 Sihui Yu
Sihui Yu Xi Li
Xi Li Shiyun Liu
Shiyun Liu Rui Yang
Rui Yang Xiangnan Liu
Xiangnan Liu Sufang Wu
Sufang Wu- 1Department of Obstetrics and Gynecology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Urology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
N6-Methyladenosine (m6A), a pervasive posttranscriptional modification which is reversible, has been among hotspot issues in the past several years. The balance of intracellular m6A levels is dynamically maintained by methyltransferase complex and demethylases. Meanwhile, m6A reader proteins specifically recognize modified residues and convey messages so as to set up an efficient and orderly network of m6A regulation. The m6A mark has proved to affect every step of RNA life cycle, from processing in nucleus to translation or degradation in cytoplasm. Subsequently, disorders in m6A methylation are directly related to aberrant RNA metabolism, which results in tumorigenesis and altered drug response. Therefore, uncovering the underlying mechanism of m6A in oncogenic transformation and tumor progression seeks opportunities for novel targets in cancer therapy. In this review, we conclude the extensive impact of m6A on RNA metabolism and highlight its relevance with human cancer, implicating the far-reaching value in clinical application.
Introduction
N6-methyladenosine (m6A), which refers to the addition of methyl groups to the N-6 position of the adenosine residue, is a pervasive posttranscriptional RNA internal modification of eukaryotes (1). Since its first discovery in the 1970s, m6A had remained an uncharted territory due to technical bottlenecks (2). The stagnation ended in 2011, when the fat mass and obesity-associated protein (FTO) was revealed to exhibit demethylation activity on m6A-modified RNAs (3). The m6A mark was thus identified as a reversible process, which generated refueled passion in this field. To date, scientists have confirmed multiple m6A regulatory enzymes and classified them as “writers,” “erasers,” and “readers” (4).
With the availability of high-throughput sequencing technique, scientists are nowadays capable of detecting m6A methylation at transcriptome-wide level (5, 6). m6A sites are mainly enriched near stop codons, in 3′-untranslated regions (3′-UTRs) and within long internal exons. Besides messenger RNAs (mRNAs) and long non-coding RNAs, a wide range of circular RNAs (circRNAs) generated by back splice events also undergoes m6A modification (7). The m6A-circRNAs frequently arise from exons that are void of m6A peaks in mRNAs. This chemical mark is evolutionarily conserved and falls within a consensus motif RRACH (R = G/A, A = m6A, H = A/C/U) (5, 6, 8). What is more, m6A RNA methylation poses a broad control on RNA metabolism including alternative splicing, subcellular localization, and translational regulation (9). The impact of m6A regulatory enzymes on RNA processes may further interplay with tumor biology, which will be respectively discussed in this review.
m6A Regulatory Enzymes Work in a Cooperative Manner
The m6A regulatory enzymes work cooperatively to maintain the balance of intracellular m6A levels (4) (Figure 1). m6A “writers” composing the methyltransferase complex catalyze this modification positively. This decoration could be reversed by m6A “erasers” harboring demethylase activity. Meanwhile, m6A “readers” specifically recognize modified residues and convey messages so as to set up an efficient and orderly network of m6A regulation.
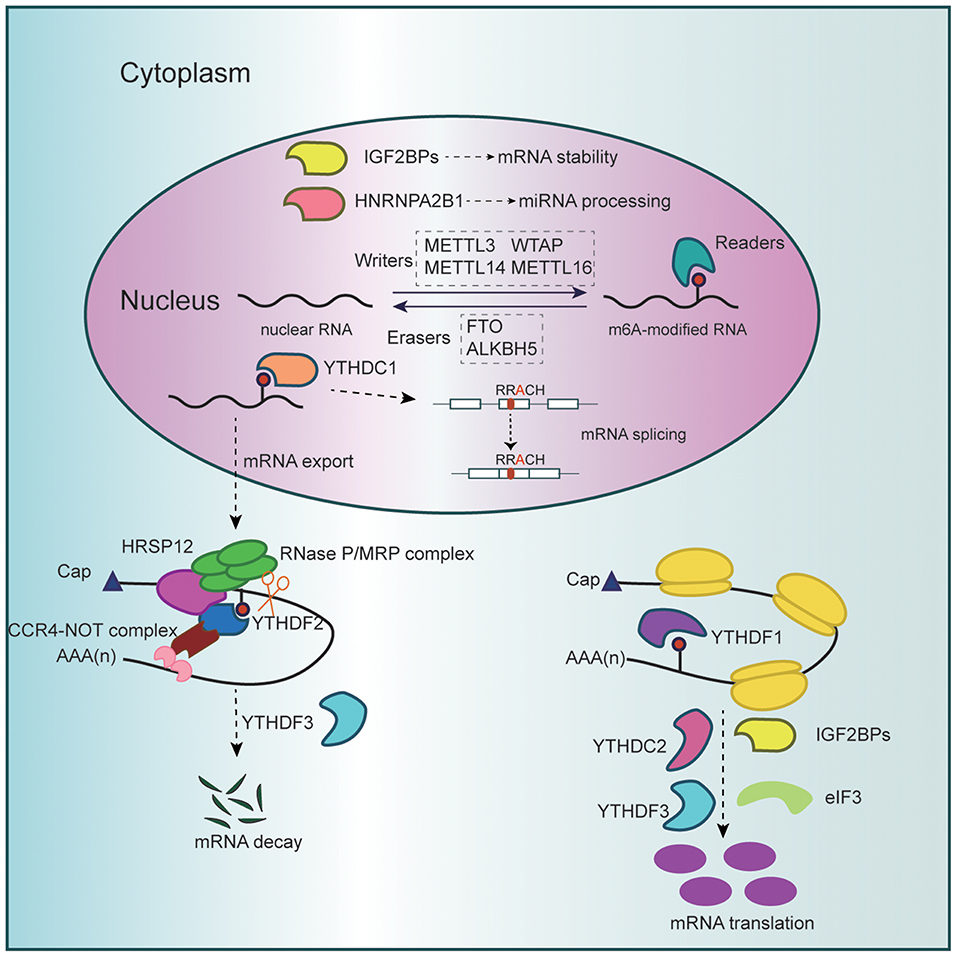
Figure 1. Schematic diagram of RNA m6A methylation. The methyltransferase complex (METTL3, METTL14, WTAP, and METTL16), which catalyzes methylation at the N6 position adenosine, and the demethylases (FTO and ALKBH5), which remove methyl groups, dynamically regulate cellular m6A levels in the nucleus. Besides, m6A modification could be specifically recognized and bound by diverse reader proteins. Nuclear m6A readers promote miRNA processing while affecting mRNA splicing, stability, and export. Cytoplasmic readers mediate m6A-marked mRNA translation and degradation.
The m6A methyltransferase complex is mainly comprised of methyltransferase-like 3 (METTL3), METTL14, and Wilms' tumor 1-associating protein (WTAP), which regulate the distribution of m6A in coordination (10). METTL3 serves as the core component, while METTL14 is integrated with METTL3 as a stable heterodimer and catalyzes m6A RNA methylation through synergistic effect (10, 11). WTAP anchors METTL3–METTL14 complex to target RNAs and promotes its accumulation in nuclear speckles (8). Since WTAP harbors no methyltransferase activity, this regulatory subunit takes effect on the premise of functional m6A methylation complex (12). Scientists have also reported some other proteins that modulate the cellular m6A landscape cooperatively. METTL16, a newly defined m6A writer targeting U6 spliceosomal small nuclear RNA, also regulates S-adenosylmethionine homeostasis by inducing the expression of S-adenosylmethionine synthetase upon methionine starvation (13–16).
The m6A erasers discovered so far involve two candidates, FTO and AlkB homolog 5 (ALKBH5). FTO was initially proved to regulate energy homeostasis and is positively related to risk of obesity (17, 18). ALKBH5 is a homolog of FTO, and they both belong to the Fe(II)- and oxoglutarate-dependent AlkB oxygenase family (17–19). In the m6A circuit, FTO and ALKBH5 identify m6A-modified nuclear RNAs as substrate and catalyze removal of m6A mark (3, 19).
The reader proteins specifically recognize m6A decoration to sort mRNAs for quicker metabolism to further perform biological functions (4). Among these readers, YT521-B homology (YTH) domain proteins are the best documented, including YTH domain family proteins (YTHDF1–3) and YTH domain containing proteins (YTHDC1–2) (20–25). YTHDF1–3 and YTHDC2 are cytoplasmic readers, while YTHDC1 mainly operates in the nucleus. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BP1–3) is a distinct family of readers with K homology (KH) domains to recognize m6A (26). More potential m6A readers are under exploration, such as heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) and eukaryotic initiation factor 3 (eIF3) (27, 28).
m6A Regulates RNA Metabolism in Physiological Conditions
The RNA life cycle comprises RNA processing, export, and translation or degradation. Formidable evidence has shown that m6A and its regulatory enzymes take part in every step of RNA metabolism. Generally speaking, the writers and erasers dictate m6A levels in specific targets, which are decoded by the readers to accelerate RNA process and translate into distinct functions.
RNA Processing in Nucleus
WTAP favors the positioning of METTL3–METTL14 complex in nuclear speckles that are sites associated with RNA processing and transcription (10). As a result, WTAP modulates alternative splicing and gene expression. Similarly, ALKBH5 colocalizes with nuclear speckles, and m6A erasure mediated by ALKBH5 is critical for correct splicing, preventing longer 3′-UTR mRNAs from quick degradation during spermiogenesis (29).
YTHDC1 binds m6A-modified pre-mRNAs and affects RNA binding affinity of splicing factors (30). Under normal circumstances, YTHDC1 promotes mRNA binding of SRSF3 while antagonizing that of SRSF10, predominantly triggering exon inclusion of targeted mRNAs (31) (Figure 1). In addition, YTHDC1 interacts with pre-mRNA 3′ end processing factors such as CPSF6 and determines the length of 3′-UTR where lie many microRNAs (miRNAs) target sites (25). Given that miRNAs pair to mRNAs of protein-coding genes and repress them at posttranscriptional level, YTHDC1 regulates mRNA stability and translation efficiency indirectly (25, 32).
Meanwhile, m6A mark induced by METTL3 on primary miRNAs could be recognized by adaptor readers such as HNRNPA2B1 (27, 33). The microprocessor protein DGCR8 is then recruited to specific precursor miRNAs and encourages their processing into mature miRNAs.
mRNA Export From Nucleus to Cytoplasm
In addition to alternative splicing and exon inclusion, YTHDC1 favors export of methylated mRNAs from nucleus to cytoplasm by the aid of SRSF3, indirectly triggering translation via increased cytoplasmic abundance of targets (34) (Figure 1).
Consistently, Alkbh5 deficiency in male mice lifts m6A levels and facilitates mRNA export to cytoplasm (35, 36). Cytoplasmic levels of mRNAs critical for proper spermatogenic maturation are altered, leading to aberrant spermatogenesis and apoptosis. Thus, it appears that m6A exerts complex roles on subsequent effect of mRNA export. In addition, the latest findings show that Fragile X mental retardation protein, a newly identified m6A reader protein, is also capable of facilitating the nuclear export of m6A-marked transcripts through directly binding to a collection of m6A sites on target mRNAs (37).
mRNA Translation or Degradation in Cytoplasm
After nuclear export, m6A-modified RNAs will be sorted into different groups depending on diverse readers and then undergo a fast-tracking metabolism for translation or dedicated degradation. This process helps to generate adequate protein for urgent demand or rapidly degrade mRNAs in necessity (2).
YTHDF1 binds m6A-modified RNAs and promotes ribosome loading via recruiting initiation factors (eIFs) that are pivotal in the rate-limiting step of translation (20) (Figure 1). Accordingly, YTHDF1 facilitates translation efficiency and protein synthesis. Other readers including YTHDC2, IGF2BPs, and eIF3 also bind m6A at its consensus motif to enhance translation efficiency (23, 24, 26, 28).
On the contrary, YTHDF2 mediates m6A-containing RNA decay, thus regulating gene expression and cell fate (21). YTHDF2 knockdown results in accumulation of untranslated target mRNAs, thereby reducing translation efficiency. Specifically speaking, carboxy-terminal domain of YTHDF2 selectively binds to m6A-modified mRNA, while amino-terminal domain localizes the YTHDF2–mRNA complex to RNA decay sites. Recently, the molecular mechanism underpinning YTHDF2-directed RNA decay has been expounded. m6A-containing linear and circular RNAs undergo endoribonucleolytic cleavage through YTHDF2-HRSP12-RNase P/MRP axis, coupled to CCR4-NOT complex-mediated deadenylating pathway (38) (Figure 1). In addition, a recent study reported that multivalent m6A-modified RNAs could promote the phase separation of YTHDFs and that phase separation of m6A and YTHDF2 might participate in cellular response to stresses, despite the uncertainty of its specific role (39).
After nuclear export of methylated RNAs, YTHDF3 tunes their delivery before YTHDF1 and YTHDF2, especially partitioning their shared targets (22). YTHDF3 accelerates translation or decay of m6A-containing mRNAs in synergy with YTHDF1 and YTHDF2. By the way, a broad range of circular RNAs generated by pre-mRNA back splicing in human transcriptome has coding potential and bears m6A modification (40). YTHDF3 drives protein translation from these circRNAs in a cap-independent fashion.
Except for the YTHDF family members, SND1, a putative m6A reader of the “royal family” also binds to m6A modification of ORF50 RNA and stabilizes the transcript, which favors the replication of Kaposi's sarcoma-associated herpesvirus (41).
m6A Poses Control on Tumorigenesis and Cancer Progression
Researchers have involved m6A decoration in the development of human diseases. Mechanistically, m6A could alter the expression of mRNAs encoding various regulators such as transcription factors and function as either barrier or facilitator of malignant transition in tumor cells. In this section, we respectively state the variable roles of m6A in tumorigenesis and cancer progression based on different m6A regulatory enzymes (Table 1).

Table 1. The summary of roles of m6A subunits in tumor formation and progression.
m6A Writers
METTL3
In glioblastoma stem cells (GSCs), Cui et al. knocked down METTL3 to hinder m6A enrichment, and they also observed enhanced growth, self-renewal of GSCs, and tumor progression (42). In this process, oncogenes such as ADAM19, EPHA3, and KLF4 were upregulated, while expression of tumor suppressors involving CDKN2A, BRCA2, and TP53I11 were impeded. On the contrary, another study argued that METTL3 plays an oncogenic role in glioblastoma via methylating 3′-UTR of SOX2 mRNA, which encodes transcription factors enabling the regain of stem-like properties and efficient DNA repair (43). The m6A modification enhances the stability of SOX2 mRNA. Accordingly, silencing of METTL3 interrupts SOX2-dependent DNA repair, impairs GSC maintenance, and delays tumor propagation in vivo. Different target mRNAs of m6A mark, genetic, and non-genetic heterogeneity of cancer stem cells (CSCs) shall account for the controversy. Studies in normal stem cells have also been performed to complement the results, which are quite different. In adult neural stem cells, depletion of METTL3 reduces m6A levels on transcripts of histone methyltransferase EZH2 and inhibits its protein expression (73). Scientists reported that m6A depletion not only suppressed cell growth but also blocked neuronal development and morphological maturation. This conclusion also implicates certain crosslink between m6A mark and histone modification.
Besides, additional studies have verified the role of METTL3 in oncogenic transformation of various tumors. For instance, METTL3 depletion sensitizes pancreatic cancer cells to anticancer agents such as gemcitabine, 5-fluorouracil, cisplatin, and irradiation (44).
The METTL3-induced m6A mark also drives malignant progression in breast tumor in aid of hepatitis B X-interacting protein (HBXIP), an oncogene in breast cancer cells (45). METTL3 lifts the mRNA and protein levels of HBXIP, which in turn promotes the expression of METTL3 and forms a positive feedback loop. The m6A regulation in mRNA stability could have a bearing on this procedure.
Scientists have also reported in bladder cancer that METTL3 accelerates the processing of pri-miR221/222 via recognition by DGCR8 (46). Subsequently, mature miR221/222 restrains the expression of the antioncogene PTEN and ultimately boosts tumor growth both in vitro and in vivo. Based on the preferential m6A recognition by YTHDF1, METTL3 also facilitates translation of oncogene CDCP1, which plays a pivotal role in bladder cancer progression (47). Simultaneously, this biological process exerts synergistic effect with chemical carcinogens in malignant transformation of uroepithelial cells.
In human lung cancer, gain-of-function study of METTL3 motivates cell growth and invasion, giving rise to tumors of larger size in mouse xenografts (48, 49). METTL3 was found to bind m6A sites near the stop codon of specific mRNAs and recruit eIF3 to translation initiation complex in the 5′ end, which mediates mRNA circularization and ribosome recycling. In this way, METTL3 directly promotes efficient translation of onco-proteins involving BRD4, EGFR, and TAZ. Notably, the methyltransferase activity and m6A-binding readers are proved to be uncoupled. This finding proposes a novel model of METTL3 in translational control, and the molecular determinants, such as the specificity of target mRNAs and localization of m6A peaks, are worth in-depth investigation.
In acute myeloid leukemia cells (AMLs), the abundance of METTL3 is elevated compared to that in normal hematopoietic stem/progenitor cells (HSPCs) (50). Cell proliferation is inhibited along with depletion of this enzyme, and leukemogenesis is also delayed in vivo. Besides, METTL3 level is negatively relevant to the status of differentiation and apoptosis in AML cells. Inactivation of AKT induced by METTL3 overexpression contributes partially to the block of differentiation in an m6A-independent manner. Further research suggests that METTL3 promotes the translation of functional proteins regulating cell cycle progression and apoptosis, such as c-MYC and BCL2. A later study instructed that METTL3 is recruited by the CAATT-box binding protein CEBPZ to promoters of active genes and mediates m6A methylation within coding regions of target transcripts (51). Translation of genes necessary for AML is thus enhanced via relieved ribosome stalling. As a result, an alternative mechanism of METTL3 in translational regulation has been put forward.
In hepatocellular carcinoma (HCC), METTL3 is significantly upregulated and indicates poor prognosis (52). Mechanistically, METTL3 promotes HCC growth and invasiveness by repressing the expression of suppressor of cytokine signaling 2 (SOCS2), a tumor suppressor in HCC, through m6A-YTHDF2-dependent mRNA degradation.
Nevertheless, expression of METTL3 is reduced in endometrial carcinoma, which stimulates AKT signaling and promotes tumor growth and invasiveness both in vitro and in vivo (53). Mechanistically, lower expression of METTL3 reduces m6A methylation, restrains YTHDF1-promoted translation of PHLPP2, a negative AKT regulator, while dampens YTHDF2-promoted decay of transcripts encoding mTORC2, which is a positive AKT regulator.
In a nutshell, METTL3 modulates the expression of oncogenes and tumor suppressor genes primarily at posttranscriptional levels, including mRNA stability and translational process. Consequently, different downstream targets of METTL3 and the dominant cancer-related pathways involved in the process bring about the discrepancy in cell fate of different tumors.
METTL14
As is the case of METTL3 in glioblastoma, METTL14 depletion facilitates the malignant phenotype, characterized by upregulated oncogenes such as ADAM19 and reduced expression of tumor suppressors such as CDKN2A (42). Meanwhile, loss-of-function mutation of METTL14 in endometrial tumor also diminishes m6A methylation, inhibits YTHDF1-mediated translation of PHLPP2, and impedes YTHDF2-related mRNA decay of mTORC2, both of which regulate AKT pathway, as aforementioned in METTL3 (53). Subsequently, cell proliferation and tumorigenicity of endometrial tumors are increased, along with AKT stimulation.
Furthermore, m6A modification is suppressed in HCC tissues, and METTL14 downregulation suggests poor prognosis for recurrence-free survival (54). In HCC, METTL14 restrains metastasis by enhancing pri-miR126 process into mature miRNA in a DGCR8-dependent manner. This result is opposite to the conclusion drawn by Chen et al. in primary HCC tissues that m6A is significantly increased and overexpression of METTL3 promotes liver carcinogenesis through m6A-YTHDF2-dependent degradation of SOCS2 mRNAs (52). The controversy may be attributed to complex factors, including different reader proteins to sort mRNA transcripts, as well as distinct tumor samples and methodology of m6A detection.
However, the opposite conclusion has been drawn in hematopoietic diseases. In normal HSPCs and AML cells carrying t(11q23), t(15;17), or t(8;21), METTL14 is overexpressed and exerts oncogenic role through m6A signal by positively manipulating the stability and translation of MYB and MYC mRNA (55). This result is partially overlapped with the impact of METTL3 in AML, and might be explained by alternative reading process mediated by IGF2BPs, for an example. METTL14 undertakes an essential role in self-renewal of leukemia stem/initiation cells (LSCs/LICs) and AML progression (55). Silencing of METTL14 facilitates differentiation of both normal HSPCs and AML cells while repressing AML cell survival.
WTAP
In cancerous tissues of glioblastoma and cholangiocarcinoma, WTAP is overexpressed and promotes cell migration and invasion (74, 75). However, the regulation WTAP exerts on cell proliferation is cell-type specific. In AML, WTAP supports tumor growth but arrests differentiation of leukemia cells (76).
In renal cell carcinoma, WTAP indicates poor survival of patients, and knockdown of WTAP impedes cell proliferation in vitro and tumorigenesis in vivo (56). Mechanistically, WTAP binds to 3′-UTR of cyclin-dependent protein kinase 2 (CDK2) mRNAs and stabilizes the transcripts, lifting CDK2 protein level. As a key regulator of cell cycle, upregulation of CDK2 enables cell to cross the G1/S limit and initiates DNA replication. Similarly, in pancreatic cancer, WTAP promotes cell migration, invasion, and chemoresistance to gemcitabine via stabilizing focal adhesion kinase (Fak) mRNA and subsequently activating Fak-PI3K-AKT and Fak-Src-GRB2-Erk1/2 pathways (57).
To the best of our knowledge, WTAP promotes tumorigenic change in a variety of tumors. However, the underlying mechanism remains elusive. Future researches are required to unveil whether the regulatory role of WTAP in m6A decoration is linked to these biological processes.
m6A Erasers
FTO
A number of studies have attested to the tumorigenic role of FTO in various sorts of cancers. In endometrial carcinoma, β-estradiol induces expression of FTO and mediates cell growth and invasion (77). In addition, FTO inhibitor has been reported to abolish the expression of oncogenes such as ADAM19 and to suppress GSC growth, self-renewal in vitro, and tumor development in vivo (42).
Silencing of FTO also attenuates cell growth and metastasis in breast cancer (58). Mechanistically, FTO disturbs the expression of BNIP3, a proapoptotic gene, both in mRNA and protein levels, via demethylating m6A residues in 3′-UTR. On the other hand, YTHDF2 binding has proved to be uncoupled.
FTO is also significantly overexpressed in AMLs with t(11q23)/MLL rearrangements, t(15;17)/PML-RARA, FLT3-ITD, or NPM1 mutations (59). Reducing m6A levels in ASB2 and RARA mRNAs, FTO destabilizes the transcripts and, as a result, enhances leukemogenesis while blocks cell differentiation induced by all-trans-retinoic acid (ATRA) in these AML subtypes. Besides, researches also precluded YTHDF1/2 as readers regulating the stability of ASB2 and RARA mRNAs.
Interestingly, R-2-hydroxyglutarate (R-2HG), a metabolic product in isocitrate dehydrogenase mutant cancers such as AML, is similar to α-KG structurally and competitively represses Fe (II)/α-KG-dependent dioxygenases (60). Thus, FTO could be suppressed by R-2HG in sensitive leukemia cells to elevate global m6A RNA modification, which destabilizes the MYC/CEBPA transcripts and reduces their expression. As a crucial transcription factor in leukemogenesis, CEBPA being inhibited further inactivates FTO as a feedback loop and reinforces the growth-suppressive effect. Compared with METTL14 which promotes MYC mRNA stability via modulating m6A abundance on 3′-terminal exons, FTO enhances MYC expression by demethylating m6A sites on 5′-terminal and internal exons, which inhibits the YTHDF2-mediated RNA decay (55, 60).
However, in epithelial ovarian cancers (EOC) with BRCA mutation, downregulation of FTO confers resistance to PARP inhibitors such as Olaparib, with m6A enrichment in 3′-UTR regions of FZD10 and increased mRNA stability (61). FZD10 positively upregulates Wnt/β-catenin pathway and further promotes activity of homologous recombination. Meanwhile, stabilization of FZD10 mRNA is mainly caused by the predominant effect of IGF2BP2, also overexpressed in resistant cells.
Obviously, when FTO mediates deprivation of m6A signaling that is previously recognized by readers promoting mRNA stability, corresponding mRNA levels would be impaired. On the contrary, protein-coding mRNAs would be upregulated if FTO prevents YTHDF2-mediated mRNA decay via m6A erasure. In this way, different binding proteins and downstream targets regulate the trend of tumor growth in coordination.
ALKBH5
Similar to FTO in BRCA-mutated EOC, expression of ALKBH5 is also inhibited, which activates Wnt/β-catenin pathway via stabilizing FZD10 mRNA and renders cell resistance to Olaparib (61).
However, all sites subject to m6A modification are not equally critical, since they are chosen to be involved in different biological pathways. ALKBH5 may play distinct roles from FTO due to their preference in molecular substrates. In hypoxic breast cancer cells, ALKBH5 demethylates NANOG mRNA and elevates the protein level via reduced mRNA decay, on the premise of hypoxia-inducible factors (HIFs) (62). As a pluripotency factor, upregulation of NANOG leads to enrichment of breast CSCs (BCSCs). Otherwise, ALKBH5 knockdown inhibits NANOG expression, reduces BCSC population, and impairs tumor formation in vivo.
Similarly, in glioblastoma, GSCs proliferation and tumor formation is disrupted upon ALKBH5 inhibition (63). Owing to enzymatic activity of ALKBH5, the nascent transcripts encoding FOXM1, a transcription factor, are stabilized and thus increases expression of relevant protein. Besides, interplay between ALKBH5 and FOXM1 can be enhanced by a non-coding RNA antisense to FOXM1 (FOXM1-AS).
m6A Readers
YTHDF1
In colorectal cancer tissues, c-Myc drives the expression of YTHDF1 transcriptionally, and high level of YTHDF1 suggests poor prognosis in patients (64). Knockdown of YTHDF1 hinders cell proliferation and renders sensitization to fluorouracil and oxaliplatin. However, the detailed mechanism remains unknown.
Notably, YTHDF1 recognizes m6A-marked transcripts of lysosomal proteases and promotes translation of lysosomal cathepsins in dendritic cells, which favors antigen degradation (78). Cross-presentation of engulfed neoantigens and cross-priming of CD8+ T cells are then suppressed, contributing to the immune evasion and incomplete tumor elimination.
On the other hand, in ocular melanoma, YTHDF1 promotes the translation of m6A-containing HINT2 mRNA, a tumor suppressor (65). Scientists reported decreased m6A levels in these tumor samples, which was significantly correlated with tumor progression both in vitro and in vivo. Therefore, specific m6A-modified targets of YTHDF1 might vary according to the cellular context, resulting in different functions of YTHDF1 in various tumors.
Interestingly, a recently released study demonstrated the critical and contradictory role of YTHDF1 in hypoxia adaptation and pathogenesis of non-small cell lung cancer (66). Under normoxia conditions, YTHDF1 depletion restrains non-small cell lung cancer tumor growth in vitro and in vivo, which resulted from reduced translational efficiency of m6A-marked transcripts such as CDK2, CDK4, and cyclin D1. On the other side, YTHDF1 deficiency renders resistance of cancer cells to cisplatin and indicates poor clinical outcome. Further study revealed that, under chemotherapy stress condition, YTHDF1 depletion leads to decreased translation of m6A-modified Keap1, which upregulates Nrf2 and AKR1C1, the clearance system of reactive oxygen species. The adverse results highlight the importance of achieving a homeostasis of YTHDF1 expression and its targets between normal and stressful conditions.
YTHDF2
In HCC cells, YTHDF2 can be specifically restricted by hypoxia and act as a tumor suppressor with inhibitory effect on tumor growth (67). Mechanistically, YTHDF2 directly binds m6A sites in 3′-UTR and mediates the degradation of EGFR mRNA, which is a main upstream regulator of extracellular-signal-regulated kinase/mitogen-activated protein kinase pathway. Hou et al. have also revealed in HCC that YTHDF2 reduction provokes inflammation and vascular reconstruction, which facilitates the progression of tumor metastasis (68). In detail, the YTHDF2-mediated decay of m6A-containing mRNAs are disrupted, such as interleukin 11 (IL11) and serpin family E member 2 (SERPINE2), which are account for the inflammation-associated malignancy and vascular abnormalization. What is more, administration of PT2385, a small molecule inhibitor targeting HIF-2α and restoring the expression of YTHDF2, also exhibits favorable effects in treating HCC cells both in vitro and in vivo.
Moreover, the roles of YTHDF2 in different context mainly depend on the degradation of respective target mRNAs. Paris et al. reported that YTHDF2 shortens half-life of m6A-modified mRNAs of TNF receptor 2 (TNFR2), which normally prevents accumulation of leukemic cells and thus facilitates AML propagation (69). Targeting YTHDF2 not only eradicates LSCs but also expands hematopoietic stem cells (HSCs) to enhance myeloid reconstitution. In consequence, YTHDF2 inhibitor is considered as a candidate strategy for AML treatment. A noteworthy phenomenon in biological condition is that YTHDF2 mediates clearance of m6A-modified mRNAs of Wnt-related genes to suppress Wnt signaling at stable state and maintain HSC quiescence (79). Upon hematological stresses, downregulation of YTHDF2 aberrantly upregulates target genes of Wnt signaling as well as survival-associated genes, which elevates not only proliferation but also regeneration capacity of HSCs synergistically, as a protective measure. Thus, we could gain a better understanding of the dual character of YTHDF2 in stem cells under physiological and pathological conditions.
YTHDC2
In colon cancer tissues, expression of YTHDC2 is positively correlated with the tumor stage (70). Further research shows that YTHDC2 unwinds highly structured 5′-UTR of mRNAs encoding transcription factors, HIF-1α and Twist1, and facilitates their translation. Notably, HIF-1α promotes epithelial-to-mesenchymal transition via the key regulator Twist1, initiating tumor metastasis.
IGF2BPs
IGF2BPs, a group of direct m6A-binding proteins, enhance mRNA stability and translation both under normal and stress conditions, which gives rise to accumulation of oncogenic products such as MYC (26). In the absence of IGF2BPs, cell proliferation and invasion are significantly repressed in cervical and liver cancer cells.
IGF2BP1 impairs miRNA-directed degradation of mRNAs and sustains expression of serum response factor in ovarian, liver, and lung cancers, potentially in an m6A dependent manner (71). This process enhances serum response factor-driven transcription and upregulates oncogenic drivers such as PDLIM7 and FOXK1.
Furthermore, IGF2BP2 has recently been proven to mediate colorectal liver metastasis, testified both in vitro and in vivo with metastasis PDX models (72). Mechanistically, the m6A modification of circNSUN2 is recognized by YTHDC1, which accelerates the cytoplasmic export and further stabilizes HMGA2 mRNA by forming a circNSUN2/IGF2BP2/HMGA2 ternary complex in the cytoplasm. This outcome suggests a brand-new role of IGF2BP2 in mRNA stabilization via an m6A-independent way and provides evidence that m6A-modified circRNAs could serve as prognostic markers.
Bioinformatics: An Emerging Series of Tools for m6A Exploration
Meanwhile, with the field of bioinformatics booming in the past several years, researchers have established a number of databases delineating m6A machinery, which provides valuable and comprehensive clues for future study (80–84). For instance, RMBase v2.0 deciphers the landscape of RNA modifications based upon epitranscriptome-sequencing data, while MODOMICS provides information regarding RNA modification pathways (80, 81).
Certainly, there exist multiple databases dedicated to the improvement of the m6A-associated knowledge. To take MeT-DB v2.0 as an example, a powerful platform for methyl-transcriptomic research, identifies m6A peaks as well as single-base sites (82). More importantly, context-specific functions of m6A are elucidated via peak distribution plot and gene expression profiles under different conditions to identify m6A-driven genes and networks. Another database, m6AVar, allows annotation and visualization of functional variants in the vicinity of m6A sites and helps interpret their impact on m6A mark by converting RNA sequences of target sites or key flanking nucleotides (83). This database also incorporates data from genome-wide association studies and ClinVar to identify disease-causing variants and explore their pathogenic molecular mechanisms. Both of the two databases intersect m6A-modified sites with functional data such as binding sites of RNA-binding proteins and splicing factors as well as miRNA target sites to obtain regulatory pairs and speculate their roles in posttranscriptional regulation (82, 83).
In addition, a study has recently reported the molecular feature and clinical relevance of m6A regulators reconstituted across 33 cancer types (84). The authors found widespread genetic alterations (mutations and copy number variations) to m6A enzymes and established the cross-talk between their expression patterns with activity of cancer hallmark-related pathways, putatively helpful in prognostic stratification. Thus, we could see that bioinformatic tools not only complements the experimental results but also expedites the discovery of unrecognized regulatory roles of m6A mark.
Clinical Relevance of m6A-Targeted Strategy
So far, small-molecule inhibitors targeting m6A regulatory enzymes are not available in clinical use. However, due to the tumorigenic role of FTO in various cancers, scientist have developed several FTO inhibitors as promising tools in antileukemia and antiglioblastoma therapies.
As mentioned above, R-2HG exhibits broad antiproliferative effects in high-FTO leukemia via targeting FTO/MYC/CEBPA signaling (60). Meanwhile, R-2HG also has synergistic effect with first-line chemotherapy drugs such as decitabine and daunorubicin, which was validated in mouse models. Later on, Huang et al. utilized structure-guided design and developed two small-molecule FTO inhibitors, FB23 and its derivative FB23-2 (85). In comparison, the latter shows significantly improved antiproliferative activity in AML cells and induces cell differentiation. The authors also observed delayed AML progression and prolonged survival in vivo, which enlightens the strategy of targeting FTO demethylase in AML treatment.
Meclofenamic acid (MA) is originally approved by the Food and Drug Administration as a non-steroidal anti-inflammatory drug (86). MA2, which refers to the ethyl ester form of MA, has been identified as a selective FTO inhibitor, increasing m6A levels in mRNAs. Application of MA2 represses GSC-initiated tumor progression and extends lifespan of xenografted mice (42).
In addition, scientist have newly identified entacapone, an inhibitor of catechol-O-methyltransferase applied for treatment of Parkinson's disease, as a chemical FTO inhibitor (87). Entacapone elicits effects on metabolic homeostasis through selectively targeting FTO activity, whereas its function in tumorigenesis remains to be elucidated.
Novel anticancer agents targeting other m6A enzymes could possibly have therapeutic value as well. For example, METTL14 inhibitors are likely to be effective strategies to treat specific AML subtypes with high METTL14 expression, especially in combination with standard agents that induce myeloid differentiation (55). Combinatorial treatment of METTL3 inhibitors plus chemo- or radiotherapy may probably display much better outcome in pancreatic cancer patients (44).
Furthermore, CSCs refer to a group of rare immortal cells that could maintain clones of continuously growing tumors (88). The stem-cell frequency in a cancer is correlated with prognosis and therefore, targeting CSCs through m6A regulation might be beneficial. For instance, competitive antagonists inhibiting ALKBH5 over other AlkB subfamily proteins such as FTO could possibly reduce the enrichment of BCSCs and impair their ability to initiate breast tumor (62). Nevertheless, m6A regulatory enzymes might exert distinct impact on stem cells in physiological and pathological conditions as mentioned above (42, 43, 69, 73, 79). It is plausible to put forward that the m6A-targeted strategy in CSCs must be conducted on the premise of distinguishing the normal stem cells from CSCs.
Conclusion
Evidently, m6A modification has tremendous influence on RNA life cycle including RNA processing, nuclear export, and translation or degradation. At the same time, m6A is involved in biological processes such as stem cell maintenance, tissue differentiation, and immune response. It seems that cellular m6A levels need to be kept within an optimal range, whereas aberrant expression of m6A factors will lead to cancer progression. Scientists have explored the impact of m6A modification on gene expression and altered cell phenotypes, in hope of presenting novel approaches to conquer diseases.
However, clinical practice of small-molecular inhibitors targeting enzymes modulating m6A levels has a great prospect but is still in its infancy. Several issues need to be tackled for the realization of its full potential. A major problem is that we need to gain a better understanding of the selectivity in transcripts and methylated sites in various tissues. Methylation patterns on transcripts might be molecular markers, which recruit distinct m6A readers to enter downstream metabolism, respectively. Subsequently, side effects caused by the complex mRNAs targeted by m6A enzymes may prevent the agents from achieving a favorable therapeutic index in the clinic. Moreover, heterogeneity in human cancer gives rise to distinct karyotypic patterns, protein and biomarker levels, and genetic profiles, which also requires consideration (89).
In conclusion, molecular mechanism of m6A regulation in cancer biology still requests further exploration. Future researches could be focused on seeking the general discipline of specific interaction between m6A mark and reader proteins as well as the heterogeneity in distinct tumor origins. Undoubtedly, m6A methylation harbors great potential in exploiting brand-new therapies for human cancers. In the future, combination of small-molecule inhibitors targeting m6A modification, biological agents, and immunotherapies may improve patient outcomes.
Author Contributions
SY and XLi wrote the first draft of the manuscript. SL organized the structure of the manuscript. RY, XLiu, and SW contributed conception of the work. All authors contributed to manucript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. (2013) 29:108–15. doi: 10.1016/j.tig.2012.11.003
2. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. (2017) 18:31–42. doi: 10.1038/nrm.2016.132
3. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687
4. Meyer KD, Jaffrey SR. Rethinking m6A readers, writers, and erasers. Annu Rev Cell Dev Biol. (2017) 33:319–42. doi: 10.1146/annurev-cellbio-100616-060758
5. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. (2012) 485:201–6. doi: 10.1038/nature11112
6. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
7. Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. (2017) 20:2262–76. doi: 10.1016/j.celrep.2017.08.027
8. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. (2014) 24:177–89. doi: 10.1038/cr.2014.3
9. Maity A, Das B. N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J. (2016) 283:1607–30. doi: 10.1111/febs.13614
10. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432
11. Bujnicki JM, Feder M, Radlinska M, Blumenthal RM. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m6A methyltransferase. J Mol Evol. (2002) 55:431–44. doi: 10.1007/s00239-002-2339-8
12. Sorci M, Ianniello Z, Cruciani S, Larivera S, Ginistrelli LC, Capuano E, et al. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. (2018) 9:796. doi: 10.1038/s41419-018-0843-z
13. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. (2017) 169:824–35.e14. doi: 10.1016/j.cell.2017.05.003
14. Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, et al. S-adenosylmethionine synthesis is regulated by selective N6-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. (2017) 21:3354–63. doi: 10.1016/j.celrep.2017.11.092
15. Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, et al. Human METTL16 is a N6-methyladenosine(m6A) (mA) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. (2017) 18:2004–14. doi: 10.15252/embr.201744940
16. Mendel M, Chen KM, Homolka D, Gos P, Pandey RR, McCarthy AA, et al. Methylation of structured RNA by the m6A writer METTL16 is essential for mouse embryonic development. Mol Cell. (2018) 71:986–1000.e11. doi: 10.1016/j.molcel.2018.08.004
17. Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Brüning JC, et al. Inactivation of the Fto gene protects from obesity. Nature. (2009) 458:894–8. doi: 10.1038/nature07848
18. Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. (2010) 42:1086–92. doi: 10.1038/ng.713
19. Tsujikawa K, Koike K, Kitae K, Shinkawa A, Arima H, Suzuki T, et al. Expression and sub-cellular localization of human ABH family molecules. J Cell Mol Med. (2007) 11:1105–16. doi: 10.1111/j.1582-4934.2007.00094.x
20. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
21. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. (2014) 505:117–20. doi: 10.1038/nature12730
22. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N-methyladenosine-modified RNA. Cell Res. (2017) 27:315–28. doi: 10.1038/cr.2017.15
23. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. (2017) 27:1115–27. doi: 10.1038/cr.2017.99
24. Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of mA transcripts by the 3′ → 5′ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. (2017) 68:374–87.e12. doi: 10.1016/j.molcel.2017.09.021
25. Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. (2018) 14:e1007412. doi: 10.1371/journal.pgen.1007412
26. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
27. Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell. (2015) 162:1299–308. doi: 10.1016/j.cell.2015.08.011
28. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5′ UTR m6A promotes cap-independent translation. Cell. (2015) 163:999–1010. doi: 10.1016/j.cell.2015.10.012
29. Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3'-UTR mRNAs in male germ cells. Proc Natl Acad Sci USA. (2018) 115:E325–33. doi: 10.1073/pnas.1717794115
30. Adhikari S, Xiao W, Zhao YL, Yang YG. m6A: signaling for mRNA splicing. RNA Biol. (2016) 13:756–9. doi: 10.1080/15476286.2016.1201628
31. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell. (2016) 61:507–19. doi: 10.1016/j.molcel.2016.01.012
32. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. (2009) 136:215–33. doi: 10.1016/j.cell.2009.01.002
33. Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. (2015) 519:482–5. doi: 10.1038/nature14281
34. Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N-methyladenosine methylated mRNAs. eLife. (2017) 6:e31311. doi: 10.7554/eLife.31311.040
35. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
36. Zheng Q, Hou J, Zhou Y, Li Z, Cao X. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat Immunol. (2017) 18:1094–103. doi: 10.1038/ni.3830
37. Hsu PJ, Shi H, Zhu AC, Lu Z, Miller N, Edens BM, et al. The RNA-binding protein FMRP facilitates the nuclear export of N6-methyladenosine-containing mRNAs. J Biol Chem. (2019). doi: 10.1074/jbc.AC119.010078. [Epub ahead of print].
38. Park OH, Ha H, Lee Y, Boo SH, Kwon DH, Song HK, et al. Endoribonucleolytic cleavage of m6A-containing RNAs by RNase P/MRP complex. Mol Cell. (2019) 74:494–507.e8. doi: 10.1016/j.molcel.2019.02.034
39. Gao Y, Pei G, Li D, Li R, Shao Y, Zhang QC, et al. Multivalent m6A motifs promote phase separation of YTHDF proteins. Cell Res. (2019). 29:767–9. doi: 10.1038/s41422-019-0210-3
40. Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N-methyladenosine. Cell Res. (2017) 27:626–41. doi: 10.1038/cr.2017.31
41. Baquero-Perez B, Antanaviciute A, Yonchev ID, Carr IM, Wilson SA, Whitehouse A. The Tudor SND1 protein is an m6A RNA reader essential for replication of Kaposi's sarcoma-associated herpesvirus. eLife. (2019) 8:e47261. doi: 10.7554/eLife.47261
42. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. (2017) 18:2622–34. doi: 10.1016/j.celrep.2017.02.059
43. Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. (2018) 37:522–33. doi: 10.1038/onc.2017.351
44. Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. (2018) 52:621–9. doi: 10.3892/ijo.2017.4219
45. Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. (2018) 415:11–9. doi: 10.1016/j.canlet.2017.11.018
46. Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. (2019) 18:110. doi: 10.1186/s12943-019-1036-9
47. Yang F, Jin H, Que B, Chao Y, Zhang H, Ying X, et al. Dynamic m6A mRNA methylation reveals the role of METTL3-m6A-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. (2019) 38:4755–72. doi: 10.1038/s41388-019-0755-0
48. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m6A Methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. (2016) 62:335–45. doi: 10.1016/j.molcel.2016.03.021
49. Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. (2018) 561:556–60. doi: 10.1038/s41586-018-0538-8
50. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. (2017) 23:1369–76. doi: 10.1038/nm.4416
51. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. (2017) 552:126–31. doi: 10.1038/nature24678
52. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. (2018) 67:2254–70. doi: 10.1002/hep.29683
53. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. (2018) 20:1074–83. doi: 10.1038/s41556-018-0174-4
54. Ma JZ, Fu Y, Chuan-chuan Z, Feng L, Ji-hang Y, Fang W, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6-methyladenosine-dependent primary MicroRNA processing. Hepatology. (2017) 65:529–43. doi: 10.1002/hep.28885
55. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. (2018) 22:191–205.e9. doi: 10.1016/j.stem.2017.11.016
56. Tang J, Wang F, Cheng G, Si S, Sun X, Han J, et al. Wilms' tumor 1-associating protein promotes renal cell carcinoma proliferation by regulating CDK2 mRNA stability. J Exp Clin Cancer Res. (2018) 37:40. doi: 10.1186/s13046-018-0706-6
57. Li BQ, Liang ZY, Seery S, Liu QF, You L, Zhang TP, et al. WT1 associated protein promotes metastasis and chemo-resistance to gemcitabine by stabilizing Fak mRNA in pancreatic cancer. Cancer Lett. (2019) 451:48–57. doi: 10.1016/j.canlet.2019.02.043
58. Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. (2019) 18:46. doi: 10.1186/s12943-019-1004-4
59. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. (2017) 31:127–41. doi: 10.1016/j.ccell.2016.11.017
60. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. (2018) 172:90–105.e23. doi: 10.1016/j.cell.2017.11.031
61. Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, et al. N-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. (2019) 79:2812–20. doi: 10.1158/0008-5472.CAN-18-3592
62. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. (2016) 113:E2047–56. doi: 10.1073/pnas.1602883113
63. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m6A Demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. (2017) 31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013
64. Nishizawa Y, Konno M, Asai A, Koseki J, Kawamoto K, Miyoshi N, et al. Oncogene c-Myc promotes epitranscriptome m6A reader YTHDF1 expression in colorectal cancer. Oncotarget. (2018) 9:7476–86. doi: 10.18632/oncotarget.23554
65. Jia R, Chai P, Wang S, Sun B, Xu Y, Yang Y, et al. m6A modification suppresses ocular melanoma through modulating HINT2 mRNA translation. Mol Cancer. (2019) 18(1):161. doi: 10.1186/s12943-019-1088-x
66. Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L, et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. (2019) 10:4892. doi: 10.1038/s41467-019-12801-6
67. Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. (2019) 442:252–61. doi: 10.1016/j.canlet.2018.11.006
68. Hou J, Zhang H, Liu J, Zhao Z, Wang J, Lu Z, et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol Cancer. (2019) 18:163. doi: 10.1186/s12943-019-1082-3
69. Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA m6A Reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. (2019) 25:137–148.e6. doi: 10.1016/j.stem.2019.03.021
70. Tanabe A, Tanikawa K, Tsunetomi M, Takai K, Ikeda H, Konno J, et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1α mRNA is translated. Cancer Lett. (2016) 376:34–42. doi: 10.1016/j.canlet.2016.02.022
71. Müller S, Glaß M, Singh AK, Haase J, Bley N, Fuchs T, et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. (2019) 47:375–90. doi: 10.1093/nar/gky1012
72. Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, et al. N-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. (2019) 10:4695. doi: 10.1038/s41467-019-12651-2
73. Chen J, Zhang YC, Huang C, Shen H, Sun B, Cheng X, et al. m6A regulates neurogenesis and neuronal development by modulating histone methyltransferase Ezh2. Genomics Proteomics Bioinformatics. (2019) 17:154–68. doi: 10.1016/j.gpb.2018.12.007
74. Jin DI, Lee SW, Han ME, Kim HJ, Seo SA, Hur GY, et al. Expression and roles of Wilms' tumor 1-associating protein in glioblastoma. Cancer Sci. (2012) 103:2102–9. doi: 10.1111/cas.12022
75. Jo HJ, Shim HE, Han ME, Kim HJ, Kim KS, Baek S, et al. WTAP regulates migration and invasion of cholangiocarcinoma cells. J Gastroenterol. (2013) 48:1271–82. doi: 10.1007/s00535-013-0748-7
76. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Proia D, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. (2014) 28:1171–4. doi: 10.1038/leu.2014.16
77. Zhang Z, Zhou D, Lai Y, Liu Y, Tao X, Wang Q, et al. Estrogen induces endometrial cancer cell proliferation and invasion by regulating the fat mass and obesity-associated gene via PI3K/AKT and MAPK signaling pathways. Cancer Lett. (2012) 319:89–97. doi: 10.1016/j.canlet.2011.12.033
78. Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature. (2019) 566:270–4. doi: 10.1038/s41586-019-0916-x
79. Wang H, Zuo H, Liu J, Wen F, Gao Y, Zhu X, et al. Loss of YTHDF2-mediated m6A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res. (2018) 28:1035–8. doi: 10.1038/s41422-018-0082-y
80. Xuan JJ, Sun WJ, Lin PH, Zhou KR, Liu S, Zheng LL, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. (2018) 46:D327–34. doi: 10.1093/nar/gkx934
81. Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. (2018) 46:D303–7. doi: 10.1093/nar/gkx1030
82. Liu H, Wang H, Wei Z, Zhang S, Hua G, Zhang SW, et al. MeT-DB V2.0: elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. (2018) 46:D281–7. doi: 10.1093/nar/gkx1080
83. Zheng Y, Nie P, Peng D, He Z, Liu M, Xie Y, et al. m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. (2018) 46:D139–45. doi: 10.1093/nar/gkx895
84. Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X, et al. Molecular characterization and clinical relevance of m6A regulators across 33 cancer types. Mol Cancer. (2019) 18:137. doi: 10.1186/s12943-019-1066-3
85. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. (2019) 35:677–91.e10. doi: 10.1016/j.ccell.2019.03.006
86. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. (2015) 43:373–84. doi: 10.1093/nar/gku1276
87. Peng S, Xiao W, Ju D, Sun B, Hou N, Liu Q, et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Science Transl Med. (2019) 11:eaau7116. doi: 10.1126/scitranslmed.aau7116
88. Clarke MF. Clinical and therapeutic implications of cancer stem cells. N Engl J Med. (2019) 380:2237–45. doi: 10.1056/NEJMra1804280
Keywords: epitranscriptome, RNA methylation, m6A, posttranscriptional control, human cancer
Citation: Yu S, Li X, Liu S, Yang R, Liu X and Wu S (2019) N6-Methyladenosine: A Novel RNA Imprint in Human Cancer. Front. Oncol. 9:1407. doi: 10.3389/fonc.2019.01407
Received: 21 October 2019; Accepted: 27 November 2019;
Published: 19 December 2019.
Edited by:
Hailong Pei, Soochow University, ChinaReviewed by:
Jia Meng, Xi'an Jiaotong-Liverpool University, ChinaKwok-Ming Yao, The University of Hong Kong, Hong Kong
Copyright © 2019 Yu, Li, Liu, Yang, Liu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sufang Wu, wsf_sfph@sjtu.edu.cn
†These authors have contributed equally to this work