 Gaurav Nayyar1†
Gaurav Nayyar1† Mitchell S. Cairo
Mitchell S. Cairo- 1Department of Pediatrics, New York Medical College, Valhalla, NY, United States
- 2Department of Cell Biology & Anatomy, New York Medical College, Valhalla, NY, United States
- 3Department of Microbiology & Immunology, New York Medical College, Valhalla, NY, United States
- 4Department of Medicine, New York Medical College, Valhalla, NY, United States
- 5Department of Pathology, New York Medical College, Valhalla, NY, United States
Despite advances in the diagnostic and therapeutic modalities, the prognosis of several solid tumor malignancies remains poor. Different factors associated with solid tumors including a varied genetic signature, complex molecular signaling pathways, defective cross talk between the tumor cells and immune cells, hypoxic and immunosuppressive effects of tumor microenvironment result in a treatment resistant and metastatic phenotype. Over the past several years, immunotherapy has emerged as an attractive therapeutic option against multiple malignancies. The unique ability of natural killer (NK) cells to target cancer cells without antigen specificity makes them an ideal candidate for use against solid tumors. However, the outcomes of adoptive NK cell infusions into patients with solid tumors have been disappointing. Extensive studies have been done to investigate different strategies to improve the NK cell function, trafficking and tumor targeting. Use of cytokines and cytokine analogs has been well described and utilized to enhance the proliferation, stimulation and persistence of NK cells. Other techniques like blocking the human leukocyte antigen-killer cell receptors (KIR) interactions with anti-KIR monoclonal antibodies, preventing CD16 receptor shedding, increasing the expression of activating NK cell receptors like NKG2D, and use of immunocytokines and immune checkpoint inhibitors can enhance NK cell mediated cytotoxicity. Using genetically modified NK cells with chimeric antigen receptors and bispecific and trispecific NK cell engagers, NK cells can be effectively redirected to the tumor cells improving their cytotoxic potential. In this review, we have described these strategies and highlighted the need to further optimize these strategies to improve the clinical outcome of NK cell based immunotherapy against solid tumors.
Introduction
Natural Killer (NK) cells are the effector cells that constitute a key part of the innate immune system. They have emerged as a promising option for immunotherapy of a variety of malignancies due to their ability to identify and kill cancer cells without any prior sensitization. NK cells have unique ability to differentiate between the normal and transformed cells. They possess a variety of activating and inhibitory receptors, and their net functional outcome is a complex integration of signals between these activating and inhibitory receptors. Over the past few decades, significant advances have been made in successfully targeting hematologic malignancies with the use of novel immunotherapeutic strategies. However, solid tumors continue to pose unique therapeutic challenges, and the conventional cytoreductive therapies have proven to be of limited efficacy. NK cell based therapeutic strategies have been applied against solid tumor with only modest success. The ability of solid tumor cells to escape the immune-surveillance, proliferate rapidly and metastasize when coupled with the abnormalities in the NK cells like decreased expression of activating receptors or overexpression of inhibitory receptors, decreased activation and persistence, defective cytokine production, abnormal intracellular signaling molecules, inefficient trafficking to the tumor site, and senescence resulting in a defective cytolytic response are likely the major contributors to the poor response of NK cells based strategies against solid tumors. In this review, we have attempted to address the unique characteristics of solid tumors and their microenvironment, mechanisms contributing to the NK cell resistance and describe the various applications that could be applied in an attempt to enhance the therapeutic potential of NK cells against solid tumors.
Challenges in Treating Solid Tumors
NK cell based immunotherapies have been used widely and successfully for different hematologic malignancies, particularly acute myeloid leukemia. One of the early studies in patients with relapsed acute myeloid leukemia, showed haploidentical NK cell infusion in combination with high dose fludarbine and cyclophosphamide caused expansion of donor NK cells, significantly increased endogenous Interleukin (IL)-15 and achieved a complete hematologic remission in 5 of 19 (26%) patients (1). More recently, NK cell based therapies have emerged as an attractive strategy for targeting solid tumors. However, there are some considerable challenges in use of NK cell based therapies against solid tumors. Solid tumors are a very heterogeneous group of malignancies that have historically been more difficult to treat even with the use of multimodal approaches. This heterogeneity could be due to differences in evolution of these tumors caused by varying gene profile signature, different mutations and involvement of different cell signaling pathways (2, 3). One of the major challenges with NK cell based therapies against solid tumors is the trafficking of these immune cells to the tumor location and infiltration into the tumor. Multiple studies have shown that the tumor progression and outcomes correlate with the presence of NK cells at the tumor site (4–6). The density of NK cells infiltrating into the tumor has been shown to be an independent predictor of the progression free survival in gastrointestinal stromal tumors, and in pulmonary adenocarcinoma (4, 7). The chemokines expressed on the surface of NK cells, and the ones secreted by the tumor cells play a central role in NK cell infiltration into the tumor (8).
It has been well established that tumor microenvironment plays a key role in the proliferation and survival of the cancerous cells. Tumor microenvironment consists of a variety of cells including tumor associated fibroblasts, tumor associated macrophages, dendritic cells, neutrophils, regulatory T cells (Tregs), myeloid derived suppressor cells (9), that provide a constant chronic inflammatory milieu leading to angiogenesis, tumor cell survival and proliferation. The presence of inhibitory signals in the tumor microenvironment and altered immunogenicity of tumor cells also leads to poor infiltration and activation of NK cells into the tumor. Furthermore, rapidly growing solid tumors create an environment of localized hypoxia (10). The low oxygen tension in the solid tumor tissue not only creates metabolic disturbances in the tumor microenvironment but also leads to generation of reactive oxygen species. This cellular environment of hypoxia is mediated by a variety of transcriptional regulators primarily, hypoxia inducible factor-1 (11). Poorly oxygenated tumor cells undergo adaptive changes at the proteomic level leading to transcriptional activity resulting in inhibition of apoptosis and promoting angiogenesis and upregulation of the tumor growth factors (12). Net result is the continued survival and proliferation of the tumor cells with an aggressive phenotype, that frequently metastasize to distant tissues, and are relatively resistance to treatment (11).
Recently, a lot of advances have been made in targeting hematologic malignancies using novel immunotherapeutic strategies like chimeric antigen receptors (CAR). However, the success stories have been less exciting against solid tumors, particularly due to lack of appropriate immunologic targets, that are highly expressed on surface of tumor tissue with relative absence on the non-vital tissues to avoid “on-target/off-tumor” effects (13). In addition, these genetically modified effector cells have to overcome the challenges posed by the physical barriers preventing infiltration into the tumor tissues and hostile tumor microenvironment (14, 15). Antigen escape phenomenon due to downregulation or loss of targetable antigen happens frequently in solid tumors rendering these CAR based therapies less effective (14, 16). However, unlike CAR T cells, NK cell based therapies have the advantage of overcoming the limitation posed by the antigen escape mechanism to a certain extent due to their inherent ability to recognize and kill tumor cells without prior sensitization. Furthermore, NK cell alloreactivity following haploidentical SCT is protective against graft vs. host disease while producing a robust graft vs. tumor/leukemia effect (17, 18).
Natural Killer Cell Biology and Target Recognition
Natural killer cells represent human body's first line of defense against tumor cells and infectious pathogens and play a key role in tumor immune surveillance. NK cells were initially identified in mice when investigators noticed a large granular subtype of lymphocytes distinct from T and B lymphocytes, and possessed cytotoxic activity against mouse tumor cell lines (19, 20). Phenotypically, NK cells lack B and T cells markers CD19/TCR/CD3 on their cell surface but they express CD16 and CD56 surface antigens. NK cells are further characterized by the degree of CD56 expression into dim and bright subsets where the subtypes have significant differences in terms of cytokine production, response to cytokines and their killing potential. Around 90% of the NK cells, including the alloreactive NK cells, express low levels of CD56 but have high expression of CD16 (CD16bright CD56dim), and are generally found in the peripheral circulation. These cells are considered to be the “mature” NK cells and have higher cytotoxic potential. The remaining 10% of the NK cells are CD16dimCD56bright with higher levels of CD56 expression, and they are considered “immature” NK cells (21, 22). These immature or unlicensed NK cells generally reside in the lymphoid tissues but they are more responsive to stimulation and respond readily by secreting a variety of cytokines including interferon γ (IFN-γ), tumor necrosis factor (TNF)-α, IL-5, IL-10, and IL-13 (4, 23). All subsets of NK cells express intermediate affinity heterodimeric IL-2 receptor. However, high affinity receptors and c-kit tyrosine kinase is only expressed by CD56bright NK cells, which gives them the unique ability to proliferate when exposed to very small concentrations of IL-2 (24, 25). In addition, there is also a differential expression of adhesion molecules between the two NK cell subsets. CD56bright cells have higher levels of expression of chemokine receptor type-7 and L-selectin which likely helps these cells to traffic to secondary lymphoid organs, whereas CD56dim cells have a higher level of expression of Leucocyte function-associated antigen-1, providing them the unique migratory properties in response to foreign pathogens (26). Therefore, CD56dim NK cells appear to have a predominantly cytotoxic function naturally, and CD56bright cells play a more immunomodulatory role. However, it is still unclear if these subsets represent just different stages in maturation of NK cells or if they are completely different cells emerging from a common hematopoietic precursor (3). Besides the circulation system, distinct subsets of NK cells also reside in tissues and organs (22). NK cells in lymph nodes, tonsils and spleen differ from NK cell subsets in peripheral blood by phenotypes and functions (27, 28).
NK cells do not require prior sensitization to target the transformed cells (4). NK cell receptor can play a stimulatory or an inhibitory role and has the unique ability to recognize major histocompatibility complex−1 (MHC-1) or MHC-1 like molecules on the target cells. The balance between the inhibitory signals received from the killer inhibitory receptors and natural killer group protein 2 family member A (NKG2A) and killer cell lectin-like receptor subfamily G member 1; and the stimulatory receptors including natural cytotoxicity receptors, NKp30, NKp44, NKp46, natural killer group protein 2 family member D (NKG2D) defines the net functional outcome of the NK cells.
NK cells recognize autologous cells that express human leucocyte antigen (HLA) Class I molecules that prevent them from attacking the host tissue, known as “tolerance to self.” During viral infections or malignant transformation, there is decreased expression of MHC class I antigens on cell surface in order to avoid recognition by the antitumor T cells. NK cells that are surveilling the tissues for a normal level of MHC class I expression, recognize this as “altered self” resulting in decreased engagement of the killer inhibitory receptors and increase expression of the stimulatory receptors resulting in effector response and cytotoxic killing of the transformed cells (29). The mechanism of target recognition by NK cells is depicted in Figure 1. There are several mechanisms by which NK cells can kill the target cells without any prior sensitization. They can exert direct cytotoxicity through release of granules containing perforin and granzyme (31). NK cells also have the unique ability exert antibody-dependent cell mediated cytotoxicity (ADCC) due to presence of Fc receptor FcγRIIIa that recognizes the Fc portion of the antibodies. In addition, they can mediate cytotoxicity via apoptotic pathways involving fas ligand or TNF-related apoptosis-inducing ligand (32, 33).
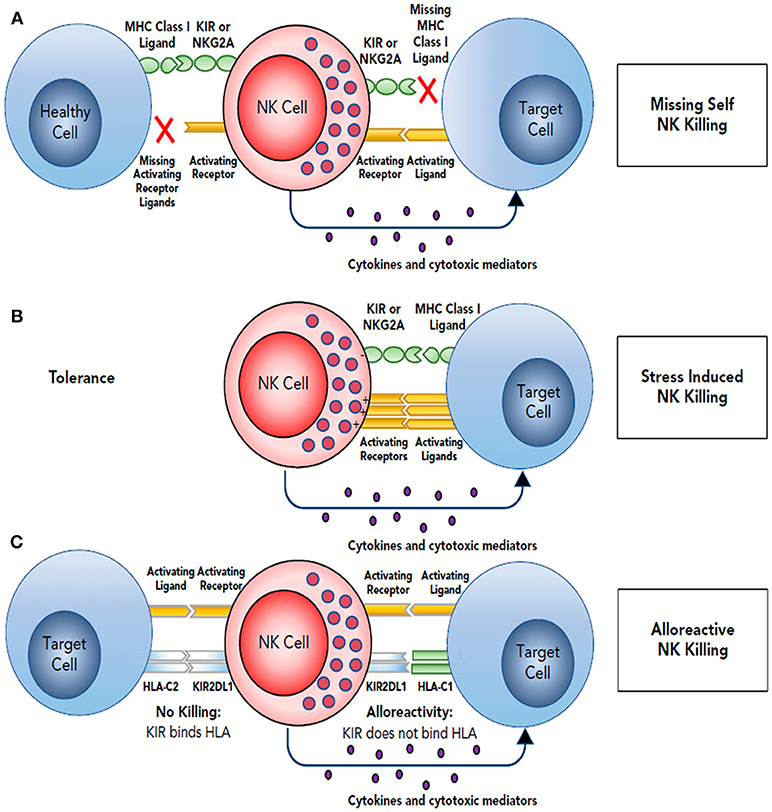
Figure 1. Target recognition, tolerance, missing self. (A) NK cells recognize and kill their targets by an integrated balance of inhibitory and activating signals to discriminate between healthy cells (tolerance) vs. elimination of transformed or virally infected targets (killing). NK-cell tolerance depends on several MHC class I inhibitory signals (either classical, HLA-A, -B, or -C, or nonclassical, HLA-E) expressed by healthy cells that engage KIR or NKG2A with minimal activation signals resulting in tolerance. Malignant transformation or viral infection promotes target cell killing by downregulation of MHC class I expression and an upregulation of signals from activating NK-cell receptors. (B) Although in some cases, MHC downregulation is variable or incomplete, target cell killing can still occur by changing the balance with activating signals upregulated by stress-induced activating receptor ligands. (C) This balance between inhibition and activation can be uniquely manipulated in the hematopoietic transplant setting by selection of donors who will respond to apparent missing self HLA class I in the HLA-mismatched recipients. For example, reconstitution with a high frequency of donor KIR2DL1+ NK cells would not be inhibited in a HLA-C1 (C1+-HLA-C) recipient (KIR ligand mismatch). Here, NK-cell alloreactivity would kill the recipient's tumor. In contrast, when the same KIR2DL1+ NK cells reconstitute in an HLA-C2 recipient (C2+-HLA-C) (KIR ligand match), the recipient's tumor would be seen as having self HLA class I and would not provoke an alloreactive NK-cell response. Reproduced with permission from Cooley et al. (30).
NK Resistance Mechanisms
NK cells have shown significant alloreactive anti-leukemic effects against liquid tumor cells especially following haploidentical SCT (stem cell transplantation) (17) and the higher NK cell immune reconstitution in the early post allogeneic SCT period has also been demonstrated to be associated with significantly improved survival and lower leukemia relapse rates (34, 35). However, the adoptive transfer of autologous NK cells showed no clinical response in patients with progressive stage IV melanoma or renal cell carcinoma (RCC) (36). In solid tumor clinical trials, NK cells often display impaired functions in patients and impaired NK cell-function is related to high disease stages and poor prognosis (37, 38). The NK resistance, on NK side, is mainly due to the small numbers of active NK cells, the short lifespan of NK cells, poor persistence and trafficking, and lack of specific tumor targeting (39). On the tumor side, tumor cells make up a microenvironment that inhibits NK cell activity by altering the balance between NK activating and inhibitory receptors such as reducing NK activating receptor NKG2D and CD16, secreting inhibitory factors such as transforming growth factor beta (TGF-β), IL-6 and IL-10, shedding NKG2D ligands such as MHC class I chain-related protein A (MICA) and MHC class I chain-related protein B (MICB), and recruiting suppressive immune cells such as Tregs and myeloid derived suppressor cells (40). Table 1 summarizes the potential mechanisms of resistance to NK cell based therapy of solid tumors.

Table 1. NK cell resistance mechanisms against solid tumors.
NK Resources for Adoptive Therapy
Four sources of active NK cells for adoptive transfer have been reported: autologous NK cells, allogeneic NK cells from donors, NK cell lines and embryo stem cell-derived/induced pluripotent stem cells -derived NK cells. Rosenberg et al. evaluated the efficacy of adoptively transferred IL-2 ex vivo activated autologous NK cells to patients with metastatic renal carcinoma and melanomas (36). Even the adoptively transferred NK cells persisted for long time, no significant clinical benefit was observed (36), indicating the limitation of utilizing patients' autologous NK cells alone as a therapeutic strategy. Due to the KIR mismatch to kill tumor cells, the adoptive transfer of allogeneic NK cells may have a superior antitumor effect compared with the approaches utilizing autologous NK cells (44). To overcome the limitation of small number of active NK cells in peripheral blood, our group and others have successfully expanded active NK cells in vitro by short term culture with cytokines alone, using cytokines and co-culture with irradiated Epstein-Barr virus-transformed lymphoblastoid cell lines as feeder cells, or cytokines and co-culture with K562 cells expressing transfected cell-membrane bound IL-15 and 4-1BBL (45–48). Lee and colleagues have developed a novel method of ex-vivo expansion of NK cells by stimulating peripheral blood mononuclear cells (PBMC) with a genetically-engineered feeder cell line, K562-mbIL21-41BBL, resulting in over 35,000-fold increase in NK cells and significant increase in NK cell functional activation (Figure 2) (49). Recently, Lee et al. used an anti-CD16 monoclonal antibody (mAb) for potent activation of resting NK cells and irradiated autologous PBMC (upregulated NKG2D ligand and CD48) for providing a suitable environment (activating receptor-ligand interactions and soluble growth factors) instead of cancer cell-based feeder cells for large-scale expansion of highly purified cytotoxic NK cells (50). These expanded NK cells showed potent cytotoxicity against various cancer cells in vitro and efficiently controlled cancer progression in severe combined immunodeficiency mouse models of human colon and lung cancer (50). Allogeneic expanded NK cells, which were expanded using CD3+ T-cell–depletion PBMCs from healthy donors with irradiated autologous PBMCs, mAb to CD3, and 500 IU/mL of IL2, were evaluated in a phase I study of adoptive transfer of these cells into patients with advanced, recurrent solid tumors besides malignant lymphoma (51). The results showed that the repetitive administration of ex-vivo expanded allogeneic NK cells was safe without any sign of graft vs. host disease or serious adverse event (51). Further studies are needed to enhance the persistence of these NK cells. Recently Jewett's group successfully expanded super-charged NK using PB-derived osteoclasts as feeder cells (52–54). These super-charged NK had superior cytotoxicity and IFN-γ secretion, survived for a longer period, and efficiently eliminated tumor growth in humanized xenografted mice (52–54). Considering more than 600,000 banked cord blood (CB) units worldwide (55), CB represents a unique opportunity as a readily available donor source with greater flexibility for the identification of HLA-compatible and KIR-mismatched lines. CB NK cells can be easily expanded with K562-mbIL21-41BBL feeder cells (18, 56) using CB mononuclear cells or they can be expanded to high log-scale with a cytokine cocktail from CD34+ CB progenitor cells (57, 58). NK cells derived from human CD34+ hematopoietic stem and progenitor cells showed efficient infiltration and killing of human ovarian cancer spheroids using an in vivo-like model system and reduced tumor progression in mice xenografted with ovarian carcinoma (59). NK cell lines also provide an unlimited source of effector cells and hold potential for development as standardized off-the-shelf therapeutics for adoptive cancer immunotherapy (60). Among different NK cell lines, NK-92 cells have been thoroughly investigated in preclinical studies and also been applied in clinical trials (61). The activated NK-92 based therapy from NanKwest was granted as the orphan drug designation against Merkel cell carcinoma. However, these NK-92 cell lines are aneuploid and must be irradiated before being administered to patients, which will limit the survival and proliferation of NK cells (62). In order to produce homogeneous and well-defined NK cells, a lot of effort has been put into generating NK cells derived from human embryonic stem cells or human induced pluripotent stem cells (iPSCs) (63–65). Most of these induced pluripotent stem cells-derived NK cells expressed no killer immunoglobulin (Ig)-like receptors (KIRs), which renders them unrestricted by recipients' HLA genotypes, and therefore they may serve as a universal “off-the-shelf” NK cell source for many recipients (64, 65).
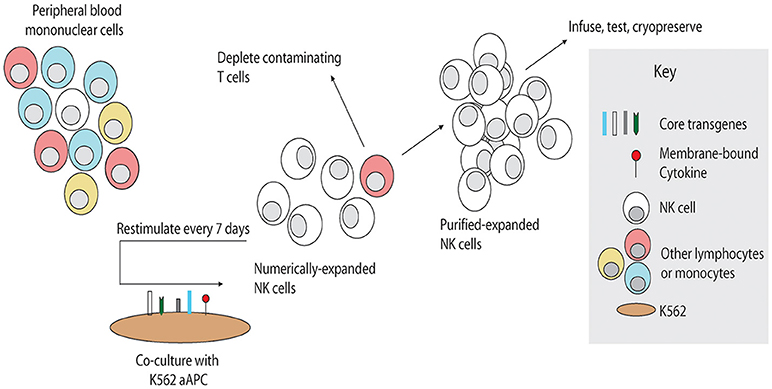
Figure 2. Schema for NK cell manufacturing with artificial antigen-presenting cells. Artificial antigen-presenting cells were produced by genetic modification of K562 to express costimulatory molecules and membrane-bound cytokines. To expand NK cells ex vivo, unfractionated PBMC are stimulated weekly with irradiated PBMC, inducing rapid proliferation of NK cells and in some cases non-specific expansion of T cells. Contaminating T cells may be depleted, and the remaining purified NK cells may be stimulated weekly by the artificial antigen-presenting cells as needed to obtain sufficient numbers. Expanded NK cells may be used directly or cryopreserved for future use. Reproduced with permission from Denman et al. (49).
Stimulation of NK Cells Effector Function Using Cytokines and Cytokine Analogs
One of the earliest and most common approaches in using stimulating and activating NK cells for cancer immunotherapy has been the use of cytokines. Multiple cytokines and other novel soluble factors have been described in the literature to enhance the number, function and persistence of NK cells in vivo. IL-2, IL-15, IL-12, IL-18, and IL-21 have all been described in regulating NK cell function, particularly their activation, maturation and survival. Two most commonly employed strategies have been either pretreatment of NK cells with cytokines before the adoptive transfer, or administration of cytokines in vivo.
IL-2
IL-2 is an immunostimulatory molecule first discovered in 1970s, and was initially described as a T cell growth factor (66). Further characterization showed the impaired NK cell function in IL-2 deficient mice, but NK cells were present in normal numbers in IL-2 null mice suggesting that IL-2 is required for modulating NK cell function but is not essential for the development and maturation (67). To date, IL-2 has been the mostly commonly used cytokine in an attempt to boost NK cells in vivo. It was also the first every cytokine approved for clinical use (68). Earlier approaches using IL-2 in patient's involved using high dose IL-2 in conjunction with adoptive transfer of autologous NK cells. However, there was severe toxicity due to capillary leak syndrome related to IL-2 with no significant improvement in clinical outcomes (69). Subsequently, IL-2 was used to generate lymphokine activated killer cells, that were infused into the patients with melanoma or advanced RCC in combination with subcutaneous doses of IL-2 (70). This approach was not only well tolerated, and but clinical response showed a trend toward improved survival in melanoma patients who received a combination of lymphokine activated killer cells with IL-2 (5). In a phase I clinical trial involving patients with metastatic/unresectable digestive tract tumors, autologous NK cells were expanded ex vivo using IL-2, OK432, and modified recombinant human fibronectin fragment FN-CH296 induced T cells, and safely infused into the patients. However, no clinical responses were observed in these patients (71). Similarly, no improvement in clinical outcomes was observed in metastatic breast cancer patients who received IL-2 with autologous NK cell infusions following autologous SCT (72). Unfortunately, despite the compelling body of evidence suggesting that successful adoptive transfer and in vivo expansion of autologous NK cells when combined with IL-2 is safe and feasible, the clinical response against solid tumors has been minimal. It is likely that these impaired responses are related to the poor functional activation of the NK cells from cancer patients (73). Another potential explanation could be that IL-2 primed NK cells are sensitized to apoptosis upon coming in contact with the vascular endothelium likely causing a reduction in migration of these cells to the tumor site and infiltration into the tumor (74). Allogeneic or haploidentical NK cells infusions supplemented with IL-2 have been shown to produce significant clinical responses in hematological malignancies (75–77). However, such studies in field of solid tumors are lacking. In patients with advanced solid tumors, infusion of irradiated NK-92 cells that were ex-vivo expanded using IL-2 resulted in clinical responses in about three fourth of the patients with advanced lung cancer (78). Another phase II clinical trial using CD3 depleted, IL-2 stimulated haploidentical PBMC infusion in patients with recurrent ovarian and breast cancer showed a partial response in 20%, and stable disease in 60% of the patients (79). One of the major limitations of using IL-2 for modulating NK cell effector function, in addition to cytokine release with high dose IL-2 infusions, has been its ability to stimulate CD25 expressing Tregs (9). These Tregs have high affinity to IL-2 receptor, and diminish the effector response to NK cells by competing with NK cells for IL-2 and via TGFβ pathway (80). In a preclinical model, researchers have developed a mutant IL-2 molecule, “super-2,” that has increased affinity to IL-2Rβ and has been shown to have superior NK cell activation and proliferation compared to wild type IL-2. Additionally, it caused selective proliferation of cytotoxic T cells but not Tregs (81). Similarly, novel fusion protein molecules that combine NK cell activating receptor ligand with IL-2 are being developed to selectively promote the in vivo expansion and activation of NK without affecting Tregs (82). It is critical for future trials using IL-2 to adopt strategies that can circumvent these inhibitory elements like Tregs and myeloid suppressor cells to improve clinical responses.
IL-15
Due to several potential drawbacks of IL-2 (as mentioned previously), IL-15 has emerged as an attractive alternative in cancer immunotherapy. IL-15 is a 15 kDa, gamma chain cytokine that possesses structural and functional similarities to IL-2, and is active in both cis- and trans- conformations (83). IL-15 receptor complex, which includes IL-15Rα/β/γ, wherein the β and γ chain receptor subunits are common to IL-2 and IL-15, and only difference exists at the α subunit. The relative high affinity between IL-15 and IL-15Rα, compared to IL-2 and IL-2Rα, results in NK cell activation at relatively lower doses. IL15Rα is expressed by a variety of immune cells like T cells, NK cells, natural killer T (NKT) cells, macrophages, and dendritic cells, and non-immune cells like skeletal muscle and endothelial cells (84, 85). IL-15 deficient mice lack NK cells, NKT cells, memory CD8+ T highlighting that IL-15 is essential for development of these immune effector cells (86). In comparison to IL-2, IL-15 has a more potent effect on NK cell expansion, and it does not upregulated the gene expressions of type 2 cytokines like IL-6, IL-10, and IL-13 (87). Similarly when compared to IL-2, soluble IL-15 does not appear to expand Tregs (88). IL-15 has been shown improve functional abilities of NK cells by inducing granzyme and perforin through mTOR pathway, resulting in enhanced cytotoxicity (89–91). The antitumor effects of IL-15 have been well established in different preclinical studies (89, 92), and in part are mediated through the activating NK cells receptor NKG2D (93). IL-15 has been shown to enhance ADCC in a murine model of colon cancer, when given in combination with anti CD40 antibody (94). Higher levels of IL-15 on Day 15 post- autologous hematopoietic stem cell transplant (HSCT) have been shown to directly correlate with improved overall survival (OS) in patients with relapsed non-Hodgkin lymphoma (NHL) (95). Early patient studies with IL-15 were done in post HSCT setting or in patients with relapsed/refractory hematologic malignancies. A phase I dose escalation study in patients with acute myeloid leukemia using recombinant IL-15 with adoptively transferred NK cells showed that it was safe and feasible to administer IL-15, and it resulted in persistence and proliferation of NK cells in vivo (96). Patients with metastatic melanoma and metastatic RCC receiving E coli derived recombinant human IL-15 for 12 consecutive days showed 10-fold expansions of NK cells and a significant efflux of NK cells and memory CD8 T cells from peripheral blood further established that IL-15 infusions are safe and feasible (97). Another phase I study established the safety and clinical efficacy of allogeneic NK cell infusions cultured with IL-15 and hydrocortisone in patients with advanced non-small cell lung cancer (98). In a different phase I/II trial, four out of six patients with refractory pediatric solid tumor who received IL-15-stimulated NK cell infusion at 30 days after haploidentical-HSCT showed a clinical response (99). Other clinical trials using IL-15 alone or in combination with other immunotherapeutic agents targeting solid tumors are currently ongoing (NCT01572493, NCT03388632).
IL-21
IL-21 is a type I cytokine synthesized by CD4+ T cells including NKT cells, T follicular helper and Th17 cells (100). It has been described to modulate both innate and adaptive immune responses, and is known to cause the lymphoid proliferation, particularly of CD8+ T cells and NK cells, and maturation of B cells (72). In addition to activating immune effector cells, IL-21 also plays a crucial role in mediating autoimmunity (101, 102). Binding of IL-21 to IL-21R primarily leads to activation of JAK1/JAK3 with subsequent phosphorylation of signal transducer and activator of transcription (STAT) (STAT3 and STAT1) signaling pathway resulting in upregulation of IFN-γ expression (103). However, IL-21 mediated activation can also occur via mitogen-activated protein kinase and phosphoinositide-3-kinase/serine/threonine kinase pathway. Combination of IL-21 and IL-15 has been shown to selectively promote the expansion of cytolytic CD56+CD16+ subtype of NK cells from human bone marrow (104). Using a K562 based antigen presenting cells genetically modified to express membrane bound IL-21 (mbIL-21), several thousand fold ex vivo expansion of NK cells can be achieved (49). Furthermore, these ex vivo expanded NK cells using mbIL-21 were found to have longer telomeres and higher expression of activating NK cell receptors. Multiple preclinical studies have established the powerful antitumor efficacy of IL-21 against solid tumors in mouse models. It has been shown to decrease tumor burden in mice bearing metastatic melanoma and RCC (105), melanoma and MethA fibrosarcoma (106), and head and neck squamous cell carcinoma (107). Several clinical trials have evaluated the safety, feasibility and antitumor effects of IL-21. Administration of recombinant IL-21 (rIL-21) has been shown to be safe with most common adverse event reporting grade1-2 toxicity, and severe toxicities requiring discontinuation being rare. A phase I study in patients with metastatic melanoma and RCC, rIL-21 at 30 μg/kg was well tolerated and shown to have antitumor activity, with about 70% patients showing some response or stable disease. One patient with melanoma achieved a complete remission (108), in a phase II study evaluating the efficacy and safety profile of IL-21 in patients, with metastatic melanoma, IL-21 was deemed safe and active against metastatic melanoma, with overall response rate being 22.5% and a favorable progression free and OS (109). Attempts to combine rIL-21 with targeted therapies have yielded mixed results. Combination of rIL-21 with sunitinib caused severe dose limiting toxicities with no clinical response resulting in early termination of the study (110). However, combining rIL-21 with sorafenib was shown to be relatively safe with mostly grade 1–2 toxicities, and was shown to have antitumor activity with objective response rate of 21% against metastatic RCC (111). Results are awaited from clinical trials evaluating the safety and efficacy of combining IL-21 with other immunotherapeutic agents (IL-21/Anti programmed cell death 1 [PD-1] against solid tumors/NCT01629758, IL-21/ipilimumab against melanoma, NCT01489059).
IL-12
IL-12 is a heterodimeric, pro-inflammatory, type I cytokine that has been shown to elicit T-helper type-1 immune responses against infectious agents and cancer cells. It is mainly secreted by antigen presenting cells (macrophages and dendritic cells) and has been shown to promote the differentiation of CD4+Th0 cells into Th1 cells. It has been shown to increase cytokine production by NK cells and T cells, particularly IFN-γ (112). IL-12 does not appear to have any direct cytotoxic properties but exerts it's effects by stimulating NK and T cell proliferation and cytolytic properties (113), and by improving ADCC (114, 115). The antitumor efficacy of IL-12 has been well established in murine models in multiple preclinical studies (116–119). Despite the initial dose escalation phase I trial using recombinant human IL-12 (rhIL-12) establishing the safety of IL-12 administration in humans (120), subsequent phase II study had to be temporarily stopped due to severe toxicities, and deaths of the 2 patients (121). Subsequent studies have focused on establishing a safe dosing regimen for IL-12 administration to optimize the dose and frequency of IL-12 in patients. It was shown that a priming dose of IL-12 2 weeks prior can significantly decrease the toxicity of subsequent relatively high doses. Intratumoral injections of rhIL-12 have been attempted in patients with head and neck squamous cell carcinoma with activation of B cell compartment, and presence of tumor infiltrating B cells, that correlated with OS (122). Other delivery methods that have been tried are electroporation of plasmid DNA coding for IL-12 in patients with melanoma (123), and PEGylated IL-12 plasmid formulations in patients with gynecologic malignancies (124). To date, clinical benefits of IL-12 administration have been modest. However, significant clinical responses with IL-12 have been reported in patients with cutaneous T cell lymphoma (125) and in patients with acquired immune deficiency syndrome associated Kaposi sarcoma (126).
IL-18
Similar to IL-12, IL-18 is another immunostimulatory cytokine belonging to IL-1 family that regulates both innate and adoptive immune responses. IL-18 is produced by monocytes, macrophages, neutrophils and dendritic cells, and is initially secreted in an inactive form pro-IL-18 which becomes biologically active upon cleavage by caspase-1 (127). IL-18 plays a key role in stimulating IFN-γ production from NK cells (128), and mice deficient in IL-18 have impaired cytotoxic responses, and decreases IFN-γ production (129). IL-18 has been shown to enhance TNF signaling in NK cells, prolonging the messenger ribonucleic acid (mRNA) expression of c-apoptosis inhibitor 2 and TNF receptor-associated factor 1 which inhibits NK cell death (130). In vivo antitumor efficacy of IL-18 has been well established in preclinical studies (131–133). However, there have only been few clinical studies evaluating its safety and efficacy in human subjects. Different phase I studies in patients with cancer have established the safety of rhIL-18 administration (134, 135). However, a subsequent study in patients with metastatic melanoma did not show any significant clinical responses as a monotherapy (136). Further studies evaluating its efficacy in combination with other cytokines and immunotherapeutic agents are required.
Cytokine Analogs
IL-15 Superagonist—ALT-803
Cytokine agonists have been well described in the literature, particularly for IL-15 (137). To further improve the biological activity and pharmacokinetics of a previously described IL-15 superagonist (IL-15N72D), investigators designed a novel molecule where IL-15N72D was fused with a dimeric IL-15 receptor a complex–(IL-15Rα/Fc). This redesigned IL-15 superagonist, ALT-803 has been shown to promote NK cell proliferation has been shown to possesses superior biological activity, higher potency and a much longer half-life (25 h vs. <40 min) compared to wild type IL-15 (138). Early preclinical studies showed that ALT-803 could upregulate the expression of NKG2D, promoted IFN-γ secretion and promoted the expansion of CD8+CD44high memory T cells in vivo in a murine multiple myeloma model (139). Several other preclinical studies have established its efficacy in animal models against bladder cancer (140), B cell lymphomas (141), glioblastoma (142), breast, and colon cancer (143), and ovarian cancer (144). These antitumor effects have been attributed to increase in specific subpopulations of NK and memory CD8+ T cells, increased IFN-γ secretion and improvement in NK cell functionality. Early successes in preclinical studies have led to further investigation of ALT-803 in multiple clinical trials. A phase I trial in relapsed hematologic malignancies following SCT, ALT-803 induced clinical responses in 19% of the patients with one patient achieving complete remission. ALT-803 also induced proliferation and expansion of NK and CD8+ T cells in these patients (145). Another phase I trial in patients with advance solid tumors has established the safety and tolerability of ALT-803 administration (146). Combination of ALT-803 with nivolumab in patients with metastatic non-small cell lung cancer showed an objective response in 29% of the patients with 76% of the patients experiencing disease control. No dose limiting toxicities were seen in this trial (147). Several other clinical trials evaluating the antitumor effects of ALT-803 are currently ongoing (NCT03228667, NCT03127098, NCT03022825, NCT02384954, NCT02138734, NCT02890758, NCT02559674, NCT03520686).
NKTR-255
NKTR-255 is another novel IL-15 analog that is currently undergoing preclinical development. NKTR-255 consists of a polymer-engineered IL-15 molecule that has been designed to optimally engage IL-15 receptor complex. In preclinical studies, it has been shown to have superior binding affinity to IL-15Ra and lower in vivo clearance (22 h vs. 1 h) in comparison to IL-15. It was also shown to induce phosphorylation of STAT5, decrease tumor burden in metastatic lung cancer mouse model and enhance the activation and proliferation of NK cells (148). The early results are exciting, and highlight its role as a promising immunotherapeutic agent. However, further studies are required at this time.
Optimizing NK Cell Mediated ADCC
One of the principle ways NK cells exert their antitumor effects is through ADCC, where Fc portion of the antitumor antibody binds to FcγRIIIA and/or FcγRIIC expressed on NK cells, leading to the NK cell activation, and initiation of a series of events like transduction of death signals via TNF family death receptor signaling, release of cytotoxic granules from NK cells, and production of inflammatory cytokines like IFN-γ causing target cell killing (149). There are wide differences in the expression of activating and inhibitory receptors profile of NK cells amongst individuals. It is also well documented that polymorphisms between FcγRIIIA and FcγRIIC can influence the Fc receptor function. These polymorphisms result in a differential activation upon binding with an antitumor antibody. Patients with higher affinity polymorphisms have been shown to have superior outcomes with mAb treatment (150, 151). In order to augment the polymorphonuclear cell mediated ADCC, investigators have attempted to design an anti-human epidermal growth factor receptor-2 with tandem IgG1/IgA2 Fc that retains IgG1 FcγR binding but also provides the benefits of FcαRI/IgA Fc interactions. Their results showed that the tandem IgG1/IgA2 approach was superior in recruiting and engaging cytotoxic polymorphonuclear cells than either the parental IgG1 or IgA2 (152). Investigators have also attempted to improve the binding affinity of mAbs to maximize the ADCC. Obinutuzumab, a glycoengineered humanized anti-CD20 antibody has been shown to be superior to chimeric anti-CD20 mAb Rituximab in preclinical studies (153). By modifying the antibody backbone, it is possible to create chimeric antibodies (Ch14.18) with significantly longer half-life compared to the murine (mouse hybridoma 3F8), and avoid the human-mouse antibody response (154). An increasing number of humanized and fully human mAbs are currently being investigated in preclinical and clinical studies. Different combination strategies have been tried to improve antitumor ADCC of mAbs. NK cells have been shown to downregulate FcγRIIIA upon activation, and this downregulation is believed to be caused by activation of matrix metalloproteinases by the target cells (155). Preclinical studies have shown that ADAM17 inhibitor inhibits FcγRIIIA shedding and increased NK cell degranulation and IFNγ production (156). Strategies to increase the target antigen density on tumor cells for more efficient targeting by mAbs have been explored. Ionizing radiation (157) and Toll like receptor-9 agonists (158) have been shown to increase the expression of certain tumor target antigens. Currently there is limited preclinical data available about the clinical efficacy of these combinations and further studies are required.
Preventing CD16 Shedding and Expressing High Affinity of CD16
CD16, also known as the human IgG Fc receptor III (FCγRIII), consists of two isoforms (CD16A and CD16B) (159). CD16A is a transmembrane protein and the only FcγR expressed by NK cells (159). It binds to IgG of an antibody and is essential for ADCC, which is a key mechanism of NK cells to lyse tumor cells (149). CD16B is mainly expressed on neutrophil cells (159). Both CD16A and CD16B are cleaved rapidly on neutrophil and NK cell activation after mitogen stimulation and co-culturing with tumor targets and the cleavage is mediated by a metalloprotease, ADAM17 (a disintegrin and metallopeptidase domain 17) (160, 161). The plasma levels of CD16 were significantly reduced in patients treated with an ADAM17 inhibitor (160, 161). The recent preclinical study demonstrated that the ADAM17 inhibitor BMS566394 significantly enhanced the expression of CD16 on NK cells and more importantly, it enhanced the cytotoxic activity and IFN-γ production of treated NK cells combined with trastuzumab against breast cancer cell lines (162). MEDI3622 is a human mAb of ADAM17 with high specificity and a potent inhibitory activity (163). The combination of MEDI3622 with anti-human epidermal growth factor receptor 2 (HER2) antibody trastuzumab greatly augmented the production of IFNγ by NK cells against ovarian cancer cell by blocking the shedding of CD16A on NK cells (164). Engineering NK cells with a CD16 mutant which has mutation(s) in the cleavage domain can also disrupt cleavage and prevent CD16 shedding. Expression high affinity CD16 FcγRIIIa in NK cells is another attractive choice. The insertion of the high affinity CD16 FcγRIIIa (158V) allele and IL-2 into NK-92 cells render NK-mediated ADCC using cetuximab, trastuzumab and pertuzumab against a variety of solid tumor cells (165). Additional strategies include engineering NK cells with chimeric receptors CD16-BB-ζ and CD64-BB-ζ (166). These engineered NK cells significantly improved cytotoxicity against CD20-positive NHL cells in the presence of rituximab (166) but their anti-tumor effects need to be evaluated for solid tumor cells with targeted antibodies.
Role of Immunocytokines in Improving NK Cell Mediated Cytotoxicity
As previously described in this review, a variety of cytokines have been utilized in an attempt to improve NK cell function and stimulation. Early clinical trials have demonstrated the improvement in outcomes in pediatric patients with neuroblastoma that received immunotherapy with anti-GD2 ch14.18 antibody in combination with IL-2 and granulocyte-macrophage colony-stimulating factor (167), whereas no clear benefit of antibody treatment without cytokine support was observed in a similar study performed by a German group suggesting a beneficial role for combining antibody therapy with cytokines (168). However, this approach has had mixed responses with limited clinical success against solid tumors. This is partly due to the challenges with systemic administration of these cytokines. Systemic cytokines have a narrow therapeutic window limiting their efficacy and they can cause severe toxicities by increasing the vascular permeability from a cytokine storm. These limitations have fueled the development of immunocytokines that are novel fusion proteins created by linking tumor specific mAbs to cytokines. The antibody component directs the cytokine molecule to the tumor location with selective activation of cytokine molecules at the site of antitumor activity. Studies have shown that treatment with immunocytokines leads to the targeted increase in the density of NK cells and lymphocytes in the tumor extracellular matrix (169, 170). Several immunocytokines molecules have shown promise in preclinical studies. Anti-GD2-IL2 fusion immunocytokine has been shown to have superior antitumor efficacy against neuroblastoma compared to both molecules administered separately at the same time. The mechanism was reported to be exclusively NK cell mediated (171). Similarly, anti-GD2-RLI (an IL-15 superagonist) fusion showed improved half-life of RLI and was effective against metastatic NXS2 neuroblastoma in a syngeneic mouse model (172). A fusion protein between tumor necrosis-targeting human IgG1 NHS76 and IL-12 (NHS-IL12) had longer half-life in vivo, stimulated lower IFN-γ release by immune cells thereby limiting the IL-12 mediated toxicity, and had superior antitumor efficacy in mouse models (173). Further modifications of IL-2 based immunocytokines have been attempted, e.g., single IL-2 variant (IL2v) moiety with loss of CD25 binding, to avoid Treg stimulation and improve the targeted biological activity (174). Several of these molecules have been tested in clinical trials. In a phase II clinical trial of hu14.18-IL2, complete resolution of bone marrow disease and metaiodobenzylguanidine avid disease was seen in 5 out of 24 Stratum-2 patients with relapsed-refractory neuroblastoma (175). Phase I/II clinical trials have established the safety of intravenous administration of TNF-IL2 fusion protein (L19-TNF) in patients with advanced solid tumors (176), and it was shown to have clinical efficacy in patients with advanced localized melanoma in combination with melphalan and mild hyperthermia (177). More recently, phase I trial of NHL-IL12 established safety in patients with metastatic solid tumors. Evaluation of peripheral immune cell subset showed an increase in activated and mature NK and NKT cells in these patients (178). These agents have shown a great promise in stimulating immune cells like NK cells and cytotoxic T cells locally at the tumor site with cytokine component while maintaining the targeted effector antibody response. Multiple ongoing clinical trials are evaluating the safety and efficacy of several other immunocytokines alone, and in combination with other therapeutic modalities like immune checkpoint inhibitors (NCT03209869, NCT03386721, NCT02627274, NCT02350673). Table 2 provides a comprehensive list of past and current clinical trials evaluating the safety and efficacy of immunocytokines against solid tumors.
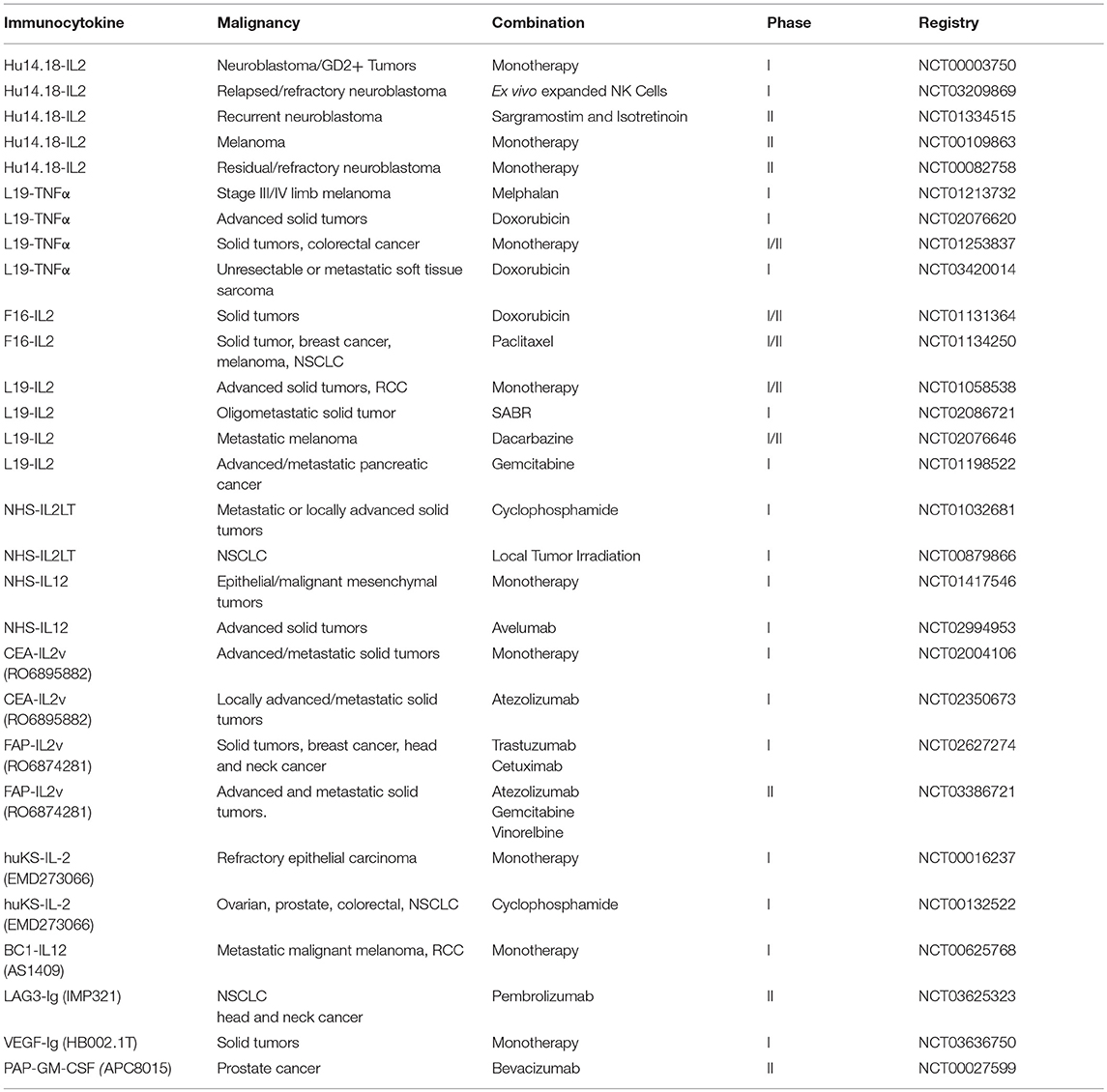
Table 2. Clinical development of immunocytokines/fusion proteins against solid tumors.
ANIT-KIR Antibodies for Improvng NK Cell Cytotoxicity
As mentioned previously, NK cells remain tolerant to cells expressing HLA class I ligands but trigger cytotoxicity against altered cells that have a decreased level of HLA expression. This distinction between self and altered cells is mediated through inhibitory KIRs on NK cell surface. KIRs can recognize HLA molecules triggering inhibitory signals and resulting in decreased ADCC by NK cells. NK cells herald the immune recovery of lymphocyte subsets following allogeneic HSCT, and have been implicated in early graft vs. malignancy effects (35). This concept has been exploited clinically in allogeneic HSCTs for hematologic malignancies where donor KIR is mismatched with recipient's tumor creating a KIR-ligand incompatibility in order to create graft vs. leukemia effect (179). Similar to the KIR-ligand mismatch concept, investigators have designed mAbs that block the HLA-KIR interactions to prevent the NK cell inhibition and trigger cytotoxicity. Phase I clinical trial with IPH2101, the first in class anti-KIR antibody that inhibits KIR2DL-1, L-2, and L-3, in patients with relapsed/refractory multiple myeloma established the safety at dose that achieve full inhibitory KIR saturation (180). In another phase I study for relapsed multiple myeloma, a combination of IPH2101 with lenalidomide resulted in objective responses in five out of 15 patients, with median progression free survival being 24 months (181). In preclinical studies, the second generation fully human IgG4 anti-KIR2DL1, -L2, -L3, -S1, -S2 antibody (IPH2102/Lirilumab) was shown to potentiate the spontaneous cytotoxicity of NK cells against lymphoma cells lines. It was also shown to augment the NK cells mediated ADCC with Rituximab against CD20 lymphoma cells, in vitro and in vivo (182). Very few studies have looked at the efficacy of these anti-KIR antibodies against solid tumors. IPH2102 was well tolerated in patients with solid tumors and hematologic malignancies, with patients experiencing only mild and transient side effects (183). A recently published study established a correlation between the expression of inhibitory KIR and PD-1 on tumor cells in patients with non-small cell lung cancer suggesting a potential benefit of combining anti-KIR antibodies with anti-PD-1 treatment to circumvent the immune escape in these patients (184). Several active studies are currently evaluating these anti-KIR antibodies against solid tumors in combination with other immune therapies (NCT03341936, NCT03203876, NCT03347123).
Re-Directing NK Cells With Chimeric Antigen Receptor (CAR)
The adoptive transfer of T cells engineered to express an artificial CAR to target a specific antigen on tumor cell surface is an exciting approach for cancer immunotherapy. CARs usually include a single-chain variable fragment from a mAb, a transmembrane hinge region, and a signaling co-stimulatory domain such as CD28, CD3-zeta, 4-1BB (CD137), or 2B4 (CD244) endodimers (185–187). The co-stimulatory components attribute greater strength of signaling, and longer in vivo T-cell persistence (39). Four generations of CAR have been developed and evaluated pre-clinically and clinically (39) (Figure 3). The advantage of the CAR strategy is that no HLA expression on the target cell is required for the epitope to be accessible to CAR+ immune cells. Thus, CAR+ immune cell application is not limited to only a subset of patients with a specific HLA type (185–187). To increase the targeting specificity of expanded NK cells, our group has investigated functional activities of peripheral blood natural killer cells modified by mRNA nucleofection with anti-CD20 CAR against CD20+ B-NHL in vitro and in xenografted NSG mice (188). Lentiviral transduced methods had been used to generate CAR expressing NK cell lines targeting solid tumor cells. The CARs have been developed and engineered in NK cells lines against several antigens for solid tumors which include epidermal growth factor receptor (EGFR), HER2, EGFRvIII, GD2, epithelial cell adhesion molecule (EpCAM) (Table 3) with efficiency in preclinical studies. Schönfeld et al. generated a stable NK92 cell line expressing a humanized anti-HER2 CAR containing CD28 and CD3ζ signaling domains and these CAR NK cells efficiently lysed HER2+ tumor cells in vitro and the specific recognition of tumor cells resulted in selective enrichment of anti-HER2 CAR NK-92 cells in orthotopic breast carcinoma xenografts and reduction of pulmonary metastasis in a RCC model, respectively (189). In another study, the repeated stereotactic injection of anti-HER2 CAR NK-92 cells improved the symptom-free survival in glioblastoma xenografted mice (190). NK-92 cells and primary NK cells were engineered to express the second generation of EGFR-CAR to target breast cancer cells (191). In vitro, compared with mock-transduced NK-92 cells or primary NK cells, EGFR-CAR-engineered NK-92 cells and primary NK cells displayed enhanced cytotoxicity and IFN-γ production when co-cultured with breast cancer cell lines (191). In the mice intracranially pre-inoculated with EGFR-expressing breast cancer cells, intratumoral administration of EGFR-CAR-transduced NK-92 cells mitigated tumor growth compared to mock NK cells (191). A human NK cell line KHYG-1 expressing anti- EGFRvIII CAR was established and exhibited the inhibition of glioblastoma cell-growth via apoptosis in an EGFRvIII-expression specific manner (192). Another group engineered NK-92 to stably express an anti-GD2 CAR and these CAR NK-92 cells facilitated tumor effective recognition and elimination of GD2+ NB cell lines and primary NB cells (193). Anti-EpCAM CAR engineered NK-92 displayed high and selective cell-killing activity against EpCAM-expressing breast carcinoma cells that were resistant to the natural cytotoxicity of unmodified NK cells (194). Additionally, our group is developing anti-ROR1 CAR engineered expanded primary NK cells through CAR mRNA electroporation technology to target ROR1+ solid tumors with promising in vitro anti-tumor effects (195). Anti-mesothelin CAR-NK cells were derived from CAR-expressing iPSCs with the optimized CAR construct (197). These CAR-NK cells showed great potent ability to kill mesothelin-expressing tumors both in vitro and in vivo, demonstrating a potential strategy to produce “off the shelf,” targeted allogeneic cell products for refractory malignancies (197). Besides designing a CAR based on the single chain variable fragment (scFv) of a mAb again an antigen on tumor cell surface, CAR can also be formed from a NK activating receptor such as NKG2D followed by transmembrane domain and signal transduction domains. Chang et al. designed a CAR termed NKG2D-DAP10-CD3ζ that was composed of the NK cell activating molecule NKG2D plus 2 key signaling molecules, DAP10 and CD3ζ (196). These NKG2D CAR engineered primary NK cells through retroviral transduction showed significantly enhanced in vitro cytotoxicity against a variety of solid tumor cell lines that express NKG2D ligands MICA/B such as the osteosarcoma cell lines U-2 OS, MG-36, HOS, the prostate carcinoma cell lines DU 145 and PC-3, and the rhabdomyosarcoma cell line RH36 (196) and significantly reduced tumor burden in osteosarcoma xenografted NSG mice compared to mock NK cells (196). Similar strategy can be applied to generate other NK activating receptor based CAR like NKp30-CAR to enhance NK cytotoxicity. The advantage of this CAR strategy is that one CAR can be applied for a variety of tumor types in a matter expressing the corresponding ligands. Considering the recent safety concerns such as cytokine release syndrome and neurotoxicity associated with infusion of CAR-modified T cells (187), a suicide gene should be incorporated into the construct as a safety measure for CAR NK therapy but it is debatable because of the short life span of NK cells compared to T cells. Additionally, IL-15 secretion CAR-NK can be generated retroviral transduction by incorporating IL-15 to CAR design (18) to enhance the CAR NK proliferation, persistence and homing in solid tumors.
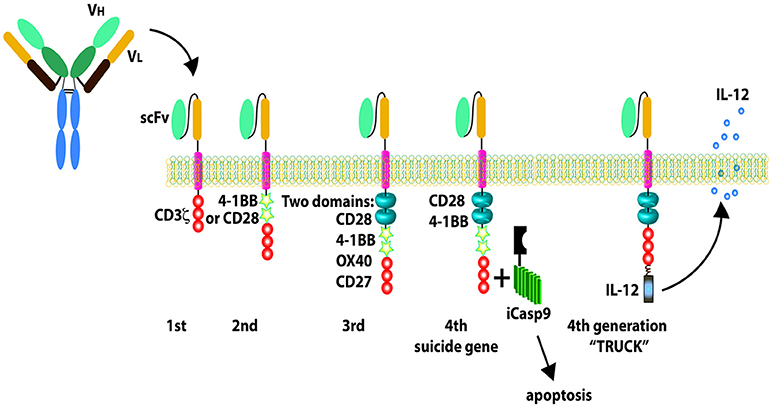
Figure 3. Chimeric antigen receptors (CARs). The first-generation CARs only have CD3 zeta signaling domain; the second-generation CARs include one CD28 or 4-1BB co-stimulatory components combined with CD3 zeta signaling domain; the third-generation CARs include two co-stimulatory domains; the fourth-generation CARs are designed with new elements including a controllable suicide gene like caspase 9 or loaded with IL-12 secretion. Reproduced with permission from: Barth et al. (39).
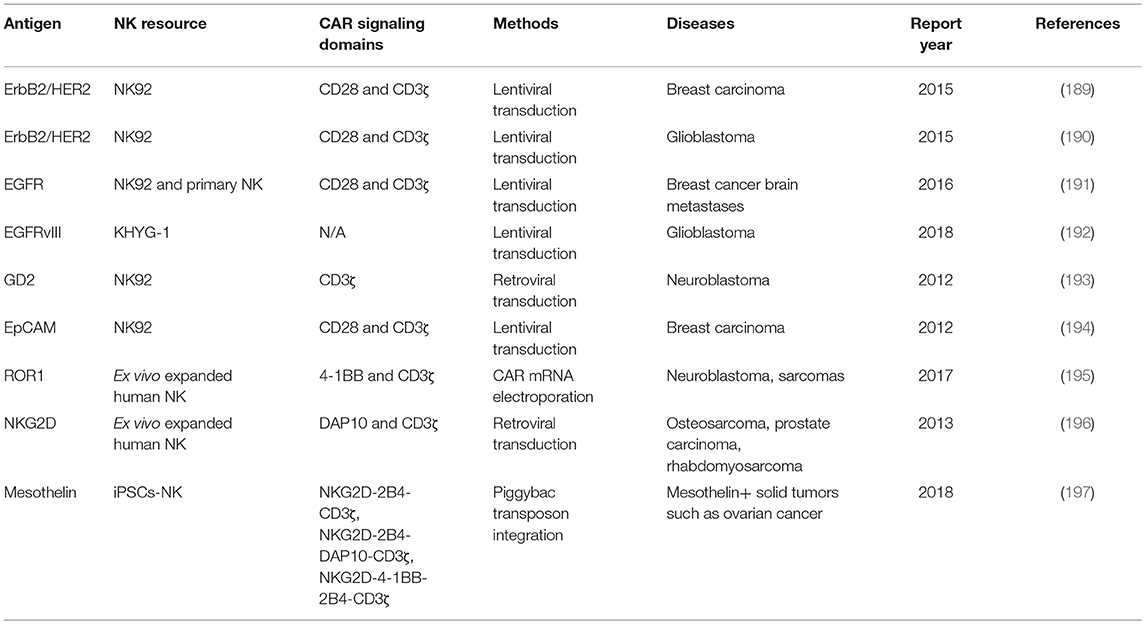
Table 3. Summary of CAR NK in preclinical studies for solid tumors.
There are currently 3 registered clinical trials testing the safety and efficacy of CAR-NK cells in patients with solid tumors. One trial is a single-center, single arm, open-label pilot study to evaluate the safety and feasibility of CAR-NK cell treatment in subjects with metastatic solid tumors using autologous or allogeneic NK cells transfected by mRNA electroporation against NKG2D-ligand expressing cancer cells (NCT03415100). Another trial is to evaluate the efficacy and safety of CAR-modified NK Cell lines in MUC1 positive advanced refractory or relapsed solid tumors (NCT02839954). These two trials are being conducted in China. The third trial is being conducted in the USA sponsored by Johann Wolfgang Goethe University Hospital to evaluate the safety and tolerability of NK-92/5.28.z (HER2.taNK) for patients with recurrent HER2-positive glioblastoma (NCT03383978). Pharmacokinetics and pharmacodynamics and potential signs of anti-tumor activity of NK-92/5.28.z cells will also be analyzed.
Bispecific Antibodies to Enhance NK Cell Killing Potential
mAbs have revolutionized the development of anticancer therapeutics over past last few decades. However, the efficacy of mAbs has been limited against solid tumors. Advances in protein engineering has made the generation of bispecific molecules possible. Bispecific antibodies are novel molecules where two antigens can be targeted at the same time by combining the specificities of two antibodies. The design of a bispecific antibody constitutes an antitumor scFv targeting a specific malignancy is linked to an anti-CD3/anti-CD16 in order to create an immune connection between cancer cell and the immune effector cells like T cell or NK cell. Recently, there has been a growing interest in development of bispecific antibodies with currently multiple studies evaluating their anti-cancer potential in preclinical and clinical studies. To date, the most success with bispecific antibodies has been seen with T cell specific bispecific molecules like catumaxomab (CD3/EpCAM) against malignant ascites, and blinatumomab (CD3/CD19) and ionotuzumab (CD3/CD22) against B cell lymphoblastic leukemia (198–200). These successes have encouraged the development of diverse bispecific antibodies with varied clinical applications besides cancer, like emicizumab/ACE910 for patients with Hemophilia A. The goal of developing these bispecific engagers is to enhance the therapeutic efficacy, improve targeted delivery to the tumor site, optimize immune cell engagement, and reduce off target effects and relative ease of administering one drug instead of two separate molecules. One major shortcoming of the T cell specific antibodies has been their potential to cause massive cytokine release causing capillary leak, hypotension and respiratory distress in a clinical setting. These shortcomings have made NK cell based bispecific NK cell engagers an attractive alternative. Bispecific NK cell based antibodies can engage the Fc portion of the antibodies through their FcγRIII (CD 16) receptor with the other portion designed to bind a specific epitope on the tumor surface. Several NK based bispecific antibodies are currently in preclinical and clinical development. Early investigations have focused on AFM13, an anti-CD30/CD16A for relapsed or refractory Hodgkin lymphoma. In Phase I trial, AFM13 was shown to be safe, caused activation of NK cells, decreased soluble CD30 in peripheral blood, was found to be active in patients resistant to brentuximab, and achieved disease control in 77% patients at doses ≥1.5 mg/kg (201). Phase II studies with AFM13 are currently ongoing. Multiple preclinical studies are evaluating different bispecific and trispecific NK cell engaging antibodies against solid tumors. A trivalent bispecific antibody targeting ErbB2 and CD16 was shown to be more potent than anti-ErbB2 single-chain variable fragment (scFv)-Fc fusion protein in vitro against breast cancer cell lines, and in vivo against breast cancer xenograft mouse model (202). Multiple other antibodies targeting the HER2- FcγRIII antigens have been described (203–205). Similarly, a completely humanized bispecific antibody targeting EpCAM and CD16 showed significant increase in ADCC, increased degranulation of NK cells with concomitant increase in IFN-g production against EpCAM positive prostate, breast, colon, and head and neck cancer cell lines (206). Modifications have been made to the dimeric structure of these bispecific antibodies to further improve the efficacy. A tribody targeting human epidermal growth factor 2 where two HER2-specific scFvs were linked to CD16 [(HER2)2xCD16] was found to be superior to trazutumab against HER2-expressing breast, pancreatic, ovarian, and esophageal tumor cells with increased NK cell degranulation and release of granzyme B (207). Insertion of a modified interleukin-15 cross-linker to an EpCAM/CD16 bispecific construct to create a trispecific construct improved NK cell proliferation and survival and showed increased ADCC (208). Table 4 highlights multiple CD16 targeting bispecific and trispecific antibodies that have undergone preclinical development. The results of these preclinical studies are encouraging and warrant further clinical development of these molecules.
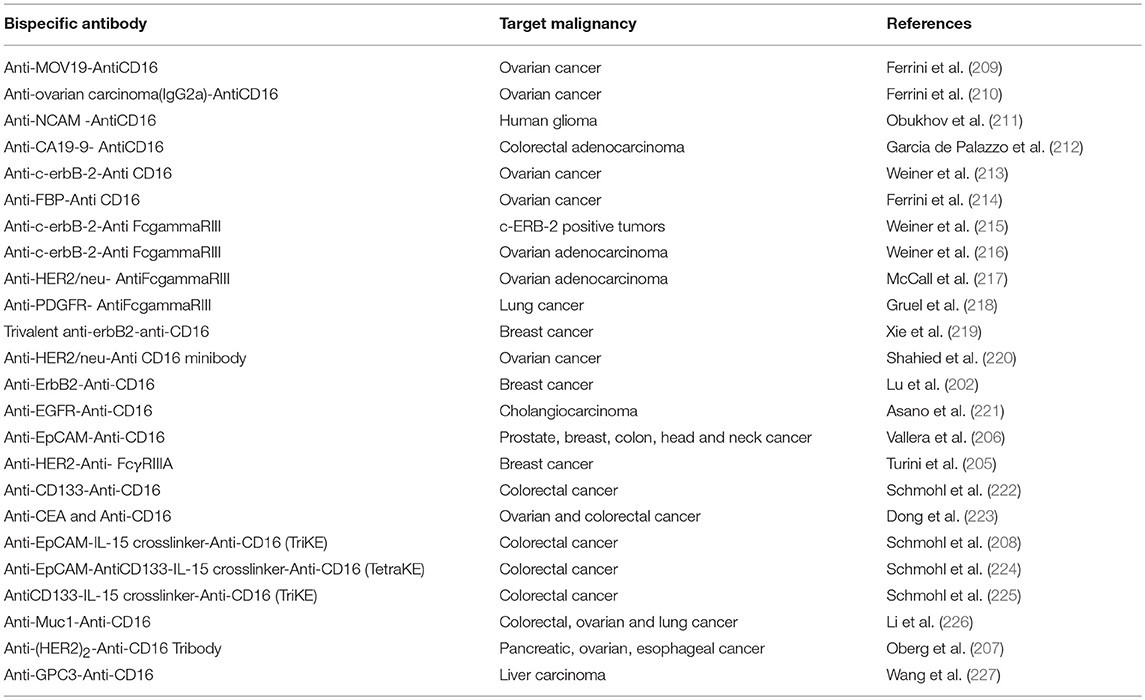
Table 4. Preclinical development of CD16 antigen based bispecific antibodies targeting solid tumors.
Targeting NK Cell Checkpoints PD-1, TIGIT, and IL-1R8
Immune checkpoints are negative regulators of immune cells, especially T cells, to help keep immune responses in check, and maintain self-tolerance during immune responses (228). Malignant cells often express high level of ligands of checkpoint inhibitory receptors, and escape from immune recognition and elimination (228). In recent years, the application of mAbs directed against immune checkpoint receptors or ligands has greatly enhanced the anti-tumor activity of the immune cells, and has resulted in remarkable clinical benefits (229, 230). Similar to T cells, NK cells also express an array of immune checkpoints which include PD-1, cytotoxic T-lymphocyte-associated protein 4, T cell immunoglobulin- and mucin-domain-containing molecule 3, T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motif (ITIM) domains (TIGIT), CD96, lymphocyte activation gene-3, and IL-1R8 besides the well-known NK inhibitor receptors: KIRs and CD94/NKG2A (231–233). The data of the cytotoxic T-lymphocyte-associated protein 4, lymphocyte activation gene 3 and mucin-domain-containing molecule 3 on NK cells functions are either scarce or controversial. But several lines of evidences strongly demonstrate the inhibitory roles of PD-1, TIGIT, and IL-1R8 on NK cells. PD-1+ NK cells are confined to CD56dimNKG2A–KIR+CD57+ mature NK population and are functionally exhausted, exhibiting reduced proliferative capability, poor cytolytic activity and impaired cytokine production as compared with the PD-1− NK cells (234, 235). A recent study demonstrated that the increased PD-1 expression on peripheral and tumor infiltrating NK cells from patients with digestive cancers indicates poorer prognosis (236). And blocking PD-1/PD-L1 signaling markedly enhances cytokines production and degranulation and suppresses apoptosis of NK cells in vitro (236). More importantly, a PD-1 blocking antibody was found to significantly suppress the growth of xenografts in nude mice, and this inhibition of tumor growth was completely abrogated by NK depletion, strongly suggesting that PD-1 is an inhibitory regulator of NK cells in digestive cancers (236). PD-1 blockade might be an efficient strategy in NK cell-based tumor immunotherapy. A phase II clinical trial is on-going to assess the effect of pembrozilumab (a humanized anti-PD-1 mAb) on NK cell function and exhaustion in melanoma (NCT03241927). TIGIT competes with the NK activating receptor DNAX Accessory Molecule-1 (CD226) for their common ligands CD112 (PVRL2) and CD155 (PVR) to directly dampen NK cell cytotoxicity (237). In vitro TIGIT blockade improves the anti-tumor effect of Trastuzumab (a recombinant humanized anti-HER2 mAb), which partially relies on NK cell-mediated ADCC (238). Recent evidence showed the upregulation of TIGIT on tumor-infiltrating NK cells in mouse models of subcutaneously administered solid tumors and the TIGIT expression on tumor-infiltrating NK cells was associated with tumor progression and was linked to functional exhaustion of NK cells (233). The blockade of TIGIT via mAbs reversed the exhaustion of anti-tumor NK cells in multiple tumor models, enhanced the infiltration of activated (CD69+) NK cells into tumors and thereafter improved the OS of the host (233). The presence of NK cells was critical for the therapeutic effects of blockade of TIGIT or the PD-1 ligand PD-L1 or combined blockade of both checkpoints (233). These findings demonstrate that the NK cell–associated TIGIT signaling pathway has a role in tumors' evasion of the immune system and that reversing NK cell exhaustion is critical for the therapeutic effects of anti-tumor immunotherapy based on the blockade of TIGIT (239). IL-1R8, also known as toll-interleukin 1 receptor or Single Ig IL-1-related receptor, is a member of interleukin-1 receptor family (IL1Rs) and acts as a negative regulator of IL1Rs and toll-like receptors (TLRs) to suppress ILR- and TLR-mediated cell activation (240). IL-1R8 is widely expressed in several epithelial tissues, in particular by epithelial cells of the kidney, digestive tract, liver, lung, lymphoid organs, and it is also expressed on monocytes, B and T lymphocytes, dendritic cells, and NK cells (240). Recently, Molgora et al. identified IL1R8 as a checkpoint protein in NK cells that regulates antitumor activity of NK cells in solid tumors (232). Utilizing IL-1R8-deficient (Il1r8–/–) mice as a study model, Molgora et al. found that IL1R8- deficient NK cells expressed significantly higher levels of the activating receptors NKG2D, DNAX Accessory Molecule-1 and Ly49H and fas ligand and produced increased levels of IFNγ and granzyme B (232). IL-1R8 partial silencing in human peripheral blood NK cells with small interfering RNA was associated with a significant increase in IFNγ production and upregulation of CD69 expression (232). In a model of sarcoma (MN/MCA1) spontaneous lung metastasis, Il1r8–/– mice showed a reduced number of hematogenous metastases, whereas primary tumor growth was unaffected and the protection was completely abolished in NK-cell-depleted Il1r8–/– mice (232). Additionally, adoptive transfer of Il1r8–/– NK cells significantly and markedly reduced the number and volume of lung and liver metastases in in the mice with MC38 colon carcinoma liver metastasis while Il1r8+/+ NK cells had no effect (232). These results suggest IL-1R8 serves as a negative regulator of NK cells and its inactivation unleashes human NK-cell effector function (232).
Focusing on NKG2D and the Ligands
NKG2D is a C-type, lectin-like, type II transmembrane glycoprotein-activating receptor expressed in humans on NK, natural killer T, activated CD8+ T cells and some CD4+ and γδ+Tcell subsets (241). In humans, NKG2D forms a hexameric structure with the adaptor molecule DNAX-activating protein of 10 kDa (DAP10) to mediate signal transduction and cellular activation upon ligand recognition (242). NKG2D ligands are structural homologs of MHC class I molecules and are upregulated on the surface of many cell types by cellular stress and viral/bacterial infections and frequently during tumorigenesis (243, 244). The currently identified human NKG2D ligands include MICA and MICB and UL16 binding protein (ULBP1–ULBP6) families (245). Several lines of evidence conclusively demonstrated that engagement of NKG2D and NKG2D ligands, such as MIC A/B elicits cytolytic responses overcoming inhibitory signals on NK cells and is sufficient to trigger cytolysis by NK cells expressing NKG2D (246–249) (Figure 4). The expression of these ligands on the tumor cell surface are regulated at multiple levels: transcriptional regulation, ribonucleic acid (RNA) splicing, posttranscriptional regulation, posttranslational regulation (245). NKG2D ligands can be cleaved from the tumor cell surface after translation by membrane matrix metalloprotease and be released as soluble ligands (245). The findings from “humanized” transgenic animal studies demonstrated the opposite roles of membrane-bound and soluble forms of NKG2D ligands (250, 251). The membrane-bound ligands binding to NKG2D play an important role in NK cell activation and tumor immune surveillance (247, 252, 253), while the soluble NKG2D ligands suppress tumor immunity by passively blocking NKG2D and inducing receptor internalization to down-regulate NKG2D on the surface of NK cells (254–256). Serum levels of soluble NKG2D ligands significantly correlate with patients prognosis and are used as prognostic markers in some tumor patients (257, 258). Therefore, therapeutic strategies have focused on enhancing NKG2D expression and signaling on NK cells such as expression of NKG2D CAR and applying IL-15 agonist as we discussed in the earlier section; enhancing the level of membrane-bound NKG2D ligand on tumor cells; and eliminating soluble NKG2D ligands (Table 5). We and others had utilized histone deacetylase inhibitors such as romidespin, entinostat, sodium valproate to enhance NKG2D ligands expression on tumor cell surface to enhance NK based immunotherapy (259, 260, 263, 264). Zhu et al. found that entinostat not only increased the expression of MICA/B on osteosarcoma cells but also simultaneously increased the expression of NKG2D on primary human NK cells to augment the activation pathways for NK cell recognition of cancer cells (259). Their results indicate that entinostat has the potential to enhance concurrent NK-cell therapy for solid tumors such as colon carcinoma and osteosarcoma (259). Proteases, such as ADAM-10, ADAM-17, and the membrane type matrix metalloproteinase 14, have been found to mediate MIC shedding through proteolytic activities (265–267). In an in vitro drug screen using a Federal Drug Administration-approved drug library, lomofungin, an antifungal drug, was found to strongly decrease ADAM17 activity in hepatocellular carcinoma cells and resulted in enhanced membrane bound MICA expression and inhibited soluble MICA production (261). Another ADAM17 inhibitor, INCB7839, was used to present HER2 cleavage and to treat patients with HER2-positive breast cancer in combination with trastuzumab and it is also in clinical trials to prevent CD20 cleavage in combination with rituximab for the treatment of diffuse large B-cell NHL (268). It would be interesting to investigate if these inhibitors prevent NKG2D ligands shedding and enhance NK-cell therapy for solid tumors. Applying neutralizing antibodies of soluble NKG2D ligands is another promising strategy to overcome immune suppressive effect of these cleaved ligands. Soluble MIC-specific mAb B10G5 was shown highly effective against primary prostate carcinoma and metastasis in the double transgenic TRAMP/MIC mouse model (262). B10G5 antibody therapy effectively induced regression of primary tumors and eliminated metastasis associated with enriched NK cell infiltration in the prostate tumor parenchyma (262). B10G5 therapy also remarkably restored NK cell pool in the periphery and the ability of NK cell homeostatic to self-renew as evidenced by bromodeoxyuridine uptake and markedly enhanced NK cell function, illustrated by increased production of IFNγ in response to mitogen stimulation and cytolytic ability against NKG2D ligand-positive target cells (262). These data conclude that targeting serum soluble MIC significantly restores NK cell homeostatic maintenance and function in MIC+ cancer host (262).
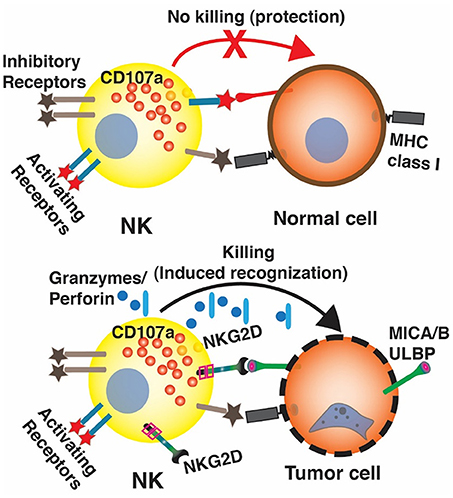
Figure 4. The interaction between NKG2D on NK cells and NKG2D ligands on tumor cells. In normal cells, NKG2D ligands express is very low. The functions of NK cells are balanced by the signals from the inhibitory and activating receptors. In humans, when normal cells are transformed into cancer cells. NKG2D ligands such as MICA/B and ULBP proteins, are often overexpressed. The engagement of NKG2D and NKG2D ligands overcomes inhibitory signals on NK cells, activates NK cells to release cytotoxic molecules such as perforin and granzyme, and trigger apoptosis of tumor cells.
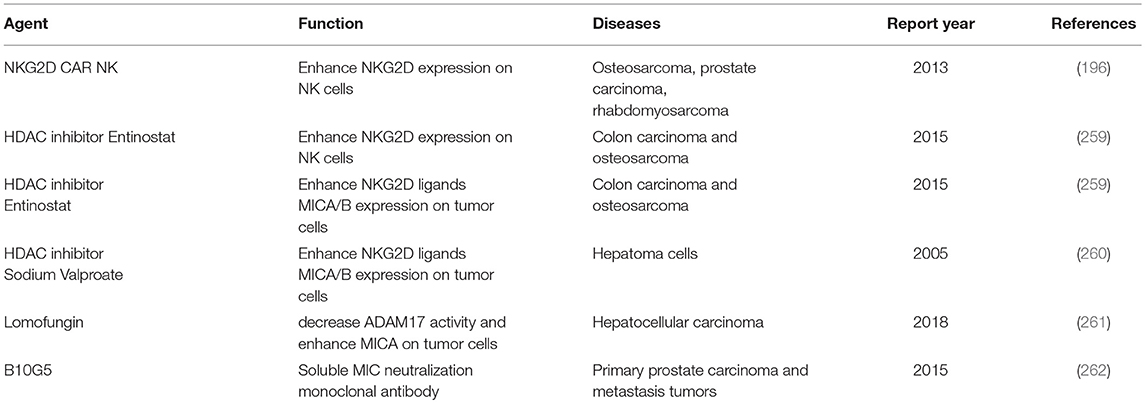
Table 5. Strategies to enhance NKG2D signaling for solid tumors.
Enhancing NK Homing and Tumor Infiltration
Several studies have shown that NK cell homing and infiltration within tumors was associated with improved tumor regression and prognosis (7, 269). The inability of NK cells to migrate to the tumor site limits the clinical outcome of adoptive NK cell infusion in patients with solid tumors (270, 271). Strategies that increase NK homing and infiltration into tumors would be plausible to enhance NK antitumor efficacy and prevent resistance and relapse. The ability of NK cells to home and infiltrate into tumors largely depends on the chemokine receptors they express as well as the chemokines secreted by the tumor cells (272). Wennerberg et al. found that ex vivo expansion NK had significantly enhanced CXCR3 expression which resulted in increased migratory capacity toward CXCL10-producing RCC and melanoma tumor cells (273). Following adoptive transfer of these ex vivo expanded human NK cells, mice bearing CXCL10+ melanoma tumors had increased intratumoral infiltration of NK cells and a significantly prolonged survival compared with mice bearing CXCL10− tumors (273). These data demonstrated the importance of CXCL10 in directing the migration and infiltration of CXCR3 human NK cells toward solid tumors (273). Prime the tumor microenvironment to secrete CXCL10 might be a good strategy to attract CXCR3 expression NK and to enhance the efficacy of NK cell-based therapy against solid tumors. Other efforts were made to genetically engineer NK cells with chemokine receptors to improve their migration toward the corresponding ligands on tumor cells surface. Various solid tumors, including RCC, secrete ligands for the chemokine receptor CXCR2 to promote angiogenesis, tumor growth and metastasis (274). Kremer et al. genetically engineered expanded human NK cells to express CXCR2 to improve their ability to specifically migrate along a tumor-derived chemokine gradient (271). CXCR2 expressing NK cells obtained increased adhesion properties and resulted in increased killing of target cells (271). Therefore, genetic engineering of ex vivo expanded NK cells to express chemokine receptor such as CXCR2 represents a novel strategy to improve anti-tumor effects following adoptive transfer of NK cells. A recent study connected the role of autophagy with CCL5-dependent NK cells infiltration in melanoma. Autophagy is a lysosomal degradation pathway for cells to self-digest their own components such as damaged organelles and misfolded proteins and such a degradation process provides nutrients to maintain cellular functions and allows survival of cancer cells under stress conditions (275, 276). Autophagy involves a Beclin-1 (BECN1)/class III phosphoinositide-3-kinase (PI3K) complex to initiate the formation of phagophore (275, 276). The previous studies from Baginska et al. demonstrated that targeting the autophagy gene BECN1 prevented the degradation of NK-derived granzyme B, and therefore restored their susceptibility to NK cell-mediated killing and significantly inhibited tumor growth in syngeneic melanoma and breast mouse models (277). A recent study from Mgrditchian et al. found that when the autophagy process was blocked in tumor cells by inhibiting the expression of BECN1, the tumor cells produced an increased amount of CCL5 to attract functional NK cells to infiltrate into the melanoma tumor (276). Consequently, this led to a significant reduction in melanoma tumor size (276). These studies highlight the importance of integrating autophagy inhibitors as an innovative strategy in enhancing NK infiltration and killing.
Targeting the Tumor Microenvironment and Blocking Transforming Growth Factor Beta (TGF-β) Pathway
It is well documented that the tumor microenvironment (TME) supports tumor growth, metastasis and suppress immune system (278). A major obstacle of ensuring high cytotoxic activity of NK cells is that these cells are surrounded by immunosuppressive cells and molecules in TME and must overcome the immunosuppressive properties from TME. One of immunosuppressive molecules is TGF-β1 (279). The increased TGF-β level was found in the plasma of advanced cancer patients such as breast cancer, ovarian cancer and neuroblastoma and correlated with worse event-free survival (280–282). Among three isoforms of TGF-β, TGF-β1 is the most abundant and widely studied isoform with 390 amino acids (283). This ligand binds to TGFβ receptor type I which results in its dimerization to TGFβ receptor type II and then phosphorylates SMAD2 and SMAD3 which complex with SMAD4 to modulate transcription of downstream genes (283, 284). TGF-β transmits biological signals to cells also through SMAD independent, alternative signaling pathways such as mitogen activated protein kinases, phosphoinositide 3′ kinase, and TNF receptor-associated factor6-TGF-β-activated kinase 1-p38/c-Jun N-terminal kinase (TRAF6-TAK1-p38/JNK) (283). TGF-β is produced by tumor cells themselves, Tregs, myeloid derived suppressor cells and other stromal cells in TME to downregulates the host immune response via driving the Th1/Th2 balance toward the Th2 immune phenotype, directly inhibiting anti-tumoral Th1-type responses and M1-type macrophages and promoting M2-type macrophages, suppressing cytotoxic CD8+ T-lymphocytes, NK, and dendritic cells functions, and stimulating CD4 + CD25+ T-regulatory cells (Treg) (285). TGF-β inhibits NK-cell proliferation and function in part by Treg cells which are known to produce high levels of TGF-β (286). One of the mechanisms by which TGF-β impairs NK cell function is by down-regulating the expression of NK activating receptors: NKp30 and NKG2D (287, 288). On the tumor side, TGF-β inhibits the transcription of the NKG2D ligands on tumor cells such as down regulation of MICA in the glioma cells, which reduced the recognition and killing by NKG2D expressing NK (289). TGF-β also represses development of NK cells from CD34+ progenitors and resulted in conversion of a minor fraction of CD56brightCD16+ cells found in peripheral blood into CD56brightCD16− cells (290). TGF-β inhibits CD16-mediated human NK cell IFN-γ production and ADCC through SMAD3 (291). Further studies demonstrated that blockade of TGF-β signaling in NK cells caused the accumulation of NK cells that produce IFN-γ (292) and neutralization of TGF-β prevented NKG2D downregulation and also restored NK cell anti-tumor reactivity (293). RNA interference of TGF-beta1 and TGF-beta2 prevented the down-regulation of NKG2D on NK cells mediated by glioma cells and strongly enhanced MICA expression in the glioma cells and promotes their recognition and lysis by NK cells (289). These evidences support an immunosuppressive effect of TGF-β on NK cells and also provide a compelling rationale for blunting the inhibitory effect of it on NK cells as an anti-cancer therapy. Some approaches aiming at decreasing circulating TGF-β, blocking ligand-receptor interactions or inhibiting TGF-β signaling pathways to enhance NK based therapies are currently under investigation pre-clinically and clinically including TGF-β neutralizing antibody, TGF-β receptor I kinase inhibitors, SMAD3-Silenced NK Cells, NK cells engineered with a dominant negative receptor II for TGF-β, NK cells engineered to express a chimeric receptor with TGF-β type II receptor extracellular and transmembrane domains and the intracellular domain of NK cell-activating receptor NKG2D (Table 6).
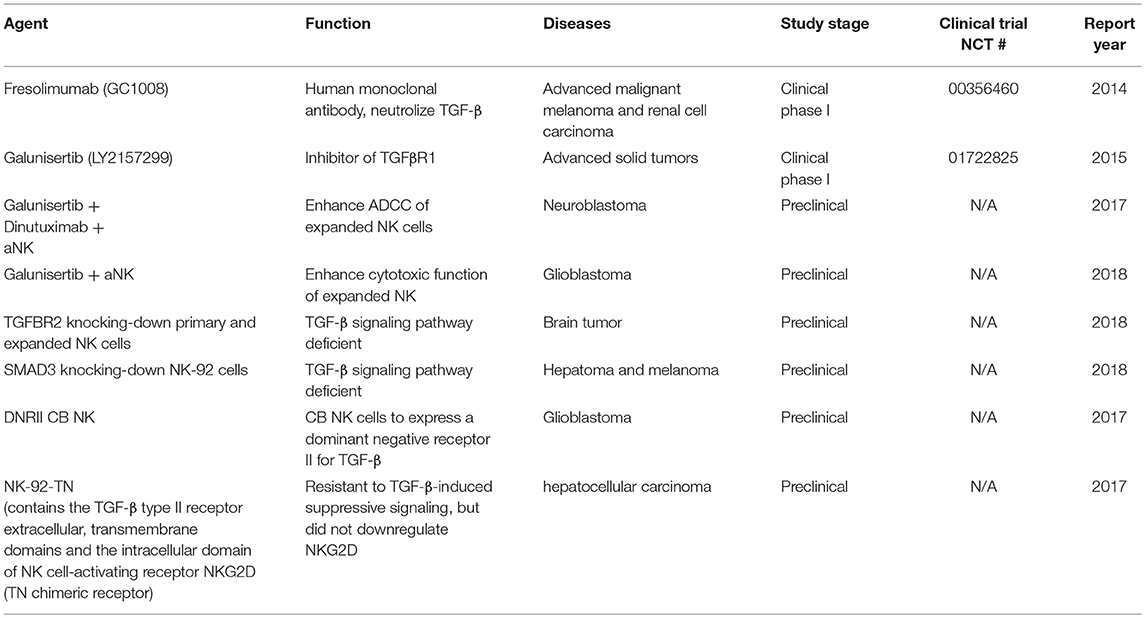
Table 6. Summary of agents utilized to block TGF-β for solid tumors.
Fresolimumab (GC1008) is a high-affinity fully human mAb that neutralizes the active form of all the three isoforms of TGF-β (294). It was designed as an IgG4 isotype to minimize immune effector function. Fresolimumab has been assessed as a potential treatment for RCC and metastatic melanoma. The safety and antitumor activity of repeated doses of fresolimumab administered to patients with advanced malignant melanoma and RCC was evaluated in a Phase I study (NCT00356460) (294). Even the study was not designed to evaluate the effect of fresolimumab on NK cells but it showed acceptable safety and displayed encouraging antitumor activity (294). The results warrant further studies of it with NK therapy.
Galunisertib (LY2157299 monohydrate) is a small-molecule inhibitor of TGFβR1 that binds antagonistically to TGFβR1 to prevent the intracellular phosphorylation of SMAD2 and SMAD3 (295). Phase I studies have demonstrated that galunisertib had an acceptable tolerability and safety profile in patients with advanced solid tumors (296). Recently the preclinical studies from Tran et al. demonstrated that galunisertib combined with anti-GD2 antibody Dinutuximab augmented the anti-tumor cytotoxicity of activated NK (aNK) cells which were activated ex vivo with K562.mbIL21 artificial antigen presenting cells (297). Galunisertib suppressed SMAD2 phosphorylation and restored the expression of DNAX Accessory Molecule-1, NKp30, NKG2D and TNF-related apoptosis-inducing ligand death ligand expression on aNK cells and also significantly enhanced the release of perforin and granzyme A from aNK cells and the direct cytotoxicity and ADCC of aNK cells against neuroblastoma cells in vitro (297). The combination of galunisertib, aNK cells plus dinutuximab reduced tumor growth and increased survival of mice xenografted with two neuroblastoma cell lines or a patient-derived xenograft (297). In another study, galunisertib was shown to preserve the cytotoxic function of ex vivo expanded, highly activated NK cells and significantly improved eradication of liver metastases of colon cancer in mice treated with adoptive NK cells compared with mice receiving NK cells or TGF beta inhibition alone (298). Overall these studies demonstrate that the therapeutic efficacy of adoptive NK cell therapy clinically will be markedly enhanced by complementary approaches targeting TGF-beta signaling in vivo.
Ex vivo manipulating NK cells by novel strategies such as knocking-down TGF-β receptor 2 (TGFBR2) and SMAD3, expressing a dominant negative receptor II for TGF-β, or engineering with a TGF-β type II receptor based chimeric receptor to block the TGF-β signaling pathway are very attractive for adoptive NK therapy for solid tumors. Kararoudi et al. knocked down TGFBR2 in human primary and expanded NK cells using the novel DNA-free Cas9 ribonucleoprotein complexes (299). TGFBR2-knockdown NK cells showed less sensitive to TGFβ (299). SMAD3 is a downstream factor in TGF-β signaling pathway and plays an essential role in TGF-β-mediated immune suppression, and in regulating transcriptional responses that are favorable to metastasis (300). SMAD3 knocked-down NK-92 cells showed enhanced cancer killing activities and enhanced IFN-γ production in vitro and better anticancer effects than NK-92 empty vector control in non-obese diabetic severe combined immunodeficiency mice bearing human hepatoma (HepG2) or melanoma (A375) in-vivo (301). Yvon et al. engineered CB NK cells to express a dominant negative receptor II for TGF-β (DNRII) (302). These CB-derived DNRII-transduced NK cells were expanded to clinically relevant numbers, retained their secretion of interferon-γ, maintained both perforin and NKG2D/DNMA1 expression, and more importantly, retained their killing ability in the presence of TGF-β for glioblastoma cells (302). NK-92 cells were engineered to express a chimeric receptor which contains the TGF-β type II receptor extracellular, transmembrane domains, and the intracellular domain of NK cell-activating receptor NKG2D (TN chimeric receptor) by Wang et al. (303). These NK-92-TN cells were resistant to TGF-β-induced suppressive signaling, did not downregulate NKG2D (303). These modified NK-92 cells had higher killing capacity and IFN-γ production against carcinoma tumor cells compared with the control cells in vitro and in in a hepatocellular carcinoma xenograft tumor model (303). More interestingly, NK-92-TN cells were better chemo-attracted to the tumor cells expressing TGF-β and their cytotoxicity was further enhanced by TGF-β (303). The presence of these modified NK-92-TN cells significantly inhibited the differentiation of human naive CD4+ T cells to regulatory T cells (303). Overall, these engineered NK cells either with SMAD3 knock-down, expressing a dominant negative receptor II for TGF-β, or with a TGF-β type II receptor based CAR should have functional advantages over unmodified NK cells in the presence of TGF-β-secreting solid tumors and will be important therapeutic approaches for NK resistance in patients with solid tumors.
Conclusion and Future Directions
NK cell based applications are a promising alternative for immunotherapy of solid tumors. Improvements in understating of NK cell biology and function are driving the further development of NK cells based novel approaches to effectively target solid tumors. We have described the multiple strategies that have been investigated for improving the cytolytic properties of NK cells (Figure 5). In the future, combinations of these approaches need to be optimized to further enhance NK efficacy in targeting solid tumors. There is also a growing need to improve the current imaging modalities to monitor the accumulation and distribution of NK cells in vivo after systemic administration, which could serve as a potential surrogate for monitoring the tumor accumulation and anti-tumor response. Improvements in manufacturing and expansion techniques are desired in order to obtain a true universal “off-the-shelf” NK cell product that is GMP-compatible, lower in cost, has a longer half-life and possesses enhanced antitumor responses.
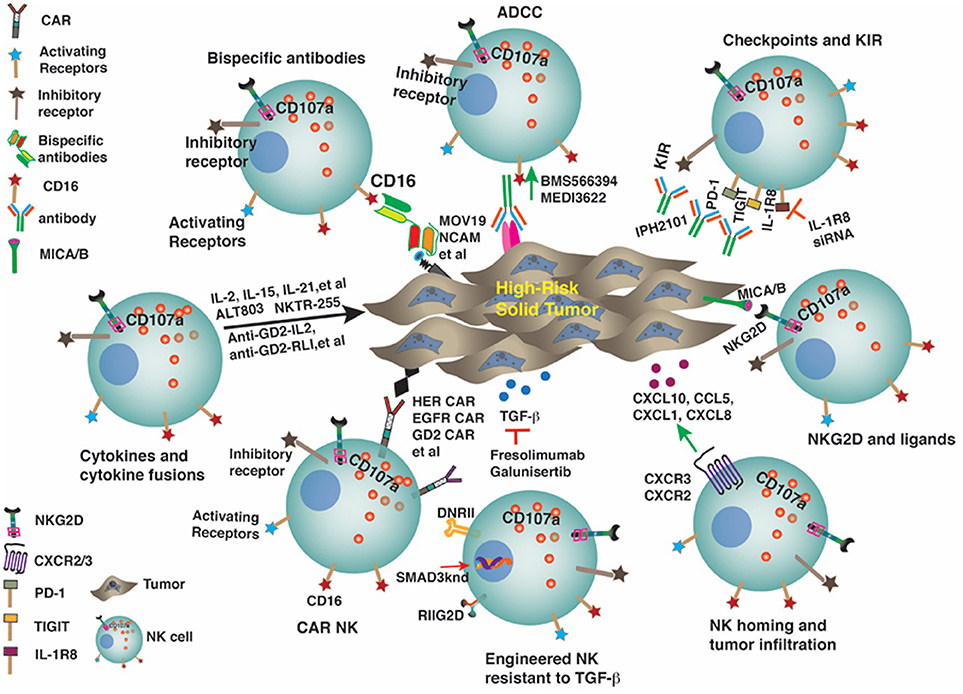
Figure 5. Strategies to overcome NK resistance in solid tumors. To enhance targeting specificity, NK cells have been engineered to express CAR such as anti-HER2 CAR, anti-EGFR CAR, anti-GD2 CAR et al. to target a specific antigen on tumor cell surface. NK cells can be activated by cytokines and cytokine fusion proteins such as IL-2, IL-15, IL-12, IL-18, IL-21, ALT-803 (an IL-15 superagonist), NKTR-255 (a polymer-engineered IL-15 molecule), anti-GD2-IL2, and anti-GD2-RLI fusions et al. Bispecific antibodies are novel molecules where two antigens can be targeted at the same time by combining the specificities of two antibodies. Bispecific antibodies can enhance NK cells targeting and killing. Preventing CD16 shedding and expressing high affinity CD16 on NK cells combined with novel engineered humanized antibodies will enhance NK mediated ADCC. The inhibitory roles of checkpoint proteins PD-1, TIGIT, IL-1R8, and KIR on NK cells are well documented. Blocking PD-1, TIGIT, and KIR with specific antibodies or knocking down IL-1R8 in NK cells unleash human NK-cell effector function. The membrane-bound ligands such as MICA/B binding to NKG2D play an important role in NK cell activation and tumor immune surveillance. Therapeutic strategies have focused on enhancing NKG2D expression and signaling on NK cells and enhancing the level of membrane-bound NKG2D ligand on tumor cells; and eliminating soluble NKG2D ligands. To enhance NK homing and tumor infiltration, NK cells can be enhanced to express chemokine receptors such as CXCR3, CXCR2 to be attracted to tumor cells that secret CXCL10, CXCL1, CXCL8, or CCL5. TGF-β plays an immunosuppressive effect of on NK cells. The approaches to block TGF-β and inhibit TGF-β pathway including TGF-β neutralizing antibody, TGF-β receptor I kinase inhibitors, SMAD3-Silenced (Smad3knd) NK Cells, NK cells engineered with a dominant negative receptor II for TGF-β (DNRII), NK cells engineered to express a chimeric receptor with TGF-β type II receptor extracellular and transmembrane domains and the intracellular domain of NK cell-activating receptor NKG2D (RIIG2D).
Author Contributions
GN and YC reviewed the literatures, developed the designed for the paper, wrote the manuscript and contributed equally. MC reviewed and approved the final manuscript.
Funding
This manuscript was supported by the grant from Department of Defense (W81XWH-17-1-0421, PI: MC), Pediatric Cancer Research Foundation (PI: MC), and New York Medical College (NYMC) School of Medicine (SOM) Translational Science Institute (TSI) Children Health Translational Research Award (PI: YC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Erin Morris, RN and Virginia Davenport, RN for their excellent assistance with the preparation of this manuscript.
Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; aNK, activated natural killer; CAR, chimeric antigen receptors; CEA, carcinoembryonic antigen; CB, cord blood; EGFR, epidermal growth factor receptor; EPCAM, epithelial cell adhesion molecule; FBP, folate binding protein; HER2, human epidermal growth factor receptor 2; HLA, human leucocyte antigen; HSCT, hematopoietic stem cell transplant; Hu, humanized; IgG, immunoglobulin g; IL, interleukin; IL1R, interleukin-1 receptor; INF-γ, interferon gamma; KIR, killer cell receptor; mAB, monoclonal antibody; MHC1, major histology antibody complex 1; MICA/B, MHC class I chain-related protein A/B; mRNA, messenger ribonucleic acid; NCAM, neural cell adhesion molecule; NK, natural killer cell; NKG2A, natural killer cell lectin-like receptor subfamily A; NKG2D, natural killer group protein 2 family member D; NKT, natural killer T cell; NSCLC, small lung cell carcinoma; PBMC, peripheral blood mononuclear cells; PD-1, programmed cell death 1; PDFGR, platelet-derived growth factor receptor; RCC, renal cell carcinoma; SABR, stereotactic ablative body radiotherapy; SCFv, single chain fragment variable region; SCT, stem cell transplantation; STAT, signal transducer and activator of transcription, TGF-β, transforming growth factor beta; TME, tumor microenviroment; TNF, tumor necrosis factor; Tregs, regulatory T cells; Trikes, trispecific killer cell engagers.
References
1. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
3. Punt CJ, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. (2017) 14:235–46. doi: 10.1038/nrclinonc.2016.171
4. Gras Navarro A, Bjorklund AT, Chekenya M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front Immunol. (2015) 6:202. doi: 10.3389/fimmu.2015.00202
5. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. (2011) 121:3609–22. doi: 10.1172/JCI45816
6. Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. (2013) 73:3499–510. doi: 10.1158/0008-5472.CAN-13-0371
7. Takanami I, Takeuchi K, Giga M. The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J Thorac Cardiovasc Surg. (2001) 121:1058–63. doi: 10.1067/mtc.2001.113026
8. Murray S, Lundqvist A. Targeting the tumor microenvironment to improve natural killer cell-based immunotherapies: on being in the right place at the right time, with resilience. Hum Vaccin Immunother. (2016) 12:607–11. doi: 10.1080/21645515.2015.1096458
9. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. (2014) 44:1582–92. doi: 10.1002/eji.201344272
10. Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. (2000) 35:71–103. doi: 10.1080/10409230091169186
11. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (2015) 3:83–92. doi: 10.2147/HP.S93413
12. Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist (2004) 9(Suppl. 5):10–7. doi: 10.1634/theoncologist.9-90005-10
13. Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric antigen receptors T cell therapy in solid tumor: challenges and clinical applications. Front Immunol. (2017) 8:1850. doi: 10.3389/fimmu.2017.01850
14. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. (2018) 9:282. doi: 10.1038/s41419-018-0278-6
15. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics (2016) 3:16006. doi: 10.1038/mto.2016.6
16. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
17. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295:2097–100. doi: 10.1126/science.1068440
18. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia (2018) 32:520–31. doi: 10.1038/leu.2017.226
19. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. (1975) 5:112–7. doi: 10.1002/eji.1830050208
20. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer (1975) 16:230–9. doi: 10.1002/ijc.2910160205
21. Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, et al. Human CD56bright NK cells: an update. J Immunol. (2016) 196:2923–31. doi: 10.4049/jimmunol.1502570
22. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
23. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. (2001) 22:633–40. doi: 10.1016/S1471-4906(01)02060-9
24. Nagler A, Lanier LL, Phillips JH. Constitutive expression of high affinity interleukin 2 receptors on human CD16-natural killer cells in vivo. J Exp Med. (1990) 171:1527–33. doi: 10.1084/jem.171.5.1527
25. Caligiuri MA, Murray C, Robertson MJ, Wang E, Cochran K, Cameron C, et al. Selective modulation of human natural killer cells in vivo after prolonged infusion of low dose recombinant interleukin 2. J Clin Invest. (1993) 91:123–32. doi: 10.1172/JCI116161
26. Frey M, Packianathan NB, Fehniger TA, Ross ME, Wang WC, Stewart CC, et al. Differential expression and function of L-selectin on CD56bright and CD56dim natural killer cell subsets. J Immunol. (1998) 161:400–8.
27. Ferlazzo G, Thomas D, Lin SL, Goodman K, Morandi B, Muller WA, et al. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J Immunol. (2004) 172:1455–62. doi: 10.4049/jimmunol.172.3.1455
28. Crinier A, Milpied P, Escaliere B, Piperoglou C, Galluso J, Balsamo A, et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity (2018) 49:971–86 e5. doi: 10.1016/j.immuni.2018.09.009
29. Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. (2015) 6:578. doi: 10.3389/fimmu.2015.00578
30. Cooley S, Parham P, Miller JS. Strategies to activate NK cells to prevent relapse and induce remission following hematopoietic stem cell transplantation. Blood (2018) 131:1053–62. doi: 10.1182/blood-2017-08-752170
31. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. (2016) 17:1025–36. doi: 10.1038/ni.3518
32. Bradley M, Zeytun A, Rafi-Janajreh A, Nagarkatti PS, Nagarkatti M. Role of spontaneous and interleukin-2-induced natural killer cell activity in the cytotoxicity and rejection of Fas+ and Fas- tumor cells. Blood (1998) 92:4248–55.
33. Kayagaki N, Yamaguchi N, Nakayama M, Takeda K, Akiba H, Tsutsui H, et al. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J Immunol. (1999) 163:1906–13.
34. Dunbar EM, Buzzeo MP, Levine JB, Schold JD, Meier-Kriesche HU, Reddy V. The relationship between circulating natural killer cells after reduced intensity conditioning hematopoietic stem cell transplantation and relapse-free survival and graft-versus-host disease. Haematologica (2008) 93:1852–8. doi: 10.3324/haematol.13033
35. Savani BN, Mielke S, Adams S, Uribe M, Rezvani K, Yong AS, et al. Rapid natural killer cell recovery determines outcome after T-cell-depleted HLA-identical stem cell transplantation in patients with myeloid leukemias but not with acute lymphoblastic leukemia. Leukemia (2007) 21:2145–52. doi: 10.1038/sj.leu.2404892
36. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. (2011) 17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347
37. Konjevic G, Jurisic V, Jovic V, Vuletic A, Mirjacic Martinovic K, Radenkovic S, et al. Investigation of NK cell function and their modulation in different malignancies. Immunol Res. (2012) 52:139–56. doi: 10.1007/s12026-012-8285-7
38. Tarle M, Kraljic I, Kastelan M. Comparison between NK cell activity and prostate cancer stage and grade in untreated patients: correlation with tumor markers and hormonal serotest data. Urol Res. (1993) 21:17–21. doi: 10.1007/BF00295186
39. Barth MJ, Chu Y, Hanley PJ, Cairo MS. Immunotherapeutic approaches for the treatment of childhood, adolescent and young adult non-Hodgkin lymphoma. Br J Haematol. (2016) 173:597–616. doi: 10.1111/bjh.14078
40. Stojanovic A, Cerwenka A. Natural killer cells and solid tumors. J Innate Immun. (2011) 3:355–64. doi: 10.1159/000325465
41. Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. (2014) 4:522–6. doi: 10.1158/2159-8290.CD-13-0985
42. Xia AL, Wang XC, Lu YJ, Lu XJ, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget (2017) 8:90521–31. doi: 10.18632/oncotarget.19361
43. Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H, et al. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res. (2008) 68:4746–53. doi: 10.1158/0008-5472.CAN-08-0054
44. Igarashi T, Wynberg J, Srinivasan R, Becknell B, McCoy JP Jr, Takahashi Y, et al. Enhanced cytotoxicity of allogeneic NK cells with killer immunoglobulin-like receptor ligand incompatibility against melanoma and renal cell carcinoma cells. Blood (2004) 104:170–7. doi: 10.1182/blood-2003-12-4438
45. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood (2005) 106:376–83. doi: 10.1182/blood-2004-12-4797
46. Ayello J, van de Ven C, Fortino W, Wade-Harris C, Satwani P, Baxi L, et al. Characterization of cord blood natural killer and lymphokine activated killer lymphocytes following ex vivo cellular engineering. Biol Blood Marrow Transplant (2006) 12:608–22. doi: 10.1016/j.bbmt.2006.01.009
47. Robinson KL, Ayello J, Hughes R, van de Ven C, Issitt L, Kurtzberg J, et al. Ex vivo expansion, maturation, and activation of umbilical cord blood-derived T lymphocytes with IL-2, IL-12, anti-CD3, and IL-7. Potential for adoptive cellular immunotherapy post-umbilical cord blood transplantation. Exp Hematol. (2002) 30:245–51. doi: 10.1016/S0301-472X(01)00781-0
48. Ayello J, van de Ven C, Cairo E, Hochberg J, Baxi L, Satwani P, et al. Characterization of natural killer and natural killer-like T cells derived from ex vivo expanded and activated cord blood mononuclear cells: implications for adoptive cellular immunotherapy. Exp Hematol. (2009) 37:1216–29. doi: 10.1016/j.exphem.2009.07.009
49. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE (2012) 7:e30264. doi: 10.1371/journal.pone.0030264
50. Lee HR, Son CH, Koh EK, Bae JH, Kang CD, Yang K, et al. Expansion of cytotoxic natural killer cells using irradiated autologous peripheral blood mononuclear cells and anti-CD16 antibody. Sci Rep. (2017) 7:11075. doi: 10.1038/s41598-017-09259-1
51. Yang Y, Lim O, Kim TM, Ahn YO, Choi H, Chung H, et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol Res. (2016) 4:215–24. doi: 10.1158/2326-6066.CIR-15-0118
52. Kozlowska AK, Kaur K, Topchyan P, Jewett A. Adoptive transfer of osteoclast-expanded natural killer cells for immunotherapy targeting cancer stem-like cells in humanized mice. Cancer Immunol Immunother. (2016) 65:835–45. doi: 10.1007/s00262-016-1822-9
53. Kaur K, Cook J, Park SH, Topchyan P, Kozlowska A, Ohanian N, et al. Novel strategy to expand super-charged NK cells with significant potential to lyse and differentiate cancer stem cells: differences in NK expansion and function between healthy and cancer patients. Front Immunol. (2017) 8:297. doi: 10.3389/fimmu.2017.00297
54. Kaur K, Topchyan P, Kozlowska AK, Ohanian N, Chiang J, Maung PO, et al. Super-charged NK cells inhibit growth and progression of stem-like/poorly differentiated oral tumors in vivo in humanized BLT mice; effect on tumor differentiation and response to chemotherapeutic drugs. Oncoimmunology (2018) 7:e1426518. doi: 10.1080/2162402X.2018.1426518
55. Lee YH. Is cord blood worth saving for public or private banking? Blood Res. (2015) 50:3–4. doi: 10.5045/br.2015.50.1.3
56. Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJ, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS ONE (2013) 8:e76781. doi: 10.1371/journal.pone.0076781
57. Cany J, van der Waart AB, Spanholtz J, Tordoir M, Jansen JH, van der Voort R, et al. Combined IL-15 and IL-12 drives the generation of CD34(+)-derived natural killer cells with superior maturation and alloreactivity potential following adoptive transfer. Oncoimmunology (2015) 4:e1017701. doi: 10.1080/2162402X.2015.1017701
58. Spanholtz J, Tordoir M, Eissens D, Preijers F, van der Meer A, Joosten I, et al. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS ONE (2010) 5:e9221. doi: 10.1371/journal.pone.0009221
59. Hoogstad-van Evert JS, Cany J, van den Brand D, Oudenampsen M, Brock R, Torensma R, et al. Umbilical cord blood CD34(+) progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rg(null) mice. Oncoimmunology (2017) 6:e1320630. doi: 10.1080/2162402X.2017.1320630
60. Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, et al. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. (2017) 8:533. doi: 10.3389/fimmu.2017.00533
61. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy (2008) 10:625–32. doi: 10.1080/14653240802301872
62. Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy - advantages of the NK-92 cell line over blood NK cells. Front Immunol. (2016) 7:91. doi: 10.3389/fimmu.2016.00091
63. Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood (2009) 113:6094–101. doi: 10.1182/blood-2008-06-165225
64. Hermanson DL, Bendzick L, Pribyl L, McCullar V, Vogel RI, Miller JS, et al. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells (2016) 34:93–101. doi: 10.1002/stem.2230
65. Zeng J, Tang SY, Toh LL, Wang S. Generation of “Off-the-Shelf” natural killer cells from peripheral blood cell-derived induced pluripotent stem cells. Stem Cell Rep. (2017) 9:1796–812. doi: 10.1016/j.stemcr.2017.10.020
66. Sim GC, Radvanyi L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. (2014) 25:377–90. doi: 10.1016/j.cytogfr.2014.07.018
67. Wu Y, Tian Z, Wei H. Developmental and functional control of natural killer cells by cytokines. Front Immunol. (2017) 8:930. doi: 10.3389/fimmu.2017.00930
68. Childs RW, Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov. (2015) 14:487–98. doi: 10.1038/nrd4506
69. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. (1985) 313:1485–92. doi: 10.1056/NEJM198512053132327
70. Rosenberg SA, Lotze MT, Yang JC, Topalian SL, Chang AE, Schwartzentruber DJ, et al. Prospective randomized trial of high-dose interleukin-2 alone or in conjunction with lymphokine-activated killer cells for the treatment of patients with advanced cancer. J Natl Cancer Inst. (1993) 85:622–32. doi: 10.1093/jnci/85.8.622
71. Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med. (2015) 13:277. doi: 10.1186/s12967-015-0632-8
72. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant (2003) 32:177–86. doi: 10.1038/sj.bmt.1704086
73. Mundy-Bosse B, Kathleen M, Mao C, Ahmed E, Chen L, Scoville SD, et al. Acute myeloid leukemia alters natural killer cell maturation and functional activation. Blood (2014) 124:754.
74. Rodella L, Zamai L, Rezzani R, Artico M, Peri G, Falconi M, et al. Interleukin 2 and interleukin 15 differentially predispose natural killer cells to apoptosis mediated by endothelial and tumour cells. Br J Haematol. (2001) 115:442–50. doi: 10.1046/j.1365-2141.2001.03055.x
75. Shaffer BC, Le Luduec JB, Forlenza C, Jakubowski AA, Perales MA, Young JW, et al. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2016) 22:705–9. doi: 10.1016/j.bbmt.2015.12.028
76. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood (2014) 123:3855–63. doi: 10.1182/blood-2013-10-532531
77. Curti A, Ruggeri L, D'Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118:3273–9. doi: 10.1182/blood-2011-01-329508
78. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy (2013) 15:1563–70. doi: 10.1016/j.jcyt.2013.06.017
79. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13:98–107. doi: 10.3109/14653249.2010.515582
80. Grossenbacher SK, Aguilar EG, Murphy WJ. Leveraging natural killer cells for cancer immunotherapy. Immunotherapy (2017) 9:487–97. doi: 10.2217/imt-2017-0013
81. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 'superkine'. Nature (2012) 484:529–U159. doi: 10.1038/nature10975
82. Ghasemi R, Lazear E, Wang X, Arefanian S, Zheleznyak A, Carreno BM, et al. Selective targeting of IL-2 to NKG2D bearing cells for improved immunotherapy. Nat Commun. (2016) 7:12878. doi: 10.1038/ncomms12878
83. Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol. (2017) 31:37–54. doi: 10.1016/j.smim.2017.07.009
84. Mortier E, Woo T, Advincula R, Gozalo S, Ma A. IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J Exp Med. (2008) 205:1213–25. doi: 10.1084/jem.20071913
85. Ochoa MC, Mazzolini G, Hervas-Stubbs S, de Sanmamed MF, Berraondo P, Melero I. Interleukin-15 in gene therapy of cancer. Curr Gene Ther. (2013) 13:15–30. doi: 10.2174/156652313804806561
86. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. (2000) 191:771–80. doi: 10.1084/jem.191.5.771
87. Sun R, Fan J, Wei H, Zhang C, Tian Z. Use of interleukin-15 for preparation of adherent NK cells from human peripheral blood: comparison with interleukin-2. J Immunol Methods (2003) 279:79–90. doi: 10.1016/S0022-1759(03)00245-X
88. Pillet AH, Theze J, Rose T. Interleukin (IL)-2 and IL-15 have different effects on human natural killer lymphocytes. Hum Immunol. (2011) 72:1013–7. doi: 10.1016/j.humimm.2011.07.311
89. Evans R, Fuller JA, Christianson G, Krupke DM, Troutt AB. IL-15 mediates anti-tumor effects after cyclophosphamide injection of tumor-bearing mice and enhances adoptive immunotherapy: the potential role of NK cell subpopulations. Cell Immunol. (1997) 179:66–73. doi: 10.1006/cimm.1997.1132
90. Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity (2007) 26:798–811. doi: 10.1016/j.immuni.2007.04.010
91. Mao Y, van Hoef V, Zhang X, Wennerberg E, Lorent J, Witt K, et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood (2016) 128:1475–89. doi: 10.1182/blood-2016-02-698027
92. Cao S, Troutt AB, Rustum YM. Interleukin 15 protects against toxicity and potentiates antitumor activity of 5-fluorouracil alone and in combination with leucovorin in rats bearing colorectal cancer. Cancer Res. (1998) 58:1695–9.
93. Jakobisiak M, Golab J, Lasek W. Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine Growth Factor Rev. (2011) 22:99–108. doi: 10.1016/j.cytogfr.2011.04.001
94. Zhang M, Yao Z, Dubois S, Ju W, Muller JR, Waldmann TA. Interleukin-15 combined with an anti-CD40 antibody provides enhanced therapeutic efficacy for murine models of colon cancer. Proc Natl Acad Sci USA. (2009) 106:7513–8. doi: 10.1073/pnas.0902637106
95. Porrata LF, Inwards DJ, Micallef IN, Johnston PB, Ansell SM, Hogan WJ, et al. Interleukin-15 affects patient survival through natural killer cell recovery after autologous hematopoietic stem cell transplantation for non-Hodgkin lymphomas. Clin Dev Immunol. (2010) 2010:914945. doi: 10.1155/2010/914945
96. Cooley S, Verneris MR, Curtsinger J, McKenna D, Weisdorf DJ, Blazar BR, et al. Recombinant human IL-15 promotes in vivo expansion of adoptively transferred NK cells in a first-in-human phase I dose escalation study in patients with AML. Blood (2012) 120:894.
97. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
98. Iliopoulou EG, Kountourakis P, Karamouzis MV, Doufexis D, Ardavanis A, Baxevanis CN, et al. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol Immunother. (2010) 59:1781–9. doi: 10.1007/s00262-010-0904-3
99. Perez-Martinez A, Fernandez L, Valentin J, Martinez-Romera I, Corral MD, Ramirez M, et al. A phase I/II trial of interleukin-15-stimulated natural killer cell infusion after haplo-identical stem cell transplantation for pediatric refractory solid tumors. Cytotherapy (2015) 17:1594–603. doi: 10.1016/j.jcyt.2015.07.011
100. Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. (2008) 26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316
101. Liu Z, Yang L, Cui Y, Wang X, Guo C, Huang Z, et al. Il-21 enhances NK cell activation and cytolytic activity and induces Th17 cell differentiation in inflammatory bowel disease. Inflamm Bowel Dis. (2009) 15:1133–44. doi: 10.1002/ibd.20923
102. Nakou M, Papadimitraki ED, Fanouriakis A, Bertsias GK, Choulaki C, Goulidaki N, et al. Interleukin-21 is increased in active systemic lupus erythematosus patients and contributes to the generation of plasma B cells. Clin Exp Rheumatol. (2013) 31:172–9.
103. Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The molecular basis of IL-21-mediated proliferation. Blood (2007) 109:4135–42. doi: 10.1182/blood-2006-10-054973
104. Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature (2000) 408:57–63. doi: 10.1038/35040504
105. Wang G, Tschoi M, Spolski R, Lou Y, Ozaki K, Feng C, et al. In vivo antitumor activity of interleukin 21 mediated by natural killer cells. Cancer Res. (2003) 63:9016–22.
106. Ma HL, Whitters MJ, Konz RF, Senices M, Young DA, Grusby MJ, et al. IL-21 activates both innate and adaptive immunity to generate potent antitumor responses that require perforin but are independent of IFN-gamma. J Immunol. (2003) 171:608–15. doi: 10.4049/jimmunol.171.2.608
107. Nakano H, Kishida T, Asada H, Shin-Ya M, Shinomiya T, Imanishi J, et al. Interleukin-21 triggers both cellular and humoral immune responses leading to therapeutic antitumor effects against head and neck squamous cell carcinoma. J Gene Med. (2006) 8:90–9. doi: 10.1002/jgm.817
108. Thompson JA, Curti BD, Redman BG, Bhatia S, Weber JS, Agarwala SS, et al. Phase I study of recombinant interleukin-21 in patients with metastatic melanoma and renal cell carcinoma. J Clin Oncol. (2008) 26:2034–9. doi: 10.1200/JCO.2007.14.5193
109. Petrella TM, Tozer R, Belanger K, Savage KJ, Wong R, Smylie M, et al. Interleukin-21 has activity in patients with metastatic melanoma: a phase II study. J Clin Oncol. (2012) 30:3396–401. doi: 10.1200/JCO.2011.40.0655
110. Grunwald V, Desar IM, Haanen J, Fiedler W, Mouritzen U, Olsen MW, et al. A phase I study of recombinant human interleukin-21 (rIL-21) in combination with sunitinib in patients with metastatic renal cell carcinoma (RCC). Acta Oncol. (2011) 50:121–6. doi: 10.3109/0284186X.2010.509104
111. Bhatia S, Curti B, Ernstoff MS, Gordon M, Heath EI, Miller WH Jr, et al. Recombinant interleukin-21 plus sorafenib for metastatic renal cell carcinoma: a phase 1/2 study. J Immunother Cancer (2014) 2:2. doi: 10.1186/2051-1426-2-2
112. Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol. (2000) 165:2665–70. doi: 10.4049/jimmunol.165.5.2665
113. Zeh HJ 3rd, Hurd S, Storkus WJ, Lotze MT. Interleukin-12 promotes the proliferation and cytolytic maturation of immune effectors: implications for the immunotherapy of cancer. J Immunother Emphasis Tumor Immunol. (1993) 14:155–61. doi: 10.1097/00002371-199308000-00012
114. Parihar R, Dierksheide J, Hu Y, Carson WE. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J Clin Invest. (2002) 110:983–92. doi: 10.1172/JCI0215950
115. Sahin U, Kraft-Bauer S, Ohnesorge S, Pfreundschuh M, Renner C. Interleukin-12 increases bispecific-antibody-mediated natural killer cell cytotoxicity against human tumors. Cancer Immunol Immunother. (1996) 42:9–14. doi: 10.1007/s002620050245
116. Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, et al. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med. (1993) 178:1223–30. doi: 10.1084/jem.178.4.1223
117. Parhar RS, Zou M, Al-Mohanna FA, Baitei EY, Assiri AM, Meyer BF, et al. IL-12 immunotherapy of Braf(V600E)-induced papillary thyroid cancer in a mouse model. Lab Invest. (2016) 96:89–97. doi: 10.1038/labinvest.2015.126
118. Teicher BA, Ara G, Buxton D, Leonard J, Schaub RG. Optimal scheduling of interleukin 12 and chemotherapy in the murine MB-49 bladder carcinoma and B16 melanoma. Clin Cancer Res. (1997) 3:1661–7.
119. Zou JP, Yamamoto N, Fujii T, Takenaka H, Kobayashi M, Herrmann SH, et al. Systemic administration of rIL-12 induces complete tumor regression and protective immunity: response is correlated with a striking reversal of suppressed IFN-gamma production by anti-tumor T cells. Int Immunol. (1995) 7:1135–45. doi: 10.1093/intimm/7.7.1135
120. Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res. (1997) 3:409–17.
121. Gollob JA, Mier JW, Veenstra K, McDermott DF, Clancy D, Clancy M, et al. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-gamma induction is associated with clinical response. Clin Cancer Res. (2000) 6:1678–92.
122. van Herpen CM, van der Voort R, van der Laak JA, Klasen IS, de Graaf AO, van Kempen LC, et al. Intratumoral rhIL-12 administration in head and neck squamous cell carcinoma patients induces B cell activation. Int J Cancer (2008) 123:2354–61. doi: 10.1002/ijc.23756
123. Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK, et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol. (2008) 26:5896–903. doi: 10.1200/JCO.2007.15.6794
124. Alvarez RD, Sill MW, Davidson SA, Muller CY, Bender DP, DeBernardo RL, et al. A phase II trial of intraperitoneal EGEN-001, an IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: a gynecologic oncology group study. Gynecol Oncol. (2014) 133:433–8. doi: 10.1016/j.ygyno.2014.03.571
125. Rook AH, Wood GS, Yoo EK, Elenitsas R, Kao DM, Sherman ML, et al. Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood (1999) 94:902–8.
126. Little RF, Pluda JM, Wyvill KM, Rodriguez-Chavez IR, Tosato G, Catanzaro AT, et al. Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood (2006) 107:4650–7. doi: 10.1182/blood-2005-11-4455
127. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature (1997) 386:619–23. doi: 10.1038/386619a0
128. Haeberlein S, Sebald H, Bogdan C, Schleicher U. IL-18, but not IL-15, contributes to the IL-12-dependent induction of NK-cell effector functions by Leishmania infantum in vivo. Eur J Immunol. (2010) 40:1708–17. doi: 10.1002/eji.200939988
129. Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity (1998) 8:383–90. doi: 10.1016/S1074-7613(00)80543-9
130. Hodge DL, Subleski JJ, Reynolds DA, Buschman MD, Schill WB, Burkett MW, et al. The proinflammatory cytokine interleukin-18 alters multiple signaling pathways to inhibit natural killer cell death. J Interferon Cytokine Res. (2006) 26:706–18. doi: 10.1089/jir.2006.26.706
131. Srivastava S, Pelloso D, Feng H, Voiles L, Lewis D, Haskova Z, et al. Effects of interleukin-18 on natural killer cells: costimulation of activation through Fc receptors for immunoglobulin. Cancer Immunol Immunother. (2013) 62:1073–82. doi: 10.1007/s00262-013-1403-0
132. Yang C, Cao H, Liu N, Xu K, Ding M, Mao LJ. Oncolytic adenovirus expressing interleukin-18 improves antitumor activity of dacarbazine for malignant melanoma. Drug Des Devel Ther. (2016) 10:3755–61. doi: 10.2147/DDDT.S115121
133. Zheng JN, Pei DS, Mao LJ, Liu XY, Sun FH, Zhang BF, et al. Oncolytic adenovirus expressing interleukin-18 induces significant antitumor effects against melanoma in mice through inhibition of angiogenesis. Cancer Gene Ther. (2010) 17:28–36. doi: 10.1038/cgt.2009.38
134. Robertson MJ, Kirkwood JM, Logan TF, Koch KM, Kathman S, Kirby LC, et al. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. (2008) 14:3462–9. doi: 10.1158/1078-0432.CCR-07-4740
135. Simpkins F, Flores A, Chu C, Berek JS, Lucci J 3rd, Murray S, et al. Chemoimmunotherapy using pegylated liposomal Doxorubicin and interleukin-18 in recurrent ovarian cancer: a phase I dose-escalation study. Cancer Immunol Res. (2013) 1:168–78. doi: 10.1158/2326-6066.CIR-13-0098
136. Tarhini AA, Millward M, Mainwaring P, Kefford R, Logan T, Pavlick A, et al. A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer (2009) 115:859–68. doi: 10.1002/cncr.24100
137. Zhu X, Marcus WD, Xu W, Lee HI, Han K, Egan JO, et al. Novel human interleukin-15 agonists. J Immunol. (2009) 183:3598–607. doi: 10.4049/jimmunol.0901244
138. Han KP, Zhu XY, Liu B, Jeng E, Kong L, Yovandich JL, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine (2011) 56:804–10. doi: 10.1016/j.cyto.2011.09.028
139. Xu W, Jones M, Liu B, Zhu X, Johnson CB, Edwards AC, et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res. (2013) 73:3075–86. doi: 10.1158/0008-5472.CAN-12-2357
140. Gomes-Giacoia E, Miyake M, Goodison S, Sriharan A, Zhang G, You L, et al. Intravesical ALT-803 and BCG treatment reduces tumor burden in a carcinogen induced bladder cancer rat model; a role for cytokine production and NK cell expansion. PLoS ONE (2014) 9:e96705. doi: 10.1371/journal.pone.0096705
141. Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, et al. The IL-15-based ALT-803 complex enhances FcgammaRIIIa-triggered NK cell responses and in vivo clearance of B cell lymphomas. Clin Cancer Res. (2016) 22:596–608. doi: 10.1158/1078-0432.CCR-15-1419
142. Mathios D, Park CK, Marcus WD, Alter S, Rhode PR, Jeng EK, et al. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int J Cancer (2016) 138:187–94. doi: 10.1002/ijc.29686
143. Kim PS, Kwilas AR, Xu WX, Alter S, Jeng EK, Wong HC, et al. IL-15 superagonist/IL-15R alpha Sushi-Fc fusion complex (IL-15SA/IL-15R alpha Su-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8(+) T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget (2016) 7:16130–45. doi: 10.18632/oncotarget.7470
144. Felices M, Chu S, Kodal B, Bendzick L, Ryan C, Lenvik AJ, et al. IL-15 super-agonist (ALT-803) enhances natural killer (NK) cell function against ovarian cancer. Gynecol Oncol. (2017) 145:453–61. doi: 10.1016/j.ygyno.2017.02.028
145. Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood (2018) 131:2515–27. doi: 10.1182/blood-2017-12-823757
146. Margolin K, Morishima C, Velcheti V, Miller JS, Lee SM, Silk AW, et al. Phase I trial of ALT-803, a novel recombinant IL15 complex, in patients with advanced solid tumors. Clin Cancer Res. (2018) 24:5552–61. doi: 10.1158/1078-0432.CCR-18-0945
147. Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. (2018) 19:694–704. doi: 10.1016/S1470-2045(18)30148-7
148. Kuo P, Maiti M, Quach P, Addepalli M, Lanka A, Mathamsetti P, et al. NKTR-255 engages the IL-15 pathway driving CD8 T cell survival and CD8 memory T cell proliferation. Cancer Research (2017) 77(13 Suppl.):1603. doi: 10.1158/1538-7445.AM2017-1603
149. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK Cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. (2015) 6:368. doi: 10.3389/fimmu.2015.00368
150. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. (2003) 21:3940–7. doi: 10.1200/JCO.2003.05.013
151. Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. (2007) 25:3712–8. doi: 10.1200/JCO.2006.08.8021
152. Borrok MJ, Luheshi NM, Beyaz N, Davies GC, Legg JW, Wu H, et al. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcalphaRI (CD89) binding. MAbs (2015) 7:743–51. doi: 10.1080/19420862.2015.1047570
153. Herter S, Herting F, Mundigl O, Waldhauer I, Weinzierl T, Fauti T, et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol Cancer Ther. (2013) 12:2031–42. doi: 10.1158/1535-7163.MCT-12-1182
154. Handgretinger R, Anderson K, Lang P, Dopfer R, Klingebiel T, Schrappe M, et al. A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur J Cancer (1995) 31A:261–7. doi: 10.1016/0959-8049(94)00413-Y
155. Grzywacz B, Kataria N, Verneris M4R. CD56(dim)CD16(+) NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia (2007) 21:356–9; author reply 9. doi: 10.1038/sj.leu.2404499
156. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood (2013) 121:3599–608. doi: 10.1182/blood-2012-04-425397
157. Voutsas IF, Mahaira LG, Fotopoulou K, Kapranos N, Reclos JG, Gritzapis AD, et al. Gamma-irradiation induces HER-2/neu overexpression in breast cancer cell lines and sensitivity to treatment with trastuzumab. Int J Radiat Biol. (2013) 89:319–25. doi: 10.3109/09553002.2013.765617
158. Jahrsdorfer B, Hartmann G, Racila E, Jackson W, Muhlenhoff L, Meinhardt G, et al. CpG DNA increases primary malignant B cell expression of costimulatory molecules and target antigens. J Leukoc Biol. (2001) 69:81–8. doi: 10.1189/jlb.69.1.81
159. Ravetch JV, Perussia B. Alternative membrane forms of Fc gamma RIII(CD16) on human natural killer cells and neutrophils. Cell type-specific expression of two genes that differ in single nucleotide substitutions. J Exp Med. (1989) 170:481–97. doi: 10.1084/jem.170.2.481
160. Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, et al. Identification of an ADAM17 cleavage region in human CD16 (FcgammaRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS ONE (2015) 10:e0121788. doi: 10.1371/journal.pone.0121788
161. Wang Y, Wu J, Newton R, Bahaie NS, Long C, Walcheck B. ADAM17 cleaves CD16b (FcgammaRIIIb) in human neutrophils. Biochim Biophys Acta (2013) 1833:680–5. doi: 10.1016/j.bbamcr.2012.11.027
162. Pham DH, Kim JS, Kim SK, Shin DJ, Uong NT, Hyun H, et al. Effects of ADAM10 and ADAM17 inhibitors on natural killer cell expansion and antibody-dependent cellular cytotoxicity against breast cancer cells in vitro. Anticancer Res. (2017) 37:5507–13. doi: 10.21873/anticanres.11981
163. Peng L, Cook K, Xu L, Cheng L, Damschroder M, Gao C, et al. Molecular basis for the mechanism of action of an anti-TACE antibody. MAbs (2016) 8:1598–605. doi: 10.1080/19420862.2016.1226716
164. Mishra HK, Pore N, Michelotti EF, Walcheck B. Anti-ADAM17 monoclonal antibody MEDI3622 increases IFNgamma production by human NK cells in the presence of antibody-bound tumor cells. Cancer Immunol Immunother. (2018) 67:1407–16. doi: 10.1007/s00262-018-2193-1
165. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM 2nd, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
166. Chen Y, You F, Jiang L, Li J, Zhu X, Bao Y, et al. Gene-modified NK-92MI cells expressing a chimeric CD16-BB-zeta or CD64-BB-zeta receptor exhibit enhanced cancer-killing ability in combination with therapeutic antibody. Oncotarget (2017) 8:37128–39. doi: 10.18632/oncotarget.16201
167. Gilman AL, Ozkaynak MF, Matthay KK, Krailo M, Yu AL, Gan J, et al. Phase I study of ch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: a report from the Children's Oncology Group. J Clin Oncol. (2009) 27:85–91. doi: 10.1200/JCO.2006.10.3564
168. Simon T, Hero B, Faldum A, Handgretinger R, Schrappe M, Niethammer D, et al. Consolidation treatment with chimeric anti-GD2-antibody ch14.18 in children older than 1 year with metastatic neuroblastoma. J Clin Oncol. (2004) 22:3549–57. doi: 10.1200/JCO.2004.08.143
169. Carnemolla B, Borsi L, Balza E, Castellani P, Meazza R, Berndt A, et al. Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood (2002) 99:1659–65. doi: 10.1182/blood.V99.5.1659
170. Halin C, Rondini S, Nilsson F, Berndt A, Kosmehl H, Zardi L, et al. Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat Biotechnol. (2002) 20:264–9. doi: 10.1038/nbt0302-264
171. Lode HN, Xiang R, Dreier T, Varki NM, Gillies SD, Reisfeld RA. Natural killer cell-mediated eradication of neuroblastoma metastases to bone marrow by targeted interleukin-2 therapy. Blood (1998) 91:1706–15.
172. Vincent M, Quemener A, Jacques Y. Antitumor activity of an immunocytokine composed of an anti-GD2 antibody and the IL-15 superagonist RLI. Oncoimmunology (2013) 2:e26441. doi: 10.4161/onci.26441
173. Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, et al. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget (2014) 5:1869–84. doi: 10.18632/oncotarget.1853
174. Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology (2017) 6:e1277306. doi: 10.1080/2162402X.2016.1277306
175. Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, et al. Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children's Oncology Group (COG) phase II study. J Clin Oncol. (2010) 28:4969–75. doi: 10.1200/JCO.2009.27.8861
176. Spitaleri G, Berardi R, Pierantoni C, De Pas T, Noberasco C, Libbra C, et al. Phase I/II study of the tumour-targeting human monoclonal antibody-cytokine fusion protein L19-TNF in patients with advanced solid tumours. J Cancer Res Clin Oncol. (2013) 139:447–55. doi: 10.1007/s00432-012-1327-7
177. Papadia F, Basso V, Patuzzo R, Maurichi A, Di Florio A, Zardi L, et al. Isolated limb perfusion with the tumor-targeting human monoclonal antibody-cytokine fusion protein L19-TNF plus melphalan and mild hyperthermia in patients with locally advanced extremity melanoma. J Surg Oncol. (2013) 107:173–9. doi: 10.1002/jso.23168
178. Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, et al. First-in-human phase I trial of a tumor-targeted cytokine (NHS-IL12) in subjects with metastatic solid tumors. Clin Cancer Res (2018) 25:99–109. doi: 10.1158/1078-0432.CCR-18-1512
179. Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood (2003) 102:814–9. doi: 10.1182/blood-2003-01-0091
180. Benson DM, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120:4324–33. doi: 10.1182/blood-2012-06-438028
181. Benson DM Jr, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res. (2015) 21:4055–61. doi: 10.1158/1078-0432.CCR-15-0304
182. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood (2014) 123:678–86. doi: 10.1182/blood-2013-08-519199
183. Vey N, Karlin L, Sadot-Lebouvier S, Broussais F, Berton-Rigaud D, Rey J, et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget (2018) 9:17675–88. doi: 10.18632/oncotarget.24832
184. He YY, Liu ST, Mattei J, Bunn PA, Zhou CC, Chan D. The combination of anti-KIR monoclonal antibodies with anti-PD-1/PD-L1 monoclonal antibodies could be a critical breakthrough in overcoming tumor immune escape in NSCLC. Drug Des Dev Ther. (2018) 12:981–6. doi: 10.2147/DDDT.S163304
185. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
186. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
187. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
188. Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, et al. Targeting CD20+ aggressive B-cell non-hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG Mice. Cancer Immunol Res. (2015) 3:333–44. doi: 10.1158/2326-6066.CIR-14-0114
189. Schonfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. (2015) 23:330–8. doi: 10.1038/mt.2014.219
190. Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. (2016) 108:djv375. doi: 10.1093/jnci/djv375
191. Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget (2016) 7:27764–77. doi: 10.18632/oncotarget.8526
192. Murakami T, Nakazawa T, Natsume A, Nishimura F, Nakamura M, Matsuda R, et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. (2018) 38:5049–56. doi: 10.21873/anticanres.12824
193. Esser R, Muller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. (2012) 16:569–81. doi: 10.1111/j.1582-4934.2011.01343.x
194. Sahm C, Schonfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. (2012) 61:1451–61. doi: 10.1007/s00262-012-1212-x
195. Park H, Awasthi A, Ayello J, Chu Y, Riddell S, Rosenblum J, et al. ROR1-specific chimeric antigen receptor (CAR) NK cell immunotherapy for high risk neuroblastomas and sarcomas. Biol Blood Marrow Transplant (2017) 23:S136–S7. doi: 10.1016/j.bbmt.2017.01.056
196. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. (2013) 73:1777–86. doi: 10.1158/0008-5472.CAN-12-3558
197. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell (2018) 23:181–92 e5.
198. Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. (2017) 376:836–47. doi: 10.1056/NEJMoa1609783
199. Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. (2016) 375:740–53. doi: 10.1056/NEJMoa1509277
200. Strohlein MA, Lordick F, Ruttinger D, Grutzner KU, Schemanski OC, Jager M, et al. Immunotherapy of peritoneal carcinomatosis with the antibody catumaxomab in colon, gastric, or pancreatic cancer: an open-label, multicenter, phase I/II trial. Onkologie (2011) 34:101–8. doi: 10.1159/000324667
201. Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood (2015) 125:4024–31. doi: 10.1182/blood-2014-12-614636
202. Lu H, Shi M, Wang M, Xie Z, Hu M, Yu M, et al. In vitro and in vivo antitumor effect of a trivalent bispecific antibody targeting ErbB2 and CD16. Cancer Biol Ther. (2008) 7:1744–50. doi: 10.4161/cbt.7.11.6725
203. Armstrong GT, Chen Y, Yasui Y, Leisenring W, Gibson TM, Mertens AC, et al. Reduction in late mortality among 5-year survivors of childhood cancer. N Engl J Med. (2016) 374:833–42. doi: 10.1056/NEJMoa1510795
204. Deng W, Liu J, Pan H, Li L, Zhou C, Wang X, et al. A bispecific antibody based on pertuzumab fab has potent antitumor activity. J Immunother. (2018) 41:1–8. doi: 10.1097/CJI.0000000000000200
205. Turini M, Chames P, Bruhns P, Baty D, Kerfelec B. A FcgammaRIII-engaging bispecific antibody expands the range of HER2-expressing breast tumors eligible to antibody therapy. Oncotarget (2014) 5:5304–19. doi: 10.18632/oncotarget.2093
206. Vallera DA, Zhang B, Gleason MK, Oh S, Weiner LM, Kaufman DS, et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother Radiopharm. (2013) 28:274–82. doi: 10.1089/cbr.2012.1329
207. Oberg HH, Kellner C, Gonnermann D, Sebens S, Bauerschlag D, Gramatzki M, et al. Tribody [(HER2)2xCD16] Is more effective than trastuzumab in enhancing gammadelta T cell and natural killer cell cytotoxicity against HER2-Expressing cancer cells. Front Immunol. (2018) 9:814. doi: 10.3389/fimmu.2018.00814
208. Schmohl JU, Felices M, Taras E, Miller JS, Vallera DA. Enhanced ADCC and NK cell activation of an anticarcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol Ther. (2016) 24:1312–22. doi: 10.1038/mt.2016.88
209. Ferrini S, Prigione I, Miotti S, Ciccone E, Cantoni C, Chen Q, et al. Bispecific monoclonal antibodies directed to CD16 and to a tumor-associated antigen induce target-cell lysis by resting NK cells and by a subset of NK clones. Int J Cancer (1991) 48:227–33. doi: 10.1002/ijc.2910480213
210. Ferrini S, Cambiaggi A, Cantoni C, Canevari S, Mezzanzanica D, Colnaghi MI, et al. Targeting of T or NK lymphocytes against tumor cells by bispecific monoclonal antibodies: role of different triggering molecules. Int J Cancer Suppl. (1992) 7:15–8.
211. Obukhov SK, Gennie MJ, Tutt AL, Kemshead JT, Coakham HB, Beverely PC. The cytotoxic action of lymphokine activated killer cells upon the human glioma cell line U251 is stimulated by bispecific monoclonal antibody (MoAb) constructs. J Neurooncol. (1992) 13:203–10. doi: 10.1007/BF00172471
212. Garcia de Palazzo I, Holmes M, Gercel-Taylor C, Weiner LM. Antitumor effects of a bispecific antibody targeting CA19–9 antigen and CD16. Cancer Res. (1992) 52:5713–9.
213. Weiner LM, Holmes M, Adams GP, LaCreta F, Watts P, Garcia de Palazzo I. A human tumor xenograft model of therapy with a bispecific monoclonal antibody targeting c-erbB-2 and CD16. Cancer Res. (1993) 53:94–100.
214. Ferrini S, Cambiaggi A, Sforzini S, Canevari S, Mezzanzanica D, Colnaghi MI, et al. Use of anti-CD3 and anti-CD16 bispecific monoclonal antibodies for the targeting of T and NK cells against tumor cells. Cancer Detect Prev. (1993) 17:295–300.
215. Weiner LM, Clark JI, Ring DB, Alpaugh RK. Clinical development of 2B1, a bispecific murine monoclonal antibody targeting c-erbB-2 and Fc gamma RIII. J Hematother. (1995) 4:453–6. doi: 10.1089/scd.1.1995.4.453
216. Weiner LM, Alpaugh RK, Amoroso AR, Adams GP, Ring DB, Barth MW. Human neutrophil interactions of a bispecific monoclonal antibody targeting tumor and human Fc gamma RIII. Cancer Immunol Immunother. (1996) 42:141–50. doi: 10.1007/s002620050264
217. McCall AM, Shahied L, Amoroso AR, Horak EM, Simmons HH, Nielson U, et al. Increasing the affinity for tumor antigen enhances bispecific antibody cytotoxicity. J Immunol. (2001) 166:6112–7. doi: 10.4049/jimmunol.166.10.6112
218. Gruel N, Fridman WH, Teillaud JL. Bypassing tumor-specific and bispecific antibodies: triggering of antitumor immunity by expression of anti-FcgammaR scFv on cancer cell surface. Gene Ther. (2001) 8:1721–8. doi: 10.1038/sj.gt.3301575
219. Xie Z, Shi M, Feng J, Yu M, Sun Y, Shen B, et al. A trivalent anti-erbB2/anti-CD16 bispecific antibody retargeting NK cells against human breast cancer cells. Biochem Biophys Res Commun. (2003) 311:307–12. doi: 10.1016/j.bbrc.2003.09.211
220. Shahied LS, Tang Y, Alpaugh RK, Somer R, Greenspon D, Weiner LM. Bispecific minibodies targeting HER2/neu and CD16 exhibit improved tumor lysis when placed in a divalent tumor antigen binding format. J Biol Chem. (2004) 279:53907–14. doi: 10.1074/jbc.M407888200
221. Asano R, Nakayama M, Kawaguchi H, Kubota T, Nakanishi T, Umetsu M, et al. Construction and humanization of a functional bispecific EGFR CD16 diabody using a refolding system. Febs J. (2012) 279:223–33. doi: 10.1111/j.1742-4658.2011.08417.x
222. Schmohl JU, Gleason MK, Dougherty PR, Miller JS, Vallera DA. Heterodimeric bispecific single chain variable fragments (scFv) killer engagers (BiKEs) enhance NK-cell activity against CD133+ colorectal cancer cells. Target Oncol. (2016) 11:353–61. doi: 10.1007/s11523-015-0391-8
223. Dong B, Zhou C, He P, Li J, Chen S, Miao J, et al. A novel bispecific antibody, BiSS, with potent anti-cancer activities. Cancer Biol Ther. (2016) 17:364–70. doi: 10.1080/15384047.2016.1139266
224. Schmohl JU, Felices M, Todhunter D, Taras E, Miller JS, Vallera DA. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget (2016) 7:73830–44. doi: 10.18632/oncotarget.12073
225. Schmohl JU, Felices M, Oh F, Lenvik AJ, Lebeau AM, Panyam J, et al. Engineering of Anti-CD133 trispecific molecule capable of inducing NK expansion and driving antibody-dependent cell-mediated cytotoxicity. Cancer Res Treat. (2017) 49:1140–52. doi: 10.4143/crt.2016.491
226. Li Y, Zhou C, Li J, Liu J, Lin L, Li L, et al. Single domain based bispecific antibody, Muc1-Bi-1, and its humanized form, Muc1-Bi-2, induce potent cancer cell killing in muc1 positive tumor cells. PLoS ONE (2018) 13:e0191024. doi: 10.1371/journal.pone.0191024
227. Wang Y, Liu J, Pan H, Xing J, Wu X, Li Q, et al. A GPC3-targeting bispecific antibody, GPC3-S-Fab, with potent cytotoxicity. J Vis Exp (2018) e57588. doi: 10.3791/57588
228. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
229. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
230. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
231. Kwon HJ, Kim N, Kim HS. Molecular checkpoints controlling natural killer cell activation and their modulation for cancer immunotherapy. Exp Mol Med. (2017) 49:e311. doi: 10.1038/emm.2017.42
232. Molgora M, Bonavita E, Ponzetta A, Riva F, Barbagallo M, Jaillon S, et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature (2017) 551:110–4. doi: 10.1038/nature24293
233. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
234. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol. (2017) 139:335–46 e3. doi: 10.1016/j.jaci.2016.04.025
235. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget (2016) 7:72961–77. doi: 10.18632/oncotarget.12150
236. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene (2017) 36:6143–53. doi: 10.1038/onc.2017.209
237. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA. (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
238. Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother. (2017) 66:1367–75. doi: 10.1007/s00262-017-2031-x
239. Stojanovic A, Cerwenka A. Checkpoint inhibition: NK cells enter the scene. Nat Immunol. (2018) 19:650–2. doi: 10.1038/s41590-018-0142-y
240. Molgora M, Barajon I, Mantovani A, Garlanda C. Regulatory role of IL-1R8 in immunity and disease. Front Immunol. (2016) 7:149. doi: 10.3389/fimmu.2016.00149
241. Lanier LL. NKG2D Receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
242. Gilfillan S, Ho EL, Cella M, Yokoyama WM, Colonna M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat Immunol. (2002) 3:1150–5. doi: 10.1038/ni857
243. Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol. (2001) 2:255–60. doi: 10.1038/85321
244. Tieng V, Le Bouguenec C, du Merle L, Bertheau P, Desreumaux P, Janin A, et al. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc Natl Acad Sci USA. (2002) 99:2977–82. doi: 10.1073/pnas.032668099
245. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “One for All, All for One”. Front Immunol. (2018) 9:476. doi: 10.3389/fimmu.2018.00476
246. Pende D, Cantoni C, Rivera P, Vitale M, Castriconi R, Marcenaro S, et al. Role of NKG2D in tumor cell lysis mediated by human NK cells: cooperation with natural cytotoxicity receptors and capability of recognizing tumors of nonepithelial origin. Eur J Immunol. (2001) 31:1076–86. doi: 10.1002/1521-4141(200104)31:4<1076::AID-IMMU1076>3.0.CO;2-Y
247. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285:727–9. doi: 10.1126/science.285.5428.727
248. Gonzalez S, Lopez-Soto A, Suarez-Alvarez B, Lopez-Vazquez A, Lopez-Larrea C. NKG2D ligands: key targets of the immune response. Trends Immunol. (2008) 29:397–403. doi: 10.1016/j.it.2008.04.007
249. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. (2003) 3:781–90. doi: 10.1038/nri1199
250. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest. (2013) 123:4410–22. doi: 10.1172/JCI69369
251. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia (2004) 6:558–68. doi: 10.1593/neo.04316
252. Obeidy P, Sharland AF. NKG2D and its ligands. Int J Biochem Cell Biol. (2009) 41:2364–7. doi: 10.1016/j.biocel.2009.07.005
253. Lanier LL. A renaissance for the tumor immunosurveillance hypothesis. Nat Med. (2001) 7:1178–80. doi: 10.1038/nm1101-1178
254. Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, et al. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. (2010) 70:481–9. doi: 10.1158/0008-5472.CAN-09-1688
255. Jinushi M, Takehara T, Tatsumi T, Hiramatsu N, Sakamori R, Yamaguchi S, et al. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J Hepatol. (2005) 43:1013–20. doi: 10.1016/j.jhep.2005.05.026
256. Song H, Kim J, Cosman D, Choi I. Soluble ULBP suppresses natural killer cell activity via down-regulating NKG2D expression. Cell Immunol. (2006) 239:22–30. doi: 10.1016/j.cellimm.2006.03.002
257. Li K, Mandai M, Hamanishi J, Matsumura N, Suzuki A, Yagi H, et al. Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: high expression of ULBP2 is an indicator of poor prognosis. Cancer Immunol Immunother. (2009) 58:641–52. doi: 10.1007/s00262-008-0585-3
258. Tamaki S, Sanefuzi N, Kawakami M, Aoki K, Imai Y, Yamanaka Y, et al. Association between soluble MICA levels and disease stage IV oral squamous cell carcinoma in Japanese patients. Hum Immunol. (2008) 69:88–93. doi: 10.1016/j.humimm.2008.01.010
259. Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, et al. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharm Res. (2015) 32:779–92. doi: 10.1007/s11095-013-1231-0
260. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. (2005) 65:6321–9. doi: 10.1158/0008-5472.CAN-04-4252
261. Arai J, Goto K, Tanoue Y, Ito S, Muroyama R, Matsubara Y, et al. Enzymatic inhibition of MICA sheddase ADAM17 by lomofungin in hepatocellular carcinoma cells. Int J Cancer (2018) 143:2575–83. doi: 10.1002/ijc.31615
262. Lu S, Zhang J, Liu D, Li G, Staveley-O'Carroll KF, Li Z, et al. Nonblocking monoclonal antibody targeting soluble MIC revamps endogenous innate and adaptive antitumor responses and eliminates primary and metastatic tumors. Clin Cancer Res. (2015) 21:4819–30. doi: 10.1158/1078-0432.CCR-15-0845
263. Chu Y, Yahr A, Huang B, Ayello J, Barth M, S Cairo M. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology (2017) 6:e1341031. doi: 10.1080/2162402X.2017.1341031
264. Satwani P, Bavishi S, Saha A, Zhao F, Ayello J, van de Ven C, et al. Upregulation of NKG2D ligands in acute lymphoblastic leukemia and non-Hodgkin lymphoma cells by romidepsin and enhanced in vitro and in vivo natural killer cell cytotoxicity. Cytotherapy (2014) 16:1431–40. doi: 10.1016/j.jcyt.2014.03.008
265. Liu G, Atteridge CL, Wang X, Lundgren AD, Wu JD. The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J Immunol. (2010) 184:3346–50. doi: 10.4049/jimmunol.0903789
266. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. (2008) 68:6368–76. doi: 10.1158/0008-5472.CAN-07-6768
267. Boutet P, Aguera-Gonzalez S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, et al. Cutting edge: the metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J Immunol. (2009) 182:49–53. doi: 10.4049/jimmunol.182.1.49
268. Moss ML, Minond D. Recent advances in ADAM17 research: a promising target for cancer and inflammation. Mediators Inflamm. (2017) 2017:9673537. doi: 10.1155/2017/9673537
269. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer (2000) 88:577–83. doi: 10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V
270. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res. (2011) 17:678–89. doi: 10.1158/1078-0432.CCR-10-2173
271. Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer (2017) 5:73. doi: 10.1186/s40425-017-0275-9
272. Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res. (2014) 2:1125–31. doi: 10.1158/2326-6066.CIR-14-0160
273. Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother. (2015) 64:225–35. doi: 10.1007/s00262-014-1629-5
274. Mestas J, Burdick MD, Reckamp K, Pantuck A, Figlin RA, Strieter RM. The role of CXCR2/CXCR2 ligand biological axis in renal cell carcinoma. J Immunol. (2005) 175:5351–7. doi: 10.4049/jimmunol.175.8.5351
275. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. (2010) 22:124–31. doi: 10.1016/j.ceb.2009.11.014
276. Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci USA. (2017) 114:E9271–9. doi: 10.1073/pnas.1703921114
277. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA. (2013) 110:17450–5. doi: 10.1073/pnas.1304790110
278. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
279. Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer (2006) 6:506–20. doi: 10.1038/nrc1926
280. Kong FM, Anscher MS, Murase T, Abbott BD, Iglehart JD, Jirtle RL. Elevated plasma transforming growth factor-beta 1 levels in breast cancer patients decrease after surgical removal of the tumor. Ann Surg. (1995) 222:155–62. doi: 10.1097/00000658-199508000-00007
281. Santin AD, Bellone S, Ravaggi A, Roman J, Smith CV, Pecorelli S, et al. Increased levels of interleukin-10 and transforming growth factor-beta in the plasma and ascitic fluid of patients with advanced ovarian cancer. BJOG (2001) 108:804–8. doi: 10.1111/j.1471-0528.2001.00206.x
282. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. (2009) 119:1524–36. doi: 10.1172/JCI37869
283. Katz LH, Li Y, Chen JS, Munoz NM, Majumdar A, Chen J, et al. Targeting TGF-beta signaling in cancer. Expert Opin Ther Targets (2013) 17:743–60. doi: 10.1517/14728222.2013.782287
284. Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature (1997) 390:465–71. doi: 10.1038/37284
285. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. (2001) 29:117–29. doi: 10.1038/ng1001-117
286. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
287. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. (2004) 172:7335–40. doi: 10.4049/jimmunol.172.12.7335
288. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
289. Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. (2004) 64:7596–603. doi: 10.1158/0008-5472.CAN-04-1627
290. Allan DS, Rybalov B, Awong G, Zuniga-Pflucker JC, Kopcow HD, Carlyle JR, et al. TGF-beta affects development and differentiation of human natural killer cell subsets. Eur J Immunol. (2010) 40:2289–95. doi: 10.1002/eji.200939910
291. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. (2008) 181:3784–92. doi: 10.4049/jimmunol.181.6.3784
292. Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. (2005) 6:600–7. doi: 10.1038/ni1197
293. Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. (2009) 69:7775–83. doi: 10.1158/0008-5472.CAN-09-2123
294. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE (2014) 9:e90353. doi: 10.1371/journal.pone.0090353
295. Yingling JM, McMillen WT, Yan L, Huang H, Sawyer JS, Graff J, et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-beta receptor type I inhibitor. Oncotarget (2018) 9:6659–77. doi: 10.18632/oncotarget.23795
296. Fujiwara Y, Nokihara H, Yamada Y, Yamamoto N, Sunami K, Utsumi H, et al. Phase 1 study of galunisertib, a TGF-beta receptor I kinase inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. (2015) 76:1143–52. doi: 10.1007/s00280-015-2895-4
297. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFbetaR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-GD2 antibody dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res. (2017) 23:804–13. doi: 10.1158/1078-0432.CCR-16-1743
298. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE (2018) 13:e0191358. doi: 10.1371/journal.pone.0191358
299. Naeimi Kararoudi M, Dolatshad H, Trikha P, Hussain SA, Elmas E, Foltz JA, et al. Generation of knock-out primary and expanded human NK cells using Cas9 ribonucleoproteins. J Vis Exp. (2018) e58237. doi: 10.3791/58237
300. Millet C, Zhang YE. Roles of Smad3 in TGF-beta signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr. (2007) 17:281–93. doi: 10.1615/CritRevEukarGeneExpr.v17.i4.30
301. Wang QM, Tang PM, Lian GY, Li C, Li J, Huang XR, et al. Enhanced cancer immunotherapy with Smad3-silenced NK-92 cells. Cancer Immunol Res. (2018) 6:965–77. doi: 10.1158/2326-6066.CIR-17-0491
302. Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy (2017) 19:408–18. doi: 10.1016/j.jcyt.2016.12.005
Keywords: natural killer cell, chimeric antigen receptor, immunotherapy, solid tumor, cytokines, tumor microenvironment, checkpoint inhibitors, bispecific antibody
Citation: Nayyar G, Chu Y and Cairo MS (2019) Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Front. Oncol. 9:51. doi: 10.3389/fonc.2019.00051
Received: 15 November 2018; Accepted: 18 January 2019;
Published: 11 February 2019.
Edited by:
Monica Thakar, Fred Hutchinson Cancer Research Center, United StatesReviewed by:
Amorette Barber, Longwood University, United StatesAnna Karolina Kozlowska, City of Hope National Medical Center, United States
Copyright © 2019 Nayyar, Chu and Cairo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitchell S. Cairo, mitchell_cairo@nymc.edu
†These authors have contributed equally to this work and share first authorship