
- The University of Queensland Diamantina Institute, Princess Alexandra Hospital, Brisbane, QLD, Australia
Conventional chemotherapeutics target the proliferating fraction of cells in the patient’s body, which will include the tumor cells, but are also toxic to actively proliferating normal tissues. Cellular stresses, such as those imposed by chemotherapeutic drugs, induce cell cycle checkpoint arrest, and currently approaches targeting these checkpoints are being explored to increase the efficacy and selectivity of conventional chemotherapeutic treatments. Loss of a checkpoint may also make cancer cells more reliant on other mechanisms to compensate for the loss of this function, and these compensatory mechanisms may be targeted using synthetic lethal approaches. Here we will discuss the utility of targeting checkpoint defects as novel anti-cancer therapies.
The Cell Cycle as an Anti-Cancer Target
Conventional chemotherapeutic agents continue to have widespread application in the treatment of a wide range of cancers. However, the obvious drawback of these agents, the normal tissue toxicity and their lack of activity in certain tumor types, is driving the development of more selective drugs that target cancer-specific defects. The success of such targeted agents as Herceptin in ErbB2 over expressing breast cancers (Esteva et al., 2010), Gleevec/Imatinib in BCR–Abl dependent CML (Druker, 2002), and Kit positive GIST (Sleijfer et al., 2008), and recently the BRaf inhibitors in melanoma (Bollag et al., 2010), has demonstrated the effectiveness of such an approach. One mechanism that is of increasing interest as an anti-cancer target is the cell cycle, specifically the cell cycle checkpoints which are commonly defective in cancers. Much of the current research employs these defects to increase the efficacy of conventional chemotherapeutics. Here we present ideas for targeting other checkpoint defects without the use of conventional chemotherapeutics, instead relying on cell damage that accumulates in absence of normal checkpoint controls to provide stress, that when correctly targeted, drives tumor cell specific cytotoxicity.
One of the hallmarks of cancer is uncontrolled proliferation, a consequence of loss of normal cell cycle control, which underlies tumor growth (Hanahan and Weinberg, 2011). The high proliferative index of cancers compared to their surrounding normal tissue was recognized very early as common feature of cancers. The higher proliferation rate of cancer cells has been used to provide a degree of targeting for conventional chemotherapeutic drugs, which preferentially kill rapidly proliferating cells. This targeting of the proliferative fraction of cells in patients is also the basis of the normal tissue toxicities commonly associated with conventional chemotherapeutics, with the drugs destroying normally proliferating tissues in the body, such as the hematopoietic system and intestinal epithelial lining, thereby limiting their clinical utility. More recently, normal cell cycle regulators, particularly the cyclin dependent kinases (cdks), a family of protein kinases controlling cell cycle progression, have been investigated as anti-cancer targets. Cdks are protein kinase subunits that form a complex with a regulatory cyclin protein, and it is the various isoforms of these cyclin/cdk complexes that regulate the ordered progression through the cell cycle (Malumbres and Barbacid, 2009). Like conventional chemotherapeutic agents, cdk directed drugs also target the proliferating fraction of cells in the body, but without the genotoxicity associated with chemotherapeutics. The cdk directed drugs are generally well tolerated with acceptable normal tissue toxicity profiles, but this approach has meet with only modest success in terms of their anti-cancer activity (Lapenna and Giordano, 2009).
Recently, attention has focused on the cell cycle mechanisms known as checkpoints. These respond to internal stresses such as incomplete replication and external stresses such as DNA damaging agents to block cell cycle progression until the stress is resolved or repaired (Medema and Macurek, 2011). Checkpoints respond to specific forms of stress to impose an arrest at specific points in the cell cycle. For example, DNA damage during G1 phase and anti-proliferative signals induce a G1 phase arrest prior to entry into S phase. Damage during S phase or stresses that affect replication forks and slow S phase progression cause an S phase checkpoint, whereas DNA damage during G2 phase or failure to completely decatenate the chromosomes can trigger a G2 phase arrest, blocking entry into mitosis. Finally, defects that threaten the fidelity of partitioning of the replicated genome, normally a consequence of failure of proper mitotic spindle function, block exit from mitosis (Figure 1). Thus checkpoints function in normal cell cycle progression to ensure the fidelity and completion of each phase of the cell cycle before progression to the subsequent phase, and in response to external stresses, to produce two genetically identical daughter cells. Many conventional chemotherapeutic agents are genotoxins and thus initiate cell cycle checkpoint responses in both normal tissue and cancer cells. For example, ionizing radiation causes double stranded DNA breaks which can trigger G1 and G2 phase checkpoint arrests, and anti-metabolites such as hydroxyurea and 5-FU block replication triggering an S phase arrest. Genotoxins such as alkylating agents or TopoII inhibitors can trigger G2 phase checkpoints, while anti-microtubule drugs trigger the mitotic checkpoint (Figure 1). Thus the presence of functional checkpoint responses in cancer cells is likely to reduce the efficacy of these drugs through the normal checkpoint role of blocking cell cycle progression and facilitating repair of the drug-induced damage (Kuntz and O’Connell, 2009; Ma et al., 2011; Medema and Macurek, 2011).
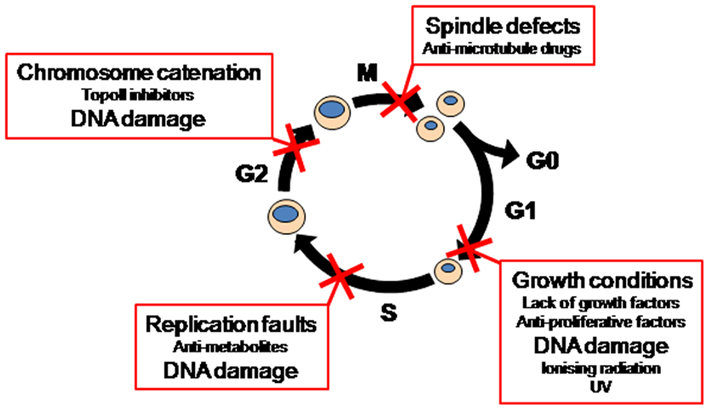
Figure 1. Cell cycle checkpoints.
Each checkpoint arrest utilizes different mechanisms that sense the stress or damage inflicted and impose the cell cycle arrest. Checkpoint mechanisms are commonly divided into two categories, p53-dependent and p53-independent mechanisms. P53 is rapidly stabilized in response to a range of genotoxic insults, and the increased levels of p53 are responsible for cell cycle arrests in G1 and G2 phases in cell lines in vitro, and for signaling apoptosis in response to excessive or persistent damage. This latter function may be its primary role in vivo (Jackson et al., 2011). Thus the frequent loss of p53 in cancers is most likely to enhance their ability to survive in the face of high levels of damage/stress. Another consequence of p53 loss is that the checkpoint response to damage is now entirely reliant on p53-independent mechanisms. These are commonly regulated through the upstream checkpoint kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 related (ATR) which in turn regulate the activity of the checkpoint kinases Chk1 and Chk2. The mitotic checkpoint utilizes a different set of genes to detect spindle defects, many localized to kinetochores, including the Mad and Bub gene families and Aurora kinases (Figure 2). Checkpoint signaling components also have roles in regulating repair mechanisms (Sorensen et al., 2005; Auclair et al., 2009; Gohler et al., 2011), thus contributing to both the arrest and repair of damage triggering the arrest.
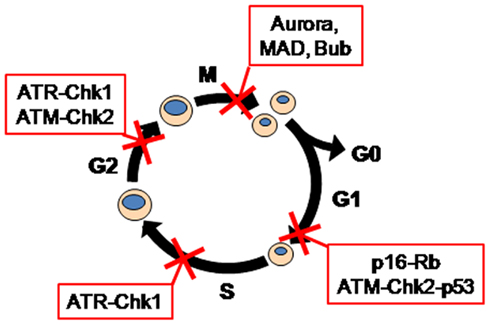
Figure 2. Checkpoint regulators.
Defective Checkpoint Control in Cancer
Many tumor suppressors are components of cell cycle checkpoints, e.g., p53, ATM, p16CDKN2A, and BRCA1, which function in checkpoint responses to a range of different stresses (Pavey et al., 1999; McDonald and El-Deiry, 2001; Stewart et al., 2003; Medema and Macurek, 2011). The frequency of tumor suppressor loss indicates that checkpoint defects are a common feature of cancers. The loss of checkpoint controls provides obvious growth advantages to cancer cells in that they are less sensitive to the normal growth signals that regulate normal cell proliferation. For example, cells defective for the p16–Rb G1 phase checkpoint are insensitive to a range of negative proliferative signals (Giacinti and Giordano, 2006). In addition, loss of checkpoint controls also increases genomic instability, another hallmark of cancer, which can provide cancer cells with an evolutionary or adaptive advantage, allowing them to modify their transcriptome and/or genome to increase their ability to thrive in new tissue environments (Hanahan and Weinberg, 2011). However, a necessary corollary of this is that because checkpoints normally have a protective role, allowing cells to repair any stress or damage before progressing in the cell cycle, defects in checkpoints must increase the burden of endogenous damage in the cancer cells. Despite this, cancer cells survive and thrive with this increased stress load (Halazonetis et al., 2008). The elevated stress levels may be a driver of the higher level of apoptosis often found in tumors compared to normal tissue. This elevated stress must be accommodated by the cells utilizing either; (1) a novel mechanism to cope with the stress, (2) an adaptation to accommodate the stress, or (3) becoming more reliant on alternative stress response mechanisms. All of these mechanisms are equally likely and it may be that cancer cells utilize a combination of all three. Whatever the mechanism, cancer cells now offer a point of divergence from normal tissue in that they are checkpoint defective and thus more reliant on alternative mechanisms to cope with a stress. The increased reliance on alternative stress response mechanisms would predict that if it is possible to disable these mechanisms, this should selectively reduce the viability of the cancer cells with normal tissue being protected by their intact checkpoint response.
Exploiting Checkpoint Defects as Anti-Cancer Targets
Targeting P53
The loss of checkpoint function is currently being exploited in preclinical models of p53 mutant tumors. Loss of p53 results in cells being reliant on the G2 phase ATM/ATR–Chk1/2 dependent checkpoint in responses to genotoxic chemotherapeutic drugs such as topoisomerase inhibitors and anti-metabolites. This checkpoint is readily bypassed using Chk1 inhibitors, resulting in cells undergoing an aberrant mitosis and eventually an increase in cell death (Mukhopadhyay et al., 2005; Dai and Grant, 2010; Morgan et al., 2010; Ma et al., 2011). The shortcoming of this approach is its continued reliance on conventional chemotherapeutic drugs with their associated normal tissue toxicities, although this may be attenuated to some extent by the reduced dose of chemotherapeutic agent required when used in combination with the Chk1 inhibitor. Doses of a range of chemotherapeutic agents can be reduced at least twofold to achieve better anti-tumor activity when combined with a range of Chk1 inhibitors in xenograft models. Combination of Chk1 inhibitors with docetaxel decreased tumor volumes a further 30–50% compared with docetaxel alone (Zhang et al., 2009), and increased anti-tumor activity by up to fourfold in combination with gemcitabine (Blasina et al., 2008; Guzi et al., 2011), and up to twofold with camptothecin (Tse et al., 2007). Chk1 inhibitors also increased the anti-tumor activity of suboptimal doses of gemcitabine and irinotecan by over fourfold (Zabludoff et al., 2008; reviewed in Garrett and Collins, 2011). Although the loss of normal p53 function may also reduce the sensitivity of cells to pro-apoptotic stimuli, this approach provides proof of principle that targeting defective checkpoints can provide tumor selective cytotoxicity in a genetically (p53 mutant) or functionally (checkpoint defective) defined subset of cancers.
Synthetic Lethal Targeting of Defective Checkpoints
The inhibition of Chk1 in p53 mutant cancers is an example of Synthetic Lethality. This is where loss of a stress response mechanism results in reduced cell viability that is tolerated by the cancer, but when combined with the loss of the compensatory mechanism results in complete loss of viability (Kaelin, 2005; Figure 3). Targeting the compensatory mechanisms to inhibit its function renders the cancer cell incapable of coping with the specific stress resulting in a dramatic loss of viability. In the case of Chk1 inhibition, the selectivity is provided by the loss of p53 function and application of an externally applied genotoxic stress (Figure 4). Synthetic lethality is exemplified in BRCA2 mutant breast cancer cells where base excision repair (BER) compensates for the loss of homologous recombination repair, a consequence of BRCA2 loss of function. Inhibiting BER using PARP inhibitors results in selective cytotoxicity of the BRCA2 mutant breast cancers, while normal tissue is protected due to the intact BRCA2 homologous recombination pathway (Farmer et al., 2005; Ashworth, 2008). In this case, the stress is an endogenous stress in the form of normally occurring DNA damage. This has the benefit of an even better toxicity profile, as no genotoxic agents are required to produce the synthetic lethality, reducing the potential for normal tissue toxicities associated with the genotoxin administration. Another example of synthetic lethality is the mechanism by which histone deacetylase inhibitors (HDACi) kill cells that are defective for a G2 phase cell cycle checkpoint triggered by HDACi. We have previously demonstrated that loss of this HDACi-sensitive checkpoint response resulted in rapid apoptosis following HDACi treatment (Qiu et al., 2000; Krauer et al., 2004). In this case, failure to checkpoint arrest results in cells undergoing an aberrant mitosis (Qiu et al., 2000; Cimini et al., 2003; Warrener et al., 2003), which should be detected by the mitotic spindle assembly checkpoint and arrest cells in mitosis until the defect is repaired (Rieder and Maiato, 2004). However, HDACi treatment rapidly overcomes this checkpoint response resulting in mitotic slippage (Warrener et al., 2003; Dowling et al., 2005; Stevens et al., 2008), and it is this slippage that triggers the apoptosis observed (Weaver and Cleveland, 2005). Thus, loss of a checkpoint control may be used as a target to selectively destroy tumors harboring that checkpoint defect. The efficacy of PARP inhibitors in BRCA2 mutant cancers and our work with HDACi and Chk1 inhibitors, demonstrate that targeting a defective checkpoint and disrupting the compensatory mechanisms can deliver tumor selective cytotoxicity by synthetic lethality. In the case of the PARP inhibitors and HDACi, the stresses that trigger the cell death observed are endogenous damage, obviating the requirement for an externally applied stressor as is required for the Chk1 inhibition in p53 mutant tumors. The reliance on endogenous stress increases the selectivity of treatment and thereby reduces the potential normal tissue toxicities. The success of this approach has resulted in an increasing number of synthetic lethality screens being performed that have identified novel drugs or targets for specific tumor types (Azorsa et al., 2009; Kaelin, 2009; Scholl et al., 2009; Astsaturov et al., 2010; Kim et al., 2010; Kuiken and Beijersbergen, 2010; Potratz et al., 2010; Martin et al., 2011). A majority of these have used siRNA libraries to identify components of the compensatory mechanism in a specific genetic or mutant pathway background. We propose that a similar approach could be used to identify synthetic lethal combinations with checkpoint defects as a functional rather than specific genetic defect. As with all targeted therapeutics, it would also be necessary to identify a molecular signature for the defective checkpoint. Our own experience from in vitro studies would suggest that targeting defective G2 phase checkpoint controls might be a more effective strategy as the consequence of a defective G2 phase checkpoint is generally cells undergoing an aberrant mitosis, the outcome of both HDACi treatment and bypassing the ATM/ATR–Chk1 dependent G2 phase checkpoint (Gabrielli et al., 2007; Stevens et al., 2008; Dai and Grant, 2010; Ma et al., 2011). Aberrant mitosis is associated with triggering of the mitotic spindle assembly checkpoint which blocks cells in mitosis until the fidelity of partitioning of the replicated chromosomes can be assured. Activation of the spindle assembly checkpoint also appears to sensitize cells to pro-apoptotic signals generated when mitosis fails (Castedo et al., 2004; Rieder and Maiato, 2004; Sudo et al., 2004; Gabrielli et al., 2007), making this a desirable outcome for a chemotherapeutic treatment. The success of anti-mitotic drugs is strong evidence that utilizing the spindle assembly checkpoint as a source of apoptotic stimulus is effective (Weaver and Cleveland, 2005; Gascoigne and Taylor, 2009; Kaestner and Bastians, 2010).
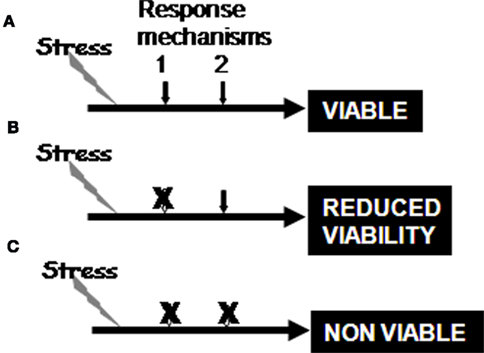
Figure 3. The stress responses are functional in normal tissue (A), and loss of a stress response will reduce cell viability in the face of that stress and the cell become reliant on a secondary mechanism to adapt to the stress (B), whereas loss of primary response and the adaptation result in complete loss of viability (C). The loss of the secondary adaptation is known as synthetic lethality.
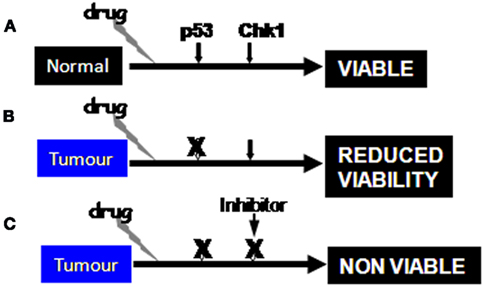
Figure 4. Administration of genotoxic drugs can be readily responded to by p53-dependent mechanisms in normal tissues (A), whereas loss of p53 function will reduce cell viability and result in cells becoming reliant on the Chk1 dependent G2 phase checkpoint (B). Inhibition of Chk1 in combination with drug treatment results in loss of viability (C). The same scenario operates with BRCA2 mutant cells where PARP inhibition blocks BER to produce synthetic lethality in response to endogenous DNA damage.
In summary, defective checkpoints offer a relatively untapped area of potentially selective cytotoxic therapies for a range of cancers. They offer selectivity by way of the defective checkpoint being an intrinsic property of the cancer, while the normal tissue is protected by their intact checkpoints, and tumor cell cytotoxicity from the loss of a protective mechanism. The challenges that lie ahead are to identify means of targeting the defective checkpoints and identifying signatures of the defects, so that patients with checkpoint defective tumors that would respond to these selective therapeutic approaches can be identified.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ashworth, A. (2008). A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 26, 3785–3790.
Astsaturov, I., Ratushny, V., Sukhanova, A., Einarson, M. B., Bagnyukova, T., Zhou, Y., Devarajan, K., Silverman, J. S., Tikhmyanova, N., Skobeleva, N., Pecherskaya, A., Nasto, R. E., Sharma, C., Jablonski, S. A., Serebriiskii, I. G., Weiner, L. M., and Golemis, E. A. (2010). Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci. Signal. 3, ra67.
Auclair, Y., Rouget, R., and Drobetsky, E. A. (2009). ATR kinase as master regulator of nucleotide excision repair during S phase of the cell cycle. Cell Cycle 8, 1865–1871.
Azorsa, D. O., Gonzales, I. M., Basu, G. D., Choudhary, A., Arora, S., Bisanz, K. M., Kiefer, J. A., Henderson, M. C., Trent, J. M., Von Hoff, D. D., and Mousses, S. (2009). Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J. Transl. Med. 7, 43.
Blasina, A., Hallin, J., Chen, E., Arango, M. E., Kraynov, E., Register, J., Grant, S., Ninkovic, S., Chen, P., Nichols, T., O’Connor, P., and Anderes, K. (2008). Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 7, 2394–2404.
Bollag, G., Hirth, P., Tsai, J., Zhang, J., Ibrahim, P. N., Cho, H., Spevak, W., Zhang, C., Zhang, Y., Habets, G., Burton, E. A., Wong, B., Tsang, G., West, B. L., Powell, B., Shellooe, R., Marimuthu, A., Nguyen, H., Zhang, K. Y., Artis, D. R., Schlessinger, J., Su, F., Higgins, B., Iyer, R., D’andrea, K., Koehler, A., Stumm, M., Lin, P. S., Lee, R. J., Grippo, J., Puzanov, I., Kim, K. B., Ribas, A., Mcarthur, G. A., Sosman, J. A., Chapman, P. B., Flaherty, K. T., Xu, X., Nathanson, K. L., and Nolop, K. (2010). Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599.
Castedo, M., Perfettini, J. L., Roumier, T., Andreau, K., Medema, R., and Kroemer, G. (2004). Cell death by mitotic catastrophe: a molecular definition. Oncogene 23, 2825–2837.
Cimini, D., Mattiuzzo, M., Torosantucci, L., and Degrassi, F. (2003). Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol. Biol. Cell 14, 3821–3833.
Dai, Y., and Grant, S. (2010). New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 16, 376–383.
Dowling, M., Voong, K. R., Kim, M., Keutmann, M. K., Harris, E., and Kao, G. D. (2005). Mitotic spindle checkpoint inactivation by trichostatin a defines a mechanism for increasing cancer cell killing by microtubule-disrupting agents. Cancer Biol. Ther. 4, 197–206.
Druker, B. J. (2002). Inhibition of the Bcr-Abl tyrosine kinase as a therapeutic strategy for CML. Oncogene 21, 8541–8546.
Esteva, F. J., Yu, D., Hung, M. C., and Hortobagyi, G. N. (2010). Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat. Rev. Clin. Oncol. 7, 98–107.
Farmer, H., Mccabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., Santarosa, M., Dillon, K. J., Hickson, I., Knights, C., Martin, N. M., Jackson, S. P., Smith, G. C., and Ashworth, A. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921.
Gabrielli, B., Chau, Y. Q., Giles, N., Harding, A., Stevens, F., and Beamish, H. (2007). Caffeine promotes apoptosis in mitotic spindle checkpoint-arrested cells. J. Biol. Chem. 282, 6954–6964.
Garrett, M. D., and Collins, I. (2011). Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol. Sci. 32, 308–316.
Gascoigne, K. E., and Taylor, S. S. (2009). How do anti-mitotic drugs kill cancer cells? J. Cell. Sci. 122, 2579–2585.
Gohler, T., Sabbioneda, S., Green, C. M., and Lehmann, A. R. (2011). ATR-mediated phosphorylation of DNA polymerase {eta} is needed for efficient recovery from UV damage. J. Cell Biol. 192, 219–227.
Guzi, T. J., Paruch, K., Dwyer, M. P., Labroli, M., Shanahan, F., Davis, N., Taricani, L., Wiswell, D., Seghezzi, W., Penaflor, E., Bhagwat, B., Wang, W., Gu, D., Hsieh, Y., Lee, S., Liu, M., and Parry, D. (2011). Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 10, 591–602.
Halazonetis, T. D., Gorgoulis, V. G., and Bartek, J. (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Jackson, J. G., Post, S. M., and Lozano, G. (2011). Regulation of tissue- and stimulus-specific cell fate decisions by p53 in vivo. J. Pathol. 223, 127–136.
Kaelin, W. G. Jr. (2005). The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 5, 689–698.
Kaelin, W. G. Jr. (2009). Synthetic lethality: a framework for the development of wiser cancer therapeutics. Genome Med. 1, 99.
Kim, S. Y., Dunn, I. F., Firestein, R., Gupta, P., Wardwell, L., Repich, K., Schinzel, A. C., Wittner, B., Silver, S. J., Root, D. E., Boehm, J. S., Ramaswamy, S., Lander, E. S., and Hahn, W. C. (2010). CK1epsilon is required for breast cancers dependent on beta-catenin activity. PLoS ONE 5, e8979. doi:10.1371/journal.pone.0008979
Krauer, K. G., Burgess, A., Buck, M., Flanagan, J., Sculley, T. B., and Gabrielli, B. (2004). The EBNA-3 gene family proteins disrupt the G2/M checkpoint. Oncogene 23, 1342–1353.
Kuiken, H. J., and Beijersbergen, R. L. (2010). Exploration of synthetic lethal interactions as cancer drug targets. Future Oncol. 6, 1789–1802.
Kuntz, K., and O’Connell, M. J. (2009). The G(2) DNA damage checkpoint: could this ancient regulator be the Achilles heel of cancer? Cancer Biol. Ther. 8, 1433–1439.
Lapenna, S., and Giordano, A. (2009). Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 8, 547–566.
Ma, C. X., Janetka, J. W., and Piwnica-Worms, H. (2011). Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends. Mol. Med. 17, 88–96.
Malumbres, M., and Barbacid, M. (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166.
Martin, S. A., Hewish, M., Sims, D., Lord, C. J., and Ashworth, A. (2011). Parallel high throughput RNA interference screens identify PINK1 as a potential therapeutic target for the treatment of DNA mismatch repair deficient cancers. Cancer Res. 71, 1836–1842.
Medema, R. H., and Macurek, L. (2011). Checkpoint control and cancer. Oncogene. doi: 10.1038/onc.2011.451
Morgan, M. A., Parsels, L. A., Zhao, L., Parsels, J. D., Davis, M. A., Hassan, M. C., Arumugarajah, S., Hylander-Gans, L., Morosini, D., Simeone, D. M., Canman, C. E., Normolle, D. P., Zabludoff, S. D., Maybaum, J., and Lawrence, T. S. (2010). Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 70, 4972–4981.
Mukhopadhyay, U. K., Senderowicz, A. M., and Ferbeyre, G. (2005). RNA silencing of checkpoint regulators sensitizes p53-defective prostate cancer cells to chemotherapy while sparing normal cells. Cancer Res. 65, 2872–2881.
Pavey, S., Conroy, S., Russell, T., and Gabrielli, B. (1999). Ultraviolet radiation induces p16CDKN2A expression in human skin. Cancer Res. 59, 4185–4189.
Potratz, J. C., Saunders, D. N., Wai, D. H., Ng, T. L., Mckinney, S. E., Carboni, J. M., Gottardis, M. M., Triche, T. J., Jurgens, H., Pollak, M. N., Aparicio, S. A., and Sorensen, P. H. (2010). Synthetic lethality screens reveal RPS6 and MST1R as modifiers of insulin-like growth factor-1 receptor inhibitor activity in childhood sarcomas. Cancer Res. 70, 8770–8781.
Qiu, L., Burgess, A., Fairlie, D. P., Leonard, H., Parsons, P. G., and Gabrielli, B. G. (2000). Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 11, 2069–2083.
Rieder, C. L., and Maiato, H. (2004). Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 7, 637–651.
Scholl, C., Frohling, S., Dunn, I. F., Schinzel, A. C., Barbie, D. A., Kim, S. Y., Silver, S. J., Tamayo, P., Wadlow, R. C., Ramaswamy, S., Dohner, K., Bullinger, L., Sandy, P., Boehm, J. S., Root, D. E., Jacks, T., Hahn, W. C., and Gilliland, D. G. (2009). Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137, 821–834.
Sleijfer, S., Wiemer, E., and Verweij, J. (2008). Drug Insight: gastrointestinal stromal tumors (GIST) – the solid tumor model for cancer-specific treatment. Nat. Clin. Pract. Oncol. 5, 102–111.
Sorensen, C. S., Hansen, L. T., Dziegielewski, J., Syljuasen, R. G., Lundin, C., Bartek, J., and Helleday, T. (2005). The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 7, 195–201.
Stevens, F. E., Beamish, H., Warrener, R., and Gabrielli, B. (2008). Histone deacetylase inhibitors induce mitotic slippage. Oncogene 27, 1345–1354.
Stewart, Z. A., Westfall, M. D., and Pietenpol, J. A. (2003). Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol. Sci. 24, 139–145.
Sudo, T., Nitta, M., Saya, H., and Ueno, N. T. (2004). Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 64, 2502–2508.
Tse, A. N., Rendahl, K. G., Sheikh, T., Cheema, H., Aardalen, K., Embry, M., Ma, S., Moler, E. J., Ni, Z. J., Lopes De Menezes, D. E., Hibner, B., Gesner, T. G., and Schwartz, G. K. (2007). CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin. Cancer Res. 13, 591–602.
Warrener, R., Beamish, H., Burgess, A., Waterhouse, N. J., Giles, N., Fairlie, D., and Gabrielli, B. (2003). Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. FASEB J. 17, 1550–1552.
Weaver, B. A., and Cleveland, D. W. (2005). Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell 8, 7–12.
Zabludoff, S. D., Deng, C., Grondine, M. R., Sheehy, A. M., Ashwell, S., Caleb, B. L., Green, S., Haye, H. R., Horn, C. L., Janetka, J. W., Liu, D., Mouchet, E., Ready, S., Rosenthal, J. L., Queva, C., Schwartz, G. K., Taylor, K. J., Tse, A. N., Walker, G. E., and White, A. M. (2008). AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 7, 2955–2966.
Zhang, C., Yan, Z., Painter, C. L., Zhang, Q., Chen, E., Arango, M. E., Kuszpit, K., Zasadny, K., Hallin, M., Hallin, J., Wong, A., Buckman, D., Sun, G., Qiu, M., Anderes, K., and Christensen, J. G. (2009). PF-00477736 mediates checkpoint kinase 1 signaling pathway and potentiates docetaxel-induced efficacy in xenografts. Clin. Cancer Res. 15, 4630–4640.
Keywords: checkpoint, targeted therapies, synthetic lethality
Citation: Gabrielli B, Brooks K and Pavey S (2012) Defective cell cycle checkpoints as targets for anti-cancer therapies. Front. Pharmacol. 3:9. doi: 10.3389/fphar.2012.00009
Received: 17 October 2011;
Paper pending published: 10 November 2011;
Accepted: 17 January 2012;
Published online: 02 February 2012.
Edited by:
Annarosa Arcangeli, University of Florence, ItalyCopyright: © 2012 Gabrielli, Brooks and Pavey. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Brian Gabrielli, The University of Queensland Diamantina Institute, Princess Alexandra Hospital, Brisbane, QLD 4102, Australia. e-mail: briang@uq.edu.au