 Mirna Swayden
Mirna Swayden Philippe Soubeyran
Philippe Soubeyran Juan Iovanna
Juan Iovanna- Centre de Recherche en Cancérologie de Marseille (CRCM), INSERM U1068, CNRS UMR 7258, Aix-Marseille Université and Institut Paoli-Calmettes, Parc Scientifique et Technologique de Luminy, Marseille, France
To date, PDAC remains the cancer having the worst prognosis with mortality rates constantly on the rise. Efficient cures are still absent, despite all attempts to understand the aggressive physiopathology underlying this disease. A major stumbling block is the outdated preclinical modeling strategies applied in assessing effectiveness of novel anticancer therapeutics. Current in vitro preclinical models have a low fidelity to mimic the exact architectural and functional complexity of PDAC tumor found in human set, due to the lack of major components such as immune system and tumor microenvironment with its associated chemical and mechanical signals. The existing PDAC preclinical platforms are still far from being reliable and trustworthy to guarantee the success of a drug in clinical trials. Therefore, there is an urgent demand to innovate novel in vitro preclinical models that mirrors with precision tumor-microenvironment interface, pressure of immune system, and molecular and morphological aspects of the PDAC normally experienced within the living organ. This review outlines the traditional preclinical models of PDAC namely 2D cell lines, genetically engineered mice, and xenografts, and describing the present famous approach of 3D organoids. We offer a detailed narration of the pros and cons of each model system. Finally, we suggest the incorporation of two off-center newly born techniques named 3D bio-printing and organs-on-chip and discuss the potentials of swine models and in silico tools, as powerful new tools able to transform PDAC preclinical modeling to a whole new level and open new gates in personalized medicine.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an exocrine malignancy which accounts for more than 90% of all cases of pancreatic cancer (1). It is a highly lethal disease exhibiting an extremely poor prognosis, where <7% of patients survive for 5 years after diagnosis (2). Currently, PDAC is the fourth leading cause of cancer related death and should become the second deadliest cancer by 2030 (3). This morbid outcome is attributed to diagnosis at late stages of the disease, its rapid dissemination and its high resistance to all conventional treatments (4). Additionally, most patients are diagnosed with metastases in liver and lung due to the absence of early screening tools, and succumb to the disease within 6–12 months thereafter (5). Surgery is potentially the only curative option available, however only 10–15% of patients are eligible for resection (6). Over the last 20 years only 3 major improvements have been introduced as treatment regimens for PDAC. In 1997, Gemcitabine emerged as an alternative to 5-Fluorouracil as a first-line therapy for PDAC, it improved overall survival by only few weeks (7). Next, Folfirinox treatment regimen (5FU, Leucovorin, Oxaliplatin, and Irinotecan) was applied in metastatic patients and contributed to a significant improvement in survival although with strong adverse side effects which is limiting its application to all non-operable patients (8). In 2013, Nab-Paclitaxel (Abraxane) in combination with Gemcitabine was approved as a first-line treatment for advance PDAC patients as it gives better efficacy than Gemcitabine alone though, with little increase of adverse effects (9). Despite of these humble progress steps, the dismal clinical situation of PDAC patients still resides, where the incidence and mortality rates are constantly on the rise. Therefore, there is an urgent need for developing new and more successful therapeutic strategies to treat PDAC patients as well as new early detection tools and diagnostic and prognostic markers.
Promising new anti-cancer compounds are tested for their pharmacokinetics, safety, toxicity, and efficacy in a preclinical phase that acts as a bridge to the clinic (Figure 1). According to the Food and Drug Administration (FDA) regulations, an affluent preclinical testing must be completed before humans are exposed to the potential drug (10). The successful evaluation of therapeutics in preclinical settings highly depends on the accuracy, reproducibility and predictive ability of the in vivo or in vitro preclinical model used. New drug development programs usually take about 12 years to transfer a compound from experimental investigation to the patient bed side (Figure 1). Additionally, it is economically challenging with a cost as high as exceeding 1.2 billion dollars (11). It is also risky in terms of economic gain since 90% of tested drugs fail under clinical trials and only 10% could finally reach the market (12). This is mainly due to inconsistencies in the experimental model utilized leading to false uncertain conclusions. Several promising drug candidates failed clinical trials after a successful preclinical testing in animal models (13) due to genetic, immunologic, physiological, and metabolic differences between humans and mouse. In order to reduce the cost and the failure rate in clinical trials, solid trustworthy preclinical models must be developed for preclinical testing. These models must be reliable allowing the prediction of drug efficacy testing in humans and capable of closely recapitulating the true PDAC pathophysiology in human body. In this review we discuss the classical, existing, and the newly emerging preclinical model systems in PDAC research (Figure 2), highlighting the strengths, and weakness of each model. Also, we offer rationales for the implementation of innovative advancement technologies newly born in the field in PDAC research, aiming to create perfect modeling approaches to ensure success of cancer therapeutics in clinical settings.
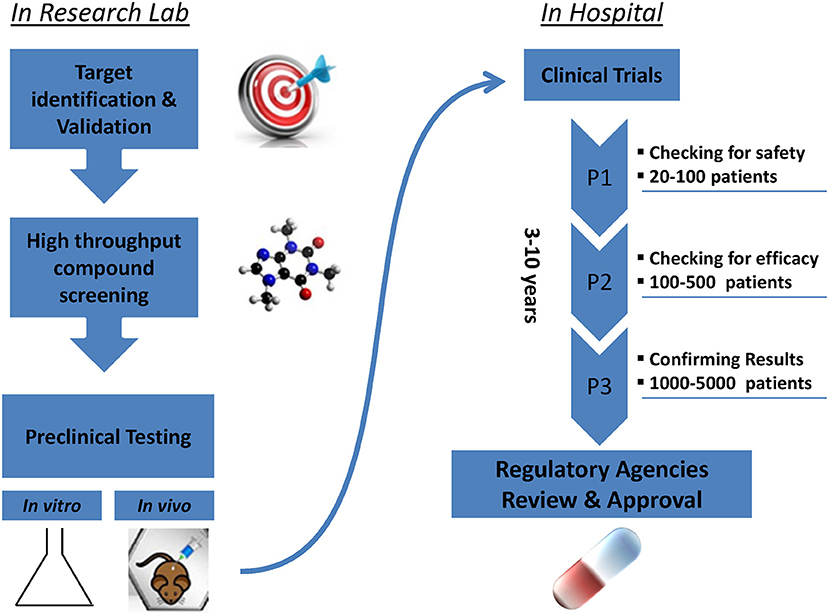
Figure 1. Steps of drug development from research lab to the patient's bed side.
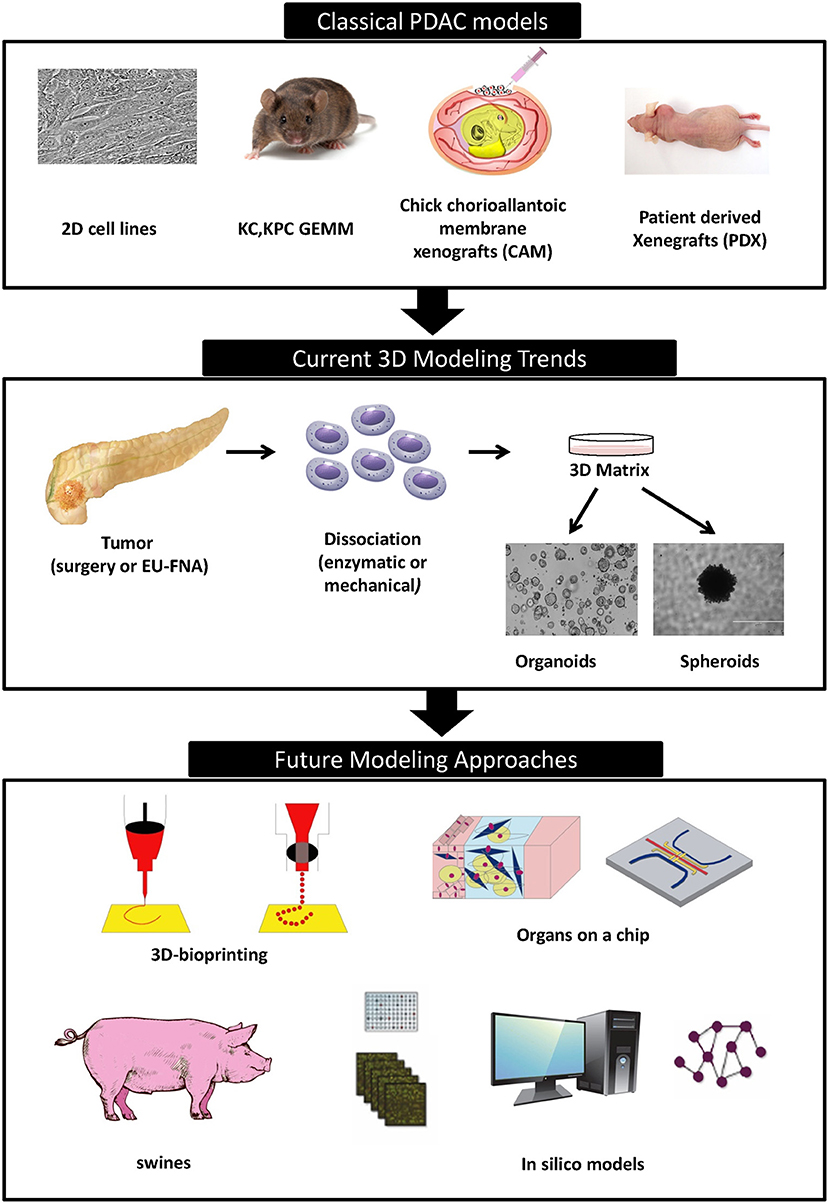
Figure 2. Timeline of different PDAC modeling approaches.
Classical Preclinical Models in PDAC Investigation
Traditional model system such as 2D cell lines, genetically engineered mice, and xenografts have shaped our current knowledge of PDAC pathology. However, the clinical relevance of these models have always been questioned. To date, the ability of these models to faithfully reflect the exact functional and structural properties of the tumor is still an unmet need. Several advantages and disadvantages characterize these models. A growing body of data urges us to develop novel preclinical testing models to bypass the pitfalls existing in current fundamental ones, able to better predict the success or failure of chemotherapeutic agents undergoing clinical trials.
PDAC Cell Lines
Human derived cell lines are the most widely used models to study the biology of cancer. The first human pancreatic cancer cell line was generated in 1963 (14), and then many PDAC cell lines from human or murine tumors have been produced. Human cell lines are easy to manipulate, they can grow indefinitely at low cost and are suitable for high throughput pharmacological screening and genetic testing. However, key limitations exist within this model. First, most cell lines are derived from resected tumors, and since most PDAC patients are ineligible to surgery, then PDAC cell lines are derived only from a small subset of patients and doesn't reflect the heterogeneity found across PDAC tumors (15). Second, the in vitro culture of normal pancreatic ductal cells is a rather difficult task, thus the comparison between normal and PDAC cells is almost impossible (16). Third, the repeated passaging of cell lines results in a genetic drift and culturing cells as monolayers in medium containing serum was shown to promote the loss of p53 function and subsequent genome instability (17). Furthermore, several studies reported significant differences in expression profiles of cell lines as compared to patient primary tumors or xenografts (18). Finally, this model is not a faithful recapitulation of the histological and biological complexity of tumor, due to the lack of tumor microenvironment mainly composed of ECM (extracellular matrix) components and several cell types such as fibroblasts, nerves, immune cells, adipocytes, and endothelial cells (19).
Genetically Engineered Mouse Models (GEMM)
Genetically engineered mice are designed by inducing specific mutations in oncogenes and/or tumor suppressor genes associated with PDAC in the mouse genome. This was firstly accomplished by introducing activating mutations in KRAS gene specifically in the pancreas which resulted in the formation of PanIN lesions capable of evolving to PDAC. The most well-studied GEMM of PDAC is surely the KPC one, which contains mutations in KRAS and TP53, both driven by the pdx1-Cre transgene which specifically expressed the Cre recombinase in all cells of pancreas starting from an early phase of embryonic development (20). In KPC model, tumors develop spontaneously with a dense desmoplasia and poor vasculature, similarly to human PDAC, thereby preserving the dynamics of tumor microenvironment (21). Therefore, it is a smart tool to study interactions between tumor and stromal cells in addition to disease progression from early stages of PanIN to primary and metastatic tumor. Moreover, GEMM intact immune system allows the study the immune response in PDAC and test novel immune-therapies (22). This model allowed scientists to reveal the complex balance between pro-malignant and tumor suppressive properties of tumor stroma (23, 24). Recently, these models were also employed to study the influence of microbiota composition and its impact on tumor development and patients survival, mainly by interacting and influencing the immune system (25, 26). GEMMs have led to pivotal strides in understanding PDAC pathogenesis by confirming causative roles of several mutant genes previously identified through human PDAC sequencing, in addition to identifying remarkable therapeutic targets such as MEK, PI3K, autophagy, and Notch pathways (27, 28). However, several pitfalls weaken this model: it is expensive due to the need of highly sophisticated imaging systems to monitor abdominal tumor growth within mice bodies. Also, it is labor intensive and time consuming to breed large number of mice colonies. Additionally, gene mutations are introduced into the germ line of the mouse, whereas they are occurring somatically and sequentially in human tumors. Several discoveries in murine models were not reproducible in human settings. For example, the anti-hedgehog therapy showed promising results in mice but failed to provide any clinical benefit in phase II study, suggesting that these models may lead to erroneous conclusions (21, 24). This can be attributed to interspecies differences in drug metabolism (29), immune function (30), and telomere activity (31) between mice and humans. Murine models have fewer mutations and less genetic complexity than human tumors. Moreover, the degree of aneuploidy in human tumors results in great variety of inter-tumoral gene modifications, in a totally different manner than it occurs in mouse (32). Taken all together, these species-related differences restrict the capacity of these models to predict the real therapeutic response of PDAC patients in clinical trial.
Xenografts Models
These methods involve the engraftment of human cell lines (cell line derived xenografts or CDX) and human tumor fragments (patient derived tumor xenografts or PDTX) into immune-compromised mice.
Cell Line Derived Xenografts (CDX)
One solution to address many of the drawbacks of 2D cultured cell lines is to transplant cell lines into immuno-deficient murine models to create a cell line derived xenografts either subcutaneously or orthotopically into the mouse pancreas. Pharmaceutical companies often use subcutaneous xenografts for drug testing due to their low cost, feasibility of rapid screening for efficiency and toxicity, and simplicity of tumor size assessment. On the other hand, orthotopic xenografts are expensive with an obligation of sacrificing the animal to detect drug response (33). However, this model is lame in predicting clinical outcomes, since xenograft murine model are immune-compromised and tumors are not subjected to pressure of immune system. Unfortunately, this model is non-feasible for evaluating immuno-therapies which is a serious need (34). Another limitation to this model is the selection of most aggressive and rapidly dividing cellular clones causing them to grow in homogeneous masses with a limited stromal infiltration, thus failing to mimic the exact morphology of the primary PDAC tumors (35). Additionally, these models are made from a limited number of cell lines therefore it fails to represent genetic and phenotypic heterogeneity existing in PDAC. Accumulating evidences show a moderate predictive values of this model and low correlation between data obtained from xenografts and the efficacy in clinic (36). For example Bruns et al. found that combining Gemcitabine with cetuximab (an EGFR inhibitor) induced 85% regression in a cell line xenografted, while proven ineffective in phase III clinical trial (37).
Patient Derived Tumor Xenografts (PDTX)
PDTXs are made by transplanting a piece of patient's tumor tissue derived from surgical resection or from tumor biopsies in immune-deficient mice. PDTXs retain the morphological characteristics of the primary tumor as well as its metastatic potential (38, 39). Importantly, PDTXs mirror response of human patients to chemotherapy (40). A study subjected 4 PDAC-derived PDTXs to 63 different drugs in different combinations to determine the most effective ones against each PDTX. While three out of the four PDTXs were sensitive to gemcitabine, one showed sensitivity to mitomycin C as well as cisplatin. The patient corresponding this PDX didn't benefit from Gemcitabine and was treated with mitomycin C and then with cisplatin. This patient was still free of disease even 50 months after diagnosis (41). Remarkably, our lab has carried out the transcriptomic analysis of several PDTX-derived from pancreatic cancer and identified specific gene signatures able to predict sensitivity to several anti-cancer drugs and clinical outcome of PDAC patients (42). For example, after performing transcriptomic profiling of 55 PDTXs, we were able to demonstrated that PDTXs with MYC-high signature are more sensitive to the BET bromo-domain inhibitor JQ1 than the PDTXs with MYC-low activity (43). Despite the promising potential of PDTX as preclinical drug testing platforms, several concerns need to be addressed such as, for example, the fact that PDTXs don't fully replicate stromal compartment of PDAC or host immune system (44). The use of immune-compromised mice limits the ability of using PDTX to examine responses to new immune-therapies. Moreover, the generation and maintenance of large colonies of immune-compromised mice for PDTX passages is extremely costly. Another drawback is that PDTX take up to 6 months to grow and waiting this long is simply untenable for most PDAC patients. To ensure a successful transplantation, a large amount of tumor tissue after surgical resection or biopsies for non-resectable patients is needed. More recently, we reported an efficient protocol to obtain PDTX directly from material recovered from endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) (45). This technical improvement must be considered as a major progress since after that, virtually all human tumors can be grown as PDTX and therefore studied. An important disadvantage of this model is that it can't precisely represent the heterogeneity of PDAC since it has been shown that most aggressive phenotypes are favored to propagate within the mouse model (46). Finally, xenografts derived from PDAC patients become infiltrated with murine stroma (45, 47), and most of human stroma that does not grow is replaced with murine cells after two or three passages (48). This mismatch between human tumor cells and mouse stroma must be taken into account when evaluating studies using PDTX model which can become an advantage as it enable the study of the interaction and communication between these two cellular compartments (45).
Creative approaches are being developed to bypass the limitations of PDTX model. One way is the co-implementation of stromal cells derived from patients (cancer associated fibroblasts, CAFs) that could prevent the invasion of the murine microenvironmental components thereby configuring the xenograft in a human manner as much as possible. In this way, a recent study showed that orthotopic co-transplantation of patient derived CAFs along with Capan-2 pancreatic cancer cells in NSG (NOD scid gamma mouse) mice revealed the tumor and metastasis-promoting ability of these patient derived CAFs (49). Importantly, CRISPR/Cas9 technology permitted the creation of syngeneic and humanized mice (50). An approach that will certainly benefit from the last advances of genome editing such as prime editing which has been described very recently and brings an important enhancement of the efficacy and specificity (51). Syngeneic/allografts transplant models are developed through the transplanting of cancer cells or solid tumors derived from the same genetic strain of mice (50). This method prevents the rejection of the transplant by the host's immune system, hence avoiding the need for immune-compromised mice, and allows the possibility of investigating immune-therapies in these models. But the major shortcoming of syngeneic mice is that the tumor cells are rodent and therefore doesn't fully mimic human PDAC (50). Syngeneic mice were successful platforms for identifying the currently approved immunotherapies such as checkpoint blockers including anti-CTLA4 (52), anti-PD-1 and anti-PDL-1 (53). Another approach is the production of humanized mice having mutations in IL-2 receptor gamma chain which allows the engraftment of hematopoietic stem cells and subsequent development of human immune systems. These mice are used to study human hematopoiesis and immunity, and can also be used to study cancers development with an immune component (54). Unfortunately, humanized murine models cannot fully replicate the human immune system due to a limited lymph node development, the HLA super families, and the appropriate immune cell trafficking (55). To date, there are limited trials of testing checkpoint inhibitors combinations in humanized mouse models. Sanmamed et al. used humanized mice model mice to test the efficacy of the combining a immune checkpoint inhibitor (nivolumab, anti-hPD-1) with an immune-stimulatory monoclonal antibody (urelumab, anti-hCD137). This study was an initial proof of concept that combination regimens can be modeled in humanized mice model. Authors were able to detect the expression of PD-1 and CD137 (Tumor Infiltrating Lymphocytes: TILs) and PD-L1 (tumor cells and antigen presenting cells) even when differences in tumor sizes were not significantly different upon the use of combination therapy vs. mono-therapy (56).
Chick Chorioallantoic Membrane Xenografts (CAM)
Chick chorioallantoic membrane is a highly vascularized extra-embryonic membrane connected to the embryo through a continuous circulatory system. It is considered easily accessible for experimental manipulation such as intravenous injection of therapeutic compounds and visualization of local response (57). It is not immune-competent until day 18, this feature makes it ideal for grafting foreign tissues without rejection ahead of this time (58). The CAM is a low cost model allowing the preclinical screening to assess the efficacy of large number of anti-cancer drugs on tumor growth (59). It is particularly faster than most mammalian models as tumor grafts become vascularized by chick vessels 2–5 days after inoculation (60). The typical CAM assay involves lowering the membrane by forming an air pocket between the shell membrane and the CAM itself, then tumor cells are grafted as an inoculum introduced through a small window made in the shell above the CAM at day 9 of embryonic development and tumors are harvested at day 16 (61). Tumor cells can be visualized in the CAM assay using diverse techniques like in vivo videomicroscopy, detection of human urokinase plasminogen activator, GFP-labeled cells, PCR amplification of human specific sequences, PET/CT imaging, viral nanoparticles, and immunohistochemistry (61). Sys et al. clearly demonstrated that tumor fragments grafted in the CAM retained the morphology of primary tumor. However, tumor-associated stroma from the human samples was largely replaced by stroma coming from chicken in the grafts (62). A potential limitation is the arising of a non-specific inflammatory response after 15 days of incubation (63). A study by Rovithi et al., provided the first evidence that primary PDAC cells transduced with firefly luciferase can form tumors on the CAM, retaining several histopathological and genetic/epigenetic characteristics of primary tumors. They also used this model for testing the modulation of key miRNAs and the activity of Gemcitabine and Crizotinib on CAM tumors. Interestingly, they showed that combined treatment resulted in 63% inhibition of tumor growth and was associated with a reduced expression of miR-21 and increased expression of miR-155 (64).
CAM is a relatively rapid, straightforward, and economical model that permits screening of a large number of pharmacological agents in a short time range. Fortunately, this model doesn't require the administrative procedures to obtain the pre-approval of animal experimentation by the ethics committee, since the chick embryo conventionally is not considered a living animal until day 17 of development. Within the CAM the vascularization network and tumor cells development occur in a quick manner, this enable the researcher to closely observe and track the real time morphological changes of cancer cells in its microcirculation (63). However, several issues were raised concerning this model, one of them being the scarce availability of commercial reagents (ex: primers, antibodies, metabolic kits, etc.) suitable for application in avian species. Another concern was related to the reliability of angiogenesis studies carried out within this platform. Timing must be an essential factor to be considered when planning angiogenesis analyses, this due to the difficulty to distinguish between the real neovascularization from a false increase in vascular density caused by remodeling of the pre-existing vessels (65). Finally, CAMs are extremely sensitive to stress induced by environmental factors such as oxygen, tension, pH, osmolarity, and keratinization levels, this may complicate the sealing process of the opening made within the shell (66).
3D Organoids: The Current Growing Trend
3D culture methods are promising tools to better mimic the tumor biology found in vivo. The main goal of 3D culture is to avoid the cells attachment to the bottom of culture dish, either by keeping cells in suspension or by culturing cells in the presence of a special matrix. 3D systems increase the number of cell-to-cell interactions and resemble more closely the architectural organization of cells in vivo. 3D cultures derived from monolayer cell lines are referred to as spheroids that share common characteristics with cells in vivo including production of ECM, increased chemo-resistance and appearance of polarized cells junctions. However, as previously described, the use of monolayer cell lines as a starting material is a limiting point to this model and practically decreases its in vivo relevance (67, 68). On the other hand, a group of cells growing in 3D culture derived directly from primary tissues, embryonic stem cells or pluripotent stem cells are termed organoids. They possess a self-renewal and self-organizing capacity, and maintain similar morphologies and functionalities as the original tissue in vivo. Interestingly, organoids can be cryo-preserved and replicated passaged indefinitely keeping their genetic stability (69). This 3D system is amenable for genetic, transcriptomic, proteomic, and biochemical analysis. Briefly, 3D organoids are created by the enzymatic and/or mechanical dissociation of the tumor (or normal tissue) into small pieces that are then embedded in a specialized matrix, usually collagen, or matrigel, with addition of specific growth factors and differentiation modulators to furnish mesenchymal-like signals, such as EGF, FGF10 (mitogens), Rspo1 (to enhance Wnt signaling), Noggin, Wnt3a, nicotinamide, N-acetylcysteine, gastrin, and A83-01 (Alk inhibitor). Prostaglandin E2 is required in the case of normal pancreas human organoids (70). In 2015, the first human and murine PDAC organoid model was established; it recapitulated successfully the physiological and morphological similarities to normal and tumor tissues from mice and human. These organoids expressed ductal epithelial markers and lacked genes representing acinar and endocrine lineages. Interestingly, when these organoids are transplanted orthotopically in to immune-deficient mice they generate pre-invasive lesions similar to PanINs able to progress in to PDAC and metastasis, thus representing an appealing model for studying PDAC progression. Additionally, it has been demonstrated by gene expression and proteomic analysis in murine pancreatic 3D organoids that both transcriptomic and proteomic profiles correlated with the original primary tumor (71). 3D organoids can potentially advance personalized medicine for PDAC; they form reliable platforms for evaluating potential diagnostic markers and wide anti-cancer drug screening. Organoids can be generated also from biopsies refined via EUS-FNA, thereby representing closely the heterogeneity of different PDAC stages and clinical conditions from both resectable and non-resectable patients. In a clinical trial setting, patient individual tumors can be utilized to form 3D organoids. A subsequent large-scale drug testing can be conducted within the next weeks after receiving the biopsy of a given patient. Such testing aims toward the identification of individual therapeutic sensitivities based on genetic alteration signatures and/or drug responses in organoids, in order to determine second-line therapies for prolonging survival and enhance quality of life in PDAC patients when the response to first-line therapy is minimal or strongly reduced. To fulfill the aim of analyzing potential biomarkers and stratifying patients based on their genetic profiles and drug response, bio-banks of 3D organoids from surgery or endoscopic ultrasound are being created in our laboratory and validated as a tool of clinical interest (72, 73). However, it is worth noting that this model is facing critical challenges to be routinely applied in clinic, since it is an expensive, time consuming, and basically lacking the components of microenvironment and immune cells normally found in vivo. Additionally, all the added external factors which are not necessarily present in parental tumor may lead to artifactual findings. Tsai et al. tried to surmount this limitation by co-culturing primary pancreatic cancer cells along with immune cells and other types of stromal cells to investigate the tumor-stroma or the tumor-immune cells interaction and assessment of immune-therapeutics using organoid models (74). Moreover, heavy chemotherapeutic pre-treatment prior to surgery or biopsy and low cancer cells content of biopsy can lead to failed organoid culture. This is only the starting point of 3D organoid model and it is crucial to standardize protocols used for isolation and passaging of 3D organoids, since currently this technique is of heavy cost and technically challenging since it requires precious patient samples. Importantly, more studies will determine if these biopsy-derived organoids faithfully represent the genetic heterogeneity and therapeutic efficacy profile of the entire primary tumor and the extent of translating its outcomes in patients.
The Dawn of Next-Generation Preclinical Models in Oncology
To date the major obstacle toward creating a perfect pre-clinical model with an absolute reliability is the poor representation of tumor microenvironment, where many studies well-demonstrated the strong influence of micro-environment components on therapeutic outcome. However, recent innovative advancements, such as 3D Bio-printing and organs-on-a-Chip, are able to meticulously simulate this tumor micro-environment. Such revolutionary models can open up a new frontier in oncology research and accelerate the development of new cancer therapeutics.
3D Bio-Printing
3D bio-printing technology generates bio-printed tissues and organs, where 3D bio-printers deposit several types of co-cultured cells in single spatial arrangement matching the natural architecture of native tissue. 3D bio-printers use various types of cells in the form of bio-inks that are mainly composed of cells suspended in a bio-compatible gel-like material. These bio-inks are deposited on a 3D scaffold after which it is gelled by polymeric inter-linking using photo or thermal activation (75). The 3D organ scaffolds are solid surfaces made up of non-toxic bio-compatible materials similar to the human ECM such as natural polymers (alginate, gelatin, collagen, chitosan, fibrin, hyaluronic acid, etc.) or synthetic molecules such as polyethylene glycol (76). Non-invasive imaging methodologies, such as computed tomography, magnetic resonance imaging, computer aided design and computer aided manufacturing tools, and mathematical modeling, are used to digitize and model the architecture of the tissues and organs in order to generate a mimicking 3D scaffold. Then, digital images are used to print tissues and organs using techniques such as laser-assisted printing (77), micro-extrusion (78), and inkjet (79). 3D bio-printed tissues or organs are novel platforms for pre-clinical anti-cancer drug testing. Organovo is a medical company in its early stages that designs functional 3D human tissues and organs for medical research aiming to accelerate the preclinical and clinical therapeutics testing at low cost and no potential risks for living patients. These bio-printed tissues and organs provide a similar micro-environment to that of native organ in the body, conserving the interaction of cells with environmental factors and biology of ECM. Importantly, this may reduce the chances of failure and costs in human clinical trials (80). 3D bio-printing technology can be utilized to produce 3D tumor models for the study of cancer biology. Several approaches such as cell seeding 3D scaffold, hydrogel embedding, multi-cellular spheroids, cell patterning, and micro-fluidic chips have been exploited to build up a 3D tumor model in vitro (81). Zhao et al. used HeLa cells in gelatin/alginate/fibrinogen hydrogels to bio-print 3D in vitro models of cervical tumors. This study revealed the increased expression of matrix metalloproteases and chemo-resistance in 3D printed tumor models when compared to 2D cell culture model (82). Similarly, it is possible to generate bio-printed PDAC tumors that can be exploited as a more transparent model of drug testing. This technology is still in its infancy and more studies must be carried out to implement this valuable platform in the preclinical modeling of PDAC as well as other tumor types. However, 3D bio-printed tumor models are a faithful match to organ in vivo and have the potential to revolutionize the entire oncology research and drug discovery.
The key advantage of bio-printing cancers cells in 3D lies it its ability to model the tumor microenvironment in vitro with highest fidelity, thereby offering a better representation of tumor formation, progression, and response to anti-cancer drugs. Several studies proved the contribution of microenvironment's components in chemoresistance. Therefore, utilization of this platform is ideal for personalized drug screening procedures (83). Despite the advancement, challenges lie ahead of this newly born technology. Current light based bio-printer can produce bio structures at a microscale resolution, where there is a great demand to achieve more sophisticated single cell structures such as networks of blood capillaries. Another factor to be enhanced is the speed of printing process since the viability of cells within the bio-ink decreases as printing time increases especially for cell types with a high metabolic profile like muscle cells for example. Additionally, no real studies investigated the effect of bio-printers on the molecular features of the cells such as gene expression or other functional aspects. Another issue lies in scaling up this technique to generate large amounts of bio-printed tumors or tissues for clinical and commercial applications. Moreover, to date, the efforts for incorporating patient's primary cells in to this platform are scarce and poorly developed. Future work is greatly needed to standardize the protocols of this technique to overcome these pitfalls, and therefore offering the best version possible for modeling diseases with accuracy. This technique, with its great innovation, holds the potential to the experimental bridge to novel clinical regimens (84).
Organ-on-a Chip
Organs on chips are micro-fluidic devices used for culturing cells, composed of plastic, glass, or polymers such as polydimethylsiloxane with hollow micro-channels populated with viable cells. These micro-fluidic devices can form tissue chips made up of a single channel lined by cells from one tissue type or organ chips with higher complexity. Organ chips combine two or more types of tissue interacting directly across a porous membrane coated with ECM or separated by an ECM gel filling the micro-channels. Cells within this system are nourished with flowing culture medium through the endothelium-lined vascular channels (85). Cultured medium can be replaced with blood for few hours of culture (86). Additionally, these devices may contain hollow side chambers of cyclic suction for the application of rhythmical stretching and relaxing of tissues interfaces and therefore replicating the organ specific mechanical signals. These organ chips devices can reproduce the organ level response to drugs (87) and mimic several types of organ specific diseases including cancer. Such technology is applied in modeling basic hallmarks of cancer including tumor growth, progression from early to late stages, invasion, angiogenesis, EMT (epithelial to mesenchymal transition), and metastasis (88). Organ-on-chips were used to generate in vitro human orthotopic cancer model in non-small cell lung carcinoma (89) and multiple myeloma (90). The major breakthrough was implementing organ-on-chips approach into modeling responses to anti-cancer therapies. One study used a micro-fluidic device created with various oxygen gradients within, to test the sensitivity of A549 lung alveolar epithelial cancer cells to tirapazamine (that is converted in to active free radical under hypoxia). The results illustrated an increased drug efficacy under low oxygen gradient (91). Another study utilized an organ-on-chip system containing 3D chambers seeded with A549 lung cancer cells connected to another chamber containing cultured CAFs and attached to a linear concentration gradient generator. Authors showed that secretion of HGF by CAFs could inhibit paclitaxel-induced death in lung cancer cells (92). Such studies are excellent examples of how precise control over chemical gradients is made feasible using this technology. In an early attempt to apply this technology in the field of personalized medicine, a team cultured lung cancer cell line, a mixture of lung cancer and stromal cell lines, or cells from fresh lung cancer tissues in 3D gels within a micro-fluidic device and treated them with different concentrations of chemotherapeutic drugs generated on-chip using a micro-fabricated concentration gradient generator. The sensitivities to different anticancer agents (Gefitinib or Cisplatin) were determined in parallel, and the doses of single drugs and combinations were optimized in eight patients (93). Another study aimed at screening for drugs able to prevent tumor dissemination. To this end, human lung adenocarcinoma or bladder carcinoma cell lines were embedded in a collagen matrix in one channel, and HUVECs (Human umbilical vein endothelial cells) were cultured in another channel where they formed vessel-like structures. Then, the entire culture was treated with different drugs known to affect EMT pathways. These drugs significantly reduced expression of EMT markers when tumor cells were cultured alone. However, these effects were diminished in the co-culture with HUVECs. These results highlighted the role of tissue-tissue interfaces and of an in vivo-like microenvironment in the evaluation of anticancer agents (94). A pioneering study by Beer et al. provides evidence that micro-fluidic chips can be applied to culture PDAC cells, while maintaining their viability, proper morphological appearance, and growth characteristics. This micro-fluidic chamber platform was used to detect the drug response of PDAC cells to the chemotherapeutic agent Cisplatin (95). This technology recapitulate successfully the tissue-tissue interface and the physiological microenvironment that are crucial for reconstituting the complex organ level architecture and function. Such privileges are not available in 2D and 3D in vitro models. Also, it can be exploited to gain new insights into fundamental processes involved in cancer biology and therapeutic targeting.
Porcine Cancer Models
Incorporating the use of swine as a large animal model in cancer research is of a important potential benefits. Swine models have several advantages as they share tremendous similarities with humans on the level of genetics, epigenetics, anatomy, size, metabolism, and pathology in addition to their reduced expenses compared to other primate models (96). Hence, disease modeling in these animals can better portray cancer development and progression as seen in humans.
Thanks to advances in genetic engineering, genetically modified pig cancer models were created and such models are able to respond to therapy in a similar fashion to humans in randomized trials (97). Current porcine models which are used for cancer research include the APC1311 model of familial adenomatous polyposis producing polyps but no tumor (98), the heterozygous TP53 knockout model of osteosarcoma (99), and a chemically induced porcine HCC (hepatocellular carcinoma) model (100). The Oncopig Cancer Model (OCM) is a novel transgenic swine model representing the next generation large animal platform for tumor studies in oncology. Like in mice, this model uses a Cre recombinase expression to induce the expression of heterozygous KRASG12D and TP53R167H transgenes (101). A porcine PDAC model is being developed using the OCM. Induction of exocrine and neuroendocrine pancreatic cancer types was demonstrated in the OCM through delivery of AdCre (Cre expressing adenovirus) in to the pancreatic duct. This method led to the development of an invasive PDAC tumor with similar histological hallmarks to human PDAC, with dense fibroblastic stroma and acinar to ductal metaplasia (102). Such PDAC porcine model can aid in better understanding of early events of carcinogenesis and facilitating earlier detection, in addition to investigating new surgical strategies as well as studying the potential of nanotechnology and localized drug delivery approaches for pancreatic cancer. Pigs have been used as a preclinical model for drugs toxicology prior to human studies. The size and easy handling of pigs allow drug administration in a similar manner to human patients. It is possible to perform longitudinal blood sampling in order to assess drug exposure kinetic data and metabolism over long periods of time (103), since studies have shown similar kinetic response to humans that can't be modeled in another animal (104). It is worth noting that OCM is considered ideal for identification of putative blood biomarkers and prognostic indicators. Porcine models are newly emerging approaches with a great potential to drive translational cancer research toward success and address the unmet clinical needs. Importantly, OCMs can be further applied to model additional cancer types such as leukemia, lymphoma, and other hematological cancers.
In silico Models
With the advancements of experimental tools for measurement of cancer genome, transcriptome, and proteome such as: microarrays platforms, next-generation sequencing, and mass spectrometry, high-throughput cancer biology data are being generated in conjunction with analytical computer-based technologies. Such information rich data permit the construction of highly sophisticated computational in silico models that aid in biological discovery and personalized medicine. Key in silico models are being utilized in oncology. They include the creation of cancer statistical models which rely on signatures of genes expression and mutation, of perturbed molecular pathways and networks, of alterations of biochemical, metabolic, and signaling, and the modeling of the interactions between tumor cells and their microenvironment (105). Cancer Statistical models based on gene mutations signatures are applied for diagnosis of cancer subtype and stratification of tumor grade as well as predicting therapeutic outcome. Relapse and overall survival were successfully modeled in non-small lung carcinoma (106), pediatric leukemia (107), and breast cancer (108). Additionally, models of transcriptional classifiers have been used to anticipate tumor response to chemotherapies in breast cancer (109, 110), colorectal cancer (111), and ovarian cancer (112). In silico approaches offer a tremendous potential in drug discovery. This process starts with target identification through chemo-informatics tools such as chemical structure similarity searching (113), data mining/machine learning (114), panel docking (115), and bioactivity spectra based algorithms (116). Once the target is identified and validated, in silico tools can initiate drug design process by the structure based or ligand based computer aided drug design (117). The Team of Ma et al. developed a computational model capable of predicting drugs to treat pancreatic cancer. Seven drugs were identified using this model, three of which are supported by literature findings and three are experimentally validated by cytotoxicity assays using cell lines (118). Moreover, in silico models are used for toxicity assessments on the level of liver, gastrointestinal tract, and blood-brain barrier (119, 120). Such computational models aim to understand the side effects of drug candidates from molecular changes to phenotypic manifestations. It has been proven effective for optimizing dose and minimizing costly phase I/II clinical trials (121, 122). Despite of confronting substantial experimental and analytical obstacles, these models are promising reliable digital representations of cancer, with the purpose of early diagnosis, prognosis, and new therapeutics innovation without exposing patients to risks.
Conclusion
Traditional preclinical models contributed to major advancements in our understanding of PDAC so far. However, they are still far of being able to ensure translational success. This is mainly due to poor representation of microenvironment of human neoplasia and absence of immune system, or the reaction of the full organism. These factors were proven to strongly affect clinical outcome and drug responses to treatments. New cancer therapeutics must be accelerated through the implementation of new sophisticated technologies able to simulate all the characteristics of PDAC within the human body (Figure 2). Such technologies will create a progressive preclinical drug screening leading to authentic conclusions highly reproducible in clinical trials and ensuring the benefit of participating patients.
Author Contributions
MS, PS, and JI wrote, read, and approved the final manuscript.
Funding
This work was supported by La Ligue Contre le Cancer to MS and JI, Fondation de France and INCa to JI, and Fondation ARC to PS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn. (2008) 10:111–22. doi: 10.2353/jmoldx.2008.070115
2. Hidalgo M. Pancreatic cancermedical progress. N Engl J Med. (2010) 362:1605–17. doi: 10.1056/NEJMra0901557
3. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. (2014) 74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155
4. Ma J, Jemal A. The rise and fall of cancer mortality in the USA: why does pancreatic cancer not follow the trend? Future Oncol. (2013) 9:917–9. doi: 10.2217/fon.13.76
5. Hidalgo M. New insights into pancreatic cancer biology. Ann Oncol. (2012) 23 (Suppl. 10):135–8. doi: 10.1093/annonc/mds313
6. Freitas D, Dos Santos Fernandes G, Hoff PM, Cunha JE. Medical management of pancreatic adenocarcinoma. Pancreatology. (2009) 9:223–32. doi: 10.1159/000199433
7. Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first- line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. (1997) 15:2403–13. doi: 10.1200/JCO.1997.15.6.2403
8. Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. (2011) 364:1817–25. doi: 10.1056/NEJMoa1011923
9. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. (2013) 369:1691–703. doi: 10.1056/NEJMoa1304369
10. Junod S. FDA and clinical drug trials: a short history. In: A Quick Guide to Clinical Trials For People Who May Not Know it All (2008). p. 25–55. Retrieved from: www.fda.gov
11. Moridani M, Harirforoosh S. Drug development and discovery: challenges and opportunities. Drug Discov Today. (2014) 19:1679–81. doi: 10.1016/j.drudis.2014.06.003
12. Toyn J. What lessons can be learned from failed Alzheimer's disease trials? Expert Rev Clin Pharmacol. (2015) 8:267–9. doi: 10.1586/17512433.2015.1034690
13. Harazono Y, Nakajima K, Raz A. Why anti-Bcl-2 clinical trials fail: a solution. Cancer Metastasis Rev. (2014) 33:285–94. doi: 10.1007/s10555-013-9450-8
14. Dobrynin Y V. Establishment and characteristics of cell strains from some epithelial tumors of human origin. J Natl Cancer Inst. (1963) 31:1173–95.
15. Baker LA, Tiriac H, Clevers H, Tuveson DA. Modeling pancreatic cancer with organoids. Trends Cancer. (2016) 2:176–90. doi: 10.1016/j.trecan.2016.03.004
16. Moreira L, Bakir B, Chatterji P, Dantes Z, Reichert M, Rustgi AK. Pancreas 3D organoids: current and future aspects as a research platform for personalized Cell Mol Gastroenterol Hepatol. (2018) 5:289–98. doi: 10.1016/j.jcmgh.2017.12.004
17. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. (2006) 9:391–403. doi: 10.1016/j.ccr.2006.03.030
18. Gadaleta E, Cutts RJ, Kelly GP, Crnogorac-Jurcevic T, Kocher HM, Lemoine NR, et al. A global insight into a cancer transcriptional space using pancreatic data: importance, findings and flaws. Nucleic Acids Res. (2011) 39:7900–07. doi: 10.1093/nar/gkr533
19. Fidler IJ. The pathogenesis of cancer metastasis: the seed and soil hypothesis revisited. Nat Rev Cancer. (2003) 3:453–8. doi: 10.1038/nrc1098
20. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. (2005) 7:469–83. doi: 10.1016/j.ccr.2005.04.023
21. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. (2009) 324:1457–61. doi: 10.1126/science.1171362
22. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–17. doi: 10.1073/pnas.1320318110
23. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
24. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
25. Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. (2019) 574:264–7. doi: 10.1038/s41586-019-1608-2
26. Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. (2019) 178:795–806.e12. doi: 10.1016/j.cell.2019.07.008
27. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. (2011) 25:717–29. doi: 10.1101/gad.2016111
28. Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res. (2015) 21:396–404. doi: 10.1158/1078-0432.CCR-14-1591
29. Uhl EW, Warner NJ. Mouse models as predictors of human responses: evolutionary medicine. Curr Pathobiol Rep. (2015) 3:219–23. doi: 10.1007/s40139-015-0086-y
30. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
31. Calado RT, Dumitriu B. Telomere dynamics in mice and humans. Semin Hematol. (2013) 50:165–74. doi: 10.1053/j.seminhematol.2013.03.030
32. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. (2012) 491:399–405. doi: 10.1038/nature11547
33. Walters DM, Stokes JB, Adair SJ, Stelow EB, Borgman CA, Lowrey BT, et al. Clinical, molecular and genetic validation of a murine orthotopic xenograft model of pancreatic adenocarcinoma using fresh human specimens. PLoS ONE. (2013) 8:e77065. doi: 10.1371/journal.pone.0077065
34. Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. (2007) 7:645–58. doi: 10.1038/nrc2192
35. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment tumor microenvironment. Clin Cancer Res. (2012) 18:4266–76. doi: 10.1158/1078-0432.CCR-11-3114
36. Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. (2001) 84:1424–31. doi: 10.1054/bjoc.2001.1796
37. Philip PA, Benedetti J, Corless CL, Wong R, O'Reilly EM, Flynn PJ, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: southwest oncology group-directed intergroup trial S0205. J Clin Oncol. (2010) 28:3605–10. doi: 10.1200/JCO.2009.25.7550
38. Loukopoulos P, Kanetaka K, Takamura M, Shibata T, Sakamoto M, Hirohashi S. Orthotopic transplantation models of pancreatic adenocarcinoma derived from cell lines and primary tumors and displaying varying metastatic activity. Pancreas. (2004) 29:193–203. doi: 10.1097/00006676-200410000-00004
39. Fu X, Guadagni F, Hoffman RM. A metastatic nude-mouse model of human pancreatic cancer constructed orthotopically with histologically intact patient specimens. Proc Natl Acad Sci USA. (2006) 89:5645–49. doi: 10.1073/pnas.89.12.5645
40. Garber K. From human to mouse and back: Tumorgraft models surge in popularity. J Natl Cancer Inst. (2009) 101:6–8. doi: 10.1093/jnci/djn481
41. Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. (2011) 10:1311–16. doi: 10.1158/1535-7163.MCT-11-0233
42. Duconseil P, Gilabert M, Gayet O, Loncle C, Moutardier V, Turrini O, et al. Transcriptomic analysis predicts survival and sensitivity to anticancer drugs of patients with a pancreatic adenocarcinoma. Am J Pathol. (2015) 185:1022–32. doi: 10.1016/j.ajpath.2014.11.029
43. Bian B, Bigonnet M, Gayet O, Loncle C, Maignan A, Gilabert M, et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: implications for individualized medicine efforts. EMBO Mol Med. (2017) 9:482–97. doi: 10.15252/emmm.201606975
44. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. (2010) 467:1114–7. doi: 10.1038/nature09515
45. Nicolle R, Blum Y, Marisa L, Loncle C, Gayet O, Moutardier V, et al. Pancreatic adenocarcinoma therapeutic targets revealed by tumor-stroma cross-talk analyses in patient-derived xenografts. Cell Rep. (2017) 21:2458–70. doi: 10.1016/j.celrep.2017.11.003.
46. Aparicio S, Hidalgo M, Kung AL. Examining the utility of patient-derived xenograft mouse models. Nat Rev Cancer. (2015) 15:311–6. doi: 10.1038/nrc3944
47. Delitto D, Pham K, Vlada AC, Sarosi GA, Thomas RM, Behrns KE, et al. Patient-derived xenograft models for pancreatic adenocarcinoma demonstrate retention of tumor morphology through incorporation of murine stromal elements. Am J Pathol. (2015) 185:1297–1303. doi: 10.1016/j.ajpath.2015.01.016
48. Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. (2014) 4:998–1013. doi: 10.1158/2159-8290.CD-14-0001
49. Knudsen ES, Balaji U, Freinkman E, McCue P, Witkiewicz AK. Unique metabolic features of pancreatic cancer stroma: relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget. (2016) 7:78396–411. doi: 10.18632/oncotarget.11893
50. Labrijn AF, Meesters JI, Bunce M, Armstrong AA, Somani S, Nesspor TC, et al. Efficient generation of bispecific murine antibodies for pre-clinical investigations in syngeneic rodent models. Sci Rep. (2017) 7:2476. doi: 10.1038/s41598-017-02823-9
51. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
52. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
53. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm0902-1039c
54. Brehm MA, Shultz LD, Greiner DL. Humanized mouse models to study human diseases. Curr Opin Endocrinol Diabetes Obes. (2010) 17:120–5. doi: 10.1097/MED.0b013e328337282f
55. Shultz LD, Brehm MA, Victor Garcia-Martinez J, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. (2012) 12:786–98. doi: 10.1038/nri3311
56. Sanmamed MF, Rodriguez I, Schalper KA, Oñate C, Azpilikueta A, Rodriguez-Ruiz ME, et al. Nivolumab and urelumab enhance antitumor activity of human T lymphocytes engrafted in Rag2-/-IL2Rγnull immunodeficient mice. Cancer Res. (2015) 75:3466–78. doi: 10.1158/1538-7445.AM2015-261
57. Pink D, Luhrs KA, Zhou L, Schulte W, Chase J, Frosch C, et al. High efficacy vasopermeability drug candidates identified by screening in an ex ovo chorioallantoic membrane model. Sci Rep. (2015) 5:15756. doi: 10.1038/srep15756
58. Ribatti D. The chick embryo chorioallantoic membrane as a model for tumor biology. Exp Cell Res. (2014) 328:314–24. doi: 10.1016/j.yexcr.2014.06.010
59. Lokman NA, Elder ASF, Ricciardelli C, Oehler MK. Chick chorioallantoic membrane (CAM) assay as an in vivo model to study the effect of newly identified molecules on ovarian cancer invasion and metastasis. Int J Mol Sci. (2012) 13:9959–70. doi: 10.3390/ijms13089959
60. Ribatti D. The chick embryo chorioallantoic membrane (CAM) assay. Reprod Toxicol. (2017) 70:97–101. doi: 10.1016/j.reprotox.2016.11.004
61. Deryugina EI, Quigley JP. Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem Cell Biol. (2008) 130:1119–30. doi: 10.1007/s00418-008-0536-2
62. Sys G, Van Bockstal M, Forsyth R, Balke M, Poffyn B, Uyttendaele D, et al. Tumor grafts derived from sarcoma patients retain tumor morphology, viability, and invasion potential and indicate disease outcomes in the chick chorioallantoic membrane model. Cancer Lett. (2012) 326:69–78. doi: 10.1016/j.canlet.2012.07.023
63. Ribatti D. The chick embryo chorioallantoic membrane (CAM). a multifaceted experimental model. Mech Dev. (2016) 141:70–7. doi: 10.1016/j.mod.2016.05.003
64. Rovithi M, Avan A, Funel N, Leon LG, Gomez VE, Wurdinger T, et al. Development of bioluminescent chick chorioallantoic membrane (CAM) models for primary pancreatic cancer cells: a platform for drug testing. Sci Rep. (2017) 7:44686. doi: 10.1038/srep44686
65. Knighton DR, Fiegel VD, Phillips GD. The assay of angiogenesis. Prog Clin Biol Res. (1991) 365:291–9.
66. Auerbach R, Akhtar N, Lewis RL, Shinners BL. Angiogenesis assays: problems and pitfalls. Cancer Metastasis Rev. (2000) 19:167–72. doi: 10.1023/a:1026574416001
67. Wen Z, Liao Q, Hu Y, You L, Zhou L, Zhao Y. A spheroid-based 3-D culture model for pancreatic cancer drug testing, using the acid phosphatase assay. Brazilian J Med Biol Res. (2013) 46:634–42. doi: 10.1590/1414-431X20132647
68. Yeon SE, No DY, Lee SH, Nam SW, Oh IH, Lee J, et al. Application of concave microwells to pancreatic tumor spheroids enabling anticancer drug evaluation in a clinically relevant drug resistance model. PLoS ONE. (2013) 8:e73345. doi: 10.1371/journal.pone.0073345
69. Blokzijl F, De Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. (2016) 538:260–4. doi: 10.1038/nature19768
70. Broutier L, Andersson-Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc. (2016) 11:1724–43. doi: 10.1038/nprot.2016.097
71. Boj SF, Hwang C, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. (2015) 160:324–38. doi: 10.1016/j.cell.2014.12.021
72. Bian B, Juiz NA, Gayet O, Bigonnet M, Brandone N, Roques J, et al. Pancreatic cancer organoids for determining sensitivity to bromodomain and extra-terminal inhibitors (BETI). Front Oncol. (2019) 9:475. doi: 10.3389/fonc.2019.00475
73. Iovanna J, Dusetti N. Speeding towards individualized treatment for pancreatic cancer by taking an alternative road. Cancer Lett. (2017) 410:63–7. doi: 10.1016/j.canlet.2017.09.016
74. Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q, et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer. (2018) 18:335. doi: 10.1186/s12885-018-4238-4
75. Stanton MM, Samitier J, Sánchez S. Bioprinting of 3D hydrogels. Lab Chip. (2015) 15:3111–5. doi: 10.1039/C5LC90069G
76. Nakamura M, Iwanaga S, Henmi C, Arai K, Nishiyama Y. Biomatrices and biomaterials for future developments of bioprinting and biofabrication. Biofabrication. (2010) 2:014110. doi: 10.1088/1758-5082/2/1/014110
77. Barron JA, Wu P, Ladouceur HD, Ringeisen BR. Biological laser printing: a novel technique for creating heterogeneous 3-dimensional cell patterns. Biomed Microdevices. (2004) 6:139–47. doi: 10.1023/B:BMMD.0000031751.67267.9f
78. Cohen DL, Malone E, Lipson H, Bonassar LJ. Direct freeform fabrication of seeded hydrogels in arbitrary geometries. Tissue Eng. (2006) 12:1325–35. doi: 10.1089/ten.2006.12.1325
79. Klebe RJ. Cytoscribing: a method for micropositioning cells and the construction of two- and three-dimensional synthetic tissues. Exp Cell Res. (1988) 179:362–73. doi: 10.1016/0014-4827(88)90275-3
80. Charbe N, McCarron PA, Tambuwala MM. Three-dimensional bio-printing: a new frontier in oncology research. World J Clin Oncol. (2017) 8:21. doi: 10.5306/wjco.v8.i1.21
81. Kim JB. Three-dimensional tissue culture models in cancer biology. Semin Cancer Biol. (2005) 15:365–77. doi: 10.1016/j.semcancer.2005.05.002
82. Zhao Y, Yao R, Ouyang L, Ding H, Zhang T, Zhang K, et al. Three-dimensional printing of Hela cells for cervical tumor model in vitro. Biofabrication. (2014) 6:035001. doi: 10.1088/1758-5082/6/3/035001
83. Knowlton S, Onal S, Yu CH, Zhao JJ, Tasoglu S. Bioprinting for cancer research. Trends Biotechnol. (2015) 33:504–13. doi: 10.1016/j.tibtech.2015.06.007
84. Ma X, Liu J, Zhu W, Tang M, Lawrence N, Yu C, et al. 3D bioprinting of functional tissue models for personalized drug screening and in vitro disease modeling. Adv Drug Deliv Rev. (2018) 132:235–51. doi: 10.1016/j.addr.2018.06.011
85. Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. (2014) 32:760–72. doi: 10.1038/nbt.2989
86. Jain A, Barrile R, van der Meer AD, Mammoto A, Mammoto T, De Ceunynck K, et al. Primary human lung alveolus-on-a-chip model of intravascular thrombosis for assessment of therapeutics. Clin Pharmacol Ther. (2018) 103:332–40. doi: 10.1002/cpt.742
87. Hassell BA, Goyal G, Lee E, Sontheimer-Phelps A, Levy O, Chen CS, et al. Human organ chip models recapitulate orthotopic lung cancer growth, therapeutic responses, and tumor dormancy in vitro. Cell Rep. (2017) 21:508–16. doi: 10.1016/j.celrep.2017.09.043
88. Sontheimer-Phelps A, Hassell BA, Ingber DE. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer. (2019) 19:65–81. doi: 10.1038/s41568-018-0104-6
89. Yousem SA, Beasley MB. Bronchioloalveolar carcinoma: a review of current concepts and evolving issues. Arch Pathol Lab Med. (2007) 131:1027–32. doi: 10.1043/1543-2165(2007)131[1027:BCAROC]2.0.CO;2
90. Abe M. Targeting the interplay between myeloma cells and the bone marrow microenvironment in myeloma. Int J Hematol. (2011) 94:334–43. doi: 10.1007/s12185-011-0949-x
91. Chen YA, King AD, Shih HC, Peng CC, Wu CY, Liao WH, et al. Generation of oxygen gradients in microfluidic devices for cell culture using spatially confined chemical reactions. Lab Chip. (2011) 11:3626–33. doi: 10.1039/c1lc20325h
92. Ying L, Zhu Z, Xu Z, He T, Li E, Guo Z, et al. Cancer associated fibroblast-derived hepatocyte growth factor inhibits the paclitaxel-induced apoptosis of lung cancer A549 cells by up-regulating the PI3K/Akt and GRP78 signaling on a microfluidic platform. PLoS ONE. (2015) 10:e0129593. doi: 10.1371/journal.pone.0129593
93. Xu Z, Gao Y, Hao Y, Li E, Wang Y, Zhang J, et al. Application of a microfluidic chip-based 3D co-culture to test drug sensitivity for individualized treatment of lung cancer. Biomaterials. (2013) 34:4109–17. doi: 10.1016/j.biomaterials.2013.02.045
94. Bai J, Tu T-Y, Kim C, Thiery JP, Kamm RD. Identification of drugs as single agents or in combination to prevent carcinoma dissemination in a microfluidic 3D environment. Oncotarget. (2015) 6:36603–14. doi: 10.18632/oncotarget.5464
95. Beer M, Kuppalu N, Stefanini M, Becker H, Schulz I, Manoli S, et al. A novel microfluidic 3D platform for culturing pancreatic ductal adenocarcinoma cells: comparison with in vitro cultures and in vivo xenografts. Sci Rep. (2017) 7:1325. doi: 10.1038/s41598-017-01256-8
96. Swindle MM, Makin A, Herron A, Clubb FJ, Frazier KS. Swine as models in biomedical research. Vet Pathol. (2012) 49:344–56. doi: 10.1177/0300985811402846
97. Flisikowska T, Kind A, Schnieke A. Pigs as models of human cancers. Theriogenology. (2016) 86:433–37. doi: 10.1016/j.theriogenology.2016.04.058
98. Flisikowska T, Merkl C, Landmann M, Eser S, Rezaei N, Cui X, et al. A porcine model of familial adenomatous polyposis. Gastroenterology. (2012) 143:1173–75.e7. doi: 10.1053/j.gastro.2012.07.110
99. Saalfrank A, Janssen KP, Ravon M, Flisikowski K, Eser S, Steiger K, et al. A porcine model of osteosarcoma. Oncogenesis. (2016) 5:e210. doi: 10.1038/oncsis.2016.19
100. Li X, Zhou X, Guan Y, Wang YXJ, Scutt D, Gong QY. N-nitrosodiethylamine-induced pig liver hepatocellular carcinoma model: radiological and histopathological studies. Cardiovasc Intervent Radiol. (2006) 29:420–28. doi: 10.1007/s00270-005-0099-8
101. Schachtschneider KM, Schwind RM, Newson J, Kinachtchouk N, Rizko M, Mendoza-Elias N, et al. The oncopig cancer model: an innovative large animal translational oncology platform. Front Oncol. (2017) 7:190. doi: 10.3389/fonc.2017.00190
102. Diaz A, Principe D, DeCant B, Grippo PJ, Rund L, Schook L. Abstract 4178: Pigs as a new weapon against cancer: modeling solid tumors in porcine. Cancer Res. (2016) 76:4178. doi: 10.1158/1538-7445
103. Ganderup NC, Harvey W, Mortensen JT, Harrouk W. The minipig as nonrodent species in toxicology - Where are we now? Int J Toxicol. (2012) 31:507–28. doi: 10.1177/1091581812462039
104. Roth WJ, Kissinger CB, McCain RR, Cooper BR, Marchant-Forde JN, Vreeman RC, et al. Assessment of juvenile pigs to serve as human pediatric surrogates for preclinical formulation pharmacokinetic testing. AAPS J. (2013) 15:763–74. doi: 10.1208/s12248-013-9482-6
105. Edelman LB, Eddy JA, Price ND. In silico models of cancer. Wiley Interdiscip Rev Syst Biol Med. (2010) 2:438–59. doi: 10.1002/wsbm.75
106. Chen H-Y, Yu S-L, Chen C-H, Chang G-C, Chen C-Y, Yuan A, et al. A five-gene signature and clinical outcome in non–small-cell lung cancer. N Engl J Med. (2007) 356:11–20. doi: 10.1056/NEJMoa060096
107. Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. (2002) 1:133–43. doi: 10.1016/S1535-6108(02)00032-6
108. Foekens JA, Atkins D, Zhang Y, Sweep FCGJ, Harbeck N, Paradiso A, et al. Multicenter validation of a gene expression-based prognostic signature in lymph node-negative primary breast cancer. J Clin Oncol. (2006) 24:1665–71. doi: 10.1200/JCO.2005.03.9115
109. Ayers M, Symmans WF, Stec J, Damokosh AI, Clark E, Hess K, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. (2004) 22:2284–93. doi: 10.1200/JCO.2004.05.166
110. Thuerigen O, Schneeweiss A, Toedt G, Warnat P, Hahn M, Kramer H, et al. Gene expression signature predicting pathologic complete response with gemcitabine, epirubicin, and docetaxel in primary breast cancer. J Clin Oncol. (2006) 24:1839–45. doi: 10.1200/JCO.2005.04.7019
111. Guinney J, Ferté C, Dry J, McEwen R, Manceau G, Kao KJ, et al. Modeling RAS phenotype in colorectal cancer uncovers novel molecular traits of RAS dependency and improves prediction of response to targeted agents in patients AC. Clin Cancer Res. (2014) 20:265–72. doi: 10.1158/1078-0432.CCR-13-1943
112. Chen P, Huhtinen K, Kaipio K, Mikkonen P, Aittomäki V, Lindell R, et al. Identification of prognostic groups in high-grade serous ovarian cancer treated with platinum-taxane chemotherapy. Cancer Res. (2015) 75:2987–98. doi: 10.1158/0008-5472.CAN-14-3242
113. Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature. (2009) 462:175–81. doi: 10.1038/nature08506
114. Nidhi, Glick M, Davies JW, Jenkins JL. Prediction of biological targets for compounds using multiple-category bayesian models trained on chemogenomics databases. J Chem Inf Model. (2006) 46:1124–33. doi: 10.1021/ci060003g
115. Li H, Gao Z, Kang L, Zhang H, Yang K, Yul K, et al. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. (2006) 34:W219–24. doi: 10.1093/nar/gkl114
116. Cheng T, Li Q, Wang Y, Bryant SH. Identifying compound-target associations by combining bioactivity profile similarity search and public databases mining. J Chem Inf Model. (2011) 51:2440–48. doi: 10.1021/ci200192v
117. Katsila T, Spyroulias GA, Patrinos GP, Matsoukas MT. Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J. (2016) 14:177–84. doi: 10.1016/j.csbj.2016.04.004
118. Ma Y, Hu J, Zhang N, Dong X, Li Y, Yang B, et al. Prediction of candidate drugs for treating pancreatic cancer by using a combined approach. PLoS ONE. (2016) 11:e0149896. doi: 10.1371/journal.pone.0149896
119. Piñero J, Furlong LI, Sanz F. In silico models in drug development: where we are. Curr Opin Pharmacol. (2018) 42:111–21. doi: 10.1016/j.coph.2018.08.007
120. Chen B, Butte AJ. Network medicine in disease analysis and therapeutics. Clin Pharmacol Ther. (2013) 94:627–9. doi: 10.1038/clpt.2013.181
Keywords: pancreatic cancer, organoids, cell culture, genetically engineered mice, 3D culture, xenografts models, 3D bio-printing, organ-on-a chip
Citation: Swayden M, Soubeyran P and Iovanna J (2020) Upcoming Revolutionary Paths in Preclinical Modeling of Pancreatic Adenocarcinoma. Front. Oncol. 9:1443. doi: 10.3389/fonc.2019.01443
Received: 28 September 2019; Accepted: 03 December 2019;
Published: 22 January 2020.
Edited by:
Marina Pajic, Garvan Institute of Medical Research, AustraliaReviewed by:
Enza Lonardo, Institute of Genetics and Biophysics (CNR), ItalyAlfredo Carrato, Ramón y Cajal University Hospital, Spain
Copyright © 2020 Swayden, Soubeyran and Iovanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe Soubeyran, Philippe.soubeyran@inserm.fr; Juan Iovanna, juan.iovanna@inserm.fr