 Alexander M. Markin
Alexander M. Markin Igor A. Sobenin
Igor A. Sobenin Andrey V. Grechko4
Andrey V. Grechko4 Dongwei Zhang
Dongwei Zhang Alexander N. Orekhov
Alexander N. Orekhov- 1Laboratory of Infection Pathology and Molecular Microecology, Institute of Human Morphology, Moscow, Russia
- 2Laboratory of Medical Genetics, Institute of Experimental Cardiology, National Medical Research Center of Cardiology, Moscow, Russia
- 3Laboratory of Angiopathology, Institute of General Pathology and Pathophysiology, Moscow, Russia
- 4Federal Research and Clinical Center of Intensive Care Medicine and Rehabilitology, Moscow, Russia
- 5Diabetes Research Centre, Traditional Chinese Medicine School, Beijing University of Chinese Medicine, Beijing, China
Atherosclerosis is one of the most common diseases of the cardiovascular system that leads to the development of life-threatening conditions, such as heart attack and stroke. Arthrosclerosis affects various arteries in the human body, but is especially dangerous in the arteries alimenting heart and brain, aorta, and arteries of the lower limbs. By its pathophysiology, atherosclerosis is an inflammatory disease. During the pathological process, lesions of arterial intima in the form of focal thickening are observed, which form atherosclerotic plaques as the disease progresses further. Given the significance of atherosclerosis for the global health, the search for novel effective therapies is highly prioritized. However, despite the constant progress, our understanding of the mechanisms of atherogenesis is still incomplete. One of the remaining puzzles in atherosclerosis development is the focal distribution of atherosclerotic lesions in the arterial wall. It implies the existence of certain mosaicism within the tissue, with some areas more susceptible to disease development than others, which may prove to be important for novel therapy development. There are many hypotheses explaining this phenomenon, for example, the influence of viruses, and the spread in the endothelium of the vessel multinucleated giant endothelial cells. We suggest the local variations of the mitochondrial genome as a possible explanation of this mosaicism. In this review, we discuss the role of genetic variations in the nuclear and mitochondrial genomes that influence the development of atherosclerosis. Changes in the mitochondrial and nuclear genome have been identified as independent factors for the development of the disease, as well as potential diagnostic markers.
Introduction
Atherosclerosis is a multifactorial disease with a complex pathogenesis. Multiple factors were shown to be involved in atherosclerotic lesion formation, and many knowledge gaps still remain. Atherosclerotic lesions can develop in any artery, but are especially dangerous in large vessels that aliment the heart, brain, and other vital organs. Macroscopically, the lesions are seen as local accumulation of fat in the arterial wall (so-called fatty streak), which is followed by local thickening of the innermost arterial wall layer called intima. At the advanced stages, the plaques acquire a thick fibrous cap, which separates them from the vessel lumen and circulating blood. Currently, several key factors in atherosclerotic lesion formation are considered. There is no doubt that impaired endothelial function and increased permeability plays an important role, especially at the initial stages of the lesion development (Mundi et al., 2018). Alterations of lipid metabolism and lipoprotein modifications are prerequisites of lipid accumulation (Summerhill et al., 2019). Chronic inflammation and immune disorders have been extensively studied in relation to atherosclerosis, which is currently regarded as an inflammatory disease (Frostegård, 2013; Rea et al., 2018). During the recent years, the prominent role of genetic factors in atherosclerosis development has been recognized, and numerous identified genes opened new possibilities for novel therapies development (Martínez et al., 2017; Wang W. et al., 2019; González-Becerra et al., 2019; Jha et al., 2019).
Numerous genetic studies were conducted to establish risk factors for the development of atherosclerosis (Björnsson et al., 2019; Li et al., 2019; Rincón et al., 2019). Researchers are constantly discovering new mutations specifically associated with cardiovascular disease. Apart from nuclear genome, mitochondrial genome variants may also be important as risk factors and disease modifiers. It is possible that mitochondrial DNA (mtDNA) mutations are responsible for predisposition to the development of atherosclerotic lesions (Sinyov et al., 2017). Mitochondria are semi-autonomous organelles that bear the genes of many, but not all mitochondrial proteins in their circular genome that resembles a bacterial chromosome. Mitochondrial dysfunction affects the cellular energy balance, metabolism and survival, and results in the development of mitochondrial cytopathies. These diseases can be caused both by nuclear genes encoding mitochondrial proteins or by mtDNA mutations that affect either mitochondrial proteins or transport RNA (tRNA). In this review, we will summarize the current knowledge on mtDNA mutations and mitochondrial dysfunction as pathophysiological factors of atherosclerosis development.
Cellular Mechanisms of Atherogenesis
Atherosclerotic lesion development takes place in the intimal layer of the arterial wall. Adult intima is a rather thick formation with complex architecture and heterogeneous cellular composition. The intima is separated from the lumen of the vessel by a monolayer of endothelial cells. Endothelium plays a key role in the transport of cells and non-cellular components of blood from the arterial bed to the vascular wall (Krüger-Genge et al., 2019). Endothelial lining is heterogeneous (Romanov et al., 1995). In addition to cells of normal shape and size, clusters of giant multinucleated endothelial cells are found in the endothelial monolayer. These giant multinucleated cells appear only during the life period, in which atherosclerosis development is most frequent, and are typically not present in young individuals. Their appearance is important for understanding the mechanisms of atherogenesis, since clusters of such cells appear to be more common in the areas predisposed to atherosclerosis. Usually these are hemodynamic stress zones (for example, bifurcation and branching of blood vessels). Presence of such zones may partially explain the mosaicism of atherosclerosis. Lesions are not diffuse, but occur locally or focally, which may be associated with a local disturbance in the permeability of giant multinucleated endothelial cells.
Under the basal membrane, intima is populated by different types of cells (Rekhter et al., 1991). Immune cells such as macrophages (3–5%), dendritic cells (0.3%), and others, are located near the endothelium. Deeper layers contain elongated smooth muscle cells (70%) and pericytes or pericyte-like cells (25–30%) (Orekhov et al., 2014). Pericyte-like cells have a stellate shape and are connected through gap junctions with each other, forming a three-dimensional network, which serves as a kind of second line of immune defense (Ivanova et al., 2015). Pericyte-like cells can perform the functions of phagocytes, are able to secrete pro-inflammatory cytokines (Hill et al., 2014), and can also act as antigen-presenting cells (Ivanova and Orekhov, 2016a). In stimulating the immune response, pericytes are inferior in effectiveness to “professional” immune cells, but due to their abundancy in the arterial wall they are able to actively participate in the innate immunity reactions.
Cellular composition of the arterial wall undergoes profound changes in atherosclerotic lesion areas (Stary, 1990; Xu et al., 1990; Padarti and Zhang, 2018). Lesion development is accompanied by a local increase of the number of cells (cellularity), especially the numbers of macrophages and hematogenous cells. Moreover, the cellular network of pericyte-like cells disintegrates, with loss of intercellular communication and changes of cellular phenotype. Pericyte-like cells, macrophages, and some smooth muscle cells accumulate lipids, turning into foam cells, which have a distinctive appearance due to the lipid droplets accumulating in their cytoplasm. Presence of such cells in the subendothelial space of the arterial wall is a known early manifestation of atherosclerosis.
The primary source of lipids that accumulate in foam cells is atherogenic modified low density lipoprotein (LDL). Particles of LDL circulate in the blood and undergo chemical modifications affecting the glycoconjugate, lipid, and protein moieties (Ivanova et al., 2017; Orekhov and Myasoedova, 2019). Noteworthy, it is the multiply modified LDL that was found in the circulation of atherosclerotic patients, and not specific LDL modifications that have been characterized in in vitro experiments, such as oxidized LDL. Atherogenic modification of LDL begins with desialylation, and continues with changes of the lipoprotein particle size, density, and electric charge resulting in the formation of small dense and electronegative LDL fraction. Oxidation is likely to occur at later stages of atherogenic LDL modification. LDL atherogenicity is enhanced by the formation of self-associates and circulating immune complexes containing modified LDL and anti-LDL autoantibodies (Kacharava et al., 1993).
Native (unmodified) LDL does not cause the accumulation of intracellular lipids in cultured cells, because LDL binds to a specialized LDL receptor (LDLR), and undergoes degradation through the receptor-mediated pathway (Zhang et al., 2016). The excess lipids are eliminated from the cell due to the efflux, in which high-density lipoprotein (HDL) and transporter proteins play a key role (Laatsch et al., 2012).
Associates of modified LDL stimulate the phagocytic activity of subendothelial macrophages and pericytes. Following phagocytosis, inflammatory cytokines are secreted, which attracts monocytes and other immune cells to the emerging site of inflammation. Inflammatory cytokines contribute to further accumulation of intracellular lipids induced by modified atherogenic LDL. Moreover, in some cases, lipid accumulation induction is observed even in absence of modified LDL (Table 1).
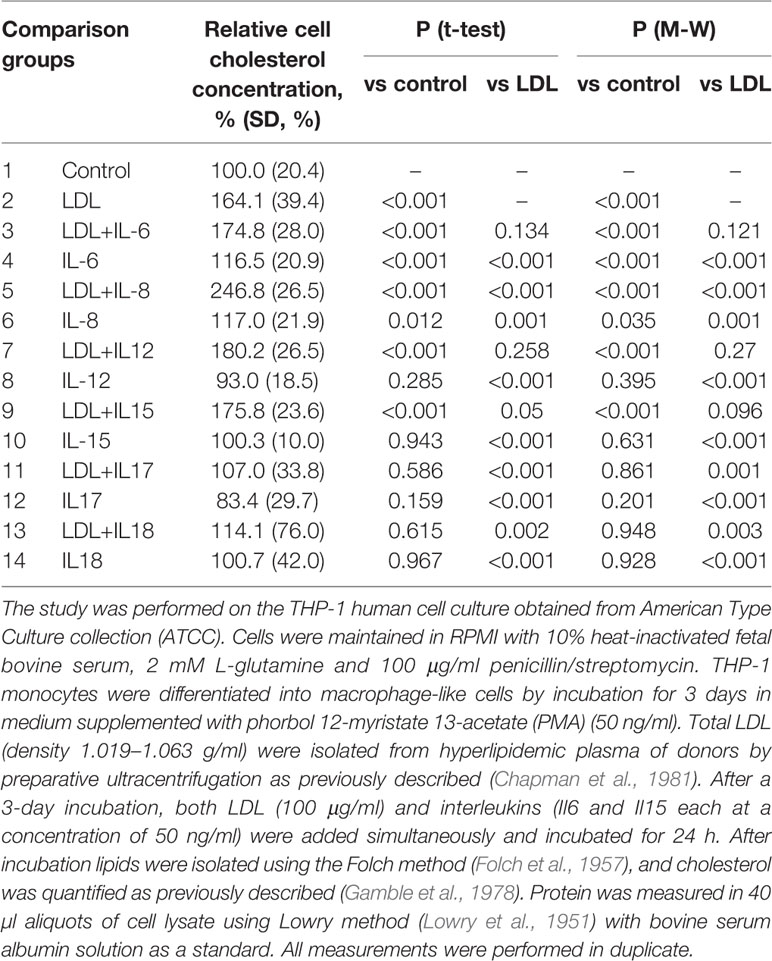
Table 1 The effect of IL-6, IL-8, IL-12, IL-15, IL-17, IL-18 on the concentration of cholesterol in THP-1 cells.
Therefore, not the accumulation of intracellular lipids caused by LDL, but the immune response to the interaction of the cell with LDL promotes or even induces the formation of foam cells. Intracellular lipid accumulation leads to the rupture of cell contacts in the three-dimensional network of pericyte-like cells (Ivanova et al., 2015). This is accompanied by increased proliferative activity and stimulation of extracellular matrix synthesis (Ponticos and Smith, 2014; Orekhov et al., 2016). Such processes are characteristic of the reparative phase of the inflammatory reaction. Normally, only a small thickening of the intimal tissue remains at the site of inflammation (Figure 1) (Orekhov and Ivanova, 2016). This process may occur in the arteries without causing visible symptoms and lesions can accumulate over time, so that focal formations become a diffuse thickening. Diffuse intimate thickening is not an atherosclerotic lesion, but is considered normal for the arteries of an adult body (Nakashima et al., 2008; Subbotin, 2016).
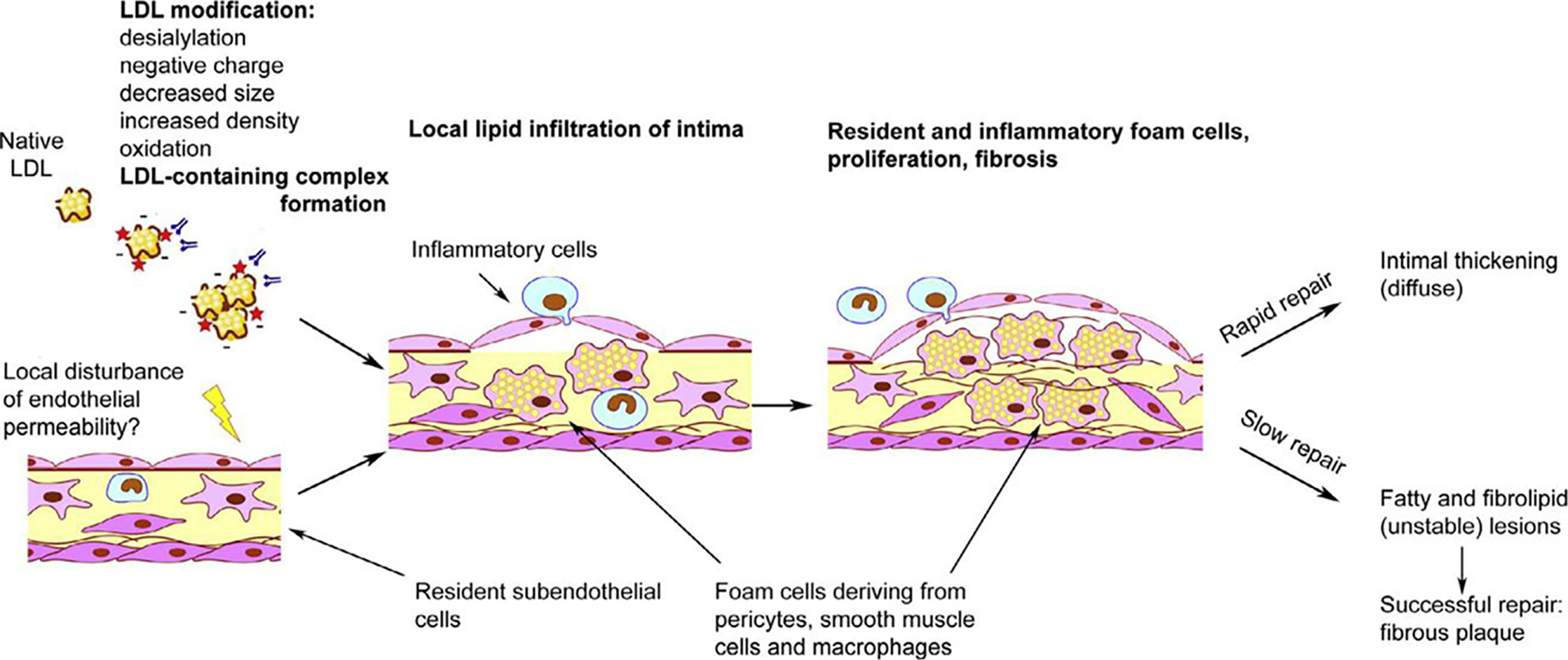
Figure 1 Schematic overview of initiation of atherosclerotic lesion formation. From Orekhov and Ivanova (2016), with permission.
According to current consensus, the primary event in atherosclerotic lesion development is local endothelial activation and increased permeability, which may be caused by hemodynamic forces occurring at the sites of blood vessel bends and bifurcations. Other known triggering factors of atherosclerosis include circulating mediators of inflammation and vasoactive substances, diet-induced alterations of the levels of circulating lipids, modified LDL, increased blood glucose level, and cigarette smoke. At the sites of local activation, the endothelial cells change their expression pattern, beginning to express cytokines and chemokines (interleukin 1 [IL-1]), tumor necrosis factor alpha (TNF-α), chemokines monocyte chemoattractant factor 1 (MCP-1), growth factors, such as platelet-derived growth factor (PDGF) and basic fibroblast growth factor (bFGF), and adhesion molecules. This leads to the increased recruitment and adhesion of circulating immune cells that interact with the endothelium and penetrate into the subendothelial space. During this process, circulating monocytes become activated and differentiate into macrophages. This further increases the pro-inflammatory signaling at the emerging lesion site. Macrophages are specialized phagocytes that actively participate in lipid uptake within the lesion and are an important source of foam cells (Koenig, 1999).
An increase in the concentration of pro-inflammatory cytokines leads to endoplasmic reticulum stress in the arterial wall cells, which in turn can lead to the initiation of apoptosis (Ivanova and Orekhov, 2016b). Cytokine-induced inflammation disrupts the normal functioning of mitochondria, their synthesis and mitophagy (Gkikas et al., 2018), which also leads to apoptosis. These effects contribute to the prolonged circulation of pro-inflammatory agents in the vascular bed, which can further stimulate the launch of a cascade of inflammatory reactions at the site of vascular damage.
It is plausible that atherosclerotic lesion development occurs when the inflammatory process cannot be resolved in a regular way and becomes chronic (Figure 1) (Orekhov and Ivanova, 2016). Thus, the response of the innate immunity is a trigger for the formation of foam cells, while violation of the normal immune response is the cause of inflammation chronification, which leads to the development of atherosclerotic lesions. It would be interesting to identify the genetic factors that regulate these processes.
Variants of the Nuclear Genome Associated with Atherosclerosis
It is generally believed that in rare cases, single nucleotide substitute in nuclear genome may greatly affect the clinical phenotype. On the opposite, as a rule, it is impossible to establish a causal relationship between a certain variant of the nuclear genome and a phenotypic manifestation of this change. This is also true for detecting genetic predisposition to chronic pathologies like atherosclerosis and related cardiovascular diseases. In the last decade, the genome-wide associations study (GWAS) approach came into play and was widely applied. In general, this is a targeted search for associations between genomic variants and phenotypic traits, i.e. for the search of associations between single nucleotide polymorphisms (SNPs) and diseases. Such studies provide a great bulk of information, which, however, can hardly be implemented into practical healthcare. On the other hand, systematic approach to the search for such associations is necessary to assess the quality of the results of numerous studies in this area and to identify the most promising sets of markers for further detailed research (Belsky et al., 2013; den Hoed et al., 2015).
Noteworthy, GWAS data are not particularly valuable without modern computational methods. An effective tool for finding the dependencies between mutations and their manifestations are systems biology methods (Huan et al., 2013; Frades et al., 2019). Analysis of molecular networks allows explaining how genetic disorders interact with the environmental factors and why this interaction manifests itself in the form of a pathological phenotype. As an example, Talukdar et al. have analyzed the genetic data and gene expression profiles of several tissues (atherosclerotic arterial wall, internal mammary artery, liver, skeletal muscle, visceral fat, subcutaneous fat, and whole blood from the late-stage patients with coronary artery disease [CAD]). As the result, 30 regulatory gene networks (RGNs) were identified being associated with CAD development and their key mechanisms of action. As a proof of concept, the researchers aimed at key drivers (AIP, DRAP1, POLR2I, and PQBP1 genes) in the RGN of the arterial wall with cross-species verification, including RNA processor genes. This RGN was re-identified in THP-1 foam cells and independently in blood-derived monocytes of CAD patients and macrophages from carotid atherosclerotic lesions. Such studies can help to better identify the candidate genes critical for the development of CAD identified in the framework of GWAS, and this is only the beginning of the path to achieving the goals of personalized medicine (Talukdar et al., 2016).
Currently, about 60 genomic regions associated with coronary heart disease have been identified. However, most cases of hereditary predisposition to atherosclerosis still cannot be explained. Thus, there exists a need for search of other loci associated with atherogenesis. An effective strategy can be a large-scale assessment of promising genomic associations, proposed as a part of various genome research projects. For example, some of the mutation changes detected are associated with endothelial cell dysfunction. Furthermore, genes regulating cellular adhesion, leucocyte migration, coagulation, inflammation, vascular smooth muscle cell differentiation and genes that regulate energy metabolism may prove to be relevant for atherosclerosis. A correlation analysis was performed that linked the identified gene regions with cell type-specific gene expression patterns and phenotypic features such as plasma protein levels (CARDIoGRAMplusC4D Consortium et al., 2013; Howson et al., 2017). Such studies may provide novel and important information that will help developing new therapies.
Numerous genetic studies have been conducted to establish the risk factors of atherosclerosis (Björnsson et al., 2019; Li et al., 2019b Rincón et al., 2019). For example, four new SNPs have recently been found recently to be specifically associated with abdominal aortic aneurysm (Marsman et al., 2019). There is evidence that an increased risk of cardiovascular disease (CVD) exists among bearers of haplogroup I1 (Y-DNA). The Y chromosome of these individuals was enriched in regulatory chromatin variants related to the development of coronary heart disease (Eales et al., 2019; Lusis, 2019). Aortic aneurysm as a whole is characterized by a pathogenesis similar to atherosclerosis, namely: infiltration by the inflammatory cells of the vessel wall, degradation of the extracellular matrix, and dysfunction of vascular smooth muscle cells (Wang Y. et al., 2019).
One way to control the CVD complications is to reduce the intake of saturated fats from food. However, if SNPs in the genes responsible for cholesterol metabolism are present, high levels of total cholesterol and LDL cholesterol and triglyceride, and low HDL cholesterol persist regardless of the diet (Walker et al., 2011).
In addition to mutations in coding regions of genes, changes in the regulatory regions are also capable of affecting gene expression. The long non-coding RNAs (lncRNAs), that are longer than 200 nucleotides, can regulate the expression of neighboring genes. Recent evidence suggests the role of lncRNAs expression on proliferation of vascular smooth muscle cells and apoptosis, and, consequently, on the risk of aortic aneurysm and atherosclerosis (Brozovich et al., 2016). The likely mechanism of such influence is overexpression of lncRNA-p21 and the increase of p53 downstream target genes Puma, Bax, Noxa, and MDM2 at mRNA and protein level. This is consistent with the involvement of these genes in the regulation of cell survival, apoptosis, and proliferation (Wu Y. et al., 2014). Importantly, the expression of this lncRNA is suppressed in atherosclerotic plaques in apoE-/- mice. In addition, several SNPs at the 9p21 locus that are known to be associated with atherosclerosis were shown to have an impact on the expression of lncRNA ANRIL, which contains at least 21 exons and has multiple linear and circular isoforms. Polymorphisms within ANRIL were shown to be associated with increased risk of different types of cancer and atherosclerosis, obesity, and type 2 diabetes (Kong et al., 2018).
One of the genes implicated in aneurism formation is LOX, which encodes for lysyl oxidase, a protein responsible for cross-linking of collagen and elastin molecules during the formation of regular collagen fibers. LOX is important for mechanical integrity of the arterial wall, and missense mutations in this gene were shown to cause aneurysm formation and aortic dissection due to insufficient crosslinking of elastin and collagen in the aortic wall. The study that linked LOX to aneurism formation offered an algorithm that could be used to search mutational changes associated with the development of coronary heart disease (Lee et al., 2016).
In most cases, familial hypercholesterolemia develops in the presence of pathogenic variants of genes encoding the low-density lipoprotein receptor (LDLR), its ligand apolipoprotein B (APOB), or the proprotein convertase subtilisin/kexin type 9 (PCSK9), which takes part in the regulation of cholesterol metabolism. Binding of PCSK9 to EGF-A (extra-cellular domain of LDLR) leads to degradation of the receptor. A decrease in LDLR causes a decreased LDL metabolism, which can lead to hypercholesterolemia. The PCSK9 is synthesized in a soluble inactive form of proenzyme, which can be activated spontaneously during intramolecular processing in the endoplasmic reticulum (Gu et al., 2013; Cariou and Dijk, 2020). Other identified genes that were less frequently detected in familial hypercholesterolemia encode apolipoprotein E (APOE) and the signal-transducing adaptor family member 1 (STAP1) (Fouchier et al., 2014). The modern classification of lipid metabolism disorders was developed by Donald Fredrickson in 1965 and is based on changes in the profile of plasma lipoproteins. This classification was adopted by the World Health Organization, although it does not take into account the level of HDL, an important factor that reduces the risk of developing atherosclerosis, as well as the role of genes that cause lipid disorders (Di Taranto et al., 2019; Gomez et al., 2019). However, the genetic disorders described above are generally not specific for typical vascular wall cells, but appear to be important at the level of the whole organism.
Variants of Mitochondrial Genome Associated With Atherosclerosis
Accumulating evidence indicates that mtDNA mutations play a role in atherosclerosis development alongside with nuclear genome polymorphisms. An important part of mitochondrial proteins that are indispensable for proper functioning of the respiratory chain and energy production are encoded by mtDNA. Therefore, mtDNA mutations can lead to serious consequences, such as altered energy homeostasis, impaired glucose and fat metabolism, elevated oxidative stress and, ultimately, cell damage and death. These processes are tightly implicated in atherosclerotic lesion development (Weakley et al., 2010).
The main function of mitochondria in the eukaryotic cell is oxidation of organic compounds and the use of the released energy for ATP synthesis and thermogenesis (Hu and Liu, 2011). The size of mtDNA, a double-stranded circular molecule, in human cells is about 16,600 nucleotide pairs. It encodes 2 rRNA, 22 tRNA, and 13 subunits of respiratory chain enzymes. Each mitochondrion can contain two to ten copies of mtDNA. If all mtDNA copies contain the same polymorphism, it is called homoplasmic, while presence of mutant and wild-type copies of the mitochondrial gene within the same cell is referred to as heteroplasmy. Mutations in mtDNA are responsible for a number of inherited human diseases. These changes are inherited almost exclusively through the maternal line (Chan, 2006; Sobenin et al., 2013; Tang et al., 2014; Strassheim et al., 2018).
Mitochondrial reactive oxygen species (ROS) generated by dysfunctional mitochondria not only contribute to cell damage during oxidative stress, but also act as intermediate signals, which are modulators of gene expression associated with the development of atherosclerosis. Mutational changes in the mitochondrial genome can partially explain the focal nature of the vascular wall damage in atherosclerosis. Cells of different sections of the vascular wall can vary significantly by the level of heteroplasmy, which leads to differences in cellular metabolism. Therefore, due to the development of mitochondrial dysfunction, and some cells become more susceptible to various pathological influences triggering the atherosclerotic process (Sobenin et al., 2013; Zorov et al., 2014).
Previous studies have revealed several atherosclerosis-associated mtDNA mutations, including m.3256C > T, m.3336T > C, m.5178C > A, m.12315G > A, m.14459G > A, m.15059G > A, and m.13513G > A. It was found that a certain spectrum of pro- and anti-atherogenic mtDNA mutations is characteristic for various types of atherosclerotic lesions of the human aortic intima (Sazonova et al., 2015).
As stated above, uncontrolled ROS production under stress of various origin changes the dynamics of mitochondrial functioning and increases mitochondrial fission and fragmentation at the expense of mitochondrial fusion. Apparently, nuclear respiratory factors (NRF1 and NRF2) play an important role in this process. These factors regulate the expression of the transcription factor of the mitochondrial genome (Tfam) and many other mitochondrial genes involved in oxidative phosphorylation. Recent studies demonstrated a link between mitochondrial dysfunction and insulin resistance through altered expression of the PPARGC1A gene in muscle and liver tissue. It was shown that the suppression of the synthesis of PGC1a protein (peroxisome proliferator-activated receptor gamma coactivator 1-alpha) impacted mitochondrial biogenesis and contributed to the induction of insulin resistance (Siasos et al., 2018). The function of PGC-1α is to stimulate mitochondrial biogenesis and promotes remodeling of muscle tissue into a fibrous composition and is also involved in the regulation of carbohydrate and lipid metabolism (Liang and Ward, 2006).
Study of the tRNAThr m.15927G> mutation revealed its association with coronary heart disease. The m.15927G > A mutation hindered the highly conserved base-pairing (28C-42G) of anticodon stem of tRNAThr. Molecular modeling study demonstrated that the m.15927G > A mutation resulted in an unstable tRNAThr structure. The study conducted on cybrid lines bearing mitochondria with the m.15927G > A mutation showed a significant decrease in the efficiency of aminoacylation, which led to a decrease in the number of polypeptides encoded by mtDNA, respiratory failure, a decrease in membrane potential, and an increase of ROS production. The increased release of cytochrome c into the cytosol and caspase 3, 7, 9 and PARP proteins indicated that the presence of this mutation contributes to the development of apoptosis. These observations confirm the data published by different groups supporting the important effect of mitochondrial mutations on the pathophysiology of coronary heart disease (Jia et al., 2019).
Mitochondrial dynamics, fission, and fusion were first detected and studied in yeast. Over the past 10 years, it has become apparent that these processes are common to all cells containing mitochondria. Impairment of fission and fusion contributes to the disease development (Diot et al., 2016). The processes of mitochondrial dynamics determine the morphology of mitochondria, their qualitative and quantitative indicators, which, as it turned out, is critical for the development of cardiovascular diseases. These processes are related to the balance between energy requirements and nutrient intake. Changes in the morphology of mitochondria can be regarded as bioenergetic adaptation during pathological remodeling of cells of the cardiovascular system (Vásquez-Trincado et al., 2016). For example, aging processes are directly related to changes of mitochondrial dynamics. It was found that in old age, the volume, integrity, and functionality of mitochondria decreases due to the accumulation of mutations in mtDNA after oxidative damage caused by ROS. In elderly organisms, mitochondria are characterized by a decrease in the efficiency of oxidative phosphorylation, ATP production, an increase in the formation of ROS and a decrease in antioxidant protection. Moreover, the regulation of mitophagy and autophagy is impaired, preventing the removal of dysfunctional mitochondria. Similar processes enhance mitochondria-mediated apoptosis (Chistiakov et al., 2014).
It is likely that a large part of mtDNA mutations occurs due to the increased ROS production in the proximity of the mitochondrial genome. Accumulation of mtDNA mutations and impaired mitochondrial function contribute to atherogenesis, as has been confirmed by several studies. That, in turn, confirms the link between increased oxidative stress and cardiovascular risk (Yu and Bennett, 2014). However, oxidative stress cannot be considered as the main mechanism of mtDNA mutagenesis (Trifunovic et al., 2005; Itsara et al., 2014). The key factors in mtDNA mutation generation are replication errors by the mitochondrial DNA polymerase γ and spontaneous base hydrolysis (Kennedy et al., 2013).
Therefore, impaired mitochondrial function, biogenesis, and dynamics disrupt the cell homeostasis. Mitochondrial damage contributes to aging and a number of age-related pathologies. Thus, in the fight against aging and age-related diseases, it is possible to use strategies that effectively improve or eliminate defects in mitochondrial dynamics. To achieve this goal, it is necessary to develop small molecules capable of enhancing mitochondrial biogenesis and inducing mitophagy for patients with age-related disorders. Consequently, effective new therapeutic strategies should include coordinated induction of both mitophagy and mitochondrial biogenesis to maintain a healthy mitochondrial population. Further intervention studies are needed to test how mitophagy and mitochondrial compounds that induce biogenesis affect human physiology. Therefore, the knowledge of the mutation spectrum of the mitochondrial genome may be useful for attending physicians and medical geneticists for early detection of atherosclerosis and analysis of predisposition to its development.
Role of Mitochondrial Mutations in Cellular Mechanism of Atherosclerosis; Chronification of Inflammation
Some of the above mentioned pro-atherogenic mtDNA mutations associated with atherosclerosis also correlated with pro-inflammatory activation of monocytes in primary culture (Orekhov et al., 2015). Two homoplasmic mutations, m.1811A > G and m.9477G > A, correlated with the degree of monocyte activation. At least three heteroplasmic mutations (m.14459G > A, m.1555A > G, and m.12315G > A), also correlated with pro-inflammatory activation of circulating human monocytes. Thus, some mutations may alter monocyte activation in atherosclerosis through mitochondrial dysfunction.
Studies on cybrid cell lines carrying variants of the mitochondrial genome obtained from atherosclerotic patients revealed the disturbances of mitochondrial functions including impaired mitophagy (Orekhov et al., 2019).
Mitophagy is involved in the innate immune response (Orekhov et al., 2020a). Inhibition of mitophagy in primary culture of human monocyte-derived macrophages increased lipopolysaccharide-induced pro-inflammatory response in the form of up-regulation of the IL-1β gene both in control cells and in the presence of mitophagy inhibitors. Repeated stimulation caused a much smaller pro-inflammatory response in control cells. When mitophagy was suppressed, re-stimulated cells continued to demonstrate a pro-inflammatory response. This means that suppression of mitophagy leads to a loss of immune tolerance and uncontrolled continuation of the pro-inflammatory response of macrophages.
These and other data allowed to formulate a hypothesis explaining the important role of mitochondrial mutations in atherogenesis (Figure 2). According to this hypothesis, circulating atherogenic multiple modified LDL induces lipid accumulation in the arterial cells (Tertov et al., 1992). Modified LDL particles form self-associates taken up by arterial cell via nonspecific phagocytosis (Tertov et al., 1989). Stimulation of phagocytosis activates the pro-inflammatory response of macrophages (Orekhov et al., 2020b). This causes accumulation of intracellular lipids (Table 1). In normal response of innate immunity, the pro-inflammatory reaction resolves quickly. However, in the case of macrophages carrying mtDNA mutations associated with defective mitophagy “non-stop” pro-inflammatory response is produced that does not arrest. Local inflammation induced by this “non-stop” response becomes chronic atherosclerotic lesion is formed.
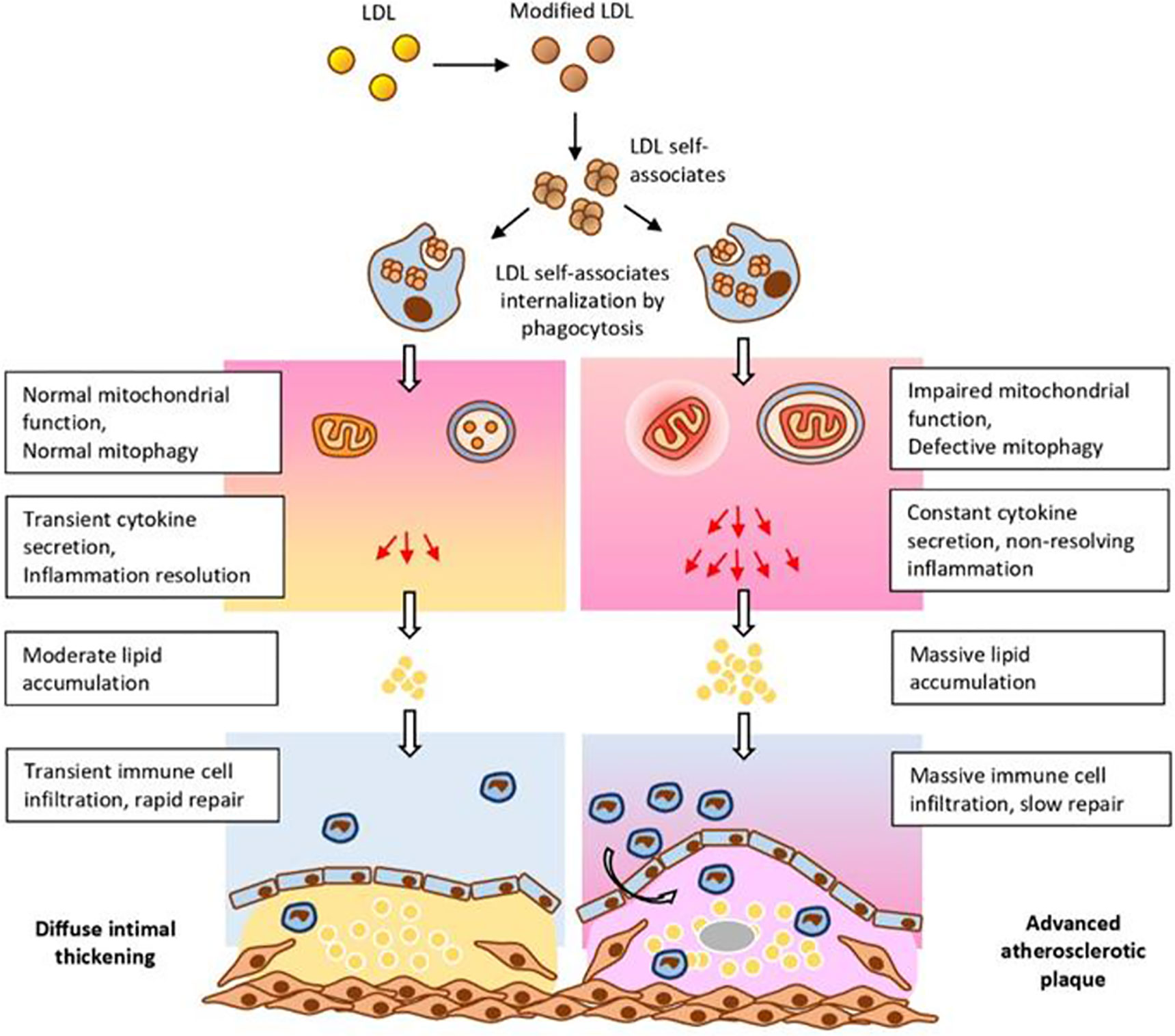
Figure 2 Impaired mitochondrial function and deficient mitophagy promote atherosclerotic lesion formation. Multiply modified LDL particles being accumulated and then internalized by macrophages are capable to alter mitochondrial function which ultimately leads to the formation of atherosclerotic plaques. From Orekhov et al. (2020a), with permission.
Conclusions
The genetic aspects of atherosclerosis attract much attention in the current research. As novel genetic methods evolve, a huge amount of data becomes available, including identification of mtDNA mutations. While genomic mutations associated with atherosclerosis are relatively well studied, mtDNA mutations provide new opportunities for development of novel diagnostic and therapeutic tools. The discovered set of mutations in the mitochondrial genome has a clear connection with the likelihood of the development of the disease, its course, and prognosis. The acquired knowledge on the mitochondrial involvement in the development of atherosclerosis and chronic inflammation will be used for designing more selective, mitochondria-targeting treatments.
Author Contributions
AM wrote the manuscript text. IS and DZ supervised the work and reviewed the manuscript. AG and AO conceptualized the work and obtained funding.
Funding
This work was supported by the Russian Science Foundation (Grant # 19-15-00297).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
bFGF, Basic fibroblast growth factor; CAD, Coronary artery disease; CVD, Cardiovascular disease; GWAS, Genome-wide associations study; HDL, High-density lipoprotein; IL, Interleukin; LDL, Low-density lipoprotein; LDLR, Low-density lipoprotein receptor; lncRNA, Long non-coding RNA; MCP-1, Monocyte chemoattractant factor 1; PDGF, Platelet-derived growth factor; RGN, Regulatory gene networks; ROS, Reactive oxygen species; SNP, Single nucleotide polymorphism; TNF, Tumor necrosis factor.
References
Belsky, D. W., Moffitt, T. E., Sugden, K., Williams, B., Houts, R., McCarthy, J., et al. (2013). Development and evaluation of a genetic risk score for obesity. Biodemography Soc. Biol. 59 (1), 85–100. doi: 10.1080/19485565.2013.774628
Björnsson, E., Thorleifsson, G., Helgadóttir, A., Guðnason, T., Guðbjartsson, T., Andersen, K., et al. (2019). Association of Genetically Predicted Lipid Levels With the Extent of Coronary Atherosclerosis in Icelandic Adults. JAMA Cardiol. 5 (1), 13–20. doi: 10.1001/jamacardio.2019.2946
Brozovich, F. V., Nicholson, C. J., Degen, C. V., Gao, Y. Z., Aggarwal, M., Morgan, K. G. (2016). Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 68 (2), 476–532. doi: 10.1124/pr.115.010652
CARDIoGRAMplusC4D Consortium, Deloukas, P., Kanoni, S., Willenborg, C., Farrall, M., Assimes, T. L., et al. (2013). Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 45 (1), 25–33. doi: 10.1038/ng.2480
Cariou, B., Dijk, W. (2020). EGF-A peptides: A promising strategy for PCSK9 inhibition. Atherosclerosis 292, 204–206. doi: 10.1016/j.atherosclerosis.2019.11.010
Chan, D. C. (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cell 125 (7), 1241–1252. doi: 10.1016/j.cell.2006.06.010
Chapman, M. J., Goldstein, S., Lagrange, D., Laplaud, P. M. (1981). A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. J. Lipid Res. 22 (2), 339–358.
Chistiakov, D. A., Sobenin, I. A., Revin, V. V., Orekhov, A. N., Bobryshev, Y. V. (2014). Mitochondrial aging and age-related dysfunction of mitochondria. BioMed. Res. Int. 2014, 238463. doi: 10.1155/2014/238463
den Hoed, M., Strawbridge, R. J., Almgren, P., Gustafsson, S., Axelsson, T., Engström, G., et al. (2015). GWAS-identified loci for coronary heart disease are associated with intima-media thickness and plaque presence at the carotid artery bulb. Atherosclerosis 239 (2), 304–310. doi: 10.1016/j.atherosclerosis.2015.01.032
Di Taranto, M. D., Giacobbe, C., Fortunato, G. (2019). Familial hypercholesterolemia: A complex genetic disease with variable phenotypes. Eur. J. Med. Genet. 25, 103831. doi: 10.1016/j.ejmg.2019.103831
Diot, A., Morten, K., Poulton, J. (2016). Mitophagy plays a central role in mitochondrial ageing. Mamm. Genome 27 (7-8), 381–395. doi: 10.1007/s00335-016-9651-x
Eales, J. M., Maan, A. A., Xu, X., Michoel, T., Hallast, P., Batini, C., et al. (2019). Human Y Chromosome Exerts Pleiotropic Effects on Susceptibility to Atherosclerosis. Arteriosclerosis Thrombosis Vasc. Biol. 39 (11), 2386–2401. doi: 10.1161/ATVBAHA.119.312405
Folch, J., Lees, M., Sloane, S. G. H. (1957). A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226 (1), 497–509.
Fouchier, S. W., Dallinga-Thie, G. M., Meijers, J. C., Zelcer, N., Kastelein, J. J., Defesche, J. C., et al. (2014). Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ. Res. 115 (6), 552–555. doi: 10.1161/CIRCRESAHA.115.304660
Frades, I., Readhead, B., Amadori, L., Koplev, S., Talukdar, H. A., Crane, H. M., et al. (2019). Systems Pharmacology Identifies an Arterial Wall Regulatory Gene Network Mediating Coronary Artery Disease Side Effects of Antiretroviral Therapy. Circ. Genom. Precis Med. 12 (6), e002390. doi: 10.1161/CIRCGEN.118.002390
Frostegård, J. (2013). Immunity, atherosclerosis and cardiovascular disease. BMC Med. 11, 117. doi: 10.1186/1741-7015-11-117
Gamble, W., Vaughan, M., Kruth, H. S., Avigan, J. (1978). Procedure for determination of free and total cholesterol in micro- or nanogram amounts suitable for studies with cultured cells. J. Lipid Res. 19 (8), 1068–1070.
Gkikas, I., Palikaras, K., Tavernarakis, N. (2018). The Role of Mitophagy in Innate Immunity. Front. Immunol. 9, 1283. doi: 10.3389/fimmu.2018.01283
Gomez, A., Colombo, R., Pontoglio, A., Helman, L., Kaeser, L., Giunta, G., et al. (2019). Functional analysis of six uncharacterised mutations in LDLR gene. Atherosclerosis 291, 44–51. doi: 10.1016/j.atherosclerosis.2019.10.013
González-Becerra, K., Ramos-Lopez, O., Barrón-Cabrera, E., Riezu-Boj, J. I., Milagro, F. I., Martínez-López, E., et al. (2019). Fatty acids, epigenetic mechanisms and chronic diseases: a systematic review. Lipids Health Dis. 18 (1), 178. doi: 10.1186/s12944-019-1120-6
Gu, H. M., Adijiang, A., Mah, M., Zhang, D. W. (2013). Characterization of the role of EGF-A of low density lipoprotein receptor in PCSK9 binding. J. Lipid Res. 54 (12), 3345–3357. doi: 10.1194/jlr.M041129
Hill, J., Rom, S., Ramirez, S. H., Persidsky, Y. (2014). Emerging roles of pericytes in the regulation of the neurovascular unit in health and disease. J. Neuroimmune Pharmacol. 9 (5), 591–605. doi: 10.1007/s11481-014-9557-x
Howson, J., Zhao, W., Barnes, D. R., Ho, W. K., Young, R., Paul, D. S., et al. (2017). Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat. Genet. 49 (7), 1113–1119. doi: 10.1038/ng.3874
Hu, F., Liu, F. (2011). Mitochondrial stress: a bridge between mitochondrial dysfunction and metabolic diseases? Cell Signal. 23 (10), 1528–1533. doi: 10.1016/j.cellsig.2011.05.008
Huan, T., Zhang, B., Wang, Z., Joehanes, R., Zhu, J., Johnson, A. D., et al. (2013). A systems biology framework identifies molecular underpinnings of coronary heart disease. Arteriosclerosis Thrombosis Vasc. Biol. 33 (6), 1427–1434. doi: 10.1161/ATVBAHA.112.300112
Itsara, L. S., Kennedy, S. R., Fox, E. J., Yu, S., Hewitt, J. J., Sanchez-Contreras, M., et al. (2014). Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PloS Genet. 10, e1003974. doi: 10.1371/journal.pgen.1003974
Ivanova, E. A., Orekhov, A. N. (2016a). Cellular Model of Atherogenesis Based on Pluripotent Vascular Wall Pericytes. Stem Cells Int. 2016, 7321404. doi: 10.1155/2016/7321404
Ivanova, E. A., Orekhov, A. N. (2016b). The Role of Endoplasmic Reticulum Stress and Unfolded Protein Response in Atherosclerosis. Int. J. Mol. Sci. 17 (2), 193. doi: 10.3390/ijms17020193
Ivanova, E. A., Bobryshev, Y. V., Orekhov, A. N. (2015). Intimal pericytes as the second line of immune defence in atherosclerosis. World J. Cardiol. 7 (10), 583–593. doi: 10.4330/wjc.v7.i10.583
Ivanova, E. A., Myasoedova, V. A., Melnichenko, A. A., Grechko, A. V., Orekhov, A. N. (2017). Small Dense Low-Density Lipoprotein as Biomarker for Atherosclerotic Diseases. Oxid. Med. Cell. Longevity 2017, 1273042. doi: 10.1155/2017/1273042
Jha, C. K., Mir, R., Banu, S., Elfaki, I., Chahal, S. M. S. (2019). Heterozygosity in LDLR rs2228671 and LDLR rs72658855 are associated with increased risk of developing Coronary artery disease in India -A case control study. Endocr. Metab. Immune Disord. Drug Targets 20 (3), 388–399. doi: 10.2174/1871530319666191015164505
Jia, Z., Zhang, Y., Li, Q., Ye, Z., Liu, Y., Fu, C., et al. (2019). A coronary artery disease-associated tRNAThr mutation altered mitochondrial function, apoptosis and angiogenesis. Nucleic Acids Res. 47 (4), 2056–2074. doi: 10.1093/nar/gky1241
Kacharava, A. G., Tertov, V. V., Orekhov, A. N. (1993). Autoantibodies against low-density lipoprotein and atherogenic potential of blood. Ann. Med. 25 (6), 551–555. doi: 10.1080/07853890.1993.12088583
Kennedy, S. R., Salk, J. J., Schmitt, M. W., Loeb, L. A. (2013). Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PloS Genet. 9, e1003794. doi: 10.1371/journal.pgen.1003794
Koenig, W. (1999). Atherosclerosis involves more than just lipids: focus on inflammation. Eur. Heart J. Suppl. 1 (suppl T), T19–T26.
Kong, Y., Hsieh, C. H., Alonso, L. C. (2018). ANRIL: A lncRNA at the CDKN2A/B Locus With Roles in Cancer and Metabolic Disease. Front. Endocrinol. (Lausanne). 9, 405. doi: 10.3389/fendo.2018.00405
Krüger-Genge, A., Blocki, A., Franke, R. P., Jung, F. (2019). Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 20 (18), 4411. doi: 10.3390/ijms20184411
Laatsch, A., Panteli, M., Sornsakrin, M., Hoffzimmer, B., Grewal, T., Heeren, J. (2012). Low density lipoprotein receptor-related protein 1 dependent endosomal trapping and recycling of apolipoprotein E. PloS One 7 (1), e29385. doi: 10.1371/journal.pone.0029385
Lee, V. S., Halabi, C. M., Hoffman, E. P., Carmichael, N., Leshchiner, I., Lian, C. G., et al. (2016). Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc. Natl. Acad. Sci. U. S. A. 113 (31), 8759–8764. doi: 10.1073/pnas.1601442113
Li, X., Meng, X., He, Y., Spiliopoulou, A., Timofeeva, M., Wei, W. Q., et al. (2019). Genetically determined serum urate levels and cardiovascular and other diseases in UK Biobank cohort: A phenome-wide mendelian randomization study. PloS Med. 16 (10), e1002937. doi: 10.1371/journal.pmed.1002937
Liang, H., Ward, W. F. (2006). PGC-1alpha: a key regulator of energy metabolism. Adv. Physiol. Educ. 30 (4), 145–151. doi: 10.1152/advan.00052.2006
Lowry, O. H., Rosebrough, N. J., Farr, A. L., Randall, R. J. (1951). Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193 (1), 265–275.
Lusis, A. J. (2019). Y-Chromosome Genetic Variation Associated With Atherosclerosis and Inflammation. Arterioscler. Thromb. Vasc. Biol. 39 (11), 2201–2202. doi: 10.1161/ATVBAHA.119.313369
Marsman, J., Gimenez, G., Day, R. C., Horsfield, J. A., Jones, G. T. (2019). A non-coding genetic variant associated with abdominal aortic aneurysm alters ERG gene regulation. Hum. Mol. Genet. 29 (4), 554–565. doi: 10.1093/hmg/ddz256
Martínez, M. S., García, A., Luzardo, E., Chávez-Castillo, M., Olivar, L. C., Salazar, J., et al. (2017). Energetic metabolism in cardiomyocytes: molecular basis of heart ischemia and arrhythmogenesis. Vessel Plus. 1, 130–141. doi: 10.20517/2574-1209.2017.34
Mundi, S., Massaro, M., Scoditti, E., Carluccio, M. A., van Hinsbergh, V. W. M., Iruela-Arispe, M. L., et al. (2018). Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review. Cardiovasc. Res. 114 (1), 35–52. doi: 10.1093/cvr/cvx226
Nakashima, Y., Wight, T. N., Sueishi, K. (2008). Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc. Res. 79 (1), 14–23. doi: 10.1093/cvr/cvn099
Orekhov, A. N., Ivanova, E. A. (2016). Cellular models of atherosclerosis and their implication for testing natural substances with anti-atherosclerotic potential. Phytomedicine 23 (11), 1190–1197. doi: 10.1016/j.phymed.2016.01.003
Orekhov, A. N., Myasoedova, V. A. (2019). Low density lipoprotein-induced lipid accumulation is a key phenomenon of atherogenesis at the arterial cell level. Vessel Plus. 2019 (3), 3. doi: 10.20517/2574-1209.2018.80
Orekhov, A. N., Bobryshev, Y. V., Chistiakov, D. A. (2014). The complexity of cell composition of the intima of large arteries: focus on pericyte-like cells. Cardiovasc. Res. 103 (4), 438–451. doi: 10.1093/cvr/cvu168
Orekhov, A. N., Zhelankin, A. V., Kolmychkova, K. I., Mitrofanov, K. Y., Kubekina, M. V., Ivanova, E. A., et al. (2015). Susceptibility of monocytes to activation correlates with atherogenic mitochondrial DNA mutations. Exp. Mol. Pathol. 99 (3), 672–676. doi: 10.1016/j.yexmp.2015.11.006
Orekhov, A. N., Andreeva, E. R., Bobryshev, Y. V. (2016). Cellular mechanisms of human atherosclerosis: Role of cell-to-cell communications in subendothelial cell functions. Tissue Cell. 48 (1), 25–34. doi: 10.1016/j.tice.2015.11.002
Orekhov, A. N., Poznyak, A. V., Sobenin, I. A., Nikifirov, N. N., Ivanova, E. A. (2019). Mitochondrion as a selective target for treatment of atherosclerosis: Role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr. Neuropharmacol. doi: 10.2174/1570159X17666191118125018
Orekhov, A. N., Nikiforov, N. G., Ivanova, E. A., Sobenin, I. A. (2020a). Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J. Clin. Med. 9, E978. doi: 10.3390/jcm9040978
Orekhov, A. N., Nikiforov, N. G., Sukhorukov, V. N., Kubekina, M. V., Sobenin, I. A., Wu, W. K., et al. (2020b). Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; a Transcriptome Analysis. Int. J. Mol. Sci. 21 (3), E817. doi: 10.3390/ijms21030817
Padarti, A., Zhang, J. (2018). Recent advances in cerebral cavernous malformation research. Vessel Plus. 2, 29. doi: 10.20517/2574-1209.2018.34
Ponticos, M., Smith, B. D. (2014). Extracellular matrix synthesis in vascular disease: hypertension, and atherosclerosis. J. Biomed. Res. 28 (1), 25–39. doi: 10.7555/JBR.27.20130064
Rea, I. M., Gibson, D. S., McGilligan, V., McNerlan, S. E., Alexander, H. D., Ross, O. A. (2018). Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 9, 586. doi: 10.3389/fimmu.2018.00586
Rekhter, M. D., Andreeva, E. R., Mironov, A. A., Orekhov, A. N. (1991). Three-dimensional cytoarchitecture of normal and atherosclerotic intima of human aorta. Am. J. Pathol. 138 (3), 569–580.
Rincón, L. M., Sanmartín, M., Alonso, G. L., Rodríguez, J. A., Muriel, A., Casas, E., et al. (2019). A genetic risk score predicts recurrent events after myocardial infarction in young adults. Rev. Esp. Cardiol. (Engl. Ed), S1885–5857 (19) 30263-4. doi: 10.1016/j.rec.2019.08.006
Romanov, Y. A., Balyasnikova, I. V., Bystrevskaya, V. B., Byzova, T. V., Ilyinskaya, O. P., Krushinsky, A. V., et al. (1995). Endothelial heterogeneity and intimal blood-borne cells. Relation Hum. Atherosclerosis. Ann. N. Y. Acad. Sci. 748, 12–37. doi: 10.1111/j.1749-6632.1994.tb17306.x
Sazonova, M., Sinyov, V., Barinova, V., Ryzhkova, A., Zhelankin, A., Postnov, A., et al. (2015). Mosaicism of Mitochondrial Genetic Variation in Atherosclerotic Lesions of the Human Aorta. BioMed. Res. Int. 825468. doi: 10.1155/2015/825468
Siasos, G., Tsigkou, V., Kosmopoulos, M., Theodosiadis, D., Simantiris, S., Tagkou, N. M., et al. (2018). Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Trans. Med. 6 (12), 256. doi: 10.21037/atm.2018.06.21
Sinyov, V. V., Sazonova, M. A., Ryzhkova, A. I., Galitsyna, E. V., Melnichenko, A. A., Postnov, A. Y., et al. (2017). Potential use of buccal epithelium for genetic diagnosis of atherosclerosis using mtDNA mutations. Vessel Plus. 1, 145–150. doi: 10.20517/2574-1209.2016.04
Sobenin, I. A., Sazonova, M. A., Postnov, A. Y., Salonen, J. T., Bobryshev, Y. V., Orekhov, A. N. (2013). Association of mitochondrial genetic variation with carotid atherosclerosis. PloS One 8 (7), e68070. doi: 10.1371/journal.pone.0068070
Stary, H. C. (1990). The sequence of cell and matrix changes in atherosclerotic lesions of coronary arteries in the first forty years of life. Eur. Heart J. Suppl. E, 3–19. doi: 10.1093/eurheartj/11.suppl_e.3
Strassheim, D., Karoor, V., Stenmark, K., Verin, A., Gerasimovskaya, E. (2018). A current view of G protein-coupled receptor-mediated signaling in pulmonary hypertension: finding opportunities for therapeutic intervention. Vessel Plus. 2, 21. doi: 10.20517/2574-1209.2018.44
Subbotin, V. M. (2016). Excessive intimal hyperplasia in human coronary arteries before intimal lipid depositions is the initiation of coronary atherosclerosis and constitutes a therapeutic target. Drug Discovery Today 21 (10), 1578–1595. doi: 10.1016/j.drudis.2016.05.017
Summerhill, V. I., Grechko, A. V., Yet, S. F., Sobenin, I. A., Orekhov, A. N. (2019). The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 20 (14), 3561. doi: 10.3390/ijms20143561
Talukdar, H. A., Foroughi Asl, H., Jain, R. K., Ermel, R., Ruusalepp, A., Franzén, O., et al. (2016). Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst. 2 (3), 196–208. doi: 10.1016/j.cels.2016.02.002
Tang, X., Luo, Y. X., Chen, H. Z., Liu, D. P. (2014). Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 5, 175. doi: 10.3389/fphys.2014.00175
Tertov, V. V., Sobenin, I. A., Gabbasov, Z. A., Popov, E. G., Orekhov, A. N. (1989). Lipoprotein aggregation as an essential condition of intracellular lipid accumulation caused by modified low density lipoproteins. Biochem. Biophys. Res. Commun. 163 (1), 489–494. doi: 10.1016/0006-291X(89)92163-3
Tertov, V. V., Sobenin, I. A., Gabbasov, Z. A., Popov, E. G., Jaakkola, O., Solakivi, T., et al. (1992). Multiple-modified desialylated low density lipoproteins that cause intracellular lipid accumulation. Isolation, fractionation and characterization. Lab. Invest. 67 (5), 665–675.
Trifunovic, A., Hansson, A., Wredenberg, A., Rovio, A. T., Dufour, E., Khvorostov, I., et al. (2005). Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. U. S. A. 102, 17993–17998. doi: 10.1073/pnas.0508886102
Vásquez-Trincado, C., García-Carvajal, I., Pennanen, C., Parra, V., Hill, J. A., Rothermel, et al. (2016). Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 594 (3), 509–525. doi: 10.1113/JP271301
Walker, C. G., Loos, R. J., Olson, A. D., Frost, G. S., Griffin, B. A., Lovegrove, J. A., et al. (2011). Genetic predisposition influences plasma lipids of participants on habitual diet, but not the response to reductions in dietary intake of saturated fatty acids. Atherosclerosis 215 (2), 421–427. doi: 10.1016/j.atherosclerosis.2010.12.039
Wang, W., Zhang, K., Zhang, H., Li, M., Zhao, Y., Wang, B., et al. (2019). Underlying Genes Involved in Atherosclerotic Macrophages: Insights from Microarray Data Mining. Med. Sci. Monit. 25, 9949–9962. doi: 10.12659/MSM.917068
Wang, Y., Jia, L., Xie, Y., Cai, Z., Liu, Z., Shen, J., et al. (2019). Involvement of macrophage-derived exosomes in abdominal aortic aneurysms development. Atherosclerosis 289, 64–72. doi: 10.1016/j.atherosclerosis.2019.08.016
Weakley, S. M., Jiang, J., Kougias, P., Lin, P. H., Yao, Q., Brunicardi, F. C., et al. (2010). Role of somatic mutations in vascular disease formation. Expert Rev. Mol. Diagn. 10 (2), 173–185. doi: 10.1586/erm.10.1
Wu, G., Cai, J., Han, Y., Chen, J., Huang, Z. P., Chen, C., et al. (2014). LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 130 (17), 1452–1465. doi: 10.1161/CIRCULATIONAHA.114.011675
Xu, Q. B., Oberhuber, G., Gruschwitz, M., Wick, G. (1990). Immunology of atherosclerosis: cellular composition and major histocompatibility complex class II antigen expression in aortic intima, fatty streaks, and atherosclerotic plaques in young and aged human specimens. Clin. Immunol. Immunopathol. 56 (3), 344–359. doi: 10.1016/0090-1229(90)90155-J
Yu, E. P., Bennett, M. R. (2014). Mitochondrial DNA damage and atherosclerosis. Trends Endocrinol. Metab. 25, 481–487. doi: 10.1016/j.tem.2014.06.008
Zhang, Y., Ma, K. L., Ruan, X. Z., Liu, B. C. (2016). Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int. J. Biol. Sci. 12 (5), 569–579. doi: 10.7150/ijbs.14027
Keywords: atherosclerosis, nuclear genome, mitochondria, mutations, inflammation, mtDNA
Citation: Markin AM, Sobenin IA, Grechko AV, Zhang D and Orekhov AN (2020) Cellular Mechanisms of Human Atherogenesis: Focus on Chronification of Inflammation and Mitochondrial Mutations. Front. Pharmacol. 11:642. doi: 10.3389/fphar.2020.00642
Received: 26 February 2020; Accepted: 22 April 2020;
Published: 14 May 2020.
Edited by:
Changhua Wang, Wuhan University, ChinaReviewed by:
Margarita Alexandrovna Sazonova, Russian Academy of Medical Sciences, RussiaMiguel Angel González-Gay, University of Cantabria, Spain
Copyright © 2020 Markin, Sobenin, Grechko, Zhang and Orekhov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Orekhov, a.h.opexob@gmail.com; Alexander M. Markin, alexander.markin.34@gmail.com