Exome Analysis Identifies a Novel Compound Heterozygous Alteration in TGM1 Gene Leading to Lamellar Ichthyosis in a Child From Saudi Arabia: Case Presentation
 Sami Raja Alallasi1
Sami Raja Alallasi1  Amal A. Kokandi2
Amal A. Kokandi2  Babajan Banagnapali1,3
Babajan Banagnapali1,3  Noor Ahmad Shaik1,3
Noor Ahmad Shaik1,3  Bandar Ali Al-Shehri3 Nuha Mohammad Alrayes4
Bandar Ali Al-Shehri3 Nuha Mohammad Alrayes4  Jumana Yousuf Al-Aama1,3
Jumana Yousuf Al-Aama1,3  Musharraf Jelani1,5*
Musharraf Jelani1,5*- 1Department of Genetic Medicine, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
- 2Department of Dermatology, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
- 3Princess Al-Jawhara Albrahim Center of Excellence in Research of Hereditary Disorders, King Abdulaziz University, Jeddah, Saudi Arabia
- 4Faculty of Applied Medical Sciences, King Abdulaziz University, Jeddah, Saudi Arabia
- 5Centre for Omic Sciences, Islamia College Peshawar, Peshawar, Pakistan
Background: Lamellar ichthyosis is an autosomal recessive type of rare skin disorders characterized with defective epidermis leading hyperkeratosis with brownish-gray scales over the body. These patients are born as collodion babies and may also exhibit additional features like erythema, ectropion, and eclabium. This disease is mainly caused by homozygous and compound heterozygous alterations in transglutaminase 1 encoding gene (TGM1), which is located on 14q12.
Case presentation: This study reports the genetic analysis of a 4-year Saudi girl presenting lamellar ichthyosis. She was the first child of unrelated parents. The family had no previous history of the disease phenotype. She was born as a collodion baby without any prenatal complications. At the time of this study she had developed rough scaly skin on her legs, arms and trunk regions with thick palms and soles. Whole exome sequencing (WES) followed by Sanger sequence validation identified a novel compound heterozygous variant in TGM1 gene. The paternal variant was a missense transition (c.1141G>A; p.Ala381Thr) present at exon 7, while maternal variant (c.758-1G>C) was present at the intron4-exon5 boundary. To the best of our knowledge these variants had not been reported before in TGM1 gene.
Conclusion: In isolated and inbred populations, homozygous variants are identified more frequently; however, our results suggest that compound heterozygous variants should also be considered especially when the marriages are not consanguineous.
Introduction
Lamellar ichthyosis (LI, MIM 242300) is one of the non-syndromic forms of ichthyoses inherited in autosomal recessive form. At birth, babies are covered with collodion-like membranes which sheds later and leaves scales all over the body (1). Severity of scaling differs from patient to patient. In general, patients have large thick scales which are either dark gray or brown in color. They may exhibit very milder form of erythema. However, palmoplantar keratoderma is more frequent while erythroderma is not consistent with LI (2). In some cases alopecia is also accompanied (3).
Genetic analyses of familial cases have reported eight genes responsible for LI phenotype. These genes include ATP binding cassette subfamily A member 12 (ABCA12, MIM 607800) at chromosome 2q35 (4), arachidonate lipoxygenase 3 (ALOXE3, MIM 607206) located at 17p13.1 (5), arachidonate 12-lipoxygenase (ALOX12B, MIM 603741) located at chromosome 17p13.1 (6), ceramide synthase 3 (CERS3, MIM 615276) located at 15q26.3 (7), cytochrome P450 family 4 subfamily F member 22 (CYP4F22, MIM 611495) located at chromosome 19p13.12 (8), NIPA like domain containing 4 (NIPAL4, MIM 609383) located at chromosome 5q33.3, patatin like phospholipase domain containing 1 (PNPLA1, MIM 612121) located at chromosome 6p21.31 (9) and transglutaminase 1 (TGM1, MIM 190195) located at chromosome 14q12 (10, 11).
Among the non-syndromic forms of autosomal recessive ichthyoses, TGM1 alterations have been identified more frequently as compared to other known genes of LI (12). Previously, it was considered that mostly homozygous variants lead to non-availability of transglutaminase enzyme which in turn disrupts cornified cell envelope formation and thus result in skin barrier defect (3, 13). However, recently, a study from Japan has shown that compound heterozygous variants also lead to severe form of LI phenotype (14).
Case Presentation
Methods
In this study, we investigated a Saudi Arabian family for the disease variant of lamellar ichthyosis phenotype. We applied a combination of strategies covering whole exome sequencing (WES) to identify list of altered genes involved in autosomal recessive ichthyosis (Table 1) and then targeted Sanger sequencing (SS) of the short listed variants retrieved from WES analysis.
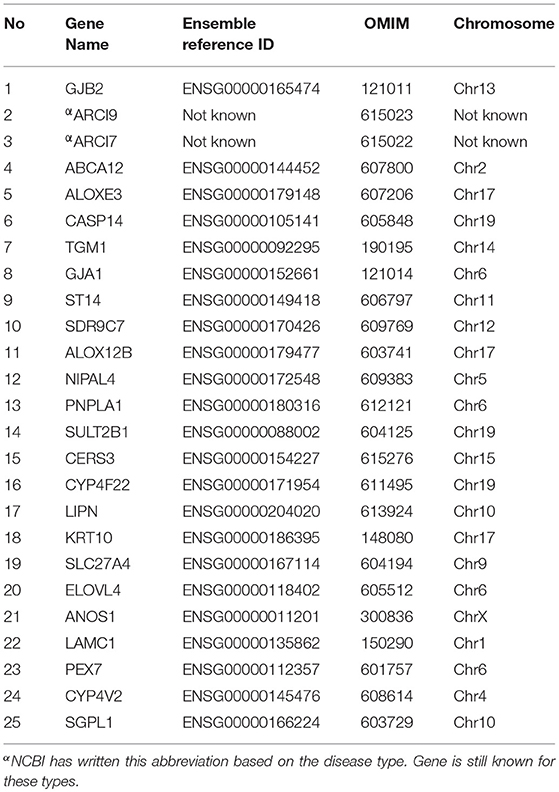
Table 1. List of candidate genes for autosomal recessive congenial ichthyosis.
Genomic DNA of the index patient (II-1) was subjected to bidirectional WES analysis at 100x resolution with 150 base pairs reads length. Sureselect target enrichment system (Agilent Technologies, USA) was used to prepare 6 Mb WES library from 2 μg genomic DNA of the patient. The 100 bases long amplified libraries were then sequenced on NOVASEQ6000 (Illumina, USA) using Macrogen Inc. South Korea NGS sequencing facility. The raw data along with the variant call files were further analyzed for the causative gene/variant(s).
The wild type genomic sequence of TGM1 gene was obtained from Ensembl Genome Browser (https://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000092295). Selected variants were then Sanger sequenced using primers (Table 2) designed by Primers3plus software (www.bioinformatics.nl/primer3plus). The target exons were PCR amplified from genomic DNA samples with GoTaq® green mater mix (Promega, USA). The single stranded sequencing products were synthesized with BigDye terminator v3.1 cycle sequencing kit and run directly on ABI 3500 Genetic Analyzer (Life Technologies, USA). The sequences files were analyzed for mismatches using BioEdit editor version 7 software (www.mbio.ncsu.edu/BioEdit/bioedit.html).

Table 2. List of primers used for the novel TGM1 variants' Sanger validation.
Variant effect predictor (VEP) toolset was used to predict the potential pathogenicity of the selected TGM1 variants. The detailed description of VEP in terms of input, output and principle is described elsewhere (15). From the output results, we selected Mendelian Clinically Applicable Pathogenicity (M-CAP) score (for missense variant) and Max Ent Scan scores (for splice site variant) to understand the variant consequences following the standard variant annotation terms of “sequence ontology.” We further tried to understand the structural consequences of p.Ala381Pro variant using 3 dimensional TGM1 protein model. Owing to the lack of experimentally solved TGM1 structure, we took the reference 3D structure (PDB ID- 1GGT) of human blood coagulation factor xiii (with a resolution of 2.65 Å, 43.6% identity, Z-score of 23.44 and 710 aa length) to create the both native and mutant 3D molecular models of TGM1 protein (NP_000350) using Swiss modeler (https://swissmodel.expasy.org/). These molecular models were analyzed for structural differences in terms of hydrogen bonding characters, solvent accessibility (16), stability (17), and structural deviations (18).
Results
Clinical History
The affected girl II-1 (4 years at the time of study), was born to a non-consanguineous union. The parents (I-1 and II-2) narrated that they had no record of similar patient in their families. She was born as collodion baby on term with a normal pregnancy. She had developed rough and dry skin with palmoplantar keratoderma (thickening of palms and soles). She had exfoliation, fissuring on the palms but hyper-linearity was not evident. Her upper back was more prominent with large thick and brownish scales (Figure 1). These scales were also present on arms and lower limbs. She had sparse hair on scalp. The primary dentition appeared normal. There was no associated anomaly of other body parts or functions.
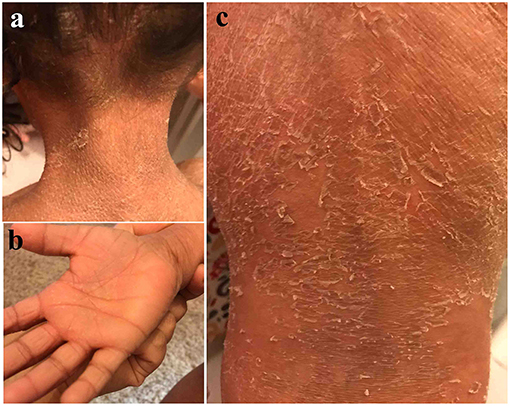
Figure 1. Clinical presentation of patient II-1 in family A. Note the rough and dry skin with over the scalp and neck (a) palmoplantar keratoderma and exfoliation, fissuring on the palms (b). The upper back area is more prominent with large thick and brownish scales (c).
Exome Sequencing
There were 103,605 variants identified in the initially analyzed variant call files of WES data. Out of these, 171 variants were present in those genes which had previously been known for autosomal recessive congenital ichthyosis (Table 1). They included five in 3′ untranslated regions, one in 5′ untranslated region, two in downstream gene position, three in intergenic regions, 119 in introns, 16 in coding exons (missense), three at splicing regions, 20 synonymous and two upstream gene variants.
Potential Candidates' Prioritization
Based on zygosity and minor alleles frequency, variants list was further analyzed. About 120 variants were in homozygous and 51 were in heterozygous conditions. Variants with minor allele frequencies equal or < 0.01 were 18 only (Table S1). Out of these 18, two variants (a splicing c.758-1G>C and a missense c.1141G>A; p. Ala381Thr of TGM1 gene NM_000359.2) were selected for Sanger validation based on clinical resemblance of our patients to the previously reported LI patients with TGM1 alterations. They were present at the splicing region exon 5 and in the coding region of exon 7 of TGM1 gene. Both these variants were heterozygous, and the compound heterozygosity was assumed in the affected sibling. Sanger sequencing was performed to validate the affected alleles segregation.
Sanger Sequencing of TGM1 Variants
Sanger sequencing confirmed that the missense variant c.1141G>A; p. Ala381Thr in exon 7 of TGM1 gene segregated from the father (I-1) and alternatively the splice site variant at exon 5 was carried by the mother (I-2) (Figure 2). Thus, the index patient had compound heterozygous status for paternal variant in exon 7 and maternal variant in exon 5. To the best of our knowledge both these variants were novel and were not reported in published literature previously. They were also not listed 1,000 genome (www.internationalgenome.org/) nor in ExAc database (exac.broadinstitute.org/) and nor greater middle eastern genome database (igm.ucsd.edu/gme/). These variants were also not found in >200 chromosomes of the ethnically matched healthy volunteers.
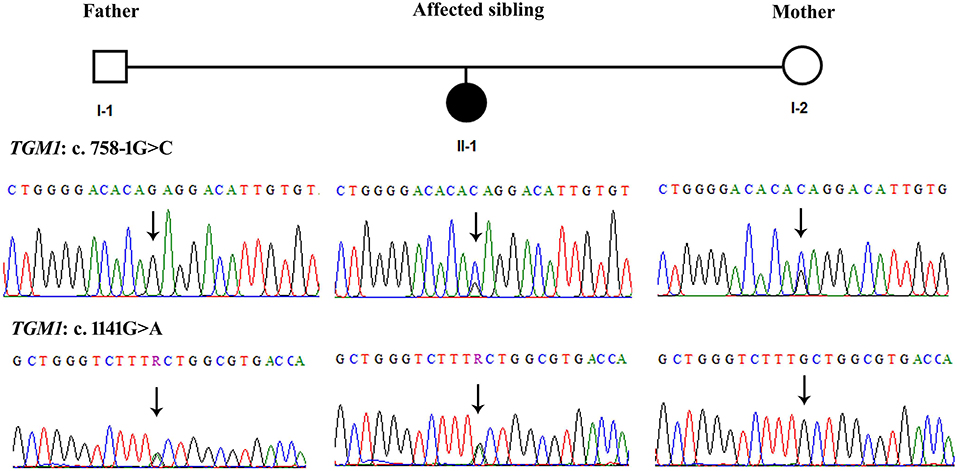
Figure 2. Sanger sequencing of the TGM1 variants showing segregation of c.758-1G>C (Intron4-Exon5 boundary) from mother and c.1141G>A (Exon 7) segregation from father. The arrows indicate respective nucleotides in the sequences. Right panels show that father is wild type for c.758-1G>A and carrier for c.1141G>A. The right panels show that mother is carrier for c.758-1G>A and wild type for c.1141G>A. The middle panels show that affected sibling has both variants in heterozygous form.
Computational Analysis of TGM1 Variants
The MaxEntScan prediction of c.758-1G>C shows that the wild type “G” and mutant “C” nucleotides possess entropy scores of 2.565 and −5.499, respectively. The “C” nucleotide is likely to disturb the splice site junction by 80.46%, hence has potential to alter the normal splicing of TGM1 transcript. Protein analysis of the missense variant (c.1141G>A; p.Ala381Pro) (Figure 3), showed an M-CAP score of 0.736, and any variant with an M-CAP score of >0.50 is considered highly deleterious to the normal protein structure and function (19). Thus, p.Ala381Pro variant is most likely pathogenic in our patient.
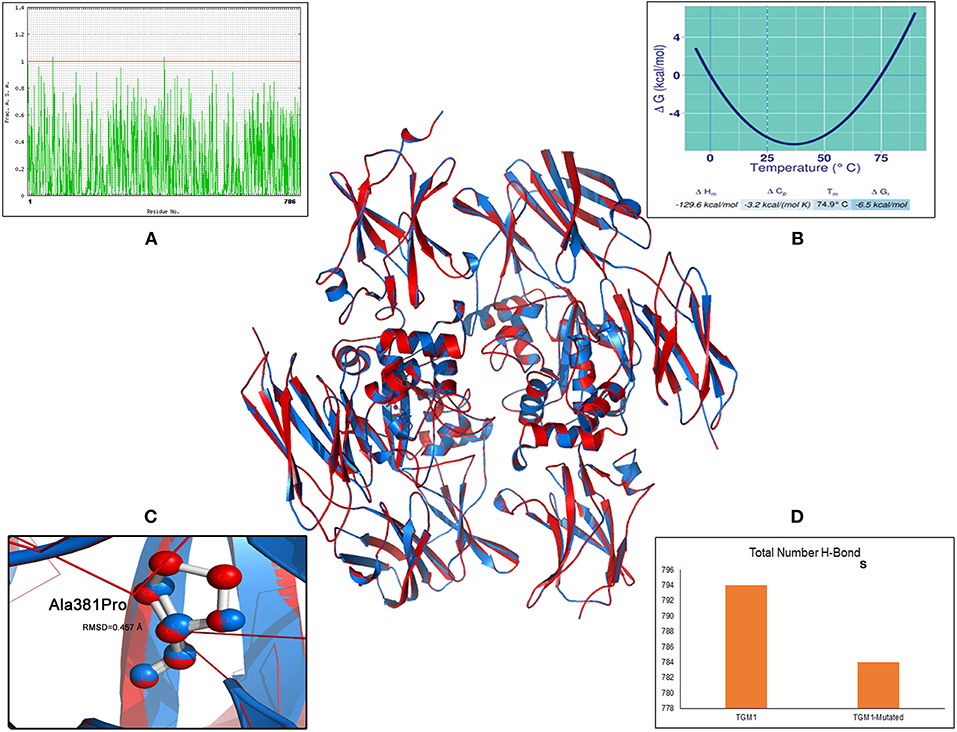
Figure 3. Molecular view of TGM1 altered (p.Ala381Pro) protein model and its biophysical characteristics. Solvent accessibility graph (A) Stability curve (B) RMSD value (C) number of hydrogen bonds (D).
The specific impact of p.Ala381Pro on TGM1 protein can be better understood by examining additional structure-based features like root mean square deviation (RMSD), stability, solvent accessibility, and hydrogen bonding. The 3-dimensional protein model analysis has suggested that the mutant Proline residue brings a RMSD value of 0.45 Å value of 381th position in the TGM1 protein. RMSD refers to the quantitative metric of structural resemblance between native and altered protein molecules. The substituted proline residue seems to bring a destabilizing effect on TGM1 protein, by shifting the Gibbs free energy values (ΔΔG) toward negative range (−6.53 Kcal/mol). The solvent accessibility value (SAV) of mutant Proline residue in TGM1 is abolished due to its buried form compared the native Alanine residue (SAV is 1.83%), which exists in exposed state. Hydrogen (H) bonding analysis revealed that the number of H-bonds are reduced in mutant state (Proline = 783 bonds) compared to its native form (Alanine = 793 bonds) (Figure 3).
Discussion
Exome sequencing analysis has widely been used as an efficient molecular diagnostic tool in single gene disorders. Marriages within closer relatives are always at high risk for autosomal recessive disorders. However, the situation becomes more worrying for those parents who are not cousins and they have first child with a life threatening autosomal recessive disorder, lamellar ichthyosis.
Three genes including TGM1 have been reported previously for LI patients from Saudi Arabia and neighboring Arab countries. First genetic analysis of Saudi patients was carried out through homozygosity mapping followed by targeted TGM1 gene's Sanger sequencing (20). More efficient approach of whole exome sequencing was carried out in United Arab Emirates families, in which three previously reported TGM1 variants were identified. The authors concluded that TGM1 was one of the most frequent cause of LI in the Middle East (20, 21). Another extensive study from Scandinavian countries has also found TGM1 alterations in most of the LI patients (22).
The transglutaminase-1 (TGM-1) enzyme is found in the epidermis, where it is responsible for cross linking the keratin precursors and to form the cornified cell envelope. This cross linking provides mechanical strength and stability to the epidermal layer of skin. The 817-aa long TGM-1 is composed of 4 functional domains including Transglut N (117–235 aa), Transglutaminase/protease-like homologs domain (TGc) (348–468 aa), Transglut C domain (587–691 aa & 699–796 aa). TGM1 protein deficiencies also have a role in the compensatory overexpression of other transcripts involved in skin barrier repair. This upregulation of autosomal recessive congenital ichthyosis genes might reflect a compensatory induction of omega-O-acyl ceramides which are part of a global barrier repair response in the patient's epidermis (23).
The p.Ala381Pro variant identified in our case lies in the main catalytic (TGc) domain of TGM1 protein. This might affect several structural disturbances by altering conformation, stability, solvent accessibility and hydrogen bonding properties of this protein. All these biophysical parameters play a very important for the maintenance of polypeptide folding and 3 dimensional conformation of protein structure, in addition to determining its metal binding characteristics (18, 24, 25). These structural disturbances might induce altered Ca2+ binding properties and misfolding of the polypeptide in the endoplasmic reticulum (ER). Which would eventually lead to ubiquitinoylation and release of TGM1 from ER instead of being trafficked to cell membrane. The abnormal ubiquitinoylation and accumulation in perinuclear structures forming aggresomes, could ultimately lead to the lower levels of transglutaminase activity in the cell membranes (26).
The c.758-1G>C splice site variation could abrogate the normal AG acceptor site at exon 5-intron 5 junction, resulting in the production of extra-long m-RNA. There are increasing evidences which have showed that point variants in 3′ donor splice site, in particular the variants involving G residue at −1 are rather rare (27). The acceptor splice variant reduces the pairing of acceptor splice site with complimentary site in U1snRNP complex, which is the first step in mRNA splicing (28). Therefore, variants acceptor splice sites might lead to intron-4 retention or exon-5 skipping or cryptic site activations as described elsewhere (29).
Conclusion
Based on our molecular genetic findings, we expect that the lamellar ichthyosis patient we studied here, has inherited two defective alleles (missense and splice site variants) from her parents. Both splice site and missense variants might have contributed to loss-of-function of transglutaminase 1 enzyme in the patient. The defective or reduced availability of TGM1 enzyme may result in the formation of an abnormally functioning stratum corneum, the outermost epidermal layer with defective intercellular lipid layers (30), thus leading to skin abnormalities and clinical manifestations of LI.
Study Limitations
This study sincerely admits that lack of functional biology assays is one of the main limitations to ascertain the causative role of TGM1 gene in this family. However, given the location of p.Ala381Pro in TGc domain, whose main role is in enzyme catalysis, we expect it to be the main causative factor for lamellar ichthyosis in this family. TGM1 molecular data produced in this study might have important implications for the prenatal diagnosis, disease prevention in risk group individuals and pave a way for future therapies aimed at personalized medicine.
Ethics Statement
Prior to start of this study an approved consent was signed by the family members. All the participants voluntarily provided their genomic DNA samples and the study was approved according to Helsinki's Declaration by ethical committee of medical and research, Faculty of Medicine, King Abdulaziz University Jeddah under the project reference number 24-14.
Author Contributions
SA performed laboratory experiments and compiled results. NS and BB performed 3D protein modeling analyses and wrote the results and discussion. AK wrote and provided clinical history of the enrolled patients. NA and BA-S collected samples and performed Sanger sequencing data analysis. JA-A critically reviewed the manuscript. MJ designed the study, analyzed WES data, wrote, and finalized the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge volunteer contribution of the family members.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2019.00044/full#supplementary-material
References
1. Small K, Ginsburg H, Greco MA, Sarita-Reyes C, Kupchik G, Blei F. More than skin deep: a case of congenital lamellar ichthyosis, lymphatic malformation, and other abnormalities. Lymphat Res Biol. (2008) 6:39–44. doi: 10.1089/lrb.2007.1020
2. Akiyama M, Sawamura D, Shimizu H. The clinical spectrum of nonbullous congenital ichthyosiform erythroderma and lamellar ichthyosis. Clin Exp Dermatol. (2003) 28:235–40. doi: 10.1046/j.1365-2230.2003.01295.x
3. Hennies HC, Raghunath M, Wiebe V, Vogel M, Velten F, Traupe H, et al. Genetic and immunohistochemical detection of mutations inactivating the keratinocyte transglutaminase in patients with lamellar ichthyosis. Hum Genet. (1998) 102:314–8.
4. Akiyama M. The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochim Biophys Acta (2014) 1841:435–40. doi: 10.1016/j.bbalip.2013.08.009
5. Sugiura K, Akiyama M. Lamellar ichthyosis caused by a previously unreported homozygous ALOXE3 mutation in East Asia. Acta Derm Venereol. (2015) 95:858–9. doi: 10.2340/00015555-2022
6. Osorio F, Leao M, Azevedo F, Magina S. Lamellar ichthyosis due to ALOX12B mutation. Actas Dermosifiliogr. (2013) 104:443–4. doi: 10.1016/j.ad.2012.07.011
7. Eckl KM, Tidhar R, Thiele H, Oji V, Hausser I, Brodesser S, et al. Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J Invest Dermatol. (2013) 133:2202–11. doi: 10.1038/jid.2013.153
8. Sugiura K, Takeichi T, Tanahashi K, Ito Y, Kosho T, Saida K, et al. Lamellar ichthyosis in a collodion baby caused by CYP4F22 mutations in a non-consanguineous family outside the Mediterranean. J Dermatol Sci. (2013) 72:193–5. doi: 10.1016/j.jdermsci.2013.06.008
9. Zimmer AD, Kim GJ, Hotz A, Bourrat E, Hausser I, Has C, et al. Sixteen novel mutations in PNPLA1 in patients with autosomal recessive congenital ichthyosis reveal the importance of an extended patatin domain in PNPLA1 that is essential for proper human skin barrier function. Br J Dermatol. (2017) 177:445–55. doi: 10.1111/bjd.15308
10. Fachal L, Rodriguez-Pazos L, Ginarte M, Beiras A, Suarez-Penaranda JM, Toribio J, et al. Characterization of TGM1 c.984+1G>A mutation identified in a homozygous carrier of lamellar ichthyosis. Int J Dermatol. (2012) 51:427–30. doi: 10.1111/j.1365-4632.2011.05171.x
11. Zhang YL, Yue ZH, Yuan P, Zhou Q, Huang WJ, Hu B, et al. Novel compound heterozygous mutations of TGM1 gene identified in a Chinese collodion baby. Zhonghua Yi Xue Yi Chuan Xue Za Zhi (2012) 29:1–4. doi: 10.3760/cma.j.issn.1003-9406.2012.01.001
12. Takeichi T, Akiyama M. Inherited ichthyosis: non-syndromic forms. J Dermatol. (2016) 43:242–51. doi: 10.1111/1346-8138.13243
13. Lugassy J, Hennies HC, Indelman M, Khamaysi Z, Bergman R, Sprecher E. Rapid detection of homozygous mutations in congenital recessive ichthyosis. Arch Dermatol Res. (2008) 300:81–5. doi: 10.1007/s00403-007-0815-0
14. Takeda M, Nomura T, Sugiyama T, Miyauchi T, Suzuki S, Fujita Y, et al. Compound heterozygous missense mutations p.Leu207Pro and p.Tyr544Cys in TGM1 cause a severe form of lamellar ichthyosis. J Dermatol. (2018) 45:1463–7. doi: 10.1111/1346-8138.14675
15. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
16. Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, et al. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. (2003) 31:3316–9. doi: 10.1093/nar/gkg565
17. Pucci F, Kwasigroch JM, Rooman M. SCooP: an accurate and fast predictor of protein stability curves as a function of temperature. Bioinformatics (2017) 33:3415–22. doi: 10.1093/bioinformatics/btx417
18. Shaik NA, Awan ZA, Verma PK, Elango R, Banaganapalli B. Protein phenotype diagnosis of autosomal dominant calmodulin mutations causing irregular heart rhythms. J Cell Biochem. (2018) 119:8233–48. doi: 10.1002/jcb.26834
19. Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. (2016) 48:1581–6. doi: 10.1038/ng.3703
20. Wakil SM, Binamer Y, Al-Dossari H, Al-Humaidy R, Thuraya RA, Khalifa O, et al. Novel mutations in TGM1 and ABCA12 cause autosomal recessive congenital ichthyosis in five Saudi families. Int J Dermatol. (2016) 55:673–9. doi: 10.1111/ijd.13279
21. Bastaki F, Mohamed M, Nair P, Saif F, Mustafa EM, Bizzari S, et al. Summary of mutations underlying autosomal recessive congenital ichthyoses (ARCI) in Arabs with four novel mutations in ARCI-related genes from the United Arab Emirates. Int J Dermatol. (2017) 56:514–23. doi: 10.1111/ijd.13568
22. Pigg MH, Bygum A, Ganemo A, Virtanen M, Brandrup F, Zimmer AD, et al. Spectrum of autosomal recessive congenital ichthyosis in scandinavia: clinical characteristics and novel and recurrent mutations in 132 patients. Acta Derm Venereol. (2016) 96:932–7. doi: 10.2340/00015555-2418
23. Zhang H, Ericsson M, Westrom S, Vahlquist A, Virtanen M, Torma H. Patients with congenital ichthyosis and TGM1 mutations overexpress other ARCI genes in the skin: part of a barrier repair response? Exp Dermatol. (2018) doi: 10.1111/exd.13813. [Epub ahead of print].
24. Banaganapalli B, Mohammed K, Khan IA, Al-Aama JY, Elango R, Shaik NA. A computational protein phenotype prediction approach to analyze the deleterious mutations of human MED12 gene. J Cell Biochem. (2016) 117:2023–35. doi: 10.1002/jcb.25499
25. Thirumal Kumar D, Eldous HG, Mahgoub ZA, George Priya Doss C, Zayed H. Computational modelling approaches as a potential platform to understand the molecular genetics association between Parkinson's and Gaucher diseases. Metab Brain Dis. (2018) 33:1835–47. doi: 10.1007/s11011-018-0286-3
26. Jiang H, Jans R, Xu W, Rorke EA, Lin CY, Chen YW, et al. Type I transglutaminase accumulation in the endoplasmic reticulum may be an underlying cause of autosomal recessive congenital ichthyosis. J Biol Chem. (2010) 285:31634–46. doi: 10.1074/jbc.M110.128645
27. Houdayer C, Dehainault C, Mattler C, Michaux D, Caux-Moncoutier V, Pages-Berhouet S, et al. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum Mutat. (2008) 29:975–82. doi: 10.1002/humu.20765
28. Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. (2005) 6:386–98. doi: 10.1038/nrm1645
29. Tian C, Yan R, Wen S, Li X, Li T, Cai Z, et al. A splice mutation and mRNA decay of EXT2 provoke hereditary multiple exostoses. PLoS ONE (2014) 9:e94848. doi: 10.1371/journal.pone.0094848
Keywords: lamellar ichthyosis, exome sequencing, transglutaminase 1 gene, novel compound heterozygous, Saudi Arabia
Citation: Alallasi SR, Kokandi AA, Banagnapali B, Shaik NA, Al-Shehri BA, Alrayes NM, Al-Aama JY and Jelani M (2019) Exome Analysis Identifies a Novel Compound Heterozygous Alteration in TGM1 Gene Leading to Lamellar Ichthyosis in a Child From Saudi Arabia: Case Presentation. Front. Pediatr. 7:44. doi: 10.3389/fped.2019.00044
Received: 03 December 2018; Accepted: 01 February 2019;
Published: 21 February 2019.
Edited by:
Haranatha R. Potteti, University of Illinois at Chicago, United StatesReviewed by:
Muhammad Umair, Quaid-i-Azam University, PakistanAshok Kumar, King Saud University, Saudi Arabia
Copyright © 2019 Alallasi, Kokandi, Banagnapali, Shaik, Al-Shehri, Alrayes, Al-Aama and Jelani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Musharraf Jelani, mjelani@kau.edu.sa