 Gaetano Gargiulo
Gaetano Gargiulo- Molecular Oncology, Max Delbrück Center for Molecular Medicine, Berlin, Germany
Animal models of human cancers played a major role in our current understanding of tumor biology. In pre-clinical oncology, animal models empowered drug target and biomarker discovery and validation. In turn, this resulted in improved care for cancer patients. In the quest for understanding and treating a diverse spectrum of cancer types, technological breakthroughs in genetic engineering and single cell “omics” offer tremendous potential to enhance the informative value of pre-clinical models. Here, I review the state-of-the-art in modeling human cancers with focus on animal models for human malignant gliomas. The review highlights the use of glioma models in dissecting mechanisms of tumor initiation, in the retrospective identification of tumor cell-of-origin, in understanding tumor heterogeneity and in testing the potential of immuno-oncology. I build on the deep review of glioma models as a basis for a more general discussion of the potential ways in which transformative technologies may shape the next-generation of pre-clinical models. I argue that refining animal models along the proposed lines will benefit the success rate of translation for pre-clinical research in oncology.
Introduction
Modeling human tumors in animals has been the leading approach to translational research in oncology over the last three decades. Successes in the field include, among others, identifying druggable targets for aggressive subtypes of leukemia, breast cancer and melanoma. In the late 1990s, the retinoic acid (RA) was successful to induce full remission in 71–91% of patients with acute promyelocytic leukemia (APL) in clinical trials that compared this treatment with standard chemotherapy (1). The first transgenic murine models for human APL were generated by inserting the oncogene PML-RAR in promyelocytes downstream the Cathepsin G or hMPR8 regulatory elements (2, 3). These models showed sensitivity to RA (4) confirming their value in the treatment of APL and making them the leading models to investigate responses to anti-cancer treatments at the cellular and molecular levels.
In solid tumors, Trastuzumab (clinical name Herceptin) was approved for treatment in Her2 positive breast cancer. Trastuzumab is an antibody binding to the EGF receptor Her2 and clinical trials showed benefit in Her2 positive breast cancer patients in terms of progression-free and overall survival (5). When the murine p185 antibody was tested in murine breast cancer xenograft models, it proved to be effective in counteracting tumor growth (6). Shortly afterwards, the murine Her2 antibody was humanized to allow its use in clinical trials (7).
Kinase inhibitors were considered the poster child of targeted therapies in the early 2000s and several were approved for treating different malignancies. Imatinib (clinical name, Gleevec) was approved to block the signaling activity of the BCR-ABL fusion protein oncogene in Chronic myeloid leukemia (CML) (8). In mice, the dependence for CML cells on BCR-ABL and the main features of response and resistance to Imatinib could be successfully demonstrated (9). In addition to mouse models, larger animal models such a spontaneous Canine B-cell Lymphoma have also been used to validate the therapeutic outlook for kinase inhibitors (10). The Ibrutinib, a Bruton's tyrosine kinase (BTK) inhibitor validated in this way, later showed a durable efficacy in relapsed or refractory mantle cell lymphoma patients as single agent (11), or in combination with an anti-CD20 antibody (clinical name, Rituximab) (12), which was later extended to chronic lymphocytic leukemia (13).
In the case of the specific BRAFV600E inhibitor Vemurafinib (14), preclinical models not only validated the response to BRAF inhibition but also revealed the RAF paradox, meaning that ERK signaling is amplified by RAF inhibition in BRAF-wild-type, RAS mutant tumors, despite RAF's position downstream RAS (15). In Non-Small Cell Lung cancer (NSCLC), Crizotinib showed great anti-tumor potential against EML4-ALK fusion positive carcinomas, and it has quickly gained momentum for treating a subset of NSCLC patients (16), after successful evaluation in a transgenic mouse model for lung adenocarcinoma (17).
Despite the great success of those compounds, which benefits many cancer patients, targeted therapies are accompanied by acquired resistance, when cancer cells experience gatekeeper mutations or activate alternative pro-tumorigenic pathways (18, 19). To overcome this limitation, targeted therapies are combined with standard chemotherapy or combinations of targeted therapies are sought in appropriate pre-clinical settings (20).
In the clinics, drug combinations often aim at extending on-target toxicity by targeting the same mechanisms (i.e., unrestricted proliferation) with multiple drugs. For instance, the combination of four different chemotherapeutic agents Doxorubicin, Bleomycin, Vinblastine, and Dacarbazine (ABVD) is the standard of care in the treatment of Hodgkin lymphoma (21). However, the use of multiple drugs is accompanied by the increased burden of side effects for patients. Testing more sophisticated approaches, such as synthetic lethality in animal models represents therefore the ideal pre-clinical development. Synthetic lethality is a concept originated from yeast biology and reflects the observation that simultaneously hitting two closely related mechanisms can lead to significant toxicity, whereas single treatments are largely well-tolerated. This offers the possibility to target cancer cells bearing specific alterations, sparing normal cells from side effects. The paradigm of a synthetic-lethal treatment that has made its way into clinical application is the use of Poly-(ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer and breast cancer (22). Targeting PARP proved to be synthetic lethal with concurrent alterations in homologous recombination (HR) DNA repair genes, such as BRCA1 and BRCA2. In pre-clinical research, BRCA-mutant mouse models have been instrumental in highlighting the strength of these treatments and have illustrated potential ways to circumvent their limitations (23). Clinical trials have led to the FDA approval of a number of PARP inhibitors showing efficacy in treatments for ovarian, breast and prostate cancer. In a BRCA-competent triple negative breast cancer/TNBC) DDR-mediated PARP antitumor activity was reinforced by concurrent PI3K-AKT-mTor pathway inhibition (24). In general, predicting long-term responders to PARP inhibitors is a critical issue that has yet to be solved by future pre-clinical breakthroughs.
In addition to their roles as discovery platforms, accurate pre-clinical models can also predict patients' response to a given treatment. In a seminal study, Singh et al. retrospectively assessed targeted therapies either alone or in combination with standard-of-care treatments thereby replicating large-scale human clinical trials (25). This was achieved by applying a similar treatment protocol and evaluating the clinical endpoints overall survival (OS) and progression free survival (PFS) for in vivo studies. Animal models for pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC) were exploited as surrogate for KRASG12D patients' response to Erlotinib and Bevacizumab, therapeutics targeting epidermal growth factor (EGFR) and vascular endothelial growth factor (VEGF), respectively. The standard of care (Carboplatin or Gemcitabine) was combined with targeted drugs thereby mimicking the original studies (25). Retrospectively, this post-clinical study obtained (largely negative) results comparable to those for human clinical trials. This set an excellent standard for future design of pre-clinical studies.
Currently, technological breakthrough in the field of genetic engineering and single cell genomics are enabling us to create ever-more sophisticated animal models of human cancers, and to exploit them in achieving a better translation of pre-clinical studies. Here, I focus on animal models for human malignant gliomas as an entry point for a retrospective review of the use of animal models in tumor biology and therapy. Thereafter, I review state-of-the-art technologies and offer future perspective to incorporating these in generating and exploiting animal models as pre-clinical tools for cancer biology and intervention, which can be valid for glioma and more in general for different types of human cancer.
Experimental Models for High-Grade Gliomas
Autochthonous Mouse Models
Autochthonous mouse models for human cancers are obtained by initiating tumors in a normal cell de novo and within the intact organism. The main advantage of these models is the pathophysiological relevance of the tumor initiation.
Mouse models for Glioblastoma Multiforme (GBM) have been systematically used to investigate tumor initiation and progression in the context of a living organism. The first example of genetically engineered mouse models (GEMMs) in modeling gliomas was developed in the Aguzzi lab. It was generated through the over-expression of the v-Src oncogene via the glial fibrillary acidic protein (GFAP) regulatory elements (26). Later, Holland and Varmus introduced to the scientific community a model based on avian retroviral gene transfer, the RCAS-TVA, which still counts as one of the most commonly used models for gliomas (27).
By far, the largest use for autochthonous models in GBM has been in the systematic dissection of the mechanisms leading to tumor initiation. Several studies have addressed the role of signaling pathways, validated the genetic dependencies on individual genes, and investigated the contribution of non-cell autonomous factors in GBM initiation (Table 1). These studies have uncovered critical pathways in tumor initiation and subsequent genetic aberrations found in high-grade lesions. The unifying conclusion from gliomagenesis in autochthonous models is that GBM is primarily driven by an intricate mix of pro-oncogenic hits cooperating with inactivation of tumor suppressive pathways. On the one hand, hyper-activation of the AKT and MAPK pathways has to occur through either supra-physiological receptor tyrosine kinase (RTK) activity (e.g., EGFR, PDGFRA) or a loss of negative regulators, such as PTEN and NF1. On the other hand, the Rb and p53 pathways must be circumvented by either direct inactivation or through deletion of the INK4A/ARF locus (also known as CDKN2A).
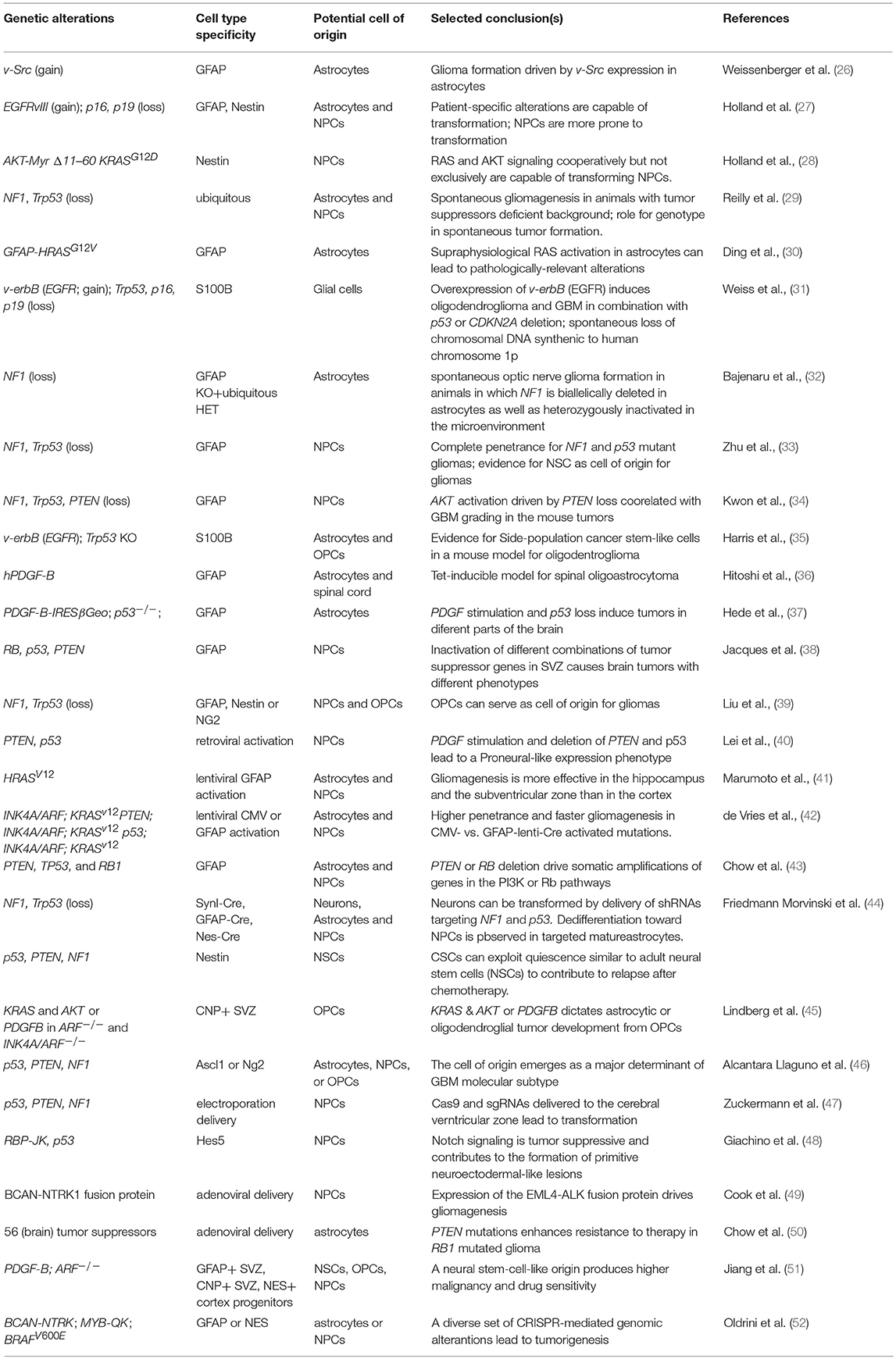
Table 1. Autochthonous mouse models for glioma.
The cellular origin of the disease is an additional intense area of research enabled by GEMM models. Mouse models are useful in this endeavor because the retrospective nature of this assessment makes it hard to precisely identify the cellular origin of cancer in humans. The identification of metastable GBM molecular subtypes and genetic biomarkers, however, provided indirect evidence that fully fledged tumors potentially bear a signature of their potential cell-of-origin (53, 54). This has been now extended to several other cancers (55). Importantly, data in GEMMs are consistent with this view. Overall, from a formal literature review it emerged that: (i) different mutations appear to dictate the cellular phenotype of the resulting tumors; (ii) tumors with similar alterations but originating in different cellular compartments have private biological properties (38, 43, 45, 46, 48, 56). Mouse models have produced overwhelming evidence that undifferentiated neural stem and progenitor cells can efficiently serve as cell-of-origin for the disease in the experimental models (Table 1). Yet it is also evident that nearly every cell in the mouse brain, including post-mitotic neurons, has a potential for transformation, if the oncogenic pressure is significant enough (44). While these studies do not necessarily address the pathophysiological relevance of the models for the origins of human gliomas, two strong messages have emerged from this research. First, neural stem cells are significantly more prone to transformation than the differentiated cells composing the brain parenchyma. Consistently, sequencing of the human SVZ appears to suggest that pre-transformation clones tend to reside in the area of the human brain containing the most undifferentiated neural progenitors (57). Second, regardless of the cell targeted by the oncogenic signaling, the formation of a glial-like malignant progenitor population appears to be an obligatory step in the malignant transformation.
Despite the sophisticated and elegant approaches that were used to generate GEMMs for glioma, the implications of these findings for therapy have so far been limited. Cell cycle regulation emerged as a major predictor of therapeutic response (51, 58), a finding that does not offer additional therapeutic options per se, but represents important ground for future glioma modeling. As discussed below, these studies provide enhanced confidence in transplantation models generated by transforming normal primary cells.
Recently, improved genetic engineering delivery and effectors, such as the CRISPR/Cas9 system have opened new routes to modeling human tumors, including gliomas. CRISPR/Cas9 operates via either non-homologous end joining (NHEJ) that generates genome deletions and insertions (indels), or by homology directed repair (HDR). This approach permits a direct genetic engineering of endogenous loci thereby avoiding random genome integration and potential genotoxicity, which is the intrinsic risk associated with retroviral delivery. For instance, in utero electroporation of gRNAs and Cas9 allowed the simultaneous in vivo deletion of the Trp53, Pten, and Nf1 tumor suppressor genes directly in the brain. This approach bypassed tedious modifications at zygote level (47). Considering that Trp53 and Nf1 are located in close proximity to each other in the genome, conventional breeding is unlikely to produce mutants through their separation onto different chromatids during chromosomal crossover without very time-consuming efforts. This illustrates one way for CRISPR/Cas9 to significantly speed up the pace at which disease models can be generated, including cancer (reviewed, among others, by Sanchez-Rivera and Jacks) (59). Most likely, other investigators will quickly adopt in vivo electroporation.
In addition to simplifying single gene modifications, CRISPR/Cas9 has permitted the modeling of complex karyotypes. For instance, CRISPR/Cas9 was instrumental to generate the mouse equivalent of fusion proteins previously discovered in human cancers (49). The intracranial adenoviral delivery of CRISPR/Cas9 directed microdeletions in the genomic loci of Bcan and Ntrk1 (Brevican and Neurotrophic Receptor Tyrosine Kinase 1, respectively), led to the generation of a mouse model for glioma in which tumorigenesis is driven by BCAN-NTRK1 fusion. This model demonstrated a good response to the kinase inhibitor Entrectinib, which preferentially targets tropomyosin receptor kinases, including NTRK1 (49). This report by the Ventura lab builds on their own pioneering work in generating an EML4-ALK fusion model for NSCLC (60), using CRISPR/Cas9 to systematically screen for fusions as oncogenic drivers in solid tumors. Using this approach, in addition to the chromosomal deletion required for the Bcan-Ntrk1 model, it has been possible to model the Myb-Qk chromosomal translocation as well as induce an equivalent of the human BRAFV600E point mutation by homology-directed-repair (52). The latter improvements in genetic engineering now make easier to generate more complex genotypes in autochthonous models. For instance, mutations in the Isocitrate dehydrogenase 1 (IDH1) are associated with a number of blood and solid tumors, including GBM. While a IDH1R132H models were generated before using classic transgenesis (61, 62), the ability of modifying single loci or create large chromosomal deletions paves the way to generate autochthonous models for human gliomas based on 1p/19q, IDH, and TERT Promoter Mutations, which represent specific entities in humans (63, 64).
Collectively, these studies support GEMMs as invaluable tools in exposing the principles underlying glioma genetics and biology. Non-autochthonous models (described below) have a different set of advantages but clearly GEMMs stand to gain even more momentum in the CRISPR/Cas9 era.
Transplantation Models for Glioma
Transplantation models represent the most widely used alternative to GEMMs, and are essentially built using cells endowed with the ability to initiate tumors in secondary recipient animals. This modeling approach offers the flexibility of spatiotemporal control on tumor initiation and of the potential to experimentally manipulate individual cells. The impact of such manipulations, in turn, can be tested competitively during tumor growth or response to treatment.
The implantation can be carried out in either the tissue in which the disease originated (orthotopic), or more accessible locations, such as the flank of a recipient animal. The choice of the tumor cell and the recipient animal defines whether a model is to be considered syngeneic, homotypic, heterotypic or xenogeneic. For instance, the transplantation of GL261 mouse glioma cells in C57BL/6 recipient mice is considered syngeneic, because GL261 also have a C57BL/6 genetic background. If the background of the donor and recipient animas is not identical, the model is defined as homotypic. A transplantation mode is defined as xenotransplantation if the genotype of the donor is from humans, whereas it is heterotypic for genomes from every species other than humans. Transplantation-based models represent a trade-off between limitations on the pathophysiological relevance and an enhanced control over the temporal initiation of the disease as well as the unique feature of permitting perturbation experiments of various types. Since the first transplantation experiments in nude mice (65, 66), transplantation has permitted testing the tumorigenic and developmental potential of glioma, dissecting its heterogeneity and characterizing underlying molecular mechanisms. Given the flexibility and scalability of this system, it is the best choice for individual target discovery and validation as well as for developing disease-relevant systematic discovery platforms in vivo.
Syngeneic Transplantation Models
Syngeneic tumor models have widely used GL261 cells, due to the fact that they exhibit key alterations in RAS, p53 and PI3K and other pathways which are commonly deregulated in human GBM. Tumors generated in this way share features with human GBM including the upregulation of VEGF and HIF-1α and a diffuse invasion pattern while retaining an intact immune system (67). Lately, syngeneic models have regained more attention with the increasing focus on cancer immunotherapy.
Evidence in cancer patients and mouse models have substantially supported that tumors can be immunogenic but also induce acquired immune tolerance (68). Thus, with the exception of tumors with high mutational and neoantigen load, such as melanoma and lung cancer (69), in heterogeneous solid tumors, the immune-checkpoint inhibitors are unlikely to be effective as single agents. Syngeneic models are well-positioned to evaluate the efficacy of combination therapies, which include the immunotherapy component. In gliomas, one viable combination therapy tested in syngeneic models is the efficacy of CAR T-cells (i.e., T-cells expressing chimeric antigen receptors) with the standard-of-care (70). In a transplantation setting, syngeneic splenocytes from C57BL/6 or VM/Dk were directed against GL-261 or SMA-497, SMA-540, and SMA-560 cells, respectively, by using the full-length NKG2D protein fused to CD3. This system has the advantage of targeting poorly expressed antigens, which is therefore better systemically tolerated. Moreover, NKG2D ligands are multiple antigens, therefore making it more difficult for the tumor to escape. Finally, NKG2D ligands expression appears to increase upon temozolomide and radiotherapy (70, 71), making these targets particularly attractive in an adjuvant setting. Future preclinical testing should include metalloproteases inhibitors, since ADAM10 and ADAM17 expression by tumor cells appear to provide a simple solution to immunevasion by producing soluble NKG2D (72), thereby dampening the γδ T-cell adaptive response (71).
Training the immune system against tumor cells using vaccines could potentially induce a long-term immune response. In a syngeneic mouse model, dendritic cell vaccination using glioma stem-like cells (GSCs) lysate resulted in a measurable response in mice (73). This approach, however, is still being perfected in the setting of human vaccination against brain tumors, as witnessed by clinical trials that failed to show an objective clinical response (74, 75).
Evidence from immunocompromised HIV/AIDS patients is compatible with the speculation that the adaptive immune system is most effective in controlling truly foreign antigens. In fact, HIV/AIDS patients largely develop virus-associated cancers with increased frequency but not other antigenically “colder” tumors (76). A compelling solution to selectively induce immune responses against foreign antigens in tumor cells is offered by oncolytic viruses (OVs). OVs with tropism for cancer cells can simultaneously act at different levels in the tumor microenvironment (TME). Local killing of tumor cells works as in situ vaccines, alerting antigen-presenting cells (APC) to multiple tumor-associated antigens (TAAs). To contribute to APCs maturation, this effect can be reinforced by OVs preloading with dedicated cargoes (e.g., GM-CSF gene as co-stimulatory treatment). OVs can promote intratumoral T-cell infiltration, for instance, by eliciting a type I interferon response. By inducing local acute inflammation, OVs can also reduce the impact of a suppressive TME. For a recent comprehensive review of the use for OVs as anti-cancer therapy, including a list of several clinical trials for OVs in cancer, I would refer the reader to Twumasi-Boateng et al. (77). Recently, Measles-based virotherapy has demonstrated synergistic activity with anti-PD-1 therapy in GBM treatment in a syngeneic C57BL/6 GL261 model (78). In a similar setting, intravenous delivery of GM-CSF/reovirus also showed synergistic activity with anti-PD-1. Moreover, intravenous reovirus delivery was also performed in human patients, and the viral payload was confirmed upon tumor resection, providing evidence of successful induction of the hallmarks of OVs activity (79). These examples are very important since checkpoint inhibition alone is insufficient to induce a response in GBM patients (80, 81). Preliminary clinical trials on OVs approaches are now completed and report on some positive indicators onto which more clinical and pre-clinical research should be designed) (82, 83), thereby underscoring the importance of animal models in testing strategies to awakening the immune system.
Thus, syngeneic models are becoming increasingly widespread. In addition to the mouse models, C6 glioma implantation in the fronto-parietal lobe of Whistar rats and 9L gliosarcoma in Fisher rats display features common to the human disease, such as proliferation, similar focal invasion as well as pseudopalisading necrosis surrounded by cells with great nuclear polymorphism (67). Interestingly, allogeneic 9L tumors in Wistar rats show a high infiltration of macrophages, microglia and CD4+/CD8+ T-cells that coexist with tumor lesions (84). This model has been successfully used to prove that dendritic cell therapy leads to enhanced tumor infiltration by CD4+/CD8+ T-cells and prolonged survival (85), and might represent a valid alternative to mouse models in the study of mechanisms of action for checkpoint inhibitors or therapeutic approaches targeting immune cells by other mechanisms, such as OVs.
Homotypic and Xenogeneic Transplantation Models
Traditionally, transplantation-based models served the purpose of testing the genetic dependence of tumor cells on individual genes or pathways (Table 2).
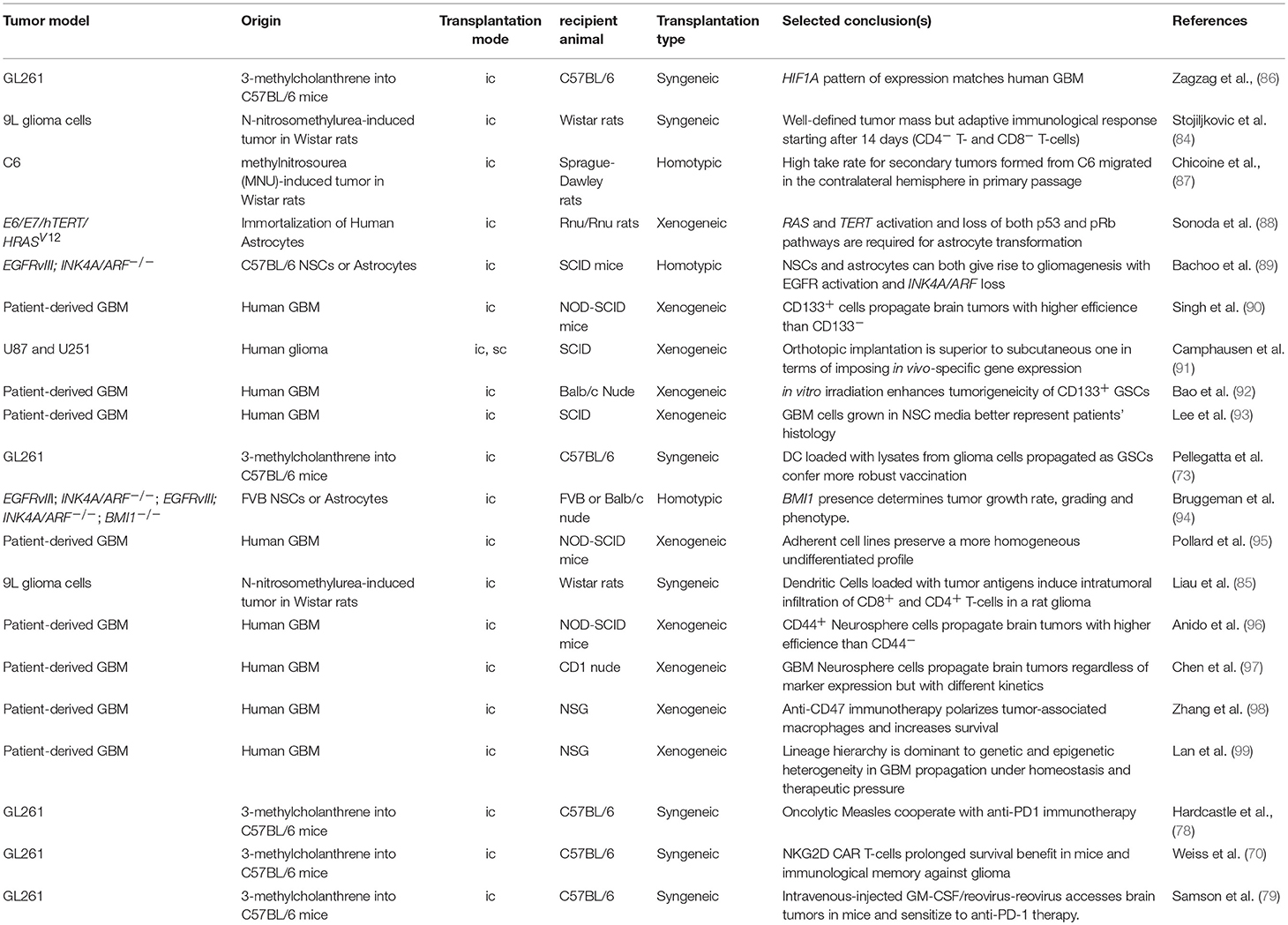
Table 2. Transplantation models for glioma.
While C6 tumors bear alterations in key tumor suppressors, such as the Cdkn2a (Ink4a/Arf) and Pten, in syngeneic as well as allogeneic transplantations, the transformation of astrocytes and neural stem cells derived from tumor suppressor mutant animals provides higher control of the tumor genotype (94), and represents a better setting for target discovery and validation (100).
A transplantation model based on immortalized astrocytes was instrumental in demonstrating that the loss of both the p53 and pRb pathways and gain of MAPK signaling and TERT-dependent telomeres protection are critical components in gliomagenesis (88). In the mouse, the minimal combination of the Ink4a/Arf locus deletion (i.e., p53 and pRb pathway inactivation) and constitutive EGFR activation by a glioma-specific mutant (i.e., MAPK activation) is dominant over the cell of origin (89). These seminal discoveries anticipated the demonstration that telomere protection can be reactivated by multiple means. TERT is reactivated, for example, in patient-derived glioma cells propagated under neural stem cell conditions, suggesting that upstream signaling can suffice (93). Moreover, there also exists a mechanism for the alternative lengthening of telomeres in GBM patients (101). Hence, while the control of telomere integrity is critical to gliomagenesis, TERT overexpression in itself may be a dispensable genetic manipulation in the process of glioma modeling in the mouse, despite promoter mutations define specific entities in human gliomas (63, 64).
Despite the fact that transplantation-induced stress can impart clonal expansion and affect gene expression, orthotopic transplantation should be considered the best approximation for a transplantation setting. In fact, subcutaneous and orthotopic growth impart very different transcriptional responses to the glioma cells' in vivo gene expression profile and response to treatment (91, 102). Consequently, orthotopic models for glioma also played a major role in the quest for potential epigenetic anti-cancer targets. The Polycomb group (PcG) gene Bmi1 was shown to have oncogenic functions in gliomagenesis as a negative regulator of the Ink4a/Arf tumor suppressor as well as Ink4a/Arf-independent functions, as found in both mouse and human cells (94, 103). Likewise, the Polycomb repressive complex enzyme EZH2 also appeared to be required for glioma cell survival and proliferation in grafting experiments (104) and might be a good target reinforcing the adjuvant chemotherapeutic agent Temozolomide (99), even though context-dependent effects in the opposite direction were observed (105). Considering that Polycomb proteins contribute to coordinate the transcriptional response to converging pathways (100), the differential response to Polycomb inhibition may reflect the contribution of context-specific stress pathways within individual experimental settings. Alternatively, or in parallel, multiple cell populations composing the intra-tumoral mass may be differentially sensitive to Polycomb inhibitors (106), and their relative abundance may determine the overall response to the single agent.
To date, the genetic pathways that have been shown to prominently contribute to glioma growth in orthotopic mouse models have been largely validated by genomic studies in human patients (107), whereas targeting the epigenetic machinery as anti-cancer strategy still awaits the identification of effective combinations to account for compensatory mechanisms including adaptive responses and intra-tumoral heterogeneity.
Dissecting the tumorigenic potential of individual cellular populations that reflect this heterogeneity have also largely relied on the use of orthotopic models (Table 2). Following pioneering studies in leukemia (108), Dirks and colleagues demonstrated that tumor initiating ability in Non-obese diabetic, severe combined immunodeficient (NOD-SCID) mice is restricted to a subset of brain tumor cells (90). These cells in human GBM could be prospectively isolated using CD133 as surface marker (90). The ability to form neurospheres in vitro (109), and to withstand ionizing radiation (92) are critical biological features associated with “cancer stem-like cells” from high-grade gliomas. Surface markers, such as CD15 and CD44 have also been used to positively enrich for brain tumor initiating cells (96, 110). However, systematic comparisons of orthotopic grafts generated in CD1 Nude mice using glioma cell populations with different profiles of surface markers revealed that the non-enriched population also has tumor-initiating ability, with delayed growth kinetics (97). The difference for heterogeneous tumors, such as GBM may reflect the trans-differentiation ability of tumor cells into a wide range of lineages (111–114). Rather, the neural stem cell growth conditions of primary GBM cells proved to be a critical determinant in how accurately the orthotopic grafts resembled the patients' tumors (93, 95, 115). Collectively, these studies made enormous contributions to uncovering the cellular components of intra-tumor heterogeneity and highlighted the importance of orthotopic xenograft models.
Xenotransplantation is becoming increasingly the setting of choice for high-throughput target discovery and validation, a research area that recently evolved around the use of orthotopic models for cancer. Several pioneering studies in various allograft transplantation models have highlighted the importance of genetic screens in relevant physiological contexts (116–118). To cope with the numerical limitations imposed by the in vivo setting (119), genetic screens using RNAi in gliomagenesis (100, 120) were performed in immunocompromised animals with small libraries, or a genome-wide CRISPR was performed first in vitro and followed later by a parallel in vivo/in vitro validation screen (121).
As inferred by these studies, the use of immunocompromised recipient animals remains an invaluable tool in investigating cell autonomous mechanisms in human cancer cells in vivo and the choice of cellular models is therefore critical. Tumor models using established tumor cell lines have enormously contributed to our knowledge of tumor biology but are increasingly being dismissed in gliomagenesis experiments (122). The state-of-the-art in the field of brain tumors is to propagate tumor cells under conditions supporting non-transformed the in vitro self-renewal of neural stem cells (93). Limitations to this approach include that: (i) ex vivo propagation remains anchored to the assumption that signaling, supplements and environmental conditions are known and can be delivered homogeneously, (ii) some low-grade tumors as well as specific genotypes systematically drop out in these conditions (123). Nonetheless, sophisticated ex vivo culture conditions represent the best approximation to preserve tumor identity while enabling experimental manipulations or in vitro screening endeavors (115). Indeed, when compared to patients' biopsies, conventional glioma cell lines fall short of representing patients' molecular profiles (53).
Patient-Derived Xenografts Models
An elegant approach to bypass cultures while focusing on patient-oriented modeling is to generate Patient-derived xenografts (PDX) or Avatar models, created by patients' biopsies without applying ex vivo culturing prior to transplantation (124). PDX models propagate the complex cellular and genetic heterogeneity of the cell surviving in the host animal, and are therefore capable of modeling responses to standard, targeted or combination therapies without forcing assumptions on the tissue sample. A variant to preserving patients' biopsy tissue structure is dissecting the tissue prior to the transplantation of live cells. This is usually exploited to bypass the low take rate for some tumors, a drawback affecting low-grade or high complexity tumors. A similar approach is used to perform limited experimental manipulations followed by serial transplantation (99).
There is a general consensus that PDX models maintain some level of concordance between patients and PDX responses to therapy (125, 126). This includes GBM, which partly preserves molecular profiles principles during xenotransplantation (53, 127). Importantly, however, the response to anti-cancer treatment largely depends on the tumor cell genotype at the time of the treatment (127). The latter piece of evidence is relevant in that both ex vivo cellular passaging and PDX intrinsically suffer a drift toward genomic instability. In a recent large-scale study, the dynamics of copy number alterations (CNAs) in 1110 patient-derived samples of different cancer types during multiple rounds of in vivo propagation have been reported. A high rate of CNAs was observed in xenografts that artificially drift away from the human counterpart. For instance, glioblastoma patients acquire extra copies of chromosome 7 during tumor evolution, whereas PDX propagation in mice results in a loss of these extra copies. While the genetic drift is not surprising for malignant gliomas given their near-complete deficiency in DNA damage checkpoint control (128), these results raise awareness of the limitations associated with the use of PDX as patients' avatars to evaluate their responses to any given therapy and call for integrating this resource with more stable models (129). It is also critical to realize that responses to therapies in PDX models will be affected by the intra-cellular heterogeneity of the transplanted tumor. Using an elegant cellular barcoding strategy, the Dirks lab has recently demonstrated that a number of different cancer cells within a tumor can contribute to its homeostasis (99). Strikingly, however, this number can change from one tumor to the next, thereby affecting the reproducibility of hypothesis testing or target discovery and validation experiments.
In immune-oncology, transplantation models are the preferred choice when testing the impact of innate immune checkpoint inhibition and CAR T-cells. For instance, disruption of the CD47/SIRPα signaling affects leukemia, glioma, melanoma and hepatocellular carcinoma growth as xenografts (98, 130–132). The efficacy of GD2-CAR T-cell against glioma cells was pre-clinically tested in NSG mice until the onset of graft-vs-host disease symptoms (GvHD; ~4 weeks), and GD2-CAR T-cells are currently tested in several clinical trials (among others, see NCT03252171).
These studies collectively highlight the continuing process of addressing the intrinsic challenges associated with transplantation models and expanding these to advanced pre-clinical settings, anticipating that this experimental system will retain its leading role in experimental medicine.
Next-Generation Autochthonous Models
In mice, the CRISPR revolution will make the generation of complex animal models increasingly easier, faster and affordable (Figure 1).
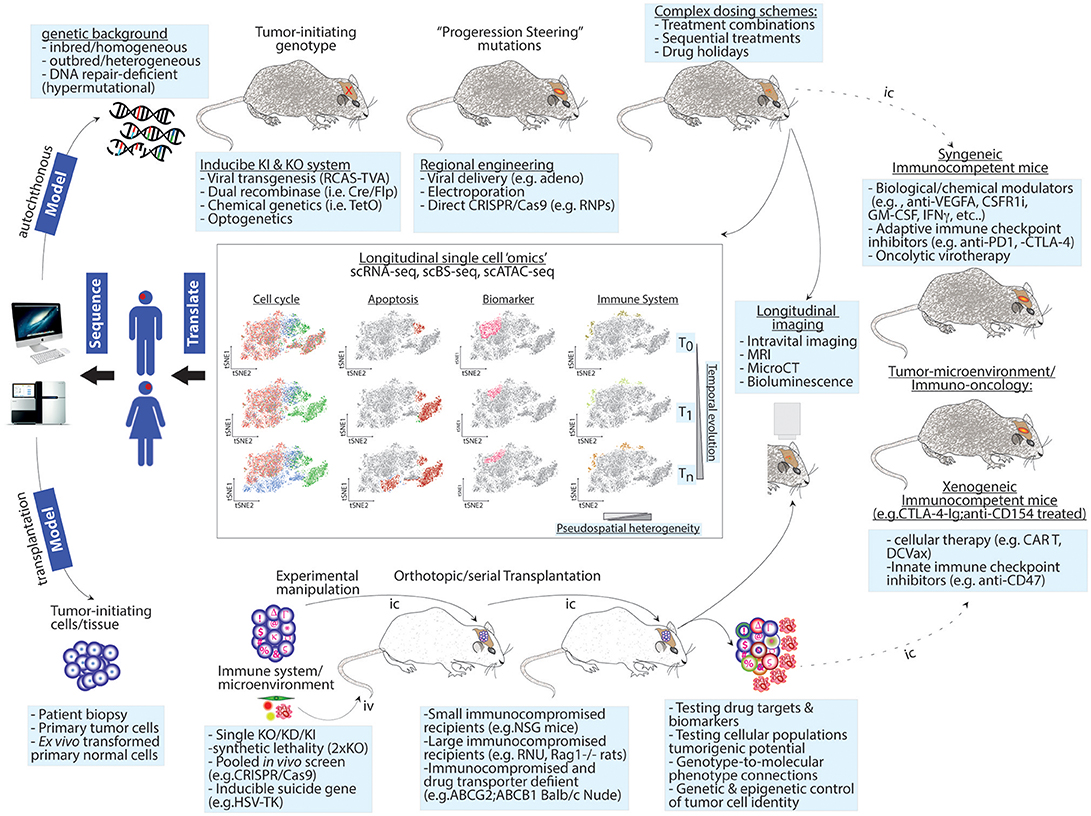
Figure 1. Next-generation modeling of human cancers: longitudinal single-cell “omics” in autochthonous and transplantation-based models. Upper panel: genetic drivers of human cancer are combined with a genetic background of choice to give rise to homogenous, heterogeneous, or hypermutational tumors. Genetic engineering can use independent genetic and/or chemical switches to permit tissue-specific and temporal control. To increase tumor heterogeneity or test the contribution of mutations identified at recurrence upon tumor formation, intratumor injections of “steering” mutations using viral or other means can be used. These sophisticated cancer GEMMs permit testing complex dosing treatments. Lower panel: to test genetic dependences or perform genetic screens, patient-derived or ex vivo transformed tumor-initiating cells are genetically modified and/or barcoded and then transplanted into recipient animals. The host background can be chosen to favor tumor take and/or drug penetration. In this setting, a co-grafting of adaptive or innate immune cells or other microenvironment players (e.g., endothelial cells, pericytes, etc.) or serial transplantation can be implemented to study non-cell autonomous mechanisms and to exacerbate competition among cancer cells. Heterogeneity increases as a function of time and intra-tumor complexity. Both experimental models can be exploited in longitudinal follow-ups using live imaging or single cell “omics” (central panels). The latter approach can simultaneously generate spatiotemporal information on changes in cell cycle progression, apoptosis, cell fate decisions/microenvironment activation (i.e., biomarker variations) and immune cell composition. Right panel: single cell preps from GEMMs or orthotopic transplantation models can be transplanted (dashed line) in the indicated immunocompetent animals thereby creating a syngeneic/xenogeneic models, respectively. Potential applications for these models are indicated. T0, T1, and Tn = time points for longitudinal analyzes; pseudospatial complexity refers to the output of tSNE maps as a surrogate for spatial information. Iv, intravenous; Ic, intracranial.
Tumor heterogeneity and evolution is a major feature of human cancers (133, 134), and these features have proven hard to model autochthonously. One important consideration for the design of future, next-generation mouse models is their genetic background. Most mouse strains are inbred and represent a great advantage in research to unravel specific targets or mechanisms in a certain type of cancer (135). Yet inbred strains are far from the real diverse setting of disease development in humans, and the genetic background of animals used as pre-clinical cancer models in research is critical to the outcome of the study. In gliomagenesis, Nf1 and p53 deletion leads to a tumor grading ranging from low-grade astrocytoma to GBM depending on the distinct mouse strain (29). Likewise, p53 heterozygosity leads to a spontaneous formation of mammary tumors in BALB/c mice but not in C57BL/6J (136), suggesting a distinct pro-tumorigenic genetic background in BALB/c mice. Moreover, the RF/J strain displays a high incidence of cancer owing to missense mutations in DNA damage repair and cancer-associated genes and may become the model of choice for genetically hypermutated tumors, such as smoking-driven lung cancer, UV-driven melanomas and DNA repair-deficient colon cancer. The choice of the background for future GEMMs may be driven by the clinical history of individual diseases and may exploit the Collaborative Cross Consortium 2012 benchmarking of several inbred mouse strains (137). In turn, this may also facilitate testing the contribution of natural variation and DNA repair in tumorigenesis and responses to treatment. This appears to be particularly important if one considers that genetic variation may not only affect tumor-intrinsic, but also and even more likely, the tumor-immune phenotype.
The recent conclusion of a colossal genomic investigation in almost 10,000 genomes spanning 33 cancer types by the Cancer Genome Atlas (TCGA) has provided an enormous dataset of cancer driver genes (138). In vivo validation and hypotheses testing follow-ups are lagging behind. Simultaneously testing dozens of putative tumor suppressors within a native and immunocompetent microenvironment is now possible. In a medium throughput in vivo screening in gliomagenesis using the stereotactic injection of a sgRNA adenoviral library for CRISPR/Cas9 mediated knock-outs, the Sidi Chen lab reported on ability for PanCancer-defined significantly mutated genes to initiate brain tumor formation (50). The combination of regional viral delivery and GFAP-Cre conditional activation of Cas9 and GFP restricted the screen to a subset of glial cells. This type of screen follows the direct RNAi delivery pioneered by the Zender lab in hepatocellular carcinoma (139). While the inability of AAV to genetically integrate into the host genome requires extensive target sequencing as direct readout, compared to earlier studies, this provided a significant higher throughput in screening for combinatorial signaling dependencies during gliomagenesis (38, 43). It also represented a more physiological setting for tumor suppressor discovery than transplantation models previously used in similar endeavors (100). Considering that adult GBM is a disease that often develops over several years and goes through regional and temporal evolution (140, 141), future studies along these lines may benefit from integrating more sophisticated, spatiotemporally precise forms of editing. Examples of the sequential delivery of cancer driver mutations exploiting classic genetic recombination techniques were previously reported (142). To enable further spatiotemporal control, biochemical, chemical or optogenetic control of tumor progression may be employed. This would be particularly important in order to better mimic disease initiation and progression (Figure 1).
An enhanced control of tumor progression using advanced genetic engineering methods may permit a validation of driver and passenger mutations as modifiers of tumor progression and responses to therapy or testing advanced genetics paradigms, such as genetic essentiality (22). To this end, while direct intracranial injection of RNP complexes has not yet been exploited in models of glioblastoma, it has been successfully applied to genome editing in the mouse brain (143). During brain tumor exposure to multimodal therapy, additional aberrations in core GBM driver pathways are acquired. Combining longitudinal intravital imaging and topical delivery of RNPs or in situ electroporation-inducing genetic engineering may help in elucidating the contribution of genetics to disease recurrence. Genetic alterations identified as critical regulators of tumor evolution could be defined as “steering” mutations, and would become an integral part of sequential modeling. In GBM modeling, two examples of such mutations may be Nf1 and Msh6, both of which are associated with recurrent tumors and TMZ-induced hypermutations (144, 145). Importantly, this approach would also permit the functional dissection of cell-autonomous mechanisms, such as tumor microtube formation (146), as well as non-cell autonomous processes, such as vessel dysmorphia (147).
Unpredictable adaptive responses to anti-cancer treatments are a hallmark of solid tumors. Cancer cells can evade chemotherapy by acquiring additional mutations, switching to a state of negligible growth, by activating survival pathways through changes in cell identity, and by other means. In modeling human cancer in mice, one should account for possible species-specific tumor genotype drifts that would not be representative of the patients'. Thus, next-generation GEM models that better represent primary tumors and their evolution at a genetic level (as depicted above) will be invaluable tools for testing complex chemotherapy-dosing schemes (Figure 1).
Longitudinal studies in humans are revealing oncogenes dominant in recurrence. In GBM, mutations in NF1 and PI3KCA appear to drive disease progression (145, 148, 149), and may be temporally controlled to mimic disease progression. In such models, complex sequential drug combinations may be tested. For instance, switching off NF1 or PI3KCA, which reinforce the RAS pathway and confer sensitivity to a combination of BRD4 and MEK inhibition (150). A neoadjuvant-like multimodal therapy followed by targeted drugs, would be prototypic examples of testing complex targeting of acquired mutations.
Metronomic chemotherapy and planned drug holidays are complex dosing schemes that aim at reversing drug tolerance. In GBM, tumor cells evade adjuvant Temozolomide using MGMT reversal and cell cycle restriction. A Temozolomide holiday may be therefore alternated with targeted therapies, such as blood-brain-barrier penetrant PI3K inhibitors (151).
An emerging paradigm in cancer biology is to drive cancer cells to acquire specific addictions and then targeting such addictions with drugs. This approach may be important in exploiting metabolic (152, 153) as well as epigenetic targets (154), or targeting collateral lethality associated with acquired resistance mechanisms [(155) and our unpublished data]. This includes also extrinsic mechanisms, such as those targeted by adaptive immune checkpoint inhibitors. Indeed, studies in syngeneic models have shown promising results in a combination or neoadjuvant setting that induced PD-L1 expression (79, 156).
While complex dosing schemes have been so far directly tested during clinical trials, the combination of accurate GEM models and complex readouts should make it standard practice at the pre-clinical level to test whether low-dose or sequential treatments are equally or even more effective than drug combinations, thereby sparing patients from the side effects of the added toxicity and possibly enhancing tumors' response. In this regard, once the specie-specific differences are accounted for, the autochthonous models are well-positioned to provide the most physiological setting for tumor initiation and organismal response to treatments.
Next-Generation Transplantation Models
Transplantation models are currently developing on parallel research lines.
To avoid the complications of genetic drifts as well as patient-specific passenger mutations, PDX xenograft models are being complemented with de novo transforming human cells derived from normal tissue with relevant cancer mutations in the projected neoplasia. Transforming human astrocytes has been instrumental in formally testing the contributions of the main pathway required for efficient gliomagenesis in human cells. Recent work using human colon organoids has shown that combining an appropriate cell of origin and set of mutations is still insufficient to recapitulate some of the biological properties of the true tumors, such as metastasis (157). Future work in this direction should be aimed at precisely dissecting genotype-to-phenotype connections in human cancers using advanced genetic screening systems. In this regard, systematically transforming normal mouse and human cells to create next-generation avatar models is expected to help address some open questions. For instance, in numerous solid tumors, the role of copy number aberrations remains to be clarified. Whereas, human GBM is clearly a disease of copy number aberrations (158), studies in autochthonous models so far fall short of clarifying whether and how this feature contributes to the disease. Moreover, emphasis should be given to developing an understanding of molecular phenotype specifications for tumors including GBM in which there is limited evidence of genetically encoded subtypes. Overall, creating reliable and homogenous tumor-initiating cells in vitro may complement PDX-models and permit testing ad hoc biological questions as well as providing a more reproducible resource for target discovery and validation (Figure 1).
The recipient animals are a critical determinant of the success of transplantation models. The informative value of such models is dependent on both cell-intrinsic and non-cell autonomous components. On the one hand, achieving the highest grafting potential through the use of severely immunocompromised animals is the essence of xenotransplantation (159). On the other, the discovery of adaptive checkpoint inhibitors and the need to identify the basis for responses to immunotherapy pose additional challenges to modeling human tumors using xenotransplantation; one solution is to reinstate the adaptive immune system in host recipients. Freshly isolated peripheral bone marrow cells (PBMCs) or specific immune cell subpopulations can be co-grafted with human cancer cells subcutaneously in immune deficient animals (e.g., NSG/NOG). Alternatively, PBMCs can be parentally infused (i.p. or i.v.) after subcutaneous or orthotopic tumor transplantation has taken place. This approach revealed, for instance, the immunogenic effect of a Carcinoembryonic Antigen CD3 T-Cell Bispecific Antibody (CEA-TCB) in promoting the infiltration of xenografts as well as adaptive PD-L1 over-expression, thereby suggesting the feasibility of combining its administration with adaptive immune-checkpoint inhibitors (160). Whereas this setting is limited to short-term studies given the potential for severe GvHD, it also enabled assessments of the efficacy of immunotherapies and the impact of T-cell subpopulations (e.g., regulatory T-cells; T-regs) or antigen-presenting cells (APC) on the activation of cancer cells (161).
The GvHD is not only a complication for the recipient animals. Immune cells that suddenly face a full-blown tumor to which they were previously naïve face an acute pathophysiological stimulation reflecting the actual tumor. Ideally, future models will feature intact innate and adaptive immune systems trained to ignore the grafting phase and triggered by pathophysiological stimuli to transiently allow grafting. An elegant example of this approach in glioma models has been achieved by blocking the T-cell activation by APC through CTL4A-IgG (clinical name: Abatacept) and anti-CD154. Immune-tolerant mice generated by this approach developed GBM lesions and these affected survival with similar kinetics as immune-deficient animals (162). As an alternative in immune-deficient backgrounds, co-grafted immune cells may be pre-exposed to tumor cells (e.g., DC loading with tumor cells lysate or RNA), thereby more closely mirroring the gradual rise of tumors in humans. Moreover, some innate immunity players may require specific co-grafting schemes. For instance, microglia cells, the resident macrophages of the central nervous system, are generated during embryonic development (163). These cells are associated with the full specification of the Mesenchymal GBM subtype identity (164–166), and most likely with responses to treatment (167). This situation calls for a reliable source of human microglia whether tumor-isolated or in vitro immortalized to serve as co-grafting partners in transplantation models.
The genetic engineering of recipient animals can also be exploited to selectively impair the tumor microenvironment. To determine the role of drug penetration and reabsorption in brain tumor models, for instance, the drug transporters ABCB1 and ABCG2 were deleted not only in wild-type glioma models, but also in immunodeficient recipients in xenotransplantation models (168, 169).
In the neurosciences, the rat is the experimental and translational model of choice wherever possible. This species is considered superior for etiological, pathophysiological, pharmacological and behavioral studies (170). The discovery of culture conditions that facilitate the expansion of rat embryonic stem cells as well as the direct use of CRISPR during zygote formation may enable extending also to rats the generation of complex models for gliomas. This would allow extending the use of these animals from a limited pharmacodynamics setting to a fully-fledged advanced modeling system.
The future development of transplantation models at the levels of both tumor cells and recipient animals will enable more sophisticated experimental dissections of cell-intrinsic and non-cell autonomous mechanisms and a more effective platform for target discovery and validation.
Outlooks and Perspectives for Single Cell “Omics”
Interrogating cell cycle responses, apoptosis and cell identity drifts at the level of single cells is a transformational change in that it permits assessing the effects of a compound and simultaneously to build hypotheses based on possible combinatorial treatments or sequential treatments (Figure 1).
Single-cell RNA sequencing has been significantly exploited in GBM to describe intratumor heterogeneity and the peculiarities of GBM subtypes and their microenvironments (164–166). This approach has also been instrumental in identifying genetic and transcriptional identities associated with tumor-specific biological properties, such as proximal and distal recurrence, infiltration and numbness to the fluorescence-guided probe for the resection of diseased tissue (5-Aminolevulinic acid, also known as 5-ALA) (149). DNA methylation is being currently implemented in clinical neuropathological practice for brain tumor classification (171). The availability of technologies enabling to simultaneously generate single cell RNA-seq (scRNA-seq) and bisulfite-converted DNA methylation (BS-seq) maps from the same cell (172) has a potential to allow the tracking of tumor responses to treatments. In turn, this will be a major driver in the development of targeted therapies in both autochthonous and transplantation models (Figure 1).
I envision that the combination of multiple autochthonous models in parallel and scRNA-seq and BS-seq may be exploited to evaluate the way the core tumor and in the infiltrating margins respond to individual treatments. Importantly, metabolic labeling of RNA in vivo now enables identifying faster and more accurately the adaptive transcriptional changes (173).
To bridge preclinical and clinical testing, longitudinal scRNA-seq in GEMMs may help assessing and improving adjuvant and second-line treatments. To some extent, autochthonous models of aggressive human cancers may well represent low Karnofsky performance score (KPS) patients or very hard-to-resect tumors, in that surgery is discouraged. In these cases, single cell profiling will be very informative in testing the consequences of standard approaches, such as radiation followed by adjuvant treatments (e.g., in GBM, Temozolomide, Bevacizumab) and provide benchmarks for new treatments. This matter has so far been restricted to advanced clinical trials in patients and may be now repositioned at the preclinical stage.
Exceptionally, single cell “omics” and autochthonous models may also help testing next-generation probes for optically guided surgery. 5-ALA is currently used in fluorescent-guided surgery to delineate tumor margins for resection, which is extremely important in preventing local recurrence in the brain parenchyma, where surgeons need to be conservative. Recent studies in GBM at the single-cell level suggest that 5-ALA appears to mark Mesenchymal subtype-specific GBM cells, leaving behind Proneural-subtyped cells (149). It will be important to test this in autochthonous models in which the two states can be modeled (56), and to compare 5-ALA to novel fluorescent probes. By resecting a tumor margin pre-labeled with such probes, single cell profiling will reveal the identity of each cell that retain or miss the labeling.
The xenogeneic transplantation setting is well-versed for testing intrinsic cellular responses to novel treatments and detecting adaptive resistance, notably in cases in which tumor perfusion is homogeneous. Indeed, the response to intracellular targets for drugs are best assessed in human cells when on-target and off-target effects need to be accounted for. Moreover, applying CRISPR screens to transplantation models using scRNA-seq readouts, such as CROP-seq (174) may improve the resolution of in vivo functional screens, thereby increasing their throughput. In the brain tumor setting, these were limited so far to a few dozen targets (100, 120), and CROP-seq may lead to more comprehensive screens without compromising the pathophysiology of the orthotopic transplantation setting (119). Importantly, even in the absence of functional perturbations, approaches like CROP-seq coupled with scRNA-seq will enable a next-generation of cellular barcoding experiments to trace in vivo tumor homeostasis and responses to treatments (99). In this area, PDX models should more systematically be complemented with de novo transforming human cells and with models better representing tumor molecular profiles.
Harnessing the therapeutic potential of the TME and immuno-oncology will significantly benefit of building experimental consensus within the community. In particular, it is critical to define the appropriateness of any given model in the assessment of the response to treatment of established tumors. Differences between animal models and humans specifically involve protein-coding genes and cis-regulatory DNA due to specie-specific adaptive selection, notably those controlling immunity (175, 176). Moreover, the consistency in the response of human cells to mouse supplements and vice versa (e.g., growth factors, cytokines, etc.) are largely anecdotal. Nevertheless, whether GEMMs, syngeneic or xenogeneic models based on orthotopic transplantation may be used for testing of TME- and immune-therapies should be systematically assessed when appropriate readouts can be faithfully reproduced in both species. This has been possible, for instance, in the quest for identifying antigen-specific TCRs. In this case, comparing immune-deficient mice reconstituted with human hematopoietic progenitors (i.e., humanized mice) and mice transgenic for the human TCRα and TCRβ loci returned similar results (177).
Despite the acknowledged pathophysiological relevance of GEMMs, syngeneic models based on established cell lines have been preferred in investigating TME- and immune-therapies (Table 2). To exploit the CRISPR/Cas9 potential in the future models, I anticipate that primary tumor cells from sophisticated GEMM models may soon replace cell lines in a secondary syngeneic orthotopic setting. This way, one could harness the genetic and spatiotemporal controls in an immune-tolerant setting (Figure 1, right panel). This approach would be the ideal development for the preclinical dissection of intrinsic, acquired and non-cell autonomous resistance mechanisms. In the example provided by the combinatorial or neoadjuvant OVs treatment, syngeneic models may be used to uncover the mechanism conferring residual resistance, which is indicated by the incomplete penetrance of the sensitization to checkpoint inhibitors (79, 156). Longitudinal scRNA-seq may reveal the optimal timing for starting the immune-checkpoint inhibition and whether cell intrinsic or other cells from the microenvironment are contributing to or preventing a fully penetrant response.
Whereas testing immunotherapy strategies in immunodeficient animals appears counterintuitive, the use of NSG mice emerges as the mainstream strain for studies involving sophisticated reagents eventually used in clinical trials. These include systems for T-cells engineering (178, 179), and innate immune checkpoint inhibitors (98, 130–132). While retaining a leading position in these experiments, a foreseeable evolution of this system will involve improving the recipient animals, including transplanting human cells into highly immunodeficient rats.
Transformative technologies, such as CRISPR/Cas9 and single-cell genomics have opened new avenues in the study of tumor biology. Here I propose that the longitudinal dissection of tumor responses in animal models can capitalize on single cell genomics approaches. The specific examples I have discussed here are cases that could clearly improve our current understanding of human cancers and their responses to treatment. Combining genetic engineering and single-cell genomics with the individual strengths of GEM and transplantation models is bringing about novel next-generation platforms for understanding tumor biology and for target discovery and validation.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I am grateful to Ulrike Brüning and to Andreas Göhrig for assistance with literature review, to Michela Serresi for critical reading and to Yuliia Dramaretska and Russel Hodge for proofreading and editing of the manuscript. I apologize for the original work I could not directly cite owing to space constraints. GG acknowledges funding from the European Research Council (ERC StG 714922), the Helmholtz Association and the MDC.
References
1. Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. (1997) 337:1021–8.
2. Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood (1997) 89:376–87.
3. Brown D, Kogan S, Lagasse E, Weissman IL, Alcalay M, Pelicci PG, et al. A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci USA. (1997) 94:2551–6.
4. Lallemand-Breitenbach V, Guillemin MC, Janin A, Daniel MT, Degos L, Kogan SC, et al. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J Exp Med. (1999) 189:1043–52.
5. Pegram MD, Lipton A, Hayes DF, Weber BL, Baselga JM, Tripathy D, et al. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. J Clin Oncol. (1998) 16:2659–71.
6. Bangalore L, Tanner AJ, Laudano AP, Stern DF. Antiserum raised against a synthetic phosphotyrosine-containing peptide selectively recognizes p185neu/erbB-2 and the epidermal growth factor receptor. Proc Natl Acad Sci USA. (1992) 89:11637–41.
7. Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA. (1992) 89:4285–9.
8. Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res. (2002) 8:935–42.
9. Dash AB, Williams IR, Kutok JL, Tomasson MH, Anastasiadou E, Lindahl K, et al. A murine model of CML blast crisis induced by cooperation between BCR/ABL and NUP98/HOXA9. Proc Natl Acad Sci USA. (2002) 99:7622–7. doi: 10.1073/pnas.102583199
10. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. (2010) 107:13075–80. doi: 10.1073/pnas.1004594107
11. Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. (2013) 369:507–16. doi: 10.1056/NEJMoa1306220
12. Wang ML, Hagemeister F, Westin JR, Fayad L. Ibrutinib and rituximab are an efficacious and safe combination in relapsed mantle cell lymphoma: preliminary results from a phase II clinical trial. Blood (2014) 124:2627.
13. Burger JA, Keating MJ, Wierda WG, Hartmann E, Hoellenriegel J, Rosin NY, et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. (2014) 15:1090–9. doi: 10.1016/S1470-2045(14)70335-3
14. Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. (2012) 11:873–86. doi: 10.1038/nrd3847
15. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature (2010) 464:431–5. doi: 10.1038/nature08833
16. Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. (2010) 363:1693–703. doi: 10.1056/NEJMoa1006448
17. Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci USA. (2008) 105:19893–7. doi: 10.1073/pnas.0805381105
18. Hochhaus A, Kreil S, Corbin AS, La Rosée P, Müller MC, Lahaye T, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia (2002) 16:2190–6. doi: 10.1038/sj.leu.2402741
19. Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell (2010) 18:683–95. doi: 10.1016/j.ccr.2010.11.023
20. Lopez JS, Banerji U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat Rev Clin Oncol. (2017) 14:57–66. doi: 10.1038/nrclinonc.2016.9621.
21. Gordon LI, Hong F, Fisher RI, Bartlett NL, Connors JM, Gascoyne RD, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol. (2013) 31:684–91. doi: 10.1200/JCO.2012.43.4803
22. O'Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. (2017) 18:613–23. doi: 10.1038/nrg.2017.47
23. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AOH, Zander SAL, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. (2008) 105:17079–84. doi: 10.1073/pnas.0806092105
24. De P, Sun Y, Carlson JH, Friedman LS, Leyland-Jones BR, Dey N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia (2014) 16:43–72. doi: 10.1593/neo.131694
25. Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. (2010) 28:585–93. doi: 10.1038/nbt.1640
26. Weissenberger J, Steinbach JP, Malin G, Spada S, Rülicke T, Aguzzi A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene (1997) 14:2005–13.
27. Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Amp Dev. (1998) 12:3675–85.
28. Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. (2000) 25:55–7. doi: 10.1038/75596
29. Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks TE. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. (2000) 26:109–13. doi: 10.1038/79075
30. Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, et al. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. (2001) 61:3826–36.
31. Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H, et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res. (2003) 63:1589–95.
32. Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. (2003) 63:8573–7.
33. Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell (2005) 8:119–30. doi: 10.1016/j.ccr.2005.07.004
34. Kwon C-H, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. (2008) 68:3286–94. doi: 10.1158/0008-5472.CAN-07-6867
35. Harris MA, Yang H, Low BE, Mukherjee J, Mukherje J, Guha A, et al. Cancer stem cells are enriched in the side population cells in a mouse model of glioma. Cancer Res. (2008) 68:10051–9. doi: 10.1158/0008-5472.CAN-08-0786
36. Hitoshi Y, Harris BT, Liu H, Popko B, Israel MA. Spinal glioma: platelet-derived growth factor B-mediated oncogenesis in the spinal cord. Cancer Res. (2008) 68:8507–15. doi: 10.1158/0008-5472.CAN-08-1063
37. Hede S-M, Hansson I, Afink GB, Eriksson A, Nazarenko I, Andrae J, et al. GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia (2009) 57:1143–53.
38. Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, O, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumor phenotypes. EMBO J. (2010) 29:222–35. doi: 10.1038/emboj.2009.327
39. Liu C, Sage JC, Miller MR, Verhaak RGW, Hippenmeyer S, Vogel H, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell (2011) 146:209–21. doi: 10.1016/j.cell.2011.06.014
40. Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, et al. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS ONE (2011) 6:e20041. doi: 10.1371/journal.pone.0020041
41. Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. (2009) 15:110–6. doi: 10.1038/nm.1863
42. de Vries NA, Bruggeman S, Hulsman D, de Vries HI, Zevenhoven J, Buckle T, et al. Rapid and robust transgenic high-grade glioma mouse models for therapy intervention studies. Clin Cancer Res. (2010) 16:3431–41. doi: 10.1158/1078-0432.CCR-09-3414
43. Chow LML, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell (2011) 19:305–16. doi: 10.1016/j.ccr.2011.01.039
44. Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science (2012) 338:1080–4. doi: 10.1126/science.1226929
45. Lindberg N, Jiang Y, Xie Y, Bolouri H, Kastemar M, Olofsson T, et al. Oncogenic signaling is dominant to cell of origin and dictates astrocytic or oligodendroglial tumor development from oligodendrocyte precursor cells. J Neurosci. (2014) 34:14644–51. doi: 10.1523/JNEUROSCI.2977-14.2014
46. Alcantara Llaguno SR, Wang Z, Sun D, Chen J, Xu J, Kim E, et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell (2015) 28:429–40. doi: 10.1016/j.ccell.2015.09.007
47. Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, Zapatka M, Northcott PA, Schramm K, et al. Somatic CRISPR/Cas9-mediated tumor suppressor disruption enables versatile brain tumor modeling. Nat Commun. (2015) 6:7391. doi: 10.1038/ncomms8391.
48. Giachino C, Boulay J-L, Ivanek R, Alvarado A, Tostado C, Lugert S, et al. A tumor suppressor function for notch signaling in forebrain tumor subtypes. Cancer Cell (2015) 28:730–42. doi: 10.1016/j.ccell.2015.10.008
49. Cook PJ, Thomas R, Kannan R, de Leon ES, Drilon A, Rosenblum MK, et al. Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat Commun. (2017) 8:15987. doi: 10.1038/ncomms15987
50. Chow RD, Guzman CD, Wang G, Schmidt F, Youngblood MW, Ye L, et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat Neurosci. (2017) 20:1329–41. doi: 10.1038/nn.4620
51. Jiang Y, Marinescu VD, Xie Y, Jarvius M, Maturi NP, Haglund C, et al. Glioblastoma Cell malignancy and drug sensitivity are affected by the cell of origin. Cell Rep. (2017) 18:977–90. doi: 10.1016/j.celrep.2017.01.003
52. Oldrini B, Curiel-Garcia A, Marques C, Matia V, Uluckan O, Graña-Castro O, et al. Somatic genome editing with the RCAS-TVA-CRISPR-Cas9 system for precision tumor modeling. Nat Commun. (2018) 9:1466. doi: 10.1038/s41467-018-03731-w
53. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
54. Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol. (2011) 29:4482–90. doi: 10.1200/JCO.2010.33.8715
55. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell (2018) 173:291–304.e6. doi: 10.1016/j.cell.2018.03.022
56. Ozawa T, Riester M, Cheng Y-K, Huse JT, Squatrito M, Helmy K, et al. Most human Non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell (2014) 26:288–300. doi: 10.1016/j.ccr.2014.06.005
57. Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature (2018) 560:243–7. doi: 10.1038/s41586-018-0389-3
58. Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature (2012) 488:522–6. doi: 10.1038/nature11287
59. Sanchez-Rivera FJ, Jacks TE. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer (2015) 15:387–95. doi: 10.1038/nrc3950
60. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han Y-C, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature (2014) 516:423–7. doi: 10.1038/nature13902
61. Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature (2012) 488:656–9. doi: 10.1038/nature11323
62. Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, et al. Expression of Idh1(R132H) in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell (2016) 30:578–94. doi: 10.1016/j.ccell.2016.08.017
63. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. (2015) 372:2499–508. doi: 10.1056/NEJMoa1407279
64. Network CGAR, Brat DJ, Verhaak RGW, Aldape KD, Yung WKA, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. (2015) 372:2481–98. doi: 10.1056/NEJMoa1402121
65. Rana MW, Pinkerton H, Thornton H, Nagy D. Heterotransplantation of human glioblastoma multiforme and meningioma to nude mice. Proc Soc Exp Biol Med. (1977) 155:85–8.
66. Shapiro WR, Basler GA, Chernik NL, Posner JB. Human brain tumor transplantation into nude mice. J Natl Cancer Inst. (1979) 62:447–53.
67. Jacobs VL, Valdes PA, Hickey WF, De Leo JA. Current review of in vivo GBM rodent models: emphasis on the CNS-1 tumor model. ASN Neuro (2011) 3:e00063. doi: 10.1042/AN20110014
68. Willimsky G, Czéh M, Loddenkemper C, Gellermann J, Schmidt K, Wust P, et al. Immunogenicity of premalignant lesions is the primary cause of general cytotoxic T lymphocyte unresponsiveness. J Exp Med. (2008) 205:1687–700. doi: 10.1084/jem.20072016
69. Schumacher TNM, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
70. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-Based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. (2018) 78:1031–43. doi: 10.1158/0008-5472.CAN-17-1788
71. Chitadze G, Lettau M, Luecke S, Wang T, Janssen O, Fürst D, et al. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human γδ T cells: modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology (2016) 5:e1093276. doi: 10.1080/2162402X.2015.1093276
72. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. (2008) 68:6368–76. doi: 10.1158/0008-5472.CAN-07-6768
73. Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez-Merino B, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. (2006) 66:10247–52. doi: 10.1158/0008-5472.CAN-06-2048
74. Finocchiaro G, Pellegatta S. Immunotherapy with dendritic cells loaded with glioblastoma stem cells: from preclinical to clinical studies. Cancer Immunol Immunother. (2016) 65:101–9. doi: 10.1007/s00262-015-1754-9
75. Srinivasan VM, Ferguson SD, Lee S, Weathers S-P, Kerrigan BCP, Heimberger AB. Tumor vaccines for malignant gliomas. Neurotherapeutics (2017) 14:345–57. doi: 10.1007/s13311-017-0522-2
76. Frisch M, Biggar RJ, Engels EA, Goedert JJ, Group A-CMRS. Association of cancer with AIDS-related immunosuppression in adults. JAMA (2001) 285:1736–45. doi: 10.1001/jama.285.13.1736
77. Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer (2018) 18:419–32. doi: 10.1038/s41568-018-0009-4
78. Hardcastle J, Mills L, Malo CS, Jin F, Kurokawa C, Geekiyanage H, et al. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. (2017) 19:493–502. doi: 10.1093/neuonc/now179
79. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. (2018) 10:eaam7577. doi: 10.1126/scitranslmed.aam7577
80. Squibb BM,. No Title. (2017). Available online at: https://news.bms.com/press-release/bmy/bristol-myers-squibb-announces-results-checkmate-143-phase-3-study-opdivo-nivoluma.
81. Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget (2017) 8:91779–94. doi: 10.18632/oncotarget.21586
82. Lang FF, Conrad C, Gomez-Manzano C, Alfred Yung WK, Sawaya R, Weinberg JS, et al. Phase I study of DNX-2401 (delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. (2018) 36:1419–27. doi: 10.1200/JCO.2017.75.8219
83. Desjardins A, Gromeier M, Herndon JE, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
84. Stojiljkovic M, Piperski V, Dacevic M, Rakic L, Ruzdijic S, Kanazir S. Characterization of 9L glioma model of the Wistar rat. J Neurooncol. (2003) 63:1–7. doi: 10.1023/A:1023732619651
85. Liau LM, Black KL, Prins RM, Sykes SN, DiPatre P-L, Cloughesy TF, et al. Treatment of intracranial gliomas with bone marrow—derived dendritic cells pulsed with tumor antigens. J Neurosurg. (2009) 90:1115–24. doi: 10.3171/jns.1999.90.6.111
86. Zagzag D, Zhong H, Scalzitti JM, Laughner E, Simons JW, Semenza GL. Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer (2000) 88:2606–18. doi: 10.1002/1097-0142(20000601)88:11<2606::AID-CNCR25>3.0.CO;2-W
87. Chicoine MR, Silbergeld DL. Invading C6 glioma cells maintaining tumorigenicity. J Neurosurg. (1995) 83:665–71. doi: 10.3171/jns.1995.83.4.0665
88. Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, et al. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. (2001) 61:4956–60.
89. Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell (2002) 1:269–77. doi: 10.1016/S1535-6108(02)00046-6
90. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumor initiating cells. Nature (2004) 432:396–401. doi: 10.1038/nature03128
91. Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF, et al. Influence of in vivo growth on human glioma cell line gene expression: convergent profiles under orthotopic conditions. Proc Natl Acad Sci USA. (2005) 102:8287–92. doi: 10.1073/pnas.0502887102
92. Bao S, Wu Q, Mclendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444:756–60. doi: 10.1038/nature05236
93. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell (2006) 9:391–403. doi: 10.1016/j.ccr.2006.03.030
94. Bruggeman S, Hulsman D, Tanger E, Buckle T, Blom M, Zevenhoven J, et al. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell (2007) 12:328–41. doi: 10.1016/j.ccr.2007.08.032
95. Pollard SM, Clarke I, Smith A. Brain cancer stem cells: a level playing field. Cell Stem Cell (2009) 5:468–9. doi: 10.1016/j.stem.2009.10.016
96. Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodón L, Folch G, Carmona MA, et al. TGF-β receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell (2010) 18:655–68. doi: 10.1016/j.ccr.2010.10.023
97. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell (2010) 17:362–75. doi: 10.1016/j.ccr.2009.12.049
98. Zhang M, Hutter G, Kahn SA, Azad TD, Gholamin S, Xu CY, et al. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages in vivo. PLoS ONE (2016) 11:e0153550. doi: 10.1371/journal.pone.0153550
99. Lan X, Jörg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature (2017) 549:227–32. doi: 10.1038/nature23666
100. Gargiulo G, Cesaroni M, Serresi M, de Vries NA, Hulsman D, Bruggeman S, et al. In vivo RNAi screen for BMI1 targets identifies TGF-β/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell (2013) 23:660–76. doi: 10.1016/j.ccr.2013.03.030
101. Hakin-Smith V, Jellinek DA, Levy D, Carroll T, Teo M, Timperley WR, et al. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet (2003) 361:836–8. doi: 10.1016/S0140-6736(03)13538-6
102. Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF, et al. Orthotopic growth of human glioma cells quantitatively and qualitatively influences radiation-induced changes in gene expression. Cancer Res. (2005) 65:10389–93. doi: 10.1158/0008-5472.CAN-05-1904
103. Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci. (2009) 29:8884–96. doi: 10.1523/JNEUROSCI.0968-09.2009
104. Suvà M-L, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle J-C, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. (2009) 69:9211–8. doi: 10.1158/0008-5472.CAN-09-1622
105. de Vries NA, Hulsman D, Akhtar W, de Jong J, Miles DC, Blom M, et al. Prolonged Ezh2 depletion in glioblastoma causes a robust switch in cell fate resulting in tumor progression. Cell Rep. (2015) 10:1–16. doi: 10.1016/j.celrep.2014.12.028
106. Jin X, Kim LJY, Wu Q, Wallace LC, Prager BC, Sanvoranart T, et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med. (2017) 23:1352–61. doi: 10.1038/nm.4415
107. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell (2016) 164:550–63. doi: 10.1016/j.cell.2015.12.028
108. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367:645–8.
109. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. (2004) 64:7011–21. doi: 10.1158/0008-5472.CAN-04-1364
110. Read T-A, Fogarty MP, Markant SL, Mclendon RE, Wei Z, Ellison DW, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell (2009) 15:135–47. doi: 10.1016/j.ccr.2008.12.016
111. Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell (2013) 153:139–52. doi: 10.1016/j.cell.2013.02.021
112. Ricci-Vitiani L, Pallini R, Larocca LM, Lombardi DG, Signore M, Pierconti F, et al. Mesenchymal differentiation of glioblastoma stem cells. Cell Death Differ. (2008) 15:1491–8. doi: 10.1038/cdd.2008.7
113. Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature (2010) 468:824–8. doi: 10.1038/nature09557
114. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Glioblastoma stem-like cells give rise to tumor endothelium. Nature (2010) 468:829–33. doi: 10.1038/nature09624
115. Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell (2009) 4:568–80. doi: 10.1016/j.stem.2009.03.014
116. Bric A, Miething C, Bialucha CU, Scuoppo C, Zender L, Krasnitz A, et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell (2009) 16:324–35. doi: 10.1016/j.ccr.2009.08.015
117. Meacham C, Ho E, Dubrovsky E, Gertler F, Hemann MT. In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat Genet. (2009) 41:1133–7. doi: 10.1038/ng.451
118. Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell (2008) 135:852–64. doi: 10.1016/j.cell.2008.09.061
119. Gargiulo G, Serresi M, Cesaroni M, Hulsman D, Van Lohuizen M. In vivo shRNA screens in solid tumors. Nat Protoc. (2014) 9:2880–902. doi: 10.1038/nprot.2014.185
120. Miller TE, Liau BB, Wallace LC, Morton AR, Xie Q, Dixit D, et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature (2017) 547:355–9. doi: 10.1038/nature23000
121. Toledo CM, Ding Y, Hoellerbauer P, Davis RJ, Basom R, Girard EJ, et al. Genome-wide CRISPR-Cas9 screens reveal loss of redundancy between PKMYT1 and WEE1 in glioblastoma stem-like cells. Cell Rep. (2015) 13:2425–39. doi: 10.1016/j.celrep.2015.11.021
122. Gengenbacher N, Singhal M, Augustin HG. Preclinical mouse solid tumor models: status quo, challenges and perspectives. Nat Rev Cancer (2017) 17:751–65. doi: 10.1038/nrc.2017.92
123. Balvers RK, Kleijn A, Kloezeman JJ, French PJ, Kremer A, van den Bent MJ, et al. Serum-free culture success of glial tumors is related to specific molecular profiles and expression of extracellular matrix-associated gene modules. Neuro Oncol. (2013) 15:1684–95. doi: 10.1093/neuonc/not116
124. Malaney P, Therapy PC. One mouse, one patient paradigm: new Avatars of personalized cancer therapy. Cancer Lett. (2015) 344:1–12. doi: 10.1016/j.canlet.2013.10.010
125. Garralda E, Paz K, López-Casas PP, Jones S, Katz A, Kann LM, et al. Integrated next-generation sequencing and avatar mouse models for personalized cancer treatment. Clin Cancer Res. (2014) 20:2476–84. doi: 10.1158/1078-0432.CCR-13-304
126. Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. (2017) 28:2595–605. doi: 10.1093/annonc/mdx416
127. Oh YT, Cho HJ, Kim J, Lee J-H, Rho K, Seo Y-J, et al. Translational validation of personalized treatment strategy based on genetic characteristics of glioblastoma. PLoS ONE (2014) 9:e103327. doi: 10.1371/journal.pone.0103327
128. Squatrito M, Holland EC. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res. (2011) 71:5945–9. doi: 10.1158/0008-5472.CAN-11-1245
129. Ben-David U, Ha G, Tseng Y-Y, Greenwald NF, Oh C, Shih J, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. (2017) 49:1567–75. doi: 10.1038/ng.3967
130. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity (2017) 47:363–373.e5. doi: 10.1016/j.immuni.2017.07.016
131. Sockolosky JT, Dougan M, Ingram JR, Ho CCM, Kauke MJ, Almo SC, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci USA. (2016) 113:E2646–54. doi: 10.1073/pnas.1604268113
132. Theocharides APA, Jin L, Cheng P-Y, Prasolava TK, Malko AV, Ho JM, et al. Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med. (2012) 209:1883–99. doi: 10.1084/jem.20120502
133. Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, et al. A Big Bang model of human colorectal tumor growth. Nat Genet. (2015) 47:209–16. doi: 10.1038/ng.3214
134. Waclaw B, Bozic I, Pittman ME, Hruban RH, Vogelstein B, Nowak MA. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature (2015) 525:261–4. doi: 10.1038/nature14971
135. Le Magnen C, Dutta A, Abate-Shen C. Optimizing mouse models for precision cancer prevention. Nat Rev Cancer (2016) 16:187–96. doi: 10.1038/nrc.2016.1
136. Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, et al. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome. Am J Pathol. (2000) 157:2151–9. doi: 10.1016/S0002-9440(10)64853-5
137. Collaborative Cross Consortium. The genome architecture of the collaborative cross mouse genetic reference population. Genetics (2012) 190:389–401. doi: 10.1534/genetics.111.132639
138. Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell (2018) 173:371–85.e18. doi: 10.1016/j.cell.2018.02.060
139. Wuestefeld T, Pesic M, Rudalska R, Dauch D, Longerich T, Kang T-W, et al. A direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell (2013) 153:389–401. doi: 10.1016/j.cell.2013.03.026
140. Kim J, Lee I-H, Cho HJ, Park C-K, Jung Y-S, Kim Y, et al. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell (2015) 28:318–28. doi: 10.1016/j.ccell.2015.07.013
141. Mazor T, Pankov A, Johnson BE, Hong C, Hamilton EG, Bell RJA, et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell (2015) 28:307–17. doi: 10.1016/j.ccell.2015.07.012
142. Schönhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, et al. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat Publ Gr. (2014) 20:1340–7. doi: 10.1038/nm.3646
143. Staahl BT, Benekareddy M, Coulon-Bainier C, Banfal AA, Floor SN, Sabo JK, et al. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat Biotechnol. (2017) 35:431–4. doi: 10.1038/nbt.3806
144. Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. (2009) 15:4622–9. doi: 10.1158/1078-0432.CCR-08-3012
145. Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DIS, Zairis S, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. (2016) 48:768–76. doi: 10.1038/ng.3590
146. Weil S, Osswald M, Solecki G, Grosch J, Jung E, Lemke D, et al. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. (2017) 19:1316–26. doi: 10.1093/neuonc/nox070
147. Mathivet T, Bouleti C, Van Woensel M, Stanchi F, Verschuere T, Phng L-K, et al. Dynamic stroma reorganization drives blood vessel dysmorphia during glioma growth. EMBO Mol Med. (2017) 9:1629–45. doi: 10.15252/emmm.201607445
148. De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martínez-Ricarte F, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumor DNA better represents the genomic alterations of brain tumors than plasma. Nat Commun. (2015) 6:8839. doi: 10.1038/ncomms9839
149. Lee J-K, Wang J, Sa JK, Ladewig E, Lee H-O, Lee I-H, et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat Genet. (2017) 49:594–9. doi: 10.1038/ng.3806
150. De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature (2014) 514:247–51. doi: 10.1038/nature13561
151. Salphati L, Alicke B, Heffron TP, Shahidi-Latham S, Nishimura M, Cao T, et al. Brain distribution and efficacy of the brain penetrant PI3K inhibitor GDC-0084 in orthotopic mouse models of human glioblastoma. Drug Metab Dispos. (2016) 44:1881–9. doi: 10.1124/dmd.116.071423
152. Dörr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Däbritz JHM, et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature (2013) 501:421–5. doi: 10.1038/nature12437
153. Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature (2013) 498:109–12. doi: 10.1038/nature12154
154. Serresi M, Gargiulo G, Proost N, Siteur B, Cesaroni M, Koppens M, et al. Polycomb repressive complex 2 is a barrier to KRAS-driven inflammation and epithelial-mesenchymal transition in non-small-cell lung cancer. Cancer Cell (2016) 29:17–31. doi: 10.1016/j.ccell.2015.12.006
155. Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell (2018) 173:1413–25.e14. doi: 10.1016/j.cell.2018.04.012
156. Bourgeois-Daigneault M-C, Roy DG, Aitken AS, El Sayes N, Martin NT, Varette O, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. (2018) 10:eaao1641. doi: 10.1126/scitranslmed.aao1641
157. Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. (2015) 21:256–62.
158. Bolouri H, Zhao LP, Holland EC. Big data visualization identifies the multidimensional molecular landscape of human gliomas. Proc Natl Acad Sci USA. (2016) 113:5394–9. doi: 10.1073/pnas.1601591113
159. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumor formation by single human melanoma cells. Nature (2008) 456:593–8. doi: 10.1038/nature07567
160. Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, et al. A Novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. (2016) 22:3286–97. doi: 10.1158/1078-0432.CCR-15-1696
161. Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood (2005) 106:1565–73. doi: 10.1182/blood-2005-02-0516
162. Semenkow S, Li S, Kahlert UD, Raabe EH, Xu J, Arnold A, et al. An immunocompetent mouse model of human glioblastoma. Oncotarget (2017) 8:61072–82. doi: 10.18632/oncotarget.17851
163. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi: 10.1126/science.1194637
164. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. (2014) 344:1396–401. doi: 10.1126/science.1254257
165. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science (2017) 355:eaai8478. doi: 10.1126/science.aai8478
166. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell (2017) 32:42–56.e6. doi: 10.1016/j.ccell.2017.06.003
167. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science (2016) 352:aad3018. doi: 10.1126/science.aad3018
168. Lin F, de Gooijer MC, Roig EM, Buil LCM, Christner SM, Beumer JH, et al. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res. (2014) 20:2703–13.
169. Zhang P, de Gooijer MC, Buil LCM, Beijnen JH, Li G, van Tellingen O. ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2-inhibitors. Int J Cancer (2015) 137:2007–18.
170. Ellenbroek B, Youn J. Rodent models in neuroscience research: is it a rat race? Dis Model Mech. (2016) 9:1079–87. doi: 10.1242/dmm.026120
171. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumors. Nature (2018) 555:469–74. doi: 10.1038/nature26000
172. Clark SJ, Smallwood SA, Lee HJ, Krueger F, Reik W, Kelsey G. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scBS-seq). Nat Protoc. (2017) 12:534–47. doi: 10.1038/nprot.2016.187
173. Matsushima W, Herzog VA, Neumann T, Gapp K, Zuber J, Ameres SL, et al. SLAM-ITseq: sequencing cell type-specific transcriptomes without cell sorting. Development (2018) 145:dev.164640. doi: 10.1242/dev.164640
174. Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods (2017) 14:297–301. doi: 10.1038/nmeth.4177
175. Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature (2014) 515:355–64. doi: 10.1038/nature13992
176. Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
177. Linnemann C, Heemskerk B, Kvistborg P, Kluin RJC, Bolotin DA, Chen X, et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Publ Gr. (2013) 19:1534–41. doi: 10.1038/nm.3359
178. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell (2016) 167:419–32.e16. doi: 10.1016/j.cell.2016.09.011
Keywords: mouse models, cancer, single-cell genomics, CRISPR/Cas9, targeted therapies, biomarker discovery, preclinical models, patient-derived xenografts (PDX)
Citation: Gargiulo G (2018) Next-Generation in vivo Modeling of Human Cancers. Front. Oncol. 8:429. doi: 10.3389/fonc.2018.00429
Received: 28 May 2018; Accepted: 13 September 2018;
Published: 10 October 2018.
Edited by:
Danilo Maddalo, Novartis, SwitzerlandReviewed by:
Giovanni Migliaccio, Consorzio per Valutazioni Biologiche e Farmacologiche, ItalyLilian Varricchio, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2018 Gargiulo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaetano Gargiulo, gaetano.gargiulo@mdc-berlin.de