 Michael Paul Kim
Michael Paul Kim Yun Zhang
Yun Zhang Guillermina Lozano
Guillermina Lozano- 1Department of Surgical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
- 2Department of Genetics, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
The functional importance of p53 as a tumor suppressor gene is evident through its pervasiveness in cancer biology. The p53 gene is the most commonly altered gene in human cancer; however, not all genetic alterations are biologically equivalent. The majority of alterations involve p53 missense mutations that result in the production of mutant p53 proteins. Such mutant p53 proteins lack normal p53 function and may concomitantly gain novel functions, often with deleterious effects. Here, we review characterized mechanisms of mutant p53 gain of function in various model systems. In addition, we review mutant p53 addiction as emerging evidence suggests that tumors may depend on sustained mutant p53 activity for continued growth. We also discuss the role of p53 in stromal elements and their contribution to tumor initiation and progression. Lastly, current genetic mouse models of mutant p53 in various organ systems are reviewed and their limitations discussed.
Mutant p53, the Elephant in the Room
Cancer is a complex disease that kills millions of people annually. Alterations in genetic and epigenetic cellular programs derail cellular controls normally responsible for maintaining homeostasis. Sequencing of human cancer genomes has identified a myriad of genomic alterations found in human cancers. Alterations in the p53 tumor suppressor gene stand out as the most common alteration in many cancers: 96% in ovarian serous carcinoma (1), 54% in invasive breast carcinomas (2), 86% in small cell lung cancer (3), and 75% in pancreas cancer (4), to name a few. Although p53 activity may be abrogated or lost through multiple mechanisms, the majority of these changes involve p53 missense mutations that result in single amino acid substitutions and expression of mutant proteins. Common mutations in the p53 gene, or “hotspots,” are present; for example, approximately 86% of mutations correspond to the DNA-binding sequence of p53 between codons 125 and 300. The predominance of mutant p53 protein expression in human cancers over the simple loss of p53 activity, in turn, suggests an inherent biologic advantage (5–7).
p53 Biological Activities in Tumor Suppression
The p53 gene encodes a transcription factor that contains a potent transcriptional activation domain, a sequence-specific DNA-binding domain, and a tetramerization domain (8). In normal cells, p53 activity is low, but in response to DNA damage and numerous other stress signals, p53 levels rise dramatically and result in the activation and transcription of hundreds of genes with important roles in cell cycle arrest, senescence, apoptosis, metabolism, and differentiation (9). The sum of these activities is to ensure that an abnormal cell fails to proliferate. Thus, tumors arise upon depletion of p53 activity through various mechanisms, including deletion or mutation of the p53 gene itself, overproduction of the p53 inhibitors, Mdm2 and Mdm4, and viral inactivation (10–12). Regardless of the mechanism of p53 loss, the downstream consequences are profound and likely due to the vast, fundamental spectrum of biologic activities in which p53 normally participates. Moreover, the loss of normal p53 function is likely coupled with the adoption of new biologic functions exerted by mutant p53 proteins with additional, deleterious effects.
Gain-of-Function Activities of Mutant p53
Single amino acid changes in the p53 gene may result in profound changes to its function. In human cancers, missense mutations comprise approximately 75% of all p53 alterations (7, 13, 14). This is in contrast to many other tumor suppressor genes that undergo deletion through the course of tumor initiation or development, such as PTEN, BRCA1, and Rb. Five arginine residues in the p53 gene are considered mutational “hotspots”; resultant mutant proteins fail to bind to sequence-specific DNA sites and therefore drastically alter the spectrum of transcriptional activity (15). Such signature mutations in the p53 gene may arise through environmental exposure to ultraviolet light or chemical carcinogens such as aflatoxins, smoking, and so on (7, 16).
The fact that most p53 alterations in tumors are missense mutations suggests that cells expressing mutant p53 have an advantage over cells lacking p53 (17). Numerous experiments have tested this hypothesis. For example, various tumor-derived human p53 mutants introduced into p53-null H1299 lung adenocarcinoma cells conferred upon tumor cells a selective survival advantage during etoposide or cisplatin treatments (18). In addition, several p53 mutants when overexpressed in Saos-2 cells, an immortalized tumor cell line that lacks p53, yielded tumors in nude mice, while the parental Saos-2 cell line did not (19). Cells expressing the most common p53 mutants, in contrast to cells lacking p53, also show increased metastatic potential and invasiveness (20, 21). Mutant p53 proteins also render some cell types more resistant to killing by therapeutic drugs such as doxorubicin, etoposide, and cisplatin (22). In Li–Fraumeni syndrome (LFS), individuals with p53 missense mutations show a higher cancer incidence and an earlier age of tumor onset (9–15 years earlier depending on the study) than individuals with other kinds of mutations (23). These novel activities of mutant p53 are referred to as gain of function (GOF).
The generation of p53 knockin alleles in mice provided direct in vivo evidence for the GOF activities of mutant p53. Knockin mouse models that express mutant p53R172H and p53R270H proteins, which mimic hot spot mutations that correspond to amino acids 175 and 273 in human cancers, respectively, develop tumors that exhibit a GOF phenotype in vivo, with high metastatic capacity compared to tumors in mice inheriting a p53-null allele (24, 25). Additionally, using autochthonous mouse models of pancreatic cancer that incorporate oncogenic K-ras, Morton et al. (26) found no metastatic burden in mice that had undergone genetic deletion of a normal p53 allele relative to a high (65%) incidence of metastasis in mice expressing a single, mutant p53 allele (26). However, other groups that have studied identical mouse models of pancreatic cancer have found cells of pancreatic origin in the bloodstream of mice that have undergone monoallelic or biallelic deletion of p53 in the pancreas, without the presence of mutant p53 (27–29). These data suggest that mutant p53 GOF activities may serve to enhance the metastatic potential and/or promote the survival and productivity of metastatic tumor cells at distant sites (26). Taken together, these studies suggest that stable mutant p53 proteins have additional activities that fuel tumor cell proliferation and metastases that are not yet fully understood.
Interestingly, the characterization of animal models containing mutant p53 alleles have demonstrated that tumor-specific events were required for the stabilization of mutant p53 in addition to its simple expression. Numerous tissues derived from mouse models with germline mutant p53 alleles failed to demonstrate detectable mutant p53 proteins, and, in some cases, tumors failed to express detectable mutant p53. Investigation into this phenomenon concluded that normal tissues failed to stabilize mutant p53 due to the presence of Mdm2 and p16INK4a. Upon loss of Mdm2 or p16INK4a, mutant p53 is stabilized and mice show decreased survival and increased metastases relative to mice with intact Mdm2 or p16INK4a alleles (30). A recent analysis of pancreatic cancer specimens demonstrated a strong correlation between p53 mutation and its stabilization through positive staining by immunohistochemistry for p53 protein expression. Such data again indicate that in patients with pancreatic cancer, mutant p53 proteins are expressed, stabilized, and play an important role in tumor development and progression (31). The GOF activity of mutant p53 therefore depends largely on multiple signals for its stabilization that may vary among normal cells and even among tumor cells.
Mechanisms of Mutant p53 GOF
Several mechanisms have now been identified that contribute to mutant p53 GOF activities. The first such mechanism discovered showed that mutant p53 proteins abrogate the tumor-suppressive activities of the p53 family members p63 and p73 (24, 25, 32–34). In addition, TGF-β and EGFR/integrin signaling pathways stabilized mutant p53 (p53R175H and p53R273H introduced into p53-null H1299 cells) and inhibited the function of p63, properties that were essential for the invasive nature of these cells (35, 36). These studies strengthened the evidence that mutant p53 proteins bind and disrupt p63 activities. However, p63 expression is limited to epithelial cells and its inhibition may therefore not explain mutant p53 GOF in tumors of mesenchymal origin. Moreover, mutant p53 was found to regulate gene expression independently of p63 and p73 in some tumors (37–40).
Using cell lines derived from these same pancreatic cancer models with Ras and p53 mutations, mutant p53 was found to drive metastasis through induction of platelet-derived growth factor receptor β (PDGFR β). Mutant p53-dependent sequestration of p73 from an NF-Y complex permits this transcriptional complex to function at the platelet-derived growth factor β promoter, resulting in expression of PDGFR β and a prometastatic phenotype (41).
Chromatin ImmunoPrecipitation (ChIP)-on-chip experiments and expression arrays using SKBR3 breast cancer cells with the p53R175H mutation identified mutant p53 complexes with the vitamin D receptor which augmented expression of survival genes and dampened expression of proapoptotic genes (42). Importantly, in these experiments, an intact transcriptional activation domain was required. Using expression arrays of MDA-MB-468 (p53R273H) breast cancer cells, Freed-Pastor et al. (43) identified increased expression of genes encoding several enzymes of the mevalonate pathway. Mutant p53 bound SREBP proteins and disrupted the acinar architecture of breast epithelial cells when grown as spheroids. In our studies, we compared primary osteosarcomas that had metastasized from p53R172H/+ mice to p53+/− tumors that lacked metastases and identified a unique set of transcriptional changes (39). In this system, mutant p53 bound the transcription factor Ets2 and enhanced expression of a phospholipase, Pla2g16, which induced migration and invasion in culture (39). Lastly, ChIP-seq experiments using LFS fibroblasts with the p53R248W mutation identified numerous promoters that contain mutant p53 (42, 44). More recently, Zhu et al. showed that p53 mutants, not wild-type (WT) p53, bind to and upregulate chromatin regulatory genes, including the methyltransferase MLL1, MLL2, and acetyltransferase MOZ, resulting in genome-wide increases of histone methylation and acetylation. Furthermore, upregulation of MLL1, MLL2, and MOZ was found in human tumors with p53 mutants, but not in WT p53 or p53-null tumors (45). In summary, these data suggest that multiple pathways contribute to the GOF phenotypes of cells with mutant p53. The emerging themes by which mutant p53 exhibits its GOF are (1) through formation of mutant p53 complexes with other proteins that modify their activities (e.g., p63 and p73) and (2) by interaction of mutant p53 with other transcription factors (e.g., SREBP and Ets2) that bring a potent transcriptional activation domain to promoters not normally regulated WT p53 (Figure 1). These mechanisms are not necessarily mutually exclusive in the genesis of different cancers and may be context dependent (46, 47).
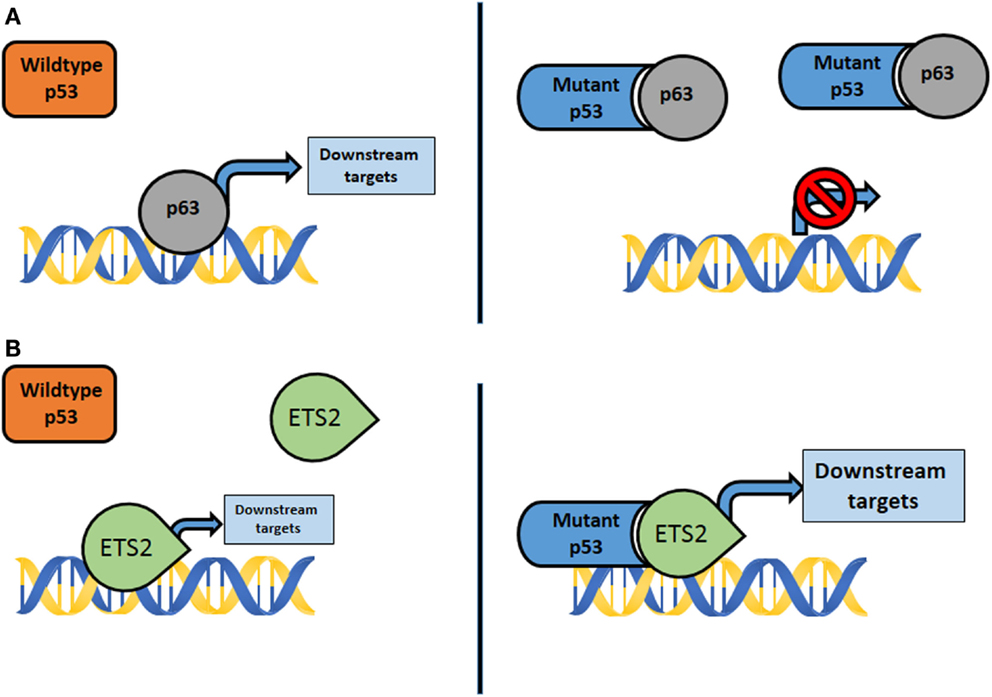
Figure 1. (A) Mutant p53 interacts with transcription factors not normally bound by wildtype p53, such as p63, p73, and Smad. The activity of downstream targets is disrupted, resulting in GOF properties. (B) Mutant p53 complexes with transcription factors, such as Ets2 and SREBP, not typically bound by wildtype p53. The results are aberrant activation of genes and downstream effectors that promote GOF properties.
Distinct Biological Activities of Different p53 Mutants
In addition to exhibiting GOF phenotypes, mutant p53 proteins exhibit intrinsic differences. Some are classified as structural mutants (e.g., p53R172H) as the mutation alters the structure of the DNA-binding domain while others are classified as DNA-binding mutants (e.g., p53R245W and p53R270H) because they alter an arginine that directly interacts with DNA. Other mutants show partial defects. For example, the p53R172P mutation, albeit rare, is able to activate the cell cycle arrest but not apoptotic programs of p53 (48). In vivo, differences in tumor spectrum were observed between p53R172H and p53R270H mice (24, 25). In addition, in humanized mutant p53 knockin models, p53R248Q/− and p53R248Q/Q, but not p53G245S/− and p53G245S/S, mice show an acceleration of tumor development and shorter survival as compared to p53−/− mice (49). Lastly, different human tumor types show different spectra of p53 mutations. For example, based on cBioPortal, mutations at the codon 248 of p53 are most prevalently observed in human pancreatic tumors, whereas in breast tumors, codons 275 and 175 are most frequently mutated, respectively (5, 6), further suggesting that different p53 mutations impart unique activities to drive development of different tumor types.
The Importance of Stroma in Tumor Suppression
The discussion has thus far focused on p53 mutations within tumor cells and has ignored a possible role of surrounding tissue on tumor evolution. Tumors are complex tissues that consist of two components: a parenchyma and stroma. The parenchyma consists of tumor cells while the stroma consists of blood and lymphatic vessels, fibroblasts, and inflammatory and immune cells (50). The importance of stromal elements in cancer development has been supported by extensive clinical and experimental evidence (51–55). The injection of human breast tumors into nude mice and subsequent analyses of copy number variations indicated that stromal cells evolved additional changes not found in the original tumor (56). In another study of human breast cancer, gene expression differences in the stroma were a better predictor of response to chemotherapy (57). Mouse models have now clearly implicated the importance of stromal alterations in tumor development. Deletion of PTEN in stromal fibroblasts accelerated initiation, progression, and malignant transformation of ErbB2/neu-driven mammary epithelial tumors, implicating a tumor-suppressive role of PTEN in stroma (58). Global gene expression profiling of stroma lacking PTEN revealed changes in the expression of genes regulating extracellular matrix (ECM) deposition, wound healing, and chronic inflammation, which were validated by staining with various markers. Lujambio and colleagues selectively deleted p53 in hepatic stellate cells, resulting in modifications to the tumor microenvironment (TME) and enhanced malignant transformation of epithelial cells (59). Mutations in p53 have also been found in the stromal component of some primary breast tumors and in carcinoma-associated fibroblasts (CAFs) (51, 60–63). Additionally, MCF7 breast tumor cells formed more aggressive tumors with shorter latency after injection into p53−/− SCID mice as compared to injection into p53+/+ SCID hosts (64). Hill et al. (65) further showed that prostate tumor cells can promote the selection and expansion of p53-deficient stromal fibroblasts through paracrine mechanisms. Highly proliferative, p53-deficient stromal cells were subsequently found to promote epithelial tumor growth and progression despite retention of WT p53. These data clearly show that changes in stroma occur and that they directly impact tumor development.
Mutant p53 Addiction
In addition to the observations that mutant p53 proteins exhibit GOF activities, a growing body of evidence suggests that tumor cells may be addicted to mutant p53 expression. Experiments using siRNA knockdown of mutant p53 in cancer cell lines showed a higher apoptotic response to drug treatment in cells with knockdown of mutant p53 (47, 66). Additional mutant p53 depletion experiments show decreases in cell growth rate, viability, replication, and clonogenicity. Constitutive inhibition of mutant p53 reduced tumor growth in nude mice and showed reduced stromal invasion and angiogenesis (67). In addition, Prives and colleagues showed that mutant p53 depletion in breast cancer cells (MDA-MB-231 cells with p53R280K and MDA-MB-468 with p53R273H) in 3D culture leads to phenotypic reversion to more normal, differentiated structures with hollow lumens (43). More recently, using a conditional mutant p53 mouse model expressing a p53R248Q hotspot mutation, Moll and colleagues showed that mutant p53 ablation restrained growth of allotransplanted and autochthonous tumors and extended animal survival, indicating that these tumors depend on sustained mutant p53 expression (68). In summary, these data suggest that tumor cells with mutant p53 may be addicted to the GOF activities of mutant p53. Tumor regression and dependence on mutant p53 is likely context dependent and the extent to which elimination of mutant p53, genetically or through pharmacologic inhibition of downstream mediators, as a viable therapy remains to be seen (69).
Mouse Models for Sporadic p53 Mutations in Cancer
Knock-in mice with germline mutations in p53 that mimic those found in LFS have more aggressive, metastatic tumors as compared to mice lacking p53 and provided convincing evidence for p53 GOF activities (24, 25). Yet germline mutations in p53 are rare in humans, and the vast majority of human cancers evolve from somatic alterations of p53. Consequently, current animal models at our disposal to study the role of p53 missense mutations in the genesis of somatic tumors are inadequate. Some involve expression of mutant p53 in breast epithelial cells using mouse mammary tumor virus (MMTV) or whey acidic protein (WAP) promoters which are hormonally regulated and therefore do not simulate expression of mutant p53 from the endogenous locus (70, 71). Currently, conditional mutant p53 alleles are only active following cre-mediated removal of a DNA “STOP” cassette flanked by LoxP sites (LSL = Lox–STOP–Lox) (25). The STOP sequence maintains the downstream gene in an unexpressed or null state in all animal cells until the STOP cassette is selectively removed and mutant p53 is expressed. Importantly, tumors in this model initiate from cells heterozygous for p53 since animal conception. Moreover, tumors under these conditions initiate and progress within a tumor microenvironment (TME) replete with p53-heterozygous stromal elements. CAFs, macrophages, T cells, neutrophils, endothelial cells, and so on remain heterozygous for p53 following conception (due to the presence of the STOP cassette) with undefined effects on tumor biology. Stroma is a requisite component of tumor initiation and growth, and as mentioned earlier, prostate tumor epithelium selects for p53-null stromal fibroblasts in p53± mice, yielding a highly proliferative stroma that contributes to tumor progression. Given the powerful roles of p53 in cellular plasticity and embryonic stem cell differentiation, tumors that develop from and under conditions of p53 heterozygosity may differ in unappreciated ways from human tumors that initiate from WT p53 cells. A model that more closely mimics the events in sporadic tumorigenesis is sorely needed.
Future Directions
Mouse models of mutant p53 carry the potential to inform us of essential mechanisms of cancer initiation and metastasis translatable to therapeutics in humans. However, many genetic mouse models used to study mutant p53 in vivo incorporate germline mutant p53 alleles that may alter normal and cancer biology in ways that compromise its relevance to human cancer. Innovation in genetic mouse models of mutant p53 is mandatory to more closely model human biology and to serve as translational biologic platforms to better evaluate and develop novel therapeutic agents in human cancers. Moreover, given the importance of the TME in cancer development and metastasis, mouse models that preserve the complex regulatory and tumor–stromal interactions are mandatory to the development of effective, translational biologic platforms to target the TME toward therapeutic ends.
Author Contributions
MK, YZ, and GL wrote and edited the manuscript. MK and YZ generated the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work is supported, in part, by grant CA082577 from the National Cancer Institute.
References
2. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature (2012) 486(7403):395–9. doi: 10.1038/nature10933
3. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet (2012) 44(10):1111–6. doi:10.1038/ng.2405
4. Yachida S, White CM, Naito Y, Zhong Y, Brosnan JA, Macgregor-Das AM, et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin Cancer Res (2012) 18(22):6339–47. doi:10.1158/1078-0432.CCR-12-1215
5. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov (2012) 2:401–4. doi:10.1158/2159-8290.CD-12-0095
6. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6:l1. doi:10.1126/scisignal.2004088
7. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol (2010) 2:a001008. doi:10.1101/cshperspect.a001008
8. Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer (2001) 1(1):68–76. doi:10.1038/35094077
9. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell (2009) 137(3):413–31. doi:10.1016/j.cell.2009.04.037
10. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature (2000) 408(6810):307–10. doi:10.1038/35042675
11. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer (2006) 6(12):909–23. doi:10.1038/nrc2012
12. Wasylishen A, Lozano G. Attenuating the p53 pathway in human cancers: Many means to the same end. In: Lozano G, Levine AJ, editors. The p53 Protein: From Cell Regulation to Cancer. Cold Spring Harbor, NY: CSH Press (in press).
13. Hamroun D, Kato S, Ishioka C, Claustres M, Beroud C, Soussi T. The UMD TP53 database and website: update and revisions. Hum Mutat (2006) 27:14–20. doi:10.1002/humu.20269
14. Soussi T, Lozano G. p53 mutation heterogeneity in cancer. Biochem Biophys Res Commun (2005) 331:834–42. doi:10.1016/j.bbrc.2005.03.190
15. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science (1994) 265:346–55. doi:10.1126/science.8023157
16. Vogelstein B, Kinzler KW. Carcinogens leave fingerprints. Nature (1992) 355:209–10. doi:10.1038/355209a0
17. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer (2009) 9(10):701–13.
18. Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene (1999) 18:477–85. doi:10.1038/sj.onc.1202314
19. Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, et al. Gain of function mutations in p53. Nat Genet (1993) 4:42–6. doi:10.1038/ng0593-42
20. Crook T, Vousden KH. Properties of p53 mutations detected in primary and secondary cervical cancers suggest mechanisms of metastasis and involvement of environmental carcinogens. EMBO J (1992) 11:3935–40.
21. Hsiao M, Low J, Dorn E, Ku D, Pattengale P, Yeargin J, et al. Gain-of-function mutations of the p53 gene induce lymphohematopoietic metastatic potential and tissue invasiveness. Am J Pathol (1994) 145:702–14.
22. Li R, Sutphin PD, Schwartz D, Matas D, Almog N, Wolkowicz R, et al. Mutant p53 protein expression interferes with p53-independent apoptotic pathways. Oncogene (1998) 16:3269–77. doi:10.1038/sj.onc.1201867
23. Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ, et al. Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene (1998) 17:1061–8. doi:10.1038/sj.onc.1202033
24. Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell (2004) 119:861–72. doi:10.1016/j.cell.2004.11.006
25. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell (2004) 119:847–60. doi:10.1016/j.cell.2004.11.004
26. Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A (2010) 107:246–51. doi:10.1073/pnas.0908428107
27. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell (2012) 148(1–2):349–61. doi:10.1016/j.cell.2011.11.025
28. Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature (2012) 487(7408):510–3. doi:10.1038/nature11217
29. Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep (2014) 8(6):1905–18. doi:10.1016/j.celrep.2014.08.029
30. Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev (2008) 22:1337–44. doi:10.1101/gad.1662908
31. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun (2015) 6:6744. doi:10.1038/ncomms7744
32. Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol (2001) 21:1874–87. doi:10.1128/MCB.21.5.1874-1887.2001
33. Marin MC, Jost CA, Brooks LA, Irwin MS, O’Nions J, Tidy JA, et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet (2000) 25:47–54. doi:10.1038/75586
34. Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y, Castagnoli L, et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem (2000) 275:29503–12. doi:10.1074/jbc.M003360200
35. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell (2009) 137:87–98. doi:10.1016/j.cell.2009.01.039
36. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell (2009) 139:1327–41. doi:10.1016/j.cell.2009.11.026
37. Scian MJ, Stagliano KE, Ellis MA, Hassan S, Bowman M, Miles MF, et al. Modulation of gene expression by tumor-derived p53 mutants. Cancer Res (2004) 64(20):7447–54. doi:10.1158/0008-5472.CAN-04-1568
38. Weisz L, Zalcenstein A, Stambolsky P, Cohen Y, Goldfinger N, Oren M, et al. Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res (2004) 64(22):8318–27. doi:10.1158/0008-5472.CAN-04-1145
39. Xiong S, Tu H, Kollareddy M, Pant V, Li Q, Zhang Y, et al. Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc Natl Acad Sci U S A (2014) 111(30):11145–50. doi:10.1073/pnas.1404139111
40. Zalcenstein A, Stambolsky P, Weisz L, Muller M, Wallach D, Goncharov TM, et al. Mutant p53 gain of function: repression of CD95(Fas/APO-1) gene expression by tumor-associated p53 mutants. Oncogene (2003) 22(36):5667–76. doi:10.1038/sj.onc.1206724
41. Weissmueller S, Manchado E, Saborowski M, Morris JPT, Wagenblast E, Davis CA, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell (2014) 157:382–94. doi:10.1016/j.cell.2014.01.066
42. Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell (2010) 17:273–85. doi:10.1016/j.ccr.2009.11.025
43. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148:244–58. doi:10.1016/j.cell.2011.12.017
44. Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V, et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev (2012) 26:830–45. doi:10.1101/gad.181685.111
45. Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature (2015) 525:206–11. doi:10.1038/nature15251
46. Jackson JG, Post SM, Lozano G. Regulation of tissue- and stimulus-specific cell fate decisions by p53 in vivo. J Pathol (2011) 223:127–36. doi:10.1002/path.2783
47. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol (2010) 2:a001107. doi:10.1101/cshperspect.a001107
48. Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet (2004) 36(1):63–8. doi:10.1038/ng1282
49. Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ (2013) 20(7):898–909. doi:10.1038/cdd.2013.17
50. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
51. Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell (2004) 6:17–32. doi:10.1016/j.ccr.2004.06.010
52. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature (2004) 432:332–7. doi:10.1038/nature03096
53. Hu M, Polyak K. Microenvironmental regulation of cancer development. Curr Opin Genet Dev (2008) 18:27–34. doi:10.1016/j.gde.2007.12.006
54. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121:335–48. doi:10.1016/j.cell.2005.02.034
55. Polyak K, Kalluri R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb Perspect Biol (2010) 2:a003244. doi:10.1101/cshperspect.a003244
56. Pelham RJ, Rodgers L, Hall I, Lucito R, Nguyen KC, Navin N, et al. Identification of alterations in DNA copy number in host stromal cells during tumor progression. Proc Natl Acad Sci U S A (2006) 103:19848–53. doi:10.1073/pnas.0609635104
57. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med (2008) 14:518–27. doi:10.1038/nm1764
58. Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature (2009) 461:1084–91. doi:10.1038/nature08486
59. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, et al. Non-cell-autonomous tumor suppression by p53. Cell (2013) 153:449–60. doi:10.1016/j.cell.2013.03.020
60. Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet (2002) 32:355–7. doi:10.1038/ng1013
61. Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL, et al. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med (2007) 357:2543–51. doi:10.1056/NEJMoa071825
62. Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet (2009) 25:30–8. doi:10.1016/j.tig.2008.10.012
63. Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet (2008) 40:650–5. doi:10.1038/ng.117
64. Kiaris H, Chatzistamou I, Trimis G, Frangou-Plemmenou M, Pafiti-Kondi A, Kalofoutis A. Evidence for nonautonomous effect of p53 tumor suppressor in carcinogenesis. Cancer Res (2005) 65:1627–30. doi:10.1158/0008-5472.CAN-04-3791
65. Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell (2005) 123(6):1001–11. doi:10.1016/j.cell.2005.09.030
66. Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene (2006) 25:304–9. doi:10.1038/sj.onc.1209026
67. Bossi G, Marampon F, Maor-Aloni R, Zani B, Rotter V, Oren M, et al. Conditional RNA interference in vivo to study mutant p53 oncogenic gain of function on tumor malignancy. Cell Cycle (2008) 7:1870–9. doi:10.4161/cc.7.12.6161
68. Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature (2015) 523:352–6. doi:10.1038/nature14430
69. Rivlin N, Katz S, Doody M, Sheffer M, Horesh S, Molchadsky A, et al. Rescue of embryonic stem cells from cellular transformation by proteomic stabilization of mutant p53 and conversion into WT conformation. Proc Natl Acad Sci U S A (2014) 111:7006–11. doi:10.1073/pnas.1320428111
70. Li B, Greenberg N, Stephens LC, Meyn R, Medina D, Rosen JM. Preferential overexpression of a 172Arg – >Leu mutant p53 in the mammary gland of transgenic mice results in altered lobuloalveolar development. Cell Growth Differ (1994) 5:711–21.
Keywords: mutant proteins, p53 mutation, gain of function, stroma, mouse models of cancer, TP53, cancer
Citation: Kim MP, Zhang Y and Lozano G (2015) Mutant p53: Multiple Mechanisms Define Biologic Activity in Cancer. Front. Oncol. 5:249. doi: 10.3389/fonc.2015.00249
Received: 13 September 2015; Accepted: 19 October 2015;
Published: 11 November 2015
Edited by:
Giovanni Blandino, Regina Elena National Cancer Institute, ItalyReviewed by:
Alessandro Rimessi, University of Ferrara, ItalySabrina Maria Strano, Regina Elena Cancer Institute, Italy
Copyright: © 2015 Kim, Zhang and Lozano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillermina Lozano, gglozano@mdanderson.org