 Chu Han1,2†
Chu Han1,2† Nafissa Ismail
Nafissa Ismail Bo Li
Bo Li- 1School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing, China
- 2Advanced Research Institute of Multidisciplinary Science, Beijing Institute of Technology, Beijing, China
- 3Beijing Key Laboratory for Separation and Analysis in Biomedicine and Pharmaceuticals, School of Life Science, Beijing Institute of Technology, Beijing, China
- 4Neuroimmunology, Stress and Endocrinology (NISE) Lab, School of Psychology, Faculty of Social Science, University of Ottawa, Ottawa, ON, Canada
- 5Brain and Mind Research Institute, University of Ottawa, Ottawa, ON, Canada
- 6School of Medicine, Nankai University, Tianjin, China
Ferroptosis is a novel regulated cell death pattern discovered when studying the mechanism of erastin-killing RAS mutant tumor cells in 2012. It is an iron-dependent programmed cell death pathway mainly caused by an increased redox imbalance but with distinct biological and morphology characteristics when compared to other known cell death patterns. Ferroptosis is associated with various diseases including acute kidney injury, cancer, and cardiovascular, neurodegenerative, and hepatic diseases. Moreover, activation or inhibition of ferroptosis using a variety of ferroptosis initiators and inhibitors can modulate disease progression in animal models. In this review, we provide a comprehensive analysis of the characteristics of ferroptosis, its initiators and inhibitors, and the potential role of its main metabolic pathways in the treatment and prevention of various diseased states. We end the review with the current knowledge gaps in this area to provide direction for future research on ferroptosis.
What Is Ferroptosis?
Definition and Discovery of Ferroptosis
Biological growth and the onset of disease are closely linked to cell death. There are two main patterns of cell death: 1) accidental cell death (ACD), and 2) regulated cell death (RCD). One type of RCD is programmed cell death or apoptosis, which occurs normally throughout development (Galluzzi et al., 2015). Apoptosis was in fact the first RCD mechanism discovered, and since then other RCD patterns have also been identified such as necrosis, pyroptosis, and, more recently, ferroptosis (Kroemer et al., 2009).
Studies on ferroptosis initiators and related mechanisms emerged several decades ago, prior to the establishment of the term “ferroptosis”. As early as 1989, Murphy's group discovered that glutamate caused neuronal cell death by inhibiting system xc- (Murphy et al., 1989), which was later named “oxytosis” by Maher's group in 2001 (Tan et al., 2001). Recent studies found that ferroptosis and oxytosis had several common characteristics, such as the role of lipoxygenase, ROS production, and gene expression (Lewerenz et al., 2018). However, there are discrepancies in a few protein-signaling pathways between ferroptosis and oxytosis (Neitemeier et al., 2017). In 2008, two compounds, ras-selective lethal small molecules 3 (RSL3) and 5 (RSL5), were screened using a high-throughput method that could selectively induce cell death in cells carrying RAS mutant subtype genes (Yang and Stockwell, 2008). The results facilitated the identification of the lethal mechanism of the compound erastin (Dolma et al., 2003). Erastin was discovered to induce cell death without nuclear morphological changes, DNA fragmentation, and caspase3 activation, and this process could not be reversed by caspase inhibitors. Therefore, it is a regulated but non-apoptotic form of cell death (Dixon et al., 2012). This erastin-induced cell death pattern is accompanied by an increasing concentration of lipid hydroperoxides (Yagoda et al., 2007) and is inhibited by iron chelators (e.g., deferoxamine mesylate) (Yang and Stockwell, 2008).
In 2012, Stockwell's group was the first to report and name this iron-dependent cell death pattern characterized by increased lipid reactive oxygen species (ROS) as “ferroptosis” (Dixon et al., 2012). In 2018, the Nomenclature Committee on Cell Death (NCCD) defined ferroptosis as “a form of regulatory cell death initiated by oxidative perturbations of the intracellular microenvironment that is under constitutive control by glutathione peroxidase 4 (GPX4) and can be inhibited by iron chelators and lipophilic antioxidants” (Galluzzi et al., 2018).
Characteristics of Ferroptosis
Ferroptosis differs from apoptosis, necrosis, and pyroptosis in morphological and physiological characteristics (Table 1) (Yagoda et al., 2007; Kroemer et al., 2009; Vandenabeele et al., 2010; Galluzzi et al., 2012; Aachoui et al., 2013; Kayagaki et al., 2015; Shi et al., 2015; Vande Walle and Lamkanfi, 2016; Wang et al., 2017; Galluzzi et al., 2018). For example, during ferroptosis, the nuclei of the cell remain intact (Friedmann Angeli et al., 2014), the chromatin is not aggregated (Xie et al., 2016a), and the plasma membrane is not broken or foamed. The shrinking mitochondria show greater inner membrane density while the outer membrane ruptures. In contrast to ferroptosis, apoptosis is characterized by cytoplasmic contraction, nuclear division, chromatin condensation, chromosomal DNA division (Galluzzi et al., 2012), and mitochondrial cytochrome c release (Galluzzi et al., 2018). In apoptotic cells, the plasma membrane foams and eventually forms a distinct intact vesicle (commonly referred to as an apoptotic body). On the other hand, mitochondrial permeability transition (MTP)-driven necrosis is characterized by cytoplasmic granulation, swelling of organelles and cells, loss of cell membrane integrity, and, ultimately, leakage of cellular content (Vandenabeele et al., 2010). In pyroptotic cells, early membrane rupture exposes the inner leaves of the plasma membrane to the extracellular surface, allowing cellular protein annexin V to bind to phosphatidylserine (PS) in the inner leaves. Pyroptotic cells are accompanied by the formation of caspase-1 activity-dependent 12-nm pores in the plasma membrane, leading to a flux of transmembrane ions, swelling of the cytoplasm, and, ultimately, the osmotic dissolution of the cell (Vande Walle and Lamkanfi, 2016). Thus, there are several key distinctions between ferroptosis and other cell death types.
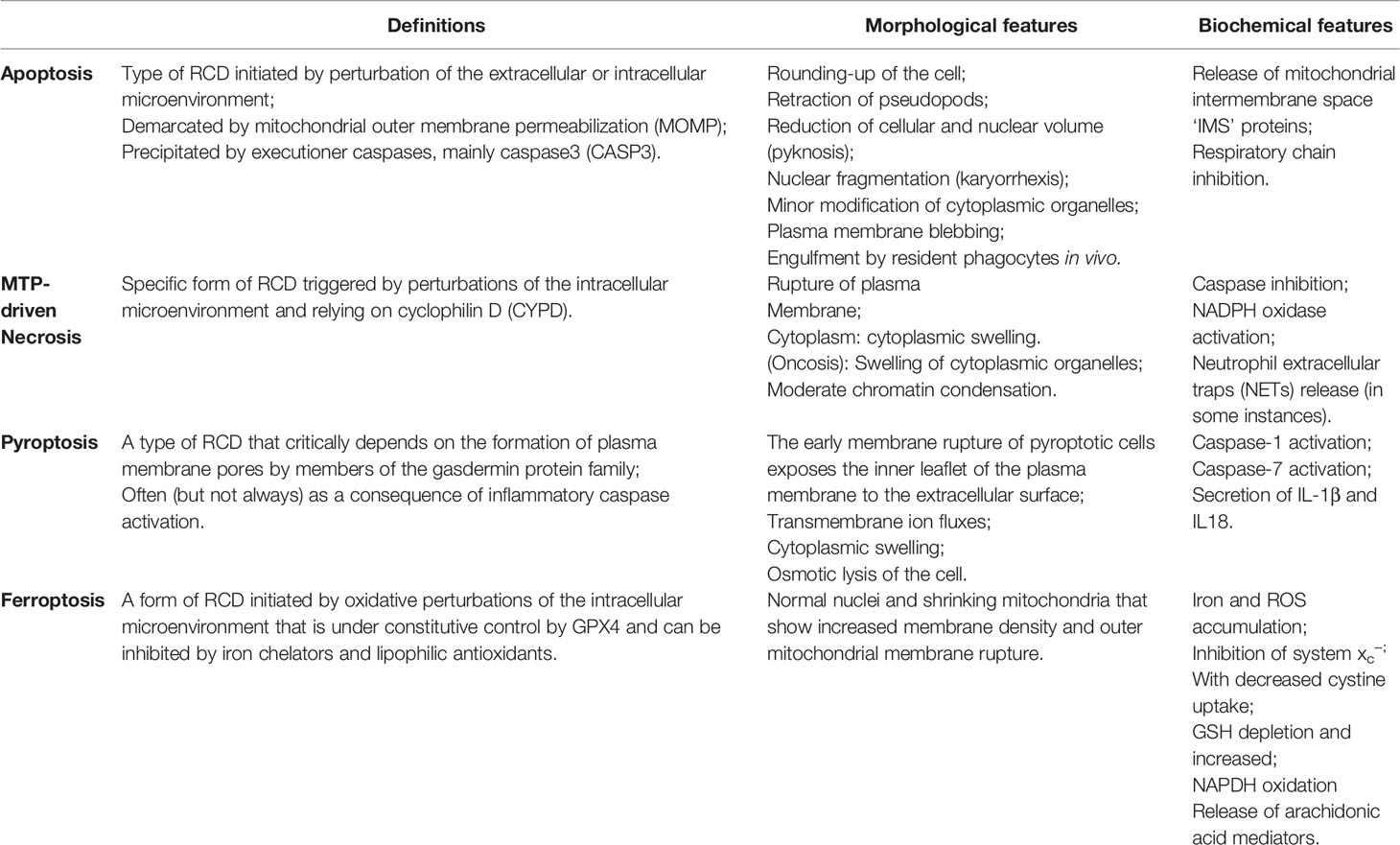
Table 1 Comparison of Characteristics of Apoptosis, Necrosis, Pyroptosis and Ferroptosis.
The most important biochemical features of ferroptosis are the elevated levels of lipid hydroperoxides (LOOH) and ferrous ion (Fe2+) concentration, as ferroptotic cells produce excessive reactive oxygen species, which initiates lipid peroxidation via Fenton Chemistry. Glutathione peroxidase 4 (GPX4), an enzyme that specifically reduces lipid peroxide to the corresponding alcohol, is another central regulator of ferroptosis (Yang et al., 2014). In addition, glutathione (GSH) acts as a GPX4 cofactor and maintains the level of GPX4 through the exchange of glutamate and cystine via the antiporter system xc- (Stockwell et al., 2017).
The genes that control ferroptosis also differ from those that control other forms of cell death. Six protein encoding genes necessary for ferroptosis were screened in HT1080 and Calu-1 cells using shRNA library targeting genes encoding predicted mitochondrial proteins, including genes encoding ribosomal protein L8 (RPL8), iron response element binding protein 2(IREB2), ATP synthase F0 complex subunit C3 (ATP5G3), citrate synthase (CS), tetratricopeptide repeat domain 35 (TTC35), and acyl-CoA synthetase family member 2 (ACSF2) proteins. In addition, TFRC, ISCU, FTH1, and FTL are key genes in ferroptosis that control iron uptake, metabolism, and storage by affecting Fe2+ levels (Dixon et al., 2012).
These genes are different from the ones that control apoptosis (e.g. BH3 interacting domain death agonist (BID), BCL2 antagonist/killer 1(BAK1), BCL2 associated X (BAX), apoptosis inducing factor mitochondria associated 1(AIFM1)) or genes that control other cell death patterns (e.g. genes peptidylprolyl isomerase F (PPIF) involved in MPT-driven necrosis) (Dixon et al., 2012; Galluzzi et al., 2018).
Regulatory Mechanisms of Ferroptosis
Lipid Oxidation Metabolism
Ferroptosis is linked to a fatal accumulation of lipid peroxidation, which is the archetype free radical chain reaction formally resulting in the insertion of O2 into a C-H bond in the oxidizable free polyunsaturated fatty acids (PUFAs) (Figure 1 Eq. 1.1-1.4). This leads to the formation and accumulation of LOOH and ROS and causes ferroptosis. Any radical that can abstract an H-atom from an oxidizable substrate like PUFAs (L-H, Figure 1 Eq. 1.1) can initiate the lipid peroxidation process in vivo. The resultant carbon-centered alkyl radical (L·) reacts with molecular O2 in the environment at (or near) the rate of diffusion, giving rise to a peroxyl radical (LOO·). (Figure 1 Eq. 1.2) (Maillard et al., 1983). The produced peroxyl radicals propagate the chain reaction at propagation rate kp by abstracting H atom from another molecule of oxidizable substrate (L-H) to yield LOOH and a new alkyl radical (L·) (Figure 1 Eq. 1.3). Radical-radical reactions are the final step resulting in non-radical products (Figure 1 Eq. 1.4) (Russell, 1956).
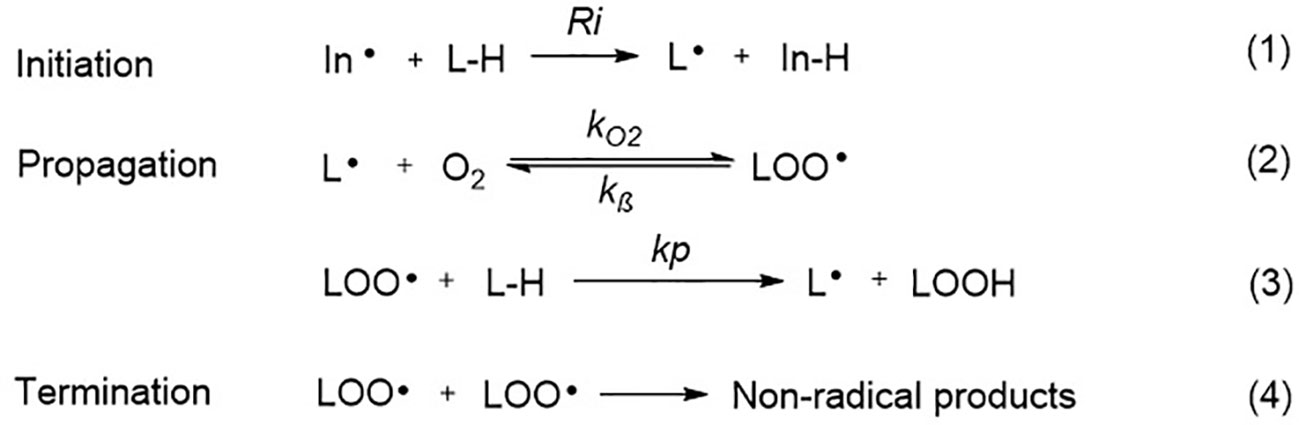
Figure 1 The free radical chain mechanism of lipid peroxidation.
The abundance and location of intracellular oxidizable substrates of lipid peroxidation determines the extent of lipid peroxidation and the extent of ferroptosis. Free polyunsaturated fatty acids are esterified into membrane phospholipids (Kagan et al., 2017) in the lipid metabolic process. Lipidomic analyses indicate that phosphatidylethanolamines (PEs) containing arachidonic acid (AA) or adrenic acid (AdA) are key membrane phospholipids and are further oxidized to phospholipid hydroperoxides (PE-AA/AdA-OOH) by non-enzymatic processes, such as the above mentioned free radical lipid peroxidation or Fenton chemistry drives ferroptosis (Doll et al., 2017; Kagan et al., 2017).
Two enzymes related to ferroptotic lipid metabolism were found through the haploid gene screening of chronic myeloid leukemia cell line KBM7 cells: acyl-CoA synthetase (a long-chain family member 4 (ACSL4) for synthesizing Pes), and lysophosphatidylcholine acyltransferase 3 (LPCAT3) for lipid remodeling. AA/AdA is acylated into membrane phospholipids by LPCAT3 and ACSL4 (PE-AA/AdA) (Figure 2) (Dixon et al., 2015; Kagan et al., 2017). The blocking of ACSL4 results in the suppression of AA or AdA esterification into PE, which reduces the sensitivity of mouse embryonic fibroblasts Pfa1 cells to ferroptosis (Kagan et al., 2017). In DU-145 cancer cells, overexpression of multiple AKR1C family members has been shown to up-regulate the aldo-keto reductase family 1 member C1-3 genes (including AKR1C1-3) and blocks ferroptosis. AKR1C1-3 encodes an aldehyde ketone reductase to reduce the final product of lipid peroxide (PE-AA/AdA-OOH) to the non-toxic corresponding lipid derived alcohol (PE-AA/AdA-OH) (Dixon et al., 2014). Together, these studies demonstrate that lipid peroxidation is a key factor in ferroptosis.
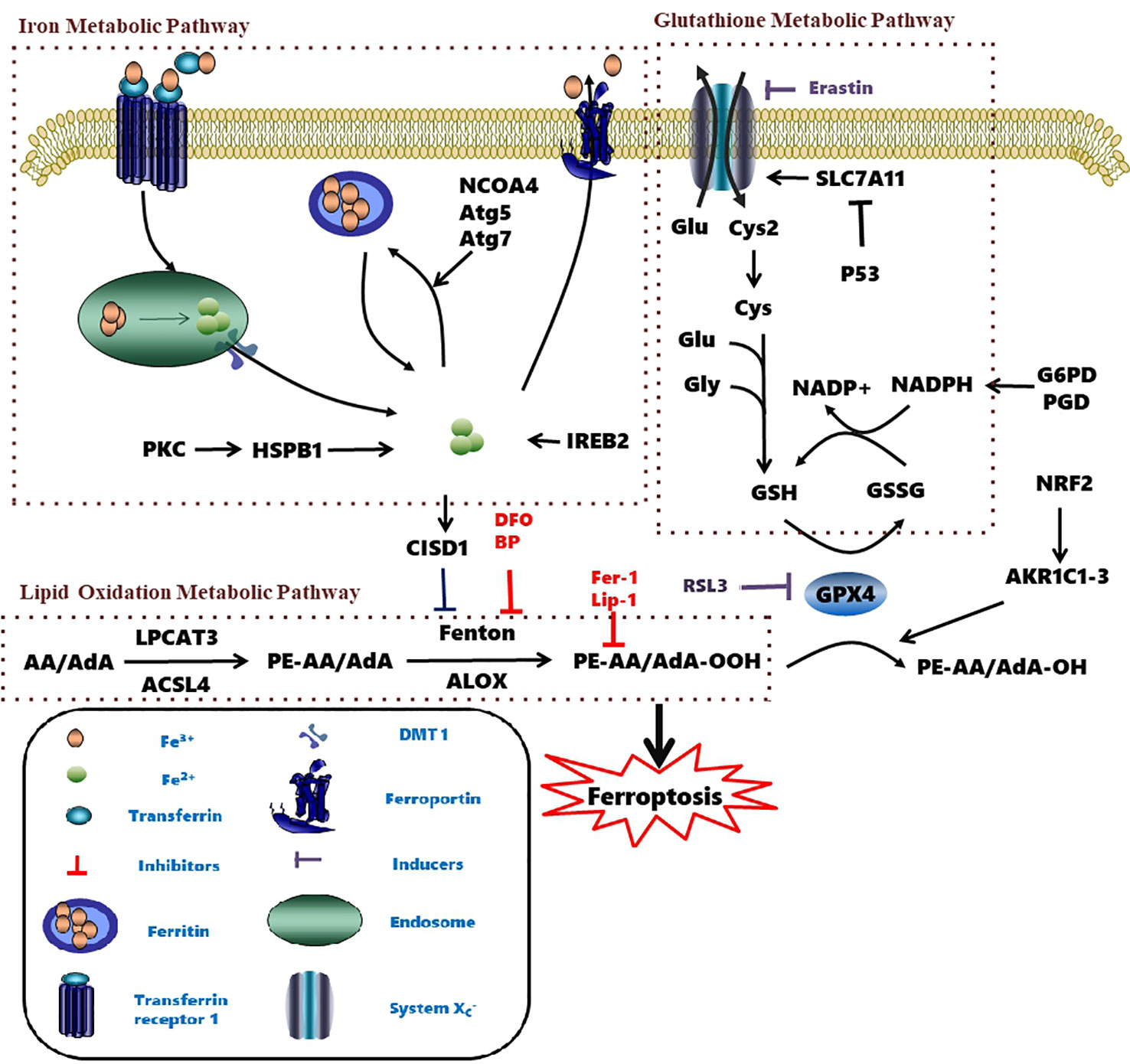
Figure 2 Metabolic pathways affecting ferroptosis. The brown box contains three currently known pathways: Lipid Oxidation Metabolism、Glutathione Metabolic Pathway、Iron Metabolic Pathway and some related mechanisms of action. Several pharmacological inducer have been shown to induce ferroptosis (eg erastin、RSL3). A variety of ferroptosis inhibitors inhibit iron death from various pathways (eg Fer-1、lip-1、BP、DFO). AA, Arachidonic acid; ACSL4, Acyl-CoA synthetase long-chain family member 4; AKR1C1-3, Aldo-keto reductase family 1 member C1-3; Atg5, autophagy-related 5; Atg7, autophagy-related 7; CISD1, CDGSH iron domain 1; Cys, cysteine; Cys2, cystine; DFO, Deferoxamine; DMT1, Divalent metal transporter 1; Fer-1, Ferrostatin-1; Glu, Glutamate; Gly, glycine; GPX4, Glutathione peroxidase 4; G6PD, Glucose-6-phosphate dehydrogenase; GSH, Glutathione; GSSH, Glutathione disulfide; IREB2, Iron-responsive element-binding protein 2; Lip-1, Liproxstatin-1; LOX, Lipoxygenase; LPCAT3, Lysophosphatidylcholine acyltransferase 3; NADPH, Nicotinamide adenine dinucleotide phosphate; NRF2, Nuclear factor erythroid 2-related factor 2; PE, Phosphatidylethanolamine; PGD, 6-Phosphogluconate dehydrogenase; PKC, Protein kinase C; RSL3, Ras-selective lethal small molecules 3; SLC7A11, Solute carrier family 7 member 11.
PUFAs could also be oxidized to the corresponding phospholipid hydroperoxides by lipoxygenases (LOXs) (Kagan et al., 2017; Raefsky et al., 2018). However, whether LOX isozymes are essential for ferroptosis remains controversial. While research using a mouse model of GPX4-induced acute renal failure showed that LOX15 is not required for ferroptosis (Friedmann Angeli et al., 2014), work on a variety of damaged cells shows that phosphatidylethanolamine binding protein 1 (PEBP1) forms a complex with ALOX15 and acts as a scaffold protein to positively regulate ferroptosis by (Wenzel et al., 2017). In addition, it was found that deuterated AA with deurerium at the 7-hydrogen atom position of bisallyl was not oxidized to reduce the sensitivity of cells to ferroptosis. These results proved that AA was involved in the lipid peroxidation during ferroptosis (Raefsky et al., 2018).
Glutathione Metabolic Pathway
The synthesis of tripeptide glutathione (GSH) appears to protect cells from ferroptotic death. The functional activity of glutathione peroxidase 4 (GPX4) is dependent on the biosynthesis of GSH (Yang et al., 2014). More specifically, depletion of GSH causes GPX4 inactivation and increases intracellular lipid peroxidation, resulting in ferroptosis (Cao and Dixon, 2016; Shimada and Stockwell, 2016).
System xc- also regulates ferroptosis (Yang and Stockwell, 2008). System xc- consists of a 12-pass transmembrane protein transporter solute carrier family 7 member 11 (SLC7A11) and a single-pass transmembrane regulatory protein solute carrier family 3 member 2 (SLC3A2). Glutamate and cystine are exchanged at a 1:1 ratio by system xc- (Sato et al., 1999). Tripeptide GSH is synthesized in two steps from cysteine, glutamic acid, and glycine (Yang et al., 2016; Stockwell et al., 2017). The efficiency of this synthetic process is limited to the concentration of cysteine in the substrate. Inhibition of systemic xc- depletes intracellular cysteine, leads to a decrease in glutathione concentration and triggers oxidative stress, and increases the sensitivity of cells to ferroptosis (Cao and Dixon, 2016). However, upregulation of SLC7A11 inhibits erastin-induced ferroptosis (Dixon et al., 2012; Shimada and Stockwell, 2016).
Moreover, some mammals use methionine as a sulfur donor to synthesize new cysteines via the trans-sulfuration pathway. Although mammals usually rely solely on extracellular uptake as the major source of cysteine, the trans-sulfuration pathway acts as a compensatory source of cysteine when system xc- uptake is inhibited (Shimada and Stockwell, 2016). A genome-wide siRNA screening of erastin-induced ferroptosis inhibitors showed that down-regulation of cysteinyl-tRNA synthase (CARS) leads to an up-regulation of the trans-sulfuration pathway and an inhibition of erastin-induced ferroptosis. This result supports the hypothesis that the trans-sulfuration pathway is a regulator of ferroptosis that compensates for cysteine depletion induced by cysteine update inhibition (Hayano et al., 2016).
Iron Metabolic Pathway
The homeostasis of intracellular iron is dependent on the balance between iron absorption, output, utilization, and storage (Galaris et al., 2019). Ferric iron (Fe3+) enters the endosome through the membrane protein transferrin receptor 1 (TFR1) and it is reduced to ferrous iron (Fe2+) by iron reductase. The unstable Fe2+ is then released into the labile iron pool in the cytoplasm by the divalent metal transporter 1 (DMT1). Excess iron ions are either stored in ferritin heteropolymers in the form of Fe3+ or are released extracellularly via the membrane protein ferroportin.
Excessive ferrous iron provides electron-promoting lipid peroxidation through the Fenton reaction (Figure 3) and produces ROS, which triggers ferroptosis. Many autophagy-related genes can also activate ferroptosis. Inhibition of autophagy-related 5 7 genes reduce the accumulation of free iron and inhibit ferroptosis (Gao et al., 2016). Down-regulation of nuclear receptor coactivator 4 (NCOA4), a ferritin phagosome receptor, also inhibits ferritin phagocytosis and reduces Fe2+ content in cells (Gao et al., 2016; Hou et al., 2016). Iron-responsive element-binding protein 2 (IREB2) encodes a major regulator of iron metabolism, and studies have shown that shRNA-mediated silencing of IREB2 alters iron uptake, metabolism, and storage-related genes like TFRC, ISCU, FTH1, and FTL expression (Dixon et al., 2012). Heat shock protein beta-1 (HSPB1) (Sun et al., 2015) and CDGSH iron domain 1 (CISD1) (Yuan et al., 2016) also affect iron metabolism and regulate ferroptosis. In Hela cells, activation of HSPB1 phosphorylation using protein kinase C (PKC) reduces iron levels and blocks ferroptosis (Sun et al., 2015). CISD1, located in the outer membrane of mitochondria, inhibits the uptake of iron ions by mitochondria and also blocks ferroptosis (Yuan et al., 2016). However, oncogenic RAS increases iron content in cells, upregulates TFR, and downregulates ferritin (Yang and Stockwell, 2008). The RAS–RAF–MEK pathway sensitizes cancer cell lines with RAS to ferroptosis via mitochondrial voltage-dependent anion channels 2/3 (VDAC2/3) (Yagoda et al., 2007). In addition, tubulin negatively regulates mitochondrial metabolism by turning off VDAC.
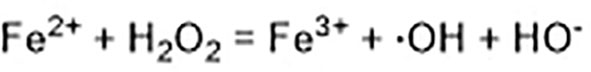
Figure 3 Fenton reaction.
Developing novel fluorescent probes which could detect labile Fe2+ levels in various cells or organs in live animals would definitely help further understanding of the role of iron in ferroptosis under various conditions. For example, an abnormal increase in Fe2+ in lysosomes and endoplasmic reticulum has been observed before the death of HT1080 cells induced by erastin using a variety of novel fluorescent probes (Figure 4) (Hirayama et al., 2019). This study further shows that iron metabolism plays a key role in the ferroptosis process (Yang and Stockwell, 2008).
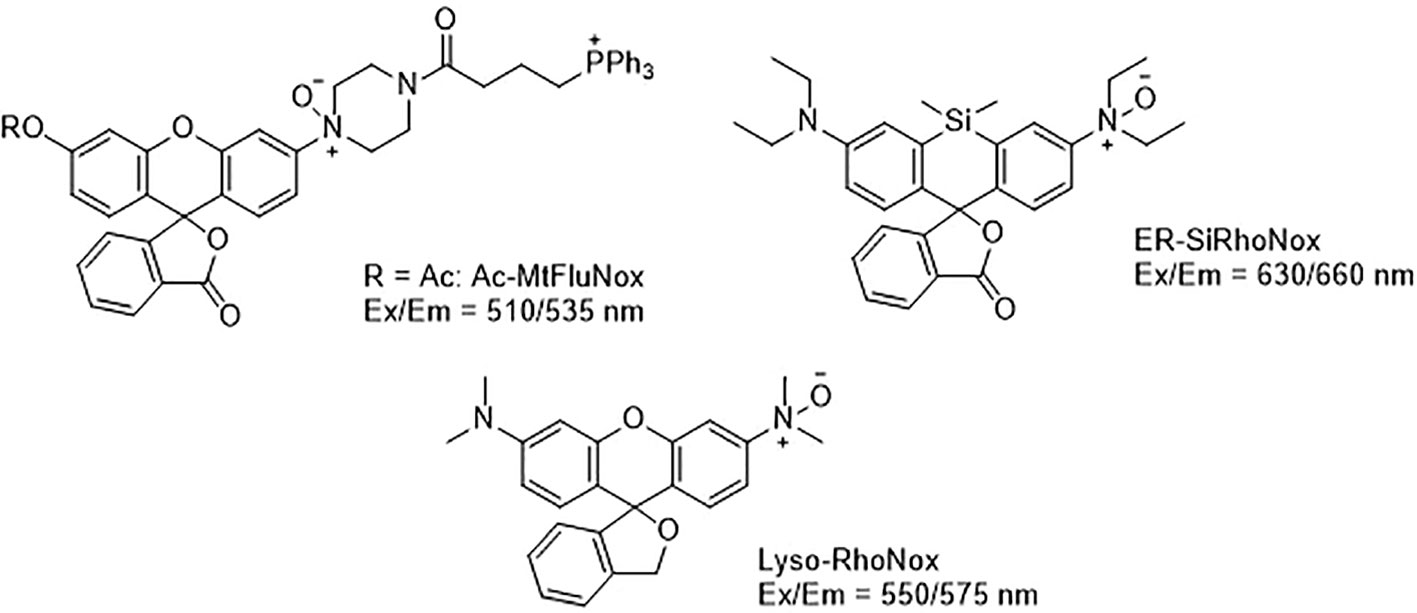
Figure 4 Structures of Ac-MtFluNox, Lyso-RhoNox, and ER-SiRhoNox.
Other Related Signaling Metabolism
In addition to the above-mentioned signaling pathways, nicotinamide adenine dinucleotide phosphate (NADPH) also impacts ferroptosis sensitivity. NADPH is one of the most important reducing agents in cells and protects from excess oxidative damage. In fact, knocking out NADPH increases the sensitivity of fibrosarcoma HT1080 cells to ferroptosis (Shimada et al., 2016a). In the presence of NADPH, glutathione reductase reduces cystine-glutathione disulfide (GSSG) to GSH and increases ferroptosis sensitivity in many cancer cell lines (Zhao et al., 2017).
The nuclear factor erythroid 2-related factor 2 (Nrf2) is a key regulator of the antioxidant response (Sun et al., 2016). Under normoxic cellular conditions, Nrf2 is bound by Kelch-like ECH-associated protein 1 (Keap1) and persists in an inactivated status through ubiquitination and degradation in the proteasome (Reisman et al., 2009). Upon oxidative or electrophilic stress, Nrf2 becomes unleashed from the Keap1 protein binding and translocates to the nucleus (Fan et al., 2017). In the nucleus, Nrf2 transcripts antioxidant responsive element (ARE)-dependent genes in order to balance oxidative mediators and maintain cellular redox homeostasis (Hayes and Dinkova-Kostova, 2014). The above-described process is also regulated by autophagy. The autophagy receptor p62 is a multifunctional protein located throughout the cell which could activate Nrf2 through inactivation of Keap1 (Komatsu et al., 2010). In 2016, Tang's group discovered that the p62-Keap1-Nrf2 antioxidative signaling pathway was involved in the ferroptosis inhibition in HCC cells (Sun et al., 2016). In addition, it had been found that the Nrf2-mediated anti-ferroptosis activity was dependent on the induction of NQO1, heme oxygenase-1 (HO-1), and ferritin heavy chain (FTH1) (Sun et al., 2016). These findings also provide a potential molecular link between ferroptosis and autophagy in the hepatocellular carcinoma cells.
With the continuous research progress on ferroptosis, research evidence has indicated that the occurrence of ferroptosis crosstalk with autophagy (Hou et al., 2016; Kang and Tang, 2017) through pathways other than p62-Keap1-Nrf2. Tang's group demonstrated that knockout or knockdown of autophagy-related 5 (Atg5) and autophagy-related 7 (Atg7) degraded ferritin in fibroblasts and cancer cells, resulting in the limitation of erastin-induced ferroptosis by decreasing intracellular ferrous iron levels and lipid peroxidation (Hou et al., 2016). Other studies also confirmed that signaling pathways closely related to the occurrence of autophagy such as GPX4, SLC7A11, Nrf2, p53, HSPB1, and ACSL4 were also involved in ferroptosis (Kang and Tang, 2017). Torii's group found that autophagy contributed to ferroptosis through the generation of lysosomal ROS in the N-Ras-mutated HT1080 cells (Torii et al., 2016). In addition, they also found that autophagy and lysosomal activity inhibitors including Baf A1, PepA-Me, and ammonium chloride were effective in the prevention of erastin-induced ferroptosis (Torii et al., 2016; Sun et al., 2018). However, the occurrence of ferroptosis is not always related to autophagy, and the relationship between ferroptosis and autophagy or other cell death pathways needs to be further clarified in future research.
P53 also mediates cell cycle inhibition, senescence, and apoptosis, and contributes to the development of tumors (Jiang et al., 2015a). P53 regulates cellular metabolism through the SLC7A11 gene (Jiang et al., 2015b). More specifically, P53 inhibits system xc- by down-regulating SLC7A11. Exposure to ROS in P53 gene-silencing human lung cancer H1299 cells does not change cell viability. However, exposure to ROS after activation of the P53 gene increases the cell death rate to 90%. This study shows that the antioxidant capacity is significantly reduced after activation of the P53 gene, and the cell death rate decreases by about 40-fold after the addition of the ferroptosis-specific inhibitor ferrostatin-1 (Jiang et al., 2015b). Therefore, it indicates that P53 plays an important role in the cellular ferroptotic-related ROS metabolic pathway.
It has been revealed that inhibition of cystine–glutamate exchange leads to the activation of endoplasmic reticulum (ER) stress response and upregulation of glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1) gene, therefore contributing to the glutathione degradation and ferroptosis execution (Rahmani et al., 2007; Dixon et al., 2014). Current research has indicated that ER stress and p53 upregulated modulator of apoptosis (PUMA) overexpression caused by ferroptosis inducers sets up an interaction between ferroptosis and apoptosis (Hong et al., 2017). It has been found that ferroptosis initiator artesunate (ART) could induce ER stress and elevate the expression of the pro-apoptotic PUMA through the ER stress–mediated PERK–eIF2a–ATF4–CHOP pathway but without inducing apoptosis (Hong et al., 2017; Lee et al., 2018). These findings provide new thought on the relationship between ferroptosis and apoptosis.
The ferroptosis suppressor protein 1 (FSP1) (formerly known as AIFM2) has recently been found to be independent of GSH and it could inhibit the transmission of lipid peroxides by reducing coenzyme Q10 to inhibit ferroptosis (Bersuker et al., 2019; Doll et al., 2019). Furthermore, N-myristoylation of FSP1 was found to be essential for ferroptosis inhibition and could provide a new target for the development of drugs targeting the inhibition of ferroptosis.
It has recently been discovered that intercellular interaction dictates ferroptosis. Epithelial extracellular E-cadherin regulates ferroptosis by intracellular NF2–YAP signaling. Antagonism of NF2 causes YAP to promote ferroptosis by up-regulating the associated signaling factors of ferroptosis, ACSL4, and TFRC. Intercellular E-cadherin interactions activate NF2 to inhibit ferroptosis in cells (Wu et al., 2019). However, whether other intercellular interactions dictate ferroptosis in other cell lines remains to be studied.
Currently, whether mitochondria are essential for ferroptosis remains controversial (Bock and Tait, 2019). On one hand, Stockwell's group found that cells lacking mitochondrial DNA were still sensitive to ferroptosis (Gaschler et al., 2018b). Therefore, their conclusion was that ROS generated by the mitochondrial electron transport chain was not essential to ferroptosis initiation. On the other hand, Jiang's group in 2019 showed that mitochondria did not play a role in GPX4 inhibition-induced ferroptosis, but it did play a crucial pro-active role in cysteine-deprivation-induced ferroptosis (Gao et al., 2019). In addition, Tang's group observed that ferroptosis initiator erastin induced the production of mitochondrial ROS (Yuan et al., 2016). They also demonstrated that intra-mitochondrial iron-mediated lipid peroxidation contributed to ferroptosis and CDGSH iron sulfur domain 1 (CISD1), an iron-containing outer mitochondrial membrane protein which limited mitochondrial iron uptake and therefore suppressed ferroptosis (Yuan et al., 2016). Based on the current research, the controversy on the role of mitochondria in ferroptosis remained to be solved.
Initiators of Ferroptosis
Diseases like cancer can be treated by regulating cell death. In some pathological processes, treatment also works by depleting the essential cytokines which then results in cell death. Numerous studies have focused on discovering novel ferroptosis inducers to treat cancer (Hassannia et al., 2019). Ferroptosis inducers can be roughly classified into three categories: (1) system xc- inhibitors, (2) GPX4 inhibitors, and (3) compounds that indirectly inhibit GPX4 activity by GSH depletion (Table 2).

Table 2 Initiators of Ferroptosis.
Erastin and RSL3 are the first ferroptosis inducers identified using high-throughput screening of small molecule libraries (Dolma et al., 2003; Yang and Stockwell, 2008). Erastin binds directly to mitochondrial voltage-dependent anion channel 2 (VDAC2) and causes ROS to be produced by NADPH-dependent pathway of mitochondrial damage (Yagoda et al., 2007). RNA interference mediates knockdown of VDAC2 or VDAC3 and leads to resistance to erastin-induced ferroptosis (Yagoda et al., 2007). In some tumor cells expressing active mutations, erastin induces cell death via the RAS-RAF-MEK pathway. In addition, erastin directly inhibits the activity of system xc-, resulting in the reduction in GSH production and inhibition of GPX4 activity, leading to accelerated production of ROS and ferroptosis (Dixon et al., 2012). Piperazine erastin (PE), a derivative of erastin, has better solubility and stability than erastin in vivo. It inhibits the proliferation of HT1080 cells by inducing ferroptosis (Yang et al., 2014). The carbonyl-containing erastin analog imidazole ketone erastin (IKE) has greater water solubility and metabolic capacity. It has been proved to be a more effective inducer of ferroptosis (Larraufie et al., 2015). It was also found that heme oxygenase-1 (HO-1) accelerates the occurrence of ferroptosis initiated by erastin through supplementing intracellular iron and producing ROS (Kwon et al., 2015). Sulfasalazine, a widely used molecule that treats chronic inflammation in the intestines, joints, and retina, can also inhibit system xc- and induce ferroptosis while inhibiting the NF-ĸB signaling pathway (Gout et al., 2001). Similarly, sorafenib inhibits system xc- and induces ferroptosis by non-kinase targets, enhanced toxicity to hepatocellular carcinoma (HCC) cells, and promoted persistent tumor regression (Louandre et al., 2013; Louandre et al., 2015).
Cell death induced by RSL3 and RSL5 shares a common feature with erastin-mediated ferroptosis. Similar to erastin, the initiation of ferroptosis induced by RSL3 and RSL5 is also dependent on the accumulation of ROS (Yang and Stockwell, 2008). The difference is that RSL3 is a direct inhibitor of GPX4, but not of system xc- (Yang et al., 2014). RSL3 inactivates GPX4 and impedes the degradation of LOOH (Yang et al., 2014). On the other hand, RSL5 acts directly with VDAC2/3 to produce ROS (Yang and Stockwell, 2008).
The stimulant buthionine sulfoximine (BSO) irreversibly inhibits the activity of γ-glutamate cysteine synthetase, which is a rate-limiting enzyme in GSH synthesis in RAS mutant cells. It reduces the synthesis of GSH and further decreases the activity of GPX4 (Yang et al., 2014). Small molecule inducer FINs (Table 2) can be divided into two categories according to their mechanism of action. The first type of FIN, like DPI2, inhibits the activity of GPX4 by depleting GSH; the second type of FIN, such as DPI7, directly inhibits the activity of GPX4 and produces lipid ROS (Yang et al., 2014). Studies show that acetaminophen (Lorincz et al., 2015) and artesunate (Eling et al., 2015) kill cancer cells, not only by autophagy and apoptosis, but also by ferroptosis. In addition, cisplatin increases the level of intracellular ROS in the treatment of tumors to induce specific morphological changes in ferroptosis in cancer cells (Yamaguchi et al., 2013).
The recent discovery of a novel mechanism by which FIN56 and FINO2 trigger cellular death provides new insights on the regulation of ferroptosis (Shimada et al., 2016b; Gaschler et al., 2018a). FINO2 promotes lipid peroxidation by inducing iron oxide and indirectly inactivating GPX4 (Gaschler et al., 2018a). FIN56-induced cell death is accompanied by lipid ROS production and can be reversed by vitamin E and iron chelator, demonstrating that they are potent inducers of ferroptosis (Shimada et al., 2016b). Unlike the previously described pathways, FIN56 stimulates ferroptosis by consuming GPX4 proteins and blocking lipophilic antioxidants, such as coenzyme Q10 (Shimada et al., 2016b).
Many traditional Chinese medicinal natural products provide significant health benefits in the prevention and/or treatment of diseases. Traditional Chinese herbal extracts such as artemisinin (from Artemisia annua) initiate ferroptosis during the process of killing cancer cells (Eling et al., 2015). It induces ferroptosis in tumor cells by increasing ROS levels, decreases GSH levels, interferes with iron metabolism, and increases Fe2+ concentration (Eling et al., 2015). However, its mechanism of action related to signaling pathways has not been elucidated entirely. In 2018, Liu and his collaborators discovered a novel antitumor compound optimized from natural saponin Albiziabioside A. It induced ferroptosis as a p53 activator through the mitochondrial pathway (Wei et al., 2018).
In summary, ferroptosis can be triggered by blocking system xc- with exogenous small molecules, interfering with GPX4, disrupting lipid metabolism balance, and iron homeostasis. Targeted therapy may be achieved by stimulating ferroptosis based on physiological differences between cancer cells and normal cells. Moreover, the rapid development of nanotechnology also provides new directions for the discovery of ferroptosis inducers.
Inhibitors of Ferroptosis
To study the potential role of ferroptosis in vivo, effective and specific small molecular ferroptosis inhibitors have been identified using either high-through put screening or by designing compounds based on the structure-activity relationship. Most ferroptosis inhibitors can be classified into lipophilic radical-trapping antioxidants (e.g. Ferrostatin-1, α-tocopherol), iron chelators (e.g. deferoxamine), and deuterated polyunsaturated fatty acid phospholipids (PUFA-PLs) (Kagan et al., 2017). They have been approved to prevent ferroptosis and regulate normal function of intracellular iron metabolism, increase GSH levels, activate GPX4, or directly inhibit lipid peroxidation (Table 3).
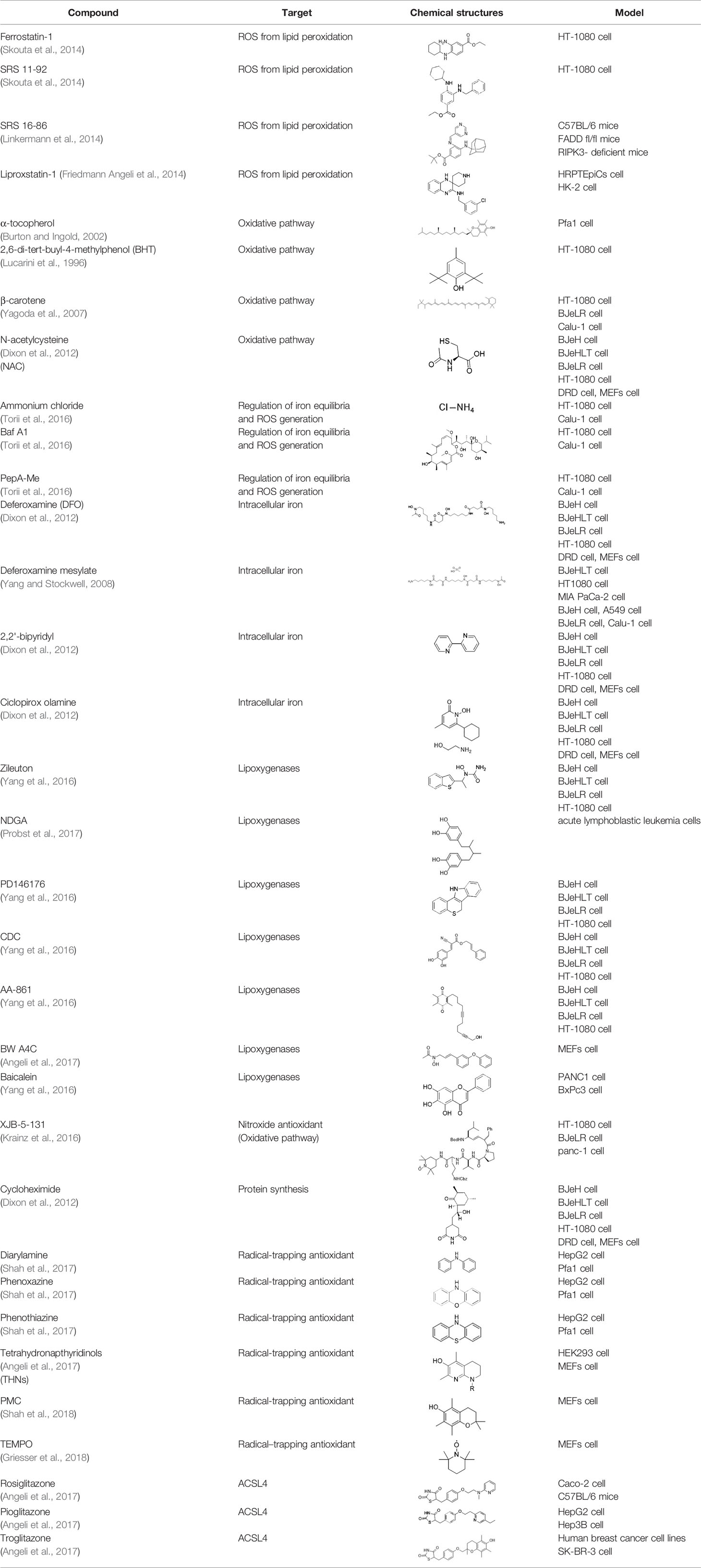
Table 3 Inhibitors of Ferroptosis.
Ferrostatins and liproxstatins act as free radical-trapping antioxidants to inhibit lipid peroxidation associated with ferroptosis (Yang and Stockwell, 2016). The first generation of ferrostatins Ferrostatin-1 (Fer-1 in Table 3) inhibits erastin and RSL3-induced ferroptosis in HT1080 cells. Their activity is dependent on the aromatic amines, which specifically inhibit lipid peroxidation (Yang and Stockwell, 2016). The second (SRS11-92) and third generation (SRS16-86) ferropstatins show significant enhancement cellular metabolism and damage prevention compared to the first generation Fer-1 (Ran et al., 2015). Fer-1 is currently considered as a probe for studying ferroptosis in different environments and as a basic potential drug molecule against lipid peroxidation-mediated tissue damage (Kabiraj et al., 2015).
Liproxstatin-1 (Lip-1) contains amide and sulfonamide subunits. It has good stability and drug absorption distribution in vivo (Hofmans et al., 2016). It can inhibit ferroptosis at low nano molar dose, but it does not interfere with other typical cell death patterns (Hofmans et al., 2016). Similar to ferrostatins and liproxstatins, the antioxidants α-tocopherol (Burton and Ingold, 2002), BHT (Lucarini et al., 1996), N-acetylcystein (NAC) (Dixon et al., 2012), etc., can block ferroptosis by inhibiting the lipid peroxidation pathway.
Free polyunsaturated fatty acids are the substrates of LOXs, which mediate ferroptotic peroxidation (Kuhn et al., 2015). Studies show that inhibitors of ferroptosis, such as tocopherols (Khanna et al., 2003) and flavonoids (Xie et al., 2016b), can also inhibit LOXs activity in some cases. However, not all lipoxygenase inhibitors inhibit ferroptosis. Effective lipoxygenase inhibitors, such as CDC and zileuton, are free radical-trapping antioxidants (Yang et al., 2016). For example, 5-lipoxygenase (5-LOX) inhibitor zileuton inhibits glutamate toxicity and ferroptosis by inhibiting cytoplasmic ROS production in HT22 cells, thereby exerting neuroprotective effects (Liu et al., 2015).
Ferroptosis can also be inhibited by iron chelators such as deferoxamine (DFO), deferoxamine mesylate, and 2, 2'-bipyridyl (Dixon et al., 2012) since they prevent the initiation of lipid peroxidation by inhibiting Fenton chemistry. Natural products have also been screened out for ferroptosis inhibitors. In 2016, Tang's lab found out baicalein was an effective ferroptosis inhibitor in pancreatic cancer cells (Xie et al., 2016b) functioned by reducing ROS, regulating iron homeostasis, chelating Fe2+, and protecting GPx4 (Xie et al., 2016b).
Moreover, inhibition of heat shock factor-1 (HSF-1) dependent heat shock protein beta 1 (HSPB1) expression can also inhibit erastin-induced ferroptosis (Sun et al., 2015). In addition, Nrf2 plays an important role in the anti-ferroptotic process in liver cancer cells. Upregulation of Nrf2 initiates transcription of antioxidant protein genes and iron metabolism protein genes, thereby inhibiting ferroptosis (Sun et al., 2016). Fms-like tyrosine kinase 3 (Flt3) and phosphoinositide 3-kinase α (PI3Kα) inhibits neuronal ferroptotic cell death, particularly in cortical neurons, providing neuroprotection (Kang et al., 2014).
Other than the screened compounds, research has also focused on designing synthetic ferroptosis inhibitors in order to provide potential health benefits to prevent and/or ease the symptom of diseases that are related to ferroptosis. Several series of effective ferroptosis inhibitors have been developed in recent years (Zilka et al., 2017). Radical-trapping antioxidants, including tetrahydronapthyridinols (THNs), phenoxazine, phenothiazine, and diarylamine (Table 3), have been shown to effectively inhibit ferroptosis in cellular models, with some exhibiting better activity than Fer-1 and Lip-1. Recently, we have designed and synthesized a series of chalcone derivatives to inhibit amyloid-β aggregation and ferroptosis at the same time in cellular models as a potential preventive and/or therapeutic agent to Alzheimer's disease (Cong et al., 2019). Together, these findings suggest the possibility for introducing multi-functional ferroptosis inhibitors to prevent and/or treat diseases that are closely associated with ferroptosis.
Potential Role of the Initiators and Inhibitors of Ferroptosis in the Treatment of Various Diseases
Neurodegenerative Diseases
Neurodegenerative diseases, such as Alzheimer's disease (AD) and Parkinson's disease (PD), are known to be associated with dysregulation of iron homeostasis and excessive ROS in the brain. Before the concept of ferroptosis, neurodegenerative diseases were thought to be caused by apoptosis (Ward et al., 2014). With the definition of ferroptosis in 2012 and iron-dependent oxidative stress as a significant marker of cellular ferroptosis, there is an increasing amount of research supporting the idea that ferroptosis is inextricably linked to neurodegenerative diseases.
Alzheimer's disease is one of the most common causes of dementia in aging individuals. It is characterized by progressive memory impairment and cognitive dysfunction. The main pathological features of AD are extracellular β-amyloid (Aβ) deposition and neurofibrillary tangles caused by abnormal phosphorylation of intracellular Tau protein. There is evidence supporting that oxidative stress and iron metabolism disorder are associated with the progression of AD (Masaldan et al., 2019).
Ferroptosis is characterized by an accumulation of lipid peroxidation and dysregulation of iron, which are precisely the hallmarks of Alzheimer's disease (Pratico and Sung, 2004; Castellani et al., 2007). Therefore, regulating ferroptosis has become a new direction for the potential treatment of Alzheimer's disease. Iron chelators also prevent the development of AD by maintaining levels of hypoxia inducible factor-1 alpha (HIF-1α) in the nerve and inhibiting neuronal death, which provides a novel neuroprotective mechanism against AD (Ashok et al., 2017). In addition to the treatment of AD by a single iron chelator, multi-target drugs by chelating Fe (II) combined with scavenging free radicals may also be effective. As a multifunctional non-toxic and brain-permeable iron chelator, M30 not only attenuates Tau phosphorylation but also activates the HIF-1α signaling pathway, showing great potential in the prevention and treatment of AD (Kupershmidt et al., 2012). Alpha-Lipoic acid not only regulates the redistribution of iron via iron chelation, but also acts as a direct free radical scavenger and an indirect antioxidant which can inhibit ferroptosis to alleviate the progression of AD (Zhang et al., 2018a). According to our understanding, the radical-trapping antioxidant α-tocopherol and the iron chelator DFO entered clinical testing to treat AD before they were discovered as inhibitors of ferroptosis (Mclachlan et al., 1991; Dysken et al., 2014). Clinical trials proved that patients with mild to moderate AD who received 2000 IU/day α-tocopherol showed a slower decline in cognitive function compared to the placebo group (Dysken et al., 2014).
Parkinson's disease is the second most common neurodegenerative disease and it is characterized by the loss of dopaminergic neurons in the substantia nigra and the formation of cytoplasmic eosinophilic inclusion bodies, i.e., Lewy bodies (Hornykiewicz, 2008). It is currently believed that lipid peroxidation of dopaminergic neurons in the substantia nigra pars compacta is important in the pathogenesis of PD (Burbulla et al., 2017). Some pathological features found in PD patients are elevated levels of free iron in the substantia nigra neurons, lipid peroxide production, and accumulation of ROS, are closely related to ferroptosis. In mammalian models, several studies have shown that iron chelators can protect against neuronal damage in PD (Kaur et al., 2003; Ayton et al., 2013; Lei et al., 2015). For example, in 2014, clinical studies of the deferiprone (DFP), an iron chelator, has shown that iron-removing treatment can alleviate the motor symptoms of early PD patients by reducing iron levels in patients (Devos et al., 2014). Iron chelator VK28, or its derivative, M30, which can penetrate the blood-brain barrier, provide significant neuroprotective effects in PD mouse models (Avramovich-Tirosh et al., 2010). In addition, genetic studies of PD have shown that PD marker α-synuclein, encoded by the SNCA gene, connect strongly with ferrous and ferric ions (Peng et al., 2010; Febbraro et al., 2012). These two forms of iron ions have been shown to accelerate the aggregation of α-syn in vitro, and the iron chelators can significantly inhibit this phenomenon (Hashimoto et al., 1999; Davies et al., 2011). For instance, deferoxamine (DFO) can be used to treat HEK293 cells, confirming that iron deficiency could inhibit the expression of α-synuclein and prevent PD-like changes in cells (Febbraro et al., 2012).
Recently, Huntington's disease (HD), a hereditary neurodegenerative disorder, has also been shown to be inextricably linked to ferroptosis. Similar to Alzheimer's and Parkinson's diseases, Huntington's disease also associates with abnormal levels in lipid peroxidation, GSH metabolism, and iron accumulation (Paul et al., 2014). Increasing lipid peroxidation was detected in cortical striatal brain sections (Skouta et al., 2014) and cerebrospinal fluid (Reddy and Shirendeb, 2012) of the mN90Q73 HD mouse model. 3-nitropropionic acid (3-NP)-induced HD mice display a decrease in GSH and GSH-S-transferase in the striatum, cortex, and hippocampus (Kumar et al., 2010). Moreover, increasing iron supplementation reduces the striatum volume and contributes to neurodegeneration (Van Bergen et al., 2016). In contrast, intracerebroventricular administration of deferoxamine (DFO) improves striatal pathology and motor phenotype in R6/2HD mice (Chen et al., 2013).
Cell stress response to ROS includes the activation of pro-survival pathways as well as the production of molecules endowed with antioxidant and anti-apoptotic activities, which is under the control of protective genes called vitagenes (Calabrese et al., 2009). Vitagene network includes members of the heat shock protein (HSP) family, such as heme oxygenase-1 (HO-1), Hsp70, sirtuins (Sirt-1), and thioredoxin/thioredoxin reductase (Trx/TrxR) (Calabrese et al., 2009). Heat shock factors (HSFs) are the master transcriptional factors that regulate the inducible synthesis of these HSPs during stress (Wu, 1995). In addition to HSF, some of the vitagenes are also upregulated as part of the phase 2 responses, a cytoprotective response that protects against various electrophiles and oxidants (Calabrese et al., 2007). Vitagene network including heme oxygenase 1, thioredoxin, and thioredoxin reductase can be upregulated by the transcription factor Nrf2 (Calabrese et al., 2010b). It has been found that GPX4, heat shock protein beta-1(HSPB1), and Nrf2 function as negative regulators of ferroptosis by limiting ROS production and reducing cellular iron uptake, respectively (Xie et al., 2016a). The protection effect of HSPs in ferroptosis has been elucidated in the past few years. In 2015, Tang's group found that inhibition of HSF-1-dependent HSPB1 expression increased ferroptosis, whereas overexpression of HSPB1 inhibited erastin-induced ferroptosis (Sun et al., 2015). They also found that the PKC-mediated HSPB1 phosphorylation in Hela cells was required for conferring resistance to erastin-induced ferroptosis, possibly through regulating iron-mediated lipid ROS production (Sun et al., 2015). In 2017, Tang's group showed that heat shock 70-kDa protein 5 (HSPA5) negatively regulated ferroptosis in human pancreatic ductal adenocarcinoma (PDAC) cells through the HSPA5-GPX4 pathway (Zhu et al., 2017). Mechanistically, activating transcription factor 4 (ATF4) resulted in the induction of HSPA5, which in turn bound glutathione peroxidase 4 (GPX4) and protected against GPX4 protein degradation and subsequent lipid peroxidation (Zhu et al., 2017). Due to the known neuron protection effect of HSPs in vitro, it provides a potential therapeutic strategy for acute injury in the nervous system.
In conclusion, current studies on the role of ferroptosis in neurodegenerative diseases mainly concentrate on studying whether ferroptosis inhibitors could slow disease progression, and mostly use animal models. Most of the experimental studies in animals have shown that effective inhibition of ferroptosis provided potential treatment. However, most clinical trials on administering iron chelators and antioxidants showed only moderate treatment effect. These results lead us to think that iron chelators and antioxidants are not sufficient to provide effective treatment. Potential molecules that regulate ferroptosis through other signaling pathways have yet to be further explored for their potential to treat neurodegenerative diseases and could provide better treatment.
Cancer
Most cancer cells exhibit elevated levels of ROS (Hassannia et al., 2019). They rely on the level of glutathione to maintain their survival and proliferation (Di Meco et al., 2017). Raising the level of ROS to a cytotoxic level can eliminate cancer cells. Endogenous cysteine produced under elevated ROS levels is not sufficient to synthesize sufficient glutathione. Therefore, extracellular cysteines need to be obtained by the reverse transporter system xc- (Dixon et al., 2014). Erastin (Dixon et al., 2012), sorafenib (Louandre et al., 2013), and sulfasalazine (Gout et al., 2001) have been explored as inhibitors of system xc- to stimulate ferroptosis in cancer cells.
Ferroptosis was first discovered in tumor cells when studying RAS mutations. In 2014, Stockwell's group (Yang et al., 2014) studied the possibility of using ferroptosis inducers for RAS mutant cancer cells by measuring erastin from 117 cancer cell lines from different tissues. Results showed that kidney cancer cells (RCCs) and leukemia (DLBCL) are more sensitive to erastin than other cancer cells, like those found in lung and ovarian cancers. They also demonstrated that RAS mutations are not associated with the efficacy of erastin. In addition, erastin can enhance the efficacy of chemotherapy drugs, such as temozolomide (Chen et al., 2015) and cisplatin (Yamaguchi et al., 2013), to treat specific cancer cells. The analogs of erastin, piperazine erastin (PE) (Yang et al., 2014), and imidazole ketone erastin (IKE) (Larraufie et al., 2015), have also been used as in vivo probes for tumor susceptibility to ferroptosis.
In 2015, Jian and colleagues (2015) elucidated a new role for P53 in the mediation of tumor suppression through ferroptosis. The authors demonstrated that P53 inhibits the uptake of cystine through the inhibition of SLC7A11 gene expression, resulting in ferroptosis. This conclusion also indicates that P53 wild-type tumors can be treated with ferroptosis inducers that inhibit system xc-. Interestingly, SLC7A11 gene-deficient mice develop normally and healthy (Sato et al., 2005), suggesting that system xc- targeted drugs with high cancer cell selectivity have few side effects in preclinical studies.
In addition, it has been found that the ACSL4 enzyme is preferentially expressed in a subset of triple-negative breast cancer cells (TNBC) and that expression of the ACSL4 enzyme is closely related to stimulating ferroptosis (Doll et al., 2017). Since triple-negative breast cancer is difficult to control, ferroptosis initiation introduces a new method for the treatment of cancer cells with ACSL4 expression (Doll et al., 2017).
In recent years, with the rapid development of nanotechnology, studies have shown that tumor xenografts in mice that use high-dose multiple intravenous injections of polyethylene nanoparticles coated with polyethylene glycol exhibit a slower growth of cancer and even signs of regression (Szwed et al., 2019). However, this phenomenon is reversed by ferroptosis inhibitor liprostatin-1 (Kim et al., 2016). This suggests that ferroptosis could have great potential for targeted cancer treatment through ultra-small silica nanoparticles.
Currently, FDA-approved drugs sorafenib (Louandre et al., 2013), sulfasalazine (Gout et al., 2001), artesunate (Eling et al., 2015), and lanperisone (Shaw et al., 2011) have been shown to induce ferroptosis in certain cancer cells. Among them, sorafenib-induced cellular ferroptosis had two different mechanisms: (1) inhibition of system xc- mediated cystine input and triggering endoplasmic reticulum stress (Dixon et al., 2014); (2) reduction of Rb protein, which is best known for its regulatory role in cell proliferation and its key role at the G1/S checkpoint accompanied by increased ROS in mitochondria (Louandre et al., 2015). However, further research is necessary to determine whether endoplasmic reticulum stress is the key initiator of cancer cell death after treatment with sorafenib. Further studies are needed to elucidate the mechanism of Rb protein production.
Ischemia Reperfusion Injury
Ferroptosis inhibitors have been used to treat a variety of kidney injuries, such as ischemia-reperfusion and oxalic acid-induced kidney damage (Gascon et al., 2016), rhabdomyolysis (Bosch et al., 2009), and acute renal failure (ARF) (Bosch et al., 2009). Ferroptosis inhibitor Fer-1 prevents cell death in an in vitro model of rhabdomyolysis-induced acute kidney injury (Skouta et al., 2014). In an in vivo model of renal ischemia-reperfusion injury, SRS16-86, a third generation ferrostatin with increased plasma and metabolic stability, protected renal function and prolonged survival after ischemia-reperfusion injury (Linkermann et al., 2014). Ferroptosis inhibitor Lip-1 can rescue acute renal failure and prolong life in mice due to GPX4 deletion (Friedmann Angeli et al., 2014). In addition, thiazolidinediones (TZDs) inhibit acyl-CoA synthase 4 and partially reduce the mortality of induced GPX4 knockout mice (Doll et al., 2017). These results reinforce the sensitivity of kidney tissue to ferroptosis and demonstrate the value of ferroptosis inhibitors in the treatment of renal damage (Sancho-Martinez et al., 2015).
When the isolated cardiac ischemia-reperfusion model of wild-type mice is treated with glutaminolysis inhibitor compound 968 and iron chelator DFO, the cardiac function is significantly enhanced when compared to the control group (Gao et al., 2015). This indicates that heart damage caused by ischemia-reperfusion can be reduced by inhibiting glutaminolysis, which is the essential component in ferroptosis. Fang and colleagues (2019) used a variety of cell death inhibitor treatments and cell death pathway-related knockout mouse models to find that only the ferroptosis-specific inhibitor Fer-1 could significantly reduce the cardiotoxicity caused by DOX, an anticancer drug. It was revealed that ferroptosis was involved in the mechanism of myocardial injury. The researchers also found the presence of ferroptosis in a mouse model of myocardial ischemia-reperfusion injury. The administration of ferroptosis inhibitors to block ferroptosis could significantly reduce acute and chronic heart damage caused by ischemia-reperfusion. These results provide new ideas and strategies for clinical heart diseases such as myocardial infarction.
Excessive acetaminophen is the most common cause of acute liver failure. Acetaminophen has been shown to induce ferroptosis in primary hepatocytes, while ferroptosis inhibitors such as Fer-1 inhibit acetaminophen-induced cell death (Lorincz et al., 2015). Moreover, Lip-1 repairs liver damage caused by ischemia-reperfusion (Friedmann Angeli et al., 2014). Collectively, these findings show the importance of ferroptosis in ischemia reperfusion injury and support the potential therapeutic application of ferroptosis inhibitors that target pathways involved in ferroptosis execution.
Other Diseases
Excessive accumulation of iron ions causes lipid peroxidation and tissue damage, leading to atherosclerosis and diabetes (Wu and Chen, 2015). Studies have shown that iron overload in the heart caused myocardial dysfunction and metabolic damage that ultimately led to heart disease (Dixon et al., 2012). In GPX4-deficient T cells, the cell membrane rapidly accumulates lipid peroxides, which induces ferroptosis. Instead, inhibiting ferroptosis promotes the survival and expansion of T cells and protects the immune function of T cells (Matsushita et al., 2015). Research also shows that ferroptosis participates in keratinocyte death due to GSH loss, and high doses of vitamin E can inhibit ferroptosis of skin keratinocytes and reduce skin damage (Wu and Chen, 2015). Recent studies show that the decreased expression of frataxin, a key protein of Friedreich's ataxia (FRDA), characterized by puberty onset, loss of tendon reflexes, and deep sensory loss, is associated with mitochondrial dysfunction, mitochondrial iron accumulation, and increased oxidative stress. Ferroptosis inhibitor SRS11-92 reduces cell death caused by FRDA (Cotticelli et al., 2019).
For a long time, researchers believed that the secondary damage caused by intracranial hemorrhage was caused by the random spread of iron ions. However, the discovery of ferroptosis has led more researchers to wonder whether the damage caused by intracranial hemorrhage is the induction of ferroptosis in cells. Alim and colleagues (2019) not only validated in hemorrhagic stroke models that stroke induces ferroptosis to some extent, but also found that the expression of the GPX4 can be driven by delivering a single dose of selenium to the brain, thereby protecting neurons and improving the behavior of mice after stroke (Green, 2018; Ingold et al., 2018). These findings provide important guidance in the nutritional care and follow-up treatment of cerebral hemorrhage.
In summary, the survival of cells is an important part of the body's normal metabolism. It is clear that ferroptosis has an intimate relationship with pathological cell death. Effective alleviation or prevention of the progression of the disease or the clinical symptoms in mice or rat models can be achieved by administering ferroptosis inhibitors or inducers. Emerging evidence also suggests that ferroptosis initiation has a potential tumor inhibitory function, which could clear tumor cells that lack key nutrients in the environment and cells that are damaged by infection (Yang et al., 2014). In-depth study and clarification of the pathophysiological mechanism of ferroptosis in related diseases will provide new ideas for discovering potential drug targets and clinical prevention methods.
Summary
Different from other cell death patterns induced by cytosolic or mitochondrial reactive oxygen species, ferroptosis is defined as a form of programmed cell death involving the accumulation of lipid hydroperoxides which can be suppressed by iron chelators and lipophilic antioxidants. It is characterized by the loss of activity of enzyme GPX4, which results in the accumulation of lethal lipid hydroperoxides. Topics on signaling pathways involved in ferroptosis, the role of ferroptosis inhibitors and initiators as well as their mechanism of action, the role of ferroptosis in disease, and the difference between ferroptosis and other cell death types involving excessive reactive oxygen species, have been widely studied in the past few years.
The difference between ferroptosis and other cell death pathways caused by excessive oxidative damage in vivo has not been elucidated yet. Recent studies indicate that ferroptosis shares a few common characteristics with several types of cell death pathways, like oxytosis (Neitemeier et al., 2017) and ferritinophagy (Zhang et al., 2018b), with a few differences in protein-signaling pathways. In cellular models, the morphology and characteristics of cells and mitochondria can be explored to provide evidence to distinguish ferroptosis from other types of cell death. However, it is challenging to do so in the diseased animal models. In addition, different cell death patterns might happen and contribute to the pathology of disease at the same time. For example, it is already known that both ferroptosis and necroptosis happen after ischemic injury (Linkermann et al., 2014) and both apoptosis and ferroptosis occur after traumatic brain damage (Raghupathi et al., 2000; Magtanong and Dixon, 2018). Therefore, detection of markers of one cell death type alone cannot indicate the lack of other cell death types in diseased model.
It is known that ROS in the biological system has hormesis feature, which means although excess ROS is harmful to keep the redox balance, small amounts of ROS, such as mitochondrial superoxide and hydrogen peroxide, play important roles in a range of cellular functions, and can also activate signaling pathways that promote cell survival and disease resistance (Calabrese et al., 2008; Mattson, 2008; Calabrese et al., 2010a). An example of cellular hormesis mediated by ROS is the study showing that oxidative stress can stimulate angiogenesis in the brain, a process that is very important in restoring blood flow to neurons after a stroke (Hougaard et al., 2013; Wei et al., 2013). To date, it is not yet clear whether ferroptosis has hormesis feature. However, some known ferroptosis inhibitors, such as the natural products curcumin and baicalein, have hormesis feature (Wang et al., 2018; Concetta Scuto et al., 2019). At low concentration, curcumin and baicalein inhibit ferroptosis in cellular models (Concetta Scuto et al., 2019; Li et al., 2019). However, at concentration above a threshold, they induced toxicities. Therefore, special precaution is necessary when applying ferroptosis inhibitors as potential protective agents.
In addition, the interpretation of cell-based data and animal-based data related to the link between ferroptosis and neurodegenerative diseases must also be carefully considered since the in vitro cellular experimental conditions are quite different from in vivo conditions. To date, there is no clinical trial using ferroptosis inhibitors or initiators to treat degenerative diseases (see www.clinicaltrials.gov for details). Further research on the mechanism through which lipid peroxidation induces ferroptotic cell death is necessary.
Author Contributions
The original idea of this study was from BL. CH and YL reviewed the literature and contributed equally to the manuscript writing and editing. BL instructed the whole manuscript formation. RD contributed to the manuscript editing and proofreading. NI and WS contributed to the manuscript revising.
Funding
The authors were supported by the grants from the Fund of Innovation Center of Radiation Application, China (KFZC2018040208), the National Natural Science Foundation of China (31971388), and the Beijing Institute of Technology Research Fund Program for Young Scholars.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to express thanks for the support from the other members in our group and the support from the Beijing Institute of Technology.
References
Aachoui, Y., Leaf, I. A., Hagar, J. A., Fontana, M. F., Campos, C. G., Zak, D. E., et al. (2013). Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978. doi: 10.1126/science.1230751
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262–1279 e1225. doi: 10.1016/j.cell.2019.03.032
Angeli, J. P. F., Shah, R., Pratt, D. A., Conrad, M. (2017). Ferroptosis inhibition: Mechanisms and opportunities. Trends Pharmacol. Sci. 38, 489–498. doi: 10.1016/j.tips.2017.02.005
Ashok, B. S., Ajith, T. A., Sivanesan, S. (2017). Hypoxia-inducible factors as neuroprotective agent in Alzheimer's disease. Clin. Exp. Pharmacol. Physiol. 44, 327–334. doi: 10.1111/1440-1681.12717
Avramovich-Tirosh, Y., Bar-Am, O., Amit, T., Youdim, M. B. H., Weinreb, O. (2010). Up-regulation of hypoxia-inducible factor (HIF)-1 alpha and HIF-target genes in cortical neurons by the novel multifunctional iron chelator anti-alzheimer drug, M30. Curr. Alzheimer Res. 7, 300–306. doi: 10.2174/156720510791162403
Ayton, S., Lei, P., Duce, J. A., Wong, B. X. W., Sedjahtera, A., Adlard, P. A., et al. (2013). Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann. Neurol. 73, 554–559. doi: 10.1002/ana.23817
Bersuker, K., Hendricks, J., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi: 10.1038/s41586-019-1705-2
Bock, F. J., Tait, S. W. G. (2019). Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100. doi: 10.1038/s41580-019-0173-8
Bosch, X., Poch, E., Grau, J. (2009). Current concepts: Rhabdomyolysis and acute kidney injury. N Engl. J. Med. 361, 62–72. doi: 10.1056/NEJMra0801327
Burbulla, L. F., Song, P. P., Mazzulli, J. R., Zampese, E., Wong, Y. C., Jeon, S., et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science 357, 1255–1261. doi: 10.1126/science.aam9080
Burton, G. W., Ingold, K. U. (2002). Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function. Acc. Chem. Res. 19, 194–201. doi: 10.1021/ar00127a001
Calabrese, V., Mancuso, C., Sapienza, M., Puleo, E., Calafato, S., Cornelius, C., et al. (2007). Oxidative stress and cellular stress response in diabetic nephropathy. Cell Stress Chaperones 12, 299–306. doi: 10.1379/CSC-270.1
Calabrese, V., Calafato, S., Puleo, E., Cornelius, C., Sapienza, M., Morganti, P., et al. (2008). Redox regulation of cellular stress response by ferulic acid ethyl ester in human dermal fibroblasts: role of vitagenes. Clin. Dermatol. 26, 358–363. doi: 10.1016/j.clindermatol.2008.01.005
Calabrese, V., Cornelius, C., Mancuso, C., Barone, E., Calafato, S., Bates, T., et al. (2009). Vitagenes, dietary antioxidants and neuroprotection in neurodegenerative diseases. Front. Bioscience-Landmark 14, 376–397. doi: 10.2741/3250
Calabrese, V., Cornelius, C., Dinkova-Kostova, A. T., Calabrese, E. J., Mattson, M. P. (2010a). Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signaling 13, 1763–1811. doi: 10.1089/ars.2009.3074
Calabrese, V., Cornelius, C., Maiolino, L., Luca, M., Chiaramonte, R., Toscano, M. A., et al. (2010b). Oxidative stress, redox homeostasis and cellular stress response in Meniere's disease: role of vitagenes. Neurochem. Res. 35, 2208–2217. doi: 10.1007/s11064-010-0304-2
Cao, J. Y., Dixon, S. J. (2016). Mechanisms of ferroptosis. Cell Mol. Life Sci. 73, 2195–2209. doi: 10.1007/s00018-016-2194-1
Castellani, R. J., Moreira, P. I., Liu, G., Dobson, J., Perry, G., Smith, M. A., et al. (2007). Iron: the redox-active center of oxidative stress in Alzheimer disease. Neurochem. Res. 32, 1640–1645. doi: 10.1007/s11064-007-9360-7
Chen, J., Marks, E., Lai, B., Zhang, Z., Duce, J. A., Lam, L. Q., et al. (2013). Iron accumulates in Huntington's disease neurons: protection by deferoxamine. PloS One 8, e77023. doi: 10.1371/journal.pone.0077023
Chen, L., Li, X., Liu, L., Yu, B., Xue, Y., Liu, Y. (2015). Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol. Rep. 33, 1465–1474. doi: 10.3892/or.2015.3712
Concetta Scuto, M., Mancuso, C., Tomasello, B., Laura Ontario, M., Cavallaro, A., Frasca, F., et al. (2019). Curcumin, hormesis and the nervous system. Nutrients 11, 2417. doi: 10.3390/nu11102417
Cong, L., Dong, X., Wang, Y., Deng, Y., Li, B., Dai, R. (2019). On the role of synthesized hydroxylated chalcones as dual functional amyloid-beta aggregation and ferroptosis inhibitors for potential treatment of Alzheimer's disease. Eur. J. Med. Chem. 166, 11–21. doi: 10.1016/j.ejmech.2019.01.039
Cotticelli, M. G., Xia, S., Lin, D., Lee, T., Terrab, L., Wipf, P., et al. (2019). Ferroptosis as a novel therapeutic target for friedreich's ataxia. J. Pharmacol. Exp. Ther. 369, 47–54. doi: 10.1124/jpet.118.252759
Davies, P., Moualla, D., Brown, D. R. (2011). Alpha-synuclein is a cellular ferrireductase. PloS One 6, e15814. doi: 10.1371/journal.pone.0015814
Devos, D., Moreau, C., Devedjian, J. C., Kluza, J., Petrault, M., Laloux, C., et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxid. Redox Signal 21, 195–210. doi: 10.1089/ars.2013.5593
Di Meco, A., Li, J. G., Blass, B. E., Abou-Gharbia, M., Lauretti, E., Pratico, D. (2017). 12/15-lipoxygenase inhibition reverses cognitive impairment, brain amyloidosis, and Tau pathology by stimulating autophagy in aged triple transgenic mice. Biol. Psychiatry 81, 92–100. doi: 10.1016/j.biopsych.2016.05.023
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., Patel, D. N., Welsch, M., Skouta, R., Lee, E. D., Hayano, M., et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. doi: 10.7554/eLife.02523
Dixon, S. J., Winter, G. E., Musavi, L. S., Lee, E. D., Snijder, B., Rebsamen, M., et al. (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609. doi: 10.1021/acschembio.5b00245
Doll, S., Proneth, B., Tyurina, Y., Panzilius, E., Kobayashi, S., Ingoid, I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. doi: 10.1038/NCHEMBIO.2239
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., Da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. doi: 10.1038/s41586-019-1707-0
Dolma, S., Lessnick, S., Hahn, W., Stockwell, B. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. doi: 10.1016/S1535-6108(03)00050-3
Dysken, M. W., Sano, M., Asthana, S., Vertrees, J. E., Pallaki, M., Llorente, M., et al. (2014). Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 311, 33–44. doi: 10.1001/jama.2013.282834
Eling, N., Reuter, L., Hazin, J., Hamacher-Brady, A., Brady, N. R. (2015). Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2, 517–532. doi: 10.18632/oncoscience.160
Fan, Z., Wirth, A. K., Chen, D., Wruck, C. J., Rauh, M., Buchfelder, M., et al. (2017). Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371. doi: 10.1038/oncsis.2017.65
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A 116, 2672–2680. doi: 10.1073/pnas.1821022116
Febbraro, F., Giorgi, M., Caldarola, S., Loreni, F., Romero-Ramos, M. (2012). alpha-Synuclein expression is modulated at the translational level by iron. Neuroreport 23, 576–580. doi: 10.1097/WNR.0b013e328354a1f0
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator GPX4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi: 10.1038/ncb3064
Galaris, D., Barbouti, A., Pantopoulos, K. (2019). Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 1866, 118535. doi: 10.1016/j.bbamcr.2019.118535
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., et al. (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120. doi: 10.1038/cdd.2011.96
Galluzzi, L., Bravo-San Pedro, J. M., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., et al. (2015). Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 22, 58–73. doi: 10.1038/cdd.2014.137
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Gao, M., Monian, P., Quadri, N., Ramasamy, R., Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308. doi: 10.1016/j.molcel.2015.06.011
Gao, M., Monian, P., Pan, Q., Zhang, W., Xiang, J., Jiang, X. (2016). Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032. doi: 10.1038/cr.2016.95
Gao, M., Yi, J., Zhu, J., Minikes, A. M., Monian, P., Thompson, C. B., et al. (2019). Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363 e353. doi: 10.1016/j.molcel.2018.10.042
Gaschler, M. M., Andia, A. A., Liu, H., Csuka, J. M., Hurlocker, B., Vaiana, C. A., et al. (2018a). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 14, 507–515. doi: 10.1038/s41589-018-0031-6
Gaschler, M. M., Hu, F., Feng, H., Linkermann, A., Min, W., Stockwell, B. R. (2018b). Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem. Biol. 13, 1013–1020. doi: 10.1021/acschembio.8b00199
Gascon, S., Murenu, E., Masserdotti, G., Ortega, F., Russo, G. L., Petrik, D., et al. (2016). Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell 18, 396–409. doi: 10.1016/j.stem.2015.12.003
Gout, P., Buckley, A., Simms, C., Bruchovsky, N. (2001). Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)(-) cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640. doi: 10.1038/sj.leu.2402238
Griesser, M., Shah, R., Van Kessel, A. T., Zilka, O., Haidasz, E. A., Pratt, D. A. (2018). The catalytic reaction of nitroxides with peroxyl radicals and its relevance to their cytoprotective properties. J. Am. Chem. Soc. 140, 3798–3808. doi: 10.1021/jacs.8b00998
Hashimoto, M., Hsu, L. J., Xia, Y., Takeda, A., Sisk, A., Sundsmo, M., et al. (1999). Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 10, 717–721. doi: 10.1097/00001756-199903170-00011
Hassannia, B., Vandenabeele, P., Vanden Berghe, T. (2019). Targeting ferroptosis to iron out cancer. Cancer Cell 35, 830–849. doi: 10.1016/j.ccell.2019.04.002
Hayano, M., Yang, W. S., Corn, C. K., Pagano, N. C., Stockwell, B. R. (2016). Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 23, 270–278. doi: 10.1038/cdd.2015.93
Hayes, J. D., Dinkova-Kostova, A. T. (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218. doi: 10.1016/j.tibs.2014.02.002
Hirayama, T., Miki, A., Nagasawa, H. (2019). Organelle-specific analysis of labile Fe(ii) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics 11, 111–117. doi: 10.1039/c8mt00212f
Hofmans, S., Vanden Berghe, T., Devisscher, L., Hassannia, B., Lyssens, S., Joossens, J., et al. (2016). Novel Ferroptosis Inhibitors with Improved Potency and ADME Properties. J. Med. Chem. 59, 2041–2053. doi: 10.1021/acs.jmedchem.5b01641
Hong, S. H., Lee, D. H., Lee, Y. S., Jo, M. J., Jeong, Y. A., Kwon, W. T., et al. (2017). Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 8, 115164–115178. doi: 10.18632/oncotarget.23046
Hornykiewicz, O. (2008). Basic research on dopamine in Parkinson's disease and the discovery of the nigrostriatal dopamine pathway: the view of an eyewitness. Neurodegener. Dis. 5, 114–117. doi: 10.1159/000113678
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, ,. H. J., 3rd, et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi: 10.1080/15548627.2016.1187366
Hougaard, K. D., Hjort, N., Zeidler, D., Sorensen, L., Norgaard, A., Thomsen, R. B., et al. (2013). Remote ischemic perconditioning in thrombolysed stroke patients: Randomized study of activating endogenous neuroprotection - design and MRI measurements. Int. J. Stroke 8, 141–146. doi: 10.1111/j.1747-4949.2012.00786.x
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2018). Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422 e421. doi: 10.1016/j.cell.2017.11.048
Jiang, L., Hickman, J. H., Wang, S. J., Gu, W. (2015a). Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 14, 2881–2885. doi: 10.1080/15384101.2015.1068479
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015b). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. doi: 10.1038/nature14344
Kabiraj, P., Valenzuela, C. A., Marin, J. E., Ramirez, D. A., Mendez, L., Hwang, M. S., et al. (2015). The neuroprotective role of ferrostatin-1 under rotenone-induced oxidative stress in dopaminergic neuroblastoma cells. Protein J. 34, 349–358. doi: 10.1007/s10930-015-9629-7
Kagan, V., Mao, G., Qu, F., Angeli, J., Doll, S., St Croix, C., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi: 10.1038/NCHEMBIO.2238
Kang, R., Tang, D. (2017). Autophagy and ferroptosis - what's the connection? Curr. Pathobiol Rep. 5, 153–159. doi: 10.1007/s40139-017-0139-5
Kang, Y., Tiziani, S., Park, G., Kaul, M., Paternostro, G. (2014). Cellular protection using Flt3 and PI3Kalpha inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat. Commun. 5, 3672. doi: 10.1038/ncomms4672
Kaur, D., Yantiri, F., Rajagopalan, S., Kumar, J., Mo, J., Boonplueang, R., et al. (2003). Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: A novel therapy for Parkinson's disease. Neuron 37, 899–909. doi: 10.1016/S0896-6273(03)00126-0
Kayagaki, N., Stowe, I. B., Lee, B. L., O'rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Khanna, S., Roy, S., Ryu, H., Bahadduri, P., Swaan, P. W., Ratan, R. R., et al. (2003). Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J. Biol. Chem. 278, 43508–43515. doi: 10.1074/jbc.M307075200
Kim, S. E., Zhang, L., Ma, K., Riegman, M., Chen, F., Ingold, I., et al. (2016). Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 11, 977–985. doi: 10.1038/nnano.2016.164
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–U217. doi: 10.1038/ncb2021
Krainz, T., Gaschler, M. M., Lim, C., Sacher, J. R., Stockwell, B. R., Wipf, P. (2016). A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent. Sci. 2, 653–659. doi: 10.1021/acscentsci.6b00199
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., et al. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11. doi: 10.1038/cdd.2008.150
Kuhn, H., Banthiya, S., Van Leyen, K. (2015). Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 1851, 308–330. doi: 10.1016/j.bbalip.2014.10.002
Kumar, P., Kalonia, H., Kumar, A. (2010). Nitric oxide mechanism in the protective effect of antidepressants against 3-nitropropionic acid-induced cognitive deficit, glutathione and mitochondrial alterations in animal model of Huntington's disease. Behav. Pharmacol. 21, 217–230. doi: 10.1097/FBP.0b013e32833a5bf4
Kupershmidt, L., Amit, T., Bar-Am, O., Weinreb, O., Youdim, M. B. H. (2012). Multi-target, neuroprotective and neurorestorative M30 improves cognitive impairment and reduces Alzheimer's-like neuropathology and age-related alterations in mice. Mol. Neurobiol. 46, 217–220. doi: 10.1007/s12035-012-8304-7
Kwon, M., Park, E., Lee, S., Chung, S. (2015). Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6, 24393–24403. doi: 10.18632/oncotarget.5162
Larraufie, M. H., Yang, W. S., Jiang, E., Thomas, A. G., Slusher, B. S., Stockwell, B. R. (2015). Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg. Med. Chem. Lett. 25, 4787–4792. doi: 10.1016/j.bmcl.2015.07.018
Lee, Y. S., Lee, D. H., Choudry, H. A., Bartlett, D. L., Lee, Y. J. (2018). Ferroptosis-induced endoplasmic reticulum stress: Cross-talk between ferroptosis and apoptosis. Mol. Cancer Res. 16, 1073–1076. doi: 10.1158/1541-7786.MCR-18-0055
Lei, P., Ayton, S., Appukuttan, A. T., Volitakis, I., Adlard, P. A., Finkelstein, D. I., et al. (2015). Clioquinol rescues Parkinsonism and dementia phenotypes of the tau knockout mouse. Neurobiol. Dis. 81, 168–175. doi: 10.1016/j.nbd.2015.03.015
Lewerenz, J., Ates, G., Methner, A., Conrad, M., Maher, P. (2018). Oxytosis/ferroptosis-(re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 12, 214. doi: 10.3389/fnins.2018.00214
Li, Q., Li, Q. Q., Jia, J. N., Sun, Q. Y., Zhou, H. H., Jin, W. L., et al. (2019). Baicalein exerts neuroprotective effects in fecl3-induced posttraumatic epileptic seizures via suppressing ferroptosis. Front. Pharmacol. 10, 638. doi: 10.3389/fphar.2019.00638
Linkermann, A., Skouta, R., Himmerkus, N., Mulay, S. R., Dewitz, C., De Zen, F., et al. (2014). Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. U. S. A 111, 16836–16841. doi: 10.1073/pnas.1415518111
Liu, Y., Wang, W., Li, Y., Xiao, Y., Cheng, J., Jia, J. (2015). The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol. Pharm. Bull. 38, 1234–1239. doi: 10.1248/bpb.b15-00048
Lorincz, T., Jemnitz, K., Kardon, T., Mandl, J., Szarka, A. (2015). Ferroptosis is involved in acetaminophen induced cell death. Pathol. Oncol. Res. 21, 1115–1121. doi: 10.1007/s12253-015-9946-3
Louandre, C., Ezzoukhry, Z., Godin, C., Barbare, J. C., Maziere, J. C., Chauffert, B., et al. (2013). Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 133, 1732–1742. doi: 10.1002/ijc.28159
Louandre, C., Marcq, I., Bouhlal, H., Lachaier, E., Godin, C., Saidak, Z., et al. (2015). The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 356, 971–977. doi: 10.1016/j.canlet.2014.11.014
Lucarini, M., Pedrielli, P., Pedulli, G. F., Cabiddu, S., Fattuoni, C. (1996). Bond dissociation energies of O–H bonds in substituted phenols from equilibration studies. J. Org. Chem. 61, 9259–9263. doi: 10.1021/jo961039i
Magtanong, L., Dixon, S. J. (2018). Ferroptosis and brain injury. Dev. Neurosci. 40, 382–395. doi: 10.1159/000496922
Maillard, B., Ingold, ,. K. U., Scaiano, J. C. (1983). Rate constants for the reactions of free radicals with oxygen in solution. J. Am. Chem. Soc. 105, 5095–5099. doi: 10.1021/ja00353a039
Masaldan, S., Belaidi, A. A., Ayton, S., Bush, A. I. (2019). Cellular senescence and iron dyshomeostasis in Alzheimer's disease. Pharm. (Basel) 12, 93. doi: 10.3390/ph12020093
Matsushita, M., Freigang, S., Schneider, C., Conrad, M., Bornkamm, G. W., Kopf, M. (2015). T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212, 555–568. doi: 10.1084/jem.20140857
Mattson, M. P. (2008). Hormesis and disease resistance: activation of cellular stress response pathways. Hum. Exp. Toxicol. 27, 155–162. doi: 10.1177/0960327107083417
Mclachlan, D. R. C., Dalton, A. J., Kruck, T. P. A., Bell, M. Y., Smith, W. L., Kalow, W., et al. (1991). Intramuscular desferrioxamine in patients with Alzheimers-disease. Lancet 337, 1304–1308. doi: 10.1016/0140-6736(91)92978-B
Murphy, T. H., Miyamoto, M., Sastre, A., Schnaar, R. L., Coyle, J. T. (1989). Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558. doi: 10.1016/0896-6273(89)90043-3
Neitemeier, S., Jelinek, A., Laino, V., Hoffmann, L., Eisenbach, I., Eying, R., et al. (2017). BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 12, 558–570. doi: 10.1016/j.redox.2017.03.007
Paul, B. D., Sbodio, J. I., Xu, R., Vandiver, M. S., Cha, J. Y., Snowman, A. M., et al. (2014). Cystathionine gamma-lyase deficiency mediates neurodegeneration in Huntington's disease. Nature 509, 96–100. doi: 10.1038/nature13136
Peng, Y., Wang, C., Xu, H. H., Liu, Y. N., Zhou, F. (2010). Binding of alpha-synuclein with Fe(III) and with Fe(II) and biological implications of the resultant complexes. J. Inorg. Biochem. 104, 365–370. doi: 10.1016/j.jinorgbio.2009.11.005
Pratico, D., Sung, S. (2004). Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer's disease. J. Alzheimers Dis. 6, 171–175. doi: 10.3233/JAD-2004-6209
Probst, L., Dachert, J., Schenk, B., Fulda, S. (2017). Lipoxygenase inhibitors protect acute lymphoblastic leukemia cells from ferroptotic cell death. Biochem. Pharmacol. 140, 41–52. doi: 10.1016/j.bcp.2017.06.112
Raefsky, S. M., Furman, R., Milne, G., Pollock, E., Axelsen, P., Mattson, M. P., et al. (2018). Deuterated polyunsaturated fatty acids reduce brain lipid peroxidation and hippocampal amyloid beta-peptide levels, without discernable behavioral effects in an APP/PS1 mutant transgenic mouse model of Alzheimer's disease. Neurobiol. Aging 66, 165–176. doi: 10.1016/j.neurobiolaging.2018.02.024
Raghupathi, R., Graham, D. I., Mcintosh, T. K. (2000). Apoptosis after traumatic brain injury. J. Neurotrauma 17, 927–938. doi: 10.1089/neu.2000.17.927
Rahmani, M., Davis, E. M., Crabtree, T. R., Habibi, J. R., Nguyen, T. K., Dent, P., et al. (2007). The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol. Cell. Biol. 27, 5499–5513. doi: 10.1128/MCB.01080-06
Ran, Q., Chen, L., Seally Hambright, W., Na, R. (2015). Ablation of GPX4 in neurons results in rapid motor neuron degeneration and paralysis. Free Radical Biol. Med. 87, s34. doi: 10.1016/j.freeradbiomed.2015.10.093
Reddy, P. H., Shirendeb, U. P. (2012). Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington's disease. Biochim. Biophys. Acta 1822, 101–110. doi: 10.1016/j.bbadis.2011.10.016
Reisman, S. A., Yeager, R. L., Yamamoto, M., Klaassen, C. D. (2009). Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol. Sci. 108, 35–47. doi: 10.1093/toxsci/kfn267
Russell, G. A. (1956). Mechanism of the termination step in autoxidation reactions. Chem. Ind., 49, 1483–1483.
Sancho-Martinez, S. M., Lopez-Novoa, J. M., Lopez-Hernandez, F. J. (2015). Pathophysiological role of different tubular epithelial cell death modes in acute kidney injury. Clin. Kidney J. 8, 548–559. doi: 10.1093/ckj/sfv069
Sato, H., Tamba, M., Ishii, T., Bannai, S. (1999). Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 274, 11455–11458. doi: 10.1074/jbc.274.17.11455
Sato, H., Shiiya, A., Kimata, M., Maebara, K., Tamba, M., Sakakura, Y., et al. (2005). Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 280, 37423–37429. doi: 10.1074/jbc.M506439200
Shah, R., Margison, K., Pratt, D. A. (2017). The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem. Biol. 12, 2538–2545. doi: 10.1021/acschembio.7b00730
Shah, R., Shchepinov, M. S., Pratt, D. A. (2018). Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 4, 387–396. doi: 10.1021/acscentsci.7b00589
Shaw, A. T., Winslow, M. M., Magendantz, M., Ouyang, C., Dowdle, J., Subramanian, A., et al. (2011). Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. U. S. A 108, 8773–8778. doi: 10.1073/pnas.1105941108
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shimada, K., Stockwell, B. R. (2016). tRNA synthase suppression activates de novo cysteine synthesis to compensate for cystine and glutathione deprivation during ferroptosis. Mol. Cell Oncol. 3, e1091059. doi: 10.1080/23723556.2015.1091059
Shimada, K., Hayano, M., Pagano, N. C., Stockwell, B. R. (2016a). Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem. Biol. 23, 225–235. doi: 10.1016/j.chembiol.2015.11.016
Shimada, K., Skouta, R., Kaplan, A., Yang, W. S., Hayano, M., Dixon, S. J., et al. (2016b). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503. doi: 10.1038/nchembio.2079
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556. doi: 10.1021/ja411006a
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Sun, X., Ou, Z., Xie, M., Kang, R., Fan, Y., Niu, X., et al. (2015). HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 34, 5617–5625. doi: 10.1038/onc.2015.32
Sun, X., Ou, Z., Chen, R., Niu, X., Chen, D., Kang, R., et al. (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. doi: 10.1002/hep.28251
Sun, Y., Zheng, Y., Wang, C., Liu, Y. (2018). Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 9, 753. doi: 10.1038/s41419-018-0794-4
Szwed, M., Sonstevold, T., Overbye, A., Engedal, N., Grallert, B., Morch, Y., et al. (2019). Small variations in nanoparticle structure dictate differential cellular stress responses and mode of cell death. Nanotoxicology 13, 761–782. doi: 10.1080/17435390.2019.1576238
Tan, S., Schubert, D., Maher, P. (2001). Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 1, 497–506. doi: 10.2174/1568026013394741
Torii, S., Shintoku, R., Kubota, C., Yaegashi, M., Torii, R., Sasaki, M., et al. (2016). An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 473, 769–777. doi: 10.1042/BJ20150658
Van Bergen, J. M., Hua, J., Unschuld, P. G., Lim, I. A., Jones, C. K., Margolis, R. L., et al. (2016). Quantitative susceptibility mapping suggests altered brain iron in premanifest Huntington disease. AJNR Am. J. Neuroradiol 37, 789–796. doi: 10.3174/ajnr.A4617
Vande Walle, L., Lamkanfi, M. (2016). Pyroptosis. Curr. Biol. 26, R568–R572. doi: 10.1016/j.cub.2016.02.019
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi: 10.1038/nrm2970
Wang, Y., Gao, W., Shi, X., Ding, J., Liu, W., He, H., et al. (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. doi: 10.1038/nature22393
Wang, D. L., Calabrese, E. J., Lian, B. L., Lin, Z. F., Calabrese, V. (2018). Hormesis as a mechanistic approach to understanding herbal treatments in traditional Chinese medicine. Pharmacol. Ther. 184, 42–50. doi: 10.1016/j.pharmthera.2017.10.013
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/s1474-4422(14)70117-6
Wei, S., Sun, J., Li, J., Wang, L., Hall, C. L., Dix, T. A., et al. (2013). Acute and delayed protective effects of pharmacologically induced hypothermia in an intracerebral hemorrhage stroke model of mice. Neuroscience 252, 489–500. doi: 10.1016/j.neuroscience.2013.07.052
Wei, G., Sun, J., Hou, Z., Luan, W., Wang, S., Cui, S., et al. (2018). Novel antitumor compound optimized from natural saponin Albiziabioside A induced caspase-dependent apoptosis and ferroptosis as a p53 activator through the mitochondrial pathway. Eur. J. Med. Chem. 157, 759–772. doi: 10.1016/j.ejmech.2018.08.036
Wenzel, S. E., Tyurina, Y. Y., Zhao, J., St. Croix, C. M., Dar, H. H., Mao, G., et al. (2017). PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641.e626. doi: 10.1016/j.cell.2017.09.044
Wu, D., Chen, L. (2015). Ferroptosis: a novel cell death form will be a promising therapy target for diseases. Acta Biochim. Biophys. Sin. (Shanghai) 47, 857–859. doi: 10.1093/abbs/gmv086
Wu, J., Minikes, A. M., Gao, M., Bian, H., Li, Y., Stockwell, B. R., et al. (2019). Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406. doi: 10.1038/s41586-019-1426-6
Wu, C. (1995). Heat shock transcription factors: Structure and regulation. Annu. Rev. Cell Dev. Biol. 11, 441–469. doi: 10.1146/annurev.cb.11.110195.002301
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016a). Ferroptosis: process and function. Cell Death Differ. 23, 369–379. doi: 10.1038/cdd.2015.158
Xie, Y., Song, X., Sun, X., Huang, J., Zhong, M., Lotze, M. T., et al. (2016b). Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem. Biophys. Res. Commun. 473, 775–780. doi: 10.1016/j.bbrc.2016.03.052
Yagoda, N., Von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. doi: 10.1038/nature05859
Yamaguchi, H., Hsu, J. L., Chen, C. T., Wang, Y. N., Hsu, M. C., Chang, S. S., et al. (2013). Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin. Cancer Res. 19, 845–854. doi: 10.1158/1078-0432.CCR-12-2621
Yang, W. S., Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245. doi: 10.1016/j.chembiol.2008.02.010
Yang, W. S., Stockwell, B. R. (2016). Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 26, 165–176. doi: 10.1016/j.tcb.2015.10.014
Yang, W. S., Sriramaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U. S. A 113, E4966–E4975. doi: 10.1073/pnas.1603244113
Yuan, H., Li, X., Zhang, X., Kang, R., Tang, D. (2016). CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 478, 838–844. doi: 10.1016/j.bbrc.2016.08.034
Zhang, Y. H., Wang, D. W., Xu, S. F., Zhang, S., Fan, Y. G., Yang, Y. Y., et al. (2018a). alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 14, 535–548. doi: 10.1016/j.redox.2017.11.001
Zhang, Z. L., Yao, Z., Wang, L., Ding, H., Shao, J. J., Chen, A. P., et al. (2018b). Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 14, 2083–2103. doi: 10.1080/15548627.2018.1503146
Zhao, Y., Hu, X., Liu, Y., Dong, S., Wen, Z., He, W., et al. (2017). ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol. Cancer 16, 79. doi: 10.1186/s12943-017-0648-1
Zhu, S., Zhang, Q., Sun, X., Zeh, ,. H. J., 3rd, Lotze, M. T., Kang, R., et al. (2017). HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 77, 2064–2077. doi: 10.1158/0008-5472.CAN-16-1979
Keywords: ferroptosis, signaling pathways, reactive oxygen species, pharmacology design, degenerative diseases
Citation: Han C, Liu Y, Dai R, Ismail N, Su W and Li B (2020) Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 11:239. doi: 10.3389/fphar.2020.00239
Received: 19 November 2019; Accepted: 21 February 2020;
Published: 17 March 2020.
Edited by:
James Connor, Penn State Milton S. Hershey Medical Center, United StatesReviewed by:
Sonia Emanuele, University of Palermo, ItalyVittorio Calabrese, University of Catania, Italy
Copyright © 2020 Han, Liu, Dai, Ismail, Su and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Li, boli@bit.edu.cn
†These authors have contributed equally to this work and share first authorship