 Roberta Buono
Roberta Buono Muneera Alhaddad1,2
Muneera Alhaddad1,2 David A. Fruman
David A. Fruman- 1Department of Molecular Biology & Biochemistry, University of California, Irvine, Irvine, CA, United States
- 2Hematology/Oncology Fellowship Program, CHOC Children's Hospital, Orange, CA, United States
High-risk subtypes of B-cell acute lymphoblastic leukemia (B-ALL) are frequently associated with aberrant activation of tyrosine kinases (TKs). These include Ph+ B-ALL driven by BCR-ABL, and Ph-like B-ALL that carries other chromosomal rearrangements and/or gene mutations that activate TK signaling. Currently, the tyrosine kinase inhibitor (TKI) dasatinib is added to chemotherapy as standard of care in Ph+ B-ALL, and TKIs are being tested in clinical trials for Ph-like B-ALL. However, growth factors and nutrients in the leukemia microenvironment can support cell cycle and survival even in cells treated with TKIs targeting the driving oncogene. These stimuli converge on the kinase mTOR, whose elevated activity is associated with poor prognosis. In preclinical models of Ph+ and Ph-like B-ALL, mTOR inhibitors strongly enhance the anti-leukemic efficacy of TKIs. Despite this strong conceptual basis for targeting mTOR in B-ALL, the first two generations of mTOR inhibitors tested clinically (rapalogs and mTOR kinase inhibitors) have not demonstrated a clear therapeutic window. The aim of this review is to introduce new therapeutic strategies to the management of Ph-like B-ALL. We discuss novel approaches to targeting mTOR in B-ALL with potential to overcome the limitations of previous mTOR inhibitor classes. One approach is to apply third-generation bi-steric inhibitors that are selective for mTOR complex-1 (mTORC1) and show preclinical efficacy with intermittent dosing. A distinct, non-pharmacological approach is to use nutrient restriction to target signaling and metabolic dependencies in malignant B-ALL cells. These two new approaches could potentiate TKI efficacy in Ph-like leukemia and improve survival.
1 Introduction
B-cell acute lymphoblastic leukemia (B-ALL) is the most common childhood cancer (1). Overall, the 5-year survival rates for children with B-ALL have increased to 90% in the last decades. Combination chemotherapy remains standard of care, with dose adjustment and adjuvant targeted therapies added to regimens based on risk stratification. Despite the increase in B-ALL survival rates, relapse still occurs in 15 to 20% of patients with poor outcomes after relapse, making B-ALL a leading cause of cancer related death among children and adults (1, 2).
Philadelphia chromosome-positive (Ph+) B-ALL was the first high-risk (HR) subtype to be defined at genetic and mechanistic levels. Characterized by the t(9;22)(q34.1;q11.2) translocation (3, 4), Ph+ B-ALL cells express the aberrantly activated tyrosine kinase (TK) fusion protein BCR-ABL1 that drives signaling pathways promoting cell proliferation and survival (5). Adding tyrosine kinase inhibitors (TKIs) such as imatinib or dasatinib to a chemotherapy backbone has contributed to improved outcomes in Ph+ B-ALL (6–9).
With the advancement of cancer genomic profiling and sequencing studies, researchers have identified different mutations and genetic rearrangements that drive other subtypes of high-risk B-ALL (1). This includes subtypes with similar signaling and transcriptomic signatures as Ph+ B-ALL but lacking the Ph chromosome, and collectively termed “Ph-like” B-ALL.
2 Ph-like ALL
Ph-like ALL was initially identified among patients with poor clinical outcomes who were found to have alterations of the IKZF1 gene (encoding the transcription factor IKAROS) that is also frequently deleted in Ph+ B-ALL (10). Based on additional similarities in tyrosine kinase-activated signaling pathways and gene expression profiles to those of patients with Ph+ B-ALL, this subtype was named Ph-like ALL (3, 10).
The genetic alterations found in Ph-like ALL have been divided into subgroups based on the kinase fusion or cytokine receptor involved. Whereas a 2016 report defined five subgroups (11), a more recent review from the Mullighan group (1) subdivided Ph-like B-ALL into four groups: (1) JAK family activating, including rearrangement of cytokine receptors (CRLF2, EPOR, IL2RB) and/or JAK mutations: (2) ABL-class including rearrangements of ABL1, ABL2, PDGFRA, PDGFRB, or CSF1R; (3) Other kinase, including FLT3 and NTRK3; (4) RAS signaling, including mutations in NRAS or KRAS. The relative frequency of genetic alterations differs between pediatric and adult patients with Ph-like B-ALL. Adults have higher rates of JAK2, EPOR, and IGH-CRLF2 rearrangements. On the other hand, pediatric as well as adolescent and young adult (AYA) populations have higher rates of other CRLF2 rearrangements (such as P2RY8-CRLF2) and ABL-class gene rearrangements (12, 13).
The prevalence of Ph-like ALL differs by age, gender, race, ethnicity, and National Cancer Institute (NCI)-defined risk groups. A higher proportion of patients with Ph-like ALL are males with a male-to-female ratio of 2:1. With regard to ethnicity, Hispanic patients have a markedly higher prevalence of Ph-like ALL, with a particularly high association of CRLF2 rearrangements (14). Ph-like B-ALL accounts for 10% of children with standard-risk (SR) B-lineage ALL and 15% of children of HR B-ALL (15, 16). The prevalence of Ph-like B-ALL increases with age, rising to 21% in adolescents and up to 27% in young adults with B-ALL, a rate almost 3 times higher than Ph+ B-ALL. In contrast to the rising incidence of Ph+ B-ALL with age, the incidence of Ph-like ALL seems to peak in the AYA population and decreases to 10% in older adults between the ages of 40 and 85 (11).
Patients with Ph-like ALL usually present with high leukocyte counts (>100,000/µL) at diagnosis and tend to have positive minimal residual disease (MRD) at the end of induction chemotherapy. Relapse rates are higher among patients with Ph-like in comparison with non-Ph-like B-ALL (17). In a completed phase 3 randomized controlled trial for HR-ALL (AALL0232) conducted by Children’s Oncology Group (COG), the 5-year event-free survival rate was lower among Ph-like B-ALL (63%) compared to non-Ph-like cases (86%) (11).
Prior to the use of TKI therapy, Ph+ B-ALL was associated with very poor survival. With the early addition of imatinib or dasatinib to intensive chemotherapy regimens in the treatment of Ph+ B-ALL, there has been a significant improvement in the survival with or without hematopoietic stem cell transplantation in first complete remission (6, 7). Given the presence of actionable TKs in many Ph-like B-ALL tumors (15, 17), and early anecdotal case studies (18, 19), researchers were prompted to launch clinical trials testing the utility of TKI in the treatment of Ph-like B-ALL. The choice of TKIs is based on the underlying genetic mutation. Thus, patients with Ph-like ALL cases are divided into two major classes for therapeutic targeting purposes: ABL class (including ABL1, ABL2, CSF1R, and PDGFRB rearrangements) and JAK pathway activating (including CRLF2, JAK2, or EPOR rearrangements, SH2B3 deletions, and IL7R indels) (Figure 1A). As described below, multiple clinical trials are currently investigating whether addition of the SRC/ABL/PDGFR inhibitor dasatinib or the JAK2 inhibitor ruxolitinib to the backbone of chemotherapy can improve the outcomes of children and adults with Ph-like ALL (13).
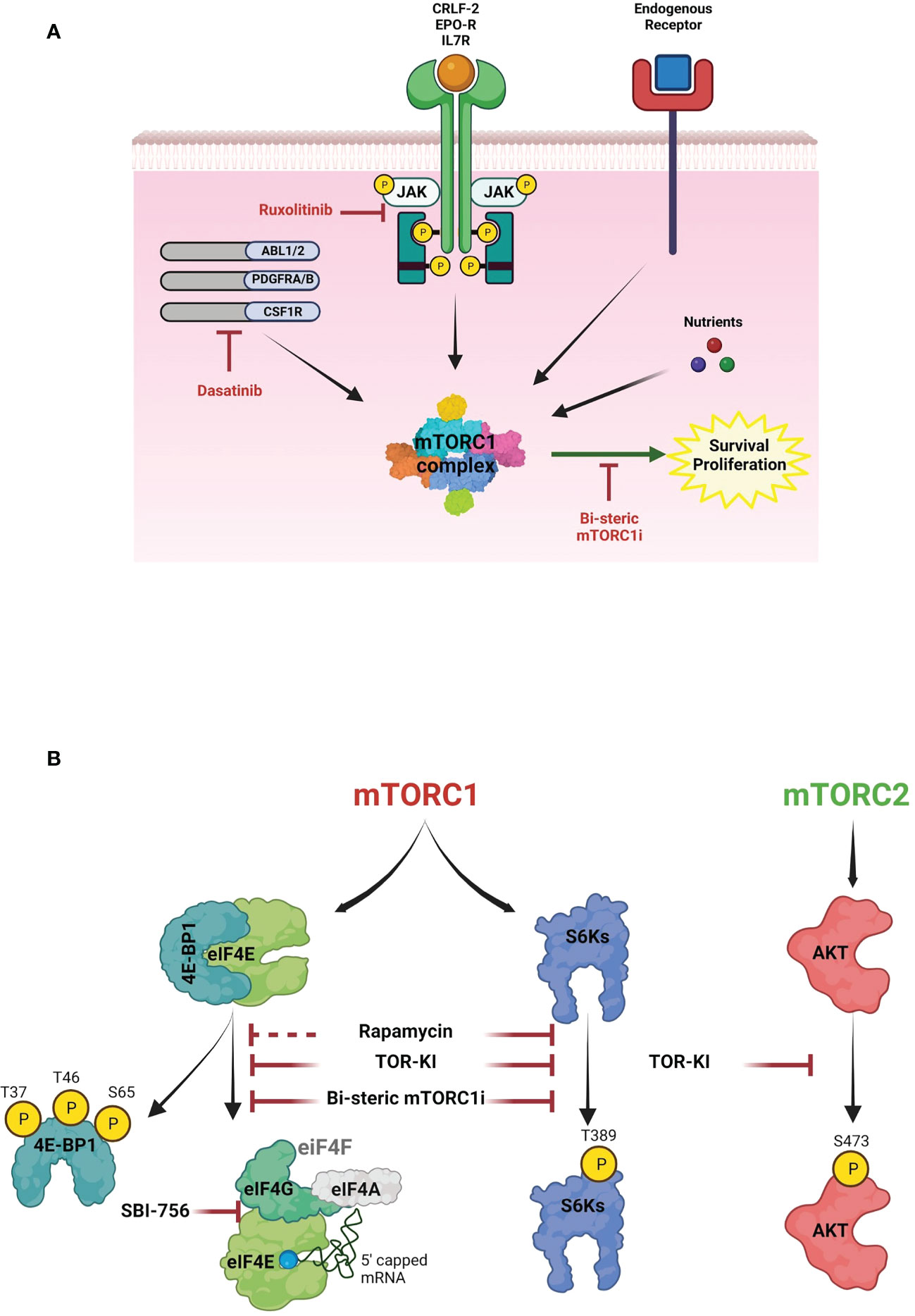
Figure 1 The role of mTOR signaling in B-ALL survival. (A) Schematic diagram of survival signaling pathways in B-ALL cells. mTORC1 contains the raptor subunit and other components that integrate signals from cytokine receptors, growth factors and nutrients to activate mTOR kinase activity. In Ph+ B-ALL, the BCR-ABL fusion tyrosine kinase (TK) increases activity of mTORC1 and other kinases. In Ph-like B-ALL, other genomic rearrangements (CRLF2, EPOR, IL7R, ABL1, ABL2, PDGFRA, PDGFRB, CSF1R) and/or JAK mutations activate TK signaling. Although clinically used TKIs (dasatinib, ruxolitinib) suppress oncogenic TK signaling, mTORC1 activity is largely maintained via nutrients and endogenous cytokine and growth factor receptors, thus promoting cell cycle progression and survival. Targeting this signaling with a selective bi-steric mTORC1 inhibitor (mTORC1i) has potential to enhance TKI cytotoxicity. (B) mTOR pathway. Two mTOR complexes, mTORC1 and mTORC2, have different substrates and together coordinate a complex network of anabolic pathways in response to signals from growth factors and oncogenes. Key substrates include ribosomal S6 kinases (S6Ks) and eIF4E-binding proteins (4E-BPs). mTORC1 phosphorylates 4E-BPs to promote eIF4F assembly. eIF4E binds to the 7-methylguanylate (m7G) cap present in most mRNAs and recruits the eIF4F complex to initiate cap-dependent translation. mTORC2 phosphorylates AKT and regulates cell proliferation and survival. First (rapamycin), second (TOR-KI) and third generation inhibitors (bisteric mTORCi) have different selectivity to inhibit the mTOR pathway. Notably, rapamycin is an allosteric inhibitor that inhibits phosphorylation of some substrates (e.g., S6Ks) more than others – particularly 4E-BP1. TOR-KIs act through an ATP-competitive mechanism to fully inhibit mTORC1 substrate phosphorylation but also inhibit mTORC2, thus suppressing AKT activity. Bi-steric mTORC1 inhibitors combine the mTORC1-selectivity of rapamycin with the complete kinase inhibition of TOR-KIs. The figures were created with BioRender.com.
2.1 Dasatinib trials
Preclinical studies showed promising anti-leukemia activity of dasatinib in ABL class Ph-like B-ALL cells in vitro and in patient derived xenograft (PDX) models (3, 17, 20). In addition, several case reports demonstrated the efficiency of adding TKIs with chemotherapy in the management of children and AYA with refractory B-ALL (4, 18, 19, 21–25). Based on these findings, multiple clinical trials proposed the use of dasatinib with chemotherapy in newly diagnosed pediatric and AYA patients with ABL-class Ph-like B-ALL: the phase 3 COG trial AALL1131 (NCT01406756) and the SJCRH Total XVII trial (NCT03117751). Additional trials are testing imatinib or ponatinib TKIs with chemotherapy (NCT03007147, NCT03911128, NCT0450161). Results from these clinical trials are pending (16).
2.2 Ruxolitinib trials
Several studies reported the efficacy of JAK inhibitors (e.g., ruxolitinib) both in CRLF2-rearranged (CRLF2-r) and other JAK activated Ph-like B-ALL cells and PDX models (20, 26, 27). A phase 1 clinical trial conducted by the COG (ADVL1011) was able to study the side effects and best dose of ruxolitinib in treating young patients with relapsed or refractory solid tumor, leukemia, or myeloproliferative disease (28). Based on the promising results, a phase 2 clinical trial was conducted to test ruxolitinib in pediatric patients (1-21 years of age) with relapsed/refractory solid tumors and hematologic malignances. A phase 2 COG AALL1521 trial (NCT02723994) and SJCRH Total XVII trial is assessing the safety and potential efficacy of ruxolitinib with chemotherapy in newly diagnosed children and AYA with CRLF2-r/JAK pathway–mutant Ph-like B-ALL (NCT03117751). Finally, a recently opened phase 1 trial at the University of Chicago and other institutions (NCT03571321) is studying ruxolitinib in combination with the pediatric-inspired CALBG 10403 chemotherapy regimen (NCT00558519) specifically in AYA patients (18-39 years of age) with newly diagnosed Ph-like B-ALL with a planned phase 2 expansion if safety is demonstrated. These clinical trials are still open and have not reported final results (13, 16).
While ongoing clinical trials indicate that adding TKIs to multi-agent chemotherapy is safe (29), the extent to which TKI addition alone will improve therapeutic outcomes is not yet clear. Indeed, a trial by MD Anderson Cancer Center (MDACC) testing the efficacy of adding dasatinib or ruxolitinib with chemotherapy in adolescents and adults with relapsed/refractory Ph-like B-ALL (NCT02420717) was terminated early due to low accrual and lack of responses. Additional combination agents might significantly increase the depth and duration of response to TKI/chemotherapy regimens. Researchers continue to study TK-independent survival pathways that could be targeted to enhance the response to TKIs.
3 Targeting the mTOR signaling network
mTOR is an evolutionarily conserved serine/threonine kinase that forms two cellular complexes with distinct functions (30). mTORC1, containing the raptor subunit, integrates both cell-intrinsic (oncogene) and extrinsic (growth factors, cytokines, nutrients) signals to promote biosynthetic processes that support cell growth and proliferation (Figure 1A). Key substrates include ribosomal S6 kinases (S6Ks) and eIF4E-binding proteins (4E-BPs) that control the efficiency of mRNA translation (Figure 1B). mTORC2, containing the rictor subunit, links growth factor receptors to key serine/threonine kinases including AKT (Figure 1B).
There is a strong mechanistic rationale for targeting mTOR in high-risk B-ALL. Many blood cancer cells exhibit high mTORC1 activity, and phosphorylation of the mTORC1 substrate 4E-BP1 correlates with poor prognosis in B-ALL (20, 31).
The first class of mTORC1 inhibitor consists of the natural product rapamycin and synthetic analogs (rapalogs). Although rapamycin is a highly potent and selective mTORC1 inhibitor, it has an allosteric mechanism that completely blocks phosphorylation of some substrates (e.g. S6Ks) but has less effect on others – especially the gatekeeper for cap-dependent translation, 4E-BP1 (32–34) (Figure 1B). Dephosphorylation of 4E-BP1 reactivates its ability to disrupt the eIF4F translation initiation complex and suppress cap-dependent translation of mRNAs with key roles in proliferation and survival (35–37). Clinical studies of rapalogs added to chemotherapy have not clearly improved outcome in B-ALL (38–41). Second generation ATP-competitive mTOR inhibitors that completely inhibit both mTORC1 and mTORC2 (mTOR kinase inhibitors; TOR-KI) are more effective at causing dephosphorylation of 4E-BP1 to restore inhibition of eIF4F assembly (Figure 1B). We and others demonstrated superior efficacy of TOR-KIs versus rapalogs in combination with TKIs in Ph+ B-ALL and Ph-like B-ALL (34, 42–48). Two investigational TOR-KIs advanced to oncology clinical trials (TAK-228 (sapanisertib), AZD2014 (vistusertib)). However, clinical activity of these TOR-KIs has again been disappointing (49–53), likely due to toxicities associated with mTORC2 inhibition.
A third generation of mTOR inhibitors, termed RapaLinks or bi-steric mTORC1 inhibitors, were described first by Shokat and colleagues (53, 54) (Figure 1B). These compounds consist of rapamycin connected by a chemical linker to an ATP-competitive TOR-KI moiety. The rapamycin piece mediates high-affinity selective binding to mTORC1, while the TOR-KI portion binds in the active site. Bi-steric inhibitors have several advantages including potency (low nM activity in cells), selectivity for mTORC1 (> 10-fold versus mTORC2), strong reactivation of 4E-BP1 (Figure 1B), and stable pathway inhibition that allows for intermittent dosing in vivo (53, 54). A biopharma company, Revolution Medicines, has reported novel bi-steric mTORC1 inhibitor molecules with ~30-fold selectivity for mTORC1 and excellent in vivo properties (55), one of which (RMC-5552) has entered oncology clinical trials (NCT04774952) (56). We demonstrated that another bi-steric mTORC1 inhibitor (RMC-4627) has anti-leukemia activity in Ph+ B-ALL (57). Notably, a single weekly dose of RMC-4627 significantly enhanced dasatinib efficacy and tolerability in a Ph+ B-ALL xenograft model (57). A recent study by Shokat and colleagues demonstrated that RapaLink molecules are brain-penetrant, and that it is possible to confine pharmacological activity to the central nervous system (CNS) by combination dosing with a brain-impermeant rapamycin analog known as RapaBlock (58). This finding has potential relevance for improving maintenance therapy in B-ALL, which usually includes CNS prophylaxis (59–61).
4 Starvation, FMD, and other nutritional approaches
Developing novel chemical agents targeting oncogenic dependencies has led to some notable successes in precision oncology but is expensive and often results in a narrow therapeutic window and/or survival benefit. A distinct, non-pharmacological approach is to use nutrient restriction to target signaling and metabolic dependencies in malignant cells. Dietary interventions including fasting and fasting mimicking diet (FMD) have been applied successfully to treat solid tumor cancers in preclinical and clinical studies (62–65).
Mechanistically, nutrient restriction inhibits the intracellular nutrient sensor mTORC1, while activating AMPK to enforce catabolic reactions (such as glycogenolysis, proteolysis and lipolysis coupled to ketogenesis) with consequent activation of adaptive cellular stress responses (DNA repair, autophagy and antioxidant defense) (66). Nutrient restriction can suppress other signals such as PI3K/AKT and RAF/MEK/ERK (MAPK) pathways that cooperate with mTORC1 to promote oncogenesis (Figure 2). Together these cellular effects of nutrient restriction limit the ability of malignant cells to thrive and proliferate.
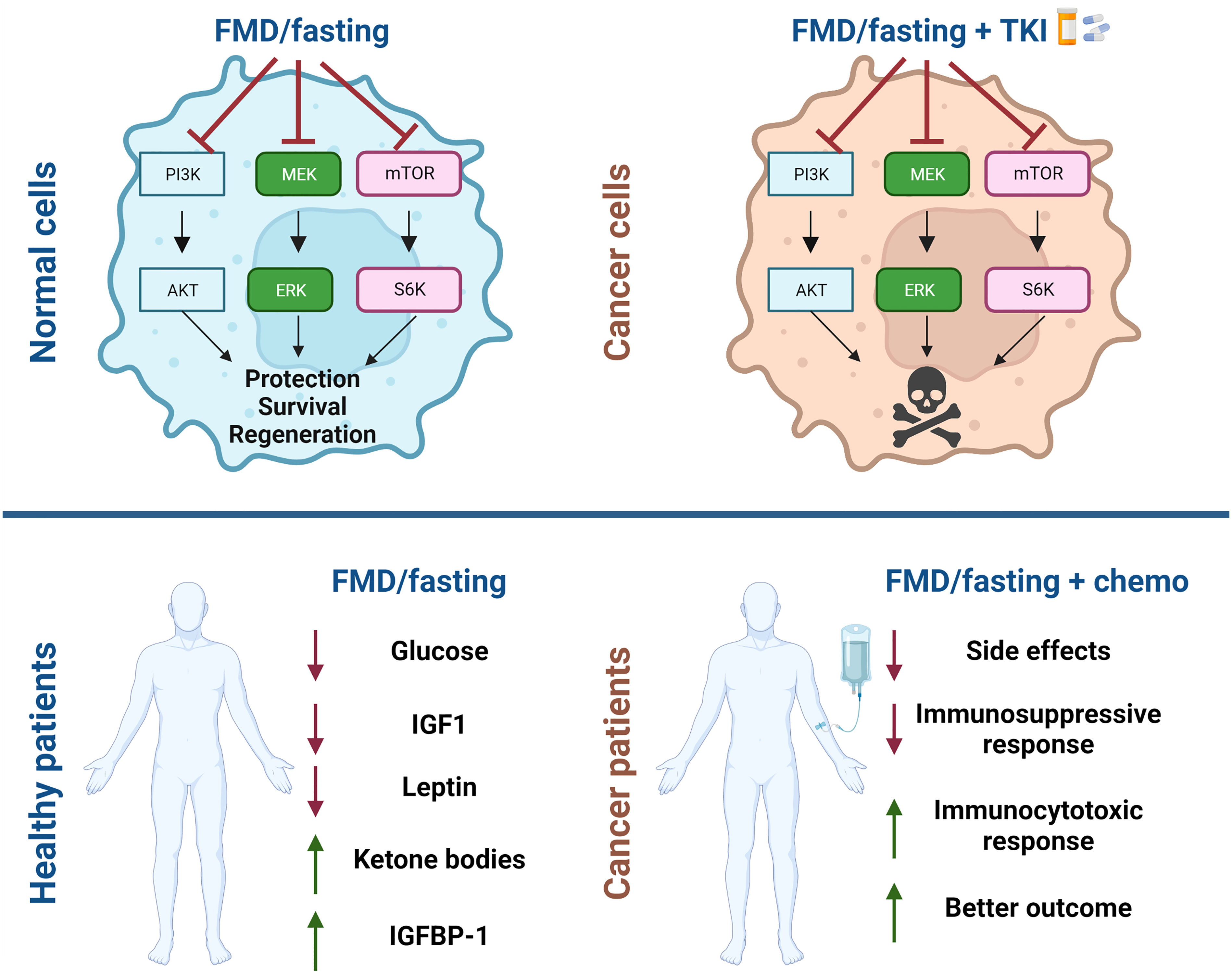
Figure 2 Schematic representation of beneficial effect of fasting and fasting mimicking diets for cancer treatment. Fasting and fasting mimicking diets (FMD) in combination with chemotherapy have a differential effect on normal cells and tumor cells. Normal cells enter in a protective mode while cancer cells are unable to survive. Systemic effects of fasting/FMD cycles are reduction in blood glucose, in circulating insulin growth factor-1 (IGF1) and leptin and increase in IGF1 binding protein-1 (IGFBP1) and serum ketone bodies. In combination with chemotherapy patients show fewer side effects, immunological changes and better outcomes. The figures were created with BioRender.com.
Fasting with water only, or chronic nutrient restriction, are dietary regimens that are difficult to sustain for long periods of time and cause nutrient deficiency with negative health consequences. For this reason, the Longo lab developed an intermittent FMD diet that can achieve the same anti-cancer benefits of fasting while providing some nutrients (67). FMD is a plant-based diet, that for each cycle of 4-7 days provides 34%–54% of normal caloric intake (11%–14% proteins, 42%–43% complex carbohydrates, 44%–46% fats) followed by FMD-free intervals (16–23 days) and has been applied successfully to treat different diseases like cancer, obesity and metabolic syndrome (68–75).
Patients undergoing FMD cycles manifest a reduction in blood glucose and in circulating insulin growth factor-1 (IGF1) and increase in IGF1 binding protein-1 (IGFBP1) and serum ketone bodies (66). In recent decades fasting and FMDs have been considered for the prevention and treatment of different solid tumors since their systemic effects have potential to enhance the efficacy of a wide variety of cancer treatments such as chemotherapy (Figure 2), immunotherapy and endocrine therapy (76). Importantly, these approaches have a differential effect on normal cells and cancer cells. Normal cells respond in ways that protect them against chemotherapy and other drugs, since in response to nutrient deprivation they arrest growth and activate stress resistance responses. This mechanism is referred to as differential stress response (DSR), while nutrient restriction weakens cancer cells by a process termed differential stress sensitization (DSS) (77). Cancer cells are sensitized by fasting/FMDs because of the constitutive activation of oncoproteins, which suppress stress resistance response and promote the generation of reactive oxygen species and induce cell death (63, 78). The effects of fasting/FMD in inducing DSS both in in vitro and in vivo models are mediated, in part, by reduction of circulating IGF-1 and glucose levels (Figure 2).
Fasting potentiates the anti-cancer activity of TKI in different types of solid tumors: breast cancer, colon rectal, and in non-small-cell lung cancer (79, 80). In vitro, short term starvation conditions increase the ability of commonly administered TKIs (erlotinib, gefitinib, lapatinib, crizotinib and regorafenib) to block cancer cell proliferation by inhibiting ERK phosphorylation and E2F-mediated transcription (79). Similar results were found by Lo Re at al., in a study showing that fasting potentiates the effect of sorafenib in a hepatocellular cancer mouse model by inhibition of MAPK signaling (80).
FMD cycles are also able to protect mice from PI3K/AKT and mTOR inhibitor-induced hyperglycemia, a common dose-limiting side effect of these inhibitor classes. In a mouse model of triple-negative breast cancer, a triple combination of FMD cycles with rapamycin and PI3K inhibitors (pictilisib, ipatasertib) resulted in long-term animal survival and reduction of side effects (71).
Among the beneficial effects of fasting and FMD is their ability to modulate immune response in the tumor site. These interventions boost tumor infiltration by CD8+ T cells and reduce immunosuppressive regulatory T cells (Treg) in syngeneic mouse models of melanoma and breast cancer. The immunomodulatory effects are mediated by downregulation of the cytoprotective enzyme heme-oxygenase HO-1 (70). A 48hr fasting period improves the efficacy of chemotherapy in a mouse model of mutant KRAS-induced lung cancers via activation of autophagy and reduction of Tregs in the tumor site (81).
Several clinical trials have evaluated the effect of fasting and FMDs in combination with chemotherapy as a therapeutic strategy in humans (73, 74, 82, 83). Patients diagnosed with different cancers that fasted voluntarily during their chemotherapy treatments reported fewer side effects and showed that fasting regimens were well tolerated. In the NCT03340935 trial that enrolled 101 patients with different type of neoplasms, five patients with advanced disease achieved complete and long-lasting tumor responses when treated with cycles of FMD plus the standard treatment (84). Another clinical trial (NCT03595540) evaluated the effect of FMD cycles on patients’ body composition and confirmed that FMD cycles are feasible and safe at low nutritional risk, with no negative impact on body composition and nutritional status in cancer patients. Some of the patients in this trial had hematological malignancies and three patients were also treated with TKI (74).
Similar to preclinical studies, FMD diet in human tumor patients reshapes anti-cancer immunity by downregulation of peripheral blood immunosuppressive myeloid cells and Tregs while increasing intratumor T-helper-1/cytotoxic responses, interferon-γ and other immune responses associated with better clinical outcomes. Together with good safety profile, the FMD had a global compliance rate of 91.8%, much higher of any other nutritional interventions (73). This high compliance rate is likely due to the short-term, intermittent regimen that includes adequate nutrition.
5 Diet and leukemia
While much of the work on dietary intervention in oncology has focused on solid tumors, there is increasing evidence for benefit in blood cancers. Fasting alone inhibits the initiation and reverses the leukemic progression of both B-cell and T-cell acute lymphoblastic leukemia (B-ALL and T-ALL, respectively), but not acute myeloid leukemia, in xenograft mouse models (85). Fasting suppresses leukemia progression in part through increased leptin receptor expression, which leads to the activation of the PRDM1 gene encoding the BLIMP1 transcription factor. The expression of leptin signaling-related genes, at early stages, prevents the development of leukemia, and at later stages, is responsible for the terminal differentiation and depletion of B and T leukemic cells. This finding provided important initial evidence that fasting could be a treatment option for patients with leukemia (85, 86).
Weight and nutritional status have a large impact on cancer survival, and obesity is a risk factor for poor outcome not only for ALL but also for other cancers (87). Notably, obese children at the time of ALL diagnosis are more likely to relapse (88, 89). Obese mice with syngeneic ALL have poorer response to chemotherapy than control mice (90). Tucci et al. demonstrated that the switching to a low-fat diet together with vincristine treatment improves ALL outcome in a syngeneic Ph+ B-ALL obese murine model (91).
The benefit of caloric restriction via diet and exercise in ALL patients was shown in a prospective, nonrandomized, controlled IDEAL (The Improving Diet and Exercise) trial. 40 patients between 10 and 21 years old during their chemotherapy treatment were subjected to 20% calorie reduction obtained by nutrient restriction together with exercise. This intervention significantly reduced MRD risk, increased circulating adiponectin and reduced insulin resistance and had the potential to augment chemotherapy efficacy in newly diagnosed with B-ALL (92). A second randomized phase 2 IDEAL trial NCT05082519 (IDEAL2) is recruiting patients and will evaluate the beneficial effect of the IDEAL intervention in reducing chemoresistance in B-ALL patients.
6 Conclusions and perspectives
The discovery of Ph-like B-ALL and its association with aberrant TK activity has led to optimism for achieving better outcomes by adding precision TKIs (dasatinib or ruxolitinib) to the backbone of chemotherapy. However, even if TKI regimens achieve regulatory approval to prevent or treat relapsed/refractory patients, we can anticipate that some patients will still fail to achieve deep and durable remissions. One biological rationale for this statement is that leukemia cells exist in a complex microenvironment that provides survival signals independent of the driving oncogene(s) (Figure 1A). This idea is supported by the finding that aggressive B-ALL cells rapidly die when cultured ex vivo, unless provided with stromal cells and cytokines (93). Thus, TK activity alone is not sufficient to drive leukemia proliferation and survival. Various lines of evidence support the hypothesis that mTORC1 activity mediates TK-independent survival signaling: (1) mTORC1 activity can be partially sustained in the presence of TKIs through signaling input from endogenous receptors for cytokines and adhesion molecules, and through direct activation by amino acids (94); (2) Elevated mTORC1 activity is associated with poor prognosis (31); (3) Three generations of mTORC1 inhibitors can all enhance responses to TKIs in preclinical models of Ph+ and Ph-like B-ALL (57, 95).
Exploiting mTORC1 dependency has proven to be a long-term challenge with the first two generations of inhibitors failing to achieve a clear therapeutic window in clinical trials. Third-generation bi-steric mTORC1 inhibitors represent a new weapon in this fight, with a clinical trial of RMC-5552 already underway in solid tumors (NCT04774952). Through potent and selective mTORC1 inhibition, bi-steric inhibitors overcome limitations of both rapamycin and TOR-KIs. Compounds of this class deserve to be evaluated in detail in preclinical models of Ph-like B-ALL, to determine whether such agents can enhance efficacy of dasatinib and/or ruxolitinib.
In parallel, we propose that dietary intervention should be evaluated as a non-pharmacological approach. In particular, FMD is a safe, well-tolerated regimen of intermittent caloric restriction that has shown impressive rates of patient compliance in clinical trials. FMD can not only suppress mTORC1 activity in tumor cells, but also PI3K/AKT and MAPK pathways through both cell-intrinsic and systemic effects. As with any dietary intervention, there may be challenges to clinical implementation of FMD – especially in children and adolescents. However, integrative hospital care with nutritionists should enable compliance to the specified diet, especially in the in-patient setting.
The history of B-ALL treatment emphasizes the benefits of therapeutic combinations. Following the original success of combination chemotherapy to achieve cures in many patients, subsequent incorporation of tyrosine kinase inhibitors, immunotherapies and stem cell transplantation have further improved prospects for patients with high-risk disease. In this spirit, it makes sense to evaluate bi-steric mTORC1 inhibitors and nutritional interventions to maximize the potential benefit of TKIs in Ph-like B-ALL.
Author contributions
RB and DF contributed to conceiving and designing the review. RB, MA and DF did the document retrieval. RB, MA, DF wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
Research on mTOR inhibitors and fasting mimicking diet in Ph-like B-ALL is supported by a Reach Grant from Alex’s Lemonade Stand Foundation (to DF 22-25509).
Acknowledgments
We thank Madhuri Paul and Dr. Van Huyhn for helpful comments on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica (2020) 105(11):2524–39. doi: 10.3324/haematol.2020.247031
2. Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J (2017) 7(6):e577. doi: 10.1038/bcj.2017.53
3. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell (2012) 22(2):153–66. doi: 10.1016/j.ccr.2012.06.005
4. Moorman AV, Schwab C, Winterman E, Hancock J, Castleton A, Cummins M, et al. Adjuvant tyrosine kinase inhibitor therapy improves outcome for children and adolescents with acute lymphoblastic leukaemia who have an ABL-class fusion. Br J Haematol (2020) 191(5):844–51. doi: 10.1111/bjh.17093
5. Foà R, Chiaretti S. Philadelphia Chromosome-positive acute lymphoblastic leukemia. N Engl J Med (2022) 386(25):2399–411. doi: 10.1056/NEJMra2113347
6. Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol (2009) 27(31):5175–81. doi: 10.1200/JCO.2008.21.2514
7. Schultz KR, Carroll A, Heerema NA, Bowman WP, Aledo A, Slayton WB, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's oncology group study AALL0031. Leukemia (2014) 28(7):1467–71. doi: 10.1038/leu.2014.30
8. Slayton WB, Schultz KR, Kairalla JA, Devidas M, Mi X, Pulsipher MA, et al. Dasatinib plus intensive chemotherapy in children, adolescents, and young adults with Philadelphia chromosome-positive acute lymphoblastic leukemia: Results of children's oncology group trial AALL0622. J Clin Oncol (2018) 36(22):2306–14. doi: 10.1200/JCO.2017.76.7228
9. Shen S, Chen X, Cai J, Yu J, Gao J, Hu S, et al. Effect of dasatinib vs imatinib in the treatment of pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: A randomized clinical trial. JAMA Oncol (2020) 6(3):358–66. doi: 10.1001/jamaoncol.2019.5868
10. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med (2009) 360(5):470–80. doi: 10.1056/NEJMoa0808253
11. Tran TH, Loh ML. Ph-like acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program. (2016) 2016(1):561–6. doi: 10.1182/asheducation-2016.1.561
12. Tasian SK, Loh ML, Hunger SP. Philadelphia Chromosome-like acute lymphoblastic leukemia. Blood (2017) 130(19):2064–72. doi: 10.1182/blood-2017-06-743252
13. Harvey RC, Tasian SK. Clinical diagnostics and treatment strategies for Philadelphia chromosome-like acute lymphoblastic leukemia. Blood Adv (2020) 4(1):218–28. doi: 10.1182/bloodadvances.2019000163
14. Harvey RC, Mullighan CG, Chen IM, Wharton W, Mikhail FM, Carroll AJ, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric b-progenitor acute lymphoblastic leukemia. Blood (2010) 115(26):5312–21. doi: 10.1182/blood-2009-09-245944
15. Pui CH, Roberts KG, Yang JJ, Mullighan CG. Philadelphia Chromosome-like acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk. (2017) 17(8):464–70. doi: 10.1016/j.clml.2017.03.299
16. Tran TH, Tasian SK. Clinical screening for ph-like ALL and the developing role of TKIs. Hematol Am Soc Hematol Educ Program. (2022) 2022(1):594–602. doi: 10.1182/hematology.2022000357
17. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in ph-like acute lymphoblastic leukemia. N Engl J Med (2014) 371(11):1005–15. doi: 10.1056/NEJMoa1403088
18. Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E. Successful tyrosine kinase inhibitor therapy in a refractory b-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica (2013) 98(11):e146–8. doi: 10.3324/haematol.2013.095372
19. Weston BW, Hayden MA, Roberts KG, Bowyer S, Hsu J, Fedoriw G, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J Clin Oncol (2013) 31(25):e413–6. doi: 10.1200/JCO.2012.47.6770
20. Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of ph-like acute lymphoblastic leukemia. Blood (2017) 129(2):177–87. doi: 10.1182/blood-2016-05-707653
21. Cario G, Leoni V, Conter V, Attarbaschi A, Zaliova M, Sramkova L, et al. Relapses and treatment-related events contributed equally to poor prognosis in children with ABL-class fusion positive b-cell acute lymphoblastic leukemia treated according to AIEOP-BFM protocols. Haematologica (2020) 105(7):1887–94. doi: 10.3324/haematol.2019.231720
22. Ding YY, Stern JW, Jubelirer TF, Wertheim GB, Lin F, Chang F, et al. Clinical efficacy of ruxolitinib and chemotherapy in a child with Philadelphia chromosome-like acute lymphoblastic leukemia with. Haematologica (2018) 103(9):e427–e31. doi: 10.3324/haematol.2018.192088
23. Tanasi I, Ba I, Sirvent N, Braun T, Cuccuini W, Ballerini P, et al. Efficacy of tyrosine kinase inhibitors in ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood (2019) 134(16):1351–5. doi: 10.1182/blood.2019001244
24. Niswander LM, Loftus JP, Lainey É, Caye-Eude A, Pondrom M, Hottman DA, et al. Therapeutic potential of ruxolitinib and ponatinib in patients with. Haematologica (2021) 106(10):2763–7. doi: 10.3324/haematol.2021.278697
25. Mayfield JR, Czuchlewski DR, Gale JM, Matlawska-Wasowska K, Vasef MA, Nickl C, et al. Integration of ruxolitinib into dose-intensified therapy targeted against a novel JAK2 F694L mutation in b-precursor acute lymphoblastic leukemia. Pediatr Blood Cancer (2017) 64(5):26328. doi: 10.1002/pbc.26328
26. Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, et al. Targeting JAK1/2 and mTOR in murine xenograft models of ph-like acute lymphoblastic leukemia. Blood (2012) 120(17):3510–8. doi: 10.1182/blood-2012-03-415448
27. Suryani S, Bracken LS, Harvey RC, Sia KC, Carol H, Chen IM, et al. Evaluation of the in vitro and in vivo efficacy of the JAK inhibitor AZD1480 against JAK-mutated acute lymphoblastic leukemia. Mol Cancer Ther (2015) 14(2):364–74. doi: 10.1158/1535-7163.MCT-14-0647
28. Loh ML, Tasian SK, Rabin KR, Brown P, Magoon D, Reid JM, et al. A phase 1 dosing study of ruxolitinib in children with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms: A children's oncology group phase 1 consortium study (ADVL1011). Pediatr Blood Cancer. (2015) 62(10):1717–24. doi: 10.1002/pbc.25575
29. K. TS, Albert A, HD S, Yining D, Mignon LL. A phase 2 study of ruxolitinib with chemotherapy in children with Philadelphia chromosome-like acute lymphoblastic leukemia (INCB18424-269/AALL1521): Dose-finding results from the part 1 safety phase. Blood (2018). doi: 10.1182/blood-2018-99-110221
30. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
31. Nemes K, Sebestyén A, Márk A, Hajdu M, Kenessey I, Sticz T, et al. Mammalian target of rapamycin (mTOR) activity dependent phospho-protein expression in childhood acute lymphoblastic leukemia (ALL). PloS One (2013) 8(4):e59335. doi: 10.1371/journal.pone.0059335
32. Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. (2008) 105(45):17414–9. doi: 10.1073/pnas.0809136105
33. Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PloS Biol (2009) 7(2):e38. doi: 10.1371/journal.pbio.1000038
34. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem (2009) 284(12):8023–32. doi: 10.1074/jbc.M900301200
35. Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discovery (2015) 14(4):261–78. doi: 10.1038/nrd4505
36. Malka-Mahieu H, Newman M, Désaubry L, Robert C, Vagner S. Molecular pathways: The eIF4F translation initiation complex-new opportunities for cancer treatment. Clin Cancer Res (2017) 23(1):21–5. doi: 10.1158/1078-0432.CCR-14-2362
37. Pal I, Safari M, Jovanovic M, Bates SE, Deng C. Targeting translation of mRNA as a therapeutic strategy in cancer. Curr Hematol Malig Rep (2019) 14(4):219–27. doi: 10.1007/s11899-019-00530-y
38. Daver N, Boumber Y, Kantarjian H, Ravandi F, Cortes J, Rytting ME, et al. A phase I/II study of the mTOR inhibitor everolimus in combination with HyperCVAD chemotherapy in patients with Relapsed/Refractory acute lymphoblastic leukemia. Clin Cancer Res (2015) 21(12):2704–14. doi: 10.1158/1078-0432.CCR-14-2888
39. Rheingold SR, Tasian SK, Whitlock JA, Teachey DT, Borowitz MJ, Liu X, et al. A phase 1 trial of temsirolimus and intensive re-induction chemotherapy for 2nd or greater relapse of acute lymphoblastic leukaemia: a children's oncology group study (ADVL1114). Br J Haematol (2017) 177(3):467–74. doi: 10.1111/bjh.14569
40. Place AE, Pikman Y, Stevenson KE, Harris MH, Pauly M, Sulis ML, et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr Blood Cancer. (2018) 65(7):e27062. doi: 10.1002/pbc.27062
41. Tasian SK, Silverman LB, Whitlock JA, Sposto R, Loftus JP, Schafer ES, et al. Temsirolimus combined with cyclophosphamide and etoposide for pediatric patients with relapsed/refractory acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia consortium trial (TACL 2014-001). Haematologica (2022) 107(10):2295–303. doi: 10.3324/haematol.2021.279520
42. Zhang Q, Shi C, Han L, Jain N, Roberts KG, Ma H, et al. Inhibition of mTORC1/C2 signaling improves anti-leukemia efficacy of JAK/STAT blockade in. Oncotarget (2018) 9(8):8027–41. doi: 10.18632/oncotarget.24261
43. Gotesman M, Vo TT, Herzog LO, Tea T, Mallya S, Tasian SK, et al. mTOR inhibition enhances efficacy of dasatinib in. Oncotarget (2018) 9(5):6562–71. doi: 10.18632/oncotarget.24020
44. Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle (2009) 8(23):3831–7. doi: 10.4161/cc.8.23.10070
45. Janes MR, Fruman DA. Targeting TOR dependence in cancer. Oncotarget (2010) 1(1):69–76. doi: 10.18632/oncotarget.110
46. Vu C, Fruman DA. Target of rapamycin signaling in leukemia and lymphoma. Clin Cancer Res (2010) 16(22):5374–80. doi: 10.1158/1078-0432.CCR-10-0480
47. Vilar E, Perez-Garcia J, Tabernero J. Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Mol Cancer Ther (2011) 10(3):395–403. doi: 10.1158/1535-7163.MCT-10-0905
48. Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med (2010) 16(2):205–13. doi: 10.1038/nm.2091
49. Eyre TA, Hildyard C, Hamblin A, Ali AS, Houlton A, Hopkins L, et al. A phase II study to assess the safety and efficacy of the dual mTORC1/2 inhibitor vistusertib in relapsed, refractory DLBCL. Hematol Oncol (2019) 37(4):352–9. doi: 10.1002/hon.2662
50. Schmid P, Zaiss M, Harper-Wynne C, Ferreira M, Dubey S, Chan S, et al. Fulvestrant plus vistusertib vs fulvestrant plus everolimus vs fulvestrant alone for women with hormone receptor-positive metastatic breast cancer: The MANTA phase 2 randomized clinical trial. JAMA Oncol (2019) 5(11):1556–64. doi: 10.1001/jamaoncol.2019.2526
51. Voss MH, Gordon MS, Mita M, Rini B, Makker V, Macarulla T, et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br J Cancer. (2020) 123(11):1590–8. doi: 10.1038/s41416-020-01041-x
52. McGregor BA, Xie W, Adib E, Stadler WM, Zakharia Y, Alva A, et al. Biomarker-based phase II study of sapanisertib (TAK-228): An mTORC1/2 inhibitor in patients with refractory metastatic renal cell carcinoma. JCO Precis Oncol (2022) 6:e2100448. doi: 10.1200/PO.21.00448
53. Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature (2016) 534(7606):272–6. doi: 10.1038/nature17963
54. Fan Q, Aksoy O, Wong RA, Ilkhanizadeh S, Novotny CJ, Gustafson WC, et al. A kinase inhibitor targeted to mTORC1 drives regression in glioblastoma. Cancer Cell (2017) 31(3):424–35. doi: 10.1016/j.ccell.2017.01.014
55. Lee BJ, Boyer JA, Burnett GL, Thottumkara AP, Tibrewal N, Wilson SL, et al. Selective inhibitors of mTORC1 activate 4EBP1 and suppress tumor growth. Nat Chem Biol (2021) 17(10):1065–74. doi: 10.1038/s41589-021-00813-7
56. Burnett GL, Yang YC, Aggen JB, Pitzen J, Gliedt MK, Semko CM, et al. Discovery of RMC-5552, a selective bi-steric inhibitor of mTORC1, for the treatment of mTORC1-activated tumors. J Med Chem (2023) 66(1):149–69. doi: 10.1021/acs.jmedchem.2c01658
57. Lee BJ, Mallya S, Dinglasan N, Fung A, Nguyen T, Herzog LO, et al. Efficacy of a novel bi-steric mTORC1 inhibitor in models of B-cell acute lymphoblastic leukemia. Front Oncol (2021) 11:673213. doi: 10.3389/fonc.2021.673213
58. Zhang Z, Fan Q, Luo X, Lou K, Weiss WA, Shokat KM. Brain-restricted mTOR inhibition with binary pharmacology. Nature (2022) 609(7928):822–8. doi: 10.1038/s41586-022-05213-y
59. Cortes J, O'Brien SM, Pierce S, Keating MJ, Freireich EJ, Kantarjian HM. The value of high-dose systemic chemotherapy and intrathecal therapy for central nervous system prophylaxis in different risk groups of adult acute lymphoblastic leukemia. Blood (1995) 86(6):2091–7. doi: 10.1182/blood.V86.6.2091.bloodjournal8662091
60. Jabbour E, Thomas D, Cortes J, Kantarjian HM, O'Brien S. Central nervous system prophylaxis in adults with acute lymphoblastic leukemia: current and emerging therapies. Cancer (2010) 116(10):2290–300. doi: 10.1002/cncr.25008
61. Wu SY, Short NJ, Nasr L, Dabaja BS, Fang PQ. Central nervous system prophylaxis and treatment in acute leukemias. Curr Treat Opt Oncol (2022) 23(12):1829–44. doi: 10.1007/s11864-022-01032-5
62. Brandhorst S, Longo VD. Fasting and caloric restriction in cancer prevention and treatment. Recent Results Cancer Res (2016) 207:241–66. doi: 10.1007/978-3-319-42118-6_12
63. Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med (2012) 4(124):124ra27. doi: 10.1126/scitranslmed.3003293
64. Mattson MP, Longo VD, Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res Rev (2017) 39:46–58. doi: 10.1016/j.arr.2016.10.005
65. Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab (2014) 19(2):181–92. doi: 10.1016/j.cmet.2013.12.008
66. Montégut L, de Cabo R, Zitvogel L, Kroemer G. Science-driven nutritional interventions for the prevention and treatment of cancer. Cancer Discovery (2022) 12(10):2258–79. doi: 10.1158/2159-8290.CD-22-0504
67. Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab (2015) 22(1):86–99. doi: 10.1016/j.cmet.2015.05.012
68. Caffa I, Spagnolo V, Vernieri C, Valdemarin F, Becherini P, Wei M, et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature (2020) 583(7817):620–4. doi: 10.1038/s41586-020-2502-7
69. Cheng CW, Villani V, Buono R, Wei M, Kumar S, Yilmaz OH, et al. Fasting-mimicking diet promotes Ngn3-driven beta-cell regeneration to reverse diabetes. Cell (2017) 168(5):775–88.e12. doi: 10.1016/j.cell.2017.01.040
70. Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell (2016) 30(1):136–46. doi: 10.1016/j.ccell.2016.06.005
71. Salvadori G, Zanardi F, Iannelli F, Lobefaro R, Vernieri C, Longo VD. Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metab (2021) 33(11):2247–59.e6. doi: 10.1016/j.cmet.2021.10.008
72. Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci Transl Med (2017) 9(377):eaai8700. doi: 10.1126/scitranslmed.aai8700
73. Vernieri C, Fucà G, Ligorio F, Huber V, Vingiani A, Iannelli F, et al. Fasting-mimicking diet is safe and reshapes metabolism and antitumor immunity in patients with cancer. Cancer Discovery (2022) 12(1):90–107. doi: 10.1158/2159-8290.CD-21-0030
74. Valdemarin F, Caffa I, Persia A, Cremonini AL, Ferrando L, Tagliafico L, et al. Safety and feasibility of fasting-mimicking diet and effects on nutritional status and circulating metabolic and inflammatory factors in cancer patients undergoing active treatment. Cancers (Basel) (2021) 13(16):4013. doi: 10.3390/cancers13164013
75. Di Tano M, Raucci F, Vernieri C, Caffa I, Buono R, Fanti M, et al. Synergistic effect of fasting-mimicking diet and vitamin c against KRAS mutated cancers. Nat Commun (2020) 11(1):2332. doi: 10.1038/s41467-020-16243-3
76. Nencioni A, Caffa I, Cortellino S, Longo VD. Fasting and cancer: molecular mechanisms and clinical application. Nat Rev Cancer. (2018) 18(11):707–19. doi: 10.1038/s41568-018-0061-0
77. Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A. (2008) 105(24):8215–20. doi: 10.1073/pnas.0708100105
78. Buono R, Longo VD. Starvation, stress resistance, and cancer. Trends Endocrinol Metab (2018) 29(4):271–80. doi: 10.1016/j.tem.2018.01.008
79. Caffa I, D'Agostino V, Damonte P, Soncini D, Cea M, Monacelli F, et al. Fasting potentiates the anticancer activity of tyrosine kinase inhibitors by strengthening MAPK signaling inhibition. Oncotarget (2015) 6(14):11820–32. doi: 10.18632/oncotarget.3689
80. Lo Re O, Panebianco C, Porto S, Cervi C, Rappa F, Di Biase S, et al. Fasting inhibits hepatic stellate cells activation and potentiates anti-cancer activity of sorafenib in hepatocellular cancer cells. J Cell Physiol (2018) 233(2):1202–12. doi: 10.1002/jcp.25987
81. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell (2016) 30(1):147–60. doi: 10.1016/j.ccell.2016.05.016
82. Raffaghello L, Safdie F, Bianchi G, Dorff T, Fontana L, Longo VD. Fasting and differential chemotherapy protection in patients. Cell Cycle (2010) 9(22):4474–6. doi: 10.4161/cc.9.22.13954
83. Safdie FM, Dorff T, Quinn D, Fontana L, Wei M, Lee C, et al. Fasting and cancer treatment in humans: A case series report. Aging (Albany NY). (2009) 1(12):988–1007. doi: 10.18632/aging.100114
84. Ligorio F, Fucà G, Provenzano L, Lobefaro R, Zanenga L, Vingiani A, et al. Exceptional tumour responses to fasting-mimicking diet combined with standard anticancer therapies: A sub-analysis of the NCT03340935 trial. Eur J Cancer. (2022) 172:300–10. doi: 10.1016/j.ejca.2022.05.046
85. Lu Z, Xie J, Wu G, Shen J, Collins R, Chen W, et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat Med (2017) 23(1):79–90. doi: 10.1038/nm.4252
86. Cheng CW, Yilmaz Ö. Starving leukemia to induce differentiation. Nat Med (2017) 23(1):14–5. doi: 10.1038/nm.4259
87. Orgel E, Tucci J, Alhushki W, Malvar J, Sposto R, Fu CH, et al. Obesity is associated with residual leukemia following induction therapy for childhood b-precursor acute lymphoblastic leukemia. Blood (2014) 124(26):3932–8. doi: 10.1182/blood-2014-08-595389
88. Dushnicky MJ, Nazarali S, Mir A, Portwine C, Samaan MC. Is there a causal relationship between childhood obesity and acute lymphoblastic leukemia? A Review. Cancers (Basel). (2020) 12(11):3082. doi: 10.3390/cancers12113082
89. Petrelli F, Cortellini A, Indini A, Tomasello G, Ghidini M, Nigro O, et al. Association of obesity with survival outcomes in patients with cancer: A systematic review and meta-analysis. JAMA Netw Open (2021) 4(3):e213520. doi: 10.1001/jamanetworkopen.2021.3520
90. Sheng X, Mittelman SD. The role of adipose tissue and obesity in causing treatment resistance of acute lymphoblastic leukemia. Front Pediatr (2014) 2:53. doi: 10.3389/fped.2014.00053
91. Tucci J, Alhushki W, Chen T, Sheng X, Kim YM, Mittelman SD. Switch to low-fat diet improves outcome of acute lymphoblastic leukemia in obese mice. Cancer Metab (2018) 6:15. doi: 10.1186/s40170-018-0189-0
92. Orgel E, Framson C, Buxton R, Kim J, Li G, Tucci J, et al. Caloric and nutrient restriction to augment chemotherapy efficacy for acute lymphoblastic leukemia: the IDEAL trial. Blood Adv (2021) 5(7):1853–61. doi: 10.1182/bloodadvances.2020004018
93. Ghamlouch H, Ouled-Haddou H, Damaj G, Royer B, Gubler B, Marolleau JP. A combination of cytokines rescues highly purified leukemic CLL b-cells from spontaneous apoptosis in vitro. PloS One (2013) 8(3):e60370. doi: 10.1371/journal.pone.0060370
94. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
Keywords: leukemia, B-ALL, tyrosine kinase inhibitors, mTOR, metabolism, nutrient restriction, fasting mimicking diet, targeted therapy
Citation: Buono R, Alhaddad M and Fruman DA (2023) Novel pharmacological and dietary approaches to target mTOR in B-cell acute lymphoblastic leukemia. Front. Oncol. 13:1162694. doi: 10.3389/fonc.2023.1162694
Received: 09 February 2023; Accepted: 17 March 2023;
Published: 14 April 2023.
Edited by:
Rozangela Curi Pedrosa, Federal University of Santa Catarina, BrazilReviewed by:
Parul Suri, St. John’s University, United StatesCopyright © 2023 Buono, Alhaddad and Fruman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Fruman, dfruman@uci.edu; Roberta Buono, buonor@uci.edu