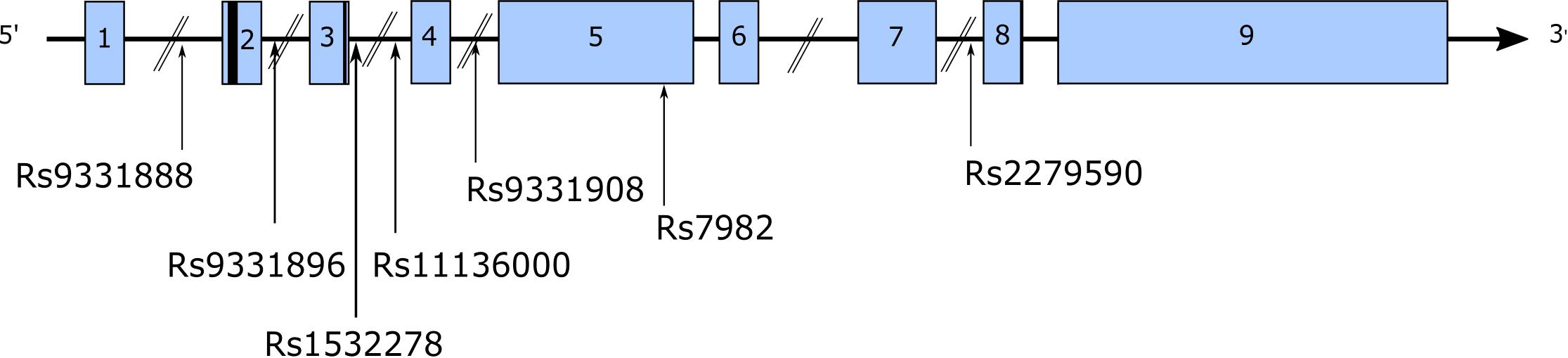
Figure 3. A multitude of SNPs have been identified in CLU, both intronic and exonic. The figure highlights a number of these SNPs identified in AD GWAS.
Rs1136000
Located in intron 3 of CLU, rs11136000 was considered the major AD risk-altering SNP in CLU (Harold et al., 2009; Lambert et al., 2009). The C allele confers an increased risk of 1.16-fold to AD (Bertram et al., 2007) and is carried by 88% of Caucasians (Lambert et al., 2009). In addition to its association with AD, rs11136000 is associated with both Mild Cognitive Impairment (MCI) (Cai et al., 2016) and the progression from MCI to AD (Carrasquillo et al., 2015). Presymptomatic C carrier individuals display increased cognitive decline (Thambisetty et al., 2013), and both AD and the non-demented elderly possessing the C allele show poorer memory scores (Pedraza et al., 2014). In comparison, the minor T allele is thought to confer protection to AD (Lin et al., 2012) and is associated with improved cognitive function in the elderly (Mengel-From and Christensen, 2011). Memory function appears to be altered by rs11136000 allelic expression, as the C allele is associated with poorer neural efficiency and increased limbic and memory area activation during working memory tasks (Lancaster et al., 2015). Although Erk et al. (2011) observed no influence of rs11136000 genotype on brain activity, they observed decoupling of the dorsolateral prefrontal cortex (DLPFC) and the hippocampus during episodic memory recall and the T allele was shown to be associated with reduced hippocampal activity during working memory tasks in healthy controls (Lancaster et al., 2011), although one study observed an increased rate of cognitive decline in individuals with the T allele of rs11136000 (Sweet et al., 2013). Imaging studies are increasingly used to identify potential correlates between genotype and structural and functional changes in the brain. Green et al. (2014) observed an additive effect of CLU genotype and APOE genotype on brain activity, observing a reduction in brain activity in the young possessing rs11136000-C and APOE E4 alleles during tasks requiring executive attention. However, results are not consistent as Biffi et al. (2010) failed to find associations between CLU rs11136000 genotype and a number of neural measures, which may be due to a small sample size. Correlations between rs11136000 and brain atrophy have been observed (Thambisetty et al., 2012; Haight et al., 2018). Even in non-demented elderly individuals, the C allele appears to influence brain structure; Qiu et al. (2016) observed altered fractional anisotropy (FA) in the left external capsule that was correlated with cognitive scores. Changes in brain structure are observed in the young too, since a reduction in white matter integrity in several areas including the corpus callosum (Braskie et al., 2011), subtle reductions in gray matter volume of the right hippocampal formation (Lancaster et al., 2015), and a bilateral increase in entorhinal cortex volume in young, healthy individuals possessing the C allele have been observed (DiBattista et al., 2014). These studies suggest brain changes occur early in life and may predispose individuals to AD at a later stage.
A functional variant of CLU in AD has not yet been identified (Bettens et al., 2010) and much effort has been placed on examining the role of intronic variants on the regulation of genes related to AD (Hardy and Singleton, 2009; Manolio et al., 2009). Research suggests that CLU SNPs may influence CLU mRNA (Szymanski et al., 2011) and protein expression (Xing et al., 2012; Cai et al., 2016; Tan et al., 2016). However, results from different studies are inconsistent. MCI patients show increased plasma clusterin in the presence of rs11136000-C allele, which negatively correlates with cognitive function scores (Cai et al., 2016). Schurmann et al. (2011) observed a reduction in clusterin plasma in a dose-dependent manner with the rs11136000 C risk allele; however, this was not observed to be significantly different between healthy individuals and AD patients. Similarly, although Mullan (2013) observed higher plasma clusterin in individuals possessing the rs11136000, TT genotype, this was observed in both AD, MCI patients and healthy controls. Ling et al. (2012) described a preferential effect of the T allele of rs11136000 on a single CLU mRNA transcript and commented that the protective T allele of rs11136000 is associated with an increased expression of one CLU transcript, which may reflect higher sCLU protein levels throughout life that may provide protection.
Other GWAS SNPs
Other CLU SNPs associated with AD risk include: rs9331888 (Lambert et al., 2009; Yu et al., 2010; Gu et al., 2011), rs2279590 (Lambert et al., 2009; Schjeide et al., 2011; Chen et al., 2012; Miyashita et al., 2013), rs7982 (Harold et al., 2009; Jun G. et al., 2011), rs9331908 (Bettens et al., 2012), and rs1532278 (Bettens et al., 2012).
Rs9331888
Rs9331888 was originally identified by Lambert et al. (2009), as an exon 1-located SNP and is associated with reduced baseline left hippocampal volume in a subset of AD patients (Tan et al., 2016). Possession of the G risk allele of rs9331888 reduces blood clusterin mRNA and protein in controls and AD patients, but this reduction is much greater in controls (Xing et al., 2012). Conversely, Szymanski et al. (2011) examined the influence of the risk G allele on the expression of CLU mRNA transcripts described at the time (NM_001831, NM_20339, and NM_001171138) and observed an increase in the relative abundance of NM_203339 in the temporal cortex of AD patients, suggesting this SNP is regulating alternative splicing of CLU. At the time, this transcript was suggested to be translated to form sCLU, but as discussed previously, NCBI annotations have been updated2 (accessed August 28th 2018), and it now appears that rs9331888 is located within an intron of the main NM_001831.3 transcript and not in exon 1 of NM_203339 as initially believed. This questions the validity of the observation of rs9331888 as a functional variant by influencing alternative splicing of CLU.
Rs1532278
Rs1532278 is located in intron 3, like rs11136000, and sits in a NKX2-5 binding motif and affects the binding of NANOG, TAF1, USF1, MAX, USF2, and GATA2. Rosenthal et al. (2014) used the RegulomeDB database to identify AD GWAS SNPs possessing regulatory functions. Out of the 34 SNPs identified in 18 genes previously linked to AD including CLU, BIN1, PICALM, and CD2AP, only three SNPs showed evidence of regulatory function, one of which was located in CLU, rs1532278. Roussotte et al. (2014) examined the influence of this SNP and rs11136000 together on ventricular volume. After both a 1-year and 2-year follow-up period, homozygous individuals for the risk alleles showed greater ventricular expansion. This was not influenced by APOE E4 genotype and regardless of dementia status. Both SNPs were also associated with longitudinal changes in ventricular volume/expansion (Roussotte et al., 2014).
Rs2279590
Rs2279590, located in intron 7, is positioned in an active regulatory region characterized by the presence of H3K27Ac, increased DNase hypersensitivity, and an eQTL for CLU (Padhy et al., 2014). The risk allele of this SNP differentially regulates CLU expression (Padhy et al., 2014). Enhancer elements flank this SNP and appear to regulate CLU expression, while deletion of these elements reduces CLU expression (Padhy et al., 2017). Additionally, the non-risk allele at this SNP site forms a transcription factor binding site for HSF-1, which is not present with the risk G allele (Padhy et al., 2017). The functional relevance of a loss of binding site for HSF-1 in the risk allele must be explored. As we have discussed, CLU expression is upregulated by a variety of signals including heat shock-induced stress. If the upregulation of CLU is protective, the loss of this binding site for HSF-1 may have a functional effect that may contribute to increased AD risk.
Rs9331896
Rs9331896, also an intronic SNP, was identified by Lambert et al. (2013) as the most significantly associated CLU SNP with AD risk and is in LD with numerous other CLU SNPs including rs11136000, rs9331888 and rs2279750 (Ward and Kellis, 2012; Lambert et al., 2013). Additionally, like other CLU SNPs, rs9331896 is significantly associated with altered CLU expression. Karch and Goate (2015) reported that this SNP is located in an eQTL for CLU and is associated with CLU expression in the white matter of the hippocampus, temporal cortex, and occipital cortex.
To date, rs11136000 is the most studied SNP found in CLU and although it was once considered to be the most influential CLU SNP affecting AD risk in Caucasian populations, it is now ranked as the second, after rs9331896. A range of CLU SNPs have been identified through GWAS, but little is known about their contribution to AD. Despite this, SNPs including rs2279590, rs9331888 and rs1532278 have been observed to have potential roles in regulating gene expression. Interestingly, most of the significant SNPs appear to exist in a linkage disequilibrium block, including rs11136000, rs9331896, and rs2279590, and may together influence risk that is not achieved when acting alone. The functional influence of CLU SNPs is unclear but several SNPs are observed to influence CLU mRNA expression and the levels of plasma clusterin. Effects are observed both in AD patients and in the young and healthy, suggesting a mechanism by which vulnerability may arise decades prior to disease onset. Although there is some conflicting evidence, replication of GWAS and patient-control studies provides strong evidence that CLU variants influence AD risk, and this is independent of APOE E4 status.
Relationships to Other AD Genes
The consensus is that GWAS SNPs and mutations have a significant but modest effect on AD risk individually. Even the APOE E4 genotype only accounts for a portion of LOAD heritability (Gatz et al., 2006), and complex genetic and environmental interactions are likely to contribute to AD development. As previously discussed, SNPs within CLU have been shown to interact with each other to influence hippocampal volume (Roussotte et al., 2014), but relationships among different AD GWAS genes and their SNPs are mostly unexplored. Identification of genotype patterns spanning multiple gene loci may be more useful than focusing on one single gene locus. Previously, studies have looked at PICALM and CLU and their individual and combined influence on brain atrophy and degeneration (Biffi et al., 2010; Erk et al., 2011; Furney et al., 2011; Melville et al., 2012). Barral et al. (2012) identified genotype patterns in PICALM and CLU that are associated with altered episodic memory performance. No SNP, when tested individually, was shown to be associated with episodic memory performance but different genotype combinations at PICALM (rs3851179), CLU (rs11136000) and APOE (E3 vs. E4) were shown to improve or worsen performance. PICALM-GG (risk) and CLU-TT (protective) had the strongest effect on memory performance, which was worsened further in the presence of APOE E4. PICALM-AG and CLU-CC showed a trend for improvement in memory which only reached significance when APOE E3 was present (Barral et al., 2012), highlighting that different genotype combinations at three AD genes produce different effects on a measurable phenotype that is AD-relevant. Interestingly, the genotype that is associated with poor memory includes a risk allele for PICALM but a protective allele for CLU (Lin et al., 2012). These observations suggest that when isolated, a SNP may be protective, but may show different effects on phenotypes in the presence of other SNPs and contribute to disease vulnerability. Similarly, Yang et al. (2016) observed that when PICALM rs3581179 (AA) protective genotype was present alongside risk genotype at CLU rs11136000 (CC) there was a detrimental effect to memory performance, which was not present when the CLU protective genotype was present (TT). It is therefore simply not enough to describe a SNP as protective or risk, without considering which other SNPs are present at different loci.
Hippocampal connectivity appears altered by both PICALM and CLU genotypes (Zhang P. et al., 2015). Zhang P. et al. (2015) observed in young, healthy Chinese Han individuals that PICALM weakened hippocampal connectivity (measured as resting state functional brain connectivity) while risk variants in CLU had a strengthening effect consistent with the reports of others (Lancaster et al., 2011). This may explain in part why PICALM risk genotype is associated with earlier cognitive decline (Sweet et al., 2013) and CLU risk genotype is associated with increased rate of decline (Sweet et al., 2013; Thambisetty et al., 2013). This interaction between PICALM and CLU genotypes is specific to hippocampal connectivity, as no influence has been observed on hippocampal volume (Bralten et al., 2011; Zhang P. et al., 2015) or pathological burden (Chibnik et al., 2011).
As well as genetic interactions between CLU and other AD-relevant genes, biological interactions have also been observed. Clusterin and APOE are both members of the apolipoprotein family of proteins, binding to lipids and cholesterol to promote their transport and processing, an important part of their homeostasis (Mahley, 1988; De Silva et al., 1990). Thus, it is not surprising that many comparisons have been made between APOE and CLU (Kounnas et al., 1995; Koch et al., 2001). The exact mechanisms by which these genes influence AD risk are unclear. It is also unclear whether they have a shared mechanism or interact to influence risk. For example, Green et al. (2014) observed that CLU variants influence brain activity in an additive manner in the presence of APOE E4 genotype, resulting in a reduction of temporal lobe brain activity in young, healthy individuals. APOE genotype is the key genetic risk factor influencing LOAD risk. There are three identified alleles of APOE (E2, E3, and E4) and while the E4 allele increases AD risk, the E2 allele reduces it. Between the alleles there are two isoform-specific amino acid differences and therefore these variants are described as missense mutations. These differences have direct effects on APOE protein structure and function and can be described as functional variants. In comparison, common variants in CLU are intronic SNPs in most cases, and therefore, unlikely to have an influence on protein structure or function. Despite their similarities, it is unlikely that the mechanism by which individual variants influence AD risk will be identical. Association signals observed between CLU and AD (Harold et al., 2009; Lambert et al., 2009) are unlikely to be due to common coding variants in CLU (Guerreiro et al., 2010) and large expression quantitative trait locus (eQTLs) are not found in CLU (Guerreiro et al., 2010; Holton et al., 2013). Some non-synonymous, rare coding mutations have been identified in AD patients, which were found clustered in exons 5–8 of the clusterin β-chain (Bettens et al., 2012). These mutations were observed to influence the secretory trafficking of clusterin and the subsequent secretion from cells in culture (Bettens et al., 2015). One such mutation (P.I03030Nfsx13), a single nucleotide insertion after amino acid residue 315, is predicted to introduce a premature stop codon resulting in total ER retention of clusterin and a loss of sCLU in HEK cells (Bettens et al., 2015). Additionally, rare mutations are observed to increase the generation of intracellular clusterin protein in cells (Zhou et al., 2014). This may have the effect of reducing the protective properties conferred by sCLU and contribute cellular vulnerability to apoptotic stress and protein aggregation.
Recently, both APOE (Atagi et al., 2015) and clusterin have been identified as triggering receptor expressed on myeloid cells (TREM2)-binding ligands and facilitate microglial uptake of Aβ (Yeh et al., 2016). TREM2 is a receptor selectively expressed by microglia in the adult CNS (Klesney-Tait et al., 2006; Zhang Y. et al., 2014) and also is an AD risk gene (Guerreiro et al., 2012; Jonsson et al., 2012). The interaction of APOE, clusterin, and TREM2 is impaired by TREM2 AD mutations and TREM2 KO (Yeh et al., 2016), including the loss-of-function R47H mutation (Wang et al., 2015), and the lipidation of Aβ with APOE and CLU facilitates uptake by microglia, which is impaired in TREM2 KO microglia (Yeh et al., 2016). There has been no indication as to whether CLU SNPs or mutations influence this relationship. Identification of a variant that would influence this relationship would be a strong functional candidate in CLU, since impairments in this would lead to a reduction of Aβ uptake and an increased probability of aggregation. Also recently, plexin A4 (PLXNA4) was identified as a novel receptor for clusterin in the adult CNS, and SNPs observed in this gene were linked to altered CSF clusterin levels (Kang et al., 2015). PLXNA4 had previously been found as an AD GWAS hit (Jun et al., 2014), and some of its genetic variants have been associated with CSF Aβ42 levels (Han et al., 2018).
After APOE, Bridging Integrator 1 or BIN1 is the second most common genetic risk factor for LOAD. Alternative splicing produces several isoforms with differing tissue expression and function (Wigge et al., 1997; Pineda-Lucena et al., 2005), and a number of neuronal specific BIN1 isoforms have been identified, which are thought to be involved in endocytosis and clathrin interactions (McMahon et al., 1997; Ramjaun and McPherson, 1998). BIN1 interacts with tau protein and its main influence on AD risk is in the modulation of tau pathology (Chapuis et al., 2013). Overexpression of intracellular clusterin and tau in cell culture results in an interaction between intracellular clusterin and tau, and between intracellular clusterin and BIN1 (Zhou et al., 2014). This interaction was deemed to be specific between intracellular clusterin and neuronal isoforms of BIN1, and is inhibited in the presence of previously identified rare AD mutations (Bettens et al., 2012). A frameshift mutation resulted in a lack of the C terminus coiled-coil motif, which is thought to be essential for the interaction of intracellular clusterin and BIN1 (Zhou et al., 2014). The exact physiological importance of the biological interactions between BIN1 and clusterin is unknown, as is the relevance of the interaction between BIN1, clusterin and tau together – is this interaction affected by more common variants and is it important in disease? It is also unclear how CLU or BIN1 SNPs or variants may influence this interaction, and although not much evidence for CLU variants and tau interactions has been observed, the rs11136000 SNP has been correlated with tau expression in the CSF of AD patients (Zhou et al., 2014).
Analyses of GWAS AD genes have highlighted a significant overrepresentation of genes found in common disease pathways such as inflammation and endocytosis (Bertram et al., 2007; Jones et al., 2010), resulting in GWAS AD genes being grouped together based on their similar functions (Rosenthal et al., 2014). For example, both APOE and CLU bind lipids and cholesterol to influence their trafficking, while clusterin and CR1 are regulators of the immune response. Clusterin and PICALM possess differing roles in Aβ metabolism: clusterin promotes clearance (Cirrito et al., 2008) while PICALM promotes deposition (Mengel-From and Christensen, 2011); however, at the genetic level their SNPs appear to interact and influence memory (Barral et al., 2012).
In summary, although its functions are still debatable, it is clear that CLU is a key genetic risk factor for LOAD, and a number of intronic and exonic SNPs have been identified, as well as several rare coding mutations. In most cases, little is known about how these SNPs influence mRNA and protein expression of clusterin and no functional variant has yet been identified. However, there is strong evidence to suggest that clusterin protein interacts with a number of other AD-relevant proteins including the main AD pathological proteins such as tau and Aβ, as well as AD risk factors such as APOE and BIN1. Although these interactions have been observed at a biological level, the influence of genetic variants in CLU on these interactions remains elusive. Investigating the influence between clusterin proteins and SNPs with other relevant risk factors and interacting proteins would provide a meaningful strategy to unravel mechanistic roles behind AD and other neurodegenerative disorders.
Conclusion
Clusterin was linked with AD shortly after its identification in 1983. Much of the subsequent focus over the last thirty years has been driven by the discoveries of elevated levels of clusterin in the AD brain, its ability to bind Aβ peptides and the latter discovery of clusterin genetic variants as AD risk factors. Despite these initial leads, insight into clusterin mechanism has been slow to unravel. These early observations have lent weight to the notion that clusterin conferred neuroprotection by acting as an Aβ chaperone leading to clearance of toxic protein fragments. However, it is clear that clusterin is a more complex protein and has both neurodegenerative and neuroprotective functions. Much of this review has concentrated on unraveling this Janus-like character. A large part of the challenge in understanding the role of clusterin in disease arises from the complexity of its biogenesis that results in multiple protein species associated with distinct cellular functions. This inherent complexity is further confounded by frequent lack of clarity and numerous discrepancies in the literature. Nonetheless, a consensus is emerging based on studies of diverse disease states including neurodegeneration, cardiovascular disease and oncology that link secreted clusterin with cytoprotection or anti-apoptosis while intracellular forms mediate apoptosis.
The observation that clusterin levels are increased in several neurodegenerative diseases, all of which share the presence of toxic protein aggregates as a common pathological hallmark, has been used to support the notion that the neuroprotective role of secreted clusterin is likely due to its chaperone function. However, increased clusterin levels are also found in non-proteotoxic neurological diseases, including schizophrenia, Rett syndrome and hypoxia-ischaemia, all of which share at least some underlying pathogenic processes with those found in AD, clearly indicating that the role of secreted clusterin in AD may be more complex than just that of a chaperone. The neuroprotective function of clusterin is evident from studies of CLU KO although constitutive KO in mouse leads to both protective and neurodegenerative effects. Given that removal of the entire gene leads to non-discriminative loss of all clusterin forms at all stages of development, such contradictory results are perhaps not so surprising. More nuanced interrogation of the roles of specific protein species will be revealed by using gene editing to control production of specific CLU variants and cellular trafficking. We have already seen how these approaches have led to identification of an intronic SNP that acts as an enhancer for clusterin and two neighboring genes, all of which are AD risk genes. Many other SNPs are associated with altered plasma clusterin expression and regulatory functions and gene-editing offers an opportunity to exploit these to identify the mechanisms by which variants influence clusterin structure and function, and LOAD risk. More recently, genetic and biological interactions between clusterin and other AD-relevant genes and proteins such as TREM2, APOE and BIN1, among others, have been discovered. The observation of a link in biological interactions between these proteins is itself interesting, although the exact physiological roles are not clearly defined, but what is more interesting is that genetic mutations in these genes that are themselves associated with AD also influence their interactions. Again, the potential of increasingly sophisticated CRISPR-based technologies to manipulate expression of multiple genes in parallel will allow exploration of gene interactions and identification of gene networks that govern the balance of protection and degeneration. We have already seen how CRISPR/Cas9 KO of clusterin in human iPSC-derived neurons leads to refractoriness to extracellular Aβ and future studies extending these approaches and interrogating function in iPSC-derived neurons, astrocytes and microglia will identify both cell autonomous and non-autonomous actions of the different clusterin species.
We have learned much about clusterin in the last 35 years but as is so often the case, the more we know, the more there is to be known. The very complexity of clusterin function may prove to be beneficial in identifying downstream pathways and searching for potential therapeutic targets that distinguish between its pro-apoptotic and anti-apoptotic actions. Although the role of clusterin has often been examined through the lens of individual disease pathologies, clusterin responds to numerous stressors including inflammation, oxidative stress, ER stress, and proteotoxic stress, all of which underlie a range of pathologies including cancer, cardiovascular disease, and neurodegeneration. There is much to be gleaned from interrogating clusterin function in these disparate disease processes that may be of direct relevance to understanding the role of clusterin in AD and which could be useful for exploiting its therapeutic potential. The growing tool box of gene editing tools, the availability of iPSCs from well characterized patient backgrounds, and increased sophistication in building molecular networks from multi-modal ‘omics’ data will all accelerate this search.
Bibliography Search and Selection Criteria
Our bibliography search was performed through Europe PMC, and included the terms (“clusterin” or “apolipoprotein j” or “apoj”) and (“Alzheimer” or “neurodegener∗”) until August 2018. It was further completed with initial key papers in the discovery and characterization of clusterin gene, protein, and function, as well as papers from other disciplines relevant to the mechanisms explored in this review, found through PubMed search and in the bibliography of selected papers. Selection was based on relevance to the scope of this review.
Author Contributions
EF and AD-V wrote the draft manuscript. NB, ER, and SL revised and edited the manuscript.
Funding
This review was supported by Wellcome Trust , The NIHR BRC for Mental Health at the South London and Maudsley NHS Foundation Trust, The Alfonso Martin Escudero Foundation, Alzheimer’s Society/BUPA Foundation, MRC Project (1246016), and AstraZeneca as part of a CASE studentship and the Valat-Jones Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Alaylloǧlu, M., Gezen-Ak, D., Dursun, E., Bilgiç, B., Hanaǧasl, H., Ertan, T., et al. (2016). The association between clusterin and APOE polymorphisms and late-onset Alzheimer disease in a Turkish cohort. J. Geriatr. Psychiatry Neurol. 29, 221–226. doi: 10.1177/0891988716640373
Alzheimer Association (2016). 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12, 459–509. doi: 10.1016/j.jalz.2016.03.001
Ammar, H., and Closset, J. L. (2008). Clusterin activates survival through the phosphatidylinositol 3-kinase/akt pathway. J. Biol. Chem. 283, 12851–12861. doi: 10.1074/jbc.M800403200
Andersen, C. L., Schepeler, T., Thorsen, K., Birkenkamp-Demtröder, K., Mansilla, F., Aaltonen, L. A., et al. (2007). Clusterin expression in normal mucosa and colorectal cancer. Mol. Cell. Proteomics 6, 1039–1048. doi: 10.1074/mcp.M600261-MCP200
Atagi, Y., Liu, C.-C., Painter, M. M., Chen, X.-F., Verbeeck, C., Zheng, H., et al. (2015). Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050. doi: 10.1074/jbc.M115.679043
Athanas, K. M., Mauney, S. L., and Woo, T. U. W. (2015). Increased extracellular clusterin in the prefrontal cortex in schizophrenia. Schizophr. Res. 169, 381–385. doi: 10.1016/j.schres.2015.10.002
Balantinou, E., Trougakos, I. P., Chondrogianni, N., Margaritis, L. H., and Gonos, E. S. (2009). Transcriptional and posttranslational regulation of clusterin by the two main cellular proteolytic pathways. Free Radic. Biol. Med. 46, 1267–1274. doi: 10.1016/j.freeradbiomed.2009.01.025
Baralla, A., Sotgiu, E., Deiana, M., Pasella, S., Pinna, S., Mannu, A., et al. (2015). Plasma Clusterin and lipid profile: a link with aging and cardiovascular diseases in a population with a consistent number of centenarians. PLoS One 10:e0128029. doi: 10.1371/journal.pone.0128029
Barral, S., Bird, T., Goate, A., Farlow, M. R., Diaz-Arrastia, R., Bennett, D. A., et al. (2012). Genotype patterns at PICALM, CR1, BIN1, CLU, and APOE genes are associated with episodic memory. Neurology 78, 1464–1471. doi: 10.1212/WNL.0b013e3182553c48
Beer, T. M., Hotte, S. J., Saad, F., Alekseev, B., Matveev, V., Fléchon, A., et al. (2017). Custirsen (OGX-011) combined with cabazitaxel and prednisone versus cabazitaxel and prednisone alone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel (AFFINITY): a randomised, open-label, international, ph. Lancet Oncol. 18, 1532–1542. doi: 10.1016/S1470-2045(17)30605-8
Bell, R. D., Sagare, A. P., Friedman, A. E., Bedi, G. S., Holtzman, D. M., Deane, R., et al. (2007). Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 27, 909–918. doi: 10.1038/sj.jcbfm.9600419
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23. doi: 10.1038/ng1934
Bertram, L., and Tanzi, R. E. (2009). Genome-wide association studies in Alzheimer’s disease. Hum. Mol. Genet. 18, 137–145. doi: 10.1093/hmg/ddp406
Bertram, L., and Tanzi, R. E. (2010). Alzheimer disease: new light on an old CLU. Nat. Rev. Neurol. 6, 11–13. doi: 10.1038/nrneurol.2009.213.ALZHEIMER
Bettens, K., Brouwers, N., Engelborghs, S., Lambert, J.-C., Rogaeva, E., Vandenberghe, R., et al. (2012). Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol. Neurodegener. 7:3. doi: 10.1186/1750-1326-7-3
Bettens, K., Sleegers, K., and Van Broeckhoven, C. (2010). Current status on alzheimer disease molecular genetics: from past, to present, to future. Hum. Mol. Genet. 19, 4–11. doi: 10.1093/hmg/ddq142
Bettens, K., Vermeulen, S., Van Cauwenberghe, C., Heeman, B., Asselbergh, B., Robberecht, C., et al. (2015). Reduced secreted clusterin as a mechanism for Alzheimer-associated CLU mutations. Mol. Neurodegener. 10:30. doi: 10.1186/s13024-015-0024-9
Biffi, A., Anderson, C., Desikan, R., Sabuncu, M., Cortellini, L., Schmansky, N., et al. (2010). Genetic variation and neuroimaging measures in AD. Arch. Neurol. 67, 677–685. doi: 10.1001/archneurol.2010.108.Genetic
Bitanihirwe, B. K. Y., and Woo, T. U. W. (2011). Oxidative stress in schizophrenia: an integrated approach. Neurosci. Biobehav. Rev. 35, 878–893. doi: 10.1016/j.neubiorev.2010.10.008
Blaschuk, O., Burdzy, K., and Fritz, I. B. (1983). Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 258, 7714–7720.
Borchardt, T., Camakaris, J., Cappai, R., Masters, C. L., Beyreuther, K., and Multhaup, G. (1999). Copper inhibits β-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor-protein secretion. Biochem. J. 344, 461–467. doi: 10.1042/0264-6021:3440461
Bralten, J., Arias-Vásquez, A., Makkinje, R., Veltman, J. A., Brunner, H. G., Fernández, G., et al. (2011). Association of the Alzheimer’s gene SORL1 with hippocampal volume in young, healthy adults. Am. J. Psychiatry 168, 1083–1089. doi: 10.1176/appi.ajp.2011.10101509
Braskie, M., Jahanshad, N., Stein, J., Barysheva, M., McMahon, K., de Zubicaray, G., et al. (2011). Common Alzheimer’s disease risk variant within the CLU gene affects white matter microstructure in young adults. J. Neurosci. 31, 6764–6770. doi: 10.1007/s11103-011-9767-z.Plastid
Bucossi, S., Polimanti, R., Mariani, S., Ventriglia, M., Bonvicini, C., Migliore, S., et al. (2012). Association of K832R and R952K SNPs of Wilson’s disease gene with Alzheimer’s disease. J. Alzheimers Dis. 29, 913–919. doi: 10.3233/JAD-2012-111997
Butterfield, D., Castegna, A., Lauderback, C., and Drake, J. (2002). Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 23, 655–664. doi: 10.1016/S0197-4580(01)00340-2
Cai, R., Han, J., Sun, J., Huang, R., Tian, S., Shen, Y., et al. (2016). Plasma clusterin and the CLU gene rs11136000 variant are associated with mild cognitive impairment in type 2 diabetic patients. Front. Aging Neurosci. 8:179. doi: 10.3389/fnagi.2016.00179
Campisi, J., and Fleshner, M. (2003). Role of extracellular HSP72 in acute stress-induced potentiation of innate immunity in active rats. J. Appl. Physiol. 94, 43–52. doi: 10.1152/japplphysiol.00681.2002
Cao, C., Shinohara, E. T., Li, H., Niermann, K. J., Kim, K. W., Sekhar, K. R., et al. (2005). Clusterin as a therapeutic target for radiation sensitization in a lung cancer model. Int. J. Radiat. Oncol. Biol. Phys. 63, 1228–1236. doi: 10.1016/j.ijrobp.2005.07.956
Caricasole, A., Copani, A., Caraci, F., Aronica, E., Rozemuller, A. J., Caruso, A., et al. (2004). Induction of dickkopf-1, a negative modulator of the wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J. Neurosci. 24, 6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004
Carrasquillo, M. M., Belbin, O., Hunter, T. A., Ma, L., Bisceglio, G. D., Zou, F., et al. (2010). Replication of CLU, CR1, and PICALM associations with alzheimer disease. Arch. Neurol. 67, 961–964. doi: 10.1001/archneurol.2010.147.Replication
Carrasquillo, M. M., Crook, J. E., Pedraza, O., Thomas, C. S., Pankratz, V. S., Allen, M., et al. (2015). Late-onset Alzheimer risk variants in memory decline, incident mild cognitive impairment and Alzheimer disease. Neurobiol. Aging 36, 60–67. doi: 10.1016/j.neurobiolaging.2014.07.042
Cascella, R., Conti, S., Tatini, F., Evangelisti, E., Scartabelli, T., Casamenti, F., et al. (2013). Extracellular chaperones prevent Aβ42-induced toxicity in rat brains. Biochim. Biophys. Acta 1832, 1217–1226. doi: 10.1016/j.bbadis.2013.04.012
Chapuis, J., Hansmannel, F., Gistelinck, M., Mounier, A., Van Cauwenberghe, C., Kolen, K. V., et al. (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. doi: 10.1038/mp.2013.1
Chen, L. H., Kao, P. Y. P., Fan, Y. H., Ho, D. T. Y., Chan, C. S. Y., Yik, P. Y., et al. (2012). Polymorphisms of CR1, CLU and PICALM confer susceptibility of Alzheimer’s disease in a southern Chinese population. Neurobiol. Aging 33, 210.e1–210.e7. doi: 10.1016/j.neurobiolaging.2011.09.016
Chi, K. N., Eisenhauer, E., Fazli, L., Jones, E. C., Goldenberg, S. L., Powers, J., et al. (2005). A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J. Natl. Cancer Inst. 97, 1287–1296. doi: 10.1093/jnci/dji252
Chi, K. N., Higano, C. S., Blumenstein, B., Ferrero, J. M., Reeves, J., Feyerabend, S., et al. (2017). Custirsen in combination with docetaxel and prednisone for patients with metastatic castration-resistant prostate cancer (SYNERGY trial): a phase 3, multicentre, open-label, randomised trial. Lancet Oncol. 18, 473–485. doi: 10.1016/S1470-2045(17)30168-7
Chibnik, L. B., Shulman, J. M., Leurgans, S. E., Schneider, J. A., Wilson, R. S., Tran, D., et al. (2011). CR1 is associated with amyloid plaque burden and age-related Cognitive Decline. Ann. Neurol. 69, 560–569. doi: 10.1002/ana.22277.CR1
Choi-Miura, N. H., and Oda, T. (1996). Relationship between multifunctional protein “clusterin” and Alzheimer disease. Neurobiol. Aging 17, 717–722. doi: 10.1016/S0197-4580(96)00106-6
Chondrogianni, N., Stratford, F. L. L., Trougakos, I. P., Friguet, B., Rivett, A. J., and Gonos, E. S. (2003). Central role of the proteasome in senescence and survival of human fibroblasts. Induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 278, 28026–28037. doi: 10.1074/jbc.M301048200
Chung, S. J., Lee, J.-H., Kim, S.-Y., You, S., Kim, M. J., Lee, J.-Y., et al. (2013). Association of GWAS top hits with late-onset Alzheimer disease in Korean population. Alzheimers Dis. Assoc. Disord. 27, 250–257. doi: 10.1097/WAD.0b013e31826d7281
Cirrito, J., Kang, J., Lee, J., Stewart, F., Verges, D., Silverio, L., et al. (2008). Endocytosis is required for synaptic activity-dependent release of amyloid-β in vivo. Neuron 58, 42–51. doi: 10.1109/TMI.2012.2196707.Separate
Cochrane, D. R., Wang, Z., Muramaki, M., Gleave, M. E., and Nelson, C. C. (2007). Differential regulation of clusterin and its isoforms by androgens in prostate cells. J. Biol. Chem. 282, 2278–2287. doi: 10.1074/jbc.M608162200
Corder, E., Saunders, A., Strittmatter, W., Schmechel, D., Gaskell, P., Small, G., et al. (1993). E type 4 allele gene dose of apolipoprotein and the risk of Alzheimer â€TM s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Criswell, T., Beman, M., Araki, S., Leskov, K., Cataldo, E., Mayo, L. D., et al. (2005). Delayed activation of insulin-like growth factor-1 receptor/Src/ MAPK/Egr-1 signaling regulates clusterin expression, a pro-survival factor. J. Biol. Chem. 280, 14212–14221. doi: 10.1074/jbc.M412569200
Criswell, T., Klokov, D., Beman, M., Lavik, J., and Bootman, D. (2003). Repression of IR-inducible clusterin expression by the p53 tumor suppressor protein. Cancer Biol. Ther. 2, 372–380. doi: 10.4161/cbt.2.4.430
Cuajungco, M. P., Goldstein, L. E., Nunomura, A., Smith, M. A., Lim, J. T., Atwood, C. S., et al. (2000). Evidence that the β-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of Aβ by zinc. J. Biol. Chem. 275, 19439–19442. doi: 10.1074/jbc.C000165200
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., et al. (2007). Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. doi: 10.1074/jbc.M607483200
De Silva, H. V., Stuart, W. D., Duvic, C. R., Wetterau, J. R., Ray, M. J., Ferguson, D. G., et al. (1990). A 70-kDa apolipoprotein designated ApoJ is a marker for subclasses of human plasma high density lipoproteins. J. Biol. Chem. 265, 13240–13247.
Deb, M., Sengupta, D., Rath, S. K., Kar, S., Parbin, S., Shilpi, A., et al. (2015). Clusterin gene is predominantly regulated by histone modifications in human colon cancer and ectopic expression of the nuclear isoform induces cell death. Biochim. Biophys. Acta 1852, 1630–1645. doi: 10.1016/j.bbadis.2015.04.021
Debure, L., Vayssiere, J.-L., Rincheval, V., Loison, F., Le Drean, Y., and Michel, D. (2003). Intracellular clusterin causes juxtanuclear aggregate formation and mitochondrial alteration. J. Cell Sci. 116, 3109–3121. doi: 10.1242/jcs.00619
Deibel, M., Ehmann, W., and Markesbery, W. (1996). Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: possible relation to oxidative stress. J. Neurol. Sci. 143, 137–142. doi: 10.1016/S0022-510X(96)00203-1
DeMattos, R. B., Cirrito, J. R., Parsadanian, M., May, P. C., O’Dell, M. A., Taylor, J. W., et al. (2004). ApoE and clusterin cooperatively suppress Aβ levels and deposition: evidence that ApoE regulates extracellular Aβ metabolism in vivo. Neuron 41, 193–202. doi: 10.1016/S0896-6273(03)00850-X
DeMattos, R. B., O’dell, M. A., Parsadanian, M., Taylor, J. W., Harmony, J. A. K., Bales, K. R., et al. (2002). Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 99, 10843–10848. doi: 10.1073/pnas.162228299
Deming, Y., Xia, J., Cai, Y., Lord, J., Holmans, P., Bertelsen, S., et al. (2016). A potential endophenotype for Alzheimer’s disease: cerebrospinal fluid clusterin. Neurobiol. Aging 37, 208.e1–208.e9. doi: 10.1038/nature12598.DNMT1-interacting
DiBattista, A. M., Stevens, B. W., Rebeck, G. W., and Green, A. E. (2014). Two Alzheimer’ disease risk genes increase entorhinal cortex volume in young adults. Front. Hum. Neurosci. 8:779. doi: 10.3389/fnhum.2014.00779
Du, W., Tan, J., Xu, W., Chen, J., and Wang, L. (2016). Association between clusterin gene polymorphism rs11136000 and late–onset Alzheimer’s disease susceptibility: a review and meta–analysis of case–control studies. Exp. Ther. Med. 12, 2915–2927. doi: 10.3892/etm.2016.3734
Dumont, P., Chainiaux, F., Petropoulou, C., Koch-brandt, C., Gonos, E. S., and Toussaint, O. (2002). Overexpression of apolipoprotein J in human fibroblasts protects against cytotoxicity and premature senescence induced by ethanol and tert-butylhydroperoxide. Cell Stress Chaperones 7, 23–35. doi: 10.1379/1466-1268(2002)007<0023:OOAJIH>2.0.CO;2
Dunckley, T., Beach, T. G., Ramsey, K. E., Grover, A., Mastroeni, D., Walker, D. G., et al. (2006). Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiol. Aging 27, 1359–1371. doi: 10.1016/j.neurobiolaging.2005.08.013
Erk, S., Meyer-Lindenberg, A., Opitz von Boberfeld, C., Esslinger, C., Schnell, K., Kirsch, P., et al. (2011). Hippocampal function in healthy carriers of the CLU Alzheimer’s disease risk variant. J. Neurosci. 31, 18180–18184. doi: 10.1523/JNEUROSCI.4960-11.2011
Filosa, S., Pecorelli, A., D’Espositoc, M., Valacchi, G., and Hajek, J. (2015). Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome. Free Radic. Biol. Med. 88, 81–90. doi: 10.1016/j.freeradbiomed.2015.04.019
Finkel, T., and Holbrook, N. J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. doi: 10.1038/35041687
Flanagan, L., Whyte, L., Chatterjee, N., and Tenniswood, M. (2010). Effects of clusterin over-expression on metastatic progression and therapy in breast cancer. BMC Cancer 10:107. doi: 10.1186/1471-2407-10-107
Foglio, E., Puddighinu, G., Fasanaro, P., D’Arcangelo, D., Perrone, G. A., Mocini, D., et al. (2015). Exosomal clusterin, identified in the pericardial fluid, improves myocardial performance following MI through epicardial activation, enhanced arteriogenesis and reduced apoptosis. Int. J. Cardiol. 197, 333–347. doi: 10.1016/j.ijcard.2015.06.008
Frippiat, C., Dewelle, J., Remacle, J., and Toussaint, O. (2002). Signal transduction in H2O2-induced senescence-like phenotype in human diploid fibroblasts. Free Radic. Biol. Med. 33, 1334–1346. doi: 10.1016/S0891-5849(02)01044-4
Fritz, I. B., and Murphy, B. (1993). Clusterin: Insights into a multifunctional protein. Trends Endocrinol. Metab. 4, 41–45. doi: 10.1016/S1043-2760(05)80013-X
Furney, S., Simmons, A., Breen, G., Pedroso, I., Lunnon, K., Proitsi, P., et al. (2011). Genome-wide association with MRI atrophy measures as a quantitative trait locus for AD. Mol. Psychiatry 16, 1130–1138. doi: 10.7205/MILMED-D-14-00168.Long-chain
Gatz, M., Reynolds, C. A., Fratiglioni, L., Johansson, B., Mortimer, J. A., Berg, S., et al. (2006). Role of genes and environments for explaining Alzheimer’s disease. Arch. Gen. Psychiatry 63, 168–174. doi: 10.1001/archpsyc.63.2.168
Gelissen, I. C., Hochgrebe, T., Wilson, M. R., Easterbrook-Smith, S. B., Jessup, W., Dean, R. T., et al. (1998). Apolipoprotein J (clusterin) induces cholesterol export from macrophage-foam cells: a potential anti-atherogenic function? Biochem. J. 331, 231–237. doi: 10.1016/S0021-9150(97)89866-8
Gemenetzi, M., and Lotery, A. J. (2014). The role of epigenetics in age-related macular degeneration. Eye 28, 1407–1417. doi: 10.1038/eye.2014.225
Ghiso, J., Matsubara, E., Koudinov, A., Choi-Miura, N. H., Tomita, M., Wisniewski, T., et al. (1993). The cerebrospinal-fluid soluble form of Alzheimer’s amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem. J. 293(Pt 1), 27–30. doi: 10.1042/bj2930027
Giannakopoulos, P., Kövari, E., French, L. E., Viard, I., Hof, P. R., and Bouras, C. (1998). Possible neuroprotective role of clusterin in Alzheimer’s disease: a quantitative immunocytochemical study. Acta Neuropathol. 95, 387–394. doi: 10.1007/s004010050815
Gibson, J. H., Slobedman, B., Harikrishnan, K. N., Williamson, S. L., Minchenko, D., El-Osta, A., et al. (2010). Downstream targets of methyl CpG binding protein 2 and their abnormal expression in the frontal cortex of the human Rett syndrome brain. BMC Neurosci. 11:53. doi: 10.1186/1471-2202-11-53
Golden, T. R., Hinerfeld, D. A., and Melov, S. (2002). Oxidative stress and aging: beyond correlation. Aging Cell 1, 117–123. doi: 10.1046/j.1474-9728.2002.00015.x
Green, A., Gray, J., DeYoung, C., Mhyre, T., Padilla, R., Dibattista, A., et al. (2014). A combined effect of two Alzheimer’s risk genes on medial temporal activity during executive attention in young adults. Neuropsychologia 56, 1–8. doi: 10.1016/j.neuropsychologia.2013.12.020.A
Green, D. R., and Kroemer, G. (2004). The pathology of mitochondrial cell death. Science 305, 626–629. doi: 10.1126/science.1099320
Greenough, M. A., Ramírez Munoz, A., Bush, A. I., and Opazo, C. M. (2016). Metallo-pathways to Alzheimer’s disease: lessons from genetic disorders of copper trafficking. Metallomics 8, 831–839. doi: 10.1039/C6MT00095A
Gregory, J. M., Whiten, D. R., Brown, R. A., Barros, T. P., Kumita, J. R., Yerbury, J. J., et al. (2017). Clusterin protects neurons against intracellular proteotoxicity. Acta Neuropathol. Commun. 5:81. doi: 10.1186/s40478-017-0481-1
Grewal, R. P., Morgan, T. E., and Finch, C. E. (1999). C1qB and clusterin mRNA increase in association with neurodegeneration in sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 271, 65–67. doi: 10.1016/S0304-3940(99)00496-6
Gu, H., Wei, X., Chen, S., Kurz, A., Müller, U., Gasser, T., et al. (2011). Association of clusterin gene polymorphisms with late-onset Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 32, 198–201. doi: 10.1159/000331276
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2012). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guerreiro, R. J., Beck, J., Gibbs, J. R., Santana, I., Rossor, M. N., Schott, J. M., et al. (2010). Genetic variability in CLU and its association with Alzheimer’s disease. PLoS One 5:e9510. doi: 10.1371/journal.pone.0009510
Haight, T., Bryan, R. N., Meirelles, O., Tracy, R., Fornage, M., Richard, M., et al. (2018). Associations of plasma clusterin and Alzheimer’s disease-related MRI markers in adults at mid-life: the CARDIA brain MRI sub-study. PLoS One 13:e0190478. doi: 10.1371/journal.pone.0190478
Hakkoum, D., Imhof, A., Vallet, P. G., Boze, H., Moulin, G., Charnay, Y., et al. (2008). Clusterin increases post-ischemic damages in organotypic hippocampal slice cultures. J. Neurochem. 106, 1791–1803. doi: 10.1111/j.1471-4159.2008.05519.x
Hallström, T., Uhde, M., Singh, B., Skerka, C., Riesbeck, K., and Zipfel, P. F. (2015). Pseudomonas aeruginosa uses Dihydrolipoamide dehydrogenase (Lpd) to bind to the human terminal pathway regulators vitronectin and clusterin to inhibit terminal pathway complement attack. PLoS One 10:e0137630. doi: 10.1371/journal.pone.0137630
Han, B. H., DeMattos, R. B., Dugan, L. L., Kim-Han, J. S., Brendza, R. P., Fryer, J. D., et al. (2001). Clusterin contributes to caspase-3-independent brain injury following neonatal hypoxia-ischemia. Nat. Med. 7, 338–343. doi: 10.1038/85487
Han, Q., Sun, Y.-A., Zong, Y., Chen, C., Wang, H.-F., and Tan, L. (2018). Common variants in PLXNA4 and correlation to CSF-related phenotypes in Alzheimer’s disease. Front. Neurosci. 12:946. doi: 10.3389/fnins.2018.00946
Hardy, J., and Singleton, A. (2009). Genomewide association studies and human disease. N. Engl. J. Med. 360, 1759–1768. doi: 10.1056/NEJMra0808700
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093. doi: 10.1038/ng.440
He, L. R., Liu, M. Z., Li, B. K., Rao, H. L., Liao, Y. J., Zhang, L. J., et al. (2009). Clusterin as a predictor for chemoradiotherapy sensitivity and patient survival in esophageal squamous cell carcinoma. Cancer Sci. 100, 2354–2360. doi: 10.1111/j.1349-7006.2009.01349.x
Hellebrekers, D. M., Melotte, V., Viré, E., Langenkamp, E., Molema, G., Fuks, F., et al. (2007). Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 67, 4138–4148. doi: 10.1158/0008-5472.CAN-06-3032
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Herault, Y., Chatelain, G., Brun, G., and Michel, D. (1992). V-src-induced-transcription of the avian clusterin gene. Nucleic Acids Res. 20, 6377–6383. doi: 10.1093/nar/20.23.6377
Hochgrebe, T. T., Humphreys, D., Wilson, M. R., and Easterbrook-Smith, S. B. (1999). A reexamination of the role of clusterin as a complement regulator. Exp. Cell Res. 249, 13–21. doi: 10.1006/excr.1999.4459
Hollander, Z., Lazarova, M., Lam, K., Ignaszewski, A., Oudit, G. Y., Dyck, J. R., et al. (2014). Biomarkers of recovered heart function. Eur. J. Heart Fail. 16, 551–559. doi: 10.1002/ejhf.65
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J. C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43, 429–436. doi: 10.1038/ng.803
Holton, P., Ryten, M., Nalls, M., Trabzuni, D., Weale, M. E., Crehan, H., et al. (2013). Initial assessment of the pathogenic mechanisms of the recently identified Alzheimer risk Loci. Ann. Hum. Genet. 77, 85–105. doi: 10.1111/ahg.12000
Holtzman, D. M., Hee Han, B., He, Y. Y., Kim, G.-M., Choi, J., and Hsu, C. (2001). Reply to “Inhibition of post-ischemic brain injury by clusterin overexpression.” Nat. Med. 7, 978–979. doi: 10.1038/nm0901-978
Honda, K., Casadesus, G., Petersen, R., Perry, G., and Smith, M. (2004). Oxidative stress and redox-active iron in AD. Ann. N. Y. Acad. Sci. 1012, 179–182. doi: 10.1196/annals.1306.015
Hu, N.-W., Klyubin, I., Anwyl, R., and Rowan, M. J. (2009). GluN2B subunit-containing NMDA receptor antagonists prevent A -mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 20504–20509. doi: 10.1073/pnas.0908083106
Huang, X., Cuajungco, M. P., Atwood, C. S., Hartshorn, M. A., Tyndall, J. D. A., Hanson, G. R., et al. (1999). Cu ( II ) potentiation of Alzheimer Aß neurotoxicity. J. Biol. Chem. 274, 37111–37116. doi: 10.1074/jbc.274.52.37111
Humphreys, D., Hochgrebe, T. T., Easterbrook-Smith, S. B., Tenniswood, M. P., and Wilson, M. R. (1997). Effects of clusterin overexpression on TNFα- and TGFβ-mediated death of L929 cells. Biochemistry 36, 15233–15243. doi: 10.1021/bi9703507
Humphreys, D. T., Carver, J. A., Easterbrook-Smith, S. B., and Wilson, M. R. (1999). Clusterin has chaperone-like activity similar to that of small heat shock proteins. J. Biol. Chem. 274, 6875–6881. doi: 10.1074/jbc.274.11.6875
Imhof, A., Charnay, Y., Vallet, P. G., Aronow, B., Kovari, E., French, L. E., et al. (2006). Sustained astrocytic clusterin expression improves remodeling after brain ischemia. Neurobiol. Dis. 22, 274–283. doi: 10.1016/j.nbd.2005.11.009
Ingram, G., Loveless, S., Howell, O. W., Hakobyan, S., Dancey, B., Harris, C. L., et al. (2014). Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol. Commun. 2:53. doi: 10.1186/2051-5960-2-53
Ishikawa, Y., Akasaka, Y., Ishii, T., Komiyama, K., Masuda, S., Asuwa, N., et al. (1998). Distribution and synthesis of apolipoprotein J in the atherosclerotic aorta. Arterioscler. Thromb. Vasc. Biol. 18, 665–672. doi: 10.1161/01.ATV.18.4.665
Itahana, Y., Piens, M., Sumida, T., Fong, S., Muschler, J., and Desprez, P.-Y. (2007). Regulation of clusterin expression in mammary epithelial cells. Exp. Cell Res. 313, 943–951. doi: 10.1016/j.yexcr.2006.12.010
James, R., Hochstrasser, A., Borghini, I., Martin, B., Pometta, D., and Hochstrasser, D. (1991). Characterization of a human high density lipoprotein-associated protein, NA1/NA2. Arterioscler. Thromb. 11, 645–653. doi: 10.1161/01.ATV.11.3.645
Jenne, D. E., and Tschopp, J. (1989). Molecular structure and functional characterization of a human complement cytolysis inhibitor found in blood and seminal plasma: identity to sulfated glycoprotein 2, a constituent of rat testis fluid. Proc. Natl. Acad. Sci. U.S.A. 86, 7123–7127. doi: 10.1073/pnas.86.18.7123
Jin, G., and Howe, P. H. (1997). Regulation of clusterin gene expression by transforming growth factor beta. J. Biol. Chem. 272, 26620–26626. doi: 10.1074/jbc.272.42.26620
Jin, G., and Howe, P. H. (1999). Transforming growth factor beta regulates clusterin gene expression via modulation of transcription factor c-Fos. Eur. J. Biochem. 263, 534–542. doi: 10.1046/j.1432-1327.1999.00533.x
Johnson, S. A., Lampert-Etchells, M., Pasinetti, G. M., Rozovsky, I., and Finch, C. E. (1992). Complement mRNA in the mammalian brain: responses to Alzheimer’s disease and experimental brain lesioning. Neurobiol. Aging 13, 641–648. doi: 10.1016/0197-4580(92)90086-D
Jones, L., Holmans, P. A., Hamshere, M. L., Harold, D., Moskvina, V., Ivanov, D., et al. (2010). Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One 5:e13950. doi: 10.1371/journal.pone.0013950
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2012). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
July, L. V., Akbari, M., Zellweger, T., Jones, E. C., Goldenberg, S. L., and Gleave, M. E. (2002). Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate 50, 179–188. doi: 10.1002/pros.10047
Jun, G., Asai, H., Zeldich, E., Drapeau, E., Chen, C., Chung, J., et al. (2014). PLXNA4 is associated with Alzheimer disease and modulates tau phosphorylation. Ann. Neurol. 76, 379–392. doi: 10.1002/ana.24219
Jun, G., Naj, A., Beecham, G., Wang, L., Buros, J., Gallins, P., et al. (2011). Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer’s disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 67, 1473–1484. doi: 10.1001/archneurol.2010.201
Jun, H. O., Kim, D. H., Lee, S. W., Lee, H. S., Seo, J. H., Kim, J. H., et al. (2011). Clusterin protects H9c2 cardiomyocytes from oxidative stress-induced apoptosis via Akt/GSK-3β signaling pathway. Exp. Mol. Med. 43, 53–61. doi: 10.3858/emm.2011.43.1.006
Kang, S. S., Kurti, A., Wojtas, A., Baker, K. E., Liu, C. C., Kanekiyo, T., et al. (2015). Identification of plexin A4 as a novel clusterin receptor links two Alzheimer’s disease risk genes. Hum. Mol. Genet. 25, 3467–3475. doi: 10.1093/hmg/ddw188
Kang, S. W., Shin, Y. J., Shim, Y. J., Jeong, S. Y., Park, I. S., and Min, B. H. (2005). Clusterin interacts with SCLIP (SCG10-like protein) and promotes neurite outgrowth of PC12 cells. Exp. Cell Res. 309, 305–315. doi: 10.1016/j.yexcr.2005.06.012
Kapron, J. T., Hilliard, G. M., Lakins, J. N., Tenniswood, M. P., West, K. A., Carr, S. A., et al. (1997). Identification and characterization of glycosylation sites in human serum clusterin. Protein Sci. 6, 2120–2133. doi: 10.1002/pro.5560061007
Karch, C. M., and Goate, A. M. (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Kelekar, A., and Thompson, C. B. (1998). Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol. 8, 324–330. doi: 10.1016/S0962-8924(98)01321-X
Killick, R., Ribe, E. M., Al-Shawi, R., Malik, B., Hooper, C., Fernandes, C., et al. (2014). Clusterin regulates beta-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol. Psychiatry 19, 88–98. doi: 10.1038/mp.2012.163
Kim, J. H., Kim, J. H., Jun, H. O., Yu, Y. S., Min, B. H., Park, K. H., et al. (2010). Protective effect of clusterin from oxidative stress-induced apoptosis in human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 51, 561–566. doi: 10.1167/iovs.09-3774
Kim, N., Yoo, J. C., Han, J. Y., Hwang, E. M., Kim, Y. S., Jeong, E. Y., et al. (2012). Human nuclear clusterin mediates apoptosis by interacting with Bcl-XL through C-terminal coiled coil domain. J. Cell. Physiol. 227, 1157–1167. doi: 10.1002/jcp.22836
Kirszbaum, L., Bozas, S. E., and Walker, I. D. (1992). SP-40,40, a protein involved in the control of the complement pathway, possesses a unique array of disulphide bridges. FEBS Lett. 297, 70–76. doi: 10.1016/0014-5793(92)80330-J
Klesney-Tait, J., Turnbull, I. R., and Colonna, M. (2006). The TREM receptor family and signal integration. Nat. Immunol. 7, 1266–1273. doi: 10.1038/ni1411
Koch, S., Donarski, N., Goetze, K., Kreckel, M., Stuerenburg, H. J., Buhmann, C., et al. (2001). Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 42, 1143–1151.
Kounnas, M. Z., Loukinova, E. B., Stefansson, S., Harmony, J. A. K., Brewer, B. H., Strickland, D. K., et al. (1995). Identification of glycoprotein 330 as an endocytic receptor for apolipoprotein J/clusterin. J. Biol. Chem. 270, 13070–13075. doi: 10.1074/jbc.270.22.13070
Kurian, K. M., and McGuone, D. (2012). Retrospective case-control study of apolipoprotein J/Clusterin protein expression in early liveborn neonatal deaths with and without pontosubicular necrosis. Patholog. Res. Int. 2012:479359. doi: 10.1155/2012/479359
Kususda, Y., Miyake, H., Gleave, M. E., and Fujisawa, M. (2012). Clusterin inhibition using OGX-011 synergistically enhances antitumour activity of sorafenib in a human renal cell carcinoma model. Br. J. Cancer 106, 1945–1952. doi: 10.1038/bjc.2012.209
Labadorf, A., Hoss, A. G., Lagomarsino, V., Latourelle, J. C., Hadzi, T. C., Bregu, J., et al. (2015). RNA sequence analysis of human huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. PLoS One 10:e0143563. doi: 10.1371/journal.pone.0143563
Lakins, J., Bennett, S. A. L., Chen, J., Arnold, J. M., Wong, P., Sullivan, J. O., et al. (1998). Clusterin biogenesis is altered during apoptosis in the regressing rat ventral prostate∗. J. Biol. Chem. 273, 27887–27895. doi: 10.1074/jbc.273.43.27887
Lambert, J., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1100. doi: 10.1038/ng.439
Lambert, J., Ibrahim-Verbaas, C., Harold, D., Naj, A., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from A 1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lamoureux, F., Thomas, C., Yin, M. J., Kuruma, H., Beraldi, E., Fazli, L., et al. (2011). Clusterin inhibition using OGX-011 synergistically enhances Hsp90 inhibitor activity by suppressing the heat shock response in castrate-resistant prostate cancer. Cancer Res. 71, 5838–5849. doi: 10.1158/0008-5472.CAN-11-0994
Lancaster, T. M., Baird, A., Wolf, C., Jackson, M. C., Johnston, S. J., Donev, R., et al. (2011). Neural hyperactivation in carriers of the Alzheimer’s risk variant on the clusterin gene. Eur. Neuropsychopharmacol. 21, 880–884. doi: 10.1016/j.euroneuro.2011.02.001
Lancaster, T. M., Brindley, L. M., Tansey, K. E., Sims, R. C., Mantripragada, K., Owen, M. J., et al. (2015). Alzheimer’s disease risk variant in CLU is associated with neural inefficiency in healthy individuals. Alzheimers Dement. 11, 1144–1152. doi: 10.1016/j.jalz.2014.10.012
Laping, N. J., Morgan, T. E., Nichols, N. R., Rozovsky, I., Young-Chan, C. S., Zarow, C., et al. (1994). Transforming growth factor-beta 1 induces neuronal and astrocyte genes: tubulin alpha 1, glial fibrillary acidic protein and clusterin. Neuroscience 58, 563–572. doi: 10.1016/0306-4522(94)90081-7
Laskin, J. J., Nicholas, G., Lee, C., Gitlitz, B., Vincent, M., Cormier, Y., et al. (2012). Phase I/II trial of custirsen (OGX-011), an inhibitor of clusterin, in combination with a gemcitabine and platinum regimen in patients with previously untreated advanced non-small cell lung cancer. J. Thorac. Oncol. 7, 579–586. doi: 10.1097/JTO.0b013e31823f459c
Lee, C., Atanelov, L., Modrek, B., and Xing, Y. (2003). ASAP: the alternative splicing annotation project. Nucleic Acids Res. 31, 101–105. doi: 10.1093/nar/gkg029
Lee, C.-H., Jin, R. J., Kwak, C., Jeong, H., Park, M. S., Lee, N. K., et al. (2002). Suppression of clusterin expression enhanced cisplatin-induced cytotoxicity on renal cell carcinoma cells. Urology 60, 516–520. doi: 10.1016/S0090-4295(02)01806-X
Leger, J. G., Montpetit, M. L., and Tenniswood, M. P. (1987). Characterization and cloning of androgen-repressed mRNAs from rat ventral prostate. Biochem. Biophys. Res. Commun. 147, 196–203. doi: 10.1016/S0006-291X(87)80106-7
Leoncini, S., De Felice, C., Signorini, C., Pecorelli, A., Durand, T., Valacchi, G., et al. (2011). Oxidative stress in Rett syndrome: natural history, genotype, and variants. Redox Rep. 16, 145–153. doi: 10.1179/1351000211Y.0000000004
Leskov, K. S., Araki, S., Lavik, J. P., Gomez, J. A., Gama, V., Gonos, E. S., et al. (2011). CRM1 protein-mediated regulation of nuclear clusterin (nCLU), an ionizing radiation-stimulated, bax-dependent pro-death factor. J. Biol. Chem. 286, 40083–40090. doi: 10.1074/jbc.M111.252957
Leskov, K. S., Criswell, T., Antonio, S., Li, J., Yang, C. R., Kinsella, T. J., et al. (2001). When X-ray-inducible proteins meet DNA double strand break repair. Semin. Radiat. Oncol. 11, 352–372. doi: 10.1053/srao.2001.26912
Leskov, K. S., Klokov, D. Y., Li, J., Kinsella, T. J., and Boothman, D. A. (2003). Synthesis and functional analyses of nuclear clusterin, a cell death protein. J. Biol. Chem. 278, 11590–11600. doi: 10.1074/jbc.M209233200
Li, N., Zoubeidi, A., Beraldi, E., and Gleave, M. E. (2013). GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene 32, 1933–1942. doi: 10.1038/onc.2012.212
Li, X., Ma, Y., Wei, X., Li, Y., Wu, H., Zhuang, J., et al. (2014). Clusterin in Alzheimer’s disease: a player in the biological behavior of amyloid-beta. Neurosci. Bull. 30, 162–168. doi: 10.1007/s12264-013-1391-2
Liao, F. T., Lee, Y. J., Ko, J. L., Tsai, C. C., Tseng, C. J., and Sheu, G. T. (2009). Hepatitis delta virus epigenetically enhances clusterin expression via histone acetylation in human hepatocellular carcinoma cells. J. Gen. Virol. 90, 1124–1134. doi: 10.1099/vir.0.007211-0
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Lin, Y.-L., Chen, S.-Y., Lai, L.-C., Chen, J.-H., Yang, S.-Y., Huang, Y.-L., et al. (2012). Genetic polymorphisms of clusterin gene are associated with a decreased risk of Alzheimer’s disease. Eur. J. Epidemiol. 27, 73–75. doi: 10.1007/s10654-012-9650-5
Ling, I. F., Bhongsatiern, J., Simpson, J. F., Fardo, D. W., and Estus, S. (2012). Genetics of clusterin isoform expression and Alzheimer’s disease risk. PLoS One 7:e33923. doi: 10.1371/journal.pone.0033923
Liu, G., Wang, H., Liu, J., Li, J., Li, H., Ma, G., et al. (2014). The CLU gene rs11136000 variant is significantly associated with Alzheimer’s disease in Caucasian and Asian populations. Neuromolecular Med. 16, 52–60. doi: 10.1007/s12017-013-8250-1
Liu, G., Zhang, H., Hao, F., Hao, J., Pan, L., Zhao, Q., et al. (2018). Clusterin reduces cold ischemia-reperfusion injury in heart transplantation through regulation of NF-kB signaling and Bax/Bcl-xL expression. Cell. Physiol. Biochem. 45, 1003–1012. doi: 10.1159/000487295
Liu, L., Lioudyno, M., Tao, R., Eriksson, P., Svensson, M., and Aldskogius, H. (1999). Hereditary absence of complement C5 in adult mice influences Wallerian degeneration, but not retrograde responses, following injury to peripheral nerve. J. Peripher. Nerv. Syst. 4, 123–133.
Liu, X., Meng, L., Li, J., Meng, J., Teng, X., Gu, H., et al. (2015). Secretory clusterin is upregulated in rats with pulmonary arterial hypertension induced by systemic-to-pulmonary shunts and exerts important roles in pulmonary artery smooth muscle cells. Acta Physiol. 213, 505–518. doi: 10.1111/apha.12352
Loison, F., Debure, L., Nizard, P., le Goff, P., Michel, D., and le Dréan, Y. (2006). Up-regulation of the clusterin gene after proteotoxic stress: implication of HSF1-HSF2 heterocomplexes. Biochem. J. 395, 223–231. doi: 10.1042/BJ20051190
Lovell, M., Robertson, J., Teesdale, W., Campbell, J., and Markesbery, W. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158, 47–52. doi: 10.1016/S0022-510X(98)00092-6
Ma, J. F., Liu, L. H., Zhang, Y., Wang, Y., Deng, Y. L., Huang, Y., et al. (2011). Association study of clusterin polymorphism rs11136000 with late onset Alzheimer’s disease in Chinese Han population. Am. J. Alzheimers Dis. Other Demen. 26, 627–630. doi: 10.1177/1533317511432735
Mackness, B., Hunt, R., Durrington, P., and Mackness, M. (1997). Increased immunolocalization of paraoxonase, clusterin, and apolipoprotein A-I in the human artery wall with the progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 17, 1233–1238. doi: 10.1161/01.ATV.17.7.1233
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport. Science 240, 622–630. doi: 10.1126/science.3283935
Manolio, T., Collins, F., Cox, N., Goldstein, D., Hindorff, L., Hunter, D., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Marti, A., Jehn, B., Costello, E., Keon, N., Ke, G., Martin, F., et al. (1994). Protein kinase A and AP-1 (c-Fos/JunD) are induced during apoptosis of mouse mammary epithelial cells. Oncogene 9, 1213–1223.
Martin-Rehrmann, M. D., Hoe, H.-S., Capuani, E. M., and Rebeck, G. W. (2005). Association of apolipoprotein J-positive beta-amyloid plaques with dystrophic neurites in Alzheimer’s disease brain. Neurotox. Res. 7, 231–242. doi: 10.1007/BF03036452
Materia, S., Cater, M. A., Klomp, L. W. J., Mercer, J. F. B., and La Fontaine, S. (2011). Clusterin (Apolipoprotein J), a molecular chaperone that facilitates degradation of the copper-ATPases ATP7A and ATP7B. J. Biol. Chem. 286, 10073–10083. doi: 10.1074/jbc.M110.190546
Materia, S., Cater, M. A., Klomp, L. W. J., Mercer, J. F. B., and La Fontaine, S. (2012). Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J. Biol. Chem. 287, 2485–2499. doi: 10.1074/jbc.M111.302216
Matsubara, E., Soto, C., Governale, S., Frangione, B., and Ghiso, J. (1996). Apolipoprotein J and Alzheimer’s amyloid beta solubility. Biochem. J. 316(Pt 2), 671–679. doi: 10.3143/geriatrics.36.110
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. doi: 10.1038/nature02621
May, P. C., Lampert-Etchells, M., Johnson, S. A., Poirier, J., Masters, J. N., and Finch, C. E. (1990). Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer’s disease and in response to experimental lesions in rat. Neuron 5, 831–839. doi: 10.1016/0896-6273(90)90342-D
May, P. C., Robison, P., Fuson, K., Smalstig, B., Stephenson, D., and Clemens, J. A. (1992). Sulfated glycoprotein-2 expression increases in rodent brain after transient global ischemia. Mol. Brain Res. 15, 33–39. doi: 10.1016/0169-328X(92)90148-5
McGeer, P. L., Akiyama, H., Itagaki, S., and McGeer, E. G. (1989). Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci. Lett. 107, 341–346. doi: 10.1016/0304-3940(89)90843-4
McGeer, P. L., Kawamata, T., and Walker, D. G. (1992). Distribution of clusterin in Alzheimer brain tissue. Brain Res. 579, 337–341. doi: 10.1016/0006-8993(92)90071-G
McLaughlin, L., Zhu, G., Mistry, M., Ley-Ebert, C., Stuart, W. D., Florio, C. J., et al. (2000). Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. J. Clin. Invest. 106, 1105–1113. doi: 10.1172/JCI9037
McMahon, H. T., Wigge, P., and Smith, C. (1997). Clathrin interacts specifically with amphiphysin and is displaced by dynamin. FEBS Lett. 413, 319–322. doi: 10.1016/S0014-5793(97)00928-9
Melville, S., Buros, J., Parrado, A., Vardarajan, B., Logue, M., Shen, L., et al. (2012). Multiple loci influencing hippocampal degeneration identified by genome scan. Ann. Neurol. 72, 65–75. doi: 10.1002/ana.23644
Mengel-From, J., and Christensen, K. (2011). Genetic variations in the CLU and PICALM genes are associated with cognitive function in the oldest old. Neurobiol. Aging 32, 554.e7–554.e11. doi: 10.1016/j.neurobiolaging.2010.07.016
Merino-Zamorano, C., Fernández-de Retana, S., Montañola, A., Batlle, A., Saint-Pol, J., Mysiorek, C., et al. (2016). Modulation of amyloid-β1–40 transport by ApoA1 and ApoJ across an in vitro model of the blood-brain barrier. J. Alzheimers Dis. 53, 677–691. doi: 10.3233/JAD-150976
Michel, D., Chatelain, G., Herault, Y., and Brun, G. (1995). The expression of the avian clusterin gene can be driven by two alternative promoters with distinct regulatory elements. Eur. J. Biochem. 229, 215–223. doi: 10.1111/j.1432-1033.1995.0215l.x
Michel, D., Chatelain, G., North, S., and Brun, G. (1997). Stress-induced transcription of the clusterin/apoJ gene. Biochem. J. 328(Pt 1), 45–50. doi: 10.1042/bj3280045
Miyake, H., Hara, I., and Gleave, M. E. (2005). Antisense oligodeoxynucleotide therapy targeting clusterin gene for prostate cancer: Vancouver experience from discovery to clinic. Int. J. Urol. 12, 785–794. doi: 10.1111/j.1442-2042.2005.01173.x
Miyashita, A., Koike, A., Jun, G., Wang, L. S., Takahashi, S., Matsubara, E., et al. (2013). SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and caucasians. PLoS One 8:e58618. doi: 10.1371/journal.pone.0058618
Miyata, M., Biro, S., Kaieda, H., Eto, H., Orihara, K., Kihara, T., et al. (2015). Muscle cells after vascular injury. Circulation 104, 1407–1413. doi: 10.1161/hc3701.095583
Moldoveanu, T., Follis, A. V., Kriwacki, R. W., and Green, D. R. (2014). Many players in BCL-2 family affairs. Trends Biochem. Sci. 39, 101–111. doi: 10.1016/j.tibs.2013.12.006
Montpetit, M., Lawless, K., and Tenniswood, M. (1986). Androgen-repressed messages in the rat ventral prostate. Prostate 8, 25–36. doi: 10.1002/pros.2990080105
Moretti, R. M., Marelli, M. M., Mai, S., Cariboni, A., Scaltriti, M., Bettuzzi, S., et al. (2007). Clusterin isoforms differentially affect growth and motility of prostate cells: possible implications in prostate tumorigenesis. Cancer Res. 67, 10325–10333. doi: 10.1158/0008-5472.CAN-07-0516
Morgan, T. E., Laping, N. J., Rozovsky, I., Oda, T., Hogan, T. H., Finch, C. E., et al. (1995). Clusterin expression by astrocytes is influenced by transforming growth factor β1 and heterotypic cell interactions. J. Neuroimmunol. 58, 101–110. doi: 10.1016/0165-5728(94)00194-S
Mulder, S. D., Nielsen, H. M., Blankenstein, M. A., Eikelenboom, P., and Veerhuis, R. (2014). Apolipoproteins E and J interfere with amyloid-beta uptake by primary human astrocytes and microglia in vitro. Glia 62, 493–503. doi: 10.1002/glia.22619
Mullan, G. (2013). Plasma clusterin levels and the rs11136000 genotype in individuals with mild cognitive impairment and Alzheimer’s disease. Curr. Alzheimers Res. 10, 973–978. doi: 10.2174/15672050113106660162
Murphy, B. F., Kirszbaum, L., Walker, I. D., and d’Apice, J. F. (1988). Sp-40,40, a newly identified normal human serum protein found in the SC5b-9 complex of complement and in the immune deposits in glomerulonephritis. J. Clin. Invest. 81, 1858–1864. doi: 10.1172/JCI113531
Murphy, B. F., Saunders, J. R., Bryan, M. K. O., Kirszbaum, L., Walker, I. D., and Apice, A. J. F. (1989). SP-40,40 is an inhibitor of C5b-6-initiated haemolysis. Int. Immunol. 1, 551–554. doi: 10.1093/intimm/1.5.551
Naj, A. C., Jun, G., Beecham, G. W., Wang, L., Narayan, B., Buros, J., et al. (2011). Common variants in MS4A4/MS4A6E, CD2uAP, CD33, and EPHA1 are associated with late-onset Alzheimer’s disease Adam. Nat. Genet. 43, 436–441. doi: 10.1038/ng.801
Nakai, K., and Horton, P. (1999). PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 24, 34–35. doi: 10.1016/S0968-0004(98)01336-X
Narayan, P., Holmström, K. M., Kim, D. H., Whitcomb, D. J., Wilson, M. R., St George-Hyslop, P., et al. (2014). Rare individual amyloid-β oligomers act on astrocytes to initiate neuronal damage. Biochemistry 53, 2442–2453. doi: 10.1021/bi401606f
Narayan, P., Orte, A., Clarke, R. W., Bolognesi, B., Hook, S., Ganzinger, K. A., et al. (2012). The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β 1-40 peptide. Nat. Struct. Mol. Biol. 19, 79–84. doi: 10.1038/nsmb.2191
Nielsen, H. M., Mulder, S. D., Beliën, J. A. M., Musters, R. J. P., Eikelenboom, P., and Veerhuis, R. (2010). Astrocytic Aβ1-42 uptake is determined by Aβ-aggregation state and the presence of amyloid-associated proteins. Glia 58, 1235–1246. doi: 10.1002/glia.21004
Nilselid, A. M., Davidsson, P., Nägga, K., Andreasen, N., Fredman, P., and Blennow, K. (2006). Clusterin in cerebrospinal fluid: analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem. Int. 48, 718–728. doi: 10.1016/j.neuint.2005.12.005
Nizard, P., Tetley, S., Le Dréan, Y., Watrin, T., Le Goff, P., Wilson, M. R., et al. (2007). Stress-induced retrotranslocation of clusterin/ApoJ into the cytosol. Traffic 8, 554–565. doi: 10.1111/j.1600-0854.2007.00549.x
Nuutinen, T., Huuskonen, J., Suuronen, T., Ojala, J., Miettinen, R., and Salminen, A. (2007). Amyloid-β 1-42 induced endocytosis and clusterin/apoJ protein accumulation in cultured human astrocytes. Neurochem. Int. 50, 540–547. doi: 10.1016/j.neuint.2006.11.002
Nuutinen, T., Suuronen, T., Kauppinen, A., and Salminen, A. (2009). Clusterin: a forgotten player in Alzheimer’s disease. Brain Res. Rev. 61, 89–104. doi: 10.1016/j.brainresrev.2009.05.007
Nuutinen, T., Suuronen, T., Kauppinen, A., and Salminen, A. (2010). Valproic acid stimulates clusterin expression in human astrocytes: implications for Alzheimer’s disease. Neurosci. Lett. 475, 64–68. doi: 10.1016/j.neulet.2010.03.041
Oda, T., Wals, P., Osterburg, H. H., Johnson, S. A., Pasinetti, G. M., Morgan, T. E., et al. (1995). Clusterin (apoJ) alters the aggregation of amyloid β-peptide (Aβ1-42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp. Neurol. 136, 22–31. doi: 10.1006/exnr.1995.1080
Ohara, T., Ninomiya, T., Hirakawa, Y., Ashikawa, K., Monji, A., Kiyohara, Y., et al. (2012). Association study of susceptibility genes for late-onset Alzheimer’s disease in the Japanese population. Psychiatr. Genet. 22, 290–293. doi: 10.1097/YPG.0b013e3283586215
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Opazo, C., Huang, X., Cherny, R. A., Moir, R. D., Roher, A. E., White, A. R., et al. (2002). Metalloenzyme-like activity of Alzheimer’s disease β-amyloid: cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J. Biol. Chem. 277, 40302–40308. doi: 10.1074/jbc.M206428200
O’Sullivan, J., Whyte, L., Drake, J., and Tenniswood, M. (2003). Alterations in the post-translational modification and intracellular trafficking of clusterin in MCF-7 cells during apoptosis. Cell Death Differ. 10, 914–927. doi: 10.1038/sj.cdd.4401254
Padhy, B., Hayat, B., Nanda, G. G., Mohanty, P. P., and Alone, D. P. (2017). Pseudoexfoliation and Alzheimer’s associated CLU risk variant, rs2279590, lies within an enhancer element and regulates CLU, EPHX2 and PTK2B gene expression. Hum. Mol. Genet. 26, 4519–4529. doi: 10.1093/hmg/ddx329
Padhy, B., Nanda, G. G., Chowdhury, M., Padhi, D., Rao, A., and Alone, D. P. (2014). Role of an extracellular chaperone, clusterin in the pathogenesis of pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Exp. Eye Res. 127, 69–76. doi: 10.1016/j.exer.2014.07.005
Parks, J. K., Smith, T. S., Trimmer, P. A., Bennett, J. P., and Davis Parker, W. J. (2001). Neurotoxic Aβ peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J. Neurochem. 76, 1050–1056. doi: 10.1046/j.1471-4159.2001.00112.x
Pasinetti, G. M., Johnson, S. A., Oda, T., Rozovsky, I., and Finch, C. E. (1994). Clusterin (SGP-2): a multifunctional glycoprotein with regional expression in astrocytes and neurons of the adult rat brain. J. Comp. Neurol. 339, 387–400. doi: 10.1002/cne.903390307
Pedraza, O., Ph, D., Allen, M., Ph, D., Jennette, K., Carrasquillo, M., et al. (2014). Evaluation of memory endophenotypes for association with CLU, CR1 and PICALM variants in African-American and Caucasian subjects. Alzheimers Dement. 10, 205–213. doi: 10.1016/j.jalz.2013.01.016
Peix, L., Evans, I. C., Pearce, D. R., Simpson, J. K., Maher, T. M., and McAnulty, R. J. (2018). Diverse functions of clusterin promote and protect against the development of pulmonary fibrosis. Sci. Rep. 8:1906. doi: 10.1038/s41598-018-20316-1
Pietersen, C. Y., Mauney, S. A., Kim, S. S., Lim, M. P., Rooney, R. J., Goldstein, J. M., et al. (2014a). Molecular profiles of pyramidal neurons in the superior temporal cortex in schizophrenia. J. Neurogenet. 28, 53–69. doi: 10.3109/01677063.2014.882918
Pietersen, C. Y., Mauney, S. A., Kim, S. S., Passeri, E., Lim, M. P., Rooney, R. J., et al. (2014b). Molecular profiles of parvalbumin-immunoreactive neurons in the superior temporal cortex in schizophrenia. J. Neurogenet. 28, 70–85. doi: 10.3109/01677063.2013.878339
Pineda-Lucena, A., Ho, C. S. W., Mao, D. Y. L., Sheng, Y., Laister, R. C., Muhandiram, R., et al. (2005). A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J. Mol. Biol. 351, 182–194. doi: 10.1016/j.jmb.2005.05.046
Poon, S., Easterbrook-Smith, S. B., Rybchyn, M. S., Carver, J. A., and Wilson, M. R. (2000). Clusterin is an ATP - Independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 39, 15953–15960. doi: 10.1021/bi002189x
Praticò, D., Clark, C. M., Liun, F., Lee, V. Y.-M., and Trojanowski, J. Q. (2002). Increase of brain oxidative stress in mild cognitive impairment. Arch. Neurol. 59, 972–976. doi: 10.1001/archneur.59.6.972
Prince, M., Wimo, A., Guerchet, M., Gemma-Claire, A., Wu, Y.-T., and Prina, M. (2015). World Alzheimer Report 2015 - The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends. London: Alzheimer’s Disease International, 84. doi: 10.1111/j.0963-7214.2004.00293.x
Prochnow, H., Gollan, R., Rohne, P., Hassemer, M., Koch-Brandt, C., and Baiersdörfer, M. (2013). Non-secreted clusterin isoforms are translated in rare amounts from distinct human mRNA variants and do not affect bax-mediated apoptosis or the NF-κB signaling pathway. PLoS One 8:e75303. doi: 10.1371/journal.pone.0075303
Pucci, S., Bonanno, E., Pichiorri, F., Angeloni, C., and Spagnoli, L. G. (2004). Modulation of different clusterin isoforms in human colon tumorigenesis. Oncogene 23, 2298–2304. doi: 10.1038/sj.onc.1207404
Purrello, M., Bettuzzi, S., Di Pietro, C., Mirabile, E., Di Blasi, M., Rimini, R., et al. (1991). The gene for SP-40,40, human homolog of rat sulfated glycoprotein 2, rat clusterin, and rat testosterone-repressed prostate message 2, maps to chromosome 8. Genomics 10, 151–156. doi: 10.1016/0888-7543(91)90495-Z
Purro, S. A., Dickins, E. M., and Salinas, P. C. (2012). The secreted wnt antagonist dickkopf-1 is required for amyloid -mediated synaptic loss. J. Neurosci. 32, 3492–3498. doi: 10.1523/JNEUROSCI.4562-11.2012
Qiu, L., He, Y., Tang, H., Zhou, Y., Wang, J., Zhang, W., et al. (2016). Genetically-mediated grey and white matter alteration in normal elderly individuals with the CLU-C allele gene. Curr. Alzheimer Res. 13, 1302–1310. doi: 10.2174/1567205013666160703
Ramjaun, A. R., and McPherson, P. S. (1998). Multiple amphiphysin II splice variants display differential clathrin binding: identification of two distinct clathrin-binding sites. J. Neurochem. 70, 2369–2376. doi: 10.1046/j.1471-4159.1998.70062369.x
Ranney, M. K., Ahmed, I. S. A., Potts, K. R., and Craven, R. J. (2007). Multiple pathways regulating the anti-apoptotic protein clusterin in breast cancer. Biochim. Biophys. Acta 1772, 1103–1111. doi: 10.1016/j.bbadis.2007.06.004
Rauhala, H. E., Porkka, K. P., Saramäki, O. R., Tammela, T. L. J., and Visakorpi, T. (2008). Clusterin is epigenetically regulated in prostate cancer. Int. J. Cancer 123, 1601–1609. doi: 10.1002/ijc.23658
Reddy, K. B., Jin, G., Karode, M. C., Harmony, J. A. K., and Howe, P. H. (1996). Transforming growth factor β (TGFβ)-induced nuclear localization of apolipoprotein J/clusterin in epithelial cells. Biochemistry 35, 6157–6163. doi: 10.1021/bi952981b
Redondo, M., Téllez, T., Roldan, M. J., Serrano, A., García-Aranda, M., Gleave, M. E., et al. (2007). Anti-clusterin treatment of breast cancer cells increases the sensitivities of chemotherapy and tamoxifen and counteracts the inhibitory action of dexamethasone on chemotherapy-induced cytotoxicity. Breast Cancer Res. 9:R86. doi: 10.1186/bcr1835
Rizzi, F., and Bettuzzi, S. (2010). The clusterin paradigm in prostate and breast carcinogenesis. Endocr. Relat. Cancer 17, R1–R17. doi: 10.1677/ERC-09-0140
Rizzi, F., Coletta, M., and Bettuzzi, S. (2009). Clusterin (CLU): from one gene and two transcripts to many proteins. Adv. Cancer Res. 104, 9–23. doi: 10.1016/S0065-230X(09)04002-0
Robbins, J. P., Perfect, L., Ribe, E. M., Maresca, M., Dangla-Valls, A., Foster, E. M., et al. (2018). Clusterin is required for β-amyloid toxicity in human iPSC-derived neurons. Front. Neurosci. 12:504. doi: 10.3389/fnins.2018.00504
Rohne, P., Prochnow, H., and Koch-Brandt, C. (2016). The CLU-files: disentanglement of a mystery. Biomol. Concepts 7, 1–15. doi: 10.1515/bmc-2015-0026
Rohne, P., Prochnow, H., Wolf, S., Renner, B., and Koch-Brandt, C. (2014). The chaperone activity of clusterin is dependent on glycosylation and redox environment. Cell. Physiol. Biochem. 34, 1626–1639. doi: 10.1159/000366365
Rönicke, R., Mikhaylova, M., Rönicke, S., Meinhardt, J., Schröder, U. H., Fändrich, M., et al. (2011). Early neuronal dysfunction by amyloid β oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 32, 2219–2228. doi: 10.1016/j.neurobiolaging.2010.01.011
Rosemblit, N., and Chen, C. C. (1994). Regulators for the rat clusterin gene: DNA methylation and cis-acting regulatory elements methylation. J. Mol. Endrocrinol. 13, 69–76. doi: 10.1677/jme.0.0130069
Rosenthal, S. L., Barmada, M. M., Wang, X., Demirci, F. Y., and Kamboh, M. I. (2014). Connecting the dots: potential of data integration to identify regulatory snps in late-onset Alzheimer’s disease GWAS findings. PLoS One 9:e95152. doi: 10.1371/journal.pone.0095152
Roussotte, F. F., Gutman, B. A., Madsen, S. K., Colby, J. B., and Thompson, P. M. (2014). Combined effects of alzheimer risk variants in the CLU and ApoE genes on ventricular expansion patterns in the elderly. J. Neurosci. 34, 6537–6545. doi: 10.1523/JNEUROSCI.5236-13.2014
Saad, F., Hotte, S., North, S., Eigl, B., Chi, K., Czaykowski, P., et al. (2011). Randomized phase II trial of custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clin. Cancer Res. 17, 5765–5773. doi: 10.1158/1078-0432.CCR-11-0859
Sabatte, J., Faigle, W., Ceballos, A., Morelle, W., Rodríguez Rodrígues, C., Remes Lenicov, F., et al. (2011). Semen clusterin is a novel DC-SIGN ligand. J. Immunol. 187, 5299–5309. doi: 10.4049/jimmunol.1101889
Sansanwal, P., Li, L., and Sarwal, M. (2015). inhibition of intracellular clusterin attenuates cell death in nephropathic cystinosis. J. Am. Soc. Nephrol. 26, 612–625. doi: 10.1681/ASN.2013060577
Santilli, G., Aronow, B. J., and Sala, A. (2003). Essential requirement of apolipoprotein J (clusterin) signaling for IκB expression and regulation of NF-κB activity. J. Biol. Chem. 278, 38214–38219. doi: 10.1074/jbc.C300252200
Sasaki, K., Doh-ura, K., Ironside, J. W., and Iwaki, T. (2002a). Increased clusterin (apolipoprotein J) expression in human and mouse brains infected with transmissible spongiform encephalopathies. Acta Neuropathol. 103, 199–208. doi: 10.1007/s004010100456
Sasaki, K., Doh-ura, K., Wakisaka, Y., and Iwaki, T. (2002b). Clusterin/apolipoprotein J is associated with cortical Lewy bodies: immunohistochemical study in cases with alpha-synucleinopathies. Acta Neuropathol. 104, 225–230. doi: 10.1007/s00401-002-0546-4
Satapathy, S. (2017). Extracellular chaperones in neuronal proteinopathies: protecting and facilitating neuronal function. Cell Commun. Insights 9, 1–13. doi: 10.1177/1179568917717952
Saunders, A. M., Strittmatter, W. J., Schmechel, D., St George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., et al. (1993). Association of apolipoprotein E allele 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1467. doi: 10.1212/WNL.43.8.1467
Saura, J., Petegnief, V., Wu, X., Liang, Y., and Paul, S. M. (2003). Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: opposite effects of lipopolysaccharide. J. Neurochem. 85, 1455–1467. doi: 10.1046/j.1471-4159.2003.01788.x
Scaltriti, M., Bettuzzi, S., Sharrard, R. M., Caporali, A., Caccamo, A. E., and Maitland, N. J. (2004a). Clusterin overexpression in both malignant and nonmalignant prostate epithelial cells induces cell cycle arrest and apoptosis. Br. J. Cancer 91, 1842–1850. doi: 10.1038/sj.bjc.6602193
Scaltriti, M., Santamaria, A., Paciucci, R., and Bettuzzi, S. (2004b). Intracellular clusterin induces G2-M phase arrest and cell death in PC-3 prostate cancer cells1. Cancer Res. 64, 6174–6182. doi: 10.1158/0008-5472.CAN-04-0920
Schepeler, T., Mansilla, F., Christensen, L. L., Ørntoft, T. F., and Andersen, C. L. (2007). Clusterin expression can be modulated by changes in TCF1-mediated Wnt signaling. J. Mol. Signal. 2:6. doi: 10.1186/1750-2187-2-6
Schjeide, B. M. M., Schnack, C., Lambert, J. C., Lill, C. M., Kirchheiner, J., Tumani, H., et al. (2011). The role of clusterin, complement receptor 1, and phosphatidylinositol binding clathrin assembly protein in Alzheimer disease risk and cerebrospinal fluid biomarker levels. Arch. Gen. Psychiatry 68, 207–213. doi: 10.1001/archgenpsychiatry.2010.196
Schurmann, B., Wiese, B., Bickel, H., Weyerer, S., Riedel-Heller, S. G., Pentzek, M., et al. (2011). Association of the Alzheimer’s disease clusterin risk allele with plasma clusterin concentration. J. Alzheimers Dis. 25, 421–424. doi: 10.3233/JAD-2011-110251
Seshadri, S., Fitzpatrick, A. L., Ikram, M. A., DeStefano, A. L., and Breteler, M. M. B. (2010). Genome-wide analysis of genetic loci associated with Alzheimer’s Disease. JAMA 303, 1832–1840. doi: 10.1001/jama.2010.574
Shelat, P. B., Chalimoniuk, M., Wang, J. H., Strosznajder, J. B., Lee, J. C., Sun, A. Y., et al. (2008). Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 106, 45–55. doi: 10.1111/j.1471-4159.2008.05347.x
Shen, Y., Lue, L.-F., Yang, L.-B., Roher, A., Kuo, Y.-M., Strohmeyer, R., et al. (2001). Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 305, 165–168. doi: 10.1016/S0304-3940(01)01842-0
Shen, Y., Yang, L., and Li, R. (2013). What does complement do in Alzheimer’s disease? Old molecules with new insights. Transl. Neurodegener. 2:21. doi: 10.1186/2047-9158-2-21
Shim, Y. J., Kang, B. H., Choi, B. K., Park, I. S., and Min, B. H. (2012). Clusterin induces the secretion of TNF-α and the chemotactic migration of macrophages. Biochem. Biophys. Res. Commun. 422, 200–205. doi: 10.1016/j.bbrc.2012.04.162
Shim, Y.-J., Kang, B.-H., Jeon, H.-S., Park, I.-S., Lee, K.-U., Lee, I.-K., et al. (2011). Clusterin induces matrix metalloproteinase-9 expression via ERK1/2 and PI3K/Akt/NF- B pathways in monocytes/macrophages. J. Leukoc. Biol. 90, 761–769. doi: 10.1189/jlb.0311110
Shiota, M., Zoubeidi, A., Kumano, M., Beraldi, E., Naito, S., Nelson, C. C., et al. (2011). Clusterin is a critical downstream mediator of stress-induced YB-1 transactivation in prostate cancer. Mol. Cancer Res. 9, 1755–1766. doi: 10.1158/1541-7786.MCR-11-0379
Shuai, P., Liu, Y., Lu, W., Liu, Q., Li, T., and Gong, B. (2015). Genetic associations of CLU rs9331888 polymorphism with Alzheimer’s disease: a meta-analysis. Neurosci. Lett. 591, 160–165. doi: 10.1016/j.neulet.2015.02.040
Sidiropoulos, K., Viteri, G., Sevilla, C., Jupe, S., Webber, M., Orlic-milacic, M., et al. (2017). Reactome enhanced pathway visualization. Bioinformatics 33, 3461–3467. doi: 10.1093/bioinformatics/btx441
Sims, R., Van Der Lee, S. J., Naj, A. C., Bellenguez, C., Badarinarayan, N., Jakobsdottir, J., et al. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384. doi: 10.1038/ng.3916
So, A., Sinneman, S., Huntsman, D., Fazli, L., and Gleave, M. (2005). Knockdown of the cytoprotective chaperone, clusterin, chemosensitizes human breast cancer cells both in vitro and in vivo. Mol. Cancer Ther. 4, 1837–1849. doi: 10.1158/1535-7163.MCT-05-0178
Sonn, C. H., Yu, Y.-B., Hong, Y.-J., Shim, Y.-J., Bluestone, J. A., Min, B.-H., et al. (2010). Clusterin synergizes with IL-2 for the expansion and IFN-γ production of natural killer cells. J. Leukoc. Biol. 88, 955–963. doi: 10.1189/jlb.0310157
Squitti, R., Polimanti, R., Bucossi, S., Ventriglia, M., Mariani, S., Manfellotto, D., et al. (2013). Linkage disequilibrium and haplotype analysis of the ATP7B gene in Alzheimer’s disease. Rejuvenation Res. 16, 3–10. doi: 10.1089/rej.2012.1357
Steve Jones, C. E., and Jomary, C. (2002). Molecules in focus. Int. J. Biochem. Cell Biol. 34, 427–431. doi: 10.1016/S1357-2725(01)00155-8
Stewart, E. M., Aquilina, J. A., Easterbrook-smith, S. B., Murphy-durland, D., Jacobsen, C., Moestrup, S., et al. (2007). Effects of glycosylation on the structure and function of the extracellular chaperone clusterin. Biochemistry 46, 1412–1422. doi: 10.1021/bi062082v
Stocker, R. (2004). Role of oxidative modifications in atherosclerosis. Physiol. Rev. 84, 1381–1478. doi: 10.1152/physrev.00047.2003
Strocchi, P., Smith, M. A., Perry, G., Tamagno, E., Danni, O., Pession, A., et al. (2006). Clusterin up-regulation following sub-lethal oxidative stress and lipid peroxidation in human neuroblastoma cells. Neurobiol. Aging 27, 1588–1594. doi: 10.1016/j.neurobiolaging.2005.09.019
Suuronen, T., Nuutinen, T., Ryhänen, T., Kaarniranta, K., and Salminen, A. (2007). Epigenetic regulation of clusterin/apolipoprotein J expression in retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 357, 397–401. doi: 10.1016/j.bbrc.2007.03.135
Sweet, R., Seltman, H., James, E., Lopez, O., Becker, J., Bis, J., et al. (2013). Effect of Alzheimer disease risk genes on trajectories of cognitive function in the cardiovascular health study. Am. J. Psychiatry 169, 954–962. doi: 10.1176/appi.ajp.2012.11121815
Swertfeger, D. K., Witte, D. P., Stuart, W. D., Rockman, H. A., and Harmony, J. A. (1996). Apolipoprotein J/clusterin induction in myocarditis: a localized response gene to myocardial injury. Am. J. Pathol. 148, 1971–1983.
Szymanski, M., Wang, R., Bassett, S. S., and Avramopoulos, D. (2011). Alzheimer’s risk variants in the clusterin gene are associated with alternative splicing. Transl. Psychiatry 1:e18. doi: 10.1038/tp.2011.17
Takase, O., Minto, A. W. M., Puri, T. S., Cunningham, P. N., Jacob, A., Hayashi, M., et al. (2008). Inhibition of NF-kappaB-dependent Bcl-xL expression by clusterin promotes albumin-induced tubular cell apoptosis. Kidney Int. 73, 567–577. doi: 10.1038/sj.ki.5002563
Tan, L., Wang, H.-F., Tan, M.-S., Tan, C.-C., Zhu, X.-C., Miao, D., et al. (2016). Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Sci. Rep. 6:26027. doi: 10.1038/srep26027
Tang, Y., Liu, F., Zheng, C., Sun, S., and Jiang, Y. (2012). Knockdown of clusterin sensitizes pancreatic cancer cells to gemcitabine chemotherapy by ERK1/2 inactivation. J. Exp. Clin. Cancer Res. 31:73. doi: 10.1186/1756-9966-31-73
Tarasoff-Conway, J. M., Carare, R. O., Osorio, R. S., Glodzik, L., Butler, T., Fieremans, E., et al. (2015). Clearance systems in the brain—implications for Alzheimer disease. Nat. Rev. Neurol. 11, 457–470. doi: 10.1038/nrneurol.2015.119
Telianidis, J., Hung, Y. H., Materia, S., and Fontaine, S. L. (2013). Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front. Aging Neurosci. 5:44. doi: 10.3389/fnagi.2013.00044
Thambisetty, M., An, Y., Kinsey, A., Koka, D., Saleem, M., Guntert, A., et al. (2012). Plasma clusterin concentration is associated with longitudinal brain atrophy in mild cognitive impairment. Neuroimage 59, 212–217. doi: 10.1016/j.neuroimage.2011.07.056
Thambisetty, M., Beason-Held, L., An, Y., Kraut, M., Nalls, M., Hernandez, D., et al. (2013). Alzheimer risk variant CLU and brain function during aging. Biol. Psychiary 73, 399–405. doi: 10.1016/j.biopsych.2012.05.026
Thambisetty, M., Simmons, A., Hye, A., Campbell, J., Westman, E., Zhang, Y., et al. (2011). Plasma biomarkers of brain atrophy in Alzheimer’s disease. PLoS One 6:e28527. doi: 10.1371/journal.pone.0028527
Thambisetty, M., Simmons, A., Velayudhan, L., Hye, A., Campbell, J., Zhang, Y., et al. (2010). Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch. Gen. Psychiatry 67, 739–748. doi: 10.1001/archgenpsychiatry.2010.78
Thomas-Salgar, S., and Millis, A. (1994). Clusterin expression in differentiating smooth muscle cells. J. Biol. Chem. 269, 17879–17885.
Trougakos, I. P., and Gonos, E. S. (2006). Regulation of clusterin/apolipoprotein J, a functional homologue to the small heat shock proteins, by oxidative stress in ageing and age-related diseases. Free Radic. Res. 40, 1324–1334. doi: 10.1080/10715760600902310
Trougakos, I. P., Lourda, M., Antonelou, M. H., Kletsas, D., Gorgoulis, V. G., Papassideri, I. S., et al. (2009). Intracellular clusterin inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizing the cytosolic Ku70-bax protein complex. Clin. Cancer Res. 15, 48–59. doi: 10.1158/1078-0432.CCR-08-1805
Trougakos, I. P., So, A., and Jansen, B. (2004). Silencing expression of the clusterin / apolipoprotein J gene in human cancer cells using small interfering RNA induces spontaneous apoptosis, reduced growth ability, and cell sensitization to genotoxic and oxidative stress silencing expression of the C. Cancer Res. 64, 1834–1842. doi: 10.1158/0008-5472.CAN-03-2664
Urban, J., Parczyk, K., Leutz, A., Kayne, M., and Kondor-Koch, C. (1987). Constitutive apical secretion of an 80-kD sulfated glycoprotein complex in the polarized epithelial Madin-Darby canine kidney cell line. J. Cell Biol. 105, 2735–2743. doi: 10.1083/jcb.105.6.2735
Urbich, C., Fritzenwanger, M., Zeiher, A. M., and Dimmeler, S. (2000). Laminar shear stress upregulates the complement-inhibitory protein clusterin. Circulation 101, 352–355. doi: 10.1161/01.CIR.101.4.352
Van Cauwenberghe, C., Van Broeckhoven, C., and Sleegers, K. (2016). The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430. doi: 10.1038/gim.2015.117
Veerhuis, R., Nielsen, H. M., and Tenner, A. J. (2011). Complement in the brain. Mol. Immunol. 48, 1592–1603. doi: 10.1016/j.molimm.2011.04.003
Viard, I., Wehrli, P., Jornot, L., Bullani, R., Vechietti, J. L., Schifferli, J. A., et al. (1999). Clusterin gene expression mediates resistance to apoptotic cell death induced by heat shock and oxidative stress. J. Invest. Dermatol. 112, 290–296. doi: 10.1046/j.1523-1747.1999.00531.x
Walton, M., Young, D., Sirimanne, E., Dodd, J., Christie, D., Williams, C., et al. (1996). Induction of clusterin in the immature brain following a hypoxic-ischemic injury. Mol. Brain Res. 39, 137–152. doi: 10.1016/0169-328X(96)00019-8
Wang, C., Jiang, K., Gao, D., Kang, X., Sun, C., Zhang, Q., et al. (2013). Clusterin protects hepatocellular carcinoma cells from endoplasmic reticulum stress induced apoptosis through GRP78. PLoS One 8:e55981. doi: 10.1371/journal.pone.0055981
Wang, C., Liu, Z., Woo, C.-W., Li, Z., Wang, L., Wei, J., et al. (2012). EZH2 mediates epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU, RUNX3 and NGFR. Cancer Res. 72, 315–324. doi: 10.1158/0008-5472.CAN-11-0961
Wang, H., and Eckel, R. H. (2014). What are lipoproteins doing in the brainα. Trends Endocrinol. Metab. 25, 8–14. doi: 10.1016/j.tem.2013.10.003
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Ward, L. D., and Kellis, M. (2012). HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, 930–934. doi: 10.1093/nar/gkr917
Wehrli, P., Charnay, Y., Vallet, P., Zhu, G., Harmony, J., Aronow, B., et al. (2001). Inhibition of post-ischemic brain injury by clusterin overexpression. Nat. Med. 7, 977–979. doi: 10.1038/nm0901-977
Wei, L., Xue, T., Wang, J., Chen, B., Lei, Y., Huang, Y., et al. (2009). Roles of clusterin in progression, chemoresistance and metastasis of human ovarian cancer. Int. J. Cancer 125, 791–806. doi: 10.1002/ijc.24316
Wigge, P., Köhler, K., Vallis, Y., Doyle, C. A., Owen, D., Hunt, S. P., et al. (1997). Amphiphysin heterodimers: potential role in clathrin-mediated endocytosis. Mol. Biol. Cell 8, 2003–2015. doi: 10.1091/mbc.8.10.2003
Wiggins, A. K., Shen, P. J., and Gundlach, A. L. (2003). Delayed, but prolonged increases in astrocytic clusterin (ApoJ) mRNA expression following acute cortical spreading depression in the rat: evidence for a role of clusterin in ischemic tolerance. Mol. Brain Res. 114, 20–30. doi: 10.1016/S0169-328X(03)00124-4
Wilson, M. R., and Easterbrook-Smith, S. B. (2000). Clusterin is a secreted mammalian chaperone. Trends Biochem. Sci. 25, 95–98. doi: 10.1016/S0968-0004(99)01534-0
Wojtas, A. M., Kang, S. S., Olley, B. M., Gatherer, M., Shinohara, M., Lozano, P. A., et al. (2017). Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl. Acad. Sci. U.S.A. 114, E6962–E6971. doi: 10.1073/pnas.1701137114
Wong, P., Pineault, J., Lakins, J., Taillefer, D., Leger, J., Wang, C., et al. (1993). Genomic organization and expression of the rat TRPM-2 (clusterin) gene, a gene implicated in apoptosis. J. Biol. Chem. 268, 5021–5031.
Wong, P., Taillefer, D., Lakins, J., Pineault, J., Chader, G., and Tenniswood, M. (1994). Molecular characterization of human TRPM-2 / clusterin, a gene associated with sperm maturation, apoptosis and neurodegeneration. Eur. J. Biochem. 221, 917–925. doi: 10.1111/j.1432-1033.1994.tb18807.x
Xie, D., Sze, H. L., Sham, J. S. T., Wu, Q. L., Fang, Y., Liang, L. Z., et al. (2005). Up-regulated expression of cytoplasmic clusterin in human ovarian carcinoma. Cancer 103, 277–283. doi: 10.1002/cncr.20765
Xie, Z., Harris-White, M. E., Wals, P. A., Frautschy, S. A., Finch, C. E., and Morgan, T. E. (2005). Apolipoprotein J (clusterin) activates rodent microglia in vivo and in vitro. J. Neurochem. 93, 1038–1046. doi: 10.1111/j.1471-4159.2005.03065.x
Xing, Y. Y., Yu, J. T., Cui, W. Z., Zhong, X. L., Wu, Z. C., Zhang, Q., et al. (2012). Blood clusterin levels, rs9331888 polymorphism, and the risk of Alzheimer’s disease. J. Alzheimers Dis. 29, 515–519. doi: 10.3233/JAD-2011-111844
Xiu, P., Dong, X., Dong, X., Xu, Z., Zhu, H., Liu, F., et al. (2013). Secretory clusterin contributes to oxaliplatin resistance by activating Akt pathway in hepatocellular carcinoma. Cancer Sci. 104, 375–382. doi: 10.1111/cas.12088
Yang, C. R., Leskov, K., Hosley-Eberlein, K., Criswell, T., Pink, J. J., Kinsella, T. J., et al. (2000). Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein that signals cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 5907–5912. doi: 10.1073/pnas.97.11.5907
Yang, C. R., Yeh, S., Leskov, K., Odegaard, E., Hsu, H. L., Chang, C., et al. (1999). Isolation of Ku70-binding proteins (KUBs). Nucleic Acids Res. 27, 2165–2174. doi: 10.1093/nar/27.10.2165
Yang, X., Li, J., Liu, B., Li, Y., and Jiang, T. (2016). Impact of PICALM and CLU on hippocampal degeneration. Hum. Brain Mapp. 37, 2419–2430. doi: 10.1002/hbm.23183
Yasuhara, O., Aimi, Y., Yamada, T., Matsuo, A., McGeer, E. G., and McGeer, P. L. (1994). Clusterin as a marker for ischaemic Purkinje cells in human brain. Neurodegeneration 3, 325–329.
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yerbury, J. J., Poon, S., Meehan, S., Thompson, B., Kumita, J. R., Dobson, C. M., et al. (2007). The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 21, 2312–2322. doi: 10.1096/fj.06-7986com
Yerbury, J. J., and Wilson, M. R. (2010). Extracellular chaperones modulate the effects of Alzheimer’s patient cerebrospinal fluid on Aβ1-42 toxicity and uptake. Cell Stress Chaperones 15, 115–121. doi: 10.1007/s12192-009-0122-0
Yom, C. K., Woo, H. Y., Min, S. Y., Kang, S. Y., and Kim, H. S. (2009). Clusterin overexpression and relapse-free survival in breast cancer. Anticancer Res. 29, 3909–3912.
Youle, R. J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59. doi: 10.1038/nrm2308
Yu, J. T., Li, L., Zhu, Q. X., Zhang, Q., Zhang, W., Wu, Z. C., et al. (2010). Implication of CLU gene polymorphisms in Chinese patients with Alzheimer’s disease. Clin. Chim. Acta 411, 1516–1519. doi: 10.1016/j.cca.2010.06.013
Zandl-Lang, M., Fanaee-Danesh, E., Sun, Y., Čančar, I., Gali, C. C., Kober, A., et al. (2017). Regulatory effects of simvastatin and apoJ on APP processing and amyloid-β clearance in blood-brain barrier endothelial cells. Biochim. Biophys. Acta 1863, 40–60. doi: 10.1016/j.bbalip.2017.09.008
Zhang, F., Kumano, M., Beraldi, E., Fazli, L., Du, C., Moore, S., et al. (2014). Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat. Commun. 5:5775. doi: 10.1038/ncomms6775
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014
Zhang, H., Kim, J. K., Edwards, C. A., Xu, Z., Taichman, R., and Wang, C.-Y. (2005). Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 7, 909–915. doi: 10.1038/ncb1291
Zhang, P., Qin, W., Wang, D., Liu, B., Zhang, Y., Jiang, T., et al. (2015). Impacts of PICALM and CLU variants associated with Alzheimer’s disease on the functional connectivity of the hippocampus in healthy young adults. Brain Struct. Funct. 220, 1463–1475. doi: 10.1007/s00429-014-0738-4
Zhang, S., Zhang, D., Jiang, Y., Wu, L., Shang, H., Liu, J., et al. (2015). CLU rs2279590 polymorphism contributes to Alzheimer’s disease susceptibility in Caucasian and Asian populations. J. Neural Transm. 122, 433–439. doi: 10.1007/s00702-014-1260-9
Zhang, S., Li, X., Ma, G., Jiang, Y., Liao, M., Feng, R., et al. (2016). CLU rs9331888 polymorphism contributes to Alzheimer’s disease susceptibility in caucasian but not east asian populations. Mol. Neurobiol. 53, 1446–1451. doi: 10.1007/s12035-015-9098-1
Zhang, X., and Le, W. (2010). Pathological role of hypoxia in Alzheimer’s disease. Exp. Neurol. 223, 299–303. doi: 10.1016/j.expneurol.2009.07.033
Zhou, Y., Hayashi, I., Wong, J., Tugusheva, K., Renger, J. J., and Zerbinatti, C. (2014). Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s Disease. PLoS One 9:e103187. doi: 10.1371/journal.pone.0103187
Zhu, B., Wang, R. M., Wang, J. T., Chen, R. L., Zheng, Y. F., Zhang, L., et al. (2017). Correlation of rs9331888 polymorphism with Alzheimer’s disease among Caucasian and Chinese populations: a meta-analysis and systematic review. Metab. Brain Dis. 32, 981–989. doi: 10.1007/s11011-017-9957-8
Zielinksi, R., and Chi, K. (2012). Custirsen (OGX-011): a second-generation antisense inhibitor of clusterin in development for the treatment of prostate cancer. Future Oncol. 8, 1239–1251. doi: 10.2217/fon.12.129
Zinkie, S., Gentil, B. J., Minotti, S., and Durham, H. D. (2013). Expression of the protein chaperone, clusterin, in spinal cord cells constitutively and following cellular stress, and upregulation by treatment with Hsp90 inhibitor. Cell Stress Chaperones 18, 745–758. doi: 10.1007/s12192-013-0427-x
Zlokovic, B. V., Martel, C. L., Matsubara, E., McComb, J. G., Zheng, G., McCluskey, R. T., et al. (1996). Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. U.S.A. 93, 4229–4234. doi: 10.1073/pnas.93.9.4229
Zoli, M., Ferraguti, F., Zini, I., Bettuzzi, S., and Agnati, L. F. (1993). Increases in sulphated glycoprotein-2 mRNA levels in the rat brain after transient forebrain ischemia or partial mesodiencephalic hemitransection. Mol. Brain Res. 18, 163–177. doi: 10.1016/0169-328X(93)90185-R
Keywords: neurodegeneration, amyloid, cell death, neuroprotection, DKK1, oxidative stress, Wnt signaling
Citation: Foster EM, Dangla-Valls A, Lovestone S, Ribe EM and Buckley NJ (2019) Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front. Neurosci. 13:164. doi: 10.3389/fnins.2019.00164
Received: 28 September 2018; Accepted: 12 February 2019;
Published: 28 February 2019.
Edited by:
Natalia N. Nalivaeva, University of Leeds, United KingdomReviewed by:
Mark Richard Wilson, University of Wollongong, AustraliaCeleste Karch, Washington University in St. Louis, United States
Copyright © 2019 Foster, Dangla-Valls, Lovestone, Ribe and Buckley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noel J. Buckley, noel.buckley@psych.ox.ac.uk
†These authors have contributed equally to this work