 Wei-long Zhang
Wei-long Zhang Zhuo-li Zhu1†
Zhuo-li Zhu1† Ya-ling Tang
Ya-ling Tang Xin-hua Liang
Xin-hua Liang- 1State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, China
- 2State Key Laboratory of Microbial Technology, Shandong University, Qingdao, China
Multiple primary cancers (MPCs) are major obstacles to long-term survival in head and neck cancer (HNSCC), however, the molecular mechanism underlying multiple carcinogenesis remains unclear. “Field cancerization” is a classical theory to elaborate the malignant progression of MPCs. Apart from environmental and immune factors, genetic factors may have great potential as molecular markers for MPCs risk prediction. This review focuses on inherited and acquired gene mutations in MPCs, including germ-line mutation, single-nucleotide polymorphism, chromosomal instability, microsatellite instability and DNA methylation. And definition and prognosis of MPCs have also been discussed. These may pave the way for the early detection, prevention and effective treatment of MPCs in HNSCC.
Introduction
Head and neck squamous cancer (HNSCC) ranks sixth among the most prevalent malignancies in the world (1). It is estimated that HNSCC accounts for 64,690 new cases and 13,740 deaths in the United states in 2018 (2). Although significant improvements have been made in the therapeutic modalities, the prognosis of HNSCC remains stagnant in the past decades, with a 5-year survival of only 50% (3). The dismal prognosis has always been attributed to local recurrences, distant metastasis, and development of multiple primary cancers (MPCs).
MPCs are defined as two or more primary cancers occurring in an individual synchronously or metachronously, neither extensions, recurrences nor metastases of each other (International Agency for Research on Cancer), which accounts for approximately one-third of deaths in HNSCC (4, 5). MPCs are also named as second primary malignancies (SPMs), secondary primary tumors (SPTs), second primary cancers (SPCs), and multiple primary tumors (MPTs) (6). Several risk factors, including smoking exposure, alcohol consumption, human papilloma virus (HPV), and hepatitis C virus (HCV), have been suggested to be associated with the development of MPCs (7–10). However, researches revealed that a major proportion of MPCs could not be fully explained by these environmental factors, and genetic factors, such as single-nucleotide polymorphism (SNP), chromosomal instability (CIN), microsatellite instability (MSI), and epigenetic alterations, may contribute to the susceptibility of MPCs in HNSCC (11).
MPCs have been considered to be a clinical quandary both in diagnosis and treatment. It is challenging for the oncologists to distinguish MPCs from a metastasis or local recurrence merely on the basis of clinical and pathological information. Furthermore, this classification leaves a profound effect on the choice of treatment as well as patients' prognosis. Recently, there are a number of novel screening strategies for MPCs prediction, such as genetic markers, and the role of genetic alterations in the development of MPCs after index HNSCC were not fully elucidated (12). Here, we conducted this review to summarize the mechanisms and relevant genetic alterations of MPCs, which might provide benefit in the detection, prevention, and treatment of MPCs in HNSCC.
Definition and Prognosis of MPCs
In 1932, Warren and Gates published the classical clinical criteria of SPT, being (1) each of the malignancies must have been verified by histologic examination, (2) the malignancies must be anatomical separate by normal mucosa, and (3) exclude the possibility that the second malignancy represents a metastasis of the index tumor (13). However, this clinical definition carries the risk of misclassification and inability to differentiate between an SPT, a recurrence and a metastasis (14). For example, what distance should lie between the malignancies? How to define the normal mucosa, by naked eye or histologic examination? How to distinguish the SPT from a metastasis or a recurrence? To solve this dilemma, Braakhuis et al. proposed a new classification on the basis of molecular profiles of the tumors and the genetically altered mucosal field between the tumors (14). Tumors displaying distinct molecular profiles or sharing a common pattern attributed to chance are defined as SPTs, while those possessing similar molecular aberration are defined as local recurrences (14).
It is widely accepted that MPCs are the leading obstacle to long-term survival among HNSCC patients (15). According to a retrospective study conducted by Shiga et al., patients with synchronous SPMs displayed a poorer 5-year overall survival rate than those with metachromous SPMs in Japan (16). In line with this result, Bugter et al. claimed that the 5-yearsurvival rate for synchronous and metachronous primary cancer patients was 25 and 85%, respectively (17). The higher proportion of high-stage tumor in the synchronous primary cancer patients and unadjusted treatment protocol may account for this discrepancy.
However, accumulating evidence has demonstrated that HPV-positive HNSCC patients were accompanied by a decreased risk for SPT than HPV-negative HNSCC patients (18–20). HPV has been established as an emerging carcinogen in a subset of HNSCCs, particularly in the oropharynx (21). HPV-positive HNSCCs differ from HPV-negative HNSCCs induced by tobacco and alcohol epidemiologically, clinically, and biologically (22–24). The putative reasons for this phenomenon are as follows: (1) HPV-positive HNSCCs exhibited higher sensitivity to radiotherapy and chemotherapy (25); (2) HPV-positive HNSCCs often arise in an environment with lower exposure to tobacco and displayed fewer smoking-related genetic abnormalities, which is less associated with smoking-related SPTs (26–28); (3) Saito et al. suggested that field cancerization effect would not be observed in HPV-positive HNSCCs, since HPV viral DNA integration was limited to the cancerous tissue (29). However, the concrete mechanisms between HPV-positive HNSCCs and SPTs are remained to be elucidated in the future.
The dismal clinical outcome of MPCs in HNSCC emphasizes on the importance of early diagnosis and prevention. These genetic alterations could serve as molecular makers to guide the early diagnosis, prevention and treatment of HNSCC patients in several aspects. Firstly, these molecular markers could be readily obtained by the primary tumor samples without bringing additional invasion to patients (30). Secondly, genetic markers could select the high-risk individuals for MPCs, which should be under strict cancer surveillance and proper preventive procedures. Given the discrepancies between HPV-positive and HPV-negative HNSCC, Jain et al. proposed that future screening procedures for MPCs may be adjusted by HPV and smoking status (31). Thirdly, in-depth understanding of the role of these molecular markers in MPCs may pave the way for targeted gene therapies. In addition, genetic markers could be utilized to distinguish MPCs from a local recurrence or a metastasis. For example, Mercer et al. showed that microsatellite PCR facilitated the discrimination between second primary cancer and metastatic HNSCC (32). Daher et al. employed combined HPV typing and TP53 mutational profiling successfully identified the accurate origin of lung tumors in 32 HNSCC patients, in which only 13 cases were diagnosed correctly on the basis of clinical and morphological data alone (33). Apart from physical and pathological information, genetic profiles analysis could be an effective tool to distinguish MPCs from a recurrence. Gasparotto et al. suggested that 3 clinically diagnosed recurrences and 2 lung lesions were actually MPTs by comparing the p53 mutation status of primary tumors and corresponding recurrences/metastases in HNSCC patients (34). Microsatellite analysis indicated that 6 tumors showing clonally-related patterns with primary tumors were recurrences, while 17 tumors with clonally-unrelated patterns were SPTs in 23 HNSCC patients with genetic changes (35).
Mechanism of MPCs
The concrete molecular mechanism underlying multiple carcinogenesis remains unclear. “Field cancerization” theory has often been applied to explain the occurrence of MPCs (Figure 1). The stem cell receives one (or more) genetic hit (Figure 1A), probably a mutation of p53 gene, and gives rise to a patch with genetically altered daughter cells [(36); Figure 1B]. Then a subsequent genetic alteration induces the patch to spread in a lateral direction and substitutes the normal epithelial cells to form a field [(36); Figure 1C]. As the field expands at the expense of normal epithelial cells, additional genetic alterations take place and promote the progression from field to an overt carcinoma (Figure 1D). The second tumor of monoclonal origin develops by implantation, intraepithelial migration or sub-mucosal spread of primary cancer cells (Figure 1E), while the polyclonal second tumor forms under the induction of final genetic hit [(14, 37); Figure 1F].
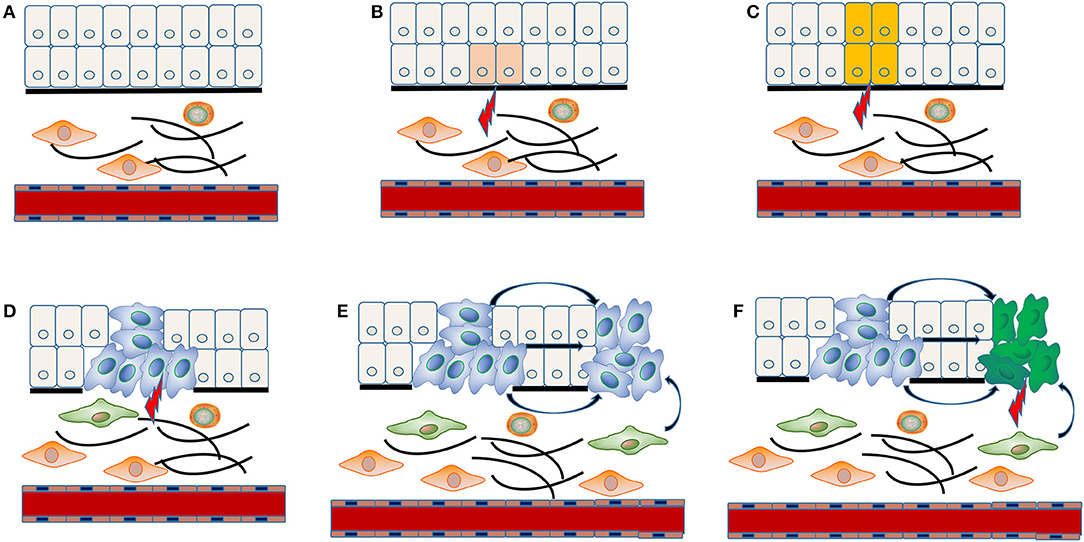
Figure 1. “Field cancerization” theory in the MPCs of HNSCC. The stem cell receives first genetic hit (A), and gives rise to a patch with genetically altered daughter cells (B). Then a patch develops into a field by expanding in a lateral direction under the indduction of a second genetic hit (C). Additional genetic alterations take place and convert the field to an overt carcinoma (D). The implantation, intraepithelial migration or sub-mucosal spread of primary cancer cells lead to the development of a SPT with monoclonal origin (E), while the final genetic hit induces the occurrence of a SPT with polyclonal origin (F).
Failure of immune surveillance also contributed to the occurrence of SPTs in HNSCC (Figure 2). Patients with decreased T-cell numbers in the circulation were predisposed to infections, disease recurrence, or a second malignancy (38). Kuss et al. reported that CD4+ and CD8+ T cells were significantly reduced in the SPT group relative to normal control group in HNSCCs (38). And patients with recurrences or SPTs showed a 25% lower number of CD4+ T cells than those with primary disease (38). The TCR associated CD3 zeta chain plays a critical role in the signal transduction of T-cell activation, the absence of which impairs T-cell signaling and consequently leads to immune dysfunction (39). Kuss et al. concluded that individuals with SPTs or recurrences exhibited lowest zeta-chain expression, which might exert long-lasting negative effects on the anti-tumor immune response (40). Decreased expression of HLA class I molecules is considered to be an effective strategy for malignant cells to evade host immunosurveillance (41). Grandis et al. suggested that the number of HLA allelic loss increased the risk of developing a new primary tumor (41). Collectively, decreased T-cell numbers, CD3 zeta chain and HLA class I molecules may be associated with the development of SPT, which may provide new opportunities for cancer immunotherapy in HNSCC.
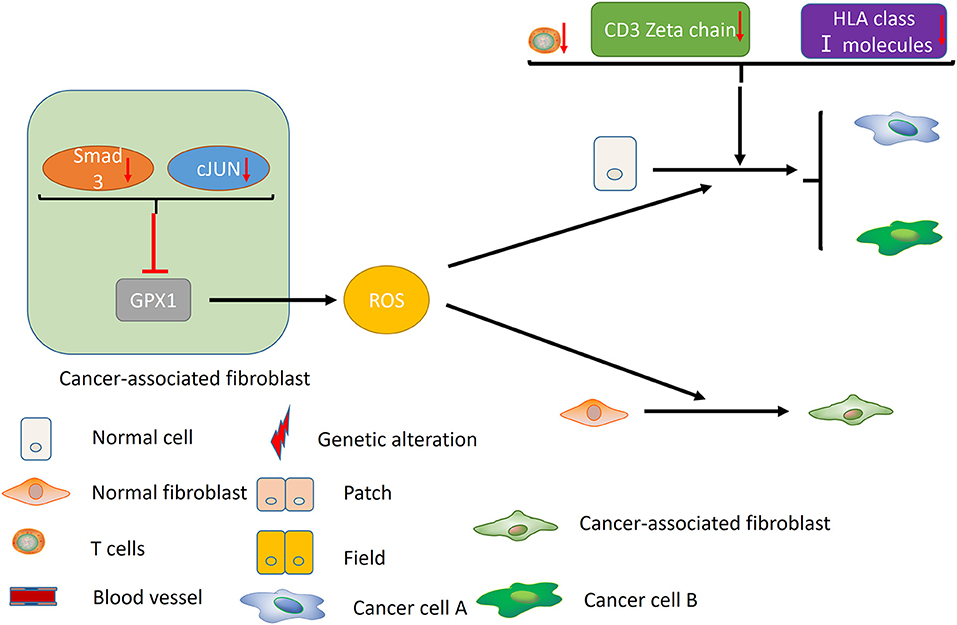
Figure 2. Immune factors and CAFs contribute to the development of MPCs. Decreased T-cell numbers, CD3 zeta chain and HLA class I molecules may promote the development of MPCs by inducing immunosuppression. In the CAFs, reduced expression of Smad3 and cJUN suppress the activity of GPX1, leading to the elevation of extracellular hydrogen peroxide. High hydrogen peroxide level in the microenvironment induces the conversion of normal fibroblast to CAF phenotype, and promotes the occurrence of MPCs.
In addition, cancer-associated fibroblasts (CAFs) may play an unneglectable role in the development of field cancerization [(42); Figure 2]. Ge et al. proposed that migratory cancer-associated fibroblasts (CAFs), also named myofibroblast, may appear beneath the cluster of genetic altered epithelial cells, and ultimately lead to the malignant transformation of these cells (42). Angadi et al. demonstrated that myofibroblasts were present in the stroma around the oral squamous cell carcinoma (OSCC) cell as well as the connective tissue below the histologically normal mucosa adjacent to OSCC by immunochemistry, which validates Ge's hypothesis further (43). Chan et al. indicated that cancer-associated fibroblasts promoted field cancerization by elevating the expression of reactive oxygen species (ROS) in the microenvironment (44). CAFs from squamous cell carcinoma reduced the expression of Smad3 and cJUN to suppress the activity of glutathione peroxidase 1 (GPX1), one key enzyme affecting hydrogen peroxide detoxification. Suppression of GPX1 leads to elevation of extracellular hydrogen peroxide, which facilitates the conversion of normal fibroblast to CAF phenotype, and promotes the tumor-forming capacity and invasiveness. Till now, there is no more available evidences on the role of tumor microenvironment in MPCs, including macrophages, myeloid-derived suppressor cells (MDSCs) and etc., which warrants further investigation in the future.
MPCs and Inherited Mutations
MPCs and Germ-Line Mutations
Germ-line mutation of tumor suppressor genes has been considered to be a potential driver of MPCs. p53 gene, known as the “the guardian of the genome,” is an well-known tumor suppressor gene which has been involved in the cell cycle control and DNA repair (45). It is estimated that p53 gene was mutated in approximately 50% of HNSCC patients (46). In 1999, Gallo and his colleagues employed polymerase chain reaction single-strand conformation polymorphism (PCR-SSCP) analysis and DNA sequencing to examine the p53 germ-line mutations in 24 HNSCC patients who developed MPCs and their first-degree relatives. As a consequence, only one missense mutation in exon 6 as well as two same-sense mutations in exon 6 and 8 have been detected (47). So the authors proposed that p53 gene might not be the only target responsible for the multiple genetic alterations of field cancerization (47).
CDKN2A, a tumor suppressor gene exerting an important role in the regulation of cell cycle, is associated with the occurrence of HNSCC. Cabanillas et al. proposed that germ-line mutations of CDKN2A gene may serve as a common feature of HNSCC (30). However, Jefferies et al. screened full coding sequence of CDKN2A gene and failed to detect any germ-line mutations in 40 HNSCC patients with a SPC, which suggested that germ-line mutations of CDKN2A contributed less to the susceptibility of MPCs (48).
Mismatch genes, such as hMLHI, contribute greatly to the MPCs of gastrointestinal cancers. Nevertheless, its germ-lime mutations don't seem to be an major event in the carcinogenesis of HNSCC or MPTs (49). Piccinin et al. analyzed the mutations of hMLHI gene by PCR-SSCP and sequencing in 67 HNSCC patients, 22 MPTs and 45 controls, and no somatic or germ-lime mutations of hMLHI have been identified (49).
Based on the above, it seems that germ-line mutations of certain tumor suppressor genes and DNA repair genes, including p53, CDKN2A, and hMLHI, exert a minor influence on the genetic predisposition of MPCs after index HNSCC.
MPCs and SNPs
It is estimated that SNP accounts for up to 90% of genetic variability (50). Researchers have demonstrated that genetic polymorphisms of various genes were correlated with the risk of MPCs in HNSCC. Based on their functions, these genes could be classified into four categories: tumor suppressor genes, oncogenes, DNA repair genes and carcinogen metabolism-related genes, which were listed in detail as follows.
SNPs of Tumor Suppressor Genes
p53 makes a substantial contribution to the regulation of cell cycle, cellular apoptosis and anti-cancer properties (45). p53 mutations have been shown to be correlated with the occurrence of primary HNSCC as well as SPMs (51). The polymorphism of p53 commonly occurred at the codon 72, with a substitution of proline for arginine, which might promote the carcinogenesis by disturbing the apoptosis process and cell cycle (52). Several studies indicated that p53 codon 72 polymorphism may play a minor role in the development of HNSCC (53, 54). However, Li et al. showed that patients with p53 72Arg/Pro and the combined p53 72Arg/Pro + Pro/Pro genotypes exhibited a significantly greater risk of SPMs in a cohort of 1271 HNSCC patients, compared with p53 72Arg/Arg genotype (51). The authors speculated that p53 codon 72 polymorphism may influence the SPM risk by modifying the response to DNA-damaging treatments (51). Despite the inherited limitations of patient selection and clinical outcome collection, these results indicated that p53 codon 72 polymorphism could serve as a genetic marker to evaluate the risk of SPMs in HNSCC.
Functionally, tumor suppressor gene p14 and p73 belong to the p53-related gene family. p14ARF gene maps to 9p21 and encodes proteins of p16INK4a and p14ARF, thus playing an important role in maintaining genomic stability via p53 and Rb pathways (6). Direct interaction between p14ARF and MDM2 impedes the protesomal degradation of p53, resulting in an abnormal cell cycle regulation and cellular apoptosis (55). Genetic Alterations of p14ARF have been considered to be rare in the development of HNSCC. Gruttgen et al. evaluated the p14 expression with immunochemistry, and concluded that loss of p14ARF merely occurred in 15 of 100 HNSCC patients (56). Zhang et al. stated that p14ARF-rs3088440 and rs3731217 polymorphisms were correlated with a moderately increased risk of SPMs in HNSCC (6). Compared with those without p14ARF variant genotypes, HNSCC patients with both variant genotypes had a 3-fold increased risk for developing SPMs (6). Therefore, p14ARF polymorphisms could serve as a risk marker for SPMs in HNSCC patients (6).
p73 displays a similar function as p53 in the regulation of cell cycle, apoptosis and DNA repair by inducing apoptosis or G1 cell cycle arrest (57). It has been assumed that p73 compensated for the absence of p53 induced by mutations (58). Previous studies have revealed that genetic abnormalities of p73, such as its G4C14-to-A4T14 polymorphism, were associated the risk of HNSCC (59, 60). Li et al. investigated the role of p73 G4C14-to-A4T14 polymorphism in SPMs in a cohort of 1384 HNSCC patients, and advocated that patients carrying p73 GC/AT heterozygotes or the combined p73 GC/AT+AT/AT genotypes had a significantly lower SPM susceptibility, compared to those with p73 GC/GC genotype. The p73 GC/AT+AT/AT genotypes conferred a pronounced protection over SPMs in several subgroups, for example, older patients, men, minorities, ever smoker, and ever drinkers, further supporting the role of p73 polymorphism as a genetic marker of MPCs in HNSCC patients (61).
FAS belongs to the death receptor family and interacts with its ligand, FASLG, to modulate the extrinsic apoptosis pathway, cellular homeostasis and immune escape of tumor cells (62, 63). Genetic alterations of the FAS/FASLG signaling pathway may lead to immune evasion, thus facilitating tumorigenesis including SPM (64). FAS-1377G>A, FAS−670A>G, FASLG-844C>T, and FASLG-124 A>G are four well-known SNPs in the FAS/FASLG signaling pathway. Zhang et al. reported that subjects with both the FAS-1377 AA and FAS-670 (GG + AG) genotypes were associated with an increased risk of HNSCC, but not for those with FASLG variant genotypes (65). But things are really different when it comes to the incidence of MPCs. It has been demonstrated that patients carrying FAS-670 AG+GG genotypes or FASLG-844 CT+TT genotypes were significantly associated with mounting risk of SPMs compared with the wild-type homozygous genotypes, which makes FAS and FASLG polymorphisms a potential marker for HNSCC patients at high SPM risk (64). Additionally, the risk of SPM was augmented in a dose-response manner for those with increasing number of risk genotypes (64).
p21 and p27 are two CDK inhibitors which participate in the regulation of DNA repair, cell cycle, and apoptosis (66). It has been demonstrated that SNPs in p21 and p27 were associated with risk of HNSCC (67, 68). HNSCC patients carrying p27 109 TG/GG, p21 98 CA/AA, and p21 70 CT/TT variant genotypes had a worse survival and an increased SPM risk than those with p27109 TT, p21 98 CC, and p21 70 CC genotypes, respectively (69). Moreover, patients with p27 (T109G) and p21 (C98A and C70T) polymorphisms were 2.4 times more susceptible to develop MPCs than those without variant genotypes (69). These results indicated that p27 T109G and p21 (C98A and C70T) polymorphisms seem to modulate the susceptibility of SPMs in HNSCC.
Accumulating evidences have established that SNPs of tumor suppressor genes made a considerable contribution to the formation of MPCs in HNSCC, which makes SNPs of tumor suppressor genes a potential molecular marker to predict the risk of MPCs in HNSCC.
SNPs of Oncogenes
Murine double minute 2 (MDM2), also known as an E3 ubiquitin ligase, is the central antagonist of the tumor suppressor p53 (70). MDM2 negatively regulates the activity of p53 by suppressing its transcriptional activity and promoting its degradation, thus contributing to the carcinogenic process (71). The overexpression and genetic alterations of MDM2 have been commonly reported in HNSCC (72). Two SNPs in its promoter region, MDM2-rs2279744 and MDM2-rs937283, may alter MDM2 expression at transcriptional level and subsequently modulate the risk of HNSCC (71). With respect to SPM, Jin et al. reported that MDM2-rs2279744 and MDM2-rs937283 increased the susceptibility of SPM in HNSCC by 90 and 20%, respectively (73).
Analogous to MDM2 in structure, murine double minute 4 (MDM4) is also a negative regulator of p53. Several studies suggested that high MDM4 expression may substitute for p53 mutations, and MDM4 overexpression was a common event in the HNSCC patients (74). Three MDM4 SNPs, rs11801299G>A, rs1380576C>G, and rs10900598G>T, have been identified in HNSCC patients. Yu et al. proposed that individuals with combined 1-3 risk genotypes of MDM4 SNPs exhibited significantly increased risk of oropharyngeal cancer (75). In a cohort of 1283 HNSCC patients, Jin et al. concluded that MDM4-rs11801299, MDM4-rs1380576, and MDM4-rs10900598 enhanced the incidence of SPMs in index HNSCC cases by 10, 10, and 40%, respectively (73). Collectively, MDM2 and MDM4 polymorphisms may increase the susceptibility of SPMs in HNSCC to some extent, which may improve the precision of risk estimates of SPMs.
SNPs of DNA Repair Genes
The MRN complex, composed of MRE11, RAD50, and Nbs1, plays a critical role in the double-strand break repair and telomere maintenance (76). To evaluate the role of MRE11 and RAD50 genes in HNSCC, Ziółkowska-Suchanek et al. conducted a case and control study of 358 HNSCC patients. Their results suggested that common variants of MRE11 and RAD50 genes contributed little to the occurrence of HNSCC and SPTs located in the head and neck region (77). XRCC3, short for X-ray repair cross-complementing group 3, is another important gene which participates in the double-strand break repair (78). Several studies revealed that XRCC3 C18067T polymorphism may play a minor role in the etiology of primary HNSCC as well as MPTs. (78, 79).
The X-ray repair cross-complementing group 1 (XRCC1) exerts on a vital role in the DNA single-strand break repair pathway (80). XRCC1 Arg194Trp, XRCC1 Arg280His, and XRCC1 Arg399Gln are the three most common examined SNPs in the XRCC1 gene (81, 82). Lou et al. suggested that XRCC1 Arg194Trp, XRCC1 Arg280His, and XRCC1 Arg399Gln posed limited effect on the HNSCC risk in a meta-analysis with 29 studies (83). Similarly, no significant associations have been presented between XRCC1 gene SNPs and the incidence of MPT in the HNSCC patients (79).
Apart from double-strand and single-strand repair pathway, the host can protect the genome from damage induced by various environmental carcinogens by means of nucleotide excision repair (NER) pathway (84). Seven SNPs of the NER genes involved in the HNSCC are listed as follows: XPC Ala499Val, XPC Lys939Gln, XPD Asp312Asn, XPD Lys751Gln, XPG His1104Asp, ERCC1 C8092A, and XPA G23A. Zafereo et al. declared that no significant association between aforementioned seven SNPs and the SPM susceptibility, independently or collectively, has been found in a recessive model (84).
All in all, no significant associations between SNPs of DNA repair genes and MPC risk have been found so far. On the basis of above evidences, it is plausible that SNPs of DNA repair genes might not play a major role in the development of MPCs in the HNSCC subjects.
SNPs of Carcinogen Metabolism-Related Genes
Glutathione peroxidase I (GPX1), a selenium-dependent enzyme, participates in the detoxification of activated oxygen species (85). Genetic alterations or polymorphism in the coding region of GPX1 gene might be involved in the development of cancer. A significant correlation has been observed between GPX1 expression and T-stage as well as index tumor sites in HNSCC patients (86). The GPX1 polymorphism represents three possible alleles, namely ALA5, ALA6, and ALA7. Jefferies et al. evaluated the association between GPX1 genetic polymorphisms and HNSCC patients who developed SPTs in a case-control study. A significant difference in allele frequencies of GPX1 ALA*6 and ALA*7 was observed between the SPT cases and controls, which indicated that polymorphisms of GPX1 gene may be a molecular marker for the development of SPTs in HNSCC (85).
CYP1A1 and CYP2E1 are two main genes associated with the carcinogen metabolic activation. Rydzanicz et al. reported that HNSCC patients with CYP1A1 genotype *1/*4 and allele *4 represented a 4.1- and 2.6-fold risk of developing MPT, respectively (79). However, no significant correlations has been established between SNPs of CYP2E1 gene and the incidence of MPT, which suggested a limited role of CYP2E1 in the susceptibility of MPTs in HNSCC (79).
Glutathione S transferase (GST) plays a critical role in the detoxication and elimination of various carcinogens (87). GSTM1, GSTT1, and GSTM3 gene are three members of the GST family in human (87). Studies reported that the GSTT1 null genotype and polymorphism in GSTM3 gene were not correlated with a statistically significant increased risk for SPTs or tobacco-related SPTs (79, 88). Inversely, a significant association was observed between the polymorphism in GSTM1 gene and development of SPTs or tobacco-related SPTs (88).
N-acetyltransferase 2 (NAT2) gene participates in the metabolism of aromatic, heterocyclic amines and hydrazines (89). Evidence from 23 case and control studies indicated that NAT2 polymorphisms could increase the incidence of HNSCC by 23% and serve as a risk factor of HNSCC in Asians (90). With respect to MPCs, NAT2*7B was significantly correlated with an increased risk for SPTs in patients after index HNSCC (79).
According to the available evidences, a significant association has been observed between MPCs and SNPs of carcinogen metabolism-related genes, such as CYP1A1, GSTM1, and NAT2. However, well-designed, large-scale, multi-center studies are still warranted to verify these conclusions in the future.
MPCs and Acquired Mutations
MPCs and CIN
CIN comprises altered DNA copy number and loss or rearrangement of the chromosomes, resulting in the loss or gain of function of certain genes (91). Piccinin et al. evaluated the LOH status at 1p, 3p, 9p, 13q, and 19p. However, no significant differences have been observed between the MPCs group and single cancer group, which suggested that chromosomal instability may not account for the propensity to develop SPMs in the upper aerodigestive tract (49).
MPCs and MSI
MSI, a major hallmark of genetic instability, originates from deficient DNA mismatch repair (92). It is mostly observed and studied in the hereditary non-polyposis colorectal cancer, which is characterized by MPCs of different organs, such as gastrointestinal, endometrial and urinary tract (93). So, MSI is considered to be a major determinant in the development of MPCs. Piccinin et al. analyzed the MSI on five chromosomes in 67 HNSCC patients, 22 MPCs and 45 controls, and revealed that no significant differences existed between MSI and MPCs cases (49). This implied that except for MSI, other systems concerning the genome integrity might be responsible for the carcinogenesis of HNSCC and tumor multiplicity of the head and neck region (49).
MPCs and DNA Methylation
DNA methylation is a well-categorized change of epigenetic alterations in tumors, which is capable of silencing the classic tumor suppressor genes (94). DNA methylation could disrupt the tumor suppressor gene function by obstructing its promoter region and impeding the transcriptional process (95). Longo et al. have detected CCNA1, DCC, and TIMP3 hypermethylation in the exfoliated cell samples of HNSCC patients (96). To investigate the relationship between hypermethylation and MPCs in HNSCC, Rettori et al. examined the methylation patterns of 19 genes in 70 HNSCC cases (97), revealing that CCNA1 and TIMP3 hypermethylation were significantly connected with formation of SPT in HNSCC. Hypermethylation of CCNA1 and TIMP3 might be a promising genetic marker to predict the incidence of SPT in HNSCC subjects, providing the basis for the use of preventive measures and adjuvant treatment.
Conclusions
The dismal prognosis of HNSCC has always been attributed to the occurrence of MPCs. “Field cancerization,” induced by carcinogens and CAFs, is proposed to elaborate the development of MPCs. Apart from environmental and immune factors, genetic factors may play a major role in the risk of MPCs. In summary, SNPs of tumor suppressor genes, oncogenes and carcinogen metabolism-related genes, together with DNA methylation, may serve as potential molecular markers of MPCs risk (Table 1). SNP chips and next-generation sequencing technology will enables us to access the strength of these “nature” components of MPCs, resulting in early diagnosis and better survival in HNSCC patients. However, there is still a long way to go before the clinical application of these genetic markers. HNSCC is a genetically heterogeneous disease with a wide range of genetic alterations (98), so a panel of genetic markers with the most accuracy and specificity need to be selected. On the other hand, large-scale, well-designed, and multi-center studies are warranted to examine their clinical relevance.
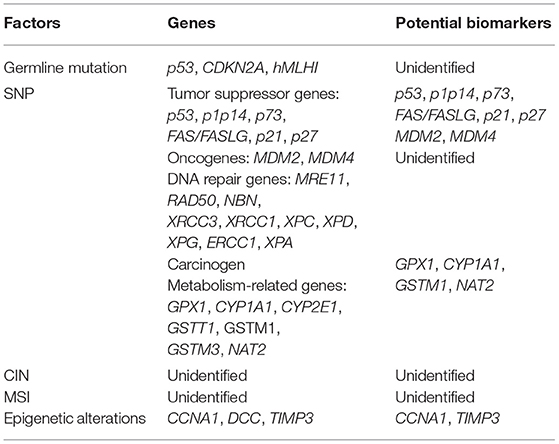
Table 1. Genetic factors, genes, and potential biomarkers of multiple primary cancer of HNSCC.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by National Natural Science Foundation of China grants (Nos. 81672672, 81572650, 81772891, 81502357, and 81972542) and by State Key Laboratory of Oral Diseases Special Funded Projects.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Whaley JT, Indelicato DJ, Morris CG, Hinerman RW, Amdur RJ, Mendenhall WM, et al. Ewing tumors of the head and neck. Am J Clin Oncol. (2010) 33:321–6. doi: 10.1097/COC.0b013e3181aaca71
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. (2018) 68:7–30. doi: 10.3322/caac.21442
3. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. (2011) 11:9–22. doi: 10.1038/nrc2982
4. Simpson MC, Massa ST, Boakye EA, Antisdel JL, Stamatakis KA, Varvares MA, et al. Primary cancer vs competing causes of death in survivors of head and neck cancer. JAMA Oncol. (2018) 4:257–9. doi: 10.1001/jamaoncol.2017.4478
5. van der Schroeff MP, van de Schans SA, Piccirillo JF, Langeveld TP, Baatenburg de Jong RJ, Janssen-Heijnen ML. Conditional relative survival in head and neck squamous cell carcinoma: permanent excess mortality risk for long-term survivors. Head Neck. (2010) 32:1613–8. doi: 10.1002/hed.21369
6. Zhang Y, Sturgis EM, Zafereo ME, Wei Q, Li G. p14ARF genetic polymorphisms and susceptibility to second primary malignancy in patients with index squamous cell carcinoma of the head and neck. Cancer. (2011) 117:1227–35. doi: 10.1002/cncr.25605
7. Do KA, Johnson MM, Lee JJ, Wu XF, Dong Q, Hong WK, et al. Longitudinal study of smoking patterns in relation to the development of smoking-related secondary primary tumors in patients with upper aerodigestive tract malignancies. Cancer. (2004) 101:2837–42. doi: 10.1002/cncr.20714
8. Leon X, Del Prado Venegas M, Orus C, Kolanczak K, Garcia J, Quer M. Metachronous second primary tumours in the aerodigestive tract in patients with early stage head and neck squamous cell carcinomas. Eur Arch Otorhinolaryngol. (2005) 262:905–9. doi: 10.1007/s00405-005-0922-5
9. Nagao Y, Sata M. High incidence of multiple primary carcinomas in HCV-infected patients with oral squamous cell carcinoma. Med Sci Monit. (2009) 15:CR453–9.
10. Xu CC, Biron VL, Puttagunta L, Seikaly H. HPV status and second primary tumours in oropharyngeal squamous cell carcinoma. J Otolaryngol Head Neck Surg. (2013) 42:36. doi: 10.1186/1916-0216-42-36
11. Foulkes WD, Brunet JS, Sieh W, Black MJ, Shenouda G, Narod SA. Familial risks of squamous cell carcinoma of the head and neck: retrospective case-control study. BMJ. (1996) 313:716–21. doi: 10.1136/bmj.313.7059.716
12. Bunbanjerdsuk S, Vorasan N, Saethang T, Pongrujikorn T, Pangpunyakulchai D, Mongkonsiri N, et al. Oncoproteomic and gene expression analyses identify prognostic biomarkers for second primary malignancy in patients with head and neck squamous cell carcinoma. Mod Pathol. (2019) 32:943–56. doi: 10.1038/s41379-019-0211-2
13. Warren S, Gates O. Multiple primary malignant tumors: a survey of the literature and statistical study. Am J Cancer. (1932) 16:1358–414.
14. Braakhuis BJ, Tabor MP, Leemans CR, van der Waal I, Snow GB, Brakenhoff RH. Second primary tumors and field cancerization in oral and oropharyngeal cancer: molecular techniques provide new insights and definitions. Head Neck. (2002) 24:198–206. doi: 10.1002/hed.10042
15. León X, Quer M, Orús C, Venegas MdP. Can cure be achieved in patients with head and neck carcinomas? The problem of second neoplasm. Expert Rev Anticancer Ther. (2001) 1:125–33. doi: 10.1586/14737140.1.1.125
16. Shiga K, Tateda M, Katagiri K, Nakanome A, Ogawa T, Asada Y, et al. Distinct features of second primary malignancies in head and neck cancer patients in Japan. Tohoku J Exp Med. (2011) 225:5–12. doi: 10.1620/tjem.225.5
17. Bugter O, van Iwaarden DLP, Dronkers EAC, de Herdt MJ, Wieringa MH, Verduijn GM, et al. Survival of patients with head and neck cancer with metachronous multiple primary tumors is surprisingly favorable. Head Neck. (2019) 41:1648–55. doi: 10.1002/hed.25595
18. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. (2010) 363:24–35. doi: 10.1056/NEJMoa0912217
19. Huang SF, Li HF, Liao CT, Wang HM, Chen IH, Chang JT, et al. Association of HPV infections with second primary tumors in early-staged oral cavity cancer. Oral Dis. (2012) 18:809–15. doi: 10.1111/j.1601-0825.2012.01950.x
20. Martel M, Alemany L, Taberna M, Mena M, Tous S, Bague S, et al. The role of HPV on the risk of second primary neoplasia in patients with oropharyngeal carcinoma. Oral Oncol. (2017) 64:37–43. doi: 10.1016/j.oraloncology.2016.11.011
21. Weinberger PM, Yu Z, Haffty BG, Kowalski D, Harigopal M, Brandsma J, et al. Molecular classification identifies a subset of human papillomavirus–associated oropharyngeal cancers with favorable prognosis. J Clin Oncol. (2006) 24:736–47. doi: 10.1200/JCO.2004.00.3335
22. D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. (2007) 356:1944–56. doi: 10.1056/NEJMoa065497
23. Benson E, Li R, Eisele D, Fakhry C. The clinical impact of HPV tumor status upon head and neck squamous cell carcinomas. Oral Oncol. (2014) 50:565–74. doi: 10.1016/j.oraloncology.2013.09.008
24. Rampias T, Sasaki C, Psyrri A. Molecular mechanisms of HPV induced carcinogenesis in head and neck. Oral Oncol. (2014) 50:356–63. doi: 10.1016/j.oraloncology.2013.07.011
25. Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. (2008) 100:261–9. doi: 10.1093/jnci/djn011
26. Licitra L, Perrone F, Bossi P, Suardi S, Mariani L, Artusi R, et al. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J Clin Oncol. (2006) 24:5630–6. doi: 10.1200/JCO.2005.04.6136
27. Klussmann JP, Mooren JJ, Lehnen M, Claessen SM, Stenner M, Huebbers CU, et al. Genetic signatures of HPV-related and unrelated oropharyngeal carcinoma and their prognostic implications. Clin Cancer Res. (2009) 15:1779–86. doi: 10.1158/1078-0432.CCR-08-1463
28. Martinez I, Wang J, Hobson KF, Ferris RL, Khan SA. Identification of differentially expressed genes in HPV-positive and HPV-negative oropharyngeal squamous cell carcinomas. Eur J Cancer. (2007) 43:415–32. doi: 10.1016/j.ejca.2006.09.001
29. Saito Y, Ebihara Y, Ushiku T, Omura G, Kobayashi K, Ando M, et al. Negative human papillomavirus status and excessive alcohol consumption are significant risk factors for second primary malignancies in Japanese patients with oropharyngeal carcinoma. Jpn J Clin Oncol. (2014) 44:564–9. doi: 10.1093/jjco/hyu042
30. Cabanillas R, Astudillo A, Valle M, de la Rosa J, Álvarez R, Durán NS, et al. Novel germline CDKN2A mutation associated with head and neck squamous cell carcinomas and melanomas. Head Neck. (2013) 35:E80–4. doi: 10.1002/hed.21911
31. Jain KS, Sikora AG, Baxi SS, Morris LG. Synchronous cancers in patients with head and neck cancer: risks in the era of human papillomavirus-associated oropharyngeal cancer. Cancer. (2013) 119:1832–7. doi: 10.1002/cncr.27988
32. Mercer RR, Lucas NC, Simmons AN, Zander DS, Tsongalis GJ, Funkhouser WK, et al. Molecular discrimination of multiple primary versus metastatic squamous cell cancers of the head/neck and lung. Exp Mol Pathol. (2009) 86:1–9. doi: 10.1016/j.yexmp.2008.11.003
33. Daher T, Tur MK, Brobeil A, Etschmann B, Witte B, Engenhart-Cabillic R, et al. Combined human papillomavirus typing and TP53 mutation analysis in distinguishing second primary tumors from lung metastases in patients with head and neck squamous cell carcinoma. Head Neck. (2018) 40:1109–19. doi: 10.1002/hed.25041
34. Gasparotto D, Maestro R, Barzan L, Vukosavljevic T, Doglioni C, Sulfaro S, et al. Recurrences and second primary tumours in the head and neck region: differentiation by p53 mutation analysis. Ann Oncol. (1995) 6:933–9. doi: 10.1093/oxfordjournals.annonc.a059362
35. Ronchetti D, Arisi E, Neri A, Pruneri G, Digiuni B, Sambataro G, et al. Microsatellite analyses of recurrence or second primary tumor in head and neck cancer. Anticancer Res. (2005) 25:2771–5.
36. Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter's concept of field cancerization: evidence and clinical implications. Cancer Res. (2003) 63:1727–30.
37. Simple M, Suresh A, Das D, Kuriakose MA. Cancer stem cells and field cancerization of oral squamous cell carcinoma. Oral Oncol. (2015) 51:643–51. doi: 10.1016/j.oraloncology.2015.04.006
38. Kuss I, Hathaway B, Ferris RL, Gooding W, Whiteside TL. Decreased absolute counts of T lymphocyte subsets and their relation to disease in squamous cell carcinoma of the head and neck. Clin Cancer Res. (2004) 10:3755–62. doi: 10.1158/1078-0432.CCR-04-0054
39. Atienza JA, Dasanu CA. Incidence of second primary malignancies in patients with treated head and neck cancer: a comprehensive review of literature. Curr Med Res Opin. (2012) 28:1899–909. doi: 10.1185/03007995.2012.746218
40. Kuss I, Saito T, Johnson JT, Whiteside TL. Clinical significance of decreased zeta chain expression in peripheral blood lymphocytes of patients with head and neck cancer. Clin Cancer Res. (1999) 5:329–34.
41. Grandis JR, Falkner DM, Melhem MF, Gooding WE, Drenning SD, Morel PA. Human leukocyte antigen class I allelic and haplotype loss in squamous cell carcinoma of the head and neck: clinical and immunogenetic consequences. Clin Cancer Res. (2000) 6:2794–802.
42. Ge L, Meng W, Zhou H, Bhowmick N. Could stroma contribute to field cancerization? Med Hypotheses. (2010) 75:26–31. doi: 10.1016/j.mehy.2010.01.019
43. Angadi PV, Patil PV, Kale AD, Hallikerimath S, Babji D. Myofibroblast presence in apparently normal mucosa adjacent to oral squamous cell carcinoma associated with chronic tobacco/areca nut use: evidence for field cancerization. Acta Odontol Scand. (2014) 72:502–8. doi: 10.3109/00016357.2013.871648
44. Chan JS, Tan MJ, Sng MK, Teo Z, Phua T, Choo CC, et al. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. (2017) 8:e2562. doi: 10.1038/cddis.2016.492
46. Brachman DG, Graves D, Vokes E, Beckett M, Haraf D, Montag A, et al. Occurrence of p53 gene deletions and human papilloma virus infection in human head and neck cancer. Cancer Res. (1992) 52:4832–6.
47. Gallo O, Sardi I, Pepe G, Franchi A, Attanasio M, Giusti B, et al. Multiple primary tumors of the upper aerodigestive tract: is there a role for constitutional mutations in the p53 gene? Int J Cancer. (1999) 82:180–6. doi: 10.1002/(SICI)1097-0215(19990719)82:2<180::AID-IJC5>3.0.CO;2-P
48. Jefferies S, Edwards S, Hamoudi R, A'Hern R, Foulkes W, Goldgar D, et al. No germline mutations in CDKN2A (p16) in patients with squamous cell cancer of the head and neck and second primary tumours. Br J Cancer. (2001) 85:1383. doi: 10.1054/bjoc.2001.2068
49. Piccinin S, Gasparotto D, Vukosavljevic T, Barzan L, Sulfaro S, Maestro R, et al. Microsatellite instability in squamous cell carcinomas of the head and neck related to field cancerization phenomena. Br J Cancer. (1998) 78:1147–51. doi: 10.1038/bjc.1998.644
50. Skierucha M, Milne AN, Offerhaus GJ, Polkowski WP, Maciejewski R, Sitarz R. Molecular alterations in gastric cancer with special reference to the early-onset subtype. World J Gastroenterol. (2016) 22:2460–74. doi: 10.3748/wjg.v22.i8.2460
51. Li F, Sturgis EM, Chen X, Zafereo ME, Wei Q, Li G. Association of p53 codon 72 polymorphism with risk of second primary malignancy in patients with squamous cell carcinoma of the head and neck. Cancer. (2010) 116:2350–9. doi: 10.1002/cncr.25072
52. Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A, et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat Genet. (2006) 38:1133–41. doi: 10.1038/ng1879
53. Mojtahedi Z, Hashemi SB, Khademi B, Karimi M, Haghshenas MR, Fattahi MJ, et al. p53 codon 72 polymorphism association with head and neck squamous cell carcinoma. Braz J Otorhinolaryngol. (2010) 76:316–20. doi: 10.1590/S1808-86942010000300008
54. Hamel N, Black MJ, Ghadirian P, Foulkes WD. No association between P53 codon 72 polymorphism and risk of squamous cell carcinoma of the head and neck. Br J Cancer. (2000) 82:757–9. doi: 10.1054/bjoc.1999.0993
55. Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Develop. (2003) 13:77–83. doi: 10.1016/S0959-437X(02)00013-8
56. Gruttgen A, Reichenzeller M, Junger M, Schlien S, Affolter A, Bosch FX. Detailed gene expression analysis but not microsatellite marker analysis of 9p21 reveals differential defects in the INK4a gene locus in the majority of head and neck cancers. J Pathol. (2001) 194:311–7. doi: 10.1002/path.906
57. Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. (2002) 416:560–4. doi: 10.1038/416560a
58. Zheng T, Wang J, Chen X, Meng X, Song X, Lu Z, et al. Disruption of p73-MDM2 binding synergizes with gemcitabine to induce apoptosis in HuCCT1 cholangiocarcinoma cell line with p53 mutation. Tumour Biol. (2010) 31:287–95. doi: 10.1007/s13277-010-0035-7
59. Li G, Sturgis EM, Wang LE, Chamberlain RM, Amos CI, Spitz MR, et al. Association of a p73 exon 2 G4C14-to-A4T14 polymorphism with risk of squamous cell carcinoma of the head and neck. Carcinogenesis. (2004) 25:1911–6. doi: 10.1093/carcin/bgh197
60. Galli P, Cadoni G, Volante M, De Feo E, Amore R, Giorgio A, et al. A case-control study on the combined effects of p53 and p73 polymorphisms on head and neck cancer risk in an Italian population. BMC Cancer. (2009) 9:137. doi: 10.1186/1471-2407-9-137
61. Li F, Sturgis EM, Zafereo ME, Liu Z, Wang LE, Wei Q, et al. p73 G4C14-to-A4T14 polymorphism and risk of second primary malignancy after index squamous cell carcinoma of head and neck. Int J Cancer. (2009) 125:2660–5. doi: 10.1002/ijc.24570
62. Blaes J, Thome CM, Pfenning PN, Rubmann P, Sahm F, Wick A, et al. Inhibition of CD95/CD95L (FAS/FASLG) signaling with APG101 prevents invasion and enhances radiation therapy for glioblastoma. Mol Cancer Res. (2018) 16:767–76. doi: 10.1158/1541-7786.MCR-17-0563
63. Reichmann E. The biological role of the Fas/FasL system during tumor formation and progression. Semin Cancer Biol. (2002) 12:309–15. doi: 10.1016/S1044-579X(02)00017-2
64. Lei D, Sturgis EM, Wang LE, Liu Z, Zafereo ME, Wei Q, et al. FAS and FASLG genetic variants and risk for second primary malignancy in patients with squamous cell carcinoma of the head and neck. Cancer Epidemiol Biomarkers Prev. (2010) 19:1484–91. doi: 10.1158/1055-9965.EPI-10-0030
65. Zhang Z, Wang LE, Sturgis EM, El-Naggar AK, Hong WK, Amos CI, et al. Polymorphisms of FAS and FAS ligand genes involved in the death pathway and risk and progression of squamous cell carcinoma of the head and neck. Clin Cancer Res. (2006) 12:5596–602. doi: 10.1158/1078-0432.CCR-05-1739
66. Llanos S, Garcia-Pedrero JM, Morgado-Palacin L, Rodrigo JP, Serrano M. Stabilization of p21 by mTORC1/4E-BP1 predicts clinical outcome of head and neck cancers. Nat Commun. (2016) 7:10438. doi: 10.1038/ncomms10438
67. Li G, Liu Z, Sturgis EM, Shi Q, Chamberlain RM, Spitz MR, et al. Genetic polymorphisms of p21 are associated with risk of squamous cell carcinoma of the head and neck. Carcinogenesis. (2005) 26:1596–602. doi: 10.1093/carcin/bgi105
68. Li G, Sturgis EM, Wang LE, Chamberlain RM, Spitz MR, El-Naggar AK, et al. Association between the V109G polymorphism of the p27 gene and the risk and progression of oral squamous cell carcinoma. Clin Cancer Res. (2004) 10:3996–4002. doi: 10.1158/1078-0432.CCR-04-0089
69. Wang Z, Sturgis EM, Zhang F, Lei D, Liu Z, Xu L, et al. Genetic variants of p27 and p21 as predictors for risk of second primary malignancy in patients with index squamous cell carcinoma of head and neck. Mol Cancer. (2012) 11:17. doi: 10.1186/1476-4598-11-17
70. Li Q, Lozano G. Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res. (2013) 19:34–41. doi: 10.1158/1078-0432.CCR-12-0053
71. Yu H, Huang YJ, Liu Z, Wang LE, Li G, Sturgis EM, et al. Effects of MDM2 promoter polymorphisms and p53 codon 72 polymorphism on risk and age at onset of squamous cell carcinoma of the head and neck. Mol Carcinog. (2011) 50:697–706. doi: 10.1002/mc.20806
72. Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. (2009) 9:95–107. doi: 10.1038/nrc2584
73. Jin L, Sturgis EM, Zhang Y, Huang Z, Wei P, Guo W, et al. Genetic variants in p53-related genes confer susceptibility to second primary malignancy in patients with index squamous cell carcinoma of head and neck. Carcinogenesis. (2013) 34:1551–7. doi: 10.1093/carcin/bgt096
74. Valentin-Vega YA, Barboza JA, Chau GP, El-Naggar AK, Lozano G. High levels of the p53 inhibitor MDM4 in head and neck squamous carcinomas. Hum. Pathol. (2007) 38:1553–62. doi: 10.1016/j.humpath.2007.03.005
75. Yu H, Wang LE, Liu Z, Wei S, Li G, Sturgis EM, et al. Polymorphisms of MDM4 and risk of squamous cell carcinoma of the head and neck. Pharmacogenet Genomics. (2011) 21:388–96. doi: 10.1097/FPC.0b013e32834632e4
76. Lamarche BJ, Orazio NI, Weitzman MD. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. (2010) 584:3682–95. doi: 10.1016/j.febslet.2010.07.029
77. Ziolkowska-Suchanek I, Mosor M, Wierzbicka M, Rydzanicz M, Baranowska M, Nowak J. The MRN protein complex genes: MRE11 and RAD50 and susceptibility to head and neck cancers. Mol Cancer. (2013) 12:113. doi: 10.1186/1476-4598-12-113
78. Shen H, Sturgis EM, Dahlstrom KR, Zheng Y, Spitz MR, Wei Q. A variant of the DNA repair gene XRCC3 and risk of squamous cell carcinoma of the head and neck: a case-control analysis. Int J Cancer. (2002) 99:869–72. doi: 10.1002/ijc.10413
79. Rydzanicz M, Wierzbicka M, Gajecka M, Szyfter W, Szyfter K. The impact of genetic factors on the incidence of multiple primary tumors (MPT) of the head and neck. Cancer Lett. (2005) 224:263–78. doi: 10.1016/j.canlet.2005.01.015
80. Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. (2001) 104:107–17. doi: 10.1016/S0092-8674(01)00195-7
81. Laantri N, Jalbout M, Khyatti M, Ayoub WB, Dahmoul S, Ayad M, et al. XRCC1 and hOGG1 genes and risk of nasopharyngeal carcinoma in North African countries. Mol Carcinog. (2011) 50:732–7. doi: 10.1002/mc.20754
82. Csejtei A, Tibold A, Koltai K, Varga Z, Szanyi I, Gobel G, et al. Association between XRCC1 polymorphisms and head and neck cancer in a Hungarian population. Anticancer Res. (2009) 29:4169–73.
83. Lou Y, Peng WJ, Cao DS, Xie J, Li HH, Jiang ZX. DNA repair gene XRCC1 polymorphisms and head and neck cancer risk: an updated meta-analysis including 16344 subjects. PLoS ONE. (2013) 8:e74059. doi: 10.1371/journal.pone.0074059
84. Zafereo ME, Sturgis EM, Liu Z, Wang LE, Wei Q, Li G. Nucleotide excision repair core gene polymorphisms and risk of second primary malignancy in patients with index squamous cell carcinoma of the head and neck. Carcinogenesis. (2009) 30:997–1002. doi: 10.1093/carcin/bgp096
85. Jefferies S, Kote-Jarai Z, Goldgar D, Houlston R, Frazer-Williams M-J, A'Hern R, et al. Association between polymorphisms of the GPX1 gene and second primary tumours after index squamous cell cancer of the head and neck. Oral Oncol. (2005) 41:455–61. doi: 10.1016/j.oraloncology.2004.09.012
86. Dequanter D, Dok R, Koolen L, Vander Poorten V, Nuyts S. Prognostic significance of glutathione peroxidase levels (GPx1) in head and neck cancers. Front Oncol. (2017) 7:84. doi: 10.3389/fonc.2017.00084
87. Singh H, Sachan R, Devi S, Pandey SN, Mittal B. Association of GSTM1, GSTT1, and GSTM3 gene polymorphisms and susceptibility to cervical cancer in a North Indian population. Am J Obstet Gynecol. (2008) 198:303 e1–6. doi: 10.1016/j.ajog.2007.09.046
88. Minard CG, Spitz MR, Wu X, Hong WK, Etzel CJ. Evaluation of glutathione S-transferase polymorphisms and mutagen sensitivity as risk factors for the development of second primary tumors in patients previously diagnosed with early-stage head and neck cancer. Cancer. (2006) 106:2636–44. doi: 10.1002/cncr.21928
89. Walker K, Ginsberg G, Hattis D, Johns DO, Guyton KZ, Sonawane B. Genetic polymorphism in N-Acetyltransferase (NAT): population distribution of NAT1 and NAT2 activity. J Toxicol Environ Health B Crit Rev. (2009) 12:440–72. doi: 10.1080/10937400903158383
90. Zhang L, Xiang Z, Hao R, Li R, Zhu Y. N-acetyltransferase 2 genetic variants confer the susceptibility to head and neck carcinoma: evidence from 23 case-control studies. Tumour Biol. (2014) 35:3585–95. doi: 10.1007/s13277-013-1473-9
91. McClelland SE. Role of chromosomal instability in cancer progression. Endocr Relat Cancer. (2017) 24:T23–31. doi: 10.1530/ERC-17-0187
92. Yamamoto H, Imai K. Microsatellite instability: an update. Arch Toxicol. (2015) 89:899–921. doi: 10.1007/s00204-015-1474-0
93. Yamashita K, Arimura Y, Kurokawa S, Itoh F, Endo T, Hirata K, et al. Microsatellite instability in patients with multiple primary cancers of the gastrointestinal tract. Gut. (2000) 46:790–4. doi: 10.1136/gut.46.6.790
94. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. (2002) 3:415–28. doi: 10.1038/nrg816
95. Garinis GA, Patrinos GP, Spanakis NE, Menounos PG. DNA hypermethylation: when tumour suppressor genes go silent. Hum Genet. (2002) 111:115–27. doi: 10.1007/s00439-002-0783-6
96. Longo AL, Rettori MM, de Carvalho AC, Kowalski LP, Carvalho AL, Vettore AL. Evaluation of the methylation profile of exfoliated cell samples from patients with head and neck squamous cell carcinoma. Head Neck. (2014) 36:631–7. doi: 10.1002/hed.23345
97. Rettori MM, de Carvalho AC, Longo AL, de Oliveira CZ, Kowalski LP, Carvalho AL, et al. TIMP3 and CCNA1 hypermethylation in HNSCC is associated with an increased incidence of second primary tumors. J Transl Med. (2013) 11:316. doi: 10.1186/1479-5876-11-316
Keywords: head and neck cancer, multiple primary cancer, field cancerization, cancer-associated fibroblasts, genetic factor, biomarker
Citation: Zhang W, Zhu Z, Huang M, Tang Y-J, Tang Y and Liang X (2019) Susceptibility of Multiple Primary Cancers in Patients With Head and Neck Cancer: Nature or Nurture? Front. Oncol. 9:1275. doi: 10.3389/fonc.2019.01275
Received: 05 September 2019; Accepted: 04 November 2019;
Published: 21 November 2019.
Edited by:
Jorge A. R. Salvador, University of Coimbra, PortugalReviewed by:
Cesare Piazza, National Tumor Institute, ItalyThorsten Fuereder, Medical University of Vienna, Austria
Copyright © 2019 Zhang, Zhu, Huang, Tang, Tang and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-ling Tang, tangyaling@scu.edu.cn; Xin-hua Liang, lxh88866@scu.edu.cn
†These authors have contributed equally to this work