 Fanny Drieux
Fanny Drieux François Lemonnier2,3
François Lemonnier2,3- 1Service d’Anatomie et de Cytologie Pathologiques, INSERM U1245, Centre Henri Becquerel, Rouen, France
- 2Unité hémopathies Lymphoïdes, Hôpitaux Universitaires Henri Mondor, Assistance Publique des Hôpitaux de Paris, Créteil, France
- 3Institut Mondor de Recherche Biomédicale, INSERM U955, Université Paris Est Créteil, Créteil, France
- 4Département de Pathologie, Hôpitaux Universitaires Henri Mondor, Assistance Publique des Hôpitaux de Paris, Créteil, France
Peripheral T-cell lymphomas (PTCL) comprised more than 30 rare heterogeneous entities, representing 10 to 15% of adult non-Hodgkin lymphomas. Although their diagnosis is still mainly based on clinical, pathological, and phenotypic features, molecular studies have allowed for a better understanding of the oncogenic mechanisms involved and the refinement of many PTCL entities in the recently updated classifications. The prognosis remains poor for most entities (5-year overall survival < 30%), with current conventional therapies based on anthracyclin-based polychemotherapy regimen, despite many years of clinical trials. The recent use of new targeted therapies appears to be promising for relapsed/refractory patients, such as demethylating agents in T-follicular helper (TFH) PTCL. However further studies are needed to evaluate the proper combination of these drugs in the setting of front-line therapy. In this review, we will summarize the oncogenic events for the main PTCL entities and report the molecular targets that have led to the development of new therapies. We will also discuss the development of innovative high throughput technologies that aid the routine workflow for the histopathological diagnosis and management of PTCL patients.
1 Introduction
Peripheral T-cell lymphomas (PTCL) represent 10 to 15% of adult non-Hodgkin lymphomas. In the latest revised WHO and ICC classifications (1, 2), more than 30 entities are described, mostly defined by their clinical and pathological and phenotypic features, with a growing element of molecular data. Indeed, molecular studies based on high-throughput technologies have allowed for a better understanding of the oncogenic mechanisms involved and have improved the characterization of several entities. Although only a few specific genomic alterations define a given entity, the use of molecular data, such as clonality assays and targeted next-generation sequencing (NGS), is now integrated into the routine diagnostic workflow of expert centers, in combination with clinical and pathological clues. However, the translation of high-throughput genomic studies to clinical practice is still limited due to the high cost of high-throughput technologies and little clinical relevance for most findings. In this review, we will detail the oncogenic mechanisms of the main non-cutaneous PTCL entities, the molecular targets that have an impact on their diagnosis or treatment, and the assays that are useful for the detection of these clinically relevant molecular alterations (3). Entities with a leukemic presentation (notably T-cell large granular lymphocytic leukemia and T-prolymphocytic leukemia) will not be detailed.
2 Biology of PTCLs
2.1 Oncogenic mechanisms
T-cell lymphomagenesis is a multistep process resulting from the accumulation of oncogenic events, such as genomic and epigenetic alterations and dysregulation of cellular signaling pathways, cell cycle, and immune surveillance (Figure 1). The microenvironment also plays a role in the initiation and maintenance of neoplastic transformation, best highlighted in angioimmunoblastic T-cell lymphoma (AITL), a disease characterized by a prominent tumor microenvironment (TME). However, the impact of the TME in other entities is still poorly understood.
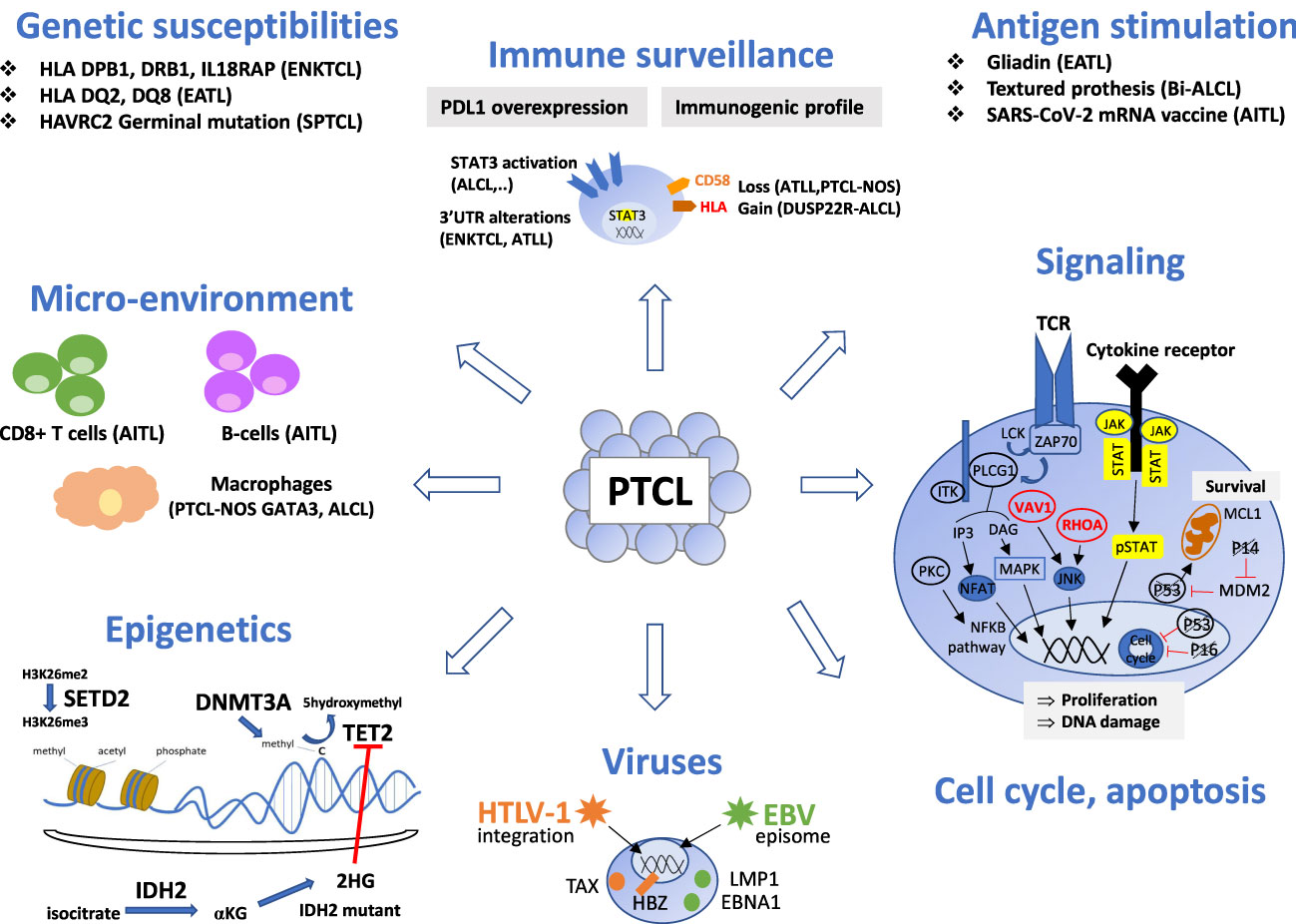
Figure 1 Oncogenic mechanisms of the main non-cutaneous PTCL entities. PTCL oncogenesis is a multistep process resulting from the accumulation of oncogenic events targeting epigenetics, signaling pathways (alterations of the TCR pathway is a common feature of TFH-PTCL, ATLL and certain PTCL-NOS, whereas alterations of the JAK/STAT pathway is shared by PTCL entities with a cytotoxic immunophenotype), cell cycle or apoptosis. Oncogenic viruses (HTLV1, EBV) are involved in a few specific entities. Chronic antigen stimulation may play a role as initiating event in several extranodal T or NK-cell lymphomas. Immune surveillance and crosstalk between neoplastic cells and reactive cells of the microenvironment is important, especially in AITL, where reactive cytotoxic CD8 T-cells and B-cells are associated with a poor and favorable outcome respectively. Genetic susceptibility is recognized in SPTCL, EATL and ENKTL. This figure depicts these events and their involvement for specific PTCL entities. Genes are crossed out when the alterations result in a loss of function. TFH, T follicular helper; ALCL, anaplastic large cell lymphoma; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified; ATLL, adult T-cell leukemia/lymphoma; ENKTCL, extra-nodal NK/T-cell lymphoma; HSTL, hepatosplenic T-cell lymphoma; EATL, enteropathy associated T-cell lymphoma; MEITL, monomorphic epitheliotropic intestinal T-cell lymphoma; SPTCL, subcutaneous panniculitis-like T cell lymphomas.
Different types of genomic alterations can modify a biological function. Chromosomal translocations, detected by cytogenetic methods (karyotype, FISH, CGH), may produce fusion transcripts, detected by various technologies such as RT-PCR, RNAseq, or ld-RTPCR. They can result in aberrant expression, detectable by immunohistochemistry (for ALK fusions), or constitutive activation of oncogenes (such as JAK2, VAV1, CD28, etc.). Mutations in coding regions (single nucleotide variations or indels), detected by targeted exome or genomic sequencing, result in the gain of function of oncogenes or the loss of function of tumor suppressor genes. Mutations in noncoding regions have also been described, but their functional consequences are unclear. Disruption of the 3’UTR of PDL1 leads to its aberrant expression in extra-nodal NK/T-cell lymphomas and nasal-type (ENKTCL) and adult T-cell leukemia/lymphoma (ATLL), thus participating in immune escape (4, 5).
Epigenetic alterations appear to be a founding event in many PTCLs, mutations of genes involved in epigenetic regulation being frequently reported among different PTCL entities. Alterations of TET2 and DNMT3A, reflecting clonal hematopoiesis (6), were initially described in tumoral and reactive cells of TFH lymphomas (7, 8), but have also been reported in other entities, such as peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), especially with a cytotoxic immunophenotype (9), or chronic lymphoproliferative disorders of NK cells (10). Although mutations of these two genes are not sufficient to induce lymphomas (11, 12), the loss of TET2 is often required in vitro and in vivo prior to the occurrence of other genomic alterations (such as RHOA G17V mutation, less frequently VAV1 alterations or FYN_TRAF3IP2 fusion) as a “second-hit” in the development of TFH-lymphoma (13–15). Recurrent mutations of IDH2 R172, responsible for the production of the oncometabolite D-2 hydroxyglutarate, measurable in the serum of patients, are confined to tumoral T-cells in AITL (16). Mutations of TET2, DNMT3A and/or IDH2 may explain the common loss of 5-hydroxymethylcytosine observed by immunohistochemistry in most PTCL entities, with the exception of hepatosplenic T-cell lymphoma (HSTL) (17), although it has been reported independently of the mutational status. Alterations of SETD2 that inactivate histone methyltransferase function are almost ubiquitous in monomorphic epitheliotropic T-cell lymphoma (MEITL) and less frequent in HSTL (18, 19). Mutations of several other epigenetic modifiers (KMT2C, KMT2D, CREBBP, EP300) have also been reported among the main PTCL entities (20–22).
T-cell lymphomagenesis also implies the deregulation of signaling pathways, which occurs in many PTCL entities. Dysregulation of the TCR pathway is a common feature of TFH-lymphoma, ATLL, and PTCL-NOS (23, 24), whereas the JAK/STAT pathway is frequently altered in PTCL with a cytotoxic immunophenotype (ALK-positive or negative anaplastic large cell lymphoma (ALCL), breast implant associated-ALCL (Bi-ALCL), cytotoxic PTCL-NOS, extra-nodal NK/T-cell lymphoma, nasal-type (ENKTCL), enteropathy-associated T-cell lymphoma (EATL) and MEITL (9, 18, 22, 25, 26).
Dysregulation of the cell cycle in cancer is mostly due to inactivation of the tumor suppressor gene TP53, which is associated with a poor prognosis. In PTCL, alterations of TP53 and CDKN2A/PTEN have been reported in GATA3-positive PTCL-NOS, associated with complex chromosomal rearrangements and genomic instability (27–29), as well as in ENKTCL (30) and EATL (31). On the contrary, these alterations appear to be infrequent in TFH-lymphoma and ATLL (24, 29). TP63 rearrangements, described in a small subset of ALK-negative ALCL, appear to correlate with a poor prognosis (32, 33).
Another mechanism involved in T-cell lymphomagenesis is immune escape. Overexpression of PD-L1, due to alterations in the 3’-UTR region, lead to the anergy of reactive intra-tumoral lymphocytes in ENKTCL and ATLL (4, 5). PD-L1 expression has also been described in ALK-positive and ALK-negative ALCL, regulated by STAT3 activation, with a debated impact on the prognosis (34–36). The loss of CD58, HLA molecules, or β2-microglobulin, observed in ATLL and PTCL NOS, impairs recognition of the tumor cells by the immune system (24, 28). By contrast, DUSP22-rearranged ALK-negative ALCL shows immunogenic cues, with overexpression of the genes of T-cell co-stimulation CD58 and CD70 and HLA class II and decreasing PDL1 expression (37).
The role of reactive immune cells and stromal cells has been highlighted in AITL, a disease in which tumor cells are commonly scarce within a prominent microenvironment, thus influencing the results of gene expression studies (38). Microenvironmental molecular signatures may have prognostic relevance: a B-cell signature is associated with a favorable outcome, whereas macrophage and CD8+ cytotoxic signatures correlate with an adverse prognosis (38–40). The presence of tumor-associated macrophages has also been reported to be associated with a poor prognosis in other PTCL entities, such as GATA3 PTCL-NOS (41), and ALK-positive anaplastic large cell lymphoma (ALCL) (42).
Viral infection (EBV and HTLV-1) is also recognized as a driver of PTCL oncogenesis.
a. HTLV-1 infection is required for the development of ATLL. This retrovirus is randomly integrated into the host DNA (43, 44), with a predilection for specific transcription factor binding sites, such as STAT1, HDAC6, and TP53 (45). While most HTLV-1 carriers are asymptomatic, with multiple clones, a dominant clone is detected in ATLL patients (46, 47). Viral replication is permitted by clonal expansion of infected CD4+ T-cells (48). Expression of the oncogenic viral proteins TAX and HBZ leads to the disruption of homeostasis of infected cells, with the modification of epigenetic processes, genetic instability, and the accumulation of mutations (49). The TAX protein is highly immunogenic and responsible for the initiation of oncogenesis through NFKB and AP-1, while HBZ is involved in tumoral maintenance (50, 51).
b. EBV infection is a pre-requisite for the development of ENKTCL and other NK/T-cell neoplasms, such as aggressive NK-cell leukemia or the rare EBV+ T/NK lymphoproliferative disorders of childhood. The mechanism for acquisition of the EBV receptor CD21 by NK and T-cells is still debated between trogocytosis and viral episome transfer (52, 53). The survival of infected cells is permitted by the type II latency pattern, with the expression of LMP1 and EBNA1 but not EBNA2. LMP1 promotes the proliferation of EBV-infected cells through deregulation of the p53, CMYC, and NF-κB pathways, in synergy with the production of cytokines (IL-2, IL-9, IL-10 et IL-15), by infected neoplastic cells and cells of the microenvironment (54).
Antigenic stimulation may also play a role in the initiation or progression of T/NK cell lymphomagenesis, as established for gliadin in EATL (55), textured breast-implants in Bi-ALCL (56), or recently suggested for the SARS-CoV-2 mRNA vaccine in AITL (57).
Finally, genetic susceptibility has been identified in several entities, notably association between the haplotypes HLA-DPB1, HLA-DRB1, and IL18RAP and ENKTCL (58, 59), HLA DQ2/DQ8 and EATL (60), and germline mutations of HAVRC2 in subcutaneous panniculitis-like T-cell lymphoma (61).
2.2 Oncogenic events of the main non-cutaneous PTCL entities
PTCL can be derived from cells of the innate or adaptative immune system. Neoplasms likely deriving from the innate immune system comprise mostly extra-nodal lymphomas (ENKTCL, EATL, MEITL, HSTL, γδ-lymphomas, and probably cases among PTCL-NOS). They share a cytotoxic phenotype, alterations of the JAK/STAT pathway, and a context suggestive of chronic antigen stimulation. PTCL derived from cells of the adaptative immune system include most lymphomas with a nodal presentation with a T helper phenotype, such as TFH-lymphomas, ATLL, and PTCL-NOS. These lymphomas often show dysregulation of the TCR signaling pathway, in addition to alterations of epigenetic modifiers. The molecular characteristics of the main non cutaneous PTCL entities are summarized in Table 1.
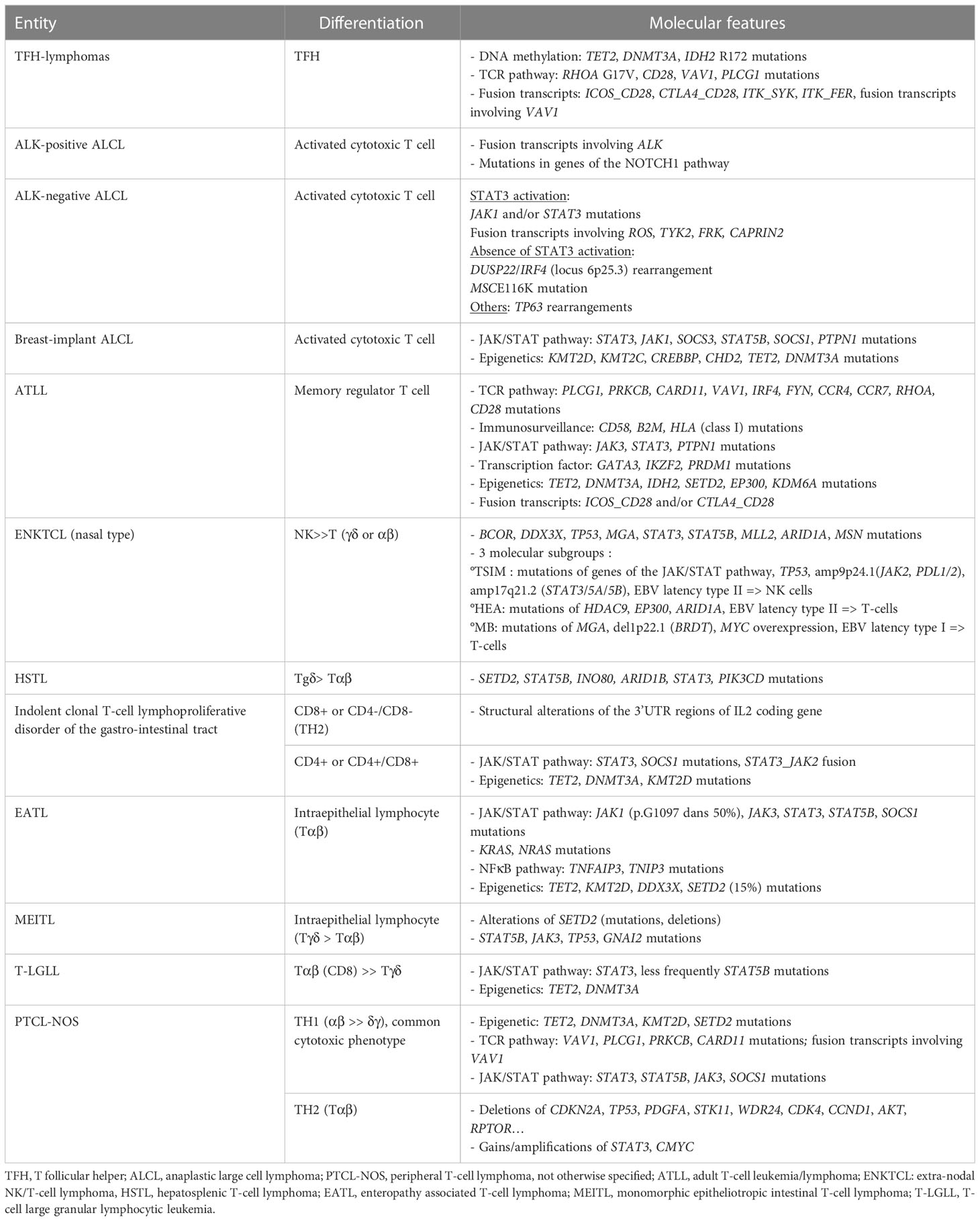
Table 1 Molecular characterization of the main non-cutaneous PTCL entities.
2.2.1 Nodal TFH lymphomas
In the revised 2022 WHO and ICC classifications, the family of lymphomas derived from TFH cells is regarded as a single disease encompassing three morphological subtypes, commonly designated angio-immunoblastic T-cell lymphoma (AITL), follicular-type, and not otherwise specified. They have distinct morphological features but share a common TFH phenotype and signature, as well as a similar molecular pattern. In routine practice, the TFH phenotype is defined by the expression of CD4, with at least two TFH markers among PD1, ICOS, CD10, CXCL13, and BCL6, although none of them, in particular PD1 and ICOS, are fully specific, as they can be expressed by non-TFH reactive cells or other non-TFH PTCLs (62–65). TFH-lymphomas show a unique mutational landscape, characterized by the accumulation of alterations in genes involved in epigenetic regulation (TET2, DNMT3A, IDH2) (7, 11, 66) and the TCR pathway (RHOA, VAV1, CD28, PLCG1, FYN, LCK) (23, 67–73). Fusion transcripts involving genes of the TCR signaling (ICOS_CD28, CTLA4_CD28, ITK_SYK or involving VAV1 with multiple partners) and NFKB (FYN_TRAF3IP2) pathways can be observed. Although mutations of TET2 and DNMT3A may be observed in tumoral and reactive cells, hotspot mutations in RHOA G17V and IDH2 R172 are thought to be restricted to the TFH tumor cells (74, 75). The recurrent RHOA G17V mutation, detected in 50 to 70% of AITL (23, 67–69, 75–78), impairs the GTPase domain, showing dominant negative activity and thus abolishing GTP binding and downstream signaling. This mutation is also responsible for VAV1 phosphorylation and TCR pathway activation (71). RHOA G17V drives TFH polarization and promotes lymphomagenesis in vivo through ICOS-PI3K-mTOR signaling (14, 15). The IDH2 R172K mutation combined with TET2 alterations modulate the tumoral microenvironment, promoting B-cell proliferation, the accumulation of plasma cells, and angiogenesis (79). Mutations in CD28, observed in 10% of TFH-PTCL, are reported to be mutually exclusive from fusion transcripts involving CD28 and other genes of the TCR pathway (23, 72, 73, 76, 80, 81). Alterations in VAV1 result in oncogenic activation of the NFAT pathway (70, 71, 82). Alterations of many other genes of the TCR pathway (FYN, PLCG1, PIK3R1, PDPK1, AKT, LCK, TRAF6) contribute to T-cell proliferation (23). The rare ITK_SYK fusion transcript has been described in follicular-type and in rare cases of AITL (83, 84). TFH lymphomas illustrate multistep oncogenesis, as shown by the development of « AITL » tumors in vivo in TET2 knock-out mice transfected with a RHOA mutated gene (13, 14), or in double-mutant mice TET2/IDH2R172K (79).
Overall, although there is no pathognomonic genomic alteration that defines the TFH category, the detection of RHOA G17V and/or IDH2 R172 mutations and, to a lesser extent, fusion transcripts involving CD28 or TRAF3IP2 constitute a supplemental clue to the diagnosis for pathologists.
2.2.2 - Anaplastic large-cell lymphomas
This category, defined by large “hallmark” cells showing strong and homogenous CD30 expression by immunohistochemistry, includes several entities based on the association of ALK-rearrangement and the clinical presentation as systemic, cutaneous, or breast implant-associated disease. Cutaneous ALCL are not considered here.
A) ALK-positive ALCL is the only entity defined by recurrent genomic translocations involving the ALK gene on chromosome 2p23 with various partners, the most frequent (~80%) being NPM1. The translocation produces an oncogenic fusion protein consisting of the association of the N-region of a partner gene with the catalytic tyrosine kinase domain of ALK, resulting in constitutive activation by dimerization. The chimeric NPM1_ALK protein triggers several oncogenic pathways (JAK/STAT, PI3K, MAPK, PLCG), leading to neoplastic transformation (85, 86), whereas TRAF1_ALK activates the NFKB pathway (87, 88). Recently, mutations of NOTCH1 and genes of the TCR pathway have also been reported (89). The diagnosis is based on the detection of aberrant ALK expression by immunohistochemistry using anti-ALK antibodies. The pattern of staining may be nuclear +/- nucleolar and/or cytoplasmic, depending on the partner gene involved in the translocation (Table 2). The disease, which mainly occurs in children and young adults, follows a generally favorable prognosis (5-year OS around 90%) after chemotherapy with CHOEP (100–102) or BV-CHP (103). The prognosis may be less favorable in cases with secondary MYC overexpression or rearrangement, in certain histologic variants (small-cell or lymphohistiocytic) occurring in children (88, 104, 105).
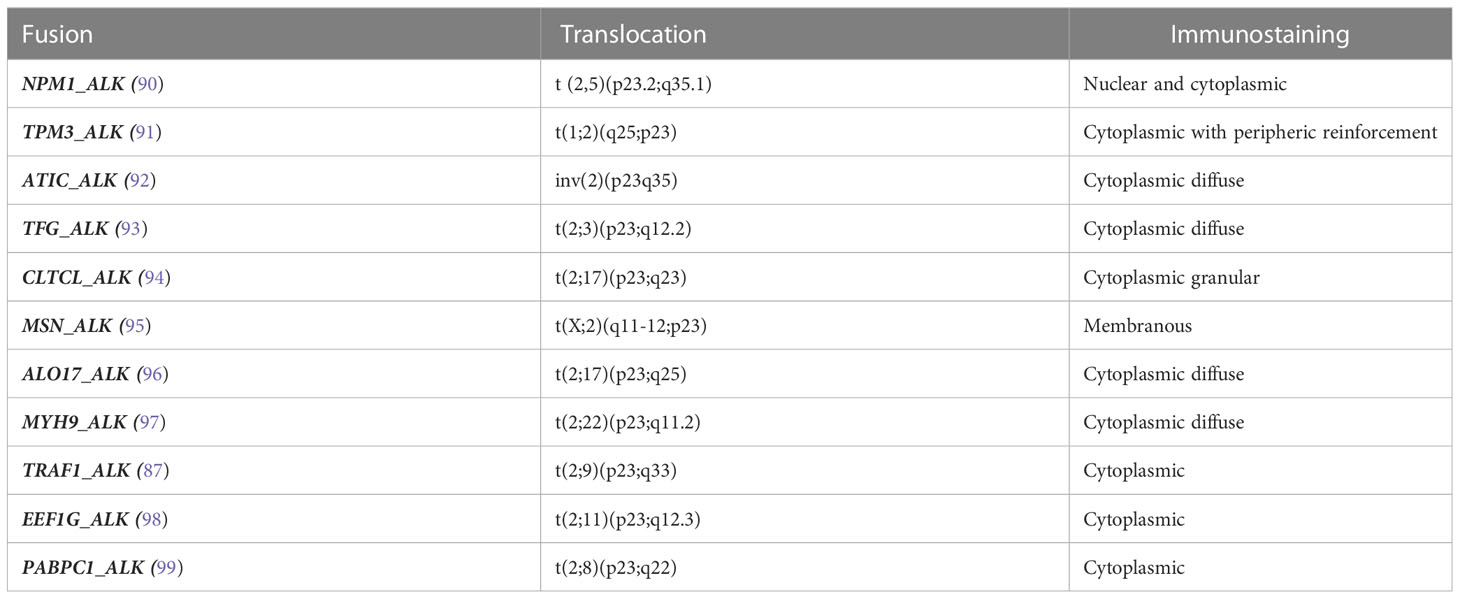
Table 2 Fusion transcripts involving ALK in ALK-positive anaplastic large cell lymphoma.
B) Systemic ALK-negative ALCL is still heterogeneous in the current classifications, gathering cases with different oncogenic pathways:
- Rearrangement of the 6p25.3 locus involving DUSP22 and IRF4 (106) defines a peculiar subgroup (approximately 25-30% of ALK-negative ALCL), characterized by a non-cytotoxic phenotype, silencing of the tumor suppressor gene DUSP22 while showing normal IRF4 expression, absence of STAT3 activation, global DNA hypomethylation, an immunogenic molecular profile (overexpression of CD58, CTA, HLA class II), and expression of LEF1 (37, 107–110). Recurrent MSC E116K mutations are responsible for activation of the CD30-IRF4-CMYC axis and the dysregulation of cell cycle arrest (111). These rearrangements were initially detected by mate-pair DNA sequencing in the context of a translocation t(6,7)(p25.3;q32.3) also involving the non-coding gene FLJ43663 at the fragile site FRA7H of chromosome 7 (106). The prognosis is debated, favorable in most studies (112, 113) but not confirmed in others (114, 115).
- Rearrangements of TP63, due to the inversion inv (3) (q26q28) or translocation t(3,6)(q28;p22.3) that produce the fusion transcripts TBL1XR1_TP63 and TP63_ATXN1 respectively, coding for oncogenic chimeric proteins, are rare and associated with a poor prognosis (32, 112, 114). The detection of P63 by immunohistochemistry may reflect P63 overexpression independently of the presence of fusion transcript (33).
- Aberrant truncated transcripts of ERBB4 was also reported in 24% of ALK-negative ALCL in one study, associated with a Hodgkin-like morphology, without clinical relevance (116).
- Expression of pSTAT3 by immunohistochemistry, reflecting activation of the JAK/STAT pathway, is common in ALK-positive and ALK-negative ALCL, with the notable exception of those cases associated with DUSP22 rearrangement. Among ALK-negative ALCLs, a recent study that excluded cases with rearranged DUSP22 suggested that positive pSTAT3 cases constitute a distinct subgroup, characterized by a cytotoxic phenotype and the expression of EMA and PDL1, that is associated with a better prognosis than negative pSTAT3 cases (117). Such constitutive phosphorylation of STAT3 has been previously shown to be related to mutations in JAK1 and/or STAT3, reported in 18% of ALK-negative ALCLs, as well as in fusion transcripts involving ROS, TYK2, and FRK (26, 118). More recently, fusion transcripts involving JAK2 with several partners (PABPC1, PCM1, ILF3, TFG, MAP7) were detected by targeted RNAseq and associated with a Hodgkin-like morphology (119).
C) Breast-implant associated ALCL is a site-specific entity that occurs after a long latency after a breast implant for reconstruction or cosmetic reasons. Most cases are non-invasive. The disease appears to be due to chronic inflammation, with possible TH2 polarization, linked to a macro-textured implant (120). High-throughput sequencing studies have highlighted alterations of genes involved in the JAK-STAT pathway (STAT3, STAT5B, JAK1, JAK3, SOCS1, SOCS3), leading to its constitutive activation, together with recurrent mutations in epigenetic modifiers (KMT2C, CREBBP) (22, 121), the loss of chromosome 20 (122), and chromosome 9p24 gains, leading to PDL1 expression (123). Recently, a STAT3_JAK2 fusion transcript was also reported (124).
Several immunohistochemical algorithms have been recently proposed to classify ALCL based onLEF1, P63, and pSTAT3 (117, 125), although this currently has no impact on the management of ALK-negative ALCL patients.
2.2.3 EBV-positive NK or T-cell neoplasms
EBV-related NK or T-cell neoplasms are heterogenous diseases derived from T or NK cells (126). The revised WHO and ICC classifications recognize ENKTCL, and primary nodal EBV-positive T/NK-cell lymphomas, characterized by nodal involvement, as distinct entities (1, 2). In addition to EBV, considered to be a driver of oncogenesis in these lymphomas, defined by EBV infection of virtually all neoplastic cells, as shown by in situ hybridization with EBER probes, the mutational landscape of ENKTCL is characterized by recurrent mutations of genes coding for RNA helicases (especially DDX3X), as well as TP53, genes of the JAK/STAT pathway (JAK3, STAT5B, STAT3) and epigenetic modifiers (MLL2, ARID1A, EP300, ASXL3) (20, 30, 127). The initial poor prognosis associated with DDX3X and TP53 mutations for patients treated with the CHOP regimen was not confirmed for patients receiving L-asparaginase treatment (20, 30, 128). Recurrent deletions of the 6q21 locus encompassing tumor suppressor genes (PRDM1, ATG5, AIM1, FOXO3 et HACE1) have been detected by CGH array (129–131). A recent large integrative analysis of genome, exome, and RNA sequencing, identified three molecular subgroups (30):
- the “TSIM (tumor suppressor and immunomodulator)” subgroup is characterized by frequent TP53 mutations, deletion of the 6q21 locus, amplification of the 9p24.1 locus containing PDL1 and PDL2, and the amplification of genes of the JAK/STAT pathway. This subgroup presents a gene expression signature enriched in NK-cell genes. There is an EBV latency II phenotype, with expression of the lytic gene BALF3, responsible for DNA damage and genomic instability.
- the “MB (MGA, BRDT)” subgroup is characterized by frequent MGA mutations, loss of heterozygosity of BRDT, and MYC overexpression, as well as activation of the MAPK, NOTCH, and WNT pathways. The EBV latency is of type I, with downregulation of LMP1.
-the “HEA (HDAC, EP300, ARID1A)” subgroup is characterized by mutations of epigenetic modifier genes (HDAC9, EP300 et ARID1A), resulting in aberrant histone acetylation. The gene expression profile is enriched in T-cell genes and shows activation of the TCR and NFKB pathways. The EBV latency is of type II, with expression of the BNRF1 lytic gene.
Although there is currently no applicability of this molecular subclassification in routine practice, the poor prognosis of the MB subgroup relative to TSIM and HEA (3-year OS rate of 38% versus 80% and 90%, respectively) may justify the evaluation of MYC expression in ENKTCL. Structural alterations of CD274, coding for PDL1, appear to confer sensitivity to immune checkpoint inhibitors (5, 132).
EBV-positive nodal T- and NK-cell lymphoma or primary nodal Epstein-Barr virus–positive T-cell/NK-cell lymphoma, is now recognized as a distinct entity in both the WHO and ICC classifications respectively, due to its differences with ENKTCL. This entity is morphologically characterized by the lack of necrosis and angiocentrism, a common CD8+ CD56- phenotype, a frequent T-cell origin, and, finally, peculiar molecular abnormalities, with frequent TET2, PIK3CD, and STAT3 mutations, activation of the NFKB, IFNγ, and JAK-STAT3 pathways, resulting in high PDL1 expression, and lower genomic instability (133). The prognosis is reported to be poorer than for ENKTCL.
The mutational landscape of ENKTCL is shared with that of other EBV-positive NK/T-cell neoplasms, in particular, aggressive NK-cell leukemia (134–136), as well as that of chronic active EBV-disease (137). This genetic landscape may be of clinical relevance in the rare cases that require a differential diagnosis from infectious mononucleosis.
2.2.4 Adult T-cell Leukemia/Lymphoma
This HTLV-1-associated T-cell neoplasm occurs after a long latency (more than 25-30 years) following infection, mainly due to prolonged breast feeding and, less frequently, sexual transmission (138). The histopathological diagnosis is challenging in the absence of information concerning the HTLV-1 status, as the pathological aspects of ATLL are highly heterogeneous. It can be evoked by the loss of CD7, together with the expression of CD25 and FOXP3, although the CD25+/FOXP3+ immunophenotype is variable and not specific to ATLL (139–142). The molecular landscape is characterized by mutations in genes of the TCR pathway (PLCG1, PRKCB, CARD11, VAV1, IRF4, FYN, CCR4, CCR7, RHOA, CD28), JAK/STAT pathway (JAK3, STAT3, PTPN1), immune surveillance (CD58, B2M, HLA class I), DNA damage (TP53, CDKN2A, POT1), epigenetic modifiers (TET2, DNMT3A, IDH2, SETD2, EP300, KDM6A), transcription factors (GATA3, IKZF2, PRDM1), and fusion transcripts involving CD28 (ICOS_CD28 and CTLA4_CD28) (24, 143). The co-expression of these two fusion transcripts can occur in patients younger than 50 years of age (144). Gene mutations of the TCR/NFKB pathway, TP53, and IRF4 are associated with an aggressive outcome, whereas STAT3 mutations are frequently observed in patients with more indolent disease (143, 145). The type of CCR4 mutation also has a specific prognostic impact (unfavorable in cases of frameshifts vs non-synonymous variations) (146).
2.2.5 Intestinal T-cell lymphomas
Enteropathy-associated T-cell lymphoma (EATL) and monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL) are two distinct entities, with different morphological and immunophenotypic features. Although both are derived from intestinal intra-epithelial lymphocytes (IEL) expressing CD103, EATL and MEITL show distinct clinico-pathological and molecular characteristics (31).
a. EATL is associated with celiac disease or gluten sensitivity. Its histopathological features include the proliferation of pleomorphic to anaplastic T-cells expressing CD3 and CD30, but lacking CD4 and CD8, despite an activated cytotoxic profile. CD103 is variably expressed. Overexpression of P53 is detectable by immunohistochemistry, independently of gene alterations (147). This entity shows frequent alterations of the JAK/STAT pathway (in particular, STAT3 and JAK1, as well as SOCS1 and SOCS3), whereas STAT5B mutations are almost constantlyabsent (18, 148, 149). TET2 and, less frequently, mutations of the RAS/MAPK pathway (149) can be observed, whereas SETD2 mutations were almost absent in most recent series (18, 148). Gene expression profiling studies have shown enrichment for genes of the JAK/STAT (STAT3, STAT5A) and IFNγ pathways (31).
b. MEITL does not associate with celiac disease and is typically characterized by the proliferation of monomorphic medium cells, showing epitheliotropism and a CD8+ CD56+ phenotype. However, approximately 25% of cases may show more pleomorphism and certain phenotypic variations associated with the prognosis, in particular, a better outcome in the presence of aberrant expression of CD20 or poor outcome in the presence of MYC expression and TP53 alterations, suggesting the utility of screening for these abnormalities in routine practice (150, 151). MEITL has a very homogeneous genetic landscape, with almost consistent alterations of SETD2 (mutation +/- deletion) associated with mutations of STAT5B (approximately 60%) or JAK3 and GNAI2, which constitute a common feature and may help pathologists in difficult cases (18, 150–152).
Indolent clonal T-cell lymphoproliferative disorder of the gastrointestinal tract (2), also designated indolent T-cell lymphoma of the gastrointestinal tract in the WHO classification (1), is now recognized as a definitive entity in both classifications due to the recent evidence of neoplastic molecular features, i.e., alterations of genes in the JAK/STAT pathway or epigenetic modifier genes and JAK2_STAT3 fusions or structural alterations of the 3’UTR of the IL2 gene, depending on the CD4 or CD8 phenotype (153, 154). Despite an indolent course, some cases may relapse, spread to other sites, or transform, indicating potential aggressiveness (155, 156).
Indolent NK-cell lymphoproliferative disorder of the gastrointestinal tract is a rare condition, and a new entity in the WHO and ICC classification. Although neoplastic molecular characteristics have also been described, in particular, recurrent deletions of STAT3, there is no extension of this lymphoproliferation beyond the gastrointestinal tract and the outcome is favorable (157).
2.2.6 Hepatosplenic T-cell lymphoma
This rare neoplasm occurs preferentially in young males but can arise at any age, with a possible context of immunosuppression. The diagnosis is based on highly characteristic pathological features, in particular sinus infiltration in the bone marrow by small to medium lymphocytes with a CD3+, CD5+, CD4-/CD8-, CD56+ phenotype, commonly TCRγδ+. The sinusal infiltration in the liver and spleen is less specific. There is typically no lymph node involvement. This entity was initially characterized by an isochromosome 7q and chromosome 8 trisomy (158, 159), but cytogenetic material is not always available in routine practice to support the diagnosis and FISH analysis can be challenging. The mutational landscape has been reported, identifying three types of mutations involving 1/epigenetic modifier genes (SETD2, ARID1B, INO80, TET3 and SMARCA2), 2/STAT5B or STAT3 that are mutually exclusive, and 3/PIK3CD (19, 160, 161). Gene expression profiling studies show a distinct signature, characterized by the overexpression of oncogenes (FOS, FOSB VAV3, MAF), NK-cell associated genes (KIR3DS1, CD244 and other KIRs), the tyrosine kinase SYK, and S1PR5, and downregulation of AIM1, which could constitute targets for therapy in this disease that has always fatal outcome (162). A recent single-cell profiling study suggested a change in the gene expression profile of the tumor cells during disease progression under the selective pressure of therapy (163).
2.2.7 PTCL-NOS
PTCL-NOS is a diagnosis of exclusion, corresponding to cases that do not fulfill the criteria for defined PTCL entities. Thus, a large panel of immunohistochemical markers and the integration of clinical and often molecular features are required to exclude any other PTCL. Gene expression profiling studies have shown two subgroups based on expression of the TBX21 and GATA3 transcription factors associated with the immunological TH1 and TH2 signatures, respectively (39, 40), confirmed by immunohistochemistry (164). The TBX21 group is enriched in genes of IFNγ and NFKB pathway signatures and shows mutations of genes involved in epigenetic regulation (TET1, TET3, DNMT3A), whereas the GATA3 group shows a cell proliferation signature driven by MYC, together with enrichment in PI3K/Akt/mTOR pathway signatures, a higher number of genomic copy number abnormalities, and a poorer outcome (27, 165). In routine practice, there is no consensus concerning the proposed thresholds of immunohistochemical markers to define these two subgroups and an understanding of the clinical relevance of such immunohistochemical algorithms requires further studies.
The mutational landscape of PTCL-NOS is currently poorly defined, likely due to the heterogeneity of this category. Only a few “omic” studies focusing on PTCL-NOS have been published to date. Targeted sequencing has shown mutations of epigenetic modulator genes, notably histone methylation (KMT2D, SETD2, KMT2A, KDM6A) or acetylation (EP300, CREBBP), as well as that of genes of the TCR pathway (TNFAIP3, TRAF3, TNFRSF14) and tumor suppressor genes (TP53, ATM, FOXO1, BCORL1) (21, 166). Recent integrative studies based on exome and RNA sequencing have confirmed mutations of genes involved in epigenetic regulation (TET2, DNMT3A, KMT2C, KMT2D, SETD2, CREBBP, ARID1A), tumor suppressor genes (TP53, TP63, ATM, FAT1, LATS1, STK3), and genes of the NOTCH pathway (NOTCH1 and 2) (28, 167). In one study, mutations in FAT1 were shown to be associated with a poor prognosis (167). RNAseq studies have shown fusion transcripts involving VAV1 with various partner genes (GSS, THAP4, MYO1F, S100, HNRNPM) and rearrangements of VAV1 were detected by FISH in 11% of PTCL-NOS (28, 70, 82). The VAV1_MYO1F transcript induces tumoral TH2 polarization and the accumulation of tumor-associated macrophages (41). Other fusion transcripts have also been reported in single cases (ITK_FER, IKZF2_ERBB4, ETV6_FGFR3) (82). A t (14, 19)(q11;q13) translocation, involving TCRA and the poliovirus receptor-related 2 gene (PVRL2), resulting in BCL3 overexpression, has also been reported in PTCL-NOS, including one case with the morphological variant of Lennert’s lymphoma (168, 169).
PTCL-NOS with a cytotoxic phenotype has been reported in 25 to 40% of cases, associated with impaired immunity and a poor prognosis (9, 170). This immunophenotypic subgroup has also been identified in gene expression studies within the PTCL-NOS TBX21 subgroup, enriched for genes of CD8/NK cells, the IFN response, and an immunosuppressive signature (39, 40). Targeted sequencing has shown recurrent mutations of genes involved in epigenetic regulation (TET2, DNMT3A), TCR (VAV1, PLCG1, PRKCB, CARD11) and the JAK/STAT pathways, as well as TP53 (9). Fusion transcripts involving VAV1 have been detected in 14% of patients. In another study, two cases of cytotoxic PTCL-NOS with diffuse cutaneous and medullary involvement showed a t (6, 14)(p25;q11.2) translocation resulting from rearrangement between the TCRα and IRF4 loci (171).
Despite these advances in our knowledge of the molecular biology of this entity, there is still an unmet need for the management of PTCL-NOS patients.
3 From biology to the diagnosis and management of PTCL patients
The diagnosis and classification of PTCLs are often challenging for pathologists, requiring experienced hematopathologists and access to molecular tests. In the absence of clear diagnostic guidelines, practices are often heterogenous between centers (172–174).
Analysis of rearrangements of the TCR loci (especially TRG or TRB) is an important element of the diagnostic process. PCR-based assays (BIOMED-2) are largely widespread in routine practice due to their reliability on FFPE samples (160–162). However, there are a number of pitfalls in the interpretation of clonality testing due to “false-negative” results in cases with low tumoral content, especially common in AITL, or due to T-cell oligoclones, as observed in AITL (175). Conversely, the presence of clonal TCR rearrangements in certain reactive conditions or even in B-cell lymphomas (notably Hodgkin lymphomas) due to TCR repertoire restriction can be misleading (176). The development of NGS-amplicon based clonality assays may improve the detection of scarce clones in a polyclonal background and allow the determination of clonotypes (177). Several authors have proposed analyzing TCR genes by whole genome sequencing, but its applicability in routine practice is still limited (178). Others have highlighted the potential interest of analyzing non-recombined T-cell receptor sequences using a digital PCR assay (179).
Recently, gene expression studies suggested molecular classifiers to discriminate the main PTCL entities, with certain limitations due to tumor cell content and the quality of the nucleic acid (40, 180, 181). Such tools should be used in routine practice with caution, as they were developed for the classification of the most common entities, their robustness has not yet been extensively evaluated, and the results need to be interpreted in the context of the histopathological analysis. Indeed, misclassification using these algorithms or discordance with the histopathological data occur for 15 to 20% of samples, likely due to a prominent microenvironment or plasticity of the tumor cells (180, 181). Sequencing of transposase-accessible chromatin (ATAC-seq) has been proposed as another innovative strategy to classify PTCL (182), but it requires fresh or frozen samples and its applicability in routine practice has not been yet evaluated.
Exome and genome sequencing studies have allowed a precise description of the mutational landscape of almost all PTCL entities. An increasing number of laboratories have developed targeted NGS panels for the molecular characterization of lymphomas or hematological neoplasms that are useful for their diagnosis and classification (183). The diagnostic performance of targeted NGS relative to that of measuring T-cell clonality by BIOMED multiplex PCR in PTCL was assessed in one study and showed similar sensitivity (approximately 95%) but significantly superior specificity (100% versus 45%) (184). However, there is currently no consensus concerning the design of the panel or the sequencing depth or coverage, which may affect the interpretation of the results. Hotspot mutations of diagnostic relevance, notably RHOA G17V or IDH2 R172 mutations, can also be detected using alternative technologies, such as allele-specific PCR, digital PCR, and RTMLPA (180, 185–188).
As described above, despite highly characteristic genetic profiles for certain PTCLs, such as TFH-lymphomas and MEITL, there is no single pathognomonic molecular alteration that can define an entity, apart from ALK-positive ALCL. However, the detection of RHOA G17V and IDH2 R172 mutations in routine practice strongly supports the diagnosis of TFH-PTCL (14, 16, 63, 64, 72). Although RHOA mutations can also be observed in 10% of ATLL, only 1% correspond to G17V (189), whereas IDH2 R172 is almost specific to AITL.
Within ALCL, the discovery of the translocation t(2,5) led to the development and use of an anti-ALK antibody in routine practice, allowing rapid and efficient determination of the ALK status by immunohistochemistry (90, 190). The identity of the ALK gene partner does not appear to be important, with no prognostic relevance, with the exception of the rare TRAF1_ALK fusion transcript, which was shown to be associated with a poor outcome in a recent study (87). In children with ALK-positive ALCL, the prognosis also correlates with the ALK antibody titer and the copy number of the ALK fusion transcript in the blood at diagnosis (MDD: minimal disseminated disease) and after treatment (MRD: minimal residual disease) (189, 191–194). The significance of these parameters is unknown in adult patients.
In routine practice, FISH is required to diagnose DUSP22-rearranged ALK-negative ALCL, a molecularly distinct subgroup that probably merits being individualized (37). Interestingly, it is also characterized by the presence of hotspot mutations of MSC E116K in 35% of DUSP22-rearranged cases, a finding currently without clinical relevance (111). In the context of intestinal T-cell lymphomas, the identification of SETD2 alterations strongly favors the diagnosis of MEITL and may be helpful in distinguishing difficult cases from EATL (18). These alterations (mutations and/or deletions) result in reduced H3K36 trimethylation, which can be detected by immunohistochemistry (195).
A number of genetic alterations may also predict the outcome of patients with a T- or NK-cell neoplasm, as observed in ENKTCL, with the poor prognosis of the MB subgroup (30), and in ATLL with CCR4 mutations or CCR7 alterations (146, 196–198). In AITL, the DNMT3AR882X mutation may be associated with a poor prognosis and resistance to anthracyclines (199), a finding that could influence the management of these patients in the future. MYC expression/rearrangement or TP53 alterations are associated with a poor prognosis in various PTCL entities, especially ALK-positive ALCL (104, 105), ENKTCL (30) and MEITL (151), but without a significant impact on the management of these patients.
The diagnosis of ATLL is challenging for pathologists without knowledge of the HTLV-1 serology status. Morphological and immunophenotypic features may be confusing for ALK-negative ALCL, GATA3 PTCL-NOS, or even TFH-lymphomas, with an impact on the appropriate management of these patients. There is an unmet need for the development of HTLV-1 biomarkers applicable to FFPE samples in routine practice. TAX is not expressed in most ATLL tumors, whereas HBZ is the only viral transcript expressed during disease progression and could be a good candidate (50, 51). In situ hybridization was proposed to detect the HBZ gene in FFPE tissues in a single study, but there has thus far been no development of this technology in routine practice (200). More recently, targeted gene expression studies have been developed to measure expression of the HBZ transcript in routinely-fixed samples (181, 201).
Thus far, the detection of fusion transcripts has not been integrated into the routine diagnosis of PTCL due to the low prevalence of known fusions (10%) and limited accessibility to available technologies. Although RNAseq is the most exhaustive technology to detect fusion transcripts, several targeted RNA sequencing alternatives have been developed (ArcherFusionPlex®, Qiaseq RNA fusion XP®, and ld-RTPCR (202)), which can be implemented in a routine laboratory at a lower cost. Despite the current lack of clinical relevance of most fusion transcripts, the recent identification of rearrangements involving JAK2 in systemic CD30-positive PTCL (119, 203), Bi-ALCL (124), in indolent clonal T-cell lymphoproliferative disorder of the gastrointestinal tract (153), and cutaneous T-cell lymphoma (204–208) opens the door to targeted therapies requiring the detection of such fusion transcripts. Furthermore, in addition to pathological features, the detection of certain transcripts may be of diagnostic value to support a diagnosis among several hypotheses. For example, ICOS_CD28, ITK_SYK, or FYN_TRAF3IP2 fusions favor a diagnosis of PTCL, especially TFH-lymphoma in difficult cases, raising the possibility of the differential diagnosis from Hodgkin lymphoma or marginal zone lymphoma.
Recent studies on a limited number of cases have demonstrated the applicability of assessing circulating tumor DNA (ctDNA) by high-throughput sequencing for PTCL. In a comparison with matched tumors, ctDNA detected by HTS-sequencing of the TCR was detected for 78% of various PTCL entities (209). The detection of hotspot mutations in AITL (RHOA and IDH2) appears to be promising and sensitive, with 100% concordance between cell-free DNA and the tumors by NGS in one study (210) and a prevalence of 70% in another using allele-specific PCR (188). In ENKTCL, a concordance of 93.5% between ctDNA and tumor biopsy sequencing was observed, with a potential prognostic significance (211–213). Beyond the potential application for the detection of minimal residual disease during follow-up or at relapse, the detection of ctDNA may also be a promising tool to help for the diagnosis of difficult cases, especially those with limited tumor material, in combination with pathological analysis.
4 From molecular targets to personalized treatment: alternatives or additive therapeutic options to standard chemotherapy
The CHO(E)P-based regimen has been the standard of care for PTCL for many decades (214). To date, most alternative therapies have failed to demonstrate a better outcome and the prognosis of patients for most PTCLs is still poor (215, 216), even for stage I-II disease (217).
4.1 Frontline targeted therapies
A recent major change in frontline therapy is the use of brentuximab-vedotin (BV), in addition to CHP chemotherapy, for patients with CD30 positive PTCL. Approval for the use of BV by the US Food and Drug Administration (FDA) followed the ECHELON-2 study, which demonstrated a significant improvement in progression free survival (PFS) (median 48 months in the BV-CHP group versus 20.8 months in the CHOP group, p=0.0110), and a reduced risk of death in the BV-CHP arm, although the median overall survival (OS) was not reached (103). However, the subgroup analyses confirmed the benefit for ALCL patients receiving BV, but not for those with AITL. For PTCL-NOS, the potential benefit is unclear, probably due to the heterogeneity of the disease with respect to the percentage of CD30-positive cells (threshold ≥10% of cells by local review). The addition of BV to standard chemotherapy has also been shown to provide an improvement in event-free survival of children with ALK-positive ALCL (218). In addition, a retrospective pooled study showed a significant improvement of OS and PFS in ALK-positive ALCL with the use of CHOEP in frontline therapy compared to CHOP, independently of age (100). To date, there has been no comparison between CHOEP and BV-CHP in the frontline management of ALK-positive ALCL patients.
A second large trial compared the addition of romidepsin to CHOP versus CHOP alone in previously untreated PTCL patients (219). Although the results of the study were negative, as PFS did not statistically increase in the romidepsin CHOP group relative to the control arm, a trend towards longer PFS was observed for TFH-lymphoma patients, suggesting susceptibility of TFH-lymphomas to drugs targeting epigenetics. A phase 2 trial combining the oral form of 5-azacytidine to CHOP in the first line for 21 PTCL patients, including 17 with TFH-lymphoma, showed promising results, with an 88% complete response (CR) rate for TFH-lymphoma patients and 69% two-year PFS. However, these promising results, based on a limited number of patients, need to be confirmed in a larger series (220).
Among ENKTCL, the introduction of asparaginase has significantly improved the prognosis of patients (221, 222). Better efficacy and tolerance have been observed with the use of pegasparaginase relative to L-asparaginase (223, 224). Although there is no international consensus concerning the treatment sequence, it is generally accepted that frontline therapy should include at least pegylated-asparaginase and gemcitabine in association with various combination of other agents or strategies (including cisplatin/oxaliplatin, dexamethasone, methotrexate, and radiotherapy), depending on the staging of the lymphoma as localized or disseminated disease (223–226).
In ATLL, the characterization of a Treg/TH2 phenotype and polarization of the tumor cells led to the development of anti-CCR4 monoclonal antibodies (227). Although this targeted therapy is currently used for refractory/relapsed patients, a recent study showed better survival of aggressive transplant-ineligible ATLL using a polychemotherapy regimen containing mogalizumab in the first line (4-year OS of 46.3% versus 20.6%, p=0.033) (228). A previous study failed to demonstrate any benefit with the addition of mogalizumab in the first line for transplant-eligible patients (229). It is still unknown whether the use of mogamulizumab could be extended in the future to other PTCLs that express CCR4, in particular, GATA3-PTCL-NOS (164).
4.2 Promising therapeutic options for relapse/refractory PTCL patients
Several ALK inhibitors have been tested in refractory/relapsed ALK-positive ALCL patients, showing an improvement in PFS and long-term complete remission (230–234). However, there are no recommendations concerning the indication or duration of treatment.
The frequent alterations of chromatin modifiers among PTCLs has led to the development of therapies to regulate epigenetic programs. Although approved by the FDA, the use of romidepsin, pralatrexate, and belinostat did not show significant efficacy in several studies, probably due to the enrollment of patients with several PTCL entities, leading to a small sample size for each (219, 235). However, subgroup analyses showed a benefit for HDAC inhibitors for TFH-lymphomas (219, 236). Prospective studies are needed to confirm these promising results for TFH-lymphoma patients and to identify predictive biomarkers of response. Several studies using hypomethylating agents, such as 5’azacytidine, have also shown promising results in AITL, usually independently of the TET2, DNMT3A, and IDH2 mutational status, although these studies had only small numbers of patients (237, 238). A phase 3 trial comparing the use of the oral form of the 5-azacytidine to investigator-choice treatment between gemcitabine, bendamustine, or romidepsin in relapsed/refractory THF-lymphoma patients was recently reported. The primary endpoint was PFS and was not met, likely due to the trial being underpowered. However, OS was longer for patients receiving 5-azacytidine, suggesting efficacy of the drug. The combination of oral 5-azacytidine and romidepsin has shown efficacy for frontline or refractory/relapsed PTCL patients, especially those with a TFH phenotype (239). The development of IDH2 Inhibitors in acute myeloid leukemia (240, 241) suggests their potential application in TFH-lymphomas with IDH2 mutations.
In AITL, the identification of gene alterations enhancing the TCR pathway paved the way for the use of dasatinib, a PKC inhibitor, which showed efficacy in vitro and in vivo in a mouse RHOA G17V mutant TET2 deleted model, as well as in a phase 1 trial for relapsed/refractory patients (71, 242).
The identification of structural alterations of PDL1 in ENKTCL led to studies to evaluate the use of immune checkpoint inhibitors, such as PD1 inhibitors, for refractory/relapsed patients (5, 132, 243). The response to these therapies may be predicted by characterization of the tumor immune microenvironment using gene expression profiling (Nanostring technology) or immunohistochemistry (anti-PDL1, anti-FOXP3, anti-CD68) (244). Surprisingly, although similar disruption of the 3’UTR of PDL1 was also detected in ATLL, the use of PD1 inhibitors in this entity led to rapid progression of the disease for at least some patients (245).
Translocations involving JAK2 leads to phosphorylation of the tyrosine kinase domain, subsequent constitutive activation, and downstream JAK/STAT pathway activation (246). This pathway is now targeted using JAK inhibitors in the clinic for myeloproliferative neoplasms and cancers with high pSTAT3 levels (247), such as ALK-negative ALCL, may be a good candidate for such targeted therapy, as suggested in vivo in a xenograft model (248). In a recent study, ruxolitinib showed some clinical activity on PTCLs, especially those with JAK or STAT mutations or activation (249).
Recently, the KIR3DL2 killer Immunoglobulin-like receptor was identified as a useful biomarker and therapeutic target among cutaneous T-cell lymphomas, including mycosis fungoides and Sezary syndrome (250, 251) and ATLL (252, 253). Its expression in other PTCL entities has been recently evaluated (254) and lacutamab, an anti KIR3DL2 antibody, is currently under investigation for KIR3DL2-positive PTCL (NCT04984837).
Given the limited efficacy of conventional chemotherapies, such as CHOP, for most PTCL patients, in the future, it may be worthwhile considering alternative treatment options that are personalized and directed according to the molecular characterization of the tumor (Table 3). However, whether the detection of actionable alterations will be clinically important for most PTCLs, which are still an unmet medical need for most, remains unknown.
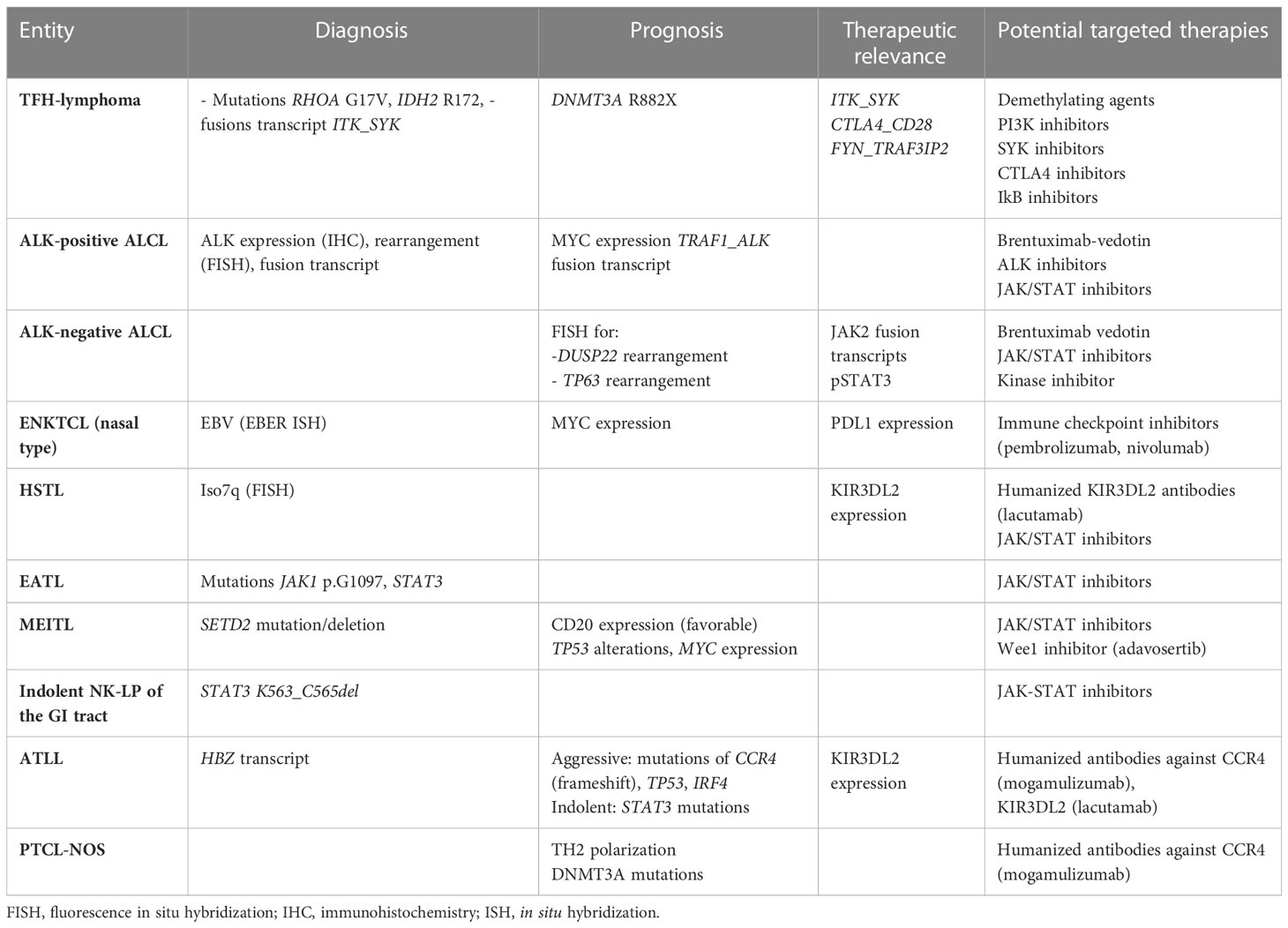
Table 3 Relevant cytogenetic or molecular findings for the management of PTCL patients.
5 Conclusion
The emergence of innovative high-throughput technologies has led to a better understanding of the pathogenesis of most PTCL entities, highlighting their diversity in terms of their biology and clinical features. A large group of TFH-lymphoma patients has emerged with a unique lymphoma oncogenesis, for which the diagnosis takes advantage of robust molecular markers and for which the treatment may benefit from the emergence of novel therapies, such as those that target epigenetics. The ALCL category is still heterogenous due to its genetic diversity, which has prognostic relevance, but may now benefit from the introduction of BV targeting CD30. The recent description of the genetic landscape of PTCL offers the rationale for an association of targeted therapies, with or without conventional chemotherapy agents, in the future, although the efficient combination for each PTCL entity or molecular subgroups still needs to be identified.
Author contributions
FD and PG wrote and supervised the manuscript. FL supervised the “oncogenic mechanisms” part, wrote and supervised the therapeutic part. All authors contributed to the article and approved the submitted version.
Funding
The authors declare that this study received funding from Force Hemato. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of the article, or the decision to submit it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IB de O, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia (2022) 36(7):1720−48. doi: 10.1038/s41375-022-01620-2
2. Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The international consensus classification of mature lymphoid neoplasms: a report from the clinical advisory committee. Blood (2022) 140(11):1229−53. doi: 10.1182/blood.2022015851
3. de Leval L, Alizadeh AA, Bergsagel PL, Campo E, Davies A, Dogan A, et al. Genomic profiling for clinical decision making in lymphoid neoplasms. Blood (2022) 140(21):2193−227. doi: 10.1182/blood.2022015854
4. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature (2016) 534(7607):402−6. doi: 10.1038/nature18294
5. Kataoka K, Miyoshi H, Sakata S, Dobashi A, Couronné L, Kogure Y, et al. Frequent structural variations involving programmed death ligands in Epstein-Barr virus-associated lymphomas. Leukemia (2019) 33(7):1687−99. doi: 10.1038/s41375-019-0380-5
6. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371(26):2477−87. doi: 10.1056/NEJMoa1409405
7. Couronné L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med (2012) 366(1):95−6. doi: 10.1056/NEJMc1111708
8. Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood (2012) 120(7):1466−9. doi: 10.1182/blood-2012-02-408542
9. Nicolae A, Bouilly J, Lara D, Fataccioli V, Lemonnier F, Drieux F, et al. Nodal peripheral T-cell lymphoma, NOS (PTCL, NOS) with a cytotoxic phenotype frequently occurs in clinical settings suggesting immune dysregulation and discloses recurrent epigenetic alterations. Mod Pathol (2022) 35(8):1126−36. doi: 10.1038/s41379-022-01022-w
10. Pastoret C, Desmots F, Drillet G, Le Gallou S, Boulland ML, Thannberger A, et al. Linking the KIR phenotype with STAT3 and TET2 mutations to identify chronic lymphoproliferative disorders of NK cells. Blood (2021) 137(23):3237−50. doi: 10.1182/blood.2020006721
11. Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell (2011) 20(1):25−38. doi: 10.1016/j.ccr.2011.06.003
12. Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer (2015) 15(3):152−65. doi: 10.1038/nrc3895
13. Zang S, Li J, Yang H, Zeng H, Han W, Zhang J, et al. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J Clin Invest (2017) 127(8):2998−3012. doi: 10.1172/JCI92026
14. Cortes JR, Ambesi-Impiombato A, Couronné L, Quinn SA, Kim CS, da Silva Almeida AC, et al. RHOA G17V induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell (2018) 33(2):259–73.e7. doi: 10.1016/j.ccell.2018.01.001
15. Ng SY, Brown L, Stevenson K, deSouza T, Aster JC, Louissaint A, et al. RhoA G17V is sufficient to induce autoimmunity and promotes T-cell lymphomagenesis in mice. Blood (2018) 132(9):935−47. doi: 10.1182/blood-2017-11-818617
16. Lemonnier F, Cairns RA, Inoue S, Li WY, Dupuy A, Broutin S, et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci (2016) 113(52):15084−9. doi: 10.1073/pnas.1617929114
17. Lemonnier F, Poullot E, Dupuy A, Couronné L, Martin N, Scourzic L, et al. Loss of 5-hydroxymethylcytosine is a frequent event in peripheral T-cell lymphomas. Haematologica (2018) 103(3):e115−8. doi: 10.3324/haematol.2017.167973
18. Roberti A, Dobay MP, Bisig B, Vallois D, Boéchat C, Lanitis E, et al. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat Commun (2016) 7(1):12602. doi: 10.1038/ncomms12602
19. McKinney M, Moffitt AB, Gaulard P, Travert M, De Leval L, Nicolae A, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov (2017) 7(4):369–79. doi: 10.1158/2159-8290.CD-16-0330
20. Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet (2015) 47(9):1061−6. doi: 10.1038/ng.3358
21. Ji MM, Huang YH, Huang JY, Wang ZF, Fu D, Liu H, et al. Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. Haematologica (2018) 103(4):679−87. doi: 10.3324/haematol.2017.182444
22. Laurent C, Nicolae A, Laurent C, Le Bras F, Haioun C, Fataccioli V, et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant-associated ALCL. Blood (2020) 135(5):360−70. doi: 10.1182/blood.2019001904
23. Vallois D, Dobay MPD, Morin RD, Lemonnier F, Missiaglia E, Juilland M, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper t-cell–derived lymphomas. Blood (2016) 128(11):1490−502. doi: 10.1182/blood-2016-02-698977
24. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga Ji, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet (2015) 47(11):1304−15. doi: 10.1038/ng.3415
25. Werner MT, Zhao C, Zhang Q, Wasik MA. Nucleophosmin-anaplastic lymphoma kinase: the ultimate oncogene and therapeutic target. Blood (2017) 129(7):823−31. doi: 10.1182/blood-2016-05-717793
26. Crescenzo R, Abate F, Lasorsa E, Tabbo’ F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic Large cell lymphoma. Cancer Cell (2015) 27(4):516−32. doi: 10.1016/j.ccell.2015.03.006
27. Heavican TB, Bouska A, Yu J, Lone W, Amador C, Gong Q, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood (2019) 133(15):1664–76. doi: 10.1182/blood-2018-09-872549
28. Watatani Y, Sato Y, Miyoshi H, Sakamoto K, Nishida K, Gion Y, et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia (2019) 33(12):2867−83. doi: 10.1038/s41375-019-0473-1
29. Maura F, Dodero A, Carniti C, Bolli N, Magni M, Monti V, et al. CDKN2A deletion is a frequent event associated with poor outcome in patients with peripheral T-cell lymphoma not otherwise specified (PTCL-NOS). Haematologica (2020) 106(11):2918−26. doi: 10.3324/haematol.2020.262659
30. Xiong J, Cui BW, Wang N, Dai YT, Zhang H, Wang CF, et al. Genomic and transcriptomic characterization of natural killer T cell lymphoma. Cancer Cell (2020) 37(3):403–419.e6. doi: 10.1016/j.ccell.2020.02.005
31. Moffitt AB, Ondrejka SL, McKinney M, Rempel RE, Goodlad JR, Teh CH, et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med (2017) 214(5):1371−86. doi: 10.1084/jem.20160894
32. Vasmatzis G, Johnson SH, Knudson RA, Ketterling RP, Braggio E, Fonseca R, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood (2012) 120(11):2280−9. doi: 10.1182/blood-2012-03-419937
33. Wang X, Boddicker RL, Dasari S, Sidhu JS, Kadin ME, Macon WR, et al. Expression of p63 protein in anaplastic large cell lymphoma: implications for genetic subtyping. Hum Pathol (2017) 64:19−27. doi: 10.1016/j.humpath.2017.01.003
34. Atsaves V, Tsesmetzis N, Chioureas D, Kis L, Leventaki V, Drakos E, et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia (2017) 31(7):1633−7. doi: 10.1038/leu.2017.103
35. Kong J, Dasari S, Feldman AL. PD-L1 expression in anaplastic large cell lymphoma. Mod Pathol (2020) 33(6):1232−3. doi: 10.1038/s41379-019-0448-9
36. Shen J, Li S, Medeiros LJ, Lin P, Wang SA, Tang G, et al. PD-L1 expression is associated with ALK positivity and STAT3 activation, but not outcome in patients with systemic anaplastic large cell lymphoma. Mod Pathol (2020) 33(3):324−33. doi: 10.1038/s41379-019-0336-3
37. Luchtel RA, Dasari S, Oishi N, Pedersen MB, Hu G, Rech KL, et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood (2018) 132(13):1386−98. doi: 10.1182/blood-2018-03-838524
38. de Leval L, Rickman DS, Thielen C, de Reynies A, Huang YL, Delsol G, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood (2007) 109(11):4952−63. doi: 10.1182/blood-2006-10-055145
39. Iqbal J, Weisenburger DD, Greiner TC, Vose JM, McKeithan T, Kucuk C, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood (2010) 115(5):1026−36. doi: 10.1182/blood-2009-06-227579
40. Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood (2014) 123(19):2915−23. doi: 10.1182/blood-2013-11-536359
41. Cortes JR, Filip I, Albero R, Patiño-Galindo JA, Quinn SA, Lin WHW, et al. Oncogenic Vav1-Myo1f induces therapeutically targetable macrophage-rich tumor microenvironment in peripheral T cell lymphoma. Cell Rep (2022) 39(3):110695. doi: 10.1016/j.celrep.2022.110695
42. Lamant L, McCarthy K, d’Amore E, Klapper W, Nakagawa A, Fraga M, et al. Prognostic impact of morphologic and phenotypic features of childhood ALK-positive anaplastic Large-cell lymphoma: results of the ALCL99 study. J Clin Oncol (2011) 29(35):4669−76. doi: 10.1200/JCO.2011.36.5411
43. Cook LB, Melamed A, Niederer H, Valganon M, Laydon D, Foroni L, et al. The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma. Blood (2014) 123(25):3925−31. doi: 10.1182/blood-2014-02-553602
44. Bangham CRM, Cook LB, Melamed A. HTLV-1 clonality in adult T-cell leukaemia and non-malignant HTLV-1 infection. Semin Cancer Biol (2014) 26:89−98. doi: 10.1016/j.semcancer.2013.11.003
45. Melamed A, Laydon DJ, Gillet NA, Tanaka Y, Taylor GP, Bangham CRM. Genome-wide determinants of proviral targeting, clonal abundance and expression in natural HTLV-1 infection. emerman m, éditeur. PloS Pathog (2013) 9(3):e1003271. doi: 10.1371/journal.ppat.1003271
46. Bangham CR, Ratner L. How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr Opin Virol (2015) 14:93−100. doi: 10.1016/j.coviro.2015.09.004
47. Bangham CRM. Human T cell leukemia virus type 1: persistence and pathogenesis. Annu Rev Immunol (2018) 36(1):43−71. doi: 10.1146/annurev-immunol-042617-053222
48. Wattel E, Vartanian JP, Pannetier C, Wain-Hobson S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol (1995) 69(5):2863−8. doi: 10.1128/jvi.69.5.2863-2868.1995
49. Giam CZ, Semmes O. HTLV-1 infection and adult T-cell Leukemia/Lymphoma–a tale of two proteins: tax and HBZ. Viruses (2016) 8(6):161. doi: 10.3390/v8060161
50. Takeda S, Maeda M, Morikawa S, Taniguchi Y, Yasunaga Ji, Nosaka K, et al. Genetic and epigenetic inactivation oftax gene in adult T-cell leukemia cells. Int J Cancer (2004) 109(4):559−67. doi: 10.1002/ijc.20007
51. Matsuoka M, Green PL. The HBZ gene, a key player in HTLV-1 pathogenesis. Retrovirology (2009) 6(1):71. doi: 10.1186/1742-4690-6-71
52. Tabiasco J, Vercellone A, Meggetto F, Hudrisier D, Brousset P, Fournié JJ. Acquisition of viral receptor by NK cells through immunological synapse. J Immunol (2003) 170(12):5993−8. doi: 10.4049/jimmunol.170.12.5993
53. Lee JH, Choi J, Ahn YO, Kim TM, Heo DS. CD21-independent Epstein-Barr virus entry into NK cells. Cell Immunol (2018) 327:21−5. doi: 10.1016/j.cellimm.2018.01.011
54. Ng SB, Selvarajan V, Huang G, Zhou J, Feldman AL, Law M, et al. Activated oncogenic pathways and therapeutic targets in extranodal nasal-type NK/T cell lymphoma revealed by gene expression profiling: gene expression profiling in NK/T lymphoma. J Pathol (2011) 223(4):496−510. doi: 10.1002/path.2823
55. Lebwohl B, Ludvigsson JF, Green PHR. Celiac disease and non-celiac gluten sensitivity. BMJ (2015) 351:h4347. doi: 10.1136/bmj.h4347
56. Deva AK, Turner SD, Kadin ME, Magnusson MR, Prince HM, Miranda RN, et al. Etiology of breast implant-associated anaplastic Large cell lymphoma (BIA-ALCL): current directions in research. Cancers (2020) 12(12):3861. doi: 10.3390/cancers12123861
57. Goldman S, Bron D, Tousseyn T, Vierasu I, Dewispelaere L, Heimann P, et al. Rapid progression of angioimmunoblastic T cell lymphoma following BNT162b2 mRNA vaccine booster shot: a case report. Front Med (2021) 8:798095. doi: 10.3389/fmed.2021.798095
58. Li Z, Xia Y, Feng LN, Chen JR, Li HM, Cui J, et al. Genetic risk of extranodal natural killer T-cell lymphoma: a genome-wide association study. Lancet Oncol (2016) 17(9):1240−7. doi: 10.1016/S1470-2045(16)30148-6
59. Lin GW, Xu C, Chen K, Huang HQ, Chen J, Song B, et al. Genetic risk of extranodal natural killer T-cell lymphoma: a genome-wide association study in multiple populations. Lancet Oncol (2020) 21(2):306−16. doi: 10.1016/S1470-2045(19)30799-5
60. Lundin EKEA, Scott H, Hansen T, Paulsen G, Halstensen TS, Fausa O, et al. Gliadin-specitlc, HLA-DQ(alpha 1*0501,beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med (1993) 178:187−96. doi: 10.1084/jem.178.1.187
61. Gayden T, Sepulveda FE, Khuong-Quang DA, Pratt J, Valera ET, Garrigue A, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet (2018) 50(12):1650−7. doi: 10.1038/s41588-018-0251-4
62. Amatore F, Ortonne N, Lopez M, Orlanducci F, Castellano R, Ingen-Housz-Oro S, et al. ICOS is widely expressed in cutaneous T-cell lymphoma, and its targeting promotes potent killing of malignant cells. Blood Adv (2020) 4(20):5203−14. doi: 10.1182/bloodadvances.2020002395
63. Luherne C, Menguy S, Ferte T, Beylot-Barry M, Seneschal J, Milpied B, et al. A high programmed cell death protein 1 hormone receptor score on skin biopsy is associated with sézary syndrome diagnosis: a study of 91 patients with erythroderma. Acta Derm Venereol (2022) 102:adv00773. doi: 10.2340/actadv.v102.1062
64. Picchio MC, Scala E, Pomponi D, Caprini E, Frontani M, Angelucci I, et al. CXCL13 is highly produced by sézary cells and enhances their migratory ability via a synergistic mechanism involving CCL19 and CCL21 chemokines. Cancer Res (2008) 68(17):7137−46. doi: 10.1158/0008-5472.CAN-08-0602
65. Masle-Farquhar E, Jeelall Y, White J, Bier J, Deenick EK, Brink R, et al. CARD11 gain-of-function mutation drives cell-autonomous accumulation of PD-1+ ICOShigh activated T cells, T-follicular, T-regulatory and T-follicular regulatory cells. Front Immunol (2023) 14:1095257. doi: 10.3389/fimmu.2023.1095257
66. Cairns RA, Iqbal J, Lemonnier F, Kucuk C, De Leval L, Jais JP, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood (2012) 119(8):1901−3. doi: 10.1182/blood-2011-11-391748
67. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(2):171−5. doi: 10.1038/ng.2872
68. Palomero T, Couronné L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet (2014) 46(2):166−70. doi: 10.1038/ng.2873
69. Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood (2014) 123(9):1293−6. doi: 10.1182/blood-2013-10-531509
70. Abate F, da Silva-Almeida AC, Zairis S, Robles-Valero J, Couronne L, Khiabanian H, et al. Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T-cell lymphomas. Proc Natl Acad Sci (2017) 114(4):764−9. doi: 10.1073/pnas.1608839114
71. Fujisawa M, Sakata-Yanagimoto M, Nishizawa S, Komori D, Gershon P, Kiryu M, et al. Activation of RHOA–VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia (2018) 32(3):694−702. doi: 10.1038/leu.2017.273
72. Lee SH, Kim JS, Kim J, Kim SJ, Kim WS, Lee S, et al. A highly recurrent novel missense mutation in CD28 among angioimmunoblastic T-cell lymphoma patients. Haematologica (2015) 100(12):e505. doi: 10.3324/haematol.2015.133074
73. Rohr J, Guo S, Huo J, Bouska A, Lachel C, Li Y, et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia (2016) 30(5):1062−70. doi: 10.1038/leu.2015.357
74. Nguyen TB, Sakata-Yanagimoto M, Asabe Y, Matsubara D, Kano J, Yoshida K, et al. Identification of cell-type-specific mutations in nodal T-cell lymphomas. Blood Cancer J (2017) 7(1):e516−e516. doi: 10.1038/bcj.2016.122
75. Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood (2015) 126(15):1741−52. doi: 10.1182/blood-2015-05-644591
76. Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(4):371−5. doi: 10.1038/ng.2916
77. Nakamoto-Matsubara R, Sakata-Yanagimoto M, Enami T, Yoshida K, Yanagimoto S, Shiozawa Y, et al. Detection of the G17V RHOA mutation in angioimmunoblastic T-cell lymphoma and related lymphomas using quantitative allele-specific PCR. PloS One (2014) 9(10):e109714. doi: 10.1371/journal.pone.0109714
78. Dobay MP, Lemonnier F, Missiaglia E, Bastard C, Vallois D, Jais JP, et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica (2017) 102(4):e148−51. doi: 10.3324/haematol.2016.158428
79. Leca J, Lemonnier F, Meydan C, Foox J, El Ghamrasni S, Mboumba DL, et al. IDH2 and TET2 mutations synergize to modulate T follicular helper cell functional interaction with the AITL microenvironment. Cancer Cell (2023) 41(2):323–39.e10. doi: 10.1016/j.ccell.2023.01.003
80. Guo. Novel fusion transcripts identified in angioimmunoblastic T cell lymphoma. Mod Pathol (2013) 26):330A.
81. Vallois D, Dupuy A, Lemonnier F, Allen G, Missiaglia E, Fataccioli V, et al. RNA Fusions involving CD28 are rare in peripheral T-cell lymphomas and concentrate mainly in those derived from follicular helper T cells. Haematologica (2018) 103(8):e360−3. doi: 10.3324/haematol.2017.186767
82. Boddicker RL, Razidlo GL, Dasari S, Zeng Y, Hu G, Knudson RA, et al. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood (2016) 128(9):1234−45. doi: 10.1182/blood-2016-03-707141
83. Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel t (5; 9)(q33; q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia (2006) 20(2):313−8. doi: 10.1038/sj.leu.2404045
84. Attygalle AD, Feldman AL, Dogan A. ITK/SYK translocation in angioimmunoblastic T-cell lymphoma. Am J Surg Pathol (2013) 37(9):1456−7. doi: 10.1097/PAS.0b013e3182991415
85. Boi M, Zucca E, Inghirami G, Bertoni F. Advances in understanding the pathogenesis of systemic anaplastic large cell lymphomas. Br J Haematol (2015) 168(6):771−83. doi: 10.1111/bjh.13265
86. Andraos E, Dignac J, Meggetto F. NPM-ALK: a driver of lymphoma pathogenesis and a therapeutic target. Cancers (2021) 13(1):144. doi: 10.3390/cancers13010144
87. Feldman AL, Vasmatzis G, Asmann YW, Davila J, Middha S, Eckloff BW, et al. Novel TRAF1-ALK fusion identified by deep RNA sequencing of anaplastic large cell lymphoma: TRAF1-ALK fusion in ALCL. Genes Chromosomes Cancer (2013) 52(11):1097−102. doi: 10.1002/gcc.22104
88. The European T-cell Lymphoma Study Group, Abate F, Todaro M, van der Krogt JA, Boi M, Landra I, et al. A novel patient-derived tumorgraft model with TRAF1-ALK anaplastic large-cell lymphoma translocation. Leukemia (2015) 29(6):1390−401. doi: 10.1038/leu.2014.347
89. Larose H, Prokoph N, Matthews JD, Schlederer M, Högler S, Alsulami AF, et al. Whole exome sequencing reveals NOTCH1 mutations in anaplastic large cell lymphoma and points to notch both as a key pathway and a potential therapeutic target. Haematologica (2021) 106(6):1693−704. doi: 10.3324/haematol.2019.238766
90. Morris S, Kirstein M, Valentine M, Dittmer K, Shapiro D, Saltman D, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-hodgkin’s lymphoma. Science (1994) 263(5151):1281−4. doi: 10.1126/science.8122112
91. Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B. A new fusion gene TPM3-ALK in anaplastic Large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood (1999) 93(9):3088−95.
92. Trinei M, Lanfrancone L, Campo E, Pulford K, Mason DY, Pelicci PG, et al. A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic Large cell lymphoma. Cancer Res (2000) 60(4):793−8.
93. Hernández L, Pinyol M, Hernández S, Beà S, Pulford K, Rosenwald A, et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic Large cell lymphoma producing two structurally DifferentTFG-ALK translocations. Blood (1999) 94(9):3265−8. doi: 10.1182/blood.V94.9.3265
94. Touriol C, Greenland C, Lamant L, Pulford K, Mason DY, Delsol G. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood (2000) 95(10):3204−7. doi: 10.1182/blood.V95.10.3204
95. Tort F, Pinyol M, Pulford K, Roncador G, Hernandez L, Nayach I, et al. Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic Large cell lymphoma. Lab Invest (2001) 81(3):419−26. doi: 10.1038/labinvest.3780249
96. Cools J, Wlodarska I, Somers R, Mentens N, Pedeutour F, Maes B, et al. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor: identification of novel ALK fusion partners. Genes Chromosomes Cancer (2002) 34(4):354−62. doi: 10.1002/gcc.10033
97. Lamant L, Gascoyne RD, Duplantier MM, Armstrong F, Raghab A, Chhanabhai M, et al. Non-muscle myosin heavy chain (MYH9): a new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer (2003) 37(4):427−32. doi: 10.1002/gcc.10232
98. Palacios G, Shaw TI, Li Y, Singh RK, Valentine M, Sandlund JT, et al. Novel ALK fusion in anaplastic large cell lymphoma involving EEF1G, a subunit of the eukaryotic elongation factor-1 complex. Leukemia (2017) 31(3):743−7. doi: 10.1038/leu.2016.331
99. Graetz D, Crews KR, Azzato EM, Singh RK, Raimondi S, Mason J, et al. Leukemic presentation of ALK-positive anaplastic large cell lymphoma with a novel partner, poly(A) binding protein cytoplasmic 1 (PABPC1), responding to single-agent crizotinib. Haematologica (2019) 104(5):e218−21. doi: 10.3324/haematol.2018.215103
100. Sibon D, Nguyen DP, Schmitz N, Suzuki R, Feldman AL, Gressin R, et al. ALK-positive anaplastic large-cell lymphoma in adults: an individual patient data pooled analysis of 263 patients. Haematologica (2019) 104(12):e562−5. doi: 10.3324/haematol.2018.213512
101. Cederleuf H, Bjerregård Pedersen M, Jerkeman M, Relander T, d’Amore F, Ellin F. The addition of etoposide to CHOP is associated with improved outcome in ALK+ adult anaplastic large cell lymphoma: a Nordic lymphoma group study. Br J Haematol (2017) 178(5):739−46. doi: 10.1111/bjh.14740
102. Brink M, Meeuwes FO, van der Poel M, Kersten M, Wondergem M, Mutsaers PGNJ, et al. Impact of etoposide and ASCT on survival among patients aged,65 years with stage II to IV PTCL: a population-based cohort study. Blood (2022) 140(9):1009−19. doi: 10.1182/blood.2021015114
103. Horwitz S, O’Connor OA, Pro B, Illidge T, Fanale M, Advani R, et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet (2019) 393(10168):229−40. doi: 10.1016/S0140-6736(18)32984-2
104. Moritake H, Shimonodan H, Marutsuka K, Kamimura S, Kojima H, Nunoi H. C-MYC rearrangement may induce an aggressive phenotype in anaplastic lymphoma kinase positive anaplastic large cell lymphoma: identification of a novel fusion gene ALO17/C-MYC. Am J Hematol (2011) 86(1):75−8. doi: 10.1002/ajh.21887
105. Raetz EA, Perkins SL, Carlson MA, Schooler KP, Carroll WL, Virshup DM. The nucleophosmin-anaplastic lymphoma kinase fusion protein induces c-myc expression in pediatric anaplastic Large cell lymphomas. Am J Pathol (2002) 161(3):875−83. doi: 10.1016/S0002-9440(10)64248-4
106. Feldman AL, Dogan A, Smith DI, Law ME, Ansell SM, Johnson SH, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood (2011) 117(3):915−9. doi: 10.1182/blood-2010-08-303305
107. King RL, Dao LN, McPhail ED, Jaffe ES, Said J, Swerdlow SH, et al. Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol (2016) 40(1):36−43. doi: 10.1097/PAS.0000000000000500
108. Mélard P, Idrissi Y, Andrique L, Poglio S, Prochazkova-Carlotti M, Berhouet S, et al. Molecular alterations and tumor suppressive function of the DUSP22 (Dual specificity phosphatase 22) gene in peripheral T-cell lymphoma subtypes. Oncotarget (2016) 7(42):68734−48. doi: 10.18632/oncotarget.11930
109. Onaindia A, de Villambrosía SG, Prieto-Torres L, Rodríguez-Pinilla SM, Montes-Moreno S, González-Vela C, et al. DUSP22 -rearranged anaplastic lymphomas are characterized by specific morphological features and a lack of cytotoxic and JAK/STAT surrogate markers. Haematologica (2019) 104(4):e158−62. doi: 10.3324/haematol.2018.205880
110. Ravindran A, Feldman AL, Ketterling RP, Dasari S, Rech KL, McPhail ED, et al. Striking association of lymphoid enhancing factor (LEF1) overexpression and DUSP22 rearrangements in anaplastic Large cell lymphoma. Am J Surg Pathol (2020) 45(4):550–7. doi: 10.1097/PAS.0000000000001614
111. Luchtel RA, Zimmermann MT, Hu G, Dasari S, Jiang M, Oishi N, et al. Recurrent MSCE116K mutations in ALK-negative anaplastic large cell lymphoma. Blood (2019) 133(26):2776−89. doi: 10.1182/blood.2019000626
112. Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood (2014) 124(9):1473−80. doi: 10.1182/blood-2014-04-571091
113. Pedersen MBa, Hamilton-Dutoit SJ, Bendix K, Ketterling RP, Bedroske PP, Luoma IM, et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: a Danish cohort study. Blood (2017) 130(4):554−7. doi: 10.1182/blood-2016-12-755496
114. Hapgood G, Ben-Neriah S, Mottok A, Lee DG, Villa D, Sehn LH, et al. Identification of high-risk DUSP22-rearranged ALK-negative anaplastic large cell lymphoma. Br J Haematol (2019) 186(3):e28−31. doi: 10.1111/bjh.15860
115. Sibon D, Bisig B, Bonnet C, Bachy E, Cavalieri D, Fataccioli V, et al. Impact du DUSP22 rearrangement on the prognosis of systemic ALK-negative anaplastic large cell lymphomas: a LYSA and TENOMIC study. Hematol Oncol (2021) 39(S2):hon.137_2880. doi: 10.1002/hon.137_2880
116. Scarfò I, Pellegrino E, Mereu E, Kwee I, Agnelli L, Bergaggio E, et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood (2016) 127(2):221−32. doi: 10.1182/blood-2014-12-614503
117. Wang Jc, Zhong Lh, Lin Wq, Zhang Wf, Xi Yf, Liu Yp, et al. JAK/STAT3 signaling activation related to distinct clinicopathologic features in systemic ALK– anaplastic Large cell lymphomas. Am J Surg Pathol (2023) 47(1):55−64. doi: 10.1097/PAS.0000000000001995
118. Hu G, Dasari S, Asmann YW, Greipp PT, Knudson RA, Benson HK, et al. Targetable fusions of the FRK tyrosine kinase in ALK-negative anaplastic large cell lymphoma. Leukemia (2018) 32(2):565−9. doi: 10.1038/leu.2017.309
119. Fitzpatrick MJ, Massoth LR, Marcus C, Vergilio JA, Severson E, Duncan D, et al. JAK2 rearrangements are a recurrent alteration in CD30+ systemic T-cell lymphomas with anaplastic morphology. Am J Surg Pathol (2021) 45(7):895−904. doi: 10.1097/PAS.0000000000001708
120. Kadin ME, Morgan J, Xu H, Epstein AL, Sieber D, Hubbard BA, et al. IL-13 is produced by tumor cells in breast implant–associated anaplastic large cell lymphoma: implications for pathogenesis. Hum Pathol (2018) 78:54−62. doi: 10.1016/j.humpath.2018.04.007
121. Blombery P, Thompson ER, Jones K, Arnau GM, Lade S, Markham JF, et al. Whole exome sequencing reveals activating JAK1 and STAT3 mutations in breast implant-associated anaplastic large cell lymphoma anaplastic large cell lymphoma. Haematologica (2016) 101(9):e387−90. doi: 10.3324/haematol.2016.146118
122. Los-de Vries GT, de Boer M, van Dijk E, Stathi P, Hijmering NJ, Roemer MGM, et al. Chromosome 20 loss is characteristic of breast implant–associated anaplastic large cell lymphoma. Blood (2020) 136(25):2927−32. doi: 10.1182/blood.2020005372
123. Tabanelli V, Corsini C, Fiori S, Agostinelli C, Calleri A, Orecchioni S, et al. Recurrent PDL1 expression and PDL1 (CD274) copy number alterations in breast implant–associated anaplastic large cell lymphomas. Hum Pathol (2019) 90:60−9. doi: 10.1016/j.humpath.2019.05.007
124. Quesada AE, Zhang Y, Ptashkin R, Ho C, Horwitz S, Benayed R, et al. Next generation sequencing of breast implant-associated anaplastic large cell lymphomas reveals a novel STAT3-JAK2 fusion among other activating genetic alterations within the JAK-STAT pathway. Breast J (2021) 27(4):314−21. doi: 10.1111/tbj.14205
125. Feldman AL, Oishi N, Ketterling RP, Ansell SM, Shi M, Dasari S. Immunohistochemical approach to genetic subtyping of anaplastic Large cell lymphoma. Am J Surg Pathol (2022) 46(11):1490−9. doi: 10.1097/PAS.0000000000001941
126. Hong M, Lee T, Young Kang S, Kim SJ, Kim W, Ko YH. Nasal-type NK/T-cell lymphomas are more frequently T rather than NK lineage based on T-cell receptor gene, RNA, and protein studies: lineage does not predict clinical behavior. Mod Pathol (2016) 29(5):430−43. doi: 10.1038/modpathol.2016.47
127. Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, et al. Janus kinase 3–activating mutations identified in natural Killer/T-cell lymphoma. Cancer Discovery (2012) 2(7):591−7. doi: 10.1158/2159-8290.CD-12-0028
128. Quintanilla-Martinez L, Kremer M, Keller G, Nathrath M, Gamboa-Dominguez A, Meneses A, et al. p53 mutations in nasal natural Killer/T-cell lymphoma from Mexico. Am J Pathol (2001) 159(6):2095−105. doi: 10.1016/S0002-9440(10)63061-1
129. Huang Y, de Reyniès A, de Leval L, Ghazi B, Martin-Garcia N, Travert M, et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood (2010) 115(6):1226−37. doi: 10.1182/blood-2009-05-221275
130. Iqbal J, Weisenburger DD, Chowdhury A, Tsai MY, Srivastava G, Greiner TC, et al. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic γδ T-cell lymphoma and is highly sensitive to a novel aurora kinase a inhibitor in vitro. Leukemia (2011) 25(2):348−58. doi: 10.1038/leu.2010.255
131. Karube K, Nakagawa M, Tsuzuki S, Takeuchi I, Honma K, Nakashima Y, et al. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood (2011) 118(12):3195−204. doi: 10.1182/blood-2011-04-346890
132. Kwong YL, Chan TSY, Tan D, Kim SJ, Poon LM, Mow B, et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood (2017) 129(17):2437−42. doi: 10.1182/blood-2016-12-756841
133. Wai CMM, Chen S, Phyu T, Fan S, Leong SM, Zheng W, et al. Immune pathway upregulation and lower genomic instability distinguish EBV-positive nodal T/NK-cell lymphoma from ENKTL and PTCL-NOS. Haematologica (2022) 107(8):1864−79. doi: 10.3324/haematol.2021.280003
134. Dufva O, Kankainen M, Kelkka T, Sekiguchi N, Awad SA, Eldfors S, et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat Commun (2018) 9(1):1567. doi: 10.1038/s41467-018-03987-2
135. Huang L, Liu D, Wang N, Ling S, Tang Y, Wu J, et al. Integrated genomic analysis identifies deregulated JAK/STAT-MYC-biosynthesis axis in aggressive NK-cell leukemia. Cell Res (2018) 28(2):172−86. doi: 10.1038/cr.2017.146
136. El Hussein S, Patel KP, Fang H, Thakral B, Loghavi S, Kanagal-Shamanna R, et al. Genomic and immunophenotypic landscape of aggressive NK-cell leukemia. Am J Surg Pathol (2020) 44(9):1235−43. doi: 10.1097/PAS.0000000000001518
137. Okuno Y, Murata T, Sato Y, Muramatsu H, Ito Y, Watanabe T, et al. Defective Epstein–Barr virus in chronic active infection and haematological malignancy. Nat Microbiol (2019) 4(3):404−13. doi: 10.1038/s41564-018-0334-0
138. Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol (2012) 3:388/abstract(388). doi: 10.3389/fmicb.2012.00388/abstract
139. Bonzheim I, Geissinger E, Tinguely M, Roth S, Grieb T, Reimer P, et al. Evaluation of FoxP3 expression in peripheral T-cell lymphoma. Am J Clin Pathol (2008) 130(4):613−9. doi: 10.1309/L65GWEQ803PP6VX1
140. Karube K, Aoki R, Sugita Y, Yoshida S, Nomura Y, Shimizu K, et al. The relationship of FOXP3 expression and clinicopathological characteristics in adult T-cell leukemia/lymphoma. Mod Pathol (2008) 21(5):617−25. doi: 10.1038/modpathol.2008.25
141. Ferreira CR, Zhao S, Sahoo MK, Pinsky B, Weber J, Lage LAPC, et al. FOXP3-positive T-cell lymphomas in non-HTLV1 carriers include ALK-negative anaplastic large cell lymphoma: expanding the spectrum of T-cell lymphomas with regulatory phenotype. Hum Pathol (2018) 80:138−44. doi: 10.1016/j.humpath.2018.06.001
142. Satou A, Asano N, Kato S, Katsuya H, Ishitsuka K, Elsayed AA, et al. FoxP3-positive T cell lymphoma arising in non-HTLV1 carrier: clinicopathological analysis of 11 cases of PTCL-NOS and 2 cases of mycosis fungoides. Histopathology (2016) 68(7):1099−108. doi: 10.1111/his.12885
143. Marçais A, Lhermitte L, Artesi M, Laurent C, Durkin K, Hahaut V, et al. Targeted deep sequencing reveals clonal and subclonal mutational signatures in adult T-cell leukemia/lymphoma and defines an unfavorable indolent subtype. Leukemia (2021) 35(3):764−76. doi: 10.1038/s41375-020-0900-3
144. Yoshida N, Shigemori K, Donaldson N, Trevisani C, Cordero NA, Stevenson KE, et al. Genomic landscape of young ATLL patients identifies frequent targetable CD28 fusions. Blood (2020) 135(17):1467−71. doi: 10.1182/blood.2019001815
145. Kataoka K, Iwanaga M, Yasunaga Ji, Nagata Y, Kitanaka A, Kameda T, et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood (2018) 131(2):215−25. doi: 10.1182/blood-2017-01-761874
146. Yoshida N, Miyoshi H, Kato T, Sakata-Yanagimoto M, Niino D, Taniguchi H, et al. CCR4 frameshift mutation identifies a distinct group of adult T cell leukaemia/lymphoma with poor prognosis: CCR4 mutation in ATLL. J Pathol (2016) 238(5):621−6. doi: 10.1002/path.4699
147. Obermann E, Diss T, Hamoudi R, Munson P, Wilkins B, Camozzi M, et al. Loss of heterozygosity at chromosome 9p21 is a frequent finding in enteropathy-type T-cell lymphoma: loss of heterozygosity at 9p21 in enteropathy-type T-cell lymphoma. J Pathol (2004) 202(2):252−62. doi: 10.1002/path.1506
148. Cording S, Lhermitte L, Malamut G, Berrabah S, Trinquand A, Guegan N, et al. Oncogenetic landscape of lymphomagenesis in coeliac disease. Gut (2021) 0):1−12. doi: 10.1136/gutjnl-2020-322935
149. Nicolae A, Xi L, Pham TH, Pham TA, Navarro W, Meeker HG, et al. Mutations in the JAK/STAT and RAS signaling pathways are common in intestinal T-cell lymphomas. Leukemia (2016) 30(11):2245−7. doi: 10.1038/leu.2016.178
150. Hang JF, Yuan CT, Chang KC, Wang RC, Chen BJ, Hsieh PP, et al. Targeted next-generation sequencing reveals a wide morphologic and immunophenotypic spectrum of monomorphic epitheliotropic intestinal T-cell lymphoma. Am J Surg Pathol (2022) 46(9):1207−18. doi: 10.1097/PAS.0000000000001914
151. Veloza L, Cavalieri D, Missiaglia E, Ledoux-Pilon A, Bisig B, Pereira B, et al. Monomorphic epitheliotropic intestinal T-cell lymphoma comprises morphologic and genomic heterogeneity impacting outcome. Haematologica (2022) 108(1):181−95. doi: 10.3324/haematol.2022.281226
152. Nairismägi ML, Tan J, Lim JQ, Nagarajan S, Ng CCY, Rajasegaran V, et al. JAK-STAT and G-protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leukemia (2016) 30(6):1311−9. doi: 10.1038/leu.2016.13
153. Sharma A, Oishi N, Boddicker RL, Hu G, Benson HK, Ketterling RP, et al. Recurrent STAT3-JAK2 fusions in indolent T-cell lymphoproliferative disorder of the gastrointestinal tract. Blood (2018) 131(20):2262−6. doi: 10.1182/blood-2018-01-830968
154. Soderquist CR, Patel N, Murty VV, Betman S, Aggarwal N, Young KH, et al. Genetic and phenotypic characterization of indolent T-cell lymphoproliferative disorders of the gastrointestinal tract. Haematologica (2020) 105(7):1895−906. doi: 10.3324/haematol.2019.230961
155. Margolskee E, Jobanputra V, Lewis SK, Alobeid B, Green PHR, Bhagat G. Indolent small intestinal CD4+ T-cell lymphoma is a distinct entity with unique biologic and clinical features. tran DQ, éditeur. PloS One (2013) 8(7):e68343. doi: 10.1371/journal.pone.0068343
156. Perry AM, Bailey NG, Bonnett M, Jaffe ES, Chan WC. Disease progression in a patient with indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Int J Surg Pathol (2019) 27(1):102−7. doi: 10.1177/1066896918785985
157. Xiao W, Gupta GK, Yao J, Jang YJ, Xi L, Baik J, et al. Recurrent somatic JAK3 mutations in NK-cell enteropathy. Blood (2019) 134(12):986−91. doi: 10.1182/blood.2019001443
158. Jonveaux P, Daniel MT, Martel V, Maarek O, Berger R. Isochromosome 7q and trisomy 8 are consistent primary, non-random chromosomal abnormalities associated with hepatosplenic T gamma/delta lymphoma. Leukemia (1996) 10(9):1453−5.
159. Wlodarska I, Martin-Garcia N, Achten R, De Wolf-Peeters C, Pauwels P, Tulliez M, et al. Fluorescence in situ hybridization study of chromosome 7 aberrations in hepatosplenic T-cell lymphoma: isochromosome 7q as a common abnormality accumulating in forms with features of cytologic progression: isochromosome 7q in hepatosplenic lymphoma. Genes Chromosomes Cancer (2002) 33(3):243−51. doi: 10.1002/gcc.10021
160. Nicolae A, Xi L, Pittaluga S, Abdullaev Z, Pack SD, Chen J, et al. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leukemia (2014) 28(11):2244−8. doi: 10.1038/leu.2014.200
161. Küçük C, Jiang B, Hu X, Zhang W, Chan JKC, Xiao W, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat Commun (2015) 6(1):6025. doi: 10.1038/ncomms7025
162. Travert M, Huang Y, de Leval L, Martin-Garcia N, Delfau-Larue MH, Berger F, et al. Molecular features of hepatosplenic T-cell lymphoma unravels potential novel therapeutic targets. Blood (2012) 119(24):5795−806. doi: 10.1182/blood-2011-12-396150
163. Song W, Zhang H, Yang F, Nakahira K, Wang C, Shi K, et al. Single cell profiling of γδ hepatosplenic T-cell lymphoma unravels tumor cell heterogeneity associated with disease progression. Cell Oncol (2023) 46(1):211−26. doi: 10.1007/s13402-022-00745-x
164. Amador C, Greiner TC, Heavican TB, Smith LM, Galvis KT, Lone W, et al. Reproducing the molecular subclassification of peripheral T-cell lymphoma–NOS by immunohistochemistry. Blood (2019) 134(24):2159−70. doi: 10.1182/blood.2019000779
165. Wang T, Feldman AL, Wada DA, Lu Y, Polk A, Briski R, et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood (2014) 123(19):3007−15. doi: 10.1182/blood-2013-12-544809
166. Schatz JH, Horwitz SM, Teruya-Feldstein J, Lunning MA, Viale A, Huberman K, et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia (2015) 29(1):237−41. doi: 10.1038/leu.2014.261
167. Laginestra MA, Cascione L, Motta G, Fuligni F, Agostinelli C, Rossi M, et al. Whole exome sequencing reveals mutations in FAT1 tumor suppressor gene clinically impacting on peripheral T-cell lymphoma not otherwise specified. Mod Pathol (2020) 33(2):179−87. doi: 10.1038/s41379-019-0279-8
168. Almire C, Bertrand P, Ruminy P, Maingonnat C, Wlodarska I, Martín-Subero JI, et al. PVRL2 is translocated to theTRA@ locus in t(14;19)(q11;q13)-positive peripheral T-cell lymphomas. Genes Chromosomes Cancer (2007) 46(11):1011−8. doi: 10.1002/gcc.20490
169. Leich E, Haralambieva E, Zettl A, Chott A, Rüdiger T, Höller S, et al. Tissue microarray-based screening for chromosomal breakpoints affecting the T-cell receptor gene loci in mature T-cell lymphomas. J Pathol (2007) 213(1):99−105. doi: 10.1002/path.2196
170. Asano N, Suzuki R, Kagami Y, Ishida F, Kitamura K, Fukutani H, et al. Clinicopathologic and prognostic significance of cytotoxic molecule expression in nodal peripheral T-cell lymphoma, unspecified. Am J Surg Pathol (2005) 29(10):1284−93. doi: 10.1097/01.pas.0000173238.17331.6b
171. Feldman AL, Law M, Remstein ED, Macon WR, Erickson LA, Grogg KL, et al. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia (2009) 23(3):574−80. doi: 10.1038/leu.2008.320
172. Hsi ED, Said J, Macon WR, Rodig SJ, Ondrejka SL, Gascoyne RD, et al. Diagnostic accuracy of a defined immunophenotypic and molecular genetic approach for peripheral T/NK-cell lymphomas: a north American PTCL study group project. Am J Surg Pathol (2014) 38(6):768−75. doi: 10.1097/PAS.0000000000000188
173. Hsi ED, Horwitz SM, Carson KR, Pinter-Brown LC, Rosen ST, Pro B, et al. Analysis of peripheral T-cell lymphoma diagnostic workup in the united states. Clin Lymphoma Myeloma Leuk (2017) 17(4):193−200. doi: 10.1016/j.clml.2016.10.001
174. Laurent C, Baron M, Amara N, Haioun C, Dandoit M, Maynadié M, et al. Impact of expert pathologic review of lymphoma diagnosis: study of patients from the French lymphopath network. J Clin Oncol (2017) 35(18):2008−17. doi: 10.1200/JCO.2016.71.2083
175. Smith JL, Hodges E, Quin CT, McCarthy KP, Wright DH. Frequent T and b cell oligoclones in histologically and immunophenotypically characterized angioimmunoblastic lymphadenopathy. Am J Pathol (2000) 156(2):661−9. doi: 10.1016/S0002-9440(10)64770-0
176. Langerak AW, Groenen PJTA, Brüggemann M, Beldjord K, Bellan C, Bonello L, et al. EuroClonality/BIOMED-2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia (2012) 26(10):2159−71. doi: 10.1038/leu.2012.246
177. on behalf of the EuroClonality-NGS Working Group, Scheijen B, Meijers RWJ, Rijntjes J, van der Klift MY, Möbs M, et al. Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: a technical feasibility study by EuroClonality-NGS. Leukemia (2019) 33(9):2227−40. doi: 10.1038/s41375-019-0508-7
178. Oon ML, Lim JQ, Lee B, Leong SM, Soon GST, Wong ZW, et al. T-Cell lymphoma clonality by copy number variation analysis of T-cell receptor genes. Cancers (2021) 13(2):340. doi: 10.3390/cancers13020340
179. Chen YL, Ho CL, Hung CY, Chen WL, Chang C, Hou YH, et al. Enhancing diagnosis of T-cell lymphoma using non-recombined T-cell receptor sequences. Front Oncol (2022) 12:1014132. doi: 10.3389/fonc.2022.1014132
180. Drieux F, Ruminy P, Abdel-Sater A, Lemonnier F, Viailly PJ, Fataccioli V, et al. Defining signatures of peripheral T-cell lymphoma with a targeted 20-marker gene expression profiling assay. Haematologica (2020) 105(6):1582−92. doi: 10.3324/haematol.2019.226647
181. Amador C, Bouska A, Wright G, Weisenburger DD, Feldman AL, Greiner TC, et al. Gene expression signatures for the accurate diagnosis of peripheral T-cell lymphoma entities in the routine clinical practice. J Clin Oncol (2022) 40(36):4261−75. doi: 10.1200/JCO.21.02707
182. Julia E, Mareschal S, Chebel A, Golfier C, Müller TA, Lours C, et al. Chromatin accessibility profiling to increase diagnostic accuracy and refine cell-of-Origin classification of mature T-cell lymphomas. Blood (2021) 138(Supplement 1):809−809. doi: 10.1182/blood-2021-150034
183. Bommier C, Mauduit C, Fontaine J, Bourbon E, Sujobert P, Huet S, et al. Real-life targeted next-generation sequencing for lymphoma diagnosis over 1 year from the French lymphoma network. Br J Haematol (2021) 193(6):1110−22. doi: 10.1111/bjh.17395
184. Syrykh C, Gorez P, Péricart S, Grand D, Escudié F, Cabarrou B, et al. Molecular diagnosis of T-cell lymphoma: a correlative study of PCR-based T-cell clonality assessment and targeted NGS. Blood Adv (2021) 5(22):4590−3. doi: 10.1182/bloodadvances.2021005249
185. Dupuy A, Lemonnier F, Fataccioli V, Martin-Garcia N, Robe C, Pelletier R, et al. Multiple ways to detect IDH2 mutations in angioimmunoblastic T cell lymphoma: from immunohistochemistry to next generation sequencing. J Mol Diagn (2018) 20(5):677−85. doi: 10.1016/j.jmoldx.2018.05.012
186. Broccoli A, Bertuzzi C, Fiorentino M, Morigi A, Stefoni V, Agostinelli C, et al. BRAF V600E-positive monomorphic epitheliotropic intestinal T-cell lymphoma complicating the course of hairy cell leukemia. OncoTargets Ther (2019) 12:4807−12. doi: 10.2147/OTT.S202061
187. Tanzima Nuhat S, Sakata-Yanagimoto M, Komori D, Hattori K, Suehara Y, Fukumoto K, et al. Droplet digital polymerase chain reaction assay and peptide nucleic acid-locked nucleic acid clamp method for RHOA mutation detection in angioimmunoblastic T-cell lymphoma. Cancer Sci (2018) 109(5):1682−9. doi: 10.1111/cas.13557
188. Hayashida M, Maekawa F, Chagi Y, Iioka F, Kobashi Y, Watanabe M, et al. Combination of multicolor flow cytometry for circulating lymphoma cells and tests for the RHOAG17V and IDH2R172 hot-spot mutations in plasma cell-free DNA as liquid biopsy for the diagnosis of angioimmunoblastic T-cell lymphoma. Leuk Lymphoma (2020) 61(10):2389−98. doi: 10.1080/10428194.2020.1768382
189. Mussolin L, Bonvini P, Ait-Tahar K, Pillon M, Tridello G, Buffardi S, et al. Kinetics of humoral response to ALK and its relationship with minimal residual disease in pediatric ALCL. Leukemia (2009) 23(2):400−2. doi: 10.1038/leu.2008.184
190. Yee H, Ponzoni M, Merson A, Goldstein M, Scarpa A, Chilosi M, et al. Molecular characterization of the t(2;5) (p23; q35) translocation in anaplastic large cell lymphoma (Ki-1) and hodgkin’s disease. Blood (1996) 87(3):1081−8. doi: 10.1182/blood.V87.3.1081.bloodjournal8731081
191. Ait-Tahar K, Damm-Welk C, Burkhardt B, Zimmermann M, Klapper W, Reiter A, et al. Correlation of the autoantibody response to the ALK oncoantigen in pediatric anaplastic lymphoma kinase–positive anaplastic large cell lymphoma with tumor dissemination and relapse risk. Blood (2010) 115(16):3314−9. doi: 10.1182/blood-2009-11-251892
192. Mussolin L, Damm-Welk C, Pillon M, Zimmermann M, Franceschetto G, Pulford K, et al. Use of minimal disseminated disease and immunity to NPM-ALK antigen to stratify ALK-positive ALCL patients with different prognosis. Leukemia (2013) 27(2):416−22. doi: 10.1038/leu.2012.205
193. Quelen C, Grand D, Sarot E, Brugières L, Sibon D, Pradines A, et al. Minimal residual disease monitoring using a 3′ALK universal probe assay in ALK-positive anaplastic Large-cell lymphoma. J Mol Diagn (2021) 23(2):131−9. doi: 10.1016/j.jmoldx.2020.11.002
194. Damm-Welk C, Kutscher N, Zimmermann M, Attarbaschi A, Schieferstein J, Knörr F, et al. Quantification of minimal disseminated disease by quantitative polymerase chain reaction and digital polymerase chain reaction for NPM-ALK as a prognostic factor in children with anaplastic large cell lymphoma. Haematologica (2020) 105(8):2141−9. doi: 10.3324/haematol.2019.232314
195. Cavalieri D, Tournilhac O, Missiglia E, Bonnet C, Ledoux-Pilon A, Bisig B, et al. Monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL): clinoci-pathological analysis of a multicenter european cohort. Hematol Oncol (2021) 39(Supplemental 2):83−4. doi: 10.1002/hon.44_2879
196. Sakamoto Y, Ishida T, Masaki A, Murase T, Yonekura K, Tashiro Y, et al. CCR4 mutations associated with superior outcome of adult T-cell leukemia/lymphoma under mogamulizumab treatment. Blood (2018) 132(7):758−61. doi: 10.1182/blood-2018-02-835991
197. Tanaka N, Mori S, Kiyotani K, Ota Y, Gotoh O, Kusumoto S, et al. Genomic determinants impacting the clinical outcome of mogamulizumab treatment for adult T-cell leukemia/lymphoma. Haematologica (2022) 107(10):2418−31. doi: 10.3324/haematol.2021.280352
198. Sakamoto Y, Ishida T, Masaki A, Murase T, Ohtsuka E, Takeshita M, et al. CCR7 alterations associated with inferior outcome of adult T-cell leukemia/lymphoma under mogamulizumab treatment. Hematol Oncol (2022) 40:876–84. doi: 10.1002/hon.3072
199. Lemonnier F, Safar V, Beldi-Ferchiou A, Cottereau AS, Bachy E, Cartron G, et al. Integrative analysis of a phase 2 trial combining lenalidomide with CHOP in angioimmunoblastic T-cell lymphoma. Blood Adv (2021) 5(2):539−48. doi: 10.1182/bloodadvances.2020003081
200. Shimizu-Kohno K, Satou Y, Arakawa F, Kiyasu J, Kimura Y, Niino D, et al. Detection of HTLV-1 by means of HBZ gene in situ hybridization in formalin-fixed and paraffin-embedded tissues. Cancer Sci (2011) 102(7):1432−6. doi: 10.1111/j.1349-7006.2011.01946.x
201. Bobée V, Drieux F, Marchand V, Sater V, Veresezan L, Picquenot JM, et al. Combining gene expression profiling and machine learning to diagnose b-cell non-Hodgkin lymphoma. Blood Cancer J (2020) 10(5):59. doi: 10.1038/s41408-020-0322-5
202. Drieux F, Ruminy P, Sater V, Marchand V, Fataccioli V, Lanic MD, et al. Detection of gene fusion transcripts in peripheral T-cell lymphoma using a multiplexed targeted sequencing assay. J Mol Diagn (2021) 23(8):929−40. doi: 10.1016/j.jmoldx.2021.04.013
203. Ehrentraut S, Nagel S, Scherr ME, Schneider B, Quentmeier H, Geffers R, et al. t(8;9)(p22;p24)/PCM1-JAK2 activates SOCS2 and SOCS3 via STAT5. climent m, éditeur. PloS One (2013) 8(1):e53767. doi: 10.1371/journal.pone.0053767
204. Panagopoulos I, Gorunova L, Spetalen S, Bassarova A, Beiske K, Micci F, et al. Fusion of the genes ataxin 2 like, ATXN2L, and janus kinase 2, JAK2, in cutaneous CD4 positive T-cell lymphoma. Oncotarget (2017) 8(61):103775−84. doi: 10.18632/oncotarget.21790
205. Ng SY, Yoshida N, Christie AL, Ghandi M, Dharia NV, Dempster J, et al. Targetable vulnerabilities in T- and NK-cell lymphomas identified through preclinical models. Nat Commun (2018) 9(1):2024. doi: 10.1038/s41467-018-04356-9
206. Rodriguez-Sevilla JJ, Salido M, Rodriguez-Rivera M, Sanchez-Gonzalez B, Gallardo F, Pujol RM, et al. PCM1::JAK2 fusion associates with an atypical form of mycosis fungoides. Virchows Arch (2022) 481(6):967−73. doi: 10.1007/s00428-022-03372-x
207. Fernandez-Pol S, Neishaboori N, Chapman CM, Khodadoust MS, Kim YH, Rieger KE, et al. Two cases of mycosis fungoides with PCM1-JAK2 fusion. JCO Precis Oncol (2021) 5):646−52. doi: 10.1200/PO.20.00366
208. Wobser M, Roth S, Appenzeller S, Kneitz H, Goebeler M, Geissinger E, et al. Oncogenic mutations and gene fusions in CD30-positive lymphoproliferations and clonally related mycosis fungoides occurring in the same patients. JID Innov (2021) 1(3):100034. doi: 10.1016/j.xjidi.2021.100034
209. Zhang W, Wang W, Han X, Gan Y, Qian L, Zhang Y, et al. Circulating tumor DNA by high-throughput sequencing of T cell receptor monitored treatment response and predicted treatment failure in T cell lymphomas. Int J Lab Hematol (2021) 43:1041−9. doi: 10.1111/ijlh.13498
210. Sakata-Yanagimoto M, Nakamoto-Matsubara R, Komori D, Nguyen TB, Hattori K, Nanmoku T, et al. Detection of the circulating tumor DNAs in angioimmunoblastic T- cell lymphoma. Ann Hematol (2017) 96(9):1471−5. doi: 10.1007/s00277-017-3038-2
211. Li Q, Zhang W, Li J, Xiong J, Liu J, Chen T, et al. Plasma circulating tumor DNA assessment reveals KMT2D as a potential poor prognostic factor in extranodal NK/T-cell lymphoma. biomark Res (2020) 8(1):27. doi: 10.1186/s40364-020-00205-4
212. Kim JJ, Kim HY, Choi Z, Hwang Sy, Jeong H, Choi JR, et al. In-depth circulating tumor DNA sequencing for prognostication and monitoring in natural killer/T-cell lymphomas. Front Oncol (2023) 13:1109715. doi: 10.3389/fonc.2023.1109715
213. Huang D, Li Q, Li X, Ma N, Duan Y, Zhu L, et al. The novel prognostic index model of combining circulating tumor DNA and PINK-e predicts the clinical outcomes for newly diagnosed extranodal NK/T-cell lymphoma. HemaSphere (2023) 7(1):e822. doi: 10.1097/HS9.0000000000000822
214. d’Amore F, Gaulard P, Trümper L, Corradini P, Kim WS, Specht L, et al. Peripheral T-cell lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2015) 26:v108−15. doi: 10.1093/annonc/mdv201
215. Mak V, Hamm J, Chhanabhai M, Shenkier T, Klasa R, Sehn LH, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J Clin Oncol (2013) 31(16):1970−6. doi: 10.1200/JCO.2012.44.7524
216. Bellei M, Foss FM, Shustov AR, Horwitz SM, Marcheselli L, Kim WS, et al. The outcome of peripheral T-cell lymphoma patients failing first-line therapy: a report from the prospective, international T-cell project. Haematologica (2018) 103(7):1191−7. doi: 10.3324/haematol.2017.186577
217. Al-Mashhadi AL, Cederleuf H, Jensen RK, Nielsen TH, Pedersen MB, Mortensenl TB, et al. Outcome of limited-stage peripheral T-cell lymphoma after CHOP(-like) therapy: a population based study of 239 patients from the Nordic lymphoma epidemiology group. Am J Hematol (2023) 98(3):388−97. doi: 10.1002/ajh.26803
218. Lowe EJ, Reilly AF, Lim MS, Gross TG, Saguilig L, Barkauskas DA, et al. Brentuximab vedotin in combination with chemotherapy for pediatric patients with ALK+ ALCL: results of COG trial ANHL12P1. Blood (2021) 137(26):3595−603. doi: 10.1182/blood.2020009806
219. Bachy E, Camus V, Thieblemont C, Sibon D, Casasnovas RO, Ysebaert L, et al. Romidepsin plus CHOP versus CHOP in patients with previously untreated peripheral T-cell lymphoma: results of the ro-CHOP phase III study (Conducted by LYSA). J Clin Oncol (2022) 40(3):242−51. doi: 10.1200/JCO.21.01815
220. Ruan J, Moskowitz AJ, Mehta-Shah N, Sokol L, Chen Z, Kotlov N, et al. Multicenter phase 2 study of oral azacitidine (CC-486) plus CHOP as initial treatment for peripheral T-cell lymphoma. Blood (2023) 141(18):2194–205. doi: 10.1182/blood.2022018254
221. Yong W, Zheng W, Zhu J, Zhang Y, Wang X, Xie Y, et al. L-asparaginase in the treatment of refractory and relapsed extranodal NK/T-cell lymphoma, nasal type. Ann Hematol (2009) 88(7):647−52. doi: 10.1007/s00277-008-0669-3
222. Jaccard A, Gachard N, Marin B, Rogez S, Audrain M, Suarez F, et al. Efficacy of l-asparaginase with methotrexate and dexamethasone (AspaMetDex regimen) in patients with refractory or relapsing extranodal NK/T-cell lymphoma, a phase 2 study. Blood (2011) 117(6):1834−9. doi: 10.1182/blood-2010-09-307454
223. Li X, Cui Y, Sun Z, Zhang L, Li L, Wang X, et al. DDGP versus SMILE in newly diagnosed advanced natural Killer/T-cell lymphoma: a randomized controlled, multicenter, open-label study in China. Clin Cancer Res (2016) 22(21):5223−8. doi: 10.1158/1078-0432.CCR-16-0153
224. Wang X, Zhang L, Liu X, Li X, Li L, Fu X, et al. Efficacy and safety of a pegasparaginase-based chemotherapy regimen vs an l-asparaginase-Based chemotherapy regimen for newly diagnosed advanced extranodal natural Killer/T-cell lymphoma: a randomized clinical trial. JAMA Oncol (2022) 8(7):1035−41. doi: 10.1001/jamaoncol.2022.1968
225. Chaubard S, Marouf A, Lavergne D, Lemonnier F, Rossignol J, Clavert A, et al. Efficacy of a short sandwich protocol, methotrexate, gemcitabine, l-asparaginase and dexamethasone chemotherapy combined with radiotherapy, in localised newly diagnosed NK/T-cell lymphoma: a French retrospective study. Br J Haematol (2023) 201(4):673–81. doi: 10.1111/bjh.18689
226. Jaccard A, Philippe L, Couronné L, Benoist JF, Hermine O. NK/T-cell lymphoma, the french experience. Haematol Oncol (2017) 35(S2):126−7. doi: 10.1002/hon.2437_116
227. Ishii T, Ishida T, Utsunomiya A, Inagaki A, Yano H, Komatsu H, et al. Defucosylated humanized anti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell Leukemia/Lymphoma. Clin Cancer Res (2010) 16(5):1520−31. doi: 10.1158/1078-0432.CCR-09-2697
228. Shichijo T, Nosaka K, Tatetsu H, Higuchi Y, Endo S, Inoue Y, et al. Beneficial impact of first-line mogamulizumab-containing chemotherapy in adult T-cell leukaemia-lymphoma. Br J Haematol (2022) 198:983−7. doi: 10.1111/bjh.18281
229. Ishida T, Jo T, Takemoto S, Suzushima H, Suehiro Y, Choi I, et al. Follow-up of a randomised phase II study of chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: impact on allogeneic haematopoietic stem cell transplantation. Br J Haematol (2019) 184:440−83. doi: 10.1111/bjh.15123
230. Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic Large-cell lymphoma. N Engl J Med (2011) 364(8):775−6. doi: 10.1056/NEJMc1013224
231. Gambacorti Passerini C, Farina F, Stasia A, Redaelli S, Ceccon M, Mologni L, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. JNCI J Natl Cancer Inst (2014) 106(2):djt378. doi: 10.1093/jnci/djt378
232. Fukano R, Mori T, Sekimizu M, Choi I, Kada A, Saito AM, et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: an open-label phase II trial. Cancer Sci (2020) 111(12):4540−7. doi: 10.1111/cas.14671
233. Bossi E, Aroldi A, Brioschi FA, Steidl C, Baretta S, Renso R, et al. Phase two study of crizotinib in patients with anaplastic lymphoma kinase ALK-positive anaplastic large cell lymphoma relapsed/refractory to chemotherapy. Am J Hematol (2020) 95(12):E319−21. doi: 10.1002/ajh.25967
234. Rindone G, Aroldi A, Bossi E, Verga L, Zambrotta G, Tarantino S, et al. A monocentric analysis of the long-term safety and efficacy of crizotinib in relapsed/refractory ALK+ lymphomas. Blood Adv (2023) 7(3):314−6. doi: 10.1182/bloodadvances.2022007538
235. Lunning MA. Treatment of peripheral T-cell lymphoma: many shades of Gray. Oncol Williston Park (2015) 29(8):545−50.
236. Rai S, Kim WS, Ando K, Choi I, Izutsu K, Tsukamoto N, et al. Oral HDAC inhibitor tucidinostat in patients with relapsed or refractory peripheral T-cell lymphoma: phase IIb results. Haematologica (2022) 108(3):811−21. doi: 10.3324/haematol.2022.280996
237. Cheminant M, Bruneau J, Kosmider O, Lefrere F, Delarue R, Gaulard P, et al. Efficacy of 5-azacytidine in a TET2 mutated angioimmunoblastic T cell lymphoma. Br J Haematol (2015) 168(6):913−6. doi: 10.1111/bjh.13170
238. Lemonnier F, Dupuis J, Sujobert P, Tournillhac O, Cheminant M, Sarkozy C, et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood (2018) 132(21):2305−9. doi: 10.1182/blood-2018-04-840538
239. Falchi L, Ma H, Klein S, Lue JK, Montanari F, Marchi E, et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: a multicenter phase 2 study. Blood (2021) 137(16):2161−70. doi: 10.1182/blood.2020009004
240. Myers R A, Wirth S, Williams S, Kiel J, Enasidenib P. An oral IDH2 inhibitor for the treatment of acute myeloid leukemia. J Adv Pract Oncol (2018) 9(4):435−40. doi: 10.6004/jadpro.2018.9.4.7
241. de Botton S, Montesinos P, Schuh AC, Papayannidis C, Vyas P, Wei AH, et al. Enasidenib vs conventional care in older patients with late-stage mutant- IDH2 relapsed/refractory AML: a randomized phase 3 trial. Blood (2023) 141(2):156−67. doi: 10.1182/blood.2021014901
242. Nguyen TB, Sakata-Yanagimoto M, Fujisawa M, Nuhat ST, Miyoshi H, Nannya Y, et al. Dasatinib is an effective treatment for angioimmunoblastic T-cell lymphoma. Cancer Res (2020) 80(9):1875−84. doi: 10.1158/0008-5472.CAN-19-2787
243. Lim JQ, Huang D, Tang T, Tan D, Laurensia Y, Peng RJ, et al. Whole-genome sequencing identifies responders to pembrolizumab in relapse/refractory natural-killer/T cell lymphoma. Leukemia (2020) 34(12):3413−9. doi: 10.1038/s41375-020-1000-0
244. Cho J, Kim SJ, Park WY, Kim J, Woo J, Kim G, et al. Immune subtyping of extranodal NK/T-cell lymphoma: a new biomarker and an immune shift during disease progression. Mod Pathol (2020) 33(4):603−15. doi: 10.1038/s41379-019-0392-8
245. Ratner L, Waldmann TA, Janakiram M, Brammer JE. Rapid progression of adult T-cell leukemia–lymphoma after PD-1 inhibitor therapy. N Engl J Med (2018) 378(20):1947−8. doi: 10.1056/NEJMc1803181
246. Hoeller S, Walz C, Reiter A, Dirnhofer S, Tzankov A. PCM1–JAK2-fusion: a potential treatment target in myelodysplastic–myeloproliferative and other hemato-lymphoid neoplasms. Expert Opin Ther Targets (2011) 15(1):53−62. doi: 10.1517/14728222.2011.538683
247. Andersson EI, Brück O, Braun T, Mannisto S, Saikko L, Lagström S, et al. STAT3 mutation is associated with STAT3 activation in CD30+ ALK– ALCL. Cancers Basel. (2020) 12(3):702. doi: 10.3390/cancers12030702
248. Chen J, Zhang Y, Petrus MN, Xiao W, Nicolae A, Raffeld M, et al. Cytokine receptor signaling is required for the survival of ALK– anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc Natl Acad Sci USA (2017) 114(15):3975−80. doi: 10.1073/pnas.1700682114
249. Moskowitz AJ, Ghione P, Jacobsen E, Ruan J, Schatz JH, Noor S, et al. A phase 2 biomarker-driven study of ruxolitinib demonstrates effectiveness of JAK/STAT targeting in T-cell lymphomas. Blood (2021) 138(26):2828−37. doi: 10.1182/blood.2021013379
250. Battistella M, Leboeuf C, Ram-Wolff C, Hurabielle C, Bonnafous C, Sicard H, et al. KIR3DL2 expression in cutaneous T-cell lymphomas: expanding the spectrum for KIR3DL2 targeting. Blood (2017) 130(26):2900−2. doi: 10.1182/blood-2017-06-792382
251. Bagot M, Porcu P, Marie-Cardine A, Battistella M, William BM, Vermeer M, et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol (2019) 20(8):1160−70. doi: 10.1016/S1470-2045(19)30320-1
252. Hurabielle C, Leboeuf C, Ram-Wolff C, Meignin V, Rivet J, Vignon-Pennamen MD, et al. KIR3DL2 expression in patients with adult T-cell lymphoma/leukaemia. Br J Dermatol (2018) 179(1):197−9. doi: 10.1111/bjd.16322
253. Cheminant M, Lhermitte L, Bruneau J, Sicard H, Bonnafous C, Touzart A, et al. KIR3DL2 contributes to the typing of acute adult T-cell leukemia and is a potential therapeutic target. Blood (2022) 140(13):1522−32. doi: 10.1182/blood.2022016765
Keywords: peripheral T-cell lymphoma, molecular diagnosis, oncogenesis, diagnosis, targeted therapy
Citation: Drieux F, Lemonnier F and Gaulard P (2023) How molecular advances may improve the diagnosis and management of PTCL patients. Front. Oncol. 13:1202964. doi: 10.3389/fonc.2023.1202964
Received: 09 April 2023; Accepted: 22 May 2023;
Published: 23 June 2023.
Edited by:
Ryan Wilcox, University of Michigan, United StatesReviewed by:
Carlos Murga-Zamalloa, University of Illinois Chicago, United StatesShih-Sung Chuang, Chi Mei Medical Center, Taiwan
Kedar Inamdar, Henry Ford Hospital, United States
Copyright © 2023 Drieux, Lemonnier and Gaulard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fanny Drieux, fanny.drieux@chb.unicancer.fr