 Elin L. Strachan1,2*
Elin L. Strachan1,2* Delphi Mac White-Begg1,3
Delphi Mac White-Begg1,3 John Crean1,2,4
John Crean1,2,4 Alison L. Reynolds1,3
Alison L. Reynolds1,3 Breandán N. Kennedy1,2
Breandán N. Kennedy1,2 Niamh C. O’Sullivan1,2
Niamh C. O’Sullivan1,2- 1UCD Conway Institute, University College Dublin, Dublin, Ireland
- 2UCD School of Biomolecular and Biomedical Science, University College Dublin, Dublin, Ireland
- 3UCD School of Veterinary Medicine, University College Dublin, Dublin, Ireland
- 4UCD Diabetes Complications Research Centre, Conway Institute of Biomolecular and Biomedical Science, University College Dublin, Dublin, Ireland
Optic atrophy (OA) with autosomal inheritance is a form of optic neuropathy characterized by the progressive and irreversible loss of vision. In some cases, this is accompanied by additional, typically neurological, extra-ocular symptoms. Underlying the loss of vision is the specific degeneration of the retinal ganglion cells (RGCs) which form the optic nerve. Whilst autosomal OA is genetically heterogenous, all currently identified causative genes appear to be associated with mitochondrial organization and function. However, it is unclear why RGCs are particularly vulnerable to mitochondrial aberration. Despite the relatively high prevalence of this disorder, there are currently no approved treatments. Combined with the lack of knowledge concerning the mechanisms through which aberrant mitochondrial function leads to RGC death, there remains a clear need for further research to identify the underlying mechanisms and develop treatments for this condition. This review summarizes the genes known to be causative of autosomal OA and the mitochondrial dysfunction caused by pathogenic mutations. Furthermore, we discuss the suitability of available in vivo models for autosomal OA with regards to both treatment development and furthering the understanding of autosomal OA pathology.
Introduction
Optic atrophy (OA) with autosomal inheritance is a heterogeneous neurodegenerative disorder primarily characterized by the bilateral degradation of axons in the optic nerve, leading to a progressive and irreversible loss of vision. OA is largely referred to in the literature as dominant optic atrophy, however, as we will discuss further below, both dominant and recessive forms of the disease are common. It is thought to be the most prevalent form of hereditary optic neuropathy, with an estimated prevalence between 1 in 25,000 to 1 in 12,000 in some regions (Toomes et al., 2001; Yu-Wai-Man et al., 2010a). Age of onset is most commonly during the first or second decade of life with diagnosis usually occurring during childhood (Lenaers et al., 2012; Ham et al., 2019). Autosomal OA has a complex symptomatology and the extent of vision loss is highly variable. Some patients experience moderate visual loss and color vision defects, while in others the visual loss is more severe resulting in blindness. Furthermore, approximately 25% of patients exhibit extra-ocular symptoms including ataxia, peripheral neuropathy, deafness, and myopathy which is referred to as syndromic-OA (Yu-Wai-Man et al., 2010b; Ham et al., 2019). As yet, there is not a clear understanding why some individuals develop syndromic-OA while others, in some instances with the same underlying genetic variations, develop non-syndromic-OA (involving visual symptoms only) (Ham et al., 2019). Although a small-scale, off-label trial has been conducted using the coenzyme-Q10 analog idebenone (Romagnoli et al., 2020), there is currently no treatment commercially available for autosomal OA, representing a significant unmet need for affected patients. Therefore, there is a clear need to further our understanding of the molecular events that give rise to this disorder so that we might develop additional treatment strategies to address autosomal OA which would have relevance for other neurodegenerative diseases. This review discusses the converging evidence for mitochondrial dysfunction underpinning axonopathy in autosomal OA. Furthermore, we examine current and potential model systems available for the study of autosomal OA, many of which would be highly amenable to adaptation for other forms of hereditary retinal and neurological disease.
The loss of sight in autosomal OA is associated with the degeneration of retinal ganglion cells (RGCs), which transmit visual information from the photoreceptors to the brain (Miyata et al., 2007). Collectively, the axons of the RGCs form the optic nerve (Figure 1). As with most neurons, RGCs have a high energy demand to ensure the continuous active transport of ions against their concentration and electrical gradients required in membrane repolarization, maintenance of calcium stores and synaptic vesicle mobilization (Wong-Riley, 2010). This energy is provided primarily through electron transport within the cristae folds of the inner mitochondrial membrane (IMM) coupled to oxidative phosphorylation (OXPHOS) to synthesize ATP. Morphologically, RGCs comprise complex dendritic arbors and long axons. Moreover, and somewhat uniquely to RGCs, the most proximal portion of the axons, within the retina, are unmyelinated (Perry and Lund, 1990). The unmyelinated region of RGC axons have a higher mitochondrial load (Figure 1; Perge et al., 2009; Wilkison et al., 2021). This is generally believed to reflect an increased energy requirement to propagate axon potentials compared to myelinated axons, however recent findings show that mitochondrial accumulation precedes RGC axon myelination bringing this supposition into question (Wilkison et al., 2021). Furthermore, while RGCs seem to be particularly vulnerable to mitochondrial defects, autosomal OA-causing mutations can result in degeneration of neurons other than RGCs as evidenced by patients with syndromic-OA. Additionally, there are several reports of the co-occurrence of autosomal OA and other neurodegenerative disorders specifically hereditary spastic paraplegia (HSP) (Yu-Wai-Man et al., 2010b; Charif et al., 2020) and Charcot-Marie-Tooth type 2 (CMT2) (Rouzier et al., 2012; Guerriero et al., 2020). Together, these provide compelling evidence that highly elongated neurons, such as RGCs, motor neurons, sensory neurons and cerebella purkinje cells, are particularly sensitive to the mutations underpinning these neurodegenerative disorders. This emphasizes the importance of comparable in vivo models so that molecular events specifically within long axons may be better understood.
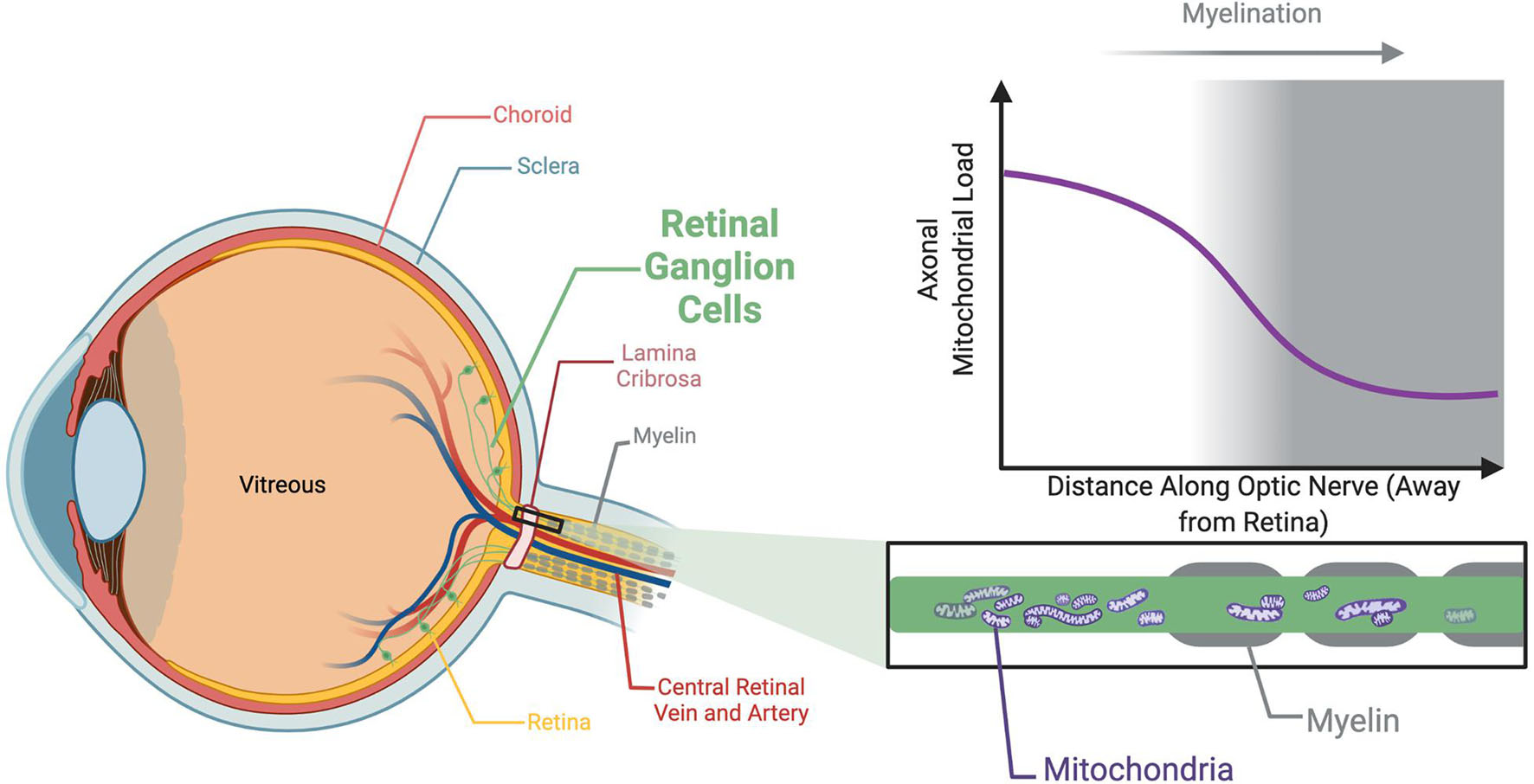
Figure 1. Unmyelinated axons within the optic nerve are associated with a greater mitochondrial load. The axons of the RGCs are unmyelinated within the retina, an adaptation which likely exists to prevent myelin from impeding light reaching the photoreceptors. Myelination initiates following the lamina cribrosa, a network of collagen fibers in the optic nerve head approximately 400 μm thick the RGC axons traverse to pass through the sclera. The unmyelinated region is associated with a higher density of mitochondria than the adjacent myelinated sections. This unique architecture of the RGCs is a possible explanation as to why the optic nerve appears so vulnerable to degeneration associated with mitochondrial dysfunction.
Autosomal OA is a genetically heterogeneous, monogenic disorder, primarily caused by mutations in nuclear genes encoding mitochondrial proteins. Mutations in OPA1 are the most common cause of autosomal OA (Almind et al., 2012; Weisschuh et al., 2021). However, as discussed in detail below, mutations in at least 10 other genes are also associated with autosomal OA. Identified disease-causing mutations run the spectrum from nonsense, missense frameshift and splice mutations, but chromosomal rearrangements including copy number variants and inversions were also identified (Fuhrmann et al., 2009; Weisschuh et al., 2021). The majority of disease-causing mutations are predicted to impair the function of the encoded protein, pointing to haploinsuffiency as the primary mechanism of pathogenicity in dominant forms of autosomal OA (Neumann et al., 2020; Sun et al., 2021). However, several cases report semi-dominant inheritance, where individuals carrying more than one OA-causing mutation present with much more severe disease than heterozygotic parents or siblings (Pesch et al., 2001; Weisschuh et al., 2021). Several forms of recessively inherited OA have also been identified (associated with mutations in TMEM126A/OPA7, SCL25A46, MCAT and RTN4IP1/OPA10) and mutations in WFS1, ACO2/OPA9 and OPA3 are associated with both recessive and dominant forms of OA (Reynier et al., 2004; Meyer et al., 2010; Kloth et al., 2019; Charif et al., 2021b). Given this genetic heterogeneity, we use the term ‘autosomal OA’ throughout this review.
All of the genes implicated in autosomal OA encode proteins associated with mitochondrial function, the majority being nuclear genes encoding mitochondrial proteins (Charif et al., 2021a). These genes have roles in mitochondrial fission or fusion, mitochondrial respiration, mitochondrial DNA replication and mitochondrial fatty acid synthesis (Figure 2). Furthermore, disrupted mitochondrial network morphology and reduced respiratory efficiency are consistently observed in autosomal OA patient fibroblasts (Kane et al., 2017; Liao et al., 2017) as well as in cell and animal models (Rahn et al., 2013; Maloney et al., 2020). Autosomal OA is not unique amongst optic neuropathies to be characterized by mitochondrial dysfunction. Mitochondrial dysfunction is a common feature of many retinal diseases including: Leber’s Hereditary Optic Neuropathy (LHON) and glaucoma (Bahr et al., 2020; Duarte, 2021). Indeed, mitochondrial dysfunction is now recognized as a common feature of axonopathies generally (Krols et al., 2016). While there is strong evidence to suggest mitochondrial dysfunction is central to neurodegeneration in autosomal OA, the underlying mechanism, and why RGCs are particularly vulnerable, remains unknown. In this article we provide a comprehensive review of studies investigating the functions of known autosomal OA-causing genes and emphasize that, despite their heterogeneity, there are just a few converging pathogenic themes emerging. Furthermore, we investigate the range of in vivo models available which offer potential to better understand the molecular and cellular events underpinning autosomal OA, and assess their suitability to uncover why RGCs may be particularly targeted in OA and to identify novel therapeutic strategies.
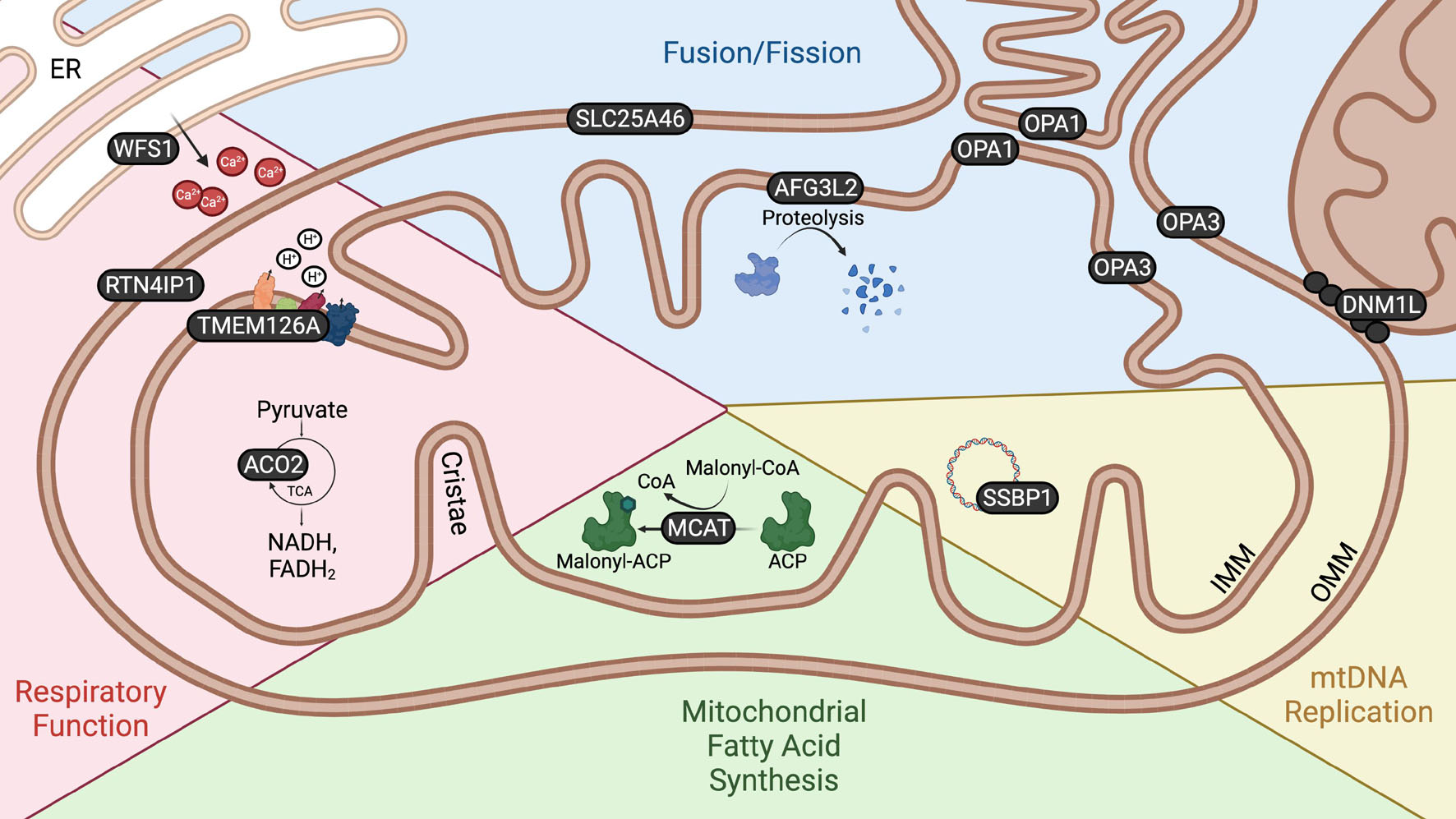
Figure 2. Mitochondrial localization and function of autosomal OA genes. Schematic demonstrating the localization and function of proteins encoded by autosomal OA-associated genes. The localization of proteins encoded by autosomal OA-associated genes within and in association with the mitochondria emphasizes the relevance of these organelles to autosomal OA. WFS1 is localized to the ER, but is enriched at MAMs. DNM1L is recruited from the cytoplasm to the OMM to facilitate mitochondrial fission. There is conflicting evidence as to whether OPA3 is localized to the IMM or OMM.
Gene Mutations Causing Autosomal Optic Atrophy
Most currently identified genes causative of autosomal OA can be broadly classified into two functional categories; mitochondrial fission/fusion and mitochondrial respiration; albeit two autosomal OA genes, with roles in mtDNA replication and mitochondrial fatty acid synthesis, do not clearly fit into either of these categories. These categories are not mutually exclusive; on the contrary, disruption of mitochondrial morphology through altered fission/fusion dynamics is known to alter mitochondrial respiration and mtDNA stability (Chen et al., 2010; Quintana-Cabrera et al., 2018; Glancy et al., 2020). Below we examine the function of proteins encoded by OA-causing genes and the mitochondrial aberrations associated with disease-causing mutations. For this review, we focus on autosomal OA-causing genes linked to more than two families where there is clear evidence that OA is the primary feature of disease (Table 1). We therefore do not include discussion on genes that cause optic atrophy as a feature of another neurological disorder such as NDUFS2 (primarily associated with Leigh syndrome) (Gerber et al., 2017a), SPG7 (primarily associated with HSP) (Charif et al., 2020), CISD2 (primarily associated with Wolfram syndrome) (Delprat et al., 2018), C19orf12 (primarily associated with MPAN syndrome) (Mignani et al., 2020) or MFN2 (primarily associated with CMT2) (Bombelli et al., 2014). The role of many of these genes in mitochondrial function has been recently reviewed elsewhere (Maresca and Carelli, 2021).
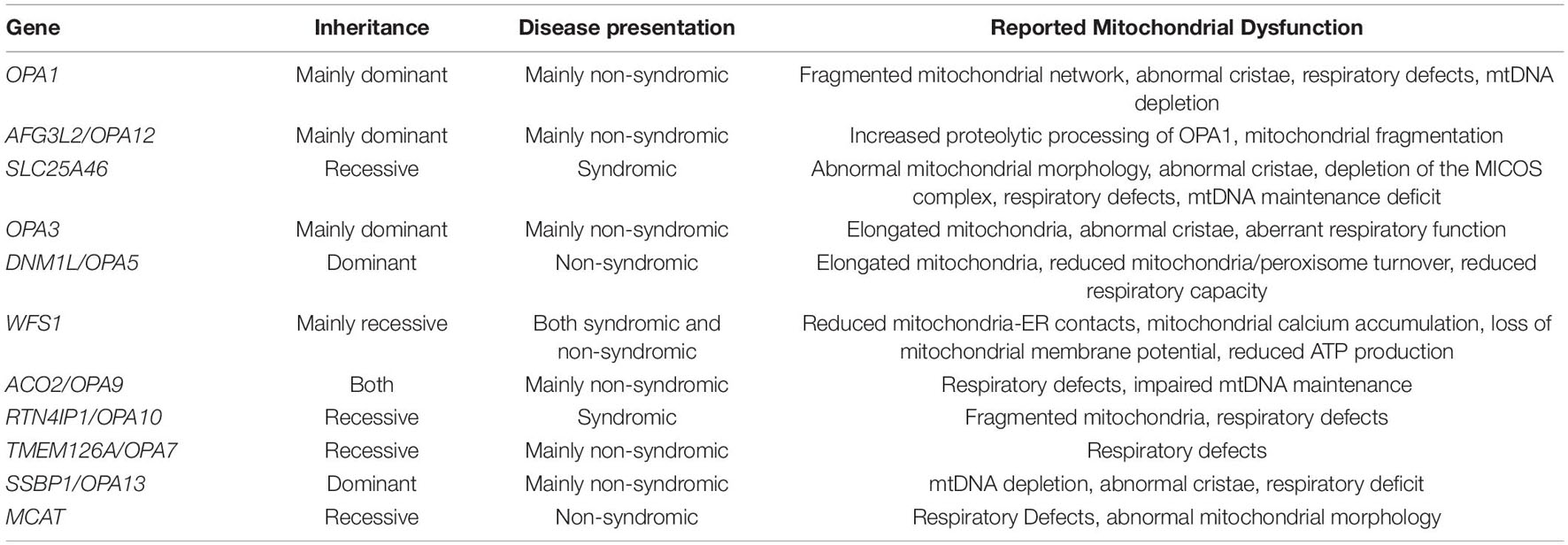
Table 1. Summary of autosomal OA-associated genes and their inheritance.
Mitochondrial Fission/Fusion
Mitochondria are highly dynamic organelles which constantly undergo fission, whereby mitochondria divide into smaller ‘daughter’ mitochondria, and fusion, whereby two or more mitochondria fuse together. These processes permit mitochondria to regulate their distribution according to metabolic demand (Wai and Langer, 2016), to communicate mtDNA and protein content (Silva Ramos et al., 2016) and to regulate mitochondrial turnover via mitophagy (Xian and Liou, 2021). To date, five genes encoding proteins which function in mitochondrial fission/fusion have been associated with autosomal OA: OPA1, AFG3L2/OPA12; SLC25A46; OPA3; DNM1L/OPA5 (Figure 2).
OPA1
Optic atrophy 1 (OPA1) is a ubiquitously expressed, highly conserved gene with OPA1 homologs present in all higher metazoans (Wong et al., 2003; Li et al., 2019). It encodes a dynamin-like GTPase localized to the inner mitochondrial membrane (IMM) and is essential for IMM fusion following the fusion of the outer mitochondrial membrane (OMM) by the mitofusins (MFN1 and MFN2) (Song et al., 2009). OPA1 is produced as a long-form (L-OPA1) which can be cleaved by either of two proteolytic enzymes, YME1L or OMA1 respectively, to produce short-form OPA1 (S-OPA1) (Anand et al., 2014). Regulation of mitochondrial fusion depends on OPA1 processing and while either L- or S-OPA1 are sufficient to induce mitochondrial fusion, optimal fusion requires the presence of equimolar concentrations of both isoforms (Anand et al., 2014; Ge et al., 2020; Ohba et al., 2020). OPA1 is the most common gene implicated in autosomal OA, accounting for roughly 70% of cases (Ferré et al., 2009). Non-syndromic-autosomal OA appears to be generally associated with mutations resulting in haploinsufficiency, whilst missense mutations are associated with syndromic-autosomal OA, and more severe ocular symptoms (Carelli et al., 2015; Ham et al., 2019). This is proposed to be due to a dominant negative effect (Amati-Bonneau et al., 2009). Approximately 20% of OPA1 patients have extra ocular symptoms, most commonly sensorineural deafness, but additionally peripheral neuropathy, ataxia, myopathy and parkinsonism (Lynch et al., 2017; Ham et al., 2019). Although the vast majority of OPA1 mutations are inherited in an autosomal dominant manner, there are examples of individuals carrying 2 apparently recessive pathogenic OPA1 mutations, usually exhibiting compound heterozygosity. These patients present with a more severe syndromic-autosomal OA, often associated with unusual features such as developmental delays, pyramidal tract abnormalities, and neuromuscular disorders (Zerem et al., 2019; Zeng et al., 2020). Some OPA1 mutations also appear to exhibit incomplete penetrance, with many examples of seemingly unaffected carriers of pathogenic mutations, as well as variable pathology within families bearing the same mutation (Toomes et al., 2001; Cohn et al., 2008). Whilst no modifying genes have been identified to explain this, there is evidence to suggest alcohol and tobacco consumption may be associated with more severe symptoms (Mei et al., 2019). Both patient fibroblasts and cell models bearing pathogenic mutations of OPA1 have a characteristic fragmented mitochondrial network, consistent with a mitochondrial fusion disorder, and highly disordered cristae (Liao et al., 2017; Del Dotto et al., 2018). Through its key role in cristae organization, changes in OPA1 expression affect the stability of the respiratory supercomplexes (Cogliati et al., 2013), quaternary structures adopted by components of the electron transport chain to carry out cellular respiration (Letts et al., 2016). Subsequently, defects in OXPHOS are often associated with insufficient OPA1 activity (Lee et al., 2017; Sun et al., 2020). Consistent with this, widespread respiratory defects, including decreased ATP synthesis, increased oxygen consumption and reduced respiration, have all been reported in OPA1 patient fibroblasts and cell models (Zanna et al., 2008; Millet et al., 2016; Zhang et al., 2017). The manner and extent to which respiration is affected appears to vary between OPA1 mutations, with some evidence to suggest that respiratory defects are associated with more severe vision loss (van Bergen et al., 2011). It is perhaps unsurprising that there is substantial variation between patients, due to incomplete penetrance and heterogeneity of symptoms.
AFG3L2/OPA12
AFG3L2/OPA12 encodes a mitochondrial AAA quality-control protease which is tethered to the IMM and exposes its enzymatic domain to the mitochondrial matrix (Puchades et al., 2019). Missense mutations, generally within the AAA domain of AFG3L2/OPA12, give rise to isolated, dominantly inherited autosomal OA, though rarer biallelic mutations have been reported which result in more severe, syndromic-autosomal OA (Colavito et al., 2017; Baderna et al., 2020; Caporali et al., 2020). Of note, missense mutations in AFG3L2/OPA12 are also known to cause the unrelated disorders spinocerebellar ataxia type 28 (SCA28) and spastic ataxia syndrome 5 (SPAX5) but these mutations are generally localized within the proteolytic domain of the protein (Caporali et al., 2020). AFG3L2 is known to have vital roles in mitochondrial maintenance including mitochondrial ribosome assembly, electron transport chain complex processing and calcium homeostasis (Puchades et al., 2019; Pareek and Pallanck, 2020) however, the mechanism by which missense mutations give rise to autosomal OA was only recently discovered. Two independent studies revealed that autosomal OA-causing mutations in AFG3L2/OPA12 cause hyperactivation of OMA1, the protease that acts on L-OPA1, resulting in increased processing of L-OPA1 to S-OPA1 (Baderna et al., 2020; Caporali et al., 2020). This imbalance in L-OPA1/S-OPA1 results in mitochondrial fragmentation which is evident in both engineered MEFs and patient fibroblasts.
SLC25A46
SLC25A46 encodes a member of the solute carrier family 25 (SLC25), a group of proteins primarily localized to the IMM containing six alpha-helical membrane spanning domains (Pebay-Peyroula et al., 2003). Many members of the SLC25 family are responsible for the transport of metabolic intermediates (such as glutamate and ADP/ATP) into the cell (Fiermonte et al., 2002; Kunji et al., 2016), with some functioning as uncoupling proteins (Fedorenko et al., 2012). SLC25A46 is unusual in the SLC25 protein family in that it localizes to the OMM (Janer et al., 2016) and has no known transporter function nor any identified substrates. Recessive, loss-of-function mutations in SLC25A46 are associated with syndromic-autosomal OA (Nguyen et al., 2017; Bitetto et al., 2020). There are a wide spectrum of reported symptoms, including Parkinsonism, Leigh syndrome, cerebellar ataxia, and congenital pontocerebellar hypoplasia, usually accompanied by optic atrophy (Wan et al., 2016; Hammer et al., 2017; Bitetto et al., 2020). Whilst the exact function of SLC25A46 is unknown, it likely has a role in mitochondrial fission/fusion dynamics. Loss of SLC25A46 function has been associated with both elongated mitochondria, suggestive of aberrant fission (Abrams et al., 2015; Wan et al., 2016) and small, circular mitochondria, which would be indicative of a fusion defect (Duchesne et al., 2017). Overexpression appears to induce fragmentation in the mitochondrial network, which would also be consistent with a role in fission (Abrams et al., 2015). Both patient samples and SLC25A46 models usually display highly abnormal mitochondrial cristae (Janer et al., 2016; Li et al., 2017; Zou et al., 2021) in some cases being completely detached from the IMM (Duchesne et al., 2017). Consistent with this, SLC25A46 is thought to operate upstream of the MICOS complex, which is significantly depleted in SLC25A46 knockout cells. The MICOS is a large protein complex localized to the IMM where it is crucial for the structural organization of the cristae (Friedman et al., 2015) and interacts with both OPA1 and MFN2 (Janer et al., 2016). Depletion of its constituents have been shown to have deleterious effects on processes reliant on the structural integrity of the cristae, such as mtDNA maintenance and respiration (John et al., 2005; Genin et al., 2016; Gödiker et al., 2018).
OPA3
OPA3 encodes a protein of unknown function localized to mitochondria, with conflicting evidence as to whether it localizes to the IMM or OMM (Da Cruz et al., 2003; Ryu et al., 2010). Most autosomal OA-causing mutations in OPA3 are dominantly inherited and result in non-syndromic disease, although hearing loss and extra-ocular neurological symptoms consistent with syndromic-autosomal OA have also been observed in some patients (Reynier et al., 2004; Sergouniotis et al., 2015). Recessive loss of function OPA3 mutations are most often associated with Costeff syndrome, which resembles syndromic-autosomal OA with some additional features such as cognitive deficit and extrapyramidal dysfunction (Grau et al., 2013). While there remains ambiguity surrounding the precise function of OPA3, overexpression and knockout studies support a role in mitochondrial fission/fusion dynamics. Overexpression of OPA3 in patient-derived or retinal pigment epithelium-cell models creates a highly fragmented mitochondrial network, suggestive of a role in mitochondrial fission (Ryu et al., 2010; Maresca et al., 2012). Consistent with this, fibroblasts from some OPA3 patients were recently reported to have enlarged mitochondria with slightly fragmented cristae (Horga et al., 2019). It is not currently clear whether the altered mitochondrial morphology resulting from OPA3 mutations disrupts mitochondrial function with conflicting reports on the effect of loss of OPA3 on ATP production and oxygen consumption (Pei et al., 2010; Meng et al., 2020).
DNM1L/OPA5
Dynamin 1-like (DNM1L)/OPA5 encodes a dynamin-like GTPase which is an essential protein in the regulation of mitochondrial fission. DNM1L is recruited to the OMM from the cytosol by mitochondrial fission factors: MFF, MID49, and MID51, where it oligomerizes forming a ring-like structure around the mitochondria (Palmer et al., 2011; Fröhlich et al., 2013). It then uses GTP hydrolysis to constrict the membrane, which can subsequently undergo scission. Presently, there are very few examples of autosomal OA patients with DNM1L/OPA5 variations (Gerber et al., 2017b). These patients, from three families, harbor 2 separate mutations within the GTPase domain, and appear to exhibit dominant inheritance. All patients present with non-syndromic-autosomal OA, except one individual who additionally had slight hearing loss. DNM1L/OPA5 mutations have previously been associated with forms of severe encephalopathy and epilepsy, often associated with infant mortality (Fahrner et al., 2016; Zaha et al., 2016). These disease-causing mutations have been reported in both the middle and GTPase domains of DNM1L and, apart from rare examples (Waterham et al., 2007; Chao et al., 2016), do not appear to be associated with optic atrophy (Fahrner et al., 2016; Ladds et al., 2018). Patient fibroblasts from individuals with non-syndromic-autosomal OA, as well as a yeast model expressing the patient mutation, display an elongated mitochondrial network, consistent with a mitochondrial fission defect (Gerber et al., 2017b). However, alterations in respiratory capacity, aberrant peroxisomes or reduced mitochondrial/peroxisomal turnover, which have been reported in samples from DNM1L-mediated encephalopathy patients, are not observed models of OA (Sheffer et al., 2016; Zaha et al., 2016; Longo et al., 2020).
Three further genes that function in the regulation of mitochondrial fission/fusion have recently been associated with autosomal OA. These include the genes encoding the IMM protease YME1L1/OPA11 and the OMM proteins mitochondrial elongation factor 1 (MIEF1) and mitochondrial fission factor (MFF). Only a few individuals or single families have been identified with these mutations thus far, hence did not fulfill our criteria for more extensive discussion, however it appears that heterozygous mutations in MIEF1 cause dominantly inherited non-syndromic-autosomal OA (Charif et al., 2021c) while homozygous mutations in YME1L1/OPA11 or MFF cause recessively inherited syndromic-autosomal OA (Shamseldin et al., 2012; Hartmann et al., 2016; Koch et al., 2016). All 3 of the proteins encoded by these genes function in the regulation of mitochondrial fission/fusion; YME1L1 via the proteolytic processing of OPA1 (Song et al., 2007; Stiburek et al., 2012), MIEF1 via the sequestration of DNM1L (Zhao et al., 2011; Yu et al., 2017) and MFF via recruitment of DNM1L to mitochondrial fission sites (Otera et al., 2010; Losón et al., 2013). Mitochondria in cells expressing disease-causing mutations or patient fibroblasts all display disrupted organization, consistent with defective regulation of fission/fusion (Koch et al., 2016; Cesnekova et al., 2018; MacVicar et al., 2019).
Mitochondrial Respiratory Function
Respiration refers to a series of reactions through which the cell breaks down macronutrients to release their energy in the form of adenosine triphosphate (ATP). Mitochondria are essential for this process, housing both the citric acid cycle and electron transport chain. The citric acid cycle is a process through which the metabolic intermediate pyruvate undergoes a series of redox reactions to reduce the electron carriers NADH and FADH2, which then pass their electrons onto the respiratory complexes of the electron transport chain, ultimately producing most of the cell’s energy through OXPHOS (Papa et al., 2012). Neurons are usually highly energetically demanding, partly due to the process of neurotransmission itself (Harris et al., 2012), but additionally due to the comparatively large distance over which cargo, including organelles and synaptic components, must be transported along axons and dendrites (Mandal and Drerup, 2019). RGCs are no exception to this, in fact they are particularly vulnerable to bioenergetic perturbation. There is evidence that RGCs may utilize glycolysis in addition to OXPHOS to meet the considerable energetic demands imposed by their unique architecture (Trevisiol et al., 2017; Casson et al., 2021). Four autosomal OA-causing genes are associated with cellular respiration: WSF1, ACO2, RTN4IP1 and TMEM126A (Figure 2).
WFS1
After OPA1, the second most common gene associated with optic atrophy is wolframin (WFS1). Mutations in WFS1 were primarily identified to cause Wolfram syndrome type 1 (WS1), a recessive condition characterized by diabetes, optic atrophy and deafness (Inoue et al., 1998; Wragg et al., 2018). However, mutations in WFS1 are recognized as a frequent cause of optic atrophy independent of other WS1 symptoms (Hogewind et al., 2010; Grenier et al., 2016; Charif et al., 2021a). In fact, a recent screen of over 1,000 autosomal OA patients identified WFS1 mutations in 12% of dominant-autosomal OA and 39% of receive-autosomal OA patients (Charif et al., 2021a). Autosomal OA mutations are most commonly missense mutations, though truncating mutations have been identified (Hogewind et al., 2010; Grenier et al., 2016). A reduction in available protein is proposed as the pathogenic mechanism, with missense mutations appearing to decrease the half-life of the resulting wolframin protein (Hofmann et al., 2003). This was supported by a recent study which found that the extent of optic atrophy progression in patients correlates with the degree of decrease in wolframin protein (Hu et al., 2021). Wolframin is a transmembrane protein enriched at mitochondria-associated ER membranes (MAM) (La Morgia et al., 2020). It is ubiquitously expressed with particularly high levels of expression in the optic-nerve (Yamamoto et al., 2006; Schmidt-Kastner et al., 2009). Wolframin has roles in many cellular pathways including ER stress (Fonseca et al., 2010; Odisho et al., 2015), calcium homeostasis (Takei et al., 2006; Nguyen et al., 2020) and mitochondrial activity (Kõks et al., 2013; Angebault et al., 2018). However, there exists good evidence that disruption of mitochondrial function by loss of WFS1 is critical to pathogenicity in OA. Mitochondrial metabolism and ATP production are tightly regulated by the proper transfer of calcium ions from the ER at MAMs (Denton, 2009; Llorente-Folch et al., 2015) and disruption of MAMs impacts mitochondrial function in several models of neurodegenerative disease (Lee et al., 2018). Wolframin localizes to MAMs and the proportion of mitochondria in contact with the ER is significantly reduced in fibroblasts from patients carrying WFS1 mutations (Angebault et al., 2018; La Morgia et al., 2020). As predicted, this MAM disruption results in reduced calcium accumulation within mitochondria and decreased mitochondrial function in patient fibroblasts (Angebault et al., 2018; La Morgia et al., 2020). Moreover, primary cortical neurons in which WFS1 has been knocked down by siRNA display decreased membrane potential and cytosolic ATP (Cagalinec et al., 2016) further supporting a role for mitochondrial dysfunction underpinning neurodegeneration in autosomal OA caused by WFS1 mutations.
ACO2/OPA9
Mutations in the gene encoding citric acid cycle enzyme aconitase 2 (ACO2)/OPA9 were until recently believed to be quite a rare cause of inherited optic atrophy. Individual families had been identified with loss-of-function ACO2 mutations leading to both dominantly inherited syndromic-autosomal OA or recessively inherited non-syndromic-autosomal OA (Metodiev et al., 2014; Kelman et al., 2018; Neumann et al., 2020). However, a recent study using next-generation sequencing to identify causative genes in a large, multicenter cohort of patients with autosomal optic neuropathies, reported that 12% of autosomal OA cases had pathogenic mutations in the gene ACO2 (Charif et al., 2021b). In this study, recessive cases displayed significantly worse vision loss than dominant cases while 12–27% displayed extraocular symptoms (Charif et al., 2021b). This implies that ACO2/OPA9 mutations are the third leading cause of autosomal OA after OPA1 and WFS1. ACO2/OPA9 encodes the mitochondrial aconitase 2 enzyme, a lyase that converts citrate to isocitrate within the citric acid cycle (Martius, 1937). The citric acid cycle consists of a series of redox reactions in the mitochondrial matrix through which pyruvate, the final product of glycolysis, is used to reduce the electron carriers NAD and FADH for the OXPHOS pathway. Through this mechanism, ACO2 is important for generating ATP through aerobic respiration. To date, disease-causing mutations have not been studied in neuronal cells, but patient-derived fibroblast consistently show reduced ACO2 enzymatic activity, impaired maintenance of mtDNA and decreased respiratory function (Neumann et al., 2020; Charif et al., 2021b). This supports the hypothesis that mitochondrial dysfunction underpins RGC degeneration in these patients, though animal models to validate and study this further are required.
RTN4IP1/OPA10
Reticulon 4 interacting protein 1 (RTN4IP1)/OPA10 encodes a widely expressed protein localized to the OMM (Hu et al., 2002; Angebault et al., 2015). It contains an oxidoreductase domain and is believed to act on Complex I of the mitochondrial respiratory chain (Hu et al., 2002). Homozygous or compound heterozygous mutations in RTN4IP1/OPA10 are found across many different populations to cause recessive forms of autosomal OA (Okamoto et al., 2017; Zou et al., 2019; D’Gama et al., 2021). Patients generally present with syndromic autosomal OA, with epilepsy, chorea and encephalopathy commonly reported, and it is suggested that more severe symptoms are associated with more deleterious mutations in the RTN4IP1/OPA10 gene (D’Gama et al., 2021). Recently, it has also been reported that some patients experience degeneration of the rods in addition to RGCs, expanding the spectrum of neurons that are disrupted by loss of this protein (Meunier et al., 2021; Rajabian et al., 2021). Autosomal OA-causing mutations result in a marked decrease or loss of RTN4IP1 protein expression in patient fibroblasts and muscle biopsies (Angebault et al., 2015; Charif et al., 2017). Moreover, there is clear evidence that mitochondrial function are disrupted by these mutations. Examination of patient fibroblasts reveals a reduction in activity of the mitochondrial respiratory chain, specifically Complex I (Angebault et al., 2015; Charif et al., 2017). Whilst the precise function of RTN4IP1 is currently unknown, Angebault et al. (2015) generated both mouse and zebrafish models with depleted RTN4IP1. Mouse pups demonstrated an increase in dendrite numbers and dendrite arborization, whilst the RGC layer was completely absent from the retina of zebrafish RTN4IP1 morphants. Whilst human samples were not available for histological analysis, patients did appear to exhibit smaller optic disks. This implies that RTN4IP1 has an important developmental role linked to the RGCs which may contribute to OA pathogenesis.
TMEM126A/OPA7
TMEM126A/OPA7 encodes a highly conserved IMM protein which functions in the mitochondrial respiratory chain Complex I assembly pathway. Homozygous mutations, both missense and nonsense mutations, are reported to cause recessive forms of autosomal OA (Hanein et al., 2009; Meyer et al., 2010; Li J. K. et al., 2020). Patients primarily present with non-syndromic autosomal OA, but auditory neuropathy has been reported in some families (Meyer et al., 2010; Désir et al., 2012; La Morgia et al., 2019). Recently, two independent studies show that TMEM126A is necessary for the biogenesis and function of mitochondrial respiratory chain Complex I (D’Angelo et al., 2021; Formosa et al., 2021). TMEM126A/OPA7 knockout cells are deficient for Complex I and display decreased respiratory capacity (D’Angelo et al., 2021; Formosa et al., 2021). Significantly, autosomal OA patients with TMEM126A/OPA7 mutations show partial Complex I deficiency in addition to increased lactic acid levels following exercise (Hanein et al., 2009; La Morgia et al., 2019), suggesting that defective OXPHOS contributes to disease pathogenesis in these patients.
Mitochondrial Fatty Acid Synthesis and mtDNA Replication
Currently, two autosomal OA-causing genes do not clearly fit into the functions contributing to mitochondrial fission/fusion or respiratory function, specifically SSBP1 and MCAT which function in the regulation of mitochondrial DNA replication and mitochondrial fatty acid synthesis, respectively. While these genes may appear to be disparate to other OA-causing genes in terms of their roles in mitochondria, there are clear similarities in the mitochondrial dysfunction observed when these genes are disrupted. Specifically mitochondrial morphology and respiratory function are impaired in patient fibroblasts or knockout cells, suggesting that the underlying pathogenic mechanism of action may be analogous to that of other OA-causing genes. Furthermore, with a growing list of novel OA-causing mutations being identified, it may be that other genes with similar functions are identified to cause OA over the coming years.
SSBP1/OPA13
Single-stranded DNA binding protein 1 (SSBP1)/OPA13 is a nuclear-encoded housekeeping gene involved in mitochondrial biogenesis. SSBP1/OPA13 encodes a constituent of the mtDNA replisome which, along with the mitochondrial DNA polymerase Pol γ and the helicase Twinkle, is one of the essential components for mtDNA replication (Korhonen et al., 2004). During mtDNA synthesis, SSBP1 binds the heavy strand, increasing the activity of Pol γ through maintaining the ssDNA in a favorable conformation and preventing the formation of secondary structure (Ciesielski et al., 2015). SSBP1 also stimulates the unwinding activity of Twinkle (Oliveira and Kaguni, 2011). Missense mutations in SSBP1/OPA13 cause autosomal OA in at least 14 different European families, though the prevalence of this mutation in other populations is not known (Jurkute et al., 2019; Del Dotto et al., 2020; Piro-Megy et al., 2020). Almost all families show dominant inheritance of non-syndromic autosomal OA and rod-cone dystrophy in addition to the optic atrophy is common, indicating that retinal neurons other than RGCs are susceptible to mitochondrial dysfunction induced by SSBP1/OPA13 mutations (Jurkute et al., 2019; Del Dotto et al., 2020). Autosomal OA-causing mutations in SSBP1/OPA13 do not affect gene expression, though stability of the SSBP1 protein, particularly protein dimers, is likely reduced by some missense mutations (Del Dotto et al., 2020; Piro-Megy et al., 2020). Moreover, patient fibroblasts consistently reveal significant depletion of mtDNA, providing strong evidence that autosomal OA-causing mutations destabilize the replication machinery and disrupt the fidelity of mtDNA replication (Del Dotto et al., 2020; Piro-Megy et al., 2020). Loss of mtDNA results in swollen mitochondria with disorganized cristae and a severe respiratory deficit, which are common in patient fibroblasts (Del Dotto et al., 2020; Piro-Megy et al., 2020). This is consistent with mitochondrial disruption observed in SSBP1 knockdown cells (Wang et al., 2017) and supports a loss of function pathogenic mechanism underpinning autosomal OA in these patients.
MCAT
Malonyl-CoA-acyl carrier protein transacylase (MCAT) is a nuclear-encoded gene involved in mitochondrial fatty acid synthesis (mtFAS), a comparatively poorly understood pathway distinct from the cytosolic equivalent. Malonyl-CoA is a key intermediate in mtFAS, transferring malonate from malonyl-CoA to the acyl carrier protein (ACP), an important step in fatty acid elongation. A rare cause of autosomal OA, MCAT mutations give rise to non-syndromic-autosomal OA, and appear to have a recessive mode of inheritance (Li H. et al., 2020; Gerber et al., 2021). In silico analysis suggests autosomal OA-causing mutations make the resulting MCAT protein unstable and less able to bind malonyl-CoA (Gerber et al., 2021), however, this has not been examined directly in patient samples or model systems. Studies in conditional Mcat knockout mice reveal that the mtFAS pathway produces the octanoyl precursor required for lipoylation of key mitochondrial proteins involved in the citric acid cycle, namely pyruvate dehydrogenase complex and α-ketoglutarate dehydrogenase (Smith et al., 2012). Depletion of Mcat, or other genes involved in mtFAS, results in a significant depletion of the respiratory complexes and limits the ability of mitochondria to respire (Smith et al., 2012; Nowinski et al., 2020). Mitochondria in cells expressing MCAT mutations are disrupted, displaying a thin, thread-like morphology with unidentified debris within the mitochondrial matrix (Li H. et al., 2020). Moreover, they show a reduced ability to uptake MitoTracker, consistent with impaired IMM function, specifically an inability to sustain its membrane potential (Keij et al., 2000). Interestingly, mutations in another mtFAS component, MECR, cause the rare neurological disorder MEPAN syndrome, which features optic atrophy (Heimer et al., 2016; Gorukmez et al., 2019). Further study is needed to characterize this pathway, in order to better understand why perturbation leads to mitochondrial dysfunction.
Taken together, it is clear that there are converging pathogenic pathways related to mitochondrial organization and function underpinning autosomally inherited OA. Going forward, we should look to studies of animal models of OA to help elucidate the particular susceptibility of RGCs and other long axons to these disruptions, as well as to identify promising targets for therapeutic interventions.
In vivo Models of Autosomal Optic Atrophy
Whilst in vitro models are invaluable for investigating the molecular underpinnings of disease, there are limitations in their applicability to studying OA. The specific architecture of RGCs, with long and heterogeneously myelinated axons, cannot yet be accurately replicated in cell culture nor in non-polarized cells such as patient fibroblasts. Moreover, as the progressive loss of visual acuity is a key unifying symptom of OA, the ability to assess visual function is paramount to both the study of OA disease pathology and assessing the value of any potential treatment. This necessitates the use of in vivo models which can be used for in situ examination of RGCs and optic nerves, as well as functional studies reflecting changes in visual function in response to OA-associated genetic mutations, and subsequently assessing treatments. This section evaluates the utility of the most commonly used animal models in the study of mitochondrial disruption in neurons of relevance to OA.
Mouse Models
Anatomically, the mouse retina is stratified in a very similar manner to that of humans, with the outermost ganglion cell layer facing the vitreous body and the innermost retinal pigment epithelium adjacent to the choroid. There are however some notable differences, for example, mice do not possess a collagenous lamina cribrosa, which is relevant to the study of OA as it surrounds and supports the unmyelinated RGC axons as they exit the sclera and form the optic nerve (Chen et al., 2017). Optic nerve density also appears to vary considerably between populations of laboratory mice, which may affect susceptibility to optic nerve atrophy, and should be considered when selecting backgrounds for autosomal OA models (Jeon et al., 1998). As with all models – gene editing – in particular CRISPR/Cas9 mediated gene editing, has become an invaluable tool for recapitulating genetic disease in mice. There are many established protocols for creating KOs, recreating patient-specific mutations and the insertion of human genes of interest into mice (Platt et al., 2014; Chen et al., 2019; Min et al., 2019). However, there are important considerations when selecting a laboratory strain to develop an autosomal OA model as several strains of laboratory mice develop age-related hearing loss, a common extra-ocular symptom of many autosomal OAs (Turner et al., 2005), and basal visual acuity varies significantly between strains (Wong and Brown, 2006), which may confound results. An advantage of mice for modeling OA is the array of assays available for examining both visual function and RGC morphology. Typically, measuring visual acuity orientates around measuring the optokinetic and optomotor reflexes and the pupillary response (Kretschmer et al., 2017). A significant obstacle is that some of these assays require some form of trained behavior. Mice are proficient learners at repetitive tasks, therefore increased task performance over time may somewhat obscure aspects of the degenerative phenotype seen in most forms of OA (Birtalan et al., 2020).
Many mouse models of autosomal OA have been reported and several common features are apparent from these studies (Table 2). Dominantly inherited autosomal OA-causing mutations are generally found to be embryonic lethal in homozygous mice and as such, many mouse studies linked to autosomal OA are conducted in heterozygotes (Wakabayashi et al., 2009; Jiang et al., 2021). Some studies report a progressive loss of visual acuity (Davies et al., 2007; Zaninello et al., 2020), although most do not investigate the presence of a visual phenotype. Many studies observe progressive degeneration of the RGCs which in some cases is accompanied by myelination defects of the optic and peripheral nerves (Li et al., 2017; Waszczykowska et al., 2020). Most models exhibit extra-ocular symptoms common to syndromic-autosomal OA, including progressive motor and hearing impairments often attributed to neuropathy (Sarzi et al., 2012; Mancini et al., 2019). Significantly, nearly all autosomal OA mouse models show some form of neuronal mitochondrial dysfunction, generally in the form of a highly fragmented mitochondrial network with a reduced number of malformed cristae (Powell et al., 2011; Sarzi et al., 2012). Elongated networks have also been observed in mice with Slc25A46 and Dnm1l mutations, consistent with observations from patient fibroblasts (Wan et al., 2016; Gerber et al., 2017b). Furthermore, several mouse models faithfully recapitulate gene-specific symptoms, such as glucose intolerance in Wfs1 mutants (Ishihara et al., 2004; Kõks et al., 2009). Taken together, it’s clear that mouse models of autosomal OA can recapitulate key characteristics of this disorder. Despite many autosomal OA-type phenotypes, there are several phenotypes which are consistently observed in mouse autosomal OA-models which are not generally associated with the corresponding human pathogenic mutations. Most commonly, mice with loss of autosomal OA-causing genes display: reduced body size and difficulty gaining weight (Smith et al., 2012; Wells et al., 2012) and cardiomyopathy (Davies et al., 2008; Jiang et al., 2021). In many cases, these symptoms are not detectable in patients carrying the comparable pathogenic mutation and whilst many autosomal OA-causing genes are thought to have a developmental role in humans, there is presently little evidence for developmental abnormalities affecting these organs in patients (Maltecca et al., 2008; Caglayan et al., 2020).

Table 2. Summary of phenotypes observed in in vivo models of autosomal OA.
Zebrafish-Danio rerio Models
Danio rerio (subsequently referred to simply as zebrafish), and particularly zebrafish larvae, are an emerging model in vision research. The stratification and function of the zebrafish retina is very similar to that of humans, with a comparable cone density. Zebrafish have four different cone types in comparison to the three cone types found in humans, conferring superior color vision, which is often affected in autosomal OA patients. Zebrafish possess RGCs which converge to form the optic nerve (also known as cranial nerve II in zebrafish), cross at the optic chiasm, and innervate the optic tectum, the visual center of the brain (Diekmann et al., 2015). Notably, RGCs in zebrafish exhibit intraretinal myelination, in contrast to rodents and primates in which myelination of the optic nerve begins a short distance outside of the globe of the eye. Unlike mammals, zebrafish can regenerate their optic nerve following chemical or mechanical damage, although this ability reduces somewhat with age (Münzel et al., 2014). It is unclear whether this will impede the pathogenic effects of autosomal OA-associated mutations. Zebrafish visual function can be readily assessed in larvae, principally through measuring the optokinetic (OKR) and visual motor responses (VMR), on a much larger scale than rodents (Emran et al., 2008; Zou et al., 2010). Zebrafish models are available for study much faster than rodent models with RGCs differentiating at 28 h post fertilization and OKR and VMR assays possible from 3 days post fertilization. At least 71% of human genes have an ortholog in zebrafish (Howe et al., 2013), with most genes demonstrating between 50 and 80% homology with their human equivalent (Broughton et al., 2001). Notably, there was a whole genome duplication event in the evolution of teleost fish (Pasquier et al., 2016) and consequently, many zebrafish homologs for mammalian genes are present in duplicate and may exhibit some degree of functional redundancy. This is the case for Wfs1 (Cairns, 2019), and may confound results when recreating pathogenic mutations (Kleinjan et al., 2008; Lu et al., 2012). Zebrafish develop externally, making them accessible to genetic manipulation through microinjection. There are 2 principal methods of genetic manipulation in zebrafish embryos, CRISPR/Cas9 and morpholinos. Morpholinos are short oligomers designed to be complementary to the transcription start site or splice site of pre-mRNAs to transiently block splicing or translation of a desired transcript, reducing the expression of the target gene. Although the typical duration of the knockdown (usually a few days post-injection) would be sufficient for studies in larvae, it would not be sustained in adult fish (Rahn et al., 2013). Whilst this could be used to look at the developmental abnormalities implicated in some of the autosomal OA-associated genes, it may be unsuitable for looking at progressive, degenerative phenotypes. It should also be noted that as much as 20% of morpholinos exhibit non-specific toxicity, particularly relating to apoptotic cell death (Ekker and Larson, 2001; Robu et al., 2007). CRISPR/Cas9 editing technology has been extensively exploited in zebrafish, with some protocols reporting editing efficiencies over 90% (Hoshijima et al., 2019). This technique can be used to create stable, heritable Kos of genes, but can additionally be utilized to insert human transgenes, or replicate patient mutations. Finally, zebrafish have many well established transgenic reporter lines, a particularly effective approach in translucent zebrafish embryos. There are several examples of lines expressing fluorescent proteins under the control of retinal ganglion cell specific promoters, for example Atonal BHLH Transcription Factor 7 (atoh7; Masai et al., 2003). Multiple transgenic lines specifically targeting mitochondria are also available, notably zebrafish with mitochondrial expression of photo-activatable fluorescent protein (mito-PAGFP) which, after photoactivation, allows for the tracking of individual mitochondria within sensory neurons (Wehnekamp et al., 2019; Mieskes et al., 2020), and MitoFish, which has been used for intravital imaging of mitochondria in immobilized larvae (Paquet et al., 2014). It would be valuable to apply available transgenic approaches to the study of retinal ganglion cell mitochondrial structure and function in zebrafish disease models of autosomal-OA.
To date, most of the published zebrafish autosomal OA models are based on morpholino-based knockdown of gene expression, many of which present with abnormalities in eye and optic nerve morphology (Pei et al., 2010; Del Dotto et al., 2020). Although vision has only been addressed in a limited number of studies, reduced visual acuity has been noted (Angebault et al., 2015; Cairns, 2019). Common secondary symptoms of autosomal OA such as motor deficits (Rahn et al., 2013; Angebault et al., 2015) and inner ear abnormalities (Cairns, 2019; Del Dotto et al., 2020) have also been observed. Models frequently exhibit mitochondrial abnormalities including disrupted mitochondrial networks, respiratory deficiency, and reduced mitochondrial trafficking (Abrams et al., 2015; Eijkenboom et al., 2019). As in the murine models, many zebrafish homozygous for autosomal OA-causing genes exhibit developmental defects of the heart and brain (Wan et al., 2016; Buglo et al., 2020; Del Dotto et al., 2020), perhaps emphasizing the need for further research into any developmental origins for autosomal OA pathogenicity. Given the relative ease of generating novel zebrafish models, there is ample opportunity for creating models for autosomal OA-causing genes, in addition to examining degenerative phenotypes in adult fish. The extant zebrafish models could be well suited to drug screening, due to the prevalence of strong visual phenotypes from the early larval stage and relative ease with which visual acuity can be determined.
Drosophila Models
The fruit fly, Drosophila melanogaster, is a well-established in vivo model system with at least 75% of disease-linked genes having Drosophila orthologs (Reiter et al., 2001). They are relatively inexpensive to rear, produce large numbers of offspring and have a maximal lifespan of approximately 3 months (Huang et al., 2020), which may prove useful in studying degenerative diseases. Of relevance to autosomal OA, flies have eyes, relatively complex brains and some organs which have comparable function to their human equivalents. A notable disadvantage of Drosophila as a model for autosomal OA is the significant disparity in eye morphology. Flies have compound eyes, comprising over 700 repeating units called ommatidia. Each ommatidium is composed of eight photoreceptors which do not converge to form a structure resembling the optic nerve (Paulk et al., 2013). Despite the lack of RGCs, Drosophila offer a strong platform for examining neurons in vivo. Their relatively simple organization allows for easily reproducible examination of the morphology and function of long axons in situ (Dolph et al., 2011). As with other models, CRISPR techniques have meant that targeted gene editing is now relatively straightforward in flies, though most published studies to date have employed knockdown of gene expression.
Efficient ubiquitous knockdown of most OA-associated genes is lethal, corroborating findings in the other animal models suggesting a developmental role for many autosomal OA genes. As such the UAS-GAL4 system, which allows for generation of tissue-specific expression of RNAis (Dolan et al., 2017; Ogienko et al., 2020), is commonly used to study the role of autosomal OA-causing genes by targeted knockdown in neurons. Locomotor issues are common, in some instances age-dependent and progressive, generally in keeping with mobility issues present in corresponding patients (Sakakibara et al., 2018; Suda et al., 2018; Pareek and Pallanck, 2020). The majority of fly autosomal OA-models do not have a gross eye phenotype, but the limited number of studies that investigated visual function have reported developmental and electrical response defects (Maier et al., 2001; Verstreken et al., 2005). Most published Drosophila models of autosomal OA-associated genes have highly abnormal mitochondria including: abnormal cristae, fragmented or aggregated mitochondrial networks, perturbed axonal trafficking, mtDNA depletion, hyperfused or elongated mitochondria, reduced ATP content, increased ROS, and reduced abundance and/or activity of complexes of the electron transport chain, generally replicating the perturbation seen in patients (Tang et al., 2009; Pareek and Pallanck, 2020). Similar to the mice, some fly models exhibit heart tube abnormalities, despite abnormal cardiac development not being prevalent in patients (Shahrestani et al., 2009; Bhandari et al., 2015). Overall, the few existing Drosophila autosomal OA models present a strong platform for examining mitochondrial and neurodegenerative phenotypes associated with autosomal OA genes.
Caenorhabditis elegans Models
Like Drosophila, the roundworm Caenorhabditis elegans has been an invaluable asset to biological research generally, as an extremely simple and abundant organism. C. elegans is inexpensive to rear, can produce up to 1,000 offspring, has an extremely short lifespan of ∼20 days, and may be kept in frozen storage over long periods of time and revived (Vita-More and Barranco, 2015). Human homologs exist for at least 80% of the C. elegans proteome (Lai et al., 2000) with known worm homologs for most autosomal OA genes, with the exception of WFS1. C. elegans does not possess eyes, retinas, or opsin pigments, and therefore has no structure that directly parallels the optic nerve or retinal ganglion cells. Instead, it perceives UV light using its distinct LITE-1 photoreceptor (Edwards et al., 2008; Gong et al., 2016), Due to this disparity between the human and nematode visual systems, they have limited use in directly modeling visual defects associated with autosomal OA. Nevertheless, C. elegans presents an interesting model for the study of mitochondrial morphology and dynamics, particularly within neurons, due to their simplistic and transparent body plan facilitating a wide array of microscopy techniques. Furthermore, as gene editing in C. elegans can be as straightforward as feeding sgRNA to transgenic animals (Liu et al., 2014), combined with the comparatively small expense of roundworms as a model organism, this could be a highly accessible avenue for expanding the field of autosomal OA research.
Many C. elegans mutants for autosomal OA-linked genes exist (Table 2), however many were created during large mutagenesis or RNAi screens. Despite this, there are several phenotypes seen within the C. elegans autosomal OA models that are conserved within higher eukaryotic organisms, notably abnormal mitochondrial morphology often accompanied by irregular cristae (Knowlton et al., 2017; Machiela et al., 2020), respiratory defects (Fujii et al., 2011; Luz et al., 2015), embryonic or larval lethality (Zubovych et al., 2010; Nargund et al., 2012), locomotor dysfunction (Machiela et al., 2020), and neuronal dysfunction (Zaninello et al., 2020). There are some notable disparities between the vertebrate and C. elegans autosomal OA models. While aberrant expression of autosomal OA genes frequently reduces maximal lifespan in Drosophila or vertebrate models, in some worm autosomal OA models lifespan is extended (Kanazawa et al., 2008; Chaudhari and Kipreos, 2017; Maglioni et al., 2019). This may be due to the ability of C. elegans to arrest development in the dauer state in response to adverse environmental conditions. Dauer larvae are highly resistant to all forms of stress, including oxidative stress, favor anaerobic respiration, and are comparatively long-lived. In at least two studies arrest in the dauer state was observed (Avery, 1993; Maglioni et al., 2019). As with all studies involving model systems, careful consideration must be given to ensure that the system used is appropriate for the research question being investigated.
Retinal Organoids
Much of our understanding of human biology has stemmed from research involving animal model organisms like those discussed previously. They have played a substantial role in informing many important scientific discoveries over the years, however, there are many fundamental differences that exist between humans and their model organism counterparts (Huch et al., 2017), especially in the context of optic neuropathies. Recent studies have highlighted significant differences in both the number and distribution of RGCs in rodents compared to primates (Peng et al., 2019) and this may explain why therapeutic approaches that appear to show great promise in animal models do not always translate effectively to human patients, highlighting the need for an accurate ex vivo model. The discovery of induced pluripotent stem cells (iPSCs) has enabled biologists to reprogram human somatic cells to a pluripotent, embryonic stem cell-like state (Takahashi et al., 2007), which, combined with recent improvements in three dimensional culture techniques has led to the emergence of organoid models as a potential avenue to remedy this issue.
An “organoid” can be simply defined as a structure that resembles an organ (Dutta et al., 2017). Derived from either pluripotent stem cells or organ progenitor cells, they undergo differentiation, forming organ-like structures in a process which appears to mimic in vivo organogenesis (Dutta et al., 2017). The first protocol for the directed differentiation of retinal organoids was published in 2012 (Nakano et al., 2012) and since then a variety of approaches have been developed (Chen et al., 2016; Chichagova et al., 2019). The differentiation process results in the formation of retinal organoids that contain all of the major retinal cell types, organized in a laminar manner which emerge at roughly the same time points as in the human retina (Capowski et al., 2019; Chichagova et al., 2019). RGCs are the first distinct cell population to emerge during retinal development and this is mirrored in organoid development (Rabesandratana et al., 2018). Differential expression of OPN4 reveals the presence of melanopsin expressing-photosensitive RGCs at as early as 5 weeks of differentiation, with positive immunostaining for the RGC-specific markers Smi32 and RBPMS indicating their orientation in the basal aspect of the organoid (Mellough et al., 2019), reminiscent of the native retina. Mitochondrial function can also be studied with relative ease in retinal organoids through the use of transmission electron microscopy (Duong et al., 2021) and the use of bioassays measuring viability (Das et al., 2020), ATP production (Duong et al., 2021) and oxygen consumption rate (Roy-Choudhury and Daadi, 2019). These organoids are capable of forming functional synapses and are responsive to light stimulation (Mellough et al., 2015), however, they still lack many features of the native retina – such as vasculature (Laschke and Menger, 2012) and the presence of microglial cells (Fathi et al., 2021) – which can somewhat limit their use in the context of modeling optic neuropathies. Retinal organoids are becoming an increasingly attractive model for the study of inherited ocular disorders as they provide researchers with the unique opportunity to model patient-specific mutations through the use of iPSCs (Bell et al., 2020). As with in vivo models, CRISPR/Cas9 gene-editing technology can also be applied, either to introduce de novo mutations or to generate isogenic controls by correcting mutations in patient-derived iPSC lines (VanderWall et al., 2020). While disease modeling through the use of retinal organoids is highly applicable, there are currently no published examples of retinal organoids being used to model OA.
The main advantage of disease modeling through the use of retinal organoids is that they provide researchers with the opportunity to study human disease within a human model system while simultaneously circumventing the ethical challenges surrounding the study of early embryonic development in humans. Organoids are highly amenable to drug screening and to genetic manipulation, meaning they have a wide range of potential applications within this field. However, there are still many issues that need to be addressed in order for retinal organoids to reach their full potential. Despite being able to produce all of the major retinal cell types (Chichagova et al., 2019), as time in culture is extended the innermost layers of the organoid containing the RGCs begin to degenerate (Fathi et al., 2021). This is likely due to the fact that there is no vasculature present in the organoid, so as it grows bigger the amount of oxygen and nutrients that can diffuse through to the inner layers is not sufficient to maintain viability (Fathi et al., 2021), while spatial constraints imposed by the culture vessels in which organoids are grown mean that RGC axons cannot extend to the length they normally would in the native retina (Ohlemacher et al., 2019). In addition to this, microglial cells, which would ordinarily interact with Müller glial cells to regulate neuronal development (Li et al., 2019) are absent (Fathi et al., 2021), while mature structures such as an optic nerve are also not present. Retinal organoids have already proven to be a valuable tool for studying development and disease, and hold huge therapeutic potential from the perspective of regenerative medicine (Kobayashi et al., 2018; Eastlake et al., 2019). They can overcome limitations of in vivo disease models in that being a human model system removes the need to account for cross-species differences and could also bypass the issue of homozygous lethality caused by the introduction of OA-causing mutations seen in many of the other models. The ability to produce fully functional, mature retinal organoids in combination with gene-editing technologies such as CRISPR could hold the key to the treatment of inherited neuropathies such as autosomal OA. Research in this field is constantly evolving with the development of co-culture methods to promote RGC axon growth (Fligor et al., 2021) and the potential use of CRISPR reporter iPSC lines making it possible to produce organoids that resemble their human organoid counterparts even more closely.
The conservation of mitochondrial aberrations and phenotypic symptoms seen in both animal models and patients supports the causative relationship between OA pathology and mitochondrial dysfunction. Additionally, as visual pathology is often able to be replicated, specifically in zebrafish and mice, through expression of OA-causing gene mutations, this indicates that RGC vulnerability likely relates to common anatomical and molecular features within these models. This is especially relevant as research is underdeveloped in this area. There is some disparity between the animal models discussed here and patients, for example heart and brain developmental abnormalities (Angebault et al., 2015). Retinal organoids derived from human cells could be a useful addition to the field for the study of human RGCs, however it is clear that there is significant refinement required before they are sufficiently able to replicate in vivo human RGC morphology and function. The model organisms discussed here already have assays and histological techniques which would be readily applicable to the study of autosomal OA: Drosophila and C. elegans are well suited to provisionally studying perturbations in mitochondrial and neuronal function, whilst mice and zebrafish are highly applicable for examining changes in RGCs and visual acuity. Lastly, there is currently a paucity of studies investigating genetic or therapeutic modifiers of OA-associated degeneration, despite the abundance of methods to do so in all species mentioned. This is of eminent importance for future development in this field due to the lack of treatment options available for patients.
Concluding Remarks
Autosomal OA is a highly genetically heterogeneous condition, with a growing list of genes which have been shown to give rise to the disorder. Despite this, as we have made clear in this review, there is an evident commonality with all presently implicated genes involved in the function and organization of mitochondria. It is unclear whether a particular aspect of mitochondrial function makes RGCs vulnerable, or induces cell degeneration, however this may be further elucidated through examining common pathways between affected genes, and common dysfunction caused by pathogenic mutations. Although various animal models bearing mutations for autosomal OA-genes exist, many recreate mutations associated with other diseases. Given the accessibility of CRISPR/Cas9 gene editing technologies, this could be easily remedied. Finally, retinal organoids present an exciting intermediary between in vitro and in vivo model systems, which could be used for the study of autosomal OA-associated pathological changes within the human eye. Any of the models discussed here could be utilized for drug screening so that we might begin to develop much needed treatment strategies for retinal disease.
Author Contributions
ES, DM, AR, BK, and NO’S wrote and edited the manuscript. ES prepared the figures. JC commented on and reviewed the manuscript prior to submission. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
This work was supported by the Irish Research Council (IRC). ES and DM are supported by IRC-Government of Ireland Ph.D. studentships (Grant Nos. GOIPG/2020/1312 and GOIPG/2020/1636). Work in the JC lab is supported by grants from Science Foundation Ireland (Grant Nos. 19/FFP/6833 and 13/RC/20733) and the European Regional Development Fund (Grant No. 13/RC/2071).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figures were created with BioRender.com.
References
Abrams, A. J., Hufnagel, R. B., Rebelo, A., Zanna, C., Patel, N., Gonzalez, M. A., et al. (2015). Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 47, 926–932. doi: 10.1038/ng.3354
Ali, M. S., Suda, K., Kowada, R., Ueoka, I., Yoshida, H., and Yamaguchi, M. (2020). Neuron-specific knockdown of solute carrier protein SLC25A46a induces locomotive defects, an abnormal neuron terminal morphology, learning disability, and shortened lifespan. IBRO Rep. 8, 65–75. doi: 10.1016/j.ibror.2020.02.001
Almind, G. J., Ek, J., Rosenberg, T., Eiberg, H., Larsen, M., LuCamp, L., et al. (2012). Dominant optic atrophy in Denmark - report of 15 novel mutations in OPA1, using a strategy with a detection rate of 90. BMC Med. Genet. 13:65. doi: 10.1186/1471-2350-13-65
Altanbyek, V., Cha, S.-J., Kang, G.-U., Im, D. S., Lee, S., Kim, H.-J., et al. (2016). Imbalance of mitochondrial dynamics in Drosophila models of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 481, 259–264. doi: 10.1016/j.bbrc.2016.10.134
Amati-Bonneau, P., Milea, D., Bonneau, D., Chevrollier, A., Ferré, M., Guillet, V., et al. (2009). OPA1-associated disorders: Phenotypes and pathophysiology. Int. J. Biochem. Cell Biol. 41, 1855–1865. doi: 10.1016/j.biocel.2009.04.012
Anand, R., Wai, T., Baker, M. J., Kladt, N., Schauss, A. C., Rugarli, E., et al. (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929. doi: 10.1083/jcb.201308006
Angebault, C., Fauconnier, J., Patergnani, S., Rieusset, J., Danese, A., Affortit, C., et al. (2018). ER-mitochondria cross-talk is regulated by the Ca 2+ sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal. 11:eaaq1380. doi: 10.1126/scisignal.aaq1380
Angebault, C., Guichet, P.-O., Talmat-Amar, Y., Charif, M., Gerber, S., Fares-Taie, L., et al. (2015). Recessive mutations in RTN4IP1 cause isolated and syndromic optic neuropathies. Am. J. Hum. Genet. 97, 769–769. doi: 10.1016/j.ajhg.2015.10.015
Avery, L. (1993). The genetics of feeding in caenorhabditis elegans. Genetics 133, 897–917. doi: 10.1093/genetics/133.4.897
Baderna, V., Schultz, J., Kearns, L. S., Fahey, M., Thompson, B. A., Ruddle, J. B., et al. (2020). A novel AFG3L2 mutation close to AAA domain leads to aberrant OMA1 and OPA1 processing in a family with optic atrophy. Acta. Neuropathol. Commun. 8, 1–93. doi: 10.1186/s40478-020-00975-w
Bahr, T., Welburn, K., Donnelly, J., and Bai, Y. (2020). Emerging model systems and treatment approaches for Leber’s hereditary optic neuropathy: Challenges and opportunities. Biochim. Biophys. Acta. Mol. Basis. Dis. 1866, 165743–165743. doi: 10.1016/j.bbadis.2020.165743
Batzir, N., Bhagwat, P. K., Eble, T. N., Liu, P., Eng, C. M., Elsea, S. H., et al. (2019). De novo missense variant in the GTPase effector domain (GED) of DNM1L leads to static encephalopathy and seizures. Cold Spring Harb. Mol. Case Stud. 5:a003673. doi: 10.1101/mcs.a003673
Bell, C. M., Zack, D. J., and Berlinicke, C. A. (2020). Human organoids for the study of retinal development and disease. Annu. Rev. Vis. Sci. 6, 91–114. doi: 10.1146/annurev-vision-121219-081855
Benedetti, C., Haynes, C. M., Yang, Y., Harding, H. P., and Ron, D. (2006). Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174, 229–239. doi: 10.1534/genetics.106.061580
Bhandari, P., Song, M., and Dorn, G. W. (2015). Dissociation of mitochondrial from sarcoplasmic reticular stress in Drosophila cardiomyopathy induced by molecularly distinct mitochondrial fusion defects. J. Mol. Cell. Cardiol. 80, 71–80. doi: 10.1016/j.yjmcc.2014.12.018
Birtalan, E., Bánhidi, A., Sanders, J. I., Balázsfi, D., and Hangya, B. (2020). Efficient training of mice on the 5-choice serial reaction time task in an automated rodent training system. Sci. Rep. 10, 22362. doi: 10.1038/s41598-020-79290-2
Bitetto, G., Malaguti, M. C., Ceravolo, R., Monfrini, E., Straniero, L., Morini, A., et al. (2020). SLC25A46 mutations in patients with Parkinson’s Disease and optic atrophy. Parkinsonism. Relat. Disord. 74, 1–5. doi: 10.1016/j.parkreldis.2020.03.018
Bombelli, F., Stojkovic, T., Dubourg, O., Echaniz-Laguna, A., Tardieu, S., Larcher, K., et al. (2014). Charcot-Marie-Tooth Disease Type 2A: From Typical to Rare Phenotypic and Genotypic Features. JAMA Neurol. 71, 1036–1042. doi: 10.1001/jamaneurol.2014.629
Bonnet-Wersinger, D., Benkafadar, N., Jagodzinska, J., Hamel, C., Tanizawa, Y., Lenaers, G., et al. (2014). Impairment of visual function and retinal ER stress activation in Wfs1-deficient mice. PloS One. 9:e97222. doi: 10.1371/journal.pone.0097222
Breckenridge, D. G., Kang, B.-H., Kokel, D., Mitani, S., Staehelin, L. A., and Xue, D. (2008). Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol. Cell. 31, 586–597. doi: 10.1016/j.molcel.2008.07.015
Broughton, R. E., Milam, J. E., and Roe, B. A. (2001). The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome. Res. 11, 1958–1967. doi: 10.1101/gr.156801
Buglo, E., Sarmiento, E., Martuscelli, N. B., Sant, D. W., Danzi, M. C., Abrams, A. J., et al. (2020). Genetic compensation in a stable slc25a46 mutant zebrafish: A case for using F0 CRISPR mutagenesis to study phenotypes caused by inherited disease. PloS. One 15:e0230566. doi: 10.1371/journal.pone.0230566
Byrne, J. J., Soh, M. S., Chandhok, G., Vijayaraghavan, T., Teoh, J.-S., Crawford, S., et al. (2019). Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell. Mol. Life Sci. 76, 1967–1985. doi: 10.1007/s00018-019-03024-5
Cagalinec, M., Liiv, M., Hodurova, Z., Hickey, M. A., Vaarmann, A., Mandel, M., et al. (2016). Role of mitochondrial dynamics in neuronal development: mechanism for wolfram syndrome. PLoS. Biol. 14:e1002511. doi: 10.1371/journal.pbio.1002511
Caglayan, S., Hashim, A., Cieslar-Pobuda, A., Jensen, V., Behringer, S., Talug, B., et al. (2020). Optic atrophy 1 controls human neuronal development by preventing aberrant nuclear DNA methylation. iScience 23:101154. doi: 10.1016/j.isci.2020.101154
Cairns, G. E. (2019). Characterisation of a zebrafish model of Wolfram Syndrome. [PhD Thesis]. Newcastle, UK: Newcastle University.
Caporali, L., Magri, S., Legati, A., Del Dotto, V., Tagliavini, F., Balistreri, F., et al. (2020). ATPase domain AFG3L2 mutations alter OPA1 processing and cause optic neuropathy. Ann. Neurol. 88, 18–32. doi: 10.1002/ana.25723
Capowski, E. E., Samimi, K., Mayerl, S. J., Phillips, M. J., Pinilla, I., Howden, S. E., et al. (2019). Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development 146:dev171686. doi: 10.1242/dev.171686
Carelli, V., Musumeci, O., Caporali, L., Zanna, C., La Morgia, C., Del Dotto, V., et al. (2015). Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 78, 21–38. doi: 10.1002/ana.24410
Casson, R. J., Chidlow, G., Crowston, J. G., Williams, P. A., and Wood, J. P. M. (2021). Retinal energy metabolism in health and glaucoma. Prog. Retin. Eye. Res. 81:100881. doi: 10.1016/j.preteyeres.2020.100881
Cesnekova, J., Rodinova, M., Hansikova, H., Zeman, J., and Stiburek, L. (2018). Loss of mitochondrial AAA proteases AFG3L2 and YME1L impairs mitochondrial structure and respiratory chain biogenesis. Int. J. Mol. Sci. 19:3930. doi: 10.3390/ijms19123930
Chao, Y.-H., Robak, L. A., Xia, F., Koenig, M. K., Adesina, A., Bacino, C. A., et al. (2016). Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila. Hum. Mol. Genet. 25, 1846–1856. doi: 10.1093/hmg/ddw059
Charif, M., Chevrollier, A., Gueguen, N., Bris, C., Goudenège, D., Desquiret-Dumas, V., et al. (2020). Mutations in the m-AAA proteases AFG3L2 and SPG7 are causing isolated dominant optic atrophy. Neurol. Genet. 6:e428. doi: 10.1212/NXG.0000000000000428
Charif, M., Bris, C., Goudenège, D., Desquiret-Dumas, V., Colin, E., Ziegler, A., et al. (2021a). Use of next-generation sequencing for the molecular diagnosis of 1,102 patients with a autosomal optic neuropathy. Front. Neurol. 12:602979. doi: 10.3389/fneur.2021.602979
Charif, M., Gueguen, N., Ferré, M., Elkarhat, Z., Khiati, S., LeMao, M., et al. (2021b). Dominant ACO2 mutations are a frequent cause of isolated optic atrophy. Brain. Commun. 3:fcab063. doi: 10.1093/braincomms/fcab063
Charif, M., Wong, Y. C., Kim, S., Guichet, A., Vignal, C., Zanlonghi, X., et al. (2021c). Dominant mutations in MIEF1 affect mitochondrial dynamics and cause a singular late onset optic neuropathy. Mol. Neurodegener. 16, 12–12. doi: 10.1186/s13024-021-00431-w
Charif, M., Nasca, A., Thompson, K., Gerber, S., Makowski, C., Mazaheri, N., et al. (2017). Neurologic phenotypes associated with mutations in RTN4IP1 (OPA10) in children and young adults. JAMA Neurol. 75, 105–113. doi: 10.1001/jamaneurol.2017.2065
Chaudhari, S. N., and Kipreos, E. T. (2017). Increased mitochondrial fusion allows the survival of older animals in diverse C. Elegans longevity pathways. Nat. Commun. 8, 182–116. doi: 10.1038/s41467-017-00274-4
Chen, H., Vermulst, M., Wang, Y. E., Chomyn, A., Prolla, T. A., McCaffery, J. M., et al. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 141, 280–289. doi: 10.1016/j.cell.2010.02.026
Chen, H. Y., Kaya, K. D., Dong, L., and Swaroop, A. (2016). Three-dimensional retinal organoids from mouse pluripotent stem cells mimic in vivo development with enhanced stratification and rod photoreceptor differentiation. Mol. Vis. 22, 1077–1094.
Chen, L., Zhao, Y., and Zhang, H. (2017). Comparative anatomy of the trabecular meshwork, the optic nerve head and the inner retina in rodent and primate models used for glaucoma research. Vision (Basel) 1:4. doi: 10.3390/vision1010004
Chen, S., Sun, S., Moonen, D., Lee, C., Lee, A. Y.-F., Schaffer, D. V., et al. (2019). CRISPR-READI: efficient generation of knockin mice by CRISPR RNP Electroporation and AAV donor infection. Cell. Rep. 27, 3780–3789. doi: 10.1016/j.celrep.2019.05.103
Chichagova, V., Dorgau, B., Felemban, M., Georgiou, M., Armstrong, L., and Lako, M. (2019). Differentiation of retinal organoids from human pluripotent stem cells. Curr. Protoc. Stem. Cell. Biol. 50:e95. doi: 10.1002/cpsc.95
Ciesielski, G. L., Bermek, O., Rosado-Ruiz, F. A., Hovde, S. L., Neitzke, O. J., Griffith, J. D., et al. (2015). Mitochondrial Single-stranded DNA-binding proteins stimulate the activity of DNA polymerase by organization of the template DNA. J. Biol. Chem. 290, 28697–28707. doi: 10.1074/jbc.M115.673707
Cogliati, S., Frezza, C., Soriano, Maria, E., Varanita, T., Quintana-Cabrera, R., et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. doi: 10.1016/j.cell.2013.08.032
Cohn, A. C., Toomes, C., Potter, C., Towns, K. V., Hewitt, A. W., Inglehearn, C. F., et al. (2008). Autosomal dominant optic atrophy: penetrance and expressivity in patients with OPA1 mutations. Am. J. Ophthalmol. 143, 656–662. doi: 10.1016/j.ajo.2006.12.038
Colavito, D., Maritan, V., Suppiej, A., Del Giudice, E., Mazzarolo, M., Miotto, S., et al. (2017). Non-syndromic isolated dominant optic atrophy caused by the p.R468C mutation in the AFG3 like matrix AAA peptidase subunit 2 gene. Biomed. Rep. 7, 451–454. doi: 10.3892/br.2017.987
Da Cruz, S., Xenarios, I., Langridge, J., Vilbois, F., Parone, P. A., and Martinou, J.-C. (2003). Proteomic analysis of the mouse liver mitochondrial inner membrane. J. Biol. Chem. 278, 41566–41571. doi: 10.1074/jbc.M304940200
D’Angelo, L., Astro, E., De Luise, M., Kurelac, I., Umesh-Ganesh, N., Ding, S., et al. (2021). NDUFS3 depletion permits complex I maturation and reveals TMEM126A/OPA7 as an assembly factor binding the ND4-module intermediate. Cell. Rep. 35:109002. doi: 10.1016/j.celrep.2021.109002
Das, A., Bell, C. M., Berlinicke, C. A., Marsh-Armstrong, N., and Zack, D. J. (2020). Programmed switch in the mitochondrial degradation pathways during human retinal ganglion cell differentiation from stem cells is critical for RGC survival. Redox. Biol. 34:101465. doi: 10.1016/j.redox.2020.101465
Davies, V. J., Hollins, A. J., Piechota, M. J., Yip, W., Davies, J. R., White, K. E., et al. (2007). Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 16, 1307–1318. doi: 10.1093/hmg/ddm079
Davies, V. J., Powell, K. A., White, K. E., Yip, W., Hogan, V., Hollins, A. J., et al. (2008). A missense mutation in the murine Opa3 gene models human Costeff syndrome. Brain 131, 368–380. doi: 10.1093/brain/awm333
Del Dotto, V., Fogazza, M., Musiani, F., Maresca, A., Aleo, S. J., Caporali, L., et al. (2018). Deciphering OPA1 mutations pathogenicity by combined analysis of human, mouse and yeast cell models. Biochim. Biophys. Acta. Mol. Basis. Dis. 1864, 3496–3514. doi: 10.1016/j.bbadis.2018.08.004
Del Dotto, V., Ullah, F., Di Meo, I., Magini, P., Gusic, M., Maresca, A., et al. (2020). SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. J. Clin. Investig. 130, 108–125. doi: 10.1172/JCI128514
Delprat, B., Maurice, T., and Delettre, C. (2018). Wolfram syndrome: MAMs’ connection? Cell Death Dis. 9:364. doi: 10.1038/s41419-018-0406-3
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta. Bioenerg. 1787, 1309–1316. doi: 10.1016/j.bbabio.2009.01.005
Désir, J., Coppieters, F., van Regemorter, N., de Baere, E., Abramowicz, M., and Cordonnier, M. (2012). TMEM126A mutation in a Moroccan family with autosomal recessive optic atrophy. Mol. Vis. 18, 1849–1857.
D’Gama, A. M., England, E., Madden, J. A., Shi, J., Chao, K. R., Wojcik, M. H., et al. (2021). Exome sequencing identifies novel missense and deletion variants in RTN4IP1 associated with optic atrophy, global developmental delay, epilepsy, ataxia, and choreoathetosis. Am. J. Med. Genet. A. 185, 203–207. doi: 10.1002/ajmg.a.61910
Diekmann, H., Kalbhen, P., and Fischer, D. (2015). Characterization of optic nerve regeneration using transgenic zebrafish. Front. Cell. Neurosci. 9:118. doi: 10.3389/fncel.2015.00118
Dolan, M.-J., Luan, H., Shropshire, W. C., Sutcliffe, B., Cocanougher, B., Scott, R. L., et al. (2017). Facilitating neuron-specific genetic manipulations in Drosophila melanogaster using a split GAL4 repressor. Genetics 206, 775–784. doi: 10.1534/genetics.116.199687
Dolph, P., Nair, A., and Raghu, P. (2011). Electroretinogram recordings of Drosophila. Cold. Spring. Harb. Protoc. 6:rot5549. doi: 10.1101/pdb.prot5549
Duarte, J. N. (2021). Neuroinflammatory mechanisms of mitochondrial dysfunction and neurodegeneration in glaucoma. J. Ophthalmol. 2021:4581909. doi: 10.1155/2021/4581909
Duchesne, A., Vaiman, A., Castille, J., Beauvallet, C., Gaignard, P., Floriot, S., et al. (2017). Bovine and murine models highlight novel roles for SLC25A46 in mitochondrial dynamics and metabolism, with implications for human and animal health. PLoS Genet. 13:e1006597. doi: 10.1371/journal.pgen.1006597
Duong, A., Evstratova, A., Sivitilli, A., Hernandez, J. J., Gosio, J., Wahedi, A., et al. (2021). Characterization of mitochondrial health from human peripheral blood mononuclear cells to cerebral organoids derived from induced pluripotent stem cells. Sci. Rep. 11:4523. doi: 10.1038/s41598-021-84071-6
Dutta, D., Heo, I., and Clevers, H. (2017). Disease modeling in stem cell-derived 3D organoid systems. Trends. Mol. Med. 23, 393–410. doi: 10.1016/j.molmed.2017.02.007
Eastlake, K., Wang, W., Jayaram, H., Murray-Dunning, C., Carr, A. J. F., Ramsden, C. M., et al. (2019). Phenotypic and functional characterization of müller glia isolated from induced pluripotent stem cell-derived retinal organoids: improvement of retinal ganglion cell function upon transplantation. Stem. Cells. Transl. Med. 8, 775–784. doi: 10.1002/sctm.18-0263
Edwards, S. L., Charlie, N. K., Milfort, M. C., Brown, B. S., Gravlin, C. N., Knecht, J. E., et al. (2008). A novel molecular solution for ultraviolet light detection in Caenorhabditis elegans. PLoS Biol 6:e60198. doi: 10.1371/journal.pbio.0060198
Eijkenboom, I., Vanoevelen, J. M., Hoeijmakers, J. G. J., Wijnen, I., Gerards, M., Faber, C. G., et al. (2019). A zebrafish model to study small-fiber neuropathy reveals a potential role for GDAP1. Mitochondrion 47, 273–281. doi: 10.1016/j.mito.2019.01.002
Ekker, S. C., and Larson, J. D. (2001). Morphant technology in model developmental systems. Genesis 30, 89–93. doi: 10.1002/gene.1038
Emran, F., Rihel, J., and Dowling, J. E. (2008). A behavioral assay to measure responsiveness of Zebrafish to changes in light intensities. J. Vis. Exp. 3:923. doi: 10.3791/923
Fahrner, J. A., Liu, R., Perry, M. S., Klein, J., and Chan, D. C. (2016). A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. A. 170A, 2002–2011. doi: 10.1002/ajmg.a.37721
Fathi, M., Ross, C. T., and Hosseinzadeh, Z. (2021). Functional 3-dimensional retinal organoids: technological progress and existing challenges. Front. Neurosci. 15:668857. doi: 10.3389/fnins.2021.668857
Fedorenko, A., Lishko, P. V., and Kirichok, Y. (2012). Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413. doi: 10.1016/j.cell.2012.09.010
Ferré, M., Bonneau, D., Milea, D., Chevrollier, A., Verny, C., Dollfus, H., et al. (2009). Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum. Mutat. 30, E692–E705. doi: 10.1002/humu.21025
Fiermonte, G., Palmieri, L., Todisco, S., Agrimi, G., Palmieri, F., and Walker, J. E. (2002). Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 277, 19289–19294. doi: 10.1074/jbc.M201572200
Fligor, C. M., Lavekar, S. S., Harkin, J., Shields, P. K., VanderWall, K. B., Huang, K. C., et al. (2021). Extension of retinofugal projections in an assembled model of human pluripotent stem cell-derived organoids. Stem Cell Rep. 16, 2228–2241. doi: 10.1016/j.stemcr.2021.05.009
Fonseca, S. G., Ishigaki, S., Oslowski, C. M., Lu, S., Lipson, K. L., Ghosh, R., et al. (2010). Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Investig. 120, 744–755. doi: 10.1172/JCI39678
Formosa, L. E., Reljic, B., Sharpe, A. J., Hock, D. H., Muellner-Wong, L., Stroud, D. A., et al. (2021). Optic atrophy-associated TMEM126A is an assembly factor for the ND4-module of mitochondrial complex I. Proc. Natl. Acad. Sci. USA. 118:e2019665118. doi: 10.1073/pnas.2019665118
Friedman, J. R., Mourier, A., Yamada, J., Michael, McCaffery, J., and Nunnari, J. (2015). MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. eLife 2015, 1–61. doi: 10.7554/eLife.07739
Fröhlich, C., Grabiger, S., Schwefel, D., Faelber, K., Rosenbaum, E., Mears, J., et al. (2013). Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 32, 1280–1292. doi: 10.1038/emboj.2013.74
Fuhrmann, N., Alavi, M. V., Bitoun, P., Woernle, S., Auburger, G., Leo-Kottler, B., et al. (2009). Genomic rearrangements in OPA1 are frequent in patients with autosomal dominant optic atrophy. J. Med. Genet. 46, 136–144. doi: 10.1136/jmg.2008.062570
Fujii, M., Yasuda, K., Hartman, P. S., Ayusawa, D., and Ishii, N. (2011). A mutation in a mitochondrial dehydrogenase/reductase gene causes an increased sensitivity to oxidative stress and mitochondrial defects in the nematode Caenorhabditis elegans. Genes. Cells. 16, 1022–1034.
Ge, Y., Shi, X., Boopathy, S., McDonald, J., Smith, A. W., and Chao, L. H. (2020). Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 9:50973. doi: 10.7554/eLife.50973
Genin, E. C., Plutino, M., Bannwarth, S., Villa, E., Cisneros-Barroso, E., Roy, M., et al. (2016). CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 8, 58–72. doi: 10.15252/emmm.201505496
Gerber, S., Ding, M. G., Gérard, X., Zwicker, K., Zanlonghi, X., Rio, M., et al. (2017a). Compound heterozygosity for severe and hypomorphic NDUFS2 mutations cause non-syndromic LHON-like optic neuropathy. J. Med. Genet. 54, 346–356. doi: 10.1136/jmedgenet-2016-104212
Gerber, S., Charif, M., Chevrollier, A., Chaumette, T., Angebault, C., Kane, M. S., et al. (2017b). Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 140, 2586–2596. doi: 10.1093/brain/awx219
Gerber, S., Orssaud, C., Kaplan, J., Johansson, C., and Rozet, J.-M. (2021). Mcat mutations cause nuclear lhon-like optic neuropathy. Genes 12:521. doi: 10.3390/genes12040521
Glancy, B., Kim, Y., Katti, P., and Willingham, T. B. (2020). The functional impact of mitochondrial structure across subcellular scales. Front. Physiol. 11:541040. doi: 10.3389/fphys.2020.541040
Gödiker, J., Grueneberg, M., DuChesne, I., Reunert, J., Rust, S., Westermann, C., et al. (2018). QIL1-dependent assembly of MICOS complex-lethal mutation in C19ORF70 resulting in liver disease and severe neurological retardation. J. Hum.Genet. 63, 707–716. doi: 10.1038/s10038-018-0442-y
Gong, J., Yuan, Y., Ward, A., Kang, L., Zhang, B., Wu, Z., et al. (2016). The C. elegans taste receptor homolog LITE-1 Is a photoreceptor. Cell 167, 1252–1263. doi: 10.1016/j.cell.2016.10.053
Gorukmez, O., Gorukmez, O., and Havalı, C. (2019). Novel MECR mutation in childhood-onset dystonia, optic atrophy, and basal ganglia signal abnormalities. Neuropediatrics 50, 336–337. doi: 10.1055/s-0039-1688767
Grau, T., Burbulla, L. F., Engl, G., Delettre, C., Delprat, B., Oexle, K., et al. (2013). A novel heterozygous OPA3 mutation located in the mitochondrial target sequence results in altered steady-state levels and fragmented mitochondrial network. J. Med. Genet. 50, 848–858. doi: 10.1136/jmedgenet-2013-101774
Grenier, J., Meunier, I., Daien, V., Baudoin, C., Halloy, F. ÇO., and Bocquet, B. É, et al. (2016). WFS1 in optic neuropathies: mutation findings in nonsyndromic optic atrophy and assessment of clinical severity. Ophthalmology 123, 1989–1998. doi: 10.1016/j.ophtha.2016.05.036
Guerriero, S., D’oria, F., Rossetti, G., Favale, R. A., Zoccolella, S., Alessio, G., et al. (2020). CMT2A harboring mitofusin 2 mutation with optic nerve atrophy and normal visual acuity. Int. Med. Case. Rep. J. 13, 41–45. doi: 10.2147/IMCRJ.S237620
Ham, M., Han, J., Osann, K., Smith, M., and Kimonis, V. (2019). Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion 46, 262–269. doi: 10.1016/j.mito.2018.07.006
Hammer, M., Ding, J., Mochel, F., Eleuch-Fayache, G., Charles, P., Coutelier, M., et al. (2017). SLC25A46 mutations associated with autosomal recessive cerebellar Ataxia in North African Families. Neurodegener. Dis. 17, 201–212. doi: 10.1159/000464445
Hanein, S., Perrault, I., Roche, O., Gerber, S., Khadom, N., Rio, M., et al. (2009). TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am. J. Hum. Genet. 84, 493–498. doi: 10.1016/j.ajhg.2009.03.003
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Hartmann, B., Wai, T., Hu, H., MacVicar, T., Musante, L., Fischer-Zirnsak, B., et al. (2016). Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. eLife 5:16078. doi: 10.7554/eLife.16078
Heimer, G., Kerätär, J. M., Riley, L. G., Balasubramaniam, S., Eyal, E., Pietikäinen, L. P., et al. (2016). MECR mutations cause childhood-onset dystonia and optic atrophy, a mitochondrial fatty acid synthesis disorder. Am. J. Hum. Genet. 99, 1229–1244. doi: 10.1016/j.ajhg.2016.09.021
Hofmann, S., Philbrook, C., Gerbitz, K.-D., and Bauer, M. F. (2003). Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum. Mol. Genet. 12, 2003–2012. doi: 10.1093/hmg/ddg214
Hogewind, B. F. T., Pennings, R. J. E., Hol, F. A., Kunst, H. P. M., Hoefsloot, E. H., Cruysberg, J. R. M., et al. (2010). Autosomal dominant optic neuropathy and sensorineual hearing loss associated with a novel mutation of WFS1. Mol. Vis. 16, 26–35.
Horga, A., Bugiardini, E., Manole, A., Bremner, F., Jaunmuktane, Z., Dankwa, L., et al. (2019). Autosomal dominant optic atrophy and cataract “plus” phenotype including axonal neuropathy. Neurol. Genet. 5:e322. doi: 10.1212/NXG.0000000000000322
Hoshijima, K., Jurynec, M. J., Klatt Shaw, D., Jacobi, A. M., Behlke, M. A., and Grunwald, D. J. (2019). Highly efficient CRISPR-Cas9-based methods for generating deletion mutations and F0 embryos that lack gene function in zebrafish. Dev. Cell. 51, 645–657. doi: 10.1016/j.devcel.2019.10.004
Howe, K., Torroja, C. F., Torrance, J., Collins, J. E., Humphray, S., McLaren, K., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503. doi: 10.1038/nature12111
Hu, K., Zatyka, M., Astuti, D., Beer, N., Dias, R. P., Kulkarni, A., et al. (2021). WFS1 protein expression correlates with clinical progression of optic atrophy in patients with Wolfram syndrome. J. Med. Genet. 2021:107257. doi: 10.1136/jmedgenet-2020-107257
Hu, W. H., Hausmann, O. N., Yan, M. S., Walters, W. M., Wong, P. K. Y., and Bethea, J. R. (2002). Identification and characterization of a novel Nogo-interacting mitochondrial protein (NIMP). J. Neurochem. 81, 36–45. doi: 10.1046/j.1471-4159.2002.00788.x
Huang, W., Campbell, T., Carbone, M. A., Jones, W. E., Unselt, D., Anholt, R. R. H., et al. (2020). Context-dependent genetic architecture of Drosophila life span. PLoS Biol. 18:e3000645. doi: 10.1371/journal.pbio.3000645
Huch, M., Knoblich, J. A., Lutolf, M. P., and Martinez-Arias, A. (2017). The hope and the hype of organoid research. Development 144, 938–941. doi: 10.1242/dev.150201
Inoue, H., Tanizawa, Y., Wasson, J., Behn, P., Kalidas, K., Bernal-Mizrachi, E., et al. (1998). A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat. Genet. 20, 143–148. doi: 10.1038/2441
Ishihara, H., Takeda, S., Tamura, A., Takahashi, R., Yamaguchi, S., Takei, D., et al. (2004). Disruption of the WFS1 gene in mice causes progressive β-cell loss and impaired stimulus–secretion coupling in insulin secretion. Hum. Mol. Genet. 13, 1159–1170. doi: 10.1093/hmg/ddh125
Janer, A., Prudent, J., Paupe, V., Fahiminiya, S., Majewski, J., Sgarioto, N., et al. (2016). SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol. Med. 8, 1019–1038. doi: 10.15252/emmm.201506159
Jeon, C.-J., Strettoi, E., and Masland, R. H. (1998). The major cell populations of the mouse retina. J. Neurosci. 18, 8936–8946. doi: 10.1523/jneurosci.18-21-08936.1998
Jiang, M., Xie, X., Zhu, X., Jiang, S., Milenkovic, D., Misic, J., et al. (2021). The mitochondrial single-stranded DNA binding protein is essential for initiation of mtDNA replication. Sci. Adv. 7:eabf8631. doi: 10.1126/sciadv.abf8631
John, G. B., Shang, Y., Li, L., Renken, C., Mannella, C. A., Selker, J. M. L., et al. (2005). The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol. Biol. Cell. 16, 1543–1554. doi: 10.1091/mbc.E04-08-0697
Jurkute, N., Leu, C., Pogoda, H. M., Arno, G., Robson, A. G., Nürnberg, G., et al. (2019). SSBP1 mutations in dominant optic atrophy with variable retinal degeneration. Ann. Neurol. 86, 368–383. doi: 10.1002/ana.25550
Kanazawa, T., Zappaterra, M. D., Hasegawa, A., Wright, A. P., Newman-Smith, E. D., Buttle, K. F., et al. (2008). The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 4:e1000022. doi: 10.1371/journal.pgen.1000022
Kane, M. S., Alban, J., Desquiret-Dumas, V., Gueguen, N., Ishak, L., Ferre, M., et al. (2017). Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J. Cell. Mol. Med. 21, 2284–2297. doi: 10.1111/jcmm.13149
Keij, J. F., Bell-Prince, C., and Steinkamp, J. A. (2000). Staining of mitochondrial membranes with 10-nonyl acridine orange, MitoFluor Green, and MitoTracker Green is affected by mitochondrial membrane potential altering drugs. Cytometry 39, 203–210. doi: 10.1002/(sici)1097-0320(20000301)39:3<203::aid-cyto5<3.0.co;2-z
Kelman, J. C., Kamien, B. A., Murray, N. C., Goel, H., Fraser, C. L., and Grigg, J. R. (2018). A sibling study of isolated optic neuropathy associated with novel variants in the ACO2 gene. Ophthalmic Genet. 39, 648–651. doi: 10.1080/13816810.2018.1509353
Kleinjan, D. A., Bancewicz, R. M., Gautier, P., Dahm, R., Schonthaler, H. B., Damante, G., et al. (2008). Subfunctionalization of duplicated zebrafish pax6 genes by cis-regulatory divergence. PLoS Genet. 4:e29. doi: 10.1371/journal.pgen.0040029
Kloth, K., Synofzik, M., Kernstock, C., Schimpf-Linzenbold, S., Schuettauf, F., Neu, A., et al. (2019). Novel likely pathogenic variants in TMEM126A identified in non-syndromic autosomal recessive optic atrophy: Two case reports. BMC Med. Genet. 20:62. doi: 10.1186/s12881-019-0795-x
Knowlton, W. M., Hubert, T., Wu, Z., Chisholm, A. D., and Jin, Y. (2017). A select subset of electron transport chain genes associated with optic atrophy link mitochondria to axon regeneration in Caenorhabditis elegans. Front. Neurosci. 11:263. doi: 10.3389/fnins.2017.00263
Kobayashi, W., Onishi, A., Tu, H. Y., Takihara, Y., Matsumura, M., Tsujimoto, K., et al. (2018). Culture systems of dissociated mouse and human pluripotent stem cell-derived retinal ganglion cells purified by two-step immunopanning. Invest. Ophthalmol. Vis. Sci. 59, 776–787. doi: 10.1167/iovs.17-22406
Koch, J., Feichtinger, R. G., Freisinger, P., Pies, M., Schrödl, F., Iuso, A., et al. (2016). Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 53, 270–278. doi: 10.1136/jmedgenet-2015-103500
Kõks, S., Overall, R. W., Ivask, M., Soomets, U., Guha, M., Vasar, E., et al. (2013). Silencing of the WFS1 gene in HEK cells induces pathways related to neurodegeneration and mitochondrial damage. Physiol. Genom. 45, 182–190. doi: 10.1152/physiolgenomics.00122.2012
Kõks, S., Soomets, U., Paya-Cano, J. L., Fernandes, C., Luuk, H., Plaas, M., et al. (2009). Wfs1 gene deletion causes growth retardation in mice and interferes with the growth hormone pathway. Physiol. Genom. 37, 249–259. doi: 10.1152/physiolgenomics.90407.2008
Korhonen, J. A., Pham, X. H., Pellegrini, M., and Falkenberg, M. (2004). Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 23, 2423–2429. doi: 10.1038/sj.emboj.7600257
Kretschmer, F., Tariq, M., Chatila, W., Wu, B., and Badea, T. C. (2017). Comparison of optomotor and optokinetic reflexes in mice. J. Neurophysiol. 118, 300–316. doi: 10.1152/jn.00055.2017
Krols, M., van Isterdael, G., Asselbergh, B., Kremer, A., Lippens, S., Timmerman, V., et al. (2016). Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 131, 505–523. doi: 10.1007/s00401-015-1528-7
Kunji, E. R. S., Aleksandrova, A., King, M. S., Majd, H., Ashton, V. L., Cerson, E., et al. (2016). The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta. Mol. Cell. Res. 1863, 2379–2393. doi: 10.1016/j.bbamcr.2016.03.015
La Morgia, C., Caporali, L., Tagliavini, F., Palombo, F., Carbonelli, M., Liguori, R., et al. (2019). First TMEM126A missense mutation in an Italian proband with optic atrophy and deafness. Neurol. Genet. 5:e329. doi: 10.1212/NXG.0000000000000329
La Morgia, C., Maresca, A., Amore, G., Gramegna, L. L., Carbonelli, M., Scimonelli, E., et al. (2020). Calcium mishandling in absence of primary mitochondrial dysfunction drives cellular pathology in Wolfram Syndrome. Sci. Rep. 10, 4785–4785. doi: 10.1038/s41598-020-61735-3
Ladds, E., Whitney, A., Dombi, E., Hofer, M., Anand, G., Harrison, V., et al. (2018). De novo DNM1L mutation associated with mitochondrial epilepsy syndrome with fever sensitivity. Neurol. Genet. 4:e258. doi: 10.1212/NXG.0000000000000258
Lai, C.-H., Chou, C.-Y., Ch’ang, L.-Y., Liu, C.-S., and Lin, W.-C. (2000). Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 10, 703–713. doi: 10.1101/gr.10.5.703
Laschke, M. W., and Menger, M. D. (2012). Vascularization in tissue engineering: angiogenesis versus inosculation. Eur. Surg. Res. 48, 85–92. doi: 10.1159/000336876
Lee, H., Smith, S. B., and Yoon, Y. (2017). The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 292, 7115–7130. doi: 10.1074/jbc.M116.762567
Lee, K.-S., Huh, S., Lee, S., Wu, Z., Kim, A.-K., Kang, H.-Y., et al. (2018). Altered ER–mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA. 115, E8844–E8853. doi: 10.1073/pnas.1721136115
Lenaers, G., Hamel, C., Delettre, C., Amati-Bonneau, P., Procaccio, V., Bonneau, D., et al. (2012). Dominant optic atrophy. Orphanet J. Rare. Dis. 7, 46–46. doi: 10.1186/1750-1172-7-46
Letts, J. A., Fiedorczuk, K., and Sazanov, L. A. (2016). The architecture of respiratory supercomplexes. Nature 537, 644–648. doi: 10.1038/nature19774
Li, F., Jiang, D., and Samuel, M. A. (2019). Microglia in the developing retina. Neural. Dev. 14:12. doi: 10.1186/s13064-019-0137-x
Li, J.-K., Li, W., Gao, F.-J., Qu, S.-F., Hu, F.-Y., Zhang, S.-H., et al. (2020). Mutation screening of mtDNA combined targeted exon sequencing in a cohort with suspected hereditary optic neuropathy. Transl. Vis. Sci. Technol. 9:11. doi: 10.1167/tvst.9.8.11
Li, H., Yuan, S., Minegishi, Y., Suga, A., Yoshitake, K., Sheng, X., et al. (2020). Novel mutations in malonyl-CoA-acyl carrier protein transacylase provoke autosomal recessive optic neuropathy. Hum. Mol. Genet. 29, 444–458. doi: 10.1093/hmg/ddz311
Li, Z., Peng, Y., Hufnagel, R. B., Hu, Y.-C., Zhao, C., Queme, L. F., et al. (2017). Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice. Hum. Mol. Genet. 26, 3776–3791. doi: 10.1093/hmg/ddx262
Liao, C., Ashley, N., Diot, A., Morten, K., Phadwal, K., Williams, A., et al. (2017). Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 88, 131–142. doi: 10.1212/WNL.0000000000003491
Liu, P., Long, L., Xiong, K., Yu, B., Chang, N., Xiong, J.-W., et al. (2014). Heritable/conditional genome editing in C. elegans using a CRISPR-Cas9 feeding system. Cell Res. 24, 886–889. doi: 10.1038/cr.2014.73
Llorente-Folch, I., Rueda, C. B., Pardo, B., Szabadkai, G., Duchen, M. R., and Satrustegui, J. (2015). The regulation of neuronal mitochondrial metabolism by calcium: Regulation of neuronal mitochondrial metabolism. J. Physiol. 593, 3447–3462. doi: 10.1113/JP270254
Longo, F., Benedetti, S., Zambon, A. A., Sora, M. G. N., Di Resta, C., De Ritis, D., et al. (2020). Impaired turnover of hyperfused mitochondria in severe axonal neuropathy due to a novel DRP1 mutation. Hum. Mol. Genet. 29, 177–188. doi: 10.1093/hmg/ddz211
Losón, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell. 24, 659–667. doi: 10.1091/mbc.E12-10-0721
Lu, J., Peatman, E., Tang, H., Lewis, J., and Liu, Z. (2012). Profiling of gene duplication patterns of sequenced teleost genomes: Evidence for rapid lineage-specific genome expansion mediated by recent tandem duplications. BMC Genom. 13:246. doi: 10.1186/1471-2164-13-246
Luz, A. L., Rooney, J. P., Kubik, L. L., Gonzalez, C. P., Song, D. H., and Meyer, J. N. (2015). Mitochondrial morphology and fundamental parameters of the mitochondrial respiratory chain are altered in caenorhabditis elegans strains deficient in mitochondrial dynamics and homeostasis processes. PloS One 10:e0130940. doi: 10.1371/journal.pone.0168738
Lynch, D. S., Loh, S. H. Y., Harley, J., Noyce, A. J., Martins, L. M., Wood, N. W., et al. (2017). Nonsyndromic Parkinson disease in a family with autosomal dominant optic atrophydue to OPA1 mutations. Neurol. Genet. 3:e188. doi: 10.1212/NXG.0000000000000188
Machiela, E., Liontis, T., Dues, D. J., Rudich, P. D., Traa, A., Wyman, L., et al. (2020). Disruption of mitochondrial dynamics increases stress resistance through activation of multiple stress response pathways. FASEB J. 34, 8475–8492. doi: 10.1096/fj.201903235R
MacVicar, T., Ohba, Y., Nolte, H., Mayer, F. C., Tatsuta, T., Sprenger, H.-G., et al. (2019). Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 575, 361–365. doi: 10.1038/s41586-019-1738-6
Maglioni, S., Mello, D. F., Schiavi, A., Meyer, J. N., and Ventura, N. (2019). Mitochondrial bioenergetic changes during development as an indicator of C. elegans health-span. Aging 11, 6535–6554. doi: 10.18632/aging.102208
Maier, D., Farr, C. L., Poeck, B., Alahari, A., Vogel, M., Fischer, S., et al. (2001). Mitochondrial single-stranded DNA-binding protein is required for mitochondrial DNA replication and development in Drosophila melanogaster. Mol. Biol. Cell. 12, 821–830. doi: 10.1091/mbc.12.4.821
Maloney, D. M., Chadderton, N., Millington-Ward, S., Palfi, A., Shortall, C., O’Byrne, J. J., et al. (2020). Optimized OPA1 Isoforms 1 and 7 provide therapeutic benefit in models of mitochondrial dysfunction. Front. Neurosci. 14:571479. doi: 10.3389/fnins.2020.571479
Maltecca, F., Aghaie, A., Schroeder, D. G., Cassina, L., Taylor, B. A., Phillips, S. J., et al. (2008). The mitochondrial protease AFG3L2 is essential for axonal development. J. Neurosci. 28, 2827–2836. doi: 10.1523/JNEUROSCI.4677-07.2008
Maltecca, F., Magnoni, R., Cerri, F., Cox, G. A., Quattrini, A., and Casari, G. (2009). Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated purkinje cell dark degeneration. J. Neurosci. 29, 9244–9254. doi: 10.1523/JNEUROSCI.1532-09.2009
Mancini, C., Hoxha, E., Iommarini, L., Brussino, A., Richter, U., Montarolo, F., et al. (2019). Mice harbouring a SCA28 patient mutation in AFG3L2 develop late-onset ataxia associated with enhanced mitochondrial proteotoxicity. Neurobiol. Dis. 124, 14–28. doi: 10.1016/j.nbd.2018.10.018
Mandal, A., and Drerup, C. M. (2019). Axonal transport and mitochondrial function in neurons. Front. Cell. Neurosci. 13:373. doi: 10.3389/fncel.2019.00373
Maresca, A., and Carelli, V. (2021). Molecular mechanisms behind inherited neurodegeneration of the optic nerve. Biomolecules 11:496. doi: 10.3390/biom11040496
Maresca, A., Zanna, C., Vidoni, S., Rugolo, M., Amati-Bonneau, P., Carelli, V., et al. (2012). OPA3, a new regulator of mitochondrial fission? Mitochondrion 12:571. doi: 10.1016/j.mito.2012.07.055
Masai, I., Lele, Z., Yamaguchi, M., Komori, A., Nakata, A., Nishiwaki, Y., et al. (2003). N-cadherin mediates retinal lamination, maintenance of forebrain compartments and patterning of retinal neurites. Development 130, 2479–2494. doi: 10.1242/dev.00465
Mei, S., Huang, X., Cheng, L., Peng, S., Zhu, T., Chen, L., et al. (2019). A missense mutation in OPA1 causes dominant optic atrophy in a chinese family. J. Ophthalmol. 2019, 1–10. doi: 10.1155/2019/1424928
Mellough, C. B., Collin, J., Khazim, M., White, K., Sernagor, E., Steel, D. H., et al. (2015). IGF-1 signaling plays an important role in the formation of three-dimensional laminated neural retina and other ocular structures from human embryonic stem cells. Stem Cells. 33, 2416–2430. doi: 10.1002/stem.2023
Mellough, C. B., Collin, J., Queen, R., Hilgen, G., Dorgau, B., Zerti, D., et al. (2019). Systematic comparison of retinal organoid differentiation from human pluripotent stem cells reveals stage specific, cell line, and methodological differences. Stem Cells Transl. Med. 8, 694–706. doi: 10.1002/sctm.18-0267
Meng, N., Glorieux, C., Zhang, Y., Liang, L., Zeng, P., Lu, W., et al. (2020). Oncogenic K-ras induces mitochondrial OPA3 expression to promote energy metabolism in pancreatic cancer cells. Cancers 12:65. doi: 10.3390/cancers12010065
Metodiev, M. D., Gerber, S., Hubert, L., Delahodde, A., Chretien, D., Gérard, X., et al. (2014). Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. J. Med. Genet. 51, 834–838. doi: 10.1136/jmedgenet-2014-102532
Meunier, I., Bocquet, B., Charif, M., Dhaenens, C.-M., Manes, G., Amati-Bonneau, P., et al. (2021). A rod-cone dystrophy is systematically associated to the RTN4IP1 recessive optic atrophy. Retina 41, 1771–1779. doi: 10.1097/IAE.0000000000003054
Meyer, E., Michaelides, M., Tee, L. J., Robson, A. G., Rahman, F., Pasha, S., et al. (2010). Nonsense mutation in TMEM126A causing autosomal recessive optic atrophy and auditory neuropathy. Mol. Vis. 16, 650–664.
Mieskes, F., Wehnekamp, F., Plucińska, G., Thong, R., Misgeld, T., and Lamb, D. C. (2020). Trajectory data of antero- and retrograde movement of mitochondria in living zebrafish larvae. Data Brief. 29:105280. doi: 10.1016/j.dib.2020.105280
Mignani, L., Zizioli, D., Borsani, G., Monti, E., and Finazzi, D. (2020). The downregulation of c19orf12 negatively affects neuronal and musculature development in zebrafish embryos. Front. Cell Dev. Biol. 8:596069. doi: 10.3389/fcell.2020.596069
Millet, A. M. C., Bertholet, A. M., Daloyau, M., Reynier, P., Galinier, A., Devin, A., et al. (2016). Loss of functional OPA1 unbalances redox state: implications in dominant optic atrophy pathogenesis. Ann. Clin. Transl. Neurol. 3, 408–421. doi: 10.1002/acn3.305
Min, Y.-L., Li, H., Rodriguez-Caycedo, C., Mireault, A. A., Huang, J., Shelton, J. M., et al. (2019). CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 5:eaav4324. doi: 10.1126/sciadv.aav4324
Miyata, K., Nakamura, M., Kondo, M., Lin, J., Ueno, S., Miyake, Y., et al. (2007). Reduction of oscillatory potentials and photopic negative response in patients with autosomal dominant optic atrophy with OPA1 mutations. Inves. Ophthalmol. Vis. Sci. 48, 820–824. doi: 10.1167/iovs.06-0845
Münzel, E. J., Becker, C. G., Becker, T., and Williams, A. (2014). Zebrafish regenerate full thickness optic nerve myelin after demyelination, but this fails with increasing age. Acta. Neuropathol. Commun. 2:77. doi: 10.1186/s40478-014-0077-y
Nakano, T., Ando, S., Takata, N., Kawada, M., Muguruma, K., Sekiguchi, K., et al. (2012). Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 10, 771–785. doi: 10.1016/j.stem.2012.05.009
Nargund, A. M., Pellegrino, M. W., Fiorese, C., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 337, 587–590. doi: 10.1126/science.1223560
Neumann, M. A.-C., Grossmann, D., Schimpf-Linzenbold, S., Dayan, D., Stingl, K., Ben-Menachem, R., et al. (2020). Haploinsufficiency due to a novel ACO2 deletion causes mitochondrial dysfunction in fibroblasts from a patient with dominant optic nerve atrophy. Sci. Rep. 10:16736. doi: 10.1038/s41598-020-73557-4
Nguyen, L. D., Fischer, T. T., Abreu, D., Arroyo, A., Urano, F., and Ehrlich, B. E. (2020). Calpain inhibitor and ibudilast rescue β cell functions in a cellular model of Wolfram syndrome. Proc. Natl. Acad. Sci. USA. 117, 17389–17398. doi: 10.1073/pnas.2007136117
Nguyen, M., Boesten, I., Hellebrekers, D. M. E. I., Mulder-den Hartog, N. M., de Coo, I. F. M., Smeets, H. J. M., et al. (2017). Novel pathogenic SLC25A46 splice-site mutation causes an optic atrophy spectrum disorder. Clin. Genet. 91, 121–125. doi: 10.1111/cge.12774
Nowinski, S. M., Solmonson, A., Rusin, S. F., Maschek, J. A., Bensard, C. L., Fogarty, S., et al. (2020). Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. eLife 9, 1–35. doi: 10.7554/ELIFE.58041
Odisho, T., Zhang, L., and Volchuk, A. (2015). ATF6β regulates the Wfs1 gene and has a cell survival role in the ER stress response in pancreatic β-cells. Exp. Cell. Res. 330, 111–122. doi: 10.1016/j.yexcr.2014.10.007
Ogienko, A. A., Andreyeva, E. N., Omelina, E. S., Oshchepkova, A. L., and Pindyurin, A. V. (2020). Molecular and cytological analysis of widely-used Gal4 driver lines for Drosophila neurobiology. BMC Genet. 21:96. doi: 10.1186/s12863-020-00895-7
Ohba, Y., MacVicar, T., and Langer, T. (2020). Regulation of mitochondrial plasticity by the i-AAA protease YME1L. Biol. Chem. 401, 877–890. doi: 10.1515/hsz-2020-0120
Ohlemacher, S. K., Langer, K. B., Fligor, C. M., Feder, E. M., Edler, M. C., and Meyer, J. S. (2019). Advances in the differentiation of retinal ganglion cells from human pluripotent stem cells. Adv. Exp. Med. Biol. 1186, 121–140. doi: 10.1007/978-3-030-28471-8_5
Okamoto, N., Miya, F., Hatsukawa, Y., Suzuki, Y., Kawato, K., Yamamoto, Y., et al. (2017). Siblings with optic neuropathy and RTN4IP1 mutation. J. Hum. Genet. 62, 927–929. doi: 10.1038/jhg.2017.68
Oliveira, M. T., and Kaguni, L. S. (2011). Reduced stimulation of recombinant DNA polymerase γ and Mitochondrial DNA (mtDNA) helicase by variants of Mitochondrial Single-Stranded DNA-binding protein (mtSSB) correlates with defects in mtDNA replication in animal cells. J. Biol. Chem. 286, 40649–40658. doi: 10.1074/jbc.M111.289983
Otera, H., Wang, C., Cleland, M. M., Setoguchi, K., Yokota, S., Youle, R. J., et al. (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158. doi: 10.1083/jcb.201007152
Palmer, C. S., Osellame, L. D., Laine, D., Koutsopoulos, O. S., Frazier, A. E., and Ryan, M. T. (2011). MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573. doi: 10.1038/embor.2011.54
Papa, S., Martino, P. L., Capitanio, G., Gaballo, A., De Rasmo, D., and Signorile, A. (2012). The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 942, 3–37. doi: 10.1007/978-94-007-2869-1_1
Paquet, D., Plucińska, G., and Misgeld, T. (2014). In vivo imaging of mitochondria in intact zebrafish larvae. Meth. Enzymol. 547, 151–164. doi: 10.1016/B978-0-12-801415-8.00009-6
Pareek, G., and Pallanck, L. J. (2020). Inactivation of the mitochondrial protease AFG3L2 results in severely diminished respiratory chain activity and widespread defects in mitochondrial gene expression. PLoS Genet. 16:e1009118. doi: 10.1371/journal.pgen.1009118
Pasquier, J., Cabau, C., Nguyen, T., Jouanno, E., Severac, D., Braasch, I., et al. (2016). Gene evolution and gene expression after whole genome duplication in fish: The PhyloFish database. BMC Genom. 17:368. doi: 10.1186/s12864-016-2709-z
Paulk, A., Millard, S. S., and van Swinderen, B. (2013). Vision in drosophila: seeing the world through a model’s eyes. Annu. Rev. Entomol. 58, 313–332. doi: 10.1146/annurev-ento-120811-153715
Pebay-Peyroula, E., Dahout-Gonzalez, C., Kahn, R., Trézéguet, V., Lauquin, G. J. M., and Brandolin, G. (2003). Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44. doi: 10.1038/nature02056
Pei, W., Kratz, L. E., Bernardini, I., Sood, R., Yokogawa, T., Dorward, H., et al. (2010). A model of Costeff Syndrome reveals metabolic and protective functions of mitochondrial OPA3. Development 137, 2587–2596. doi: 10.1242/dev.043745
Peng, Y. R., Shekhar, K., Yan, W., Herrmann, D., Sappington, A., Bryman, G. S., et al. (2019). Molecular classification and comparative taxonomics of foveal and peripheral cells in primate retina. Cell 176, 1222–1237. doi: 10.1016/j.cell.2019.01.004
Perge, J. A., Koch, K., Miller, R., Sterling, P., and Balasubramanian, V. (2009). How the optic nerve allocates space, energy capacity, and information. J. Neurosci. 29, 7917–7928. doi: 10.1523/JNEUROSCI.5200-08.2009
Perry, V. H., and Lund, R. D. (1990). Evidence that the lamina cribrosa prevents intraretinal myelination of retinal ganglion cell axons. J. Neurocytol. 19, 265–272. doi: 10.1007/BF01217304
Pesch, U. E. A., Leo-Kottler, B., Mayer, S., Jurklies, B., Kellner, U., Apfelstedt-Sylla, E., et al. (2001). OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum. Mol. Genet. 10, 1359–1368. doi: 10.1093/hmg/10.13.1359
Piro-Megy, C., Sarzi, E., Tarres-Sole, A., Pequignot, M., Hensen, F., Quiles, M., et al. (2020). Dominant mutations in mtDNA maintenance gene SSBP1 cause optic atrophy and foveopathy. J. Clin. Investig. 130, 143–156. doi: 10.1172/JCI128513
Platt, R. J., Chen, S., Zhou, Y., Yim, M. J., Swiech, L., Kempton, H. R., et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. doi: 10.1016/j.cell.2014.09.014
Powell, K. A., Davies, J. R., Taylor, E., Wride, M. A., and Votruba, M. (2011). Mitochondrial localization and ocular expression of mutant Opa3 in a mouse model of 3-Methylglutaconicaciduria type III. Investig. Ophthalmol. Vis. Sci. 52, 4369–4380. doi: 10.1167/iovs.10-6406
Puchades, C., Ding, B., Song, A., Wiseman, R. L., Lander, G. C., and Glynn, S. E. (2019). Unique structural features of the mitochondrial AAA+ Protease AFG3L2 reveal the molecular basis for activity in health and disease. Mol. Cell. 75, 1073–1085. doi: 10.1016/j.molcel.2019.06.016
Quintana-Cabrera, R., Quirin, C., Glytsou, C., Corrado, M., Urbani, A., Pellattiero, A., et al. (2018). The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 9:3399. doi: 10.1038/s41467-018-05655-x
Rabesandratana, O., Goureau, O., and Orieux, G. (2018). Pluripotent stem cell-based approaches to explore and treat optic neuropathies. Front. Neurosci. 12:651. doi: 10.3389/fnins.2018.00651
Rahn, J. J., Stackley, K. D., and Chan, S. S. L. (2013). Opa1 is required for proper mitochondrial metabolism in early development. PloS One 8:e59218. doi: 10.1371/journal.pone.0059218
Rajabian, F., Manitto, M. P., Palombo, F., Caporali, L., Grazioli, A., Starace, V., et al. (2021). Combined optic atrophy and rod-cone dystrophy expands the RTN4IP1 (Optic Atrophy 10) phenotype. J. Neuroophthalmol. 41, e290–e292. doi: 10.1097/WNO.0000000000001124
Reiter, L. T., Potocki, L., Chien, S., Gribskov, M., and Bier, E. (2001). A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 11, 1114–1125. doi: 10.1101/gr.169101
Reynier, P., Amati-Bonneau, P., Verny, C., Olichon, A., Simard, G., Guichet, A., et al. (2004). OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J. Med. Genet. 41:e110. doi: 10.1136/jmg.2003.016576
Robu, M. E., Larson, J. D., Nasevicius, A., Beiraghi, S., Brenner, C., Farber, S. A., et al. (2007). p53 activation by knockdown technologies. PLoS Genet 3:e78. doi: 10.1371/journal.pgen.0030078
Romagnoli, M., La Morgia, C., Carbonelli, M., Di Vito, L., Amore, G., Zenesini, C., et al. (2020). Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann. Clin. Transl. Neurol. 7, 590–594. doi: 10.1002/acn3.51026
Rouzier, C., Bannwarth, S., Chaussenot, A., Chevrollier, A., Verschueren, A., Bonello-Palot, N., et al. (2012). The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain 135, 23–34.
Roy-Choudhury, G., and Daadi, M. M. (2019). Assay for assessing mitochondrial function in iPSC-derived neural stem cells and dopaminergic neurons. Methods. Mol. Biol. 1919, 161–173. doi: 10.1007/978-1-4939-9007-8_12
Ryu, S.-W., Jeong, H. J., Choi, M., Karbowski, M., and Choi, C. (2010). Optic atrophy 3 as a protein of the mitochondrial outer membrane induces mitochondrial fragmentation. Cell. Mol. Life Sci. 67, 2839–2850. doi: 10.1007/s00018-010-0365-z
Sakakibara, Y., Sekiya, M., Fujisaki, N., Quan, X., and Iijima, K. M. (2018). Knockdown of wfs1, a fly homolog of Wolfram syndrome 1, in the nervous system increases susceptibility to age- and stress-induced neuronal dysfunction and degeneration in Drosophila. PLoS Genet 14:e1007196. doi: 10.1371/journal.pgen.1007196
Sarzi, E., Angebault, C., Seveno, M., Gueguen, N., Chaix, B., Bielicki, G., et al. (2012). The human OPA1delTTAG mutation induces premature age-related systemic neurodegeneration in mouse. Brain 135, 3599–3613. doi: 10.1093/brain/aws303
Schmidt-Kastner, R., Kreczmanski, P., Preising, M., Diederen, R., Schmitz, C., Reis, D., et al. (2009). Expression of the diabetes risk gene wolframin (WFS1) in the human retina. Exp. Eye Res. 89, 568–574. doi: 10.1016/j.exer.2009.05.007
Scholtes, C., Bellemin, S., Martin, E., Carre-Pierrat, M., Mollereau, B., Gieseler, K., et al. (2018). DRP-1-mediated apoptosis induces muscle degeneration in dystrophin mutants. Sci. Rep. 8:7354. doi: 10.1038/s41598-018-25727-8
Sergouniotis, P. I., Perveen, R., Thiselton, D. L., Giannopoulos, K., Sarros, M., Davies, J. R., et al. (2015). Clinical and molecular genetic findings in autosomal dominant OPA3-related optic neuropathy. Neurogenetics 16, 69–75. doi: 10.1007/s10048-014-0416-y
Shahrestani, P., Leung, H.-T., Le, P. K., Pak, W. L., Tse, S., Ocorr, K., et al. (2009). Heterozygous mutation of Drosophila Opa1 causes the development of multiple organ abnormalities in an age-dependent and organ-specific manner. PloS One 4:e6867. doi: 10.1371/journal.pone.0006867
Shamseldin, H. E., Alshammari, M., Al-Sheddi, T., Salih, M. A., Alkhalidi, H., Kentab, A., et al. (2012). Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 49, 234–241. doi: 10.1136/jmedgenet-2012-100836
Sheffer, R., Douiev, L., Edvardson, S., Shaag, A., Tamimi, K., Soiferman, D., et al. (2016). Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Gent. A. 170A, 1603–1607. doi: 10.1002/ajmg.a.37624
Silva Ramos, E., Larsson, N.-G., and Mourier, A. (2016). Bioenergetic roles of mitochondrial fusion. Biochim. Biophys. Acta. 1857, 1277–1283. doi: 10.1016/j.bbabio.2016.04.002
Simmer, F., Moorman, C., Van Der Linden, A. M., Kuijk, E., Van Den Berghe, P. V. E., Kamath, R. S., et al. (2003). Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 1, 77–84. doi: 10.1371/journal.pbio.0000012
Smith, S., Witkowski, A., Moghul, A., Yoshinaga, Y., Nefedov, M., de Jong, P., et al. (2012). Compromised mitochondrial fatty acid synthesis in transgenic mice results in defective protein lipoylation and energy disequilibrium. PloS One 7:e47196. doi: 10.1371/journal.pone.0047196
Song, Z., Chen, H., Fiket, M., Alexander, C., and Chan, D. C. (2007). OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 178, 749–755. doi: 10.1083/jcb.200704110
Song, Z., Ghochani, M., McCaffery, J. M., Frey, T. G., and Chan, D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell. 20, 3525–3532. doi: 10.1091/mbc.E09-03-0252
Stiburek, L., Cesnekova, J., Kostkova, O., Fornuskova, D., Vinsova, K., Wenchich, L., et al. (2012). YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell. 23, 1010–1023. doi: 10.1091/mbc.E11-08-0674
Suda, K., Ueoka, I., Azuma, Y., Muraoka, Y., Yoshida, H., and Yamaguchi, M. (2018). Novel Drosophila model for mitochondrial diseases by targeting of a solute carrier protein SLC25A46. Brain Res. 1689, 30–44. doi: 10.1016/j.brainres.2018.03.028
Sugimoto, T., Mori, C., Takanami, T., Sasagawa, Y., Saito, R., Ichiishi, E., et al. (2008). Caenorhabditis elegans par2.1/mtssb-1 is essential for mitochondrial DNA replication and its defect causes comprehensive transcriptional alterations including a hypoxia response. Exp. Cell Res. 314, 103–114. doi: 10.1016/j.yexcr.2007.08.015
Sun, C., Wu, X., Bai, H.-X., Wang, C., Liu, Z., Yang, C., et al. (2021). OPA1 haploinsufficiency due to a novel splicing variant resulting in mitochondrial dysfunction without mitochondrial DNA depletion. Ophthalmic Genet. 42, 45–52. doi: 10.1080/13816810.2020.1849313
Sun, S., Erchova, I., Sengpiel, F., and Votruba, M. (2020). Opa1 deficiency leads to diminished mitochondrial bioenergetics with compensatory increased mitochondrial motility. Investig. Ophthalmol. Vis. Sci. 61:42. doi: 10.1167/IOVS.61.6.42
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Takei, D., Ishihara, H., Yamaguchi, S., Yamada, T., Tamura, A., Katagiri, H., et al. (2006). WFS1 protein modulates the free Ca2+ concentration in the endoplasmic reticulum. FEBS Lett. 580, 5635–5640. doi: 10.1016/j.febslet.2006.09.007
Tang, S., Le, P. K., Tse, S., Wallace, D. C., and Huang, T. (2009). Heterozygous mutation of Opa1 in Drosophila shortens lifespan mediated through increased reactive oxygen species production. PloS One 4:e4492. doi: 10.1371/journal.pone.0004492
Toomes, C., Marchbank, N. J., Mackey, D. A., Craig, J. E., Newbury-Ecob, R. A., Bennett, C. P., et al. (2001). Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum. Mol. Genet. 10, 1369–1378. doi: 10.1093/hmg/10.13.1369
Trevisiol, A., Saab, A. S., Winkler, U., Marx, G., Imamura, H., Möbius, W., et al. (2017). Monitoring ATP dynamics in electrically active white matter tracts. eLife 6:e24241. doi: 10.7554/eLife.24241
Turner, J. G., Parrish, J. L., Hughes, L. F., Toth, L. A., and Caspary, D. M. (2005). Hearing in laboratory animals: strain differences and nonauditory effects of noise. Comp. Med. 55, 12–23.
van Bergen, N. J., Crowston, J. G., Kearns, L. S., Staffieri, S. E., Hewitt, A. W., Cohn, A. C., et al. (2011). Mitochondrial oxidative phosphorylation compensation may preserve vision in patients with OPA1-linked autosomal dominant optic atrophy. PloS One 6:e21347. doi: 10.1371/journal.pone.0021347
VanderWall, K. B., Huang, K. C., Pan, Y., Lavekar, S. S., Fligor, C. M., Allsop, A. R., et al. (2020). Retinal ganglion cells with a glaucoma OPTN(E50K) mutation exhibit neurodegenerative phenotypes when derived from three-dimensional retinal organoids. Stem Cell Rep. 15, 52–66. doi: 10.1016/j.stemcr.2020.05.009
Verstreken, P., Ly, C. V., Venken, K. J. T., Koh, T.-W., Zhou, Y., and Bellen, H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at drosophila neuromuscular junctions. Neuron 47, 365–378. doi: 10.1016/j.neuron.2005.06.018
Vita-More, N., and Barranco, D. (2015). Persistence of long-term memory in vitrified and revived caenorhabditis elegans. Rejuvenation Res. 18, 458–463. doi: 10.1089/rej.2014.1636
Wai, T., and Langer, T. (2016). Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 27, 105–117. doi: 10.1016/j.tem.2015.12.001
Wakabayashi, J., Zhang, Z., Wakabayashi, N., Tamura, Y., Fukaya, M., Kensler, T. W., et al. (2009). The dynamin-related Gtpase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 186, 805–816. doi: 10.1083/jcb.200903065
Wan, J., Steffen, J., Yourshaw, M., Mamsa, H., Andersen, E., Rudnik-Schöneborn, S., et al. (2016). Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain 139, 2877–2890. doi: 10.1093/brain/aww212
Wang, Y., Hu, L., Zhang, X., Zhao, H., Xu, H., Wei, Y., et al. (2017). Downregulation of mitochondrial single stranded DNA binding protein (SSBP1) induces mitochondrial dysfunction and increases the radiosensitivity in non-small cell lung cancer cells. J. Cancer 8, 1400–1409. doi: 10.7150/jca.18170
Waszczykowska, A., Zmysłowska, A., Braun, M., Ivask, M., Koks, S., Jurowski, P., et al. (2020). Multiple retinal anomalies in Wfs1-deficient mice. Diagnostics 10:607. doi: 10.3390/diagnostics10090607
Waterham, H. R., Koster, J., van Roermund, C. W. T., Mooyer, P. A. W., Wanders, R. J. A., and Leonard, J. V. (2007). A lethal defect of mitochondrial and peroxisomal fission. New Eng. J. Med. 356, 1736–1741. doi: 10.1056/NEJMoa064436
Wehnekamp, F., Plucińska, G., Thong, R., Misgeld, T., and Lamb, D. C. (2019). Nanoresolution real-time 3D orbital tracking for studying mitochondrial trafficking in vertebrate axons in vivo. Elife 8:e46059. doi: 10.7554/eLife.46059
Weisschuh, N., Schimpf-Linzenbold, S., Mazzola, P., Kieninger, S., Xiao, T., Kellner, U., et al. (2021). Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: Identification and classification of 48 novel variants. PloS One 16:e0253987. doi: 10.1371/journal.pone.0253987
Wells, T., Davies, J. R., Guschina, I. A., Ball, D. J., Davies, J. S., Davies, V. J., et al. (2012). Opa3, a novel regulator of mitochondrial function, controls thermogenesis and abdominal fat mass in a mouse model for costeff syndrome. Hum. Mol. Genet. 21, 4836–4844. doi: 10.1093/hmg/dds315
Wilkison, S. J., Bright, C. L., Vancini, R., Song, D. J., Bomze, H. M., and Cartoni, R. (2021). Local accumulation of axonal mitochondria in the optic nerve glial lamina precedes myelination. Front. Neuroanat. 15:678501. doi: 10.3389/fnana.2021.678501
Wong, A. A., and Brown, R. E. (2006). Visual detection, pattern discrimination and visual acuity in 14 strains of mice. Genes Brain Behav. 5, 389–403. doi: 10.1111/j.1601-183X.2005.00173.x
Wong, E. D., Wagner, J. A., Scott, S. V., Okreglak, V., Holewinske, T. J., Cassidy-Stone, A., et al. (2003). The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J. Cell Biol. 160, 303–311. doi: 10.1083/jcb.200209015
Wong-Riley, M. T. T. (2010). Energy metabolism of the visual system. Eye Brain 2, 99–116. doi: 10.2147/EB.S9078
Wragg, R., Dias, R. P., Barrett, T., and McCarthy, L. (2018). Bladder dysfunction in Wolfram syndrome is highly prevalent and progresses to megacystis. J. Pediatr. Surg. 53, 321–325. doi: 10.1016/j.jpedsurg.2017.11.025
Xian, H., and Liou, Y.-C. (2021). Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 28, 827–842. doi: 10.1038/s41418-020-00657-z
Yamamoto, H., Hofmann, S., Hamasaki, D. I., Yamamoto, H., Kreczmanski, P., Schmitz, C., et al. (2006). Wolfram syndrome 1 (WFS1) protein expression in retinal ganglion cells and optic nerve glia of the cynomolgus monkey. Exp. Eye Res. 83, 1303–1306. doi: 10.1016/j.exer.2006.06.010
Yu, R., Liu, T., Jin, S.-B., Ning, C., Lendahl, U., Nistér, M., et al. (2017). MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Sci. Rep. 7, 880–816. doi: 10.1038/s41598-017-00853-x
Yu-Wai-Man, P., Griffiths, P., Burke, A., Sellar, P., Clarke, M., Gnanaraj, L., et al. (2010a). The prevalence and natural history of dominant optic atrophy due to OPA1 Mutations. Ophthalmology 117, 1538–1546. doi: 10.1016/j.ophtha.2009.12.038
Yu-Wai-Man, P., Griffiths, P. G., Gorman, G. S., Lourenco, C. M., Wright, A. F., Auer-Grumbach, M., et al. (2010b). Multi-system neurological disease is common in patients with OPA1 mutations. Brain 133, 771–786. doi: 10.1093/brain/awq007
Zaha, K., Matsumoto, H., Itoh, M., Saitsu, H., Kato, K., Kato, M., et al. (2016). DNM1L-related encephalopathy in infancy with Leigh syndrome-like phenotype and suppression-burst. Clin. Genet. 90, 472–474. doi: 10.1111/cge.12805
Zaninello, M., Palikaras, K., Naon, D., Iwata, K., Herkenne, S., Quintana-Cabrera, R., et al. (2020). Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat. Commun. 11:4029. doi: 10.1038/s41467-020-17821-1
Zanna, C., Ghelli, A., Porcelli, A. M., Karbowski, M., Youle, R. J., Schimpf, S., et al. (2008). OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 131, 352–367. doi: 10.1093/brain/awm335
Zeng, T., Liao, L., Guo, Y., Liu, X., Xiong, X., Zhang, Y., et al. (2020). Concurrent OPA1 mutation and chromosome 3q deletion leading to Behr syndrome: A case report. BMC Pediatr. 20:420. doi: 10.1186/s12887-020-02309-0
Zerem, A., Yosovich, K., Rappaport, Y. C., Libzon, S., Blumkin, L., Ben-Sira, L., et al. (2019). Metabolic stroke in a patient with bi-allelic OPA1 mutations. Metab. Brain Dis. 34, 1043–1048. doi: 10.1007/s11011-019-00415-2
Zhang, J., Liu, X., Liang, X., Lu, Y., Zhu, L., Fu, R., et al. (2017). A novel ADOA-associated OPA1 mutation alters the mitochondrial function, membrane potential, ROS production and apoptosis. Sci. Rep. 7, 5704–5713. doi: 10.1038/s41598-017-05571-y
Zhao, J., Liu, T., Jin, S., Wang, X., Qu, M., Uhlén, P., et al. (2011). Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 30, 2762–2778. doi: 10.1038/emboj.2011.198
Zou, S.-Q., Yin, W., Zhang, M.-J., Hu, C.-R., Huang, Y.-B., and Hu, B. (2010). Using the optokinetic response to study visual function of zebrafish. J. Vis. Exp. 36:1742. doi: 10.3791/1742
Zou, W., Chen, Q., Slone, J., Yang, L., Lou, X., Diao, J., et al. (2021). Nanoscopic quantification of sub-mitochondrial morphology, mitophagy and mitochondrial dynamics in living cells derived from patients with mitochondrial diseases. J. Nanobiotechnol. 19:136. doi: 10.1186/s12951-021-00882-9
Zou, X.-H., Guo, X.-X., Su, H.-Z., Wang, C., Dong, E.-L., Wang, N., et al. (2019). Whole exome sequencing identifies two novel mutations in the Reticulon 4–interacting protein 1 gene in a Chinese family with autosomal recessive optic neuropathies. J. Mol. Neurosci. 68, 640–646. doi: 10.1007/s12031-019-01319-7
Keywords: mitochondria, retinal ganglion cells (RGC), optic atrophy, in vivo models, retinal organoids
Citation: Strachan EL, Mac White-Begg D, Crean J, Reynolds AL, Kennedy BN and O’Sullivan NC (2021) The Role of Mitochondria in Optic Atrophy With Autosomal Inheritance. Front. Neurosci. 15:784987. doi: 10.3389/fnins.2021.784987
Received: 28 September 2021; Accepted: 22 October 2021;
Published: 15 November 2021.
Edited by:
Alvaro Sanchez-Martinez, University of Cambridge, United KingdomReviewed by:
Jennifer E. Bestman, College of William and Mary, United StatesGuy Perkins, University of California, San Diego, United States
Copyright © 2021 Strachan, Mac White-Begg, Crean, Reynolds, Kennedy and O’Sullivan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elin L. Strachan, elin.strachan@ucdconnect.ie